
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In situ electrochemical characterisation of graphene and various carbon-based electrode materials: an internal standard approach†
Dale A. C.
Brownson
,
Peter J.
Kelly
and
Craig E.
Banks
*
Faculty of Science and Engineering, School of Science and the Environment, Division of Chemistry and Environmental Science, Manchester Metropolitan University, Chester Street, Manchester M1 5GD, UK. E-mail: c.banks@mmu.ac.uk; Web: http://www.craigbanksresearch.com Fax: +44 (0)1612476831; Tel: +44 (0)1612471196
First published on 23rd April 2015
Abstract
We employ an internal standard protocol to simultaneously characterise and utilise electrode materials during their electrochemical implementation. The proposed approach involves ‘spiking’ a solution containing the analyte of interest (namely, β-nicotinamide adenine dinucleotide (NADH)) with a common electrochemical redox probe (such as hexaammine-ruthenium(III) chloride), which consequently allows information on the electrochemical properties of the electrode being utilised to be obtained and monitored throughout its application. This approach is explored using a range of commonly encountered carbonaceous electrode materials, including various graphene configurations, such as monolayer, double- and few-layered graphene electrodes – the latter is reported for the first time. The variability in structural quality and stability of the graphene electrodes used (particularly between batches) highlights the necessity for implementation of such approaches within the literature. This work provides a simple, yet effective option for the in situ electrochemical characterisation of various electrode materials, essential where the quality and composition of a ‘reported’ electrode material can vary greatly depending on its fabrication (batch-to-batch quality) or during the course of experimental use.
Introduction
Electrochemists are constantly searching for new electrode materials that can give rise to improvements in the performance of various electrochemical devices,1,2 for example in the fabrication of electroanalytical sensors and in a multitude of energy storage and generation applications.3–7 Carbon based electrode materials are widely used in both analytical and industrial electrochemistry, where in many areas they have outperformed the more traditional ‘noble metal’ based alternatives.5,8 Most recently, the classical carbon materials based on graphite, glassy carbon, diamond and carbon black have, reportedly, been outperformed by the distinctive properties of micro-fabricated carbon structures,8–10 such as carbon nanotubes and graphene, which are now fore-fronting innovation and dominating the field.1,2,11A major problem in electrochemistry, typically encountered when employing and exploring the performance of various novel electrode substrates, particularly in graphene research, is the lack of standardisation and appropriate characterisation relating to the electrode materials used; whether supplied externally or fabricated in-house. The most common approach when utilising graphene is to characterise (typically using transmission electron microscopy (TEM), Raman spectroscopy, X-ray photoelectron spectroscopy (XPS), electrochemistry and so on)12,13 specific macro- and/or micro- domains across the surface of an electrode prior to its use (ex situ).1,11,14 Whilst this approach provides useful information regarding the composition of the electrode material in question, it does not account for the occurrence of any surface change or deterioration that may or may not occur during the application of said material. Furthermore, researchers often only characterise a representative sample from a given batch of electrodes (or from a specific site on an individual electrode), in which case the electrochemistry reported is seldom from the same material used for characterisation (or is not entirely representative of the complete electrode surface) and thus one does not account for possible inter- and intra-batch variation of the electrode material. The latter point is a critical oversight, particularly in terms of graphene electrochemical research, where too often electrochemists report the ‘outstanding’ attributes and performance of a ‘graphene’ electrode, which is not the specific electrode utilised to obtain the material's characterisation.1,2 In such cases one cannot be certain that the response observed originates due to the specific features identified during characterisation.
The above issue is widespread within electrochemistry and is applicable to all commercial and non-commercially sourced electrode materials. However, the problem is particularly prevalent with graphene based electrodes given the high variability in structural composition which arises due to the large number of available fabrication routes (each producing graphene with shifting characteristics, such as the number of layers, lateral grain size, presence/absence of functionalities and overall structural ‘quality’).1,11,15 This, in addition to the use of ‘graphene modified’ electrodes (where one simply immobilises a pre-characterised graphene ‘material’ onto a supporting electrode surface in order to electrically connect to and use the graphene in question),13,16 results in literature reports whereby various comparable research groups claiming to be utilising graphene (or any widely available electrode material) of identical ‘quality’ obtain incomparable electrochemical data, leading in some cases to the inability to repeat reported literature values obtained whilst utilising ‘graphene’ electrodes.1,2,11,17–20 In light of the aforementioned problems encountered when employing novel electrode materials, where it is not always possible to fully characterise the specific electrode being utilised (pre- or post-use), it is clear that a standardised approach is required that can clarify the overall surface-state of a specific electrode at the exact time of its employment. An approach to alleviate such issues, as noted above, is to utilise an internal standard within electrochemical measurements.
Internal standards are regularly used in analytical chemistry to aid in the quantification of analyte signals, for example in chromatographic techniques.21 In terms of electrochemistry, internal standards (in the form of a well-defined redox couple, such as the ferrocene/ferrocenium couple)22 have been employed as a reference potential standard in circumstances where the use of a reference electrode alone does not provide an adequately known reference potential in a particular system, such that the potential (oxidation/reduction peak positions) of the chosen internal standard can be compared to literature values and the experimental data adjusted/corrected appropriately.21–23 Furthermore, internal standards have been utilised in electrochemical measurements as a means of quantifying analytical signals based on comparative analysis of the respective peak currents between the analyte of interest and the chosen standard.24,25 It is important to note however, the internal standard approach has not previously been implemented to allow the simultaneous characterisation and utilisation of novel electrode materials during electrochemical measurements, realising an in situ electrochemical characterisation technique.
Inspired by the limitations encountered when employing novel electrode materials within fundamental electrochemical studies and applications, we explore, for the first time, the use of an in situ electrochemical characterisation technique, namely an internal standard approach, to simultaneously characterise and utilise electrode materials of various carbonaceous forms during their implementation as sensor substrates.
Experimental section
All chemicals used were of analytical grade and were used as received from Sigma-Aldrich without any further purification. All solutions were prepared with deionised water of resistivity not less than 18.2 MΩ cm and were vigorously degassed prior to electrochemical measurements with high purity, oxygen free nitrogen.Voltammetric measurements were carried out using an ‘Autolab PGSTAT 101’ (Metrohm Autolab, The Netherlands) potentiostat. All measurements were conducted using a three electrode system. The edge plane pyrolytic graphite (EPPG) working electrode (Le Carbone, Ltd. Sussex, UK) was machined into a 4.9 mm diameter, with the disc face parallel to the edge plane as required from a slab of highly ordered pyrolytic graphite (HOPG: highest grade available; SPI-1, equivalent to Union Carbide's ZYA grade, with a lateral grain size, La of 1–10 μm and 0.4 ± 0.1° mosaic spread); alternatively, the basal plane pyrolytic graphite (BPPG) working electrode (4.9 mm diameter, Le Carbone, Ltd. Sussex, UK) was machined as above however with the disc face parallel to the basal plane as required; a glassy carbon (GC) electrode (3 mm diameter, BAS, USA) and a boron-doped diamond (BDD) working electrode (3 mm diameter, BAS, USA) were also utilised. A platinum wire and a saturated calomel electrode (SCE) were used as counter and reference electrodes respectively.
The range of ‘graphene’ working electrodes utilised herein were commercially obtained from ‘Graphene Supermarket’ (Reading, MA, USA)26 and consist of various configurations of chemical vapour deposition (CVD) grown ‘graphene’ films provided supported on oxidised silicon wafers. Variables include a monolayer graphene film, a double-layer (with a high level of surface/structural defects) graphene film and a few-layer graphene film (comprising on average 4-graphene-layers, termed quasi-graphene).2,27,28 Full details of the CVD fabrication process employed for each of the graphene samples used are provided in the ESI† along with the accompanying physicochemical characterisation (atomic force microscopy (AFM), Raman spectroscopy and XPS) for each of the graphene samples. For employment of the ‘CVD grown graphene’ working electrodes a specialised electrochemical cell was utilised as described previously by our group.27,29 Essentially, the CVD graphene film was secured into a polytetrafluoroethylene (PTFE) housing unit with a silicone O-ring defining the working surface (diameter, 4.9 mm) and a steel contact making connection to the back of the electrode, which via the use of silver conductive paint (applied to cover the back and sides of the graphene chip in their entirety) ensures electrical conductivity from the front ‘working surface’ of the electrode to the electrode connector (to which a lead for the working electrode can be attached). Full details along with figures depicting the experimental set-up are provided in the ESI.† This unique cell design ensures that the graphene film is the only electrochemically active surface that is in contact with the solution during electrochemical measurements.
Where stated, the effective heterogeneous electron transfer (HET) rate constant, koeff, was determined utilising a method developed by Nicholson,30 applicable for quasi-reversible systems using the following equation, ψ = koeff[πDnυF/(RT)]−1/2, where ψ is a kinetic parameter, D is the diffusion coefficient (D = 9.1 × 10−6 cm2 s−1;27,31 for Ru(NH3)62+/3+ in 0.1 M KCl supporting electrolyte), n is the number of electrons involved in the process, F is the Faraday constant, R the gas constant and T the temperature. The kinetic parameter, ψ, is tabulated as a function of peak-to-peak separation (ΔEP) at a set temperature (298 K) for a one-step, one electron process (where the transfer coefficient, α = 0.5).1,2,30 The function of ψ(ΔEP), which fits Nicholson's data, for practical usage (rather than producing a working curve) is given by:2,32ψ = (−0.6288 + 0.0021X)/(1 − 0.017X) where X = ΔEP is used to determine ψ as a function of ΔEP from the experimentally recorded voltammetry. From this, a plot of ψ against [πDnυF/(RT)]−1/2 allows koeff to be readily determined. Note that all koeff values were deduced over the scan rate range of 15–400 mV s−1.
Throughout the entirety of this work Ru(NH3)62+/3+ is utilised as the internal standard. Ru(NH3)62+/3+ was chosen due to it being an outer-sphere electron transfer redox probe, which is dependent only on the electronic structure (DoS) of carbon based electrode materials and thus is the best probe to use when characterising carbon surfaces.2,27 A method for deducing/relating the % edge plane coverage of a given carbon/graphitic based electrode relative to the koeff value obtained can be applied when utilising outer-sphere systems (such as that used herein, where the response is clearly dependent only on the population of edge plane like- sites/defects (DoS)) and has been reported previously.1,2,18,27,33 Note that other outer-sphere systems can be utilised in accordance with the suggested considerations detailed in the conclusions. Furthermore, if required, inner-sphere redox probes that are dependent upon both the electronic structure and carbon–oxygen groups present (surface micro-structure) can also be utilised when adhering to the stated considerations (for example, if interested in the influence of specific oxygen content upon the electrochemical response), however in such cases the electrochemical response is not easy to de-convolute.2,27
Results and discussion
In order to benchmark our approach we first consider the voltammetric response of various commonly encountered carbonaceous electrode materials, namely those comprised of EPPG, BPPG, GC and BDD, towards the electrochemical oxidation of NADH. NADH is of great significance since it is utilised in a diverse range of over 300 dehydrogenase-based biosensors, which are dependent on the coenzyme (NADH) and its oxidised form (NAD+).29Fig. 1 depicts typical cyclic voltammetric profiles obtained at each of the electrode materials of interest towards the detection of 0.5 mM NADH in pH 7 phosphate buffer solution (PBS, 0.1 M KCl), incorporating 0.5 mM hexaammine-ruthenium(III) chloride (Ru(NH3)62+/3+) as an internal standard. Of note is the voltammetric potential and relative current intensity at which the electrochemical oxidation peak (relating to the detection of NADH) occurs. Peak potentials of ca. +0.45, +0.49, +0.61 and +0.72 V (vs. SCE, 100 mV s−1) are evident for the EPPG, GC, BDD and BPPG electrodes respectively, which are in good agreement with previous literature reports.29,34–36 Furthermore, current densities of ca. 1.55 × 10−4, 1.46 × 10−4, 1.38 × 10−4 and 0.94 × 10−4 A cm−2 (vs. SCE, 100 mV s−1) are observed at the respective electrodes, supporting the above correlation. Clearly, there is a dependence on the electronic structure of the carbon-based electrodes.29 The electrochemical oxidation of NADH occurs at the lowest over-potential and exhibits the largest current density (most beneficial electrochemical response) at the EPPG electrode, which, according to previous literature reports,1,27,37 has a large global coverage of edge plane like- sites/defects giving rise to fast electron transfer properties/kinetics in comparison to the alternative electrodes.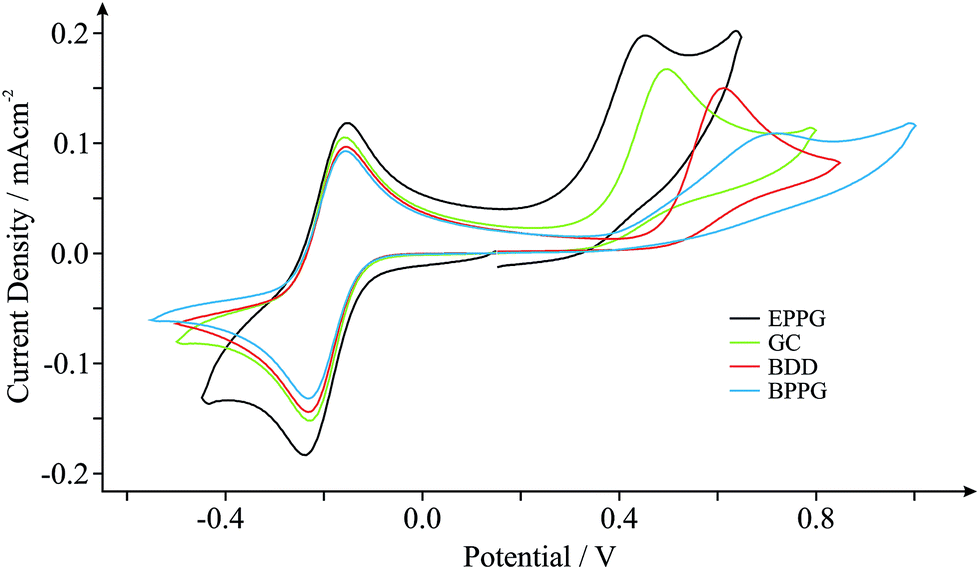 | ||
| Fig. 1 Cyclic voltammograms obtained in pH 7 PBS (with 0.1 M KCl) containing 0.5 mM NADH and 0.5 mM Ru(NH3)62+/3+ (internal standard) at EPPG (black), GC (green), BDD (red) and BPPG (blue) electrodes. Scan rate shown: 100 mV s−1 (vs. SCE). | ||
The inclusion of an internal standard during electrochemical measurements allows such inferences (vide supra) to be further investigated. Scan rate studies were performed during experimental exploration such that the specific electrochemical properties, koeff, of each electrode material can be determined through in situ electrochemical characterisation using the Nicholson method (see Experimental section). Utilising the Ru(NH3)62+/3+ redox probe (internal standard) koeff was estimated to correspond to ca. 4.08 × 10−2, 3.32 × 10−2, 2.15 × 10−2 and 1.39 × 10−2 cm s−1 at the EPPG, GC, BDD and BPPG electrodes respectively. Indeed the EPPG electrode exhibits favourable electrochemical properties (a high density of electronic states, DoS) and consequently possesses fast HET kinetics when compared to the alternative electrodes, resulting in the improved voltammetry that is observed towards NADH in Fig. 1. This conclusion highlights the importance and usefulness of being able to determine the electronic state of specific electrode materials during their implementation via the proposed in situ internal standard electrochemical characterisation method.
Graphene, one of the world's thinnest electrode materials,1,27 has in recent years begun attracting enormous interest from the electrochemical community and is now widely considered ‘the electrode material of choice’ for implementation in numerous electrochemical applications.2 With this in mind, the electrochemical response of different ‘graphene’ based electrodes is next explored. Fig. 2 shows the typical cyclic voltammograms obtained utilising monolayer, double- and few-layered graphene electrodes towards the electrochemical detection of 0.5 mM NADH (pH 7 PBS, 0.1 M KCl) in the presence of 0.5 mM Ru(NH3)62+/3+ (internal standard). Given the incorporation of our internal standard during electrochemical measurements, we are first able to determine the specific surface state (electrochemical properties, koeff) of our ‘graphene’ electrodes at the time of their implementation, allowing comparisons to be made with the graphene literature. Scan rate studies were performed as described in the Experimental section, enabling the determination of koeff, corresponding to the estimated electron transfer kinetics of ca. 2.05 × 10−3, 13.4 × 10−3 and 6.50 × 10−3 cm s−1 at the monolayer, double-layered (defect-abundant graphene) and few-layered (ca. 4-graphene-layers, thus termed quasi-graphene)1,2,27,28 graphene electrodes respectively (utilising Ru(NH3)62+/3+). Analysis of the HET kinetics reveals a trend that corresponds to that previously reported:27 slow and unfavourable electron transfer rates are evident at the monolayer graphene electrode, where it is apparent that increasing the number of graphene layers to ca. 4 (quasi-graphene) results in improved HET properties, which has previously been attributed to the respective coverage (and accessibility) of reactive edge plane sites on these graphitic materials; with increased edge plane content resulting in an improved density of electronic states, DoS, and thus increased HET rates and a beneficial electrochemical response.1,2,18,27 The response of the double-layer graphene, which by its nature possesses a high level of edge plane like structural defects (see Experimental section), conforms to the aforementioned trend, clearly exhibiting the most favourable HET properties of the ‘graphene’ electrodes given that it possesses the highest global coverage of ‘reactive’ edge plane sites.27 However, of note is that the performance of said ‘graphene’ electrodes does not surpass that observed at the HOPG electrodes (vide supra).
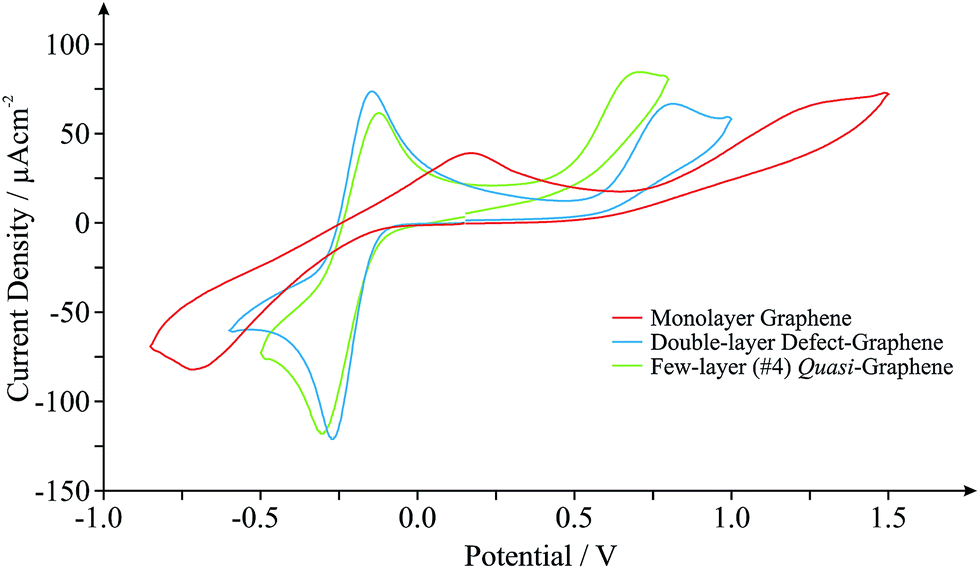 | ||
| Fig. 2 Cyclic voltammograms obtained in pH 7 PBS (with 0.1 M KCl) containing 0.5 mM NADH and 0.5 mM Ru(NH3)62+/3+(internal standard) at monolayer graphene (red), double-layer defect-graphene (blue) and quasi-graphene (green) electrodes. Scan rate shown: 100 mV s−1 (vs. SCE). | ||
Interestingly, when comparison with previous literature is sought, although similar trends are observed, the exact koeff values reported in each case can vary significantly.1,27,38,39 If we consider the response of monolayer graphene for example, a recent report utilising a graphene electrode (obtained and fabricated via identical means) has determined a koeff value of ca. 1.11 × 10−3 (utilising Ru(NH3)62+/3+),27 which is 54% less than that reported herein. Given the high variability in structural composition of various graphene samples (likely resulting from the fabrication)15,40 resulting in inconsistent quality and hence experimental discrepancies, it is clear that in situ electrochemical characterisation is required to standardise the field and to aid in de-convoluting the literature.
Returning to analysis of Fig. 2 and the response of NADH, an electrochemical oxidation peak is evident at ca. +1.13, +0.79 and +0.64 V at the monolayer, double- and few-layered graphene electrodes respectively (vs. SCE, 100 mV s−1), with current densities corresponding to ca. 0.40 × 10−4, 0.51 × 10−4 and 0.46 × 10−4 A cm−2. Inspection of the current densities reveals that the relative performance of each electrode corresponds to the HET properties identified earlier, with a larger koeff value resulting in improved electroanalytical credentials. Analysis of the over-potential at which the NADH oxidation occurs also follows the general trend as identified earlier at ‘graphene’ electrodes, however, the slight deviation evident in this case is likely a result of specific oxygenated species present on the quasi-graphene electrode that are absent (or at different levels) on the other ‘graphene’ electrodes (see Experimental section). This observation follows that the electrochemical behaviour of NADH has been shown to be predominantly dominated by an electrode's DoS and % edge plane content (graphitic materials) but is also influenced by specific oxygenated species.29,41
As highlighted in the introduction (and noted briefly above), a major issue in graphene research is the variation in the quality and geometry of fabricated ‘graphene’ samples used by various research groups which claim to be using, principally, the ‘same’ material. With this in mind we next consider the inter-repeatability of three double-layered graphene electrodes, each acquired and prepared for use as an electrode as indicated in the Experimental section. Fig. 3 shows the electrochemical responses obtained at each of the double-layer graphene electrodes towards the detection of 0.5 mM NADH (pH 7 PBS, 0.1 M KCl) with 0.5 mM Ru(NH3)62+/3+ present (internal standard). It is clear from inspection of Fig. 3 that distinct electrochemical properties are observed, with varying HET kinetics existing. Indeed, analysis of the peak height and position relating to the oxidation of NADH revealed a % Relative Standard Deviation (RSD, N = 3) of 6.28 and 10.7% respectively, with 46.3% RSD calculated in the koeff values. Such large variations in the % RSD of the electrochemical response observed at ‘graphene’ samples (obtained from the same source) highlights the issues faced when this variation is applied to graphenes utilised by numerous research groups distributed worldwide, incorporating differing fabrication routes. Furthermore, similarly high % RSD values (as high as 60.8% RSD)27 have been reported previously for the intra-repeatability of monolayer graphene samples. The high % RSD was attributed to the degradation of the monolayer graphene film during electrochemical experimental use.27 If indeed the electrode surface undergoes alterations during its application, it is clear that full physical characterisation pre- or post-use (ex situ) will not suffice and that the proposed in situ electrochemical characterisation approach should be implemented, allowing electrochemists to observe such changes (if present) in real-time and to accurately report their findings (and the implications of this) accordingly. Again, the variability in structural quality and stability of the ‘graphene’ electrodes used highlights the necessity for implementation of such internal standard approaches within the literature in order to avoid the potential misinterpretation of results. Interestingly, variation in the calculated koeff of the EPPG electrode was determined to be ca. 4.57% (N = 3). The statistics (and comparisons presented) show that the EPPG is a reproducible electrode surface (unlike that of the graphene alternatives) and that the reported internal standard approach sufficiently captures these variations.27,37,42 Using the in situ approach is clearly useful for characterising reproducible electrode surfaces and for those where variation is present is due to the surface.1,27,42
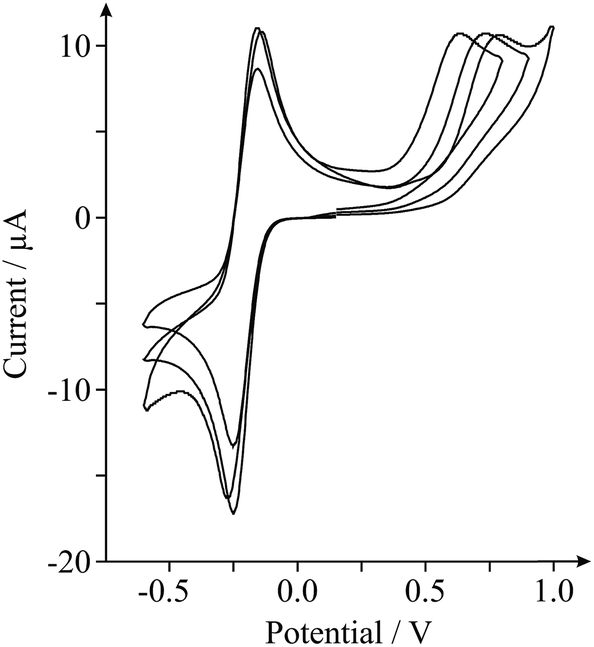 | ||
| Fig. 3 Cyclic voltammograms obtained in pH 7 PBS (with 0.1 M KCl) containing 0.5 mM NADH and 0.5 mM Ru(NH3)62+/3+ (internal standard) at three different double-layered defect-abundant graphene electrodes to investigate the inter-repeatability. Scan rate shown: 100 mV s−1 (vs. SCE). | ||
Last, we show proof of concept for the application of our internal standard in situ electrochemical characterisation technique during the electrochemical sensing of our target analyte, NADH. Analysis of the current densities reported above indicates that the EPPG electrode possesses the most favourable electroanalytical credentials towards the sensing of NADH (the highest current density), hence we utilise this electrode for the latter part. Fig. 4 shows the response arising from successive additions of NADH into a PBS (pH 7, containing 0.1 M KCl and 0.5 mM Ru(NH3)62+/3+). A clear linear response is evident over the concentration range studied (10–100 μM) and the analytical sensitivity of our system was calculated at ca. 64.4 mA M−1, which agrees well with the sensitivity calculated in the absence of the internal standard (ca. 51.2 mA M−1, see inset of Fig. 4). Evidently, there are no obvious inherent disadvantages of incorporating the internal standard, Ru(NH3)62+/3+, into electrochemical measurements and in analytical applications towards NADH. The proposed internal standard approach allows the real-time in situ electrochemical characterisation of a range of electrode materials, such that the surface state of an electrode is known during testing. This information allows the comparison and potential worldwide repetition of research (acting as a form of standardisation), which is currently lacking in the graphene research domain.
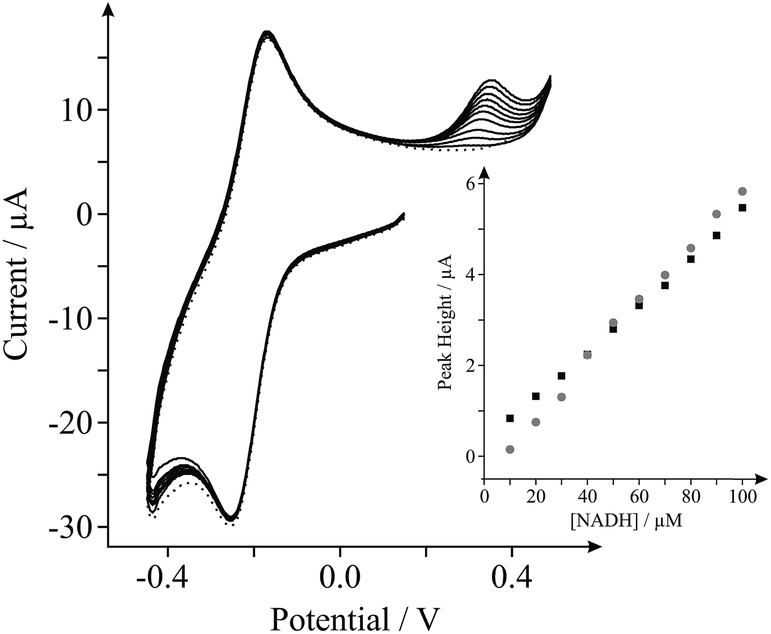 | ||
| Fig. 4 Cyclic voltammograms obtained utilising an EPPG electrode in pH 7 PBS (with 0.1 M KCl) containing 0.5 mM Ru(NH3)62+/3+ (internal standard); ‘blank’ (dotted line) and following successive additions of NADH in the range of 10 to 100 μM (solid lines). Inset: calibration plots of NADH concentration versus the respective voltammetric peak height, obtained using cyclic voltammetry at the EPPG electrode with the internal standard utilised (grey-circles) and without incorporation of the internal standard (black-squares, thus using only pH 7 PBS with 0.1 M KCl as the supporting buffer). All measurements performed at a scan rate of 100 mV s−1 (vs. SCE). | ||
Conclusions
The use of an in situ electrochemical characterisation technique has been explored for the first time. Using an internal standard protocol we have simultaneously reported the electrochemical characterisation and analytical utilisation of various carbon based electrode materials. This approach has particular implications within graphene research, where graphene electrodes are variable and require time consuming surface characterisation both prior to and post use in order to confirm the specific ‘quality’, ‘geometry’ and ‘surface state’ of the particular ‘graphene’ electrode utilised. The approach reported herein overcomes the former issue by providing real time electrochemical characterisation of the chosen electrode material, allowing electrochemists to accurately report the surface state/condition of their electrode during measurements, minimising any potential misinterpretation of results and allowing reliable data to be obtained for standardised and clear comparisons to be drawn with literature; thus allowing the worldwide repetition of such fundamental research.We propose the following considerations when an in situ internal standard approach is used within electrochemistry:
(i) The voltammetric signatures of the target analyte and the internal standard probe are to be sufficiently separated, in terms of peak potential (E0, E1/2etc.), at the chosen experimental scan rate;
(ii) The in situ redox probe and target analyte should not chemically react, either in the non-electrochemical activated or the electrochemical activated states;
(iii) The internal standard must remain stable for the duration of the measurement and must exhibit a repeatable response in the system utilised;
(iv) The in situ probe must not induce fouling or inactivation of the electrode surface;
(v) The internal standard must be incorporated at an appropriate concentration relative to that of the target analyte, such that it does not interfere with or inhibit the targeted response, for example masking the changes of the target analyte.
We recommend that the in situ protocol is employed in all electrochemical experiments where graphene is utilised.
References
- D. A. C. Brownson, D. K. Kampouris and C. E. Banks, Chem. Soc. Rev., 2012, 41, 6944–6976 RSC.
- D. A. C. Brownson and C. E. Banks, The Handbook of Graphene Electrochemistry, Springer, London, 2014 Search PubMed.
- A. T. Lawal, Talanta, 2015, 131, 424–443 CrossRef CAS PubMed.
- A. Merkoçi, M. Pumera, X. Llopis, B. Pérez, M. Valle and S. Alegret, TrAC, Trends Anal. Chem., 2005, 24, 826–838 CrossRef PubMed.
- D. A. C. Brownson and C. E. Banks, Analyst, 2010, 135, 2768–2778 RSC.
- Y.-G. Guo, J.-S. Hu and L.-J. Wan, Adv. Mater., 2008, 20, 2878–2887 CrossRef CAS PubMed.
- D. A. C. Brownson, D. K. Kampouris and C. E. Banks, J. Power Sources, 2011, 196, 4873–4885 CrossRef CAS PubMed.
- R. L. McCreery, Chem. Rev., 2008, 108, 2646–2687 CrossRef CAS PubMed.
- J. Liu, Z. Liu, C. J. Barrow and W. Yang, Anal. Chim. Acta, 2015, 859, 1–19 CrossRef CAS PubMed.
- J. Zhu, D. Yang, Z. Yin, Q. Yan and H. Zhang, Small, 2014, 10, 3480–3498 CrossRef CAS PubMed.
- A. Ambrosi, C. K. Chua, A. Bonanni and M. Pumera, Chem. Rev., 2014, 114, 7150–7188 CrossRef CAS PubMed.
- C. N. R. Rao and K. Biswas, Annu. Rev. Anal. Chem., 2009, 2, 435–462 CrossRef CAS PubMed.
- E. P. Randviir, D. A. C. Brownson, J. P. Metters, R. O. Kadara and C. E. Banks, Phys. Chem. Chem. Phys., 2014, 16, 4598–4611 RSC.
- X. Huang, Z. Yin, S. Wu, X. Qi, Q. He, Q. Zhang, Q. Yan, F. Boey and H. Zhang, Small, 2011, 7, 1876–1902 CrossRef CAS PubMed.
- D. A. C. Brownson and C. E. Banks, Phys. Chem. Chem. Phys., 2012, 14, 8264–8281 RSC.
- M. M. Lounasvuori, M. Rosillo-Lopez, C. G. Salzmann, D. J. Caruana and K. B. Holt, Faraday Discuss., 2014, 172, 293–310 RSC.
- M. S. Goh and M. Pumera, Anal. Chem., 2010, 82, 8367–8370 CrossRef CAS PubMed.
- D. A. C. Brownson, L. J. Munro, D. K. Kampouris and C. E. Banks, RSC Adv., 2011, 1, 978–988 RSC.
- N. L. Ritzert, J. Rodriguez-Lopez, C. Tan and H. D. Abruna, Langmuir, 2013, 29, 1683–1694 CrossRef CAS PubMed.
- G. P. Keeley, N. McEvoy, H. Nolan, M. Holzinger, S. Cosnier and G. S. Duesberg, Chem. Mater., 2014, 26, 1807–1812 CrossRef CAS.
- C. G. Zoski, Handbook of Electrochemistry, Elsevier, London, 2007 Search PubMed.
- R. R. Gagne, C. A. Koval and G. C. Lisensky, Inorg. Chem., 1980, 19, 2854–2855 CrossRef CAS.
- M. E. Morgan and C. R. Freed, J. Pharmacol. Exp. Ther., 1981, 219, 49–53 CAS.
- J. C. Chen, J. L. Shih, C. H. Liu, M. Y. Kuo and J. M. Zen, Anal. Chem., 2006, 78, 3752–3757 CrossRef CAS PubMed.
- J.-A. Annie-Ho, W.-L. Hsu, W.-C. Liao, J.-K. Chiu, M.-L. Chen, H.-C. Chang and C.-C. Li, Biosens. Bioelectron., 2010, 26, 1021–1027 CrossRef PubMed.
- Graphene-Supermarket, http://www.graphene-supermarket.com, accessed 1st September 2014.
- D. A. C. Brownson, S. A. Varey, F. Hussain, S. J. Haigh and C. E. Banks, Nanoscale, 2014, 6, 1607–1621 RSC.
- D. A. C. Brownson, L. C. S. Figueiredo-Filho, X. Ji, M. Gomez-Mingot, J. Iniesta, O. Fatibello-Filho, D. K. Kampouris and C. E. Banks, J. Mater. Chem. A, 2013, 1, 5962–5972 CAS.
- D. A. C. Brownson, R. V. Gorbachev, S. J. Haigh and C. E. Banks, Analyst, 2012, 137, 833–839 RSC.
- R. S. Nicholson, Anal. Chem., 1965, 37, 1351–1355 CrossRef CAS.
- C. E. Banks, R. G. Compton, A. C. Fisher and I. E. Henley, Phys. Chem. Chem. Phys., 2004, 6, 3147–3152 RSC.
- I. Lavagnini, R. Antiochia and F. Magno, Electroanalysis, 2004, 16, 505–506 CrossRef CAS PubMed.
- P. M. Hallam and C. E. Banks, Electrochem. Commun., 2011, 13, 8–11 CrossRef CAS PubMed.
- L. C. S. Figueiredo-Filho, D. A. C. Brownson, O. Fatibello-Filho and C. E. Banks, Analyst, 2013, 138, 4436–4442 RSC.
- M. Musameh, J. Wang, A. Merkoci and Y. Lin, Electrochem. Commun., 2002, 4, 743–746 CrossRef CAS.
- T. N. Rao, I. Yagi, T. Miwa, D. A. Tryk and A. Fujishima, Anal. Chem., 1999, 71, 2506–2511 CrossRef CAS PubMed.
- C. E. Banks and R. G. Compton, Analyst, 2006, 131, 15–21 RSC.
- A. G. Güell, N. Ebejer, M. E. Snowden, J. V. Macpherson and P. R. Unwin, J. Am. Chem. Soc., 2012, 134, 7258–7261 CrossRef PubMed.
- P. S. Toth, A. T. Valota, M. Velicky, I. A. Kinloch, K. S. Novoselov, E. W. Hill and R. A. W. Dryfe, Chem. Sci., 2014, 5, 582–589 RSC.
- F. Bonaccorso, A. Lombardo, T. Hasan, Z. Sun, L. Colombo and A. C. Ferrari, Mater. Today, 2012, 15, 564–589 CrossRef CAS.
- D. A. C. Brownson, M. Gomez-Mingot and C. E. Banks, Phys. Chem. Chem. Phys., 2011, 13, 20284–20288 RSC.
- D. A. C. Brownson, C. W. Foster and C. E. Banks, Analyst, 2012, 137, 1815–1823 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra03049h |
| This journal is © The Royal Society of Chemistry 2015 |