DOI:
10.1039/C5QI00239G
(Research Article)
Inorg. Chem. Front., 2016,
3, 370-375
Electrochemical fabrication of one-dimensional porphyrinic wires on electrodes†
Received
5th November 2015
, Accepted 23rd December 2015
First published on 12th January 2016
Abstract
[5,15-Di(4-aminophenyl)-10,20-diphenylporphyrinato]zinc(II) was found to electropolymerize on electrodes such as glassy carbon (GC), indium tin oxide (ITO), and tin oxide, to form a redox-active, stable, and reproducible π-conjugated polymer. Electrochemical and UV/vis spectroscopic studies showed that the electropolymerization involves azobenzene linkage formation via radical intermediates. The electrode modified with the porphyrinic wire featured strong light absorptions in the visible region, thereby serving as a photoanode for a photocurrent generation system.
 Ryota Sakamoto | Ryota Sakamoto was born in Yamagata Village, Nagano, Japan in 1980. He graduated from The University of Tokyo (Japan) in 2002, and received his Ph.D. degree from the same university in 2007 under the supervision of Prof. Hiroshi Nishihara. Then he was appointed as an assistant professor at Tokyo University of Science (Japan), working with Prof. Takeshi Yamamura. In 2010 he moved to The University of Tokyo, joining Prof. Nishihara's group again. His current research interest includes the construction of molecule-based nanostructures, and photonic and electronic devices thereof. |
Introduction
A series of valuable characteristics, such as developed π-conjugation, intense color, rigid structure, and high chemical stability, allows porphyrins to be utilized in broad application areas like electronics,1–4 medicine,5–7 catalysis8–10 and so on. In particular, because of their versatility in light absorptions and redox activities, porphyrins are frequently used in optoelectronics and photovoltaics applications. With adequate electron transfer ability and capability to acquire light in wide wavelengths, porphyrins become eminent in light-induced electron donating systems, leading to applications in dye-sensitized solar cells11–13 and bulk heterojunction solar cells.14–16 Moreover, the redox properties of porphyrins can be tuned at will by introducing various types of substituents and metal centres.
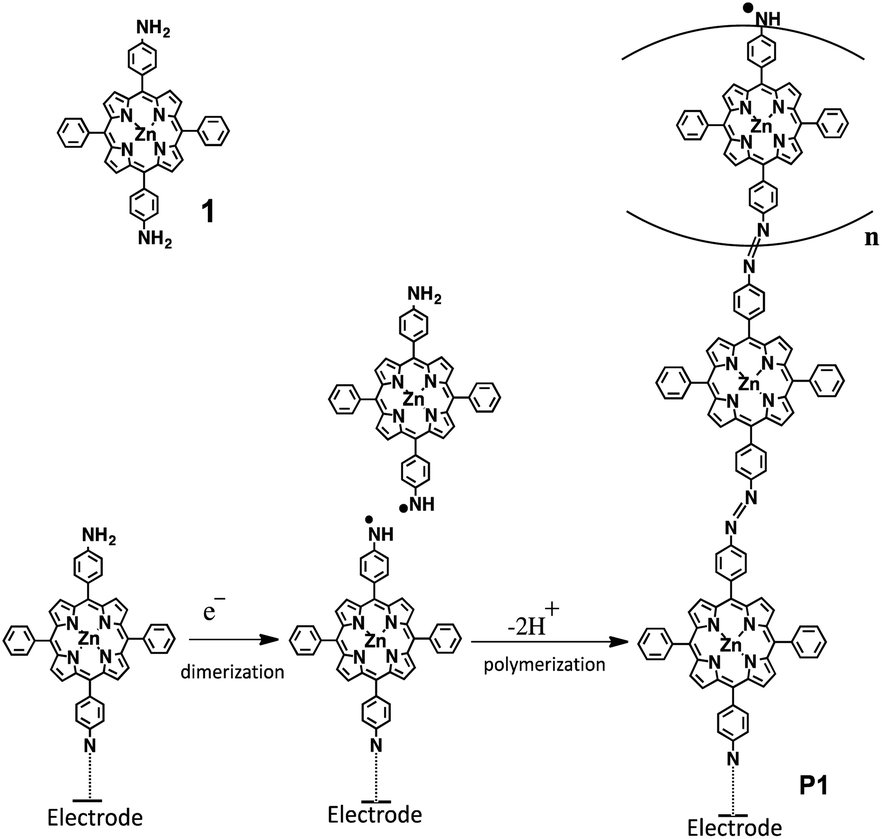
Synthesizing porphyrinic polymers on electrode surfaces is an important technique for applications.17 Such polymer structures may be accomplished by coordination reactions between porphyrin-containing bridging ligand molecules and metal ions,18 or by oxidative radical coupling of functionalized porphyrins.19 The electropolymerizable substituents include vinyl, amino, hydroxylphenyl, pyrrolyl,20 carbazolyl21 and thienyl22 groups. In the present work, we show a new procedure to form a porphyrinic polymer on various electrode surfaces, including glassy carbon (GC), indium tin oxide (ITO), and tin oxide (SnO2). [5,15-Di(4-aminophenyl)-10,20-diphenylporphyrinato]zinc(II) (1, Scheme 1 and Fig. 1) is electrooxidized to form an N-radical, such that porphyrinic oligomers linked by azobenzene (P1) are attached on an electrode surface (Scheme 1). The polymer film is characterized by means of electrochemistry, Raman, and UV/vis spectroscopy. The photoelectric conversion ability of the modified electrode is also reported.
 |
| | Scheme 1 Proposed mechanism of the oxidative electropolymerization of 1 to form P1. | |
 |
| | Fig. 1 Molecular size of 1 estimated by means of DFT calculation. | |
Experimental section
Reagents and instruments
All chemicals were purchased from Kanto Chemical Co., Tokyo Chemical Industry Co., Ltd, or Wako Pure Chemical Industries, Ltd, were used as received, unless otherwise stated. 1 was synthesized according to previous literature studies23–26 with a few modifications. Tetra-n-butylammonium perchlorate, Bu4NClO4, employed as a supporting electrolyte was purified by recrystallization from ethanol, and dried in vacuo. A Milli-Q purification system (Merck KGaA) was used to obtain pure water. ITO was treated before use upon sonication in acetone (10 min) and nonionic detergent in water (30 min × 2) and, and purified water (10 min). The cleaned substrate was stored in water, and dried by nitrogen blow just prior to use. All 1H NMR data were recorded on a Bruker-DRX500 using CDCl3 as the solvent and tetramethylsilane (δH = 0.00) as an internal standard. High-resolution fast atom bombardment mass spectrometry (HR-FAB-MS) was conducted using a JEOL JMS-700 MStation mass spectrometer. UV/vis spectra were obtained with a JASCO V-570 spectrometer. Raman spectra were recorded using a JASCO NRS-5100. AFM measurements were carried out using an Agilent Technologies 5500 scanning probe microscope in the high-amplitude mode (tapping mode) with a silicon cantilever Nano World PPP-NCL probe. FE-SEM images were recorded using JEOL JSM-7400FNT with an acceleration voltage of 1.5 kV. All experiments were carried out under an ambient condition unless otherwise stated. The molecular size of 1 was determined by DFT calculation. The DFT calculation was carried out using a Gaussian09 Revision D.01 program package. The geometry optimization was performed using B3LYP functional with the LANL2DZ basis set for Zn and 6-31g(d) basis set for the other atoms and the result was visualized using GaussView 5.0.8 software.27
Photoelectric conversion measurement
The modified ITO electrode was assigned as a working electrode (photoanode) with an Ag+/Ag electrode (0.01 M AgClO4 in 0.1 M Bu4NClO4/acetonitrile) acting as a reference electrode and a Pt wire as a counter electrode. All three electrodes were engaged in an acetonitrile solution of 0.1 M Bu4NClO4 containing triethanolamine (TEOA, 0.05 M) as a sacrificial donor reagent. The cell was sealed and deoxygenized by argon bubbling for 5 min.28 Monochromatic light for an action spectrum (400–500 nm in every 10 nm) was extracted from a xenon lamp (MAX-302, Asahi Spectra Co., Ltd) and a monochromator (CT-10, JASCO Corporation) was utilized to monochromate the photon flux while the size of the electrode active area was approximated from a fluorocarbon rubber o-shaped ring; 0.264 cm2. An electrochemical analyser (ALS 750A, BAS Inc.) was used to control the electrode potential and photocurrent acquisition of the photoelectric conversion system. A photon counter (8230E and 82311B, ADC Corporation) was engaged in quantifying the photon flux of the incident light. The insignificant dark current was observed when the potential of the photoanode fixed close to the open circuit potential (−0.22 V vs. Ag/Ag+). The calculation for quantum yield of photocurrent generation was made according to a previous literature.29
Results and discussion
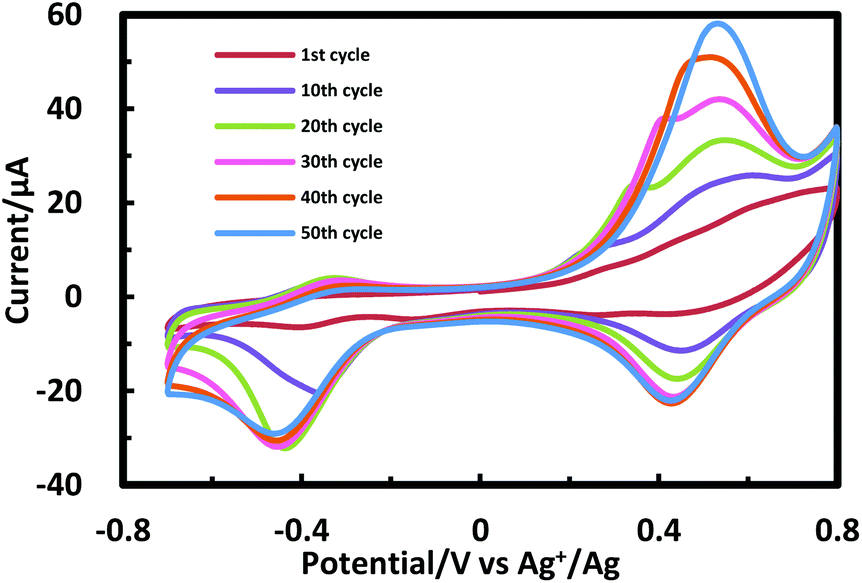
Porphyrinic polymer wire P1 was synthesized by the electrochemical oxidation method. Fig. 2 shows a cyclic voltammogram of 1 (2.0 mM) at GC in 0.1 M Bu4NClO4–CH2Cl2 in a repetitive redox process. An anodic peak appears at around 0.52 V vs. Ag+/Ag, corresponding to the oxidations of both amino group30 and porphyrin moiety.31 This argument is strongly supported by the disappearance of the shoulder peaks in Fig. 2 after the electropolymerization was complete (Fig. 4). Its peak current as shown in Fig. 3 increases with the number of oxidation cycles, indicative of the formation and growth of an electroactive film on the GC surface.32 In general, the oxidation of aromatic amines is likely to afford either polyaniline or azobenzene.33 In the present case, the azobenzene linkage is responsible to elongate porphyrinic polymer wire P1 (Scheme 1). Fig. 4 shows a cyclic voltammogram of P1 on GC in 0.1 M Bu4NClO4–CH2Cl2, displaying a quasi-reversible redox wave due to the [porphyrinato]zinc(II) moiety at ca. 0.52 V. The surface coverage of the porphyrin unit was calculated to be 2.7 × 10−9 mol cm−2 from the area of the anodic redox wave.
 |
| | Fig. 2 Cyclic voltammogram for 1 (2 mM) upon repetitive scans (from 1st to 50th cycles) in 0.1 M Bu4NClO4–CH2Cl2 at GC. | |
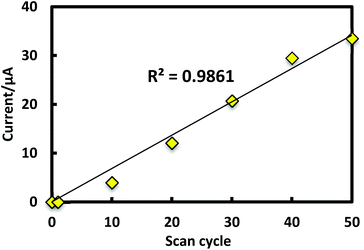 |
| | Fig. 3 Plot of anodic peak current vs. scan cycles. | |
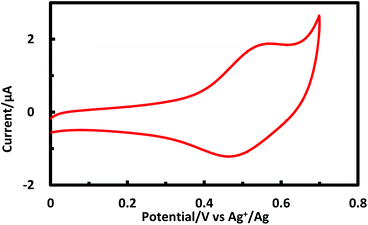 |
| | Fig. 4 Cyclic voltammogram of P1 on GC in 0.1 M Bu4NClO4–CH2Cl2. | |
The electropolymerization of 1 to form P1 was also conducted on transparent electrodes such as indium tin oxide (ITO) and tin oxide (SnO2) in order to allow spectroscopic studies. Practically identical electrochemical behaviour was observed (Fig. S1†).
The morphology of P1 was investigated by AFM and SEM. In order to observe the edge of the electropolymerized film, the modified electrode was scratched using a metal spatula, and the border of the scratched region was inspected. From the cross-sectional profile in AFM (Fig. 5), the average thickness of P1 was calculated to be 112 nm. The surface coverage of the [porphyrnato]zinc(II) unit was estimated from the average thickness and size of 1 (Fig. 1), giving a value of 3.6 × 10−9 mol cm−2. This value was consistent with the calculated value from the cyclic voltammetry (Fig. 3). On the other hand, SEM also gave a concrete evidence for the polymer formation, showing distinguishable domains (Fig. 6). The bare ITO surface as a lower part was rather smooth, while the area modified with P1 as the higher part displayed a creased texture.
 |
| | Fig. 5 (a, b) AFM height (3D and normal) images of an ITO electrode modified with the porphyrin polymer. (c) AFM cross-sectional profile along the blue line. | |
 |
| | Fig. 6 SEM image an ITO electrode modified with the porphyrin polymer. | |
Fig. 7 shows the absorption spectra of 1 in dichloromethane and the P1 film on ITO. Note that P1 deposited on ITO exhibited a greenish yellow color (Fig. 8). The spectrum of the P1 film features absorption bands typical of porphyrin;34 the Soret band (435 nm) and Q-bands (563 and 607 nm). Moreover, the spectrum of P1 (the Soret band at 424 nm and Q-bands at 554 and 588 nm) is quite similar to that of 1. This series of facts indicates that the porphyrinic skeleton was maintained in P1 in the course of the electropolymerization process.
 |
| | Fig. 7 Absorption spectra of 1 in dichloromethane and P1 on ITO. | |
 |
| | Fig. 8 Photographs of an ITO electrode before and after the modification with P1. | |
The Raman spectra of 1 and P1 were measured using probe light at 532 nm, and the result is depicted in Fig. 9. In addition to a common band at around 1460–1470 cm−1, P1 showed a characteristic band at 1446 cm−1, whereas no band was detected in 1. This band can be assigned to the stretching vibration of the azo group.35–37 This result supports our claim that the azobenzene linkage was generated upon the electrooxidative polymerization process.
 |
| | Fig. 9 Raman spectra of 1 and P1. | |
As a functionality of P1, photoelectric conversion ability was examined. P1 on an ITO electrode was employed as a working electrode, and the whole setup of the photoelectric conversion system is described in the Experimental section. Upon irradiation with visible light to the working electrode applied with a bias potential (−0.22 V) led to generate an anodic current as shown in Fig. 8. The action spectrum of the photocurrent generation was coincident with the UV-vis absorption spectrum of P1 thin films on an ITO modified electrode in the 400–500 nm range. This indicates that light absorption by the porphyrinic moiety is responsible for the photocurrent generation. The quantum efficiency of the system was calculated to be 0.005% (Fig. 10).38
 |
| | Fig. 10 Action spectrum for the photocurrent generation (black dots) and the absorption spectrum of P1 on an ITO substrate (black solid line with orange dots). | |
Proposed polymerization mechanism
Cyclic voltammetry disclosed that P1 is attached on electrode surfaces, Raman spectroscopy confirmed the presence of the azobenzene linkage in P1, and UV/vis absorption spectroscopy indicated that the porphyrinic moiety was preserved. These studies help us propose a possible polymerization mechanism (Scheme 1). The electrooxidation of P1 leads to the formation of a reactive anilino radical form,39 which can be applied for the modification of the glassy carbon surface. In the case of ITO and SnO2, although the detail chemical structure is unclear, an electron pathway is formed via the possible interactions such as coordination bonds and hydrogen bonds. On the other hand, the growth of P1 is enabled by the coupling between two anilino radical species: one is tethered on the electrode surface, and the other is in the solution phase. The coupling results in the formation of a hydrazine linkage, which undergoes electrochemical oxidation to the azobenzene linkage. In fact, the repetitive cyclic voltammetry scan in Fig. 2 shows that the anodic peak current for the oxidation process is greater than the cathodic one. The additional anodic current corresponds to the irreversible oxidation of the hydrazine linkage.
Conclusion
In summary, 5,15-di(4-aminophenyl)-10,20-diphenylporphyrinatozinc(II) monomer 1 was successfully electropolymerized on GC, ITO and SnO2 electrodes to give porphyrin polymer P1 tethered with azobenzene linkages. A possible oxidative polymerization mechanism involves the dimerization of 1 through the formation of an anilino radical form. The presence of azobenzene linkage in P1 was confirmed by a series of electrochemical and spectroscopic studies. The polymer displays photocurrent generation ability.
Acknowledgements
The authors acknowledge Grants-in-Aid from MEXT of Japan (no. 26708005, 26107510, 26620039, 15H00862, 26220801, 26110505, areas 2406 [All Nippon Artificial Photosynthesis Project for Living Earth], 2506 [Science of Atomic Layers] and 2509 [Molecular Architectonics]), and JSPS fellowship for young scientists. R. S. is grateful to Ogasawara Foundation for the Promotion of Science & Engineering, The Kao Foundation for Arts and Sciences, The Asahi Glass Foundation, The Noguchi Institute, Japan Association for Chemical Innovation, The Mikiya Science and Technology Foundation, Yazaki Memorial Foundation for Science and Technology, Shorai Foundation for Science and Technology, The Hitachi Global Foundation, Iketani Science and Technology Foundation, Kumagai Foundation for Science and Technology, The Foundation for Interaction in Science & Technology, The Foundation for the Promotion of Ion Engineering, The Foundation Advanced Technology Institute, Izumi Science and Technology Foundation, LIXIL JS Foundation, and The Iwatani Naoji Foundation for financial support. S. M. is grateful to Monbukagakusho scholarship.
Notes and references
- W. J. Park, S. H. Chae, J. Shin, D. H. Choi and S. J. Lee, Synthetic Metals, 2015, 205, 206–211 CrossRef CAS.
- X. Zhang and Y. Chen, Inorg. Chem. Commun., 2014, 39, 79–82 CrossRef.
- M. Miyachi, Y. Yamanoi, K. Nakazato and H. Nishihara, Biochim. Biophys. Acta, 2013, 1837, 1567–1571 CrossRef PubMed.
- M. Vasilopoulou, A. M. Douvas, D. G. Georgiadou, V. Constantoudis, D. Davazoglou, S. Kennou, L. C. Palilis, D. Daphnomili, A. G. Coutsolelos and P. Argitis, Nano Res., 2014, 7, 679–693 CrossRef.
- X. Deng, Y. Liang, X. Peng, T. Su, S. Luo, J. Cao, Z. Gu and B. He, Chem. Commun., 2015, 51, 4271–4274 RSC.
- K. Lu, C. He and W. Lin, J. Am. Chem. Soc., 2015, 137, 7600–7603 CrossRef CAS PubMed.
- T. Zhao, Y.-L. Wang, L.-N. Zhu, Y.-F. Huo, Y.-J. Wang and D.-M. Kong, RSC Adv., 2015, 5, 47709–47717 RSC.
- G. Tseberlidis, P. Zardi, A. Caselli, D. Cancogni, M. Fusari, L. Lay and E. Gallo, Organometallics, 2015, 34, 3774–3781 CrossRef CAS.
- C.-H. Hung, B. B. Beyene and S. B. Mane, Chem. Commun., 2015, 51, 15067–15070 RSC.
- S. Kumar, M. Y. Wani, C. T. Arranja, J. D. a. e Silva, B. Avula and A. J. F. N. Sobral, J. Mater. Chem. A, 2015, 3, 19615–19637 CAS.
- J. Gasiorowski, N. Pootrakulchote, C. Reanprayoon, K. Jaisabuy, P. Vanalabhpatana, N. S. Sariciftci and P. Thamyongkit, RSC Adv., 2015, 5, 72900–72906 RSC.
- C.-L. Mai, T. Moehl, C.-H. Hsieh, J.-D. Décoppet, S. M. Zakeeruddin, M. Grätzel and C.-Y. Yeh, ACS Appl. Mater. Interfaces, 2015, 7, 14975–14982 CAS.
- J. Chen, Y. Sheng, S. Ko, L. Liu, H. Han and X. Li, New J. Chem., 2015, 39, 5231–5239 RSC.
- C. V. Kumar, L. Cabau, E. N. Koukaras, A. Sharma, G. D. Sharma and E. J. Palomares, J. Mater. Chem. A, 2015, 3, 16287–16301 CAS.
- K. Gao, L. Li, T. Lai, L. Xiao, Y. Huang, F. Huang, J. Peng, Y. Cao, F. Liu, T. P. Russel, R. A. J. Janssen and X. Peng, J. Am. Chem. Soc., 2015, 137, 7282–7285 CrossRef CAS PubMed.
- J. Kesters, P. Verstappen, M. Kelchtermans, L. Lutsen, D. Vanderzande and W. Maes, Adv. Energy Mater., 2015, 5, 1500218 Search PubMed.
- L. Zhang, K. Wang, X. Qian, H. Liu and Z. Shi, ACS Appl. Mater. Interfaces, 2013, 5, 2761–2766 CAS.
- S. A. Ikbal, S. Brahma and S. P. Rath, Inorg. Chem., 2012, 51, 9666–9676 CrossRef CAS PubMed.
- P. A. Liddell, M. Gervaldo, J. W. Bridgewater, A. E. Keirstead, S. Lin, T. A. Moore, A. L. Moore and D. Gust, Chem. Mater., 2008, 20, 135–142 CrossRef CAS.
- F. Bedioui, J. Devynck and B.-D. Claude, Acc. Chem. Res., 1995, 28, 30–36 CrossRef CAS.
- J. Durantini, L. Otero, M. Funes, E. N. Durantini, F. Fungo and M. Gervaldo, Electrochim. Acta, 2011, 56, 4126–4134 CrossRef CAS.
- K. Takechi, N. Takahashi, T. Shiga, T. Akiyama and S. Yamada, Mol. Cryst. Liq. Cryst., 2011, 538, 10–14 CrossRef CAS.
- T. Lin, X. S. Shang, J. Adisoejoso, P. N. Liu and N. Lin, J. Am. Chem. Soc., 2013, 17, 3576–3582 CrossRef PubMed.
- M. Boccalon, E. Iengo and P. Tecilla, Org. Biomol. Chem., 2013, 11, 4056–4067 CAS.
- S. M. S. Chauhan and N. G. Giri, Supramol. Chem., 2008, 20, 743–752 CrossRef CAS.
- C. He, Q. He, C. Deng, L. Shi, D. Zhu, Y. Fu, H. Cao and J. Cheng, Chem. Commun., 2010, 46, 7536–7538 RSC.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Wallingford, CT, 2009.
- R. Matsuoka, R. Toyoda, R. Sakamoto, M. Tsuchiya, K. Hoshiko, T. Nagayama, Y. Nonoguchi, K. Sugimoto, E. Nishibori, T. Kawaii and H. Nishihara, Chem. Sci., 2015, 6, 2853–2858 RSC.
- H. Yamada, H. Imahori, S. Fukuzumi, Y. Nishimura and I. Yamazaki, Chem. Commun., 2000, 1921–1922 RSC.
- R. L. Hand and R. F. Nelson, J. Am. Chem. Soc., 1974, 96, 850–860 CrossRef.
- K. M. Kadish and L. R. Shiue, Inorg. Chem., 1982, 21, 3623–3630 CrossRef CAS.
- H. Nishihara, M. Noguchi and K. Aramaki, Inorg. Chem., 1987, 26, 2862–2869 CrossRef.
- D. A. Buttry, J. C. M. Peng, J.-B. Donnet and S. Rebouillat, Carbon, 1999, 37, 1929–1940 CrossRef CAS.
- M. Gouterman, J. Mol. Spectrosc., 1961, 6, 138–163 CrossRef CAS.
- Z. Meić, G. Baranovic, V. Smrečki, P. Novak, G. Keresztury and S. Holly, J. Mol. Struct., 1997, 408–409, 399–403 CrossRef.
- Y. C. Liu and R. L. McCreery, Anal. Chem., 1997, 69, 2091–2097 CrossRef CAS PubMed.
- S. Koide, Y. Udagawa, N. Mikami, K. Kaya and M. Ito, Bul. Chem. Soc. Jpn., 1972, 45, 3542–3543 CrossRef CAS.
- R. Sakamoto, K. Hoshiko, Q. Liu, T. Yagi, T. Nagayama, S. Kusaka, M. Tsuchiya, Y. Kitagawa, W.-Y. Wong and H. Nishihara, Nat. Commun., 2015, 6, 6713 CrossRef CAS PubMed.
- R. S. Deinhammer, M. Ho, J. W. Anderegg and M. D. Porter, Langmuir, 1994, 10, 1306–1313 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of 1, electrochemical polymerization of 1 on ITO and SnO2 electrodes. See DOI: 10.1039/c5qi00239g |
|
| This journal is © the Partner Organisations 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.