
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fabrication of calcium phosphate microcapsules using emulsion droplets stabilized with branched copolymers as templates†
Robert V.
Bell
ab,
Luke A.
Rochford
a,
Rafael T. M.
de Rosales
 c,
Molly
Stevens
b,
Jonathan V. M.
Weaver‡
b and
Stefan A. F.
Bon
*a
c,
Molly
Stevens
b,
Jonathan V. M.
Weaver‡
b and
Stefan A. F.
Bon
*a
aDepartment of Chemistry, University of Warwick, Coventry CV4 7AL, UK. E-mail: s.bon@warwick.ac.uk; Web: http://www.bonlab.info
bDepartment of Materials, Imperial College London, Exhibition Road, London SW7 2AZ, UK
cDivision of Imaging Sciences & Biomedical Engineering, King's College London, 4th Floor, Lambeth Wing, St. Thomas' Hospital, London SE1 7EH, UK
First published on 9th June 2015
Abstract
We report on a versatile and time-efficient method to fabricate calcium phosphate (CaP) microcapsules by utilizing oil-in-water emulsion droplets stabilized with synthetic branched copolymer (BCP) as templates. The BCP was designed to provide a suitable architecture and functionality to produce stable emulsion droplets, and to permit the mineralization of CaP at the surface of the oil droplet when incubated in a solution containing calcium and phosphate ions. The CaP shells of the microcapsules were established to be calcium deficient hydroxyapatite with incorporated chlorine and carbonate species. These capsule walls were made fluorescent by decoration with a fluorescein–bisphosphonate conjugate.
Introduction
Calcium phosphate (CaP) materials have drawn particular interest in the areas of medicine and health care due to the similarities of composition and structure to the inorganic constituents of hard tissues in vertebrates, such as bone and teeth.1 The physicochemical properties of CaP, as in its bioactivity, biocompatibility, biodegradability and osteoconductivity,2 allow it to be widely utilized in bone fillers,3,4 bone tissue engineering scaffolds,5–7 bioactive coatings and composites,8 and drug/protein/gene delivery systems.9–16 The specific structure of CaP can be tuned to optimize the desired material properties for a specific end application. The chemical composition of CaP can be modified with relative ease by using the ability of the apatite structure to accept numerous ionic substituents. CaP, in particular hydroxyapatite (Ca10(PO4)6(OH)2 (HA)), has been used in fluorescence labeling or imaging by doping its crystal structure with lanthanides.17–20 Furthermore, it has been demonstrated that the incorporation of silver or fluoride ions provides anti-bacterial properties.21,22 Next to applications related to health care, HA has also been used as a low cost absorbent for heavy metal pollutants because of its high sorption efficiency for divalent heavy metals ions, low water solubility, and high stability under reducing and oxidizing conditions.23–27 The surface of CaP-based materials can be functionalized through chemi- or physisorption. CaP possesses three main sorption sites on its apatite structure, which are, positively charged calcium sites, negatively charged phosphate sites, and hydroxyl sites. CaP therefore has been used in column chromatography for separation of biomolecules,28–30 in catalysis,31,32 as organic absorbent,33 and in imaging.34–36We have a general interest in the synthesis of microcapsules, as they find use in a wide range of applications.37,38 Capsule walls made from CaP are attractive for reasons mentioned above. Capsules with a CaP shell have been produced using various synthesis routes. One of the approaches widely used is to use solid particles and objects as a template, around which the CaP capsule wall is being constructed. Solid templates are readily available in a wide range of sizes and shapes, with a narrow size distribution if required. Solid templates, such as glass microspheres,39–42 calcium carbonate,43–45 polymeric nanoparticles46 have been used to fabricate CaP capsules. A substantial drawback is the removal of the solid core template, as it often requires the use of high temperatures and/or acid. These harsh conditions can lead to deterioration of mechanical robustness of the capsule and alter the permeability properties of the wall. Furthermore, refilling the hollow core can only be achieved once the solid template has been removed, and loading the hollow core in an efficient way may arise to some challenges. To minimize the drawbacks of using solid templates, routes based on using soft templates have been explored. Organized systems such as yeast cells,47 micelles,10,48–51 liposomes,52–55 and colloidosomes56 have been employed for preparing CaP capsules. Template free routes have also been presented to produce hollow CaP capsules.57–61 The latter system relies on CaP nanoparticles as building blocks to arrange and self-assemble into supracolloidal structures.
One of the soft templating routes which is especially attractive is the use of emulsion droplets as templates. Liquid cores are easily removed by gentle evaporation or dissolution in a common solvent after the CaP shell formation. More importantly, the emulsion droplet can be pre-loaded with the desired active materials efficiently and with ease, prior to the formation of the CaP shell. Furthermore, the production of emulsion droplets is straightforward and the droplet size distributions can be controlled. Böker and co-workers have presented an emulsion templating route to produce CaP capsules by incubating oil droplets stabilized with protein (that is ferritin or hydrophobin) in an aqueous calcium-phosphate solution.62–64 The incubation process allows mineralization of CaP at the surface of the oil droplets, resulting in CaP capsules. The procedure is straightforward, requires mild conditions with no organic solvents needed, and all reactants are non-toxic. However, the mineralization process to produce the CaP capsules required a long incubation period (24–28 days), in which the mineralization solution had to be exchanged regularly. Furthermore, the use of ferritin and hydrophobin proteins as stabilizer, and the potential addition of an enzyme to reduce the mineralization period, is costly.
Herein, we demonstrate the fabrication of CaP capsules by utilizing oil-in-water emulsions stabilized with a synthetic branched copolymer (BCP) made by free radical polymerization. The BCP has the right architecture and functionality to produce stable oil droplets, and allows formation of the CaP mineral at the surface of the oil droplets. We believe our method provides a versatile and robust platform in the fabrication of CaP capsules, as the BCP is readily synthesized in large quantities at low cost, and incubation periods are reduced to 72 hours. In addition we demonstrate a simple post-treatment of the CaP capsules, to render them fluorescent upon sorption with a bisphosphonate conjugate containing fluorescein.
Results and discussion
The amphiphilic BCP based on methacrylic acid (MAA) with hydrophobic dodecane chain ends was synthesized by thiol-regulated free radical polymerization, as previously reported by Weaver and coworkers.65 MAA functionality was chosen to provide electrostatic stabilization in basic solution for the emulsion droplets, and its ability to chelate to calcium ions, which in turn allows the mineralization of CaP mineral at the surface of the oil droplets. 1-Dodecanethiol (DDT) was employed as a chain transfer agent to provide hydrophobic chain ends, and to mimic the oil phase of the emulsion droplet, here dodecane, for strong oil-droplet surface adsorption. The branched architecture of the BCP ensures multiple potential points of attachment to the droplet surface (see ESI,† Fig. S1A and B). The 1H NMR spectrum and information on the molar mass distribution of the BCP are provided in the ESI† (Fig. S1C, D, S2A, and Table S1). Oil-in-water emulsions (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 vol%) were prepared by homogenizing dodecane in aqueous BCP solutions at pH 10, when MAA is in its anionic form. Stable oil droplets were produced, which creamed after 24 hours due to the lower oil density, but did not coalesce. The emulsion droplets can be simply re-dispersed with gentle agitation. Laser diffraction measurements revealed a monomodal population of emulsion droplets ranging from 3.3 μm to 35.0 μm, with an average droplet diameter of 13.3 μm. The emulsions showed no significant change in droplet size or size distribution for 30 days, as measured with laser diffraction, and no oil separation (as a result of coalescence) over a five months period was observed, proving their colloidal stability (see ESI,† Fig. S4). The protocol for the formation of the CaP capsules is illustrated in Fig. 1. The main domain of the BCP, MAA, has a high density of carboxylate groups and they act as nucleating sites for CaP. The carboxylate groups, located at the surface of the droplet, chelate with calcium ions located in the aqueous phase. Phosphate ions condense with the calcium ions via electrostatic interaction. This increases the ionic concentration above the critical saturation point and triggers the nucleation and growth of CaP minerals.
:50 vol%) were prepared by homogenizing dodecane in aqueous BCP solutions at pH 10, when MAA is in its anionic form. Stable oil droplets were produced, which creamed after 24 hours due to the lower oil density, but did not coalesce. The emulsion droplets can be simply re-dispersed with gentle agitation. Laser diffraction measurements revealed a monomodal population of emulsion droplets ranging from 3.3 μm to 35.0 μm, with an average droplet diameter of 13.3 μm. The emulsions showed no significant change in droplet size or size distribution for 30 days, as measured with laser diffraction, and no oil separation (as a result of coalescence) over a five months period was observed, proving their colloidal stability (see ESI,† Fig. S4). The protocol for the formation of the CaP capsules is illustrated in Fig. 1. The main domain of the BCP, MAA, has a high density of carboxylate groups and they act as nucleating sites for CaP. The carboxylate groups, located at the surface of the droplet, chelate with calcium ions located in the aqueous phase. Phosphate ions condense with the calcium ions via electrostatic interaction. This increases the ionic concentration above the critical saturation point and triggers the nucleation and growth of CaP minerals.
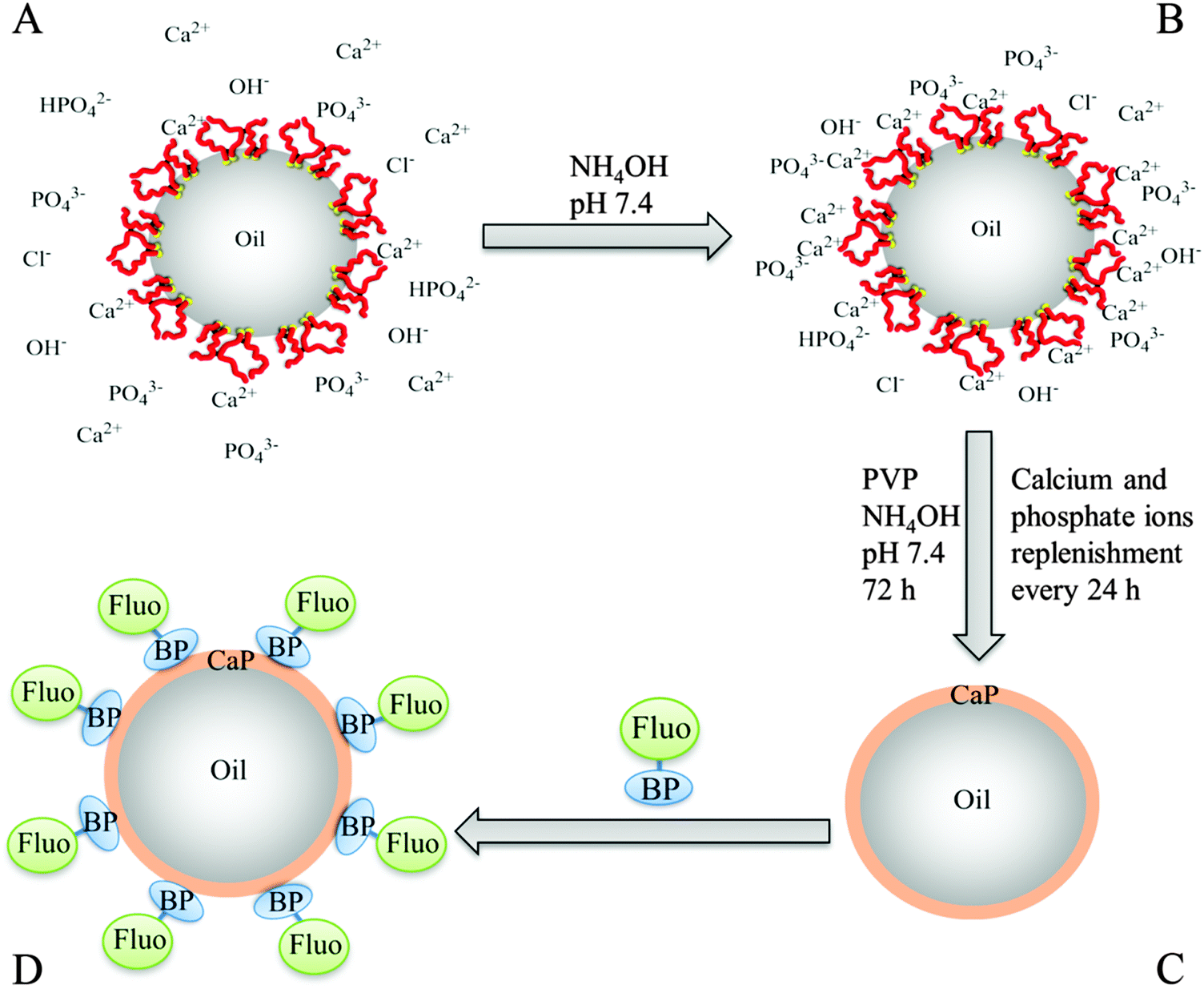 | ||
| Fig. 1 Schematic representation of the production of CaP capsule and the surface sorption of fluorescein–bisphosphonate conjugate on the surface of the CaP shell. (A) Oil-in-water emulsion droplet stabilized with branched copolymer (BCP) in a slightly acidic calcium-phosphate solution (BCP: red = methacrylic acid (MAA) domain, and yellow = hydrophobic dodecanethiol chain ends). The MAA domain of the BCP is located at the interface of the droplet and acts as a nucleating site for calcium phosphate (CaP) deposition. (B) The pH of the calcium-phosphate solution is raised to pH 7.4 with NH4OH to increase the number of nucleation sites for CaP, and to initiate CaP formation at the surface of the emulsion droplet. (C) The calcium phosphate solution is replenished twice (every 24 h) either by solvent replacement or addition of extra calcium and phosphate ions. The solution pH was maintained at pH 7.4 with the addition of NH4OH, and poly(vinyl pyrrolidone) (PVP) was added to the water phase to reduce the aggregation of the droplets. (D) Fluorescein–bisphosphonate conjugate (Fluo–BP) is added to the CaP capsule and attaches itself to the CaP shell to yield a fluorescent capsule. | ||
An aliquot of the creamed emulsion was pipetted into a calcium-phosphate solution to initiate the mineralization of CaP at the surface of the oil droplets. The calcium-phosphate solution has an initial pH of around 6.40. At this pH, the emulsion droplets have a calculated percentage of 37% carboxylate anion nucleation sites at their interface for CaP. Note that for this calculation the input value for the pKa of the BCP was determined to be 6.63 (see ESI,† Fig. S3). The pH was raised to pH 7.4 to increase the available nucleation sites from 37% to 86%. The reason why the pH was capped at 7.4, and not raised further to fully deprotonate all MAA sites, was to prevent the formation of CaP particles in the water phase as a competing event, and hence to allow for a 100% CaP deposition efficiency onto the surface of the droplets. No mineralization of CaP at the surface of the emulsion droplets was noticed after 24 hours of incubation in the calcium-phosphate solution. After 48 hours of incubation, the formations of small clusters of CaP were observed at the surface of the emulsion droplets. These droplets flattened when they were dried for imaging by SEM because they possess low structural stability (Fig. 2B). Unwanted aggregation of the oil droplets was noticed after 48 hours of incubation. This is due to the compression of the electric double layer and a potential reduction in surface charge density by the chelating action of the carboxylate groups with calcium ions. Poly(vinyl pyrrolidone) (PVP, 5 wt%), a water soluble non-ionic polymer, was added into the calcium-phosphate solution to reduce the extent of this aggregation process after the initial 24 hours of incubation. In addition, PVP aids the formation of CaP at the interface of the droplets and hence, reduce the incubation period.66 After incubating for a total period of 2 days, another aliquot of calcium and phosphate ions were added. At 60 hours of incubation period, the individual clusters of CaP, which were observed after 48 hours, had started to merge and cover the entire surface of the droplet to form a thin CaP shell. At this instance, these CaP capsules were still flexible and buckled upon drying (Fig. 2C). After 72 hours of incubation, the CaP shell had grown thick, strong and rigid, providing structural stability for the CaP capsules (Fig. 2D and E). The CaP capsules have a rough surface morphology, which can be seen in Fig. 2F. The CaP capsules were annealed at 600 °C, in order to increase crystallinity of the CaP and at the same time burn off the polymer. The resulting microcapsules can be observed in Fig. 2G. Most of the capsules remained intact, especially the ones with smaller sizes, which is logical because of the correlation between wall thickness and curvature. The annealing process changed the surface morphology of the CaP shell considerably, with more pronounced morphology in comparison to the un-annealed CaP capsules (Fig. 2H). This is due to the fact that HA crystals grow together to form a rodlike and open structure upon exposure to high temperature, which in turn changes the permeability properties of the shell. A cross-sectional view of the CaP capsule revealed a shell thickness of approximately 400 nm (Fig. 2I and J).
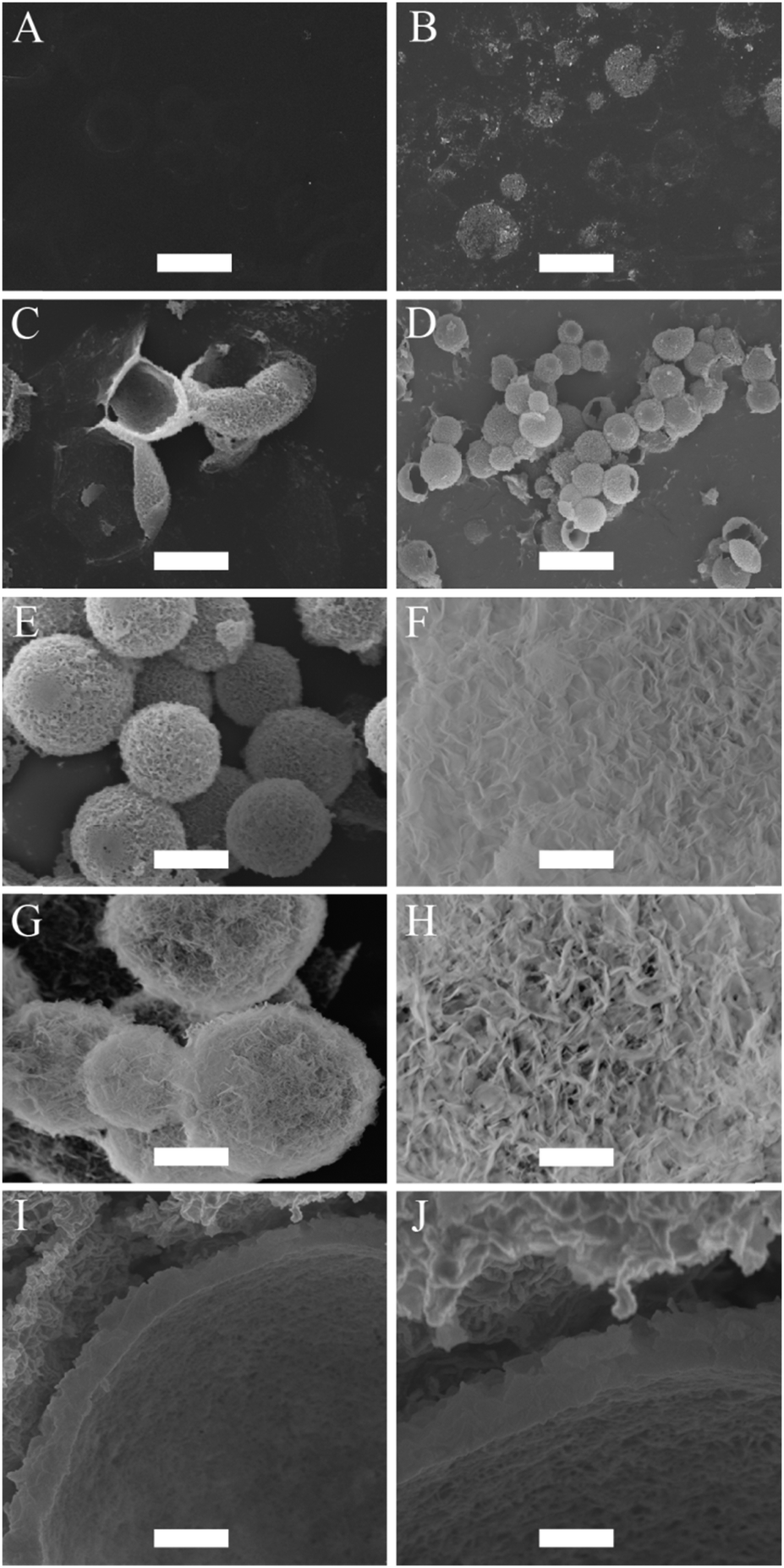 | ||
| Fig. 2 SEM micrographs illustrating the mineralization of CaP at the surface of oil droplets stabilized with BCP. (A) Incubation periods of 0 hours (scale bar = 37 μm), (B) 48 hours (scale bar = 16 μm), (C) 60 hours (scale bar = 7 μm), (D and E) 72 hours (scale bars = 23 μm and 7 μm, respectively), (F) surface morphology of CaP capsule (scale bar = 704 nm), (G) CaP capsules annealed at 600 °C (scale bar = 2 μm), (H) surface morphology of CaP capsule after annealing at 600 °C (scale bar = 648 nm), and (I and J) shell thickness of the CaP capsules before annealing (scale bars = 1 μm and 540 nm, respectively). | ||
The Fourier transform infrared (FTIR) spectra of the CaP capsules and the CaP capsules annealed at 1000 °C are shown in Fig. 3A. The reason for this annealing at a more elevated temperature was to confirm the potential incorporation of carbonate species within the CaP matrix. We first look at the spectrum of the CaP capsules before annealing. The appearance of various vibration modes of the P–O bond of the phosphate group confirms the existence of CaP in the shell. The characteristic degenerated asymmetric stretching mode (ν3) (at 1046 cm−1), non-generated symmetric stretching mode (ν1) (at 962 cm−1), and degenerated bending mode (ν4) (at 562 cm−1 and 574 cm−1) can be found in the spectrum. However, ν3 and ν4 are not distinctly split into a triple peak, suggesting the formation of amorphous CaP. The broad band around 3450 cm−1 is due to the water molecules adsorbed onto the CaP surface, whereas the other water molecule peak at around 1650 cm−1 is overlapped by the BCP or some PVP residue (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond stretch). The hydroxyl group's stretching (νs) and librational (νL) modes, around 3572 cm−1 and 631 cm−1, respectively, are not clearly seen in the CaP spectrum. This is maybe due to the amorphous nature of the CaP or complete substitution of the OH group with Cl−. The small bands at 1410 cm−1 and 875 cm−1 are due to ν3 CO3 and ν2 CO3, respectively. This indicates that carbonate species was incorporated into the CaP crystal structure. Carbon dioxide is reversibly soluble in water to form H2CO3 and the source of the CO32− in the mineralization process. The CaP capsules were annealed at 1000 °C to see whether the degree of crystallinity of the amorphous CaP would increase. The splitting of the vibration bands ν3 (ν3a at 1087 cm−1, ν3b at 1046 cm−1, and ν3c at 1032 cm−1) and ν4 (ν4a at 600 cm−1, ν4b at 574 cm−1, and ν4c at 564 cm−1) into distinct triplet peaks suggests the recrystallization and stabilization of the CaP lattice. Also, the characteristic double degenerated bending mode (ν2) (at 472 cm−1) can be clearly seen in the annealed sample. The carbonate peaks are lost when annealed at 1000 °C because CO32− is liberated as CO2(g) from the CaP crystal structure at 550–1000 °C.67 This is confirmed by the mass loss between 550–1000 °C from the thermogravimetric analysis (TGA) (Fig. 3B). On the TGA curve of the CaP capsules, there are three regions of mass losses. The mass loss of 13% between 30–275 °C is characteristic for the physisorbed water release and evaporation of dodecane residue, 10% mass loss between 275–550 °C represents the decomposition of BCP and PVP residue. The 2.6% mass loss in the region from 550 °C to 1000 °C is characteristic for the CO2(g) release from the decomposition of carbonates from the CaP structure.
O bond stretch). The hydroxyl group's stretching (νs) and librational (νL) modes, around 3572 cm−1 and 631 cm−1, respectively, are not clearly seen in the CaP spectrum. This is maybe due to the amorphous nature of the CaP or complete substitution of the OH group with Cl−. The small bands at 1410 cm−1 and 875 cm−1 are due to ν3 CO3 and ν2 CO3, respectively. This indicates that carbonate species was incorporated into the CaP crystal structure. Carbon dioxide is reversibly soluble in water to form H2CO3 and the source of the CO32− in the mineralization process. The CaP capsules were annealed at 1000 °C to see whether the degree of crystallinity of the amorphous CaP would increase. The splitting of the vibration bands ν3 (ν3a at 1087 cm−1, ν3b at 1046 cm−1, and ν3c at 1032 cm−1) and ν4 (ν4a at 600 cm−1, ν4b at 574 cm−1, and ν4c at 564 cm−1) into distinct triplet peaks suggests the recrystallization and stabilization of the CaP lattice. Also, the characteristic double degenerated bending mode (ν2) (at 472 cm−1) can be clearly seen in the annealed sample. The carbonate peaks are lost when annealed at 1000 °C because CO32− is liberated as CO2(g) from the CaP crystal structure at 550–1000 °C.67 This is confirmed by the mass loss between 550–1000 °C from the thermogravimetric analysis (TGA) (Fig. 3B). On the TGA curve of the CaP capsules, there are three regions of mass losses. The mass loss of 13% between 30–275 °C is characteristic for the physisorbed water release and evaporation of dodecane residue, 10% mass loss between 275–550 °C represents the decomposition of BCP and PVP residue. The 2.6% mass loss in the region from 550 °C to 1000 °C is characteristic for the CO2(g) release from the decomposition of carbonates from the CaP structure.
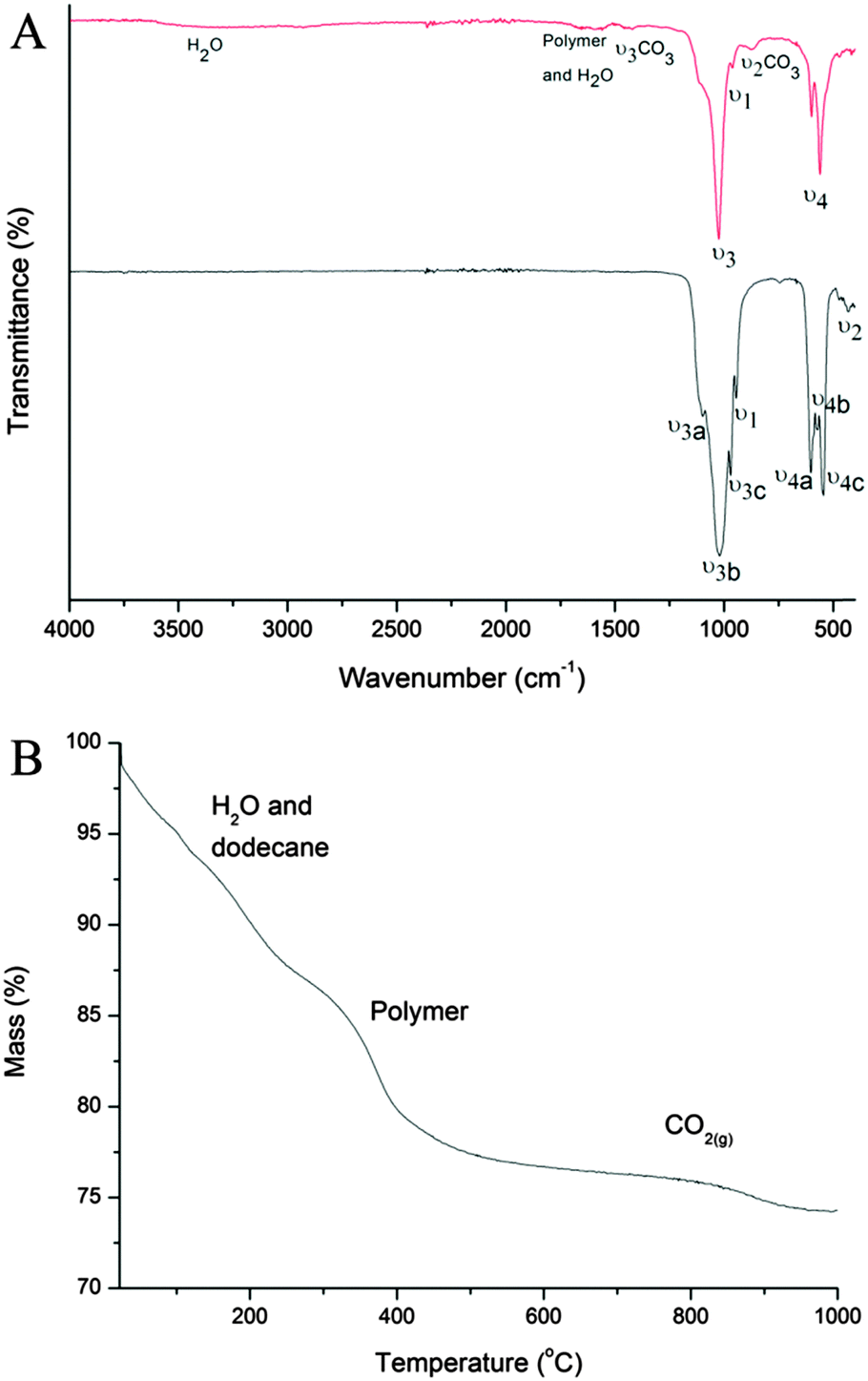 | ||
| Fig. 3 (A) FTIR spectra of CaP capsules (red line), and CaP capsules annealed at 1000 °C (black line). (B) TGA curve of CaP capsules. | ||
The elemental composition of the CaP capsules was studied with an energy dispersive X-ray (EDX) spectrometer. The EDX spectrum (Fig. 4A) of the CaP capsules comprises of calcium, phosphorous, and oxygen peaks, confirming the presence of CaP. Chlorine was also detected suggesting that chlorine was incorporated in the CaP crystal structure, which originated from CaCl2 as a calcium source, and HCl(aq) which was used to acidify the system. The carbon peak originates from the carbon coating of the CaP capsules used for the EDX analysis, and also from the BCP. The calcium-to-phosphorous (Ca/P) atomic ratio of the CaP capsules was established with an inductively coupled plasma-optical emission spectrometry (ICP-OES) to determine the type of CaP mineral phase produced in the mineralization process. The ICP-OES analysis shows the Ca/P atomic ratio is around 1.58 (see ESI,† Table S2). We can therefore propose that the shell of the CaP capsule is made of calcium deficient hydroxyapatite.2 X-ray diffraction (XRD) patterns of the CaP capsules as synthesized and after annealing at 600 °C are shown in Fig. 4B along with an indexed simulated powder pattern of single crystal hydroxyapatite (ICSD number 22059). In both cases, patterns demonstrate very broad peaks indicative of nano-scale crystal sizes. Average size determination using Scherrer analysis was not possible due to the low intensities of the peaks. The CaP capsules sample demonstrates two broad features at around 26° and 32.5° but their intensities are too low to enable indexing of individual Bragg peak contributions. In the annealed sample features corresponding to peaks from the single crystal pattern with qualitatively correct intensity ratios can be identified suggesting crystallinity is induced by the annealing process, or amorphous material contributing to the total scattering of the sample has been removed. Our final task was to modify the CaP capsules with a fluorescent marker. Bisphosphonates (BPs) are stable synthetic analogues of inorganic pyrophosphate characterized by a non-hydrolysable P–C–P structure. BPs have been used to treat and image bone disorders, such as osteoporosis, because BPs present high affinity to adsorb onto bone mineral, and therefore prevent mineral dissolution and bone resorption, and inhibiting osteoclast activity. BP was chosen as the base material to decorate the CaP capsules because the phosphonate groups have a high affinity to CaP. In addition BPs can be modified with additional substituents adjacent to the central carbon atom to introduce a desired functionality.68 De Rosales et al. have recently used this strategy to introduce metallic radionuclides and fluorophores to BPs for PET/SPECT nuclear imaging and fluorescence detection of calcium phosphate and iron oxide nanomaterials in vivo/vitro.69–71 We now demonstrate that fluorescein–bisphosphonate conjugate (Fluo–BP) (Fig. 5A) can be attached to the CaP capsule walls, to produce fluorescent CaP capsules (Fig. 1D).72 The fluorescence properties arise from the fluorescein group, and the adsorption mode of Fluo–BP on to the CaP shell is due to the coordinate covalent interaction between the phosphonate groups in the BP and the calcium atoms in the CaP. The Fluo–BP labeled CaP capsules were imaged with confocal microscopy, see Fig. 5B. As a control experiment Fluo–BP was added to the dodecane droplets stabilized with BCP. No adhesion of the Fluo–BP was observed onto the surface of the emulsion droplets, which confirmed the affinity of the fluorescent labeling agent for the CaP (see ESI,† Fig. S10).
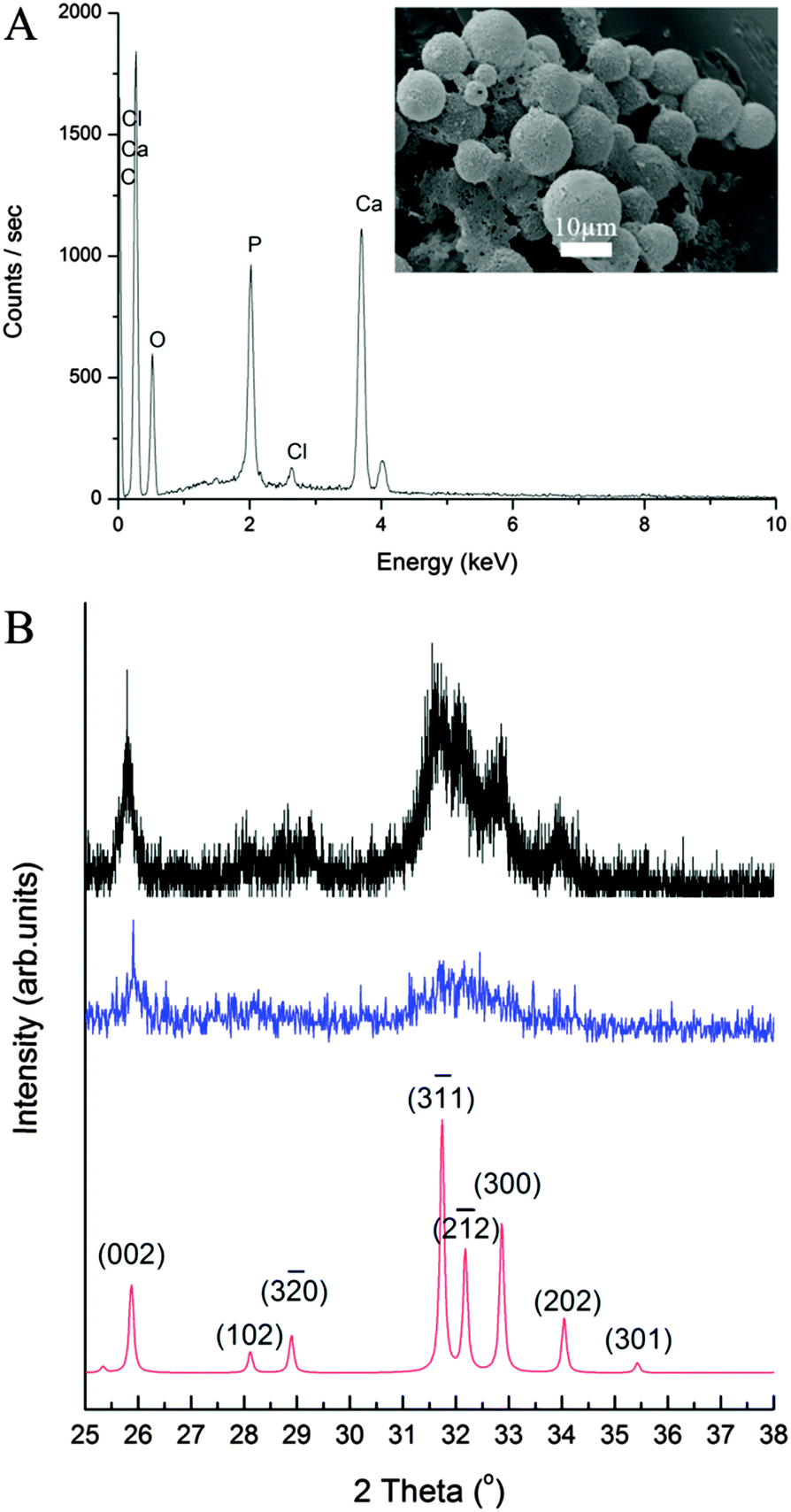 | ||
| Fig. 4 (A) EDX spectrum and SEM micrograph of CaP capsules. (B) XRD patterns of CaP capsules as synthesized (blue line), CaP capsules annealed at 600 °C (black line), and simulated powder pattern of single crystal hydroxyapatite (red line). | ||
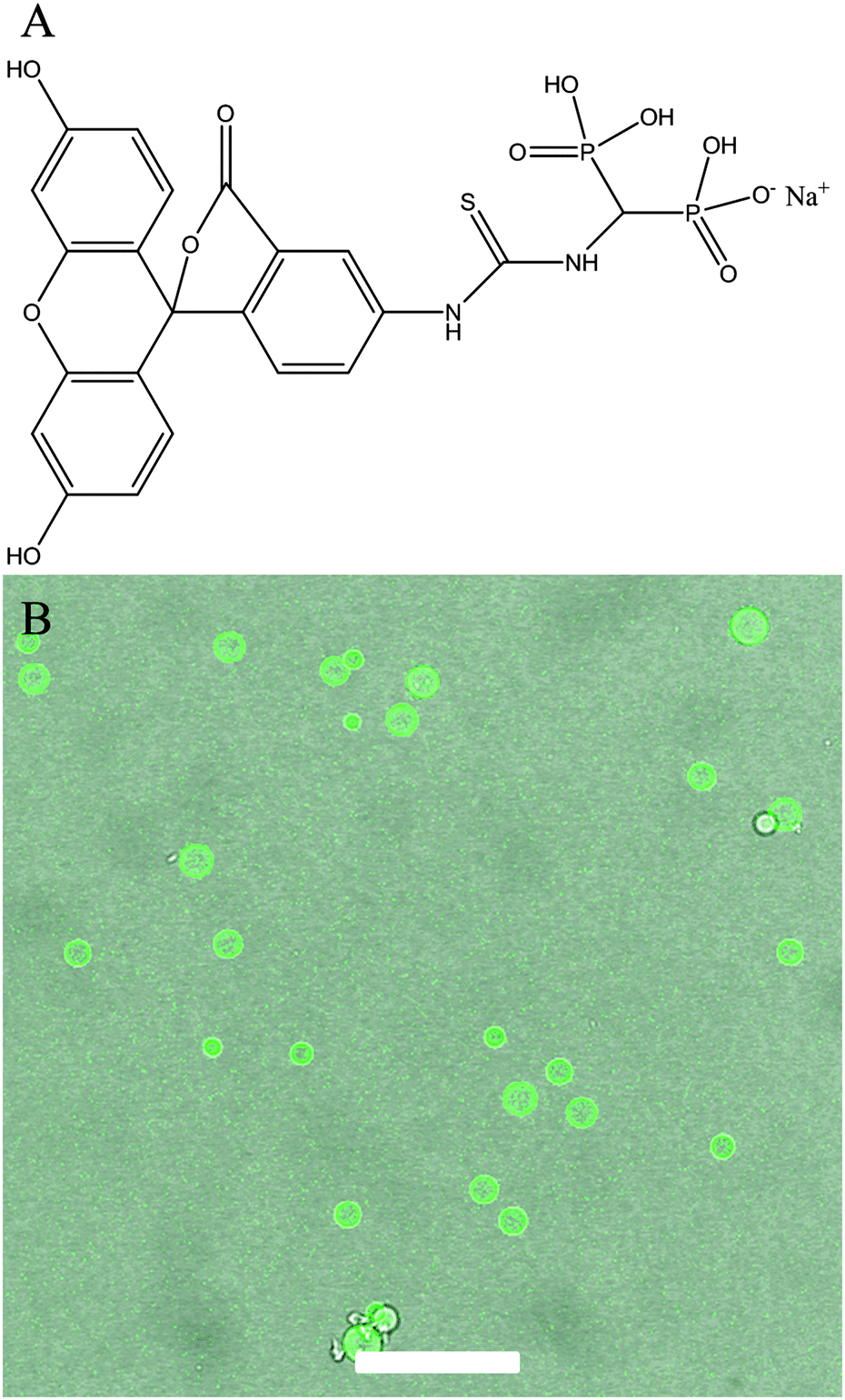 | ||
| Fig. 5 (A) Chemical structure of Fluo–BP. (B) An overlay confocal micrograph of CaP capsules with Fluo–BP. Scale bar represents 50 μm. | ||
Conclusions
We have demonstrated an effective and efficient synthesis method to produce CaP microcapsules by templating oil-in-water emulsion droplets stabilized with synthetic BCP. The process is simple, requires mild conditions, high yielding, low cost, and short incubation time of 72 hours. The BCP provided the right architecture and functionalities to produce stable emulsion droplets, and allows the mineralization of CaP mineral at the surface of the droplet. The CaP wall was established to be calcium deficient hydroxyapatite with chlorine and carbonate species. The oil phase of the CaP capsules can act as a compartment for hydrophobic materials and can be release on demand with crushing, sonication or acidification. We successfully attached a BP conjugate containing a fluorescein fluorophore on the CaP capsules to yield fluorescent capsules. The BP moiety allows for variation in chemical structure opening up the potential to provide a specific functional group as part of the capsule wall. Interestingly the BP molecules can be recovered through capsule wall dissolution upon acidification, potentially of use for diagnostics. We foresee that the ease and versatility of our method to manufacture CaP capsules will lead to exploration of CaP capsules in a wide variety of applications, such as delivery system, micro reactors, imaging, catalysis, bioactive composites and cosmetics.Experimental
Materials
Methacrylic acid (MAA, 99%, Sigma-Aldrich), ethyleneglycol dimethacrylate (EGDMA, 98%, Sigma-Aldrich), 1-dodecanethiol (DDT, ≥99%, Sigma-Aldrich), (trimethylsilyl)diazomethane (2 M in hexanes, Sigma-Aldrich), dodecane (≥99%, Sigma-Aldrich), calcium chloride dihydrate (Analar Normapur), ammonium dihydrogen phosphate (+99%, Across Organics), synthetic hydroxyapatite (≥99.995%, Sigma-Aldrich), poly(vinyl pyrrolidone) (PVP, Mw = 360000 g mol−1, Fluka Analytical), hydrochloric acid (37%, Analar Normapur), sodium hydroxide pellets (Fisher chemical), ammonium hydroxide (35%, Fisher chemical) and distilled water (Milli-Q). Azobis(isobutyronitrile) (AIBN) was purchased from BDH and recrystallized from methanol prior to use. Deuterated chloroform and methanol were purchased from Sigma Aldrich. All organic solvents were standard laboratory grade. Fluorescein–bisphosphonate conjugate (Fluo–BP) was synthesized according to Kapustin et al.72
Synthesis of branched copolymer (BCP)
BCP was synthesized via thiol-regulated free radical polymerization. A 250 mL round bottom flask was charged with MAA (10 g, 116.2 mmol), EGDMA (2.303 g, 11.6 mmol), DDT (2.351 g, 11.6 mmol) and ethanol (147 mL). The reaction mixture was degassed with nitrogen gas for 20 min, and subsequently immersed into a preheated oil bath at 70 °C. The polymerization was initiated by addition of AIBN (0.147 g, 0.89 mmol) and was left stirring for 48 h. The reaction mixture was concentrated under vacuum and the polymer was purified by precipitation in cold diethyl ether (−20 °C). The polymer was left to dry under vacuum overnight.Characterization of BCP
1H NMR spectra were recorded on 1 wt% branched copolymer solutions in deuterated solvents using Bruker Specrospin 400 Ultrashield™ operating at 400 MHz. Molar mass, molar mass distribution (Đ), and Mark–Houwink (α) values were measured by triple-detection gel permeation chromatography (TD-GPC) using an Agilent 390-MDS Multi Detector Suite equipped with refractive index, viscometer, and UV detectors. A mobile phase of DMF with 5 mM NH3BF4 was employed with a flow rate of 1 mL min−1. Two PL gel Mixed-D columns and an additional guard column were used with an oven temperature of 50 °C. Specific calibration was carried out using poly(methyl methacrylate) standards. The BCP was esterified and the 1H NMR spectrum of the esterified-BCP was used to determine the relative molar ratios of the co-monomers and chain transfer agent in BCP. BCP was esterified by firstly dissolving the BCP (50 mg) in methanol (0.2 mL). Toluene (0.2 mL) was added and agitated to obtain a homogeneous solution. (Trimethylsilyl)diazomethane was then added drop wise until a yellow colour appeared and did not become colourless upon agitation. The resulting solution was purified via evaporation, and left to dry in the vacuum oven overnight at 60 °C.Potentiometric titration of BCP
A potentiometric titration curve of BCP was generated by dissolving BCP (0.1 wt%) in a basic solution of pH ∼11.8 using NaOH (0.5 M) to ensure complete dissolution of BCP. Changes in pH values were monitored (with an Oakton pH meter) following incremental additions of HCl (0.1 M) to the basic BCP solution. The degree of protonation (α) of BCP with pH was calculated using the Henderson–Hasselbalch equation.Emulsion preparation and characterization
An aqueous solution containing 3 wt% of BCP was prepared and the pH was adjusted to pH 10 (1 M NH4OH). An equal amount of dodecane was added and this biphasic mixture was homogenized at 24 000 rpm for a minute to produce an oil-in-water emulsion. The emulsion was left for 24 h to equilibrate and the resulting emulsion formed a creamed layer (localized oil volume fraction, φoil = 0.64). The size, span and stability of the emulsion were recoded using laser diffraction (Mastersizer 2000 equipped with a Hydro 2000S dispersion unit). 10 μL of creamed emulsion was added to the dispersion unit containing 100 mL of basic water (pH adjusted to pH 10 with 1 M NH4OH) with a stirring rate of 1000 rpm. The volume average droplet diameters (D4/3) mentioned were obtained from at least 5 repeat runs (D4/3 = ∑Di4Ni/∑Di3Ni). The span is a measure of the distribution of the droplet size distribution and is expressed mathematically as (D(0.9) − D(0.1))/D(0.5), where D(0.9) is the diameter under which 90% of the particles fall, D(0.5) is the diameter under which 50% of the particles fall, and D(0.1) is the diameter under which 10% of the particles fall. Light micrographs were taken using a Leica DM 2500M microscope equipped with a Nikon D5.100 digital camera (see, ESI,† Fig. S4 and S5).Fabrication of calcium phosphate (CaP) capsules
To a slightly acidic solution containing calcium and phosphate ions (pH ∼ 6.4, [calcium] = 2.5 mM, and [phosphate] = 1 mM), creamed emulsion (0.1 vt%) was added and dispersed into the solution. The pH was adjusted to 7.4 with the addition of NH4OH. After 24 hours, the old calcium-phosphate solution was renewed with a fresh calcium-phosphate solution containing PVP (pH ∼ 7.4, [calcium] = 2.5 mM, [phosphate] = 1 mM, and 10 wt% PVP), and emulsion droplets were left to incubate with gentle stirring. At 48 hours, calcium-phosphate solution (100 vt% (with respect to the solution volume at 24 h), pH ∼ 6.7, [calcium] = 5 mM, and [phosphate] = 2 mM) was added to the mineralization solution at 5 mL h−1. The pH was adjusted to pH 7.4 and left to incubate for another 24 hours. The pH of the solution was maintained at pH 7.4 throughout the mineralization process. Prior to characterization, CaP capsules were washed three times by centrifugation with distilled water to remove the PVP. Samples were dried under vacuum at 60 °C overnight.Characterization of CaP capsules
Production of fluorescent CaP capsules
An aqueous solution of fluorescein–bisphosphonate conjugate (Fluo–BP) (200 μm, 0.1 wt%) was added to the CaP capsules solution and left for 1 h in the dark with stirring. CaP capsules were washed twice by centrifugation with distilled water to remove excess Fluo–BP. Confocal imaging was performed on a Zeiss SM 510 confocal microscope. A laser with a wavelength of 490 nm was used to excite Fluo–BP labelled CaP capsules.Author contributions
R.V.B. conducted experiments and wrote the manuscript. R.T.M.R. designed and synthesized Fluo–BP. XRD analysis was performed by L.A.R. S.A.F.B., J.V.M.W., and M.S. oversaw and contributed to the research.Acknowledgements
The authors would like to dedicate this paper in memory of Dr Jonathan V. M. Weaver. Part of the equipment used in this research was supported by Birmingham Science City: Innovative Uses for Advanced Materials in the Modern World (West Midlands Centre for Advanced Materials Project 2), with support from Advantage West Midlands (AWM) and part funded by the European Regional Development Fund (ERDF). L.A.R. acknowledges support from the EPSRC, UK (Grant no. EP/H021388/1), and the PANalytical MPD diffractometer used in this research was obtained through the Science City Energy Futures Project: Hydrogen Energy, with support from Advantage West Midlands. The synthesis of Fluo–BP was funded by The Centre of Excellence in Medical Engineering at King's College London funded by the Welcome Trust and EPSRC (Grant no. WT088641/Z/09/Z).Notes and references
- A. K. Nair, A. Gautieri, S.-W. Chang and M. J. Buehler, Nat. Commun., 2013, 4, 1724 CrossRef PubMed.
- S. V. Dorozhkin, Acta Biomater., 2010, 6, 715 CrossRef CAS PubMed.
- M. Vallet-Regi, Prog. Solid State Chem., 2004, 32, 1 CrossRef CAS PubMed.
- S. V. Dorozhkin, Materials, 2009, 2, 1975 CrossRef CAS PubMed.
- S. P. Nukavarapu, S. G. Kumbar, J. L. Brown, N. R. Krogman, A. L. Weikel, M. D. Hindenlang, L. S. Nair, H. R. Allcock and C. T. Laurencin, Biomacromolecules, 2008, 9, 1818 CrossRef CAS PubMed.
- L. Xia, K. Lin, X. Jiang, Y. Xu, M. Zhang, J. Chang and Z. Zhang, J. Mater. Chem. B, 2013, 1, 5403 RSC.
- Y. Hu, H. Gao, Z. Du, Y. Liu, Y. Yang and C. Wang, J. Mater. Chem. B, 2015, 3, 3848 RSC.
- Y. Fan, K. Duan and R. Wang, Biomaterials, 2005, 26, 1623 CrossRef CAS PubMed.
- J. Li, Y. Yang and L. Huang, J. Controlled Release, 2012, 158, 108 CrossRef CAS PubMed.
- F. Ye, H. Guo, H. Zhang and X. He, Acta Biomater., 2010, 6, 2212 CrossRef CAS PubMed.
- F. Shao, L. Liu, K. Fan, Y. Cai and J. Yao, J. Mater. Sci., 2011, 47, 1054 CrossRef.
- Y.-P. Guo, L.-H. Guo, Y. Yao, C.-Q. Ning and Y.-J. Guo, Chem. Commun., 2011, 47, 12215 RSC.
- Y.-P. Guo, Y.-B. Yao, Y.-J. Guo and C.-Q. Ning, Microporous Mesoporous Mater., 2012, 155, 245 CrossRef CAS PubMed.
- E. V Giger, J. Puigmartí-Luis, R. Schlatter, B. Castagner, P. S. Dittrich and J.-C. Leroux, J. Controlled Release, 2011, 150, 87 CrossRef PubMed.
- L. Wu, Y. Dou, K. Lin, W. Zhai, W. Cui and J. Chang, Chem. Commun., 2011, 47, 11674 RSC.
- C. Zhang, C. Li, S. Huang, Z. Hou, Z. Cheng, P. Yang, C. Peng and J. Lin, Biomaterials, 2010, 31, 3374 CrossRef CAS PubMed.
- A. Lebugle, F. Pellé, C. Charvillat, I. Rousselot and J. Y. Chane-Ching, Chem. Commun., 2006, 606 RSC.
- S. P. Mondéjar, A. Kovtun and M. Epple, J. Mater. Chem., 2007, 17, 4153 RSC.
- D. E. Wagner, K. M. Eisenmann, A. L. Nestor-Kalinoski and S. B. Bhaduri, Acta Biomater., 2013, 9, 8422 CrossRef CAS PubMed.
- A. Doat, F. Pellé, N. Gardant and A. Lebugle, J. Solid State Chem., 2004, 177, 1179 CrossRef CAS PubMed.
- V. Stanić, D. Janaćković, S. Dimitrijević, S. B. Tanasković, M. Mitrić, M. S. Pavlović, A. Krstić, D. Jovanović and S. Raičević, Appl. Surf. Sci., 2011, 257, 4510 CrossRef PubMed.
- X. Ge, Y. Leng, C. Bao, S. L. Xu, R. Wang and F. Ren, J. Biomed. Mater. Res., Part A, 2010, 95, 588 CrossRef PubMed.
- L. Cui, L. Hu, X. Guo, Y. Zhang, Y. Wang, Q. Wei and B. Du, J. Mol. Liq., 2014, 198, 157 CrossRef CAS PubMed.
- A. Corami, S. Mignardi and V. Ferrini, J. Colloid Interface Sci., 2008, 317, 402 CrossRef CAS PubMed.
- A. Corami, S. Mignardi and V. Ferrini, J. Hazard. Mater., 2007, 146, 164 CrossRef CAS PubMed.
- S. Saber-samandari, S. Saber-samandari, N. Nezafati and K. Yahya, J. Environ. Manage., 2014, 146, 481 CrossRef CAS PubMed.
- M. Vila, S. Sánchez-Salcedo, M. Cicuéndez, I. Izquierdo-Barba and M. Vallet-Regí, J. Hazard. Mater., 2011, 192, 71 CAS.
- K. Ohta, H. Monma and S. Takahashi, J. Biomed. Mater. Res., 2001, 55, 409 CrossRef CAS.
- R. Freitag and F. Hilbrig, Biotechnol. J., 2012, 7, 90 CrossRef PubMed.
- C. J. Morrison, P. Gagnon and S. M. Cramer, Biotechnol. Bioeng., 2011, 108, 813 CrossRef CAS PubMed.
- J. Xu, T. White, P. Li, C. He and Y.-F. Han, J. Am. Chem. Soc., 2010, 132, 13172 CrossRef CAS PubMed.
- M. Vukomanović, V. Žunič, M. Otoničar, U. Repnik, B. Turk, S. D. Škapin and D. Suvorov, J. Mater. Chem., 2012, 22, 10571 RSC.
- K. Lin, J. Pan, Y. Chen, R. Cheng and X. Xu, J. Hazard. Mater., 2009, 161, 231 CrossRef CAS PubMed.
- K. Ganesan, A. Kovtun, S. Neumann, R. Heumann and M. Epple, J. Mater. Chem., 2008, 18, 3655 RSC.
- M. Epple, K. Ganesan, R. Heumann, J. Klesing, A. Kovtun, S. Neumann and V. Sokolova, J. Mater. Chem., 2010, 20, 18 RSC.
- E. I. Altinoglu, T. J. Russin, J. M. Kaiser, B. M. Barth, P. C. Eklund, M. Kester and J. H. Adair, ACS Nano, 2008, 2, 2075 CrossRef CAS PubMed.
- J. Liu, F. Liu, K. Gao, J. Wu and D. Xue, J. Mater. Chem., 2009, 19, 6073 RSC.
- X. W. Lou, L. A. Archer and Z. Yang, Adv. Mater., 2008, 20, 3987 CrossRef CAS PubMed.
- H. Fu, M. N. Rahaman, R. F. Brown and D. E. Day, Mater. Sci. Eng., C, 2013, 33, 2245 CrossRef CAS PubMed.
- H. Fu, M. N. Rahaman, D. E. Day and R. F. Brown, J. Mater. Sci.: Mater. Med., 2011, 22, 579 CrossRef CAS PubMed.
- Y. Wang, A. Yao, W. Huang, D. Wang and J. Zhou, J. Cryst. Growth, 2011, 327, 245 CrossRef CAS PubMed.
- Q. Wang, W. Huang, D. Wang, B. W. Darvell, D. E. Day and M. N. Rahaman, J. Mater. Sci.: Mater. Med., 2006, 17, 641 CrossRef CAS PubMed.
- K. Lin, X. Liu, J. Chang and Y. Zhu, Nanoscale, 2011, 3, 3052 RSC.
- Y.-H. Yang, C.-H. Liu, Y.-H. Liang, F.-H. Lin and K. C.-W. Wu, J. Mater. Chem. B, 2013, 1, 2447 RSC.
- M.-Y. Ma, Y.-J. Zhu, L. Li and S.-W. Cao, J. Mater. Chem., 2008, 18, 2722 RSC.
- A. Ethirajan, U. Ziener and K. Landfester, Chem. Mater., 2009, 21, 2218 CrossRef CAS.
- M. Huang and Y. Wang, J. Mater. Chem., 2012, 22, 626 RSC.
- B. P. Bastakoti, M. Inuoe, S. Yusa, S.-H. Liao, K. C.-W. Wu, K. Nakashima and Y. Yamauchi, Chem. Commun., 2012, 48, 6532 RSC.
- K. K. Perkin, J. L. Turner, K. L. Wooley and S. Mann, Nano Lett., 2005, 5, 1457 CrossRef CAS.
- H. J. Lee and S. C. Lee, Polym. Bull., 2010, 65, 743 CrossRef CAS.
- W. Tjandra, P. Ravi, J. Yao and K. C. Tam, Nanotechnology, 2006, 17, 5988 CrossRef CAS.
- H. T. Schmidt, B. L. Gray, P. A. Wingert and A. E. Ostafin, Chem. Mater., 2004, 16, 4942 CrossRef CAS.
- Y. Fukui and K. Fujimoto, Chem. Mater., 2011, 23, 4701 CrossRef CAS.
- Q. Xu, Y. Tanaka and J. T. Czernuszka, Biomaterials, 2007, 28, 2687 CrossRef CAS PubMed.
- H. T. Schmidt and A. E. Ostafin, Adv. Mater., 2002, 14, 532 CrossRef CAS.
- J. S. Sander and a. R. Studart, Langmuir, 2011, 27, 3301–3307 CrossRef CAS PubMed.
- C. Qi, Y.-J. Zhu, B.-Q. Lu, X.-Y. Zhao, J. Zhao, F. Chen and J. Wu, Chem. – Eur. J., 2013, 19, 5332 CrossRef CAS PubMed.
- C. Qi, Y.-J. Zhu, B.-Q. Lu, X.-Y. Zhao, J. Zhao and F. Chen, J. Mater. Chem., 2012, 22, 22642 RSC.
- K.-W. Wang, Y.-J. Zhu, F. Chen, G.-F. Cheng and Y.-H. Huang, Mater. Lett., 2011, 65, 2361 CrossRef CAS PubMed.
- S.-D. Jiang, Q.-Z. Yao, G.-T. Zhou and S.-Q. Fu, J. Phys. Chem. C, 2012, 116, 4484 CAS.
- M.-G. Ma and J.-F. Zhu, Eur. J. Inorg. Chem., 2009, 5522 CAS.
- G. Jutz and A. Böker, J. Mater. Chem., 2010, 20, 4299 RSC.
- A. Schulz, B. M. Liebeck, D. John, A. Heiss, T. Subkowski and A. Böker, J. Mater. Chem., 2011, 21, 9731 RSC.
- A. Schulz, B. Varnholt, B. M. Liebeck, M. J. Richter, K. Kreuels, T. Subkowski and A. Böker, J. Mater. Chem. B, 2013, 1, 1190 RSC.
- R. T. Woodward and J. V. M. Weaver, Polym. Chem., 2011, 2, 403 RSC.
- Y. Zhang and J. Lu, Cryst. Growth Des., 2008, 8, 2101 CAS.
- S. Liao, F. Watari, M. Uo, S. Ohkawa, K. Tamura, W. Wang and F. Cui, J. Biomed. Mater. Res., Part B, 2005, 74, 817 CrossRef PubMed.
- P. Pascaud, F. Errassifi, F. Brouillet, S. Sarda, A. Barroug, A. Legrouri and C. Rey, J. Mater. Sci.: Mater. Med., 2014, 25, 2373 CrossRef CAS PubMed.
- R. Torres Martin de Rosales, R. Tavaré, R. L. Paul, M. Jauregui-Osoro, A. Protti, A. Glaria, G. Varma, I. Szanda and P. J. Blower, Angew. Chem., Int. Ed., 2011, 50, 5509 CrossRef PubMed.
- L. Sandiford, A. Phinikaridou, A. Protti, L. K. Meszaros, X. Cui, Y. Yan, G. Frodsham, P. A. Williamson, N. Gaddum, R. M. Botnar, P. J. Blower, M. A. Green and R. T. M. De Rosales, ACS Nano, 2013, 7, 500 CrossRef CAS PubMed.
- R. T. M. De Rosales, R. Tavaré, A. Glaria, G. Varma, A. Protti and P. J. Blower, Bioconjugate Chem., 2011, 22, 455 CrossRef PubMed.
- A. N. Kapustin, M. L. L. Chatrou, I. Drozdov, Y. Zheng, S. M. Davidson, D. Soong, M. Furmanik, P. Sanchis, R. Torres Martin de Rosales, D. Alvarez-Hernandez, R. Shroff, X. Yin, K. Muller, J. N. Skepper, M. Mayr, C. P. Reutelingsperger, A. Chester, S. Bertazoo, L. J. Schurgers and C. M. Shanahan, Circ. Res., 2015, 116, 1312 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5tb00893j |
| ‡ Deceased. |
| This journal is © The Royal Society of Chemistry 2015 |