
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chromatographic methods for the isolation, separation and characterisation of dissolved organic matter†
Sara
Sandron
a,
Alfonso
Rojas
a,
Richard
Wilson
b,
Noel W.
Davies
b,
Paul R.
Haddad
a,
Robert A.
Shellie
a,
Pavel N.
Nesterenko
a,
Brian P.
Kelleher
c and
Brett
Paull
*a
aAustralian Centre for Research on Separation Sciences (ACROSS), University of Tasmania, Private Bag 75, Hobart, Tasmania, Australia 7001. E-mail: Brett.Paull@utas.edu.au; Fax: +61 03 6226 2858; Tel: +61 03 6226 6680
bCentral Science Laboratory (CSL), University of Tasmania, Private Bag 74, Hobart, Tasmania, Australia 7001
cIrish Separation Science Cluster, National Centre for Sensor Research, School of Chemical Sciences, Dublin City University, Glasnevin, Dublin 9, Ireland
First published on 10th August 2015
Abstract
This review presents an overview of the separation techniques applied to the complex challenge of dissolved organic matter characterisation. The review discusses methods for isolation of dissolved organic matter from natural waters, and the range of separation techniques used to further fractionate this complex material. The review covers both liquid and gas chromatographic techniques, in their various modes, and electrophoretic based approaches. For each, the challenges that the separation and fractionation of such an immensely complex sample poses is critically reviewed.
 Sara Sandron | Dr Sara Sandron received her Ph.D in Analytical Chemistry, from Dublin City University (Ireland). Her work focused on the application of multi-dimensional and multimodal chromatography to the separation of Dissolved Organic Matter (DOM). At present, she is undergoing a postdoctoral fellowship at the Australian Centre for Research on Separation Science (ACROSS) at the University of Tasmania, Hobart, Australia. The project also focuses on the use of novel multidimensional separation approaches to resolve DOM. Her current research interests include sample preparation techniques, liquid chromatography, gas chromatography, high-performance counter current chromatography, mass spectrometry and nuclear magnetic resonance. |
 Richard Wilson | Dr Richard Wilson graduated from the University of Manchester with a Ph.D in biochemistry in 1998. His post-doctoral studies on the function of extracellular matrix proteins in the pathology of inherited skeletal diseases led to an interest in proteomics, and the discovery of novel proteins involved in cartilage development and joint disease. Since 2011 Dr Wilson has been manager of the University of Tasmania high resolution mass spectrometry facility and supports research in diverse areas including proteomics, metabolomics and the separation and analysis of other complex sample matrices. |
 Noel W. Davies | Prof. Noel Davies is Principal Research Fellow and OIC of the Organic Mass Spectrometry Facility of the University of Tasmania's Central Science Laboratory. Noel has 40 years' experience in mass spectrometry and associated GC and LC separation methods for the analysis of mixtures of organic compounds. He has published 170 refereed journal articles which in turn have received around 4000 citations. He was awarded the Analytical Division Medal of the Royal Australian Chemical Institute in 2012 and the Morrison Medal of the Australian and New Zealand Society for Mass Spectrometry in 2013. |
 Pavel N. Nesterenko | Prof. Pavel Nesterenko received his M.Sc in Petrochemistry and Organic Catalysis, Ph.D and D.Sc degrees in Analytical Chemistry from the Department of Chemistry, M.V. Lomonosov Moscow State University (Moscow, Russian Federation). At present, Prof. Nesterenko holds a New Stars Professor appointment within the Australian Centre for Research on Separation Science (ACROSS) at the University of Tasmania, Hobart, Australia. Author of more than 300 scientific publications, including 3 monographs, 8 Chapters in books, 23 reviews, 250 regular papers and 12 patents. Member of advisory and editorial boards for 6 international journals in the field of analytical chemistry and separation sciences. Editor-in-Chief of the journal Current Chromatography. |
 Brett Paull | Prof. Brett Paull is a B.Sc, Ph.D and D.Sc graduate University of Plymouth, and an RSC Fellow. His first lectureship was at the University of Tasmania (1995 to 1997), before moving to Dublin City University (1998–2011). In 2011 Brett rejoined the University of Tasmania as Professor, and is currently Director of the Australian Centre for Research on Separation Science (ACROSS). Brett's research interests lie in the fields of analytical/bioanalytical chemistry, and materials science. His work is documented in over 200 publications, including 170 peer review journal articles. Within ACROSS research focusses upon production and characterisation of new materials and platforms for application within the analytical/bioanalytical sciences, and advanced inorganic and organic materials for selective extraction and separation purposes. |
Environmental impactThis critical review paper has been produced to aid those working the fields of marine science and environmental geoscience, and related areas investigating carbon cycles, sources and fate. The authors are aware of the importance of separation science to the molecular characterisation and understanding of this important and highly complex environmental system, yet no definitive review in the literature focused on this subject exists. This review compliments a recent review published by Minor et al. on the structural characterisation of DOM, with greater focus on spectroscopic analysis and characterisation. We believe that together the two reviews cover the essential pairing of ‘detection’ and ‘separation’ and collectively offer researchers a substantial resource to help them with their research. |
1. Introduction
1.1. Dissolved organic matter
In simplest terms, the organic matter held within the global water system can be classified as either dissolved or particulate matter. Present within all marine and freshwater sources, dissolved organic matter (DOM) constitutes one of the Earth's largest carbon reservoirs, comparable to atmospheric CO2 (624 and 750 gT, respectively).1 Indeed, atmospheric CO2 is directly influenced by these global DOM reservoirs, as CO2 is itself both a primary source of DOM via the activity of phytoplankton, and a primary product of DOM mineralisation. As DOM is an important component within the global carbon cycle, long term changes in environmental conditions and global systems, for example increasing levels of atmospheric CO2, ocean acidification, and global warming, could potentially affect those complex processes responsible for DOM production and removal.2–11Freshwater aquatic systems can also affect the global carbon balance by transporting terrestrially derived organic matter from land to the sea.12–19 The input of terrestrial DOM represents 2–3% of the total DOM pool, however this percentage can increase when DOM from coastal areas is considered.20 Up to 0.9 gT of carbon per year leaves the terrestrial environment and, of this, 0.25–0.7 gT is delivered from rivers to the sea, whereas 0.2 gT are from ground waters, discharging to the sea without entering rivers.12,21
DOM is often sub-classified as either labile (bioavailable) or refractory. The origins of refractory DOM have been the subject of debate for many decades, although primary sources of seawater or freshwater DOM, such as from soil, vegetation, oil seepages and wildfires are well documented.1,22–24 More recently, the role of microbes in the conversion of labile DOM to the refractory form via the so-called ‘microbial carbon pump’ has been reported.1 Microbial systems are able to metabolise and transform labile DOM from phytoplankton photosynthesis, viral lysis of bacteria and phytoplankton, and protozoan and zooplankton grazing.1,25–28 The bulk of the refractory moieties produced via this process persist within the water column, potentially for periods of several thousand years, without further transformation or digestion.1
Key to a greater understanding of the complex system of biogeochemical processes involved in the formation and removal of DOM is an understanding of the exact nature of DOM itself. Investigations into the content and nature of extracted DOM date back over a century, and research effort in this area increases annually (Fig. 1 shows the research papers published annually based upon an article title search (Scopus) using the term ‘dissolved organic matter’). Traditional definitions of what constitutes DOM, of which most are based on filtration, are now being challenged through increasingly powerful (in terms of resolution) molecular studies. Such studies have pointed to what is more accurately described as “an organic matter continuum”,1,29 with materials ranging in size from the diverse mass of small organic molecules (<1 kDa), to organic colloids, to sub-micron particles, to large and structurally diverse natural polymers. Indeed, the complexity of DOM is such that no reliable estimates of the number of classes of compounds present are available, let alone firm ideas on the number of individual compounds. A further source of complexity, in terms of molecular resolution (physical separation) of this immensely complex material, is the issue of concentration, with compounds present within the range of micromolar to sub-picomolar levels.30
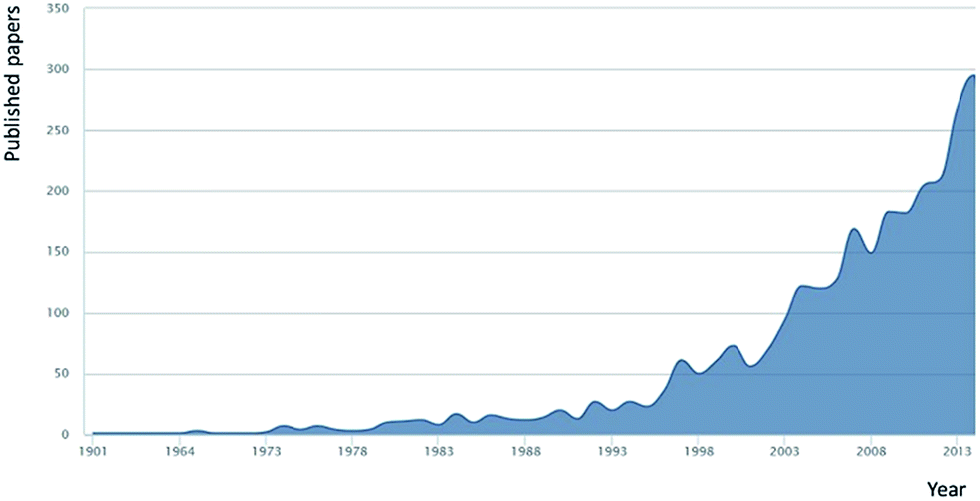 | ||
| Fig. 1 Annual research publications with the term ‘dissolved organic matter’ within the article title (source Scopus Jan 2015). | ||
Compounds present within DOM can also be classified according to polarity, which ranges from high to very low. Within this polarity spectrum, the following functional groups can be found in abundance: substituted alkyl carbons, unsaturated carbons, amides, carboxylic groups, aldehydes and ketones, amino groups and phosphate esters.30,31 Hertkorn et al. utilised NMR to characterise seawater DOM, reporting the following prominent features: aliphatic C–H and C–C bonds, C–N carbon linkages, aliphatic C–O linkages typical of alcohols, esters, ethers and anomeric carbons, aromatic and olefinic carbon linkages, carbonyl groups of amides, carboxylic acids, esters and ketones, with less significant phenol peaks coming from tannin and lignin-like materials.32 Flavonoids and simple phenolics add to this complex mix. These classifications are commonly supported with data obtained from high resolution mass spectrometry (HR-MS).22,32–46
The above functional groups are found within major classes of compounds, such as amino acids, proteins, peptides, sugars, amino-sugars, carboxylic rich alicyclic molecules (CRAM), materials derived from linear terpenoids (MDLT), neutral lipids, DNA, RNA, and sterols.30–33,47–50
Despite the wealth of literature on the nature and classes of compounds present within DOM, there remains a great deal to be revealed regarding its exact composition, how such complex material and chemical systems interact, and how composition varies between seawater and freshwater, geographically and seasonally. Two analytical approaches are used for the chemical characterisation of DOM, methods either based upon the direct analysis within the water sample itself (e.g. bulk measurement, such as fluorescence or nuclear magnetic resonance spectroscopy (NMR)51), or upon the analysis of extracted DOM.30,31 The former potentially avoids contamination and artefacts, but is generally low resolution and not suited to the identification of organic compounds at nano- or picomolar level, particularly when present in saline samples.31,51–53 The latter approach is restricted by the limited availability of well-characterised extraction techniques available for DOM isolation.
Even with a ‘standard method’ for obtaining DOM (for which there is currently none), such diversity in structure, size and concentration would present a considerable analytical challenge, with the need for ultra-high resolution analytical technology to mine such samples for molecular definition. Such advanced instrumental approaches to DOM, predominantly mass spectrometry (MS) and NMR based methods, were reviewed in 2007 by Mopper et al., together with discussion on DOM extraction techniques applied to marine samples.31 Later, in 2011, both Hutta et al., and Duarte et al., critically outlined the most prominent methods to analyse, fractions of DOM, such as humic substances and water soluble organic matter from atmospheric aerosols.54,55 Within both cases, the importance of chromatographic methods prior to advanced detection and identification methods was strongly emphasised, but not reviewed in detail. A more recent review by Minor et al., focussed on the structural characterisation of DOM, approaches to DOM extraction, and bulk characterisation using spectrophotometry, MS and HR-MS, NMR and Fourier transform-infrared spectroscopy (FT-IR).56
Mostly absent in each of the above excellent review papers, is a detailed analysis of significant role separation science has played, and continues to play, in the molecular characterisation of DOM. This aspect of the published literature on DOM characterisation has yet to be the subject of a dedicated review and is certainly worthy of critical discussion. This review therefore selectively covers DOM extraction, fractionation and high-performance separation methods, including both liquid and gas phase chromatography and highlights aspects where advances in separation sciences have had, and will have, a major impact in helping to resolve such complex organic mixtures.
1.2. Isolation of dissolved organic matter
Scheme 1 shows the range of separation methods used in the isolation and separation of DOM, in what often involves 3, 4 or 5 separate procedures/dimensions. In each step the critical role selectivity plays in any final analytical characterisation is very clear. Table 1 includes each of these methodologies and summarises the main purpose of the process, the inherent selectivity (or lack of) and examples of particular applications. Sampling and isolation of DOM represents the first step in all published analytical studies, and it is this first step which possibly presents the biggest challenge in understanding the exact composition of DOM, namely achieving efficient, reproducible extraction of representative, uncontaminated samples, with acceptable recoveries. The first stage of this process involves initial sample filtration to remove particulate matter. This filtering steps applied define DOM according to the porosity of the filter itself. Initial work in this area in the 1970s, applied glass fibre filters (GF-F) filters with pore size ranging from 0.45 to 1.0 μm for the isolation of DOM.30 Nowadays, the filters used to separate POM from DOM have pore size ranging from 0.2 to 0.1 μm. According to this size-based fractionation, POM commonly includes pollen and small organisms such as zooplankton, phytoplankton and bacteria, whereas DOM comprises classes of compounds such as viruses, macromolecules and small molecules (1000–0.1 nm).30 Filters applied to DOM isolation have been traditionally heat treated (calcined) to remove organic contamination, and solvent washed prior to use.30,31,56,57 However, clearly given the idea of the “organic matter continuum”, the current definition of DOM on the basis of filter porosity is an imperfect one. All the compounds considered to constitute DOM pass through these filters, while those classified as particulate organic matter (POM) do not. | ||
| Scheme 1 Analytical approaches to DOM isolation and separation. Abbreviations: extraction: UF – ultrafiltration, RO + ED – reverse osmosis coupled to electrodialysis, PS – passive sampling, SPE – solid phase extraction. Sample treatment: BSTFA – bis-trimethylsilyl trifluoroacetamide, SPME – solid phase microextraction TMAH – tetramethylammonium hydroxide, TMAAc – tetramethylammonium acetate. Separation: RP-LC – reversed-phase liquid chromatography, HPCCC – high performance counter current chromatography, HILIC – hydrophilic interaction liquid chromatography, SEC – size exclusion chromatography, NP-LC – normal phase liquid chromatography, IEC – ion exchange chromatography, IMAC – immobilised metal affinity chromatography, IC – ion chromatography. Detection: FID – flame ionisation detector, TOC – total organic carbon, MS – mass spectrometry, UV – UV absorbance, CD – conductivity detection, RI – refractive index, FT-IR – Fourier transform infrared, DAD – diode array absorbance detection, NMR – nuclear magnetic resonance, PAD – pulsed amperometric detection. | ||
| Techniquea | Purpose | Selectivity/applicationb | Ref. |
|---|---|---|---|
| a Abbreviations as in Scheme 1, SPME: solid phase micro-extraction. b FW: freshwater, SW: seawater. | |||
| Sample extraction | |||
| UF | Extraction/concentration/desalination | Size-selective/FW, SW | 52, 59, 60, 76, 77, 81–83 and 193 |
| SPE | Extraction/concentration/desalination/solvent exchange | Variable selectivity (variable sorbents)/low-mid-polarity compounds/FW, SW | 57, 59–61, 67, 77, 90, 92–95, 98–100, 102, 109, 110, 309, 310 and 311 |
| RO | Extraction/concentration/desalination | Non-selective/FW, SW | 70–72, 74, 114, 117 and 312 |
| PS | Extraction/concentration/desalination | Variable selectivity (variable sorbents)/FW, SW | 119 and 123 |
| SPME | Compound extraction/compound concentration | Phenols | 110, 308 and 319 |
| Polycyclic aromatic hydrocarbons | |||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||
| Sample treatment | |||
| TMAH | Compound alkylation/increase volatility | Non selective/fatty acids | 206, 254, 257, 264, 265 and 267–269 |
| Lignin | |||
| Terrigenous DOM | |||
| Aromatic acids | |||
| TMAAc | Compound acylation/increase volatility | Sugars | 206, 242 and 270 |
| Humic substances | |||
| Pyrolysis | Compound degradation: oxidation/reduction/increase volatility | Non selective/lignin | 260, 266, 273 and 318 |
| Humic acids | |||
| Fulvic acids | |||
| Terrigenous DOM | |||
| Wet oxidation | Compound oxidation | Sugars | 270–272 |
| Lipids | |||
| Lignin | |||
| Terrigenous DOM | |||
| BSTFA | Compound silylation/increase volatility | Sugars | 261, 268, 270 and 272 |
| Lipids | |||
| Humic substances | |||
|
|||
| Separation method | |||
| RP-LC | General fractionation/compound group/class isolation | Mid-low polarity: decreasing polarity | 16, 22, 42, 98, 99, 140, 141, 143, 157, 162, 177, 205 and 227 |
| Isotope separation | Terrigenous DOM | ||
| Humic substances | |||
| Fulvic acids | |||
| Aromatics | |||
| Aliphatics | |||
| Metal complexes | |||
| Lignin | |||
| SEC | Fractionation/compound screening | Size-selective: decreasing molecular size | 59, 64, 76, 107, 132, 189, 190, 193, 197–201, 204–206, 208–212, 214, 217, 300 and 315 |
| Terrigenous DOM | |||
| Organic acids | |||
| Humic substances | |||
| Fulvic acids | |||
| Carbohydrates | |||
| Proteins | |||
| Amino acids | |||
| Metal complexes | |||
| HILIC | Fractionation | Hydrophilic compounds: decreasing hydrophobicity | 133 and 134 |
| Compound screening | |||
| IEC | Fractionation | Charged/polar species | 229, 230, 241, 242, 320 and 321 |
| Specific compound isolation | Sugars | ||
| IMAC | Fractionation | Organic ligands | 231–234 and 322 |
| Specific compound isolation | |||
| HPCCC | Fractionation | Non-selective: partition based | 246 |
| GC | Fractionation | Volatile species: decreasing polarity | 110, 206, 242, 254, 257, 260, 261, 264–273 and 318 |
| Specific compound isolation | Terrigenous DOM | ||
| Compound screening | Humic substances | ||
| Fulvic acids | |||
| Aromatics | |||
| Lignin | |||
| Sugars | |||
| Lipids | |||
| Phenols | |||
| Polycyclic aromatic hydrocarbons | |||
|
|||
| Detection | |||
| MS, HR-MS | Compound screening | Mass selective | 33, 34, 39, 101, 141, 152, 162, 184, 323 and 324 |
| m/z information | Targeted analysis | ||
| Structural information | Ionisation mode dependent | ||
| Quantitative analysis | |||
| NMR, 2D-NMR, 3D-NMR | Structural information | Non selective/functional group | 33, 36, 51, 73, 80, 100, 133, 134, 140 and 325–327 |
| Intra-molecule interaction | |||
| UV, DAD, fluorescence | Qualitative analysis | Selective for chromophoric compounds | 199, 208, 217, 289, 304, 314, 316, 328 and 329 |
| Analysis of chromophores | |||
| IR | Isotope analysis | Functional group selectivity | 63, 273 and 330 |
| Quantitative analysis | |||
| TOC, elemental analysis | Bulk chemical characteristics | Non-selective | 111, 199, 208, 217, 313 and 331 |
| Quantitative analysis | |||
| RI | Qualitative analysis | Non-selective | 317 and 332 |
| CD | Qualitative analysis | Organic acids | 333 and 334 |
| Quantitative analysis | Inorganic ions | ||
| PAD | Qualitative analysis | Sugars/organic acids | 223, 225, 230 and 335 |
| Quantitative analysis | |||
| FID | Qualitative analysis | Non-selective-organic molecules | 336 and 337 |
| Compound screening | Lipids | ||
Second to exactly what is being extracted, is how much can be extracted, given the need to obtain sufficient sample for subsequent analysis. This itself is challenging when considering the volume required (often obtaining, handling and storing 25 to > 100 L) and varying degrees of sample salinity, which in the case of seawater contains 20–35 g L−1 of inorganic salts, compared to 1–3 mg L−1 of DOM (thus selective desalting is of primary importance, particularly prior to MS analysis).30 Preservation of collected water samples prior to DOM extraction should minimise loss of sample integrity, which is not trivial, given the chemical heterogeneity of DOM. For example, the acidification of a water sample to pH 2 can degrade the sample, denature proteins and peptides, and change the reactivity of some classes of compounds within DOM. However, it is very difficult to understand any such changes that DOM may undergo after extraction and practically impossible to compare the chemical characteristics of the original liquid sample with those of the solid/reconstituted material recovered after isolation. Further, any precise evaluation of extraction procedures is hampered by a lack of reference materials. As the composition of DOM is dependent upon the sampling location and season, it is not possible to obtain a universal reference DOM standard.31 Reproducibility studies on DOM samples obtained from analogous locations and times are also unavailable, which further underlines our lack of knowledge regarding inter-sample variability. From an operational point of view, one is unlikely to obtain identical samples from the same location at different time-points, as currents, seasonal variability and weather conditions affect sample reproducibility.
However, despite the above challenges, several widely-accepted protocols for DOM extraction have been developed, some now viewed as pseudo-standard methods. In addition, the International Humic Substances Society (IHSS) now provides reference materials, which are commonly utilised as standards for method development and validation.58 The most widely used reference standard is Suwannee River DOM, with organic carbon concentrations from 25–75 mg L−1 and pH of approximately 4.0. However, the IHSS does not guarantee that successive collected batches are fully identical, and given its freshwater nature, Suwannee River material is not ideally representative of seawater DOM.
Ultrafiltration (UF) and solid-phase extraction (SPE) are the most commonly used techniques applied for DOM extraction (see Tables 2 and 3), and are in detail discussed separately below. The two approaches differ significantly, not least as UF is a physical process (based on mass discrimination), whilst SPE is based on the solute partition coefficient between sorbent and aqueous phases, and hence greatly dependent upon solute and phase chemistries. Unsurprisingly, the fundamental differences between these techniques can produce several compositional differences within the extracted DOM.31,57,59–63 For both UF and SPE, it would appear that recoveries for marine DOM can be highly variable, and thus it is questionable if the extracted DOM can be regarded as being truly representative.31 In addition, when applying these extraction procedures, retentates are often freeze-dried to facilitate sample storage,64 which for labile materials within DOM (i.e. proteins) presents the additional risk of structural damage from ice crystal growth if the rate of freezing is too fast and large crystals are formed. Further limitations of these methods include, contamination due to bleeding/leaching of polymeric material (e.g. from polymer resins or membranes), side reactions with DOM functional groups and the irreversible adsorption of DOM components from the solid support, particularly in the case of SPE.65–69 Due to the large volumes of water that are commonly extracted, SPE is usually used in off-line modes, however this procedure is time consuming and often requires many steps before obtaining a sufficiently concentrated sample, increasing the risk of contamination, sample loss and degradation.
| Methoda | Membrane specifications/cut-offb | Water source | Recovery, % | Comments | Characterisation method(s)c | Ref. |
|---|---|---|---|---|---|---|
| a Abbreviations as in Scheme 1 and Table 1, NF: nanofiltration membranes, GAC: granular-activated carbon. b Membrane details such as material, pore size and molecular cut-off, which is indicative of the size range of the extracted sample, ODS: octadecyl silica, MMA: methyl methacrylate copolymer, PS-DVB: polystyrene divinylbenzene. c DT-MS: direct temperature-mass spectrometry, NDIR: non-dispersive infrared, other abbreviations as in Scheme 1. | ||||||
| UF | Diaflo UM-05: 0.5 kDa cut-off; Diaflo UM-10: 10 kDa cut-off; Diaflo XF-100: 100 kDa cut-off | Seawater | Up to 23 | — | Wet combustion | 83 |
| RO + cation exchange resin | Filmtec CrW30-4619 A membrane: cross-linked aromatic polyamide skin; Dowex 50: cation exchange resin | Freshwater | 90 | Reverse osmosis followed by retentate treatment through cation exchange resin and subsequent lyophilisation. | Elemental analysis | 114 |
| UF | Amicon spiralwound: 1 kDa cut-off | Seawater | 24–55 | Both retentate and filtrate were studied (ref. 82) | Elemental analysis, NMR (ref. 52), IEC-PAD (ref. 81) TOC, elemental analysis, IRMS (ref. 82) | 52, 81 and 82 |
| XAD™, UF | S1N1 spiralwound: 1 kDa cut-off; XAD™: no details specified | Freshwater | Up to 43 | Both retentate and filtrate were studied | TOC, UV, SEC-UV | 193 |
| RO, NF | Fluid systems CA-SD: 0.1 kDa cut-off; fluid systems TFCS: 0.2 kDa cut-off | Drinking water | 97 | — | TOC, NDIR, UV, FT-IR, Zeta potential | 117 |
| SPE, UF + cation exchange resin, XAD™ | S1N1 spiralwound: 1 kDa cut-off; BIORAD GX50: cation exchange resin; C18 BOND ELUT: ODS; Amberlite XAD-8™: MMA; XAD-4™: PS-DVB | Freshwater | Up to 50 | Ultrafiltration followed by retentate treatment through cation exchange resin | Fluorescence, UV, NMR, TOC | 59 |
| UF, SPE | Amicon 8400: 1 kDa cut-off; 3M C18 SPE DISK: ODS | Seawater | Up to 70 | Both retentate and filtrate were studied | FT-IR, DT-MS | 60 |
| PS + anion exchange resin | Membranes used in the PS preparation: diethylaminoethylcellulose-cellulose (DEAE): anion exchange media; polyvinylidene fluoride porous membrane, 1 kDa cut-off; Amberjet 1200H plus: PS-DVB | Freshwater | Up to 89 | Filtrate treatment through anion exchange resin | NMR | 119 |
| RO + ED | Dow FilmTec TW30-4021: polyamide composite; Neosepta AMX: anion exchange membrane; Neosepta CMX: cation exchange membrane | Freshwater (ref. 70) seawater (ref. 71) | 92–93 | — | TOC | 70 and 71 |
| GAC, RO, XAD-4™, XAD-8™ | F-300, Chemviron GAC: Bitumenic Norit GAC; dow FilmTec TW30-2514: spiral module; XAD-8™: MMA; XAD-4™: PS-DVB | Wastewater | Up to 90 | Sample treated through GAC prior to RO | TOC, NDIR, UV | 312 |
| SPE, UF | Amicon 375 mL: 1 kDa cut-off; 3M C18 SPE DISK: ODS | Freshwater | Up to 69 | — | UV, TOC | 77 |
| Cascade UF | Fisherbrand: prefiltration nylon net; Nalgene 250 mL polycarbonate cell and osmosis nylon membranes: 20 to 0.1 μm pore size; Amicon 8400 mL: 0.1 to 1 kDa cut-off | Freshwater | 80 | Retentates and filtrates were studied | UV, TOC | 76 |
| Adsorbent typea | Water source (pH) | Recovery, % | Comments | Characterisation method(s)b | Ref. |
|---|---|---|---|---|---|
| a DEAE: diethylaminoethyl cellulose, MWCNTs: multi walled carbon nanotubes, other abbreviations as in Scheme 1 and Tables 1 and 2. b GPC-UV: gel permeation chromatography with UV detection, ESI-MS: electrospray ionisation-mass spectrometry, other abbreviations as in Scheme 1 and Tables 1 and 2. | |||||
| Amberlite XAD-8™: MMA; Bio-Rad Ag-MP-50: PS-DVB cation exchange resin; Duolite A-7: phenol-formaldehyde-based anion-exchange resin | Freshwater (2) | 81 | Adsorbents in series | IR | 94 |
| (Ref. 98) C18 SEP-PAK: ODS; (ref. 56) C18 BOND ELUT: ODS; (ref. 99) C2 BOND ELUT: C2-functionalised silica adsorbent; (ref. 99) phenyl BOND ELUT: phenyl-functionalised silica adsorbent | Seawater (3–8) | Up to 30 | Comparison of adsorbents (ref. 99) | TOC, RP-LC-UV | 98 and 99 |
| Bio-Rad Ag-MP-50: PS-DVB cation exchange resin; Amberlite XAD-8™: MMA; XAD-4™: PS-DVB | Freshwater (2) | Up to 58 | Comparison of adsorbents | Elemental analysis, molecular weight, titration, NMR | 310 |
| XAD-8™: MMA; XAD-4™: PS-DVB | Freshwater (2) | Up to 85 | Adsorbents in series | TOC, elemental analysis | 95 |
| XAD-8™: MMA; Dowex 50W-8X: cation exchange resin | Freshwater (2) | 87 | Adsorbents in series | GPC-UV | 67 |
| XAD-2™: PS-DVB | Seawater (2) | Up to 67 | — | Radiolabelling, scintillation | 93 |
| XAD-8™: MMA; XAD-4™: PS-DVB | Freshwater (3–3.5) | Up to 85 | Adsorbents in series | TOC, NMR | 92 |
| SUPERCLEAN LC-18: ODS; SUPELCLEAN ENVI-Chrom P: PS-DVB | Freshwater (4.1–7.8) | Up to 132 | STUF coupled to SPE comparison of adsorbents | TOC | 90 |
| Supelco polyacrylate-coated fibre | Aldrich humic acid mixture (7.3) | 40 | SPME | TOC, MS | 109 |
| 3M C18 SPE DISK: ODS | Freshwater (2–2.5) | 60 | — | ESI-MS, NMR, TOC, UV | 100 |
| C18 BOND ELUT: ODS; Amberlite XAD-8™: MMA; XAD-4™: PS-DVB | Freshwater (3–8) | Up to 50 | Technique and comparison of adsorbents | Fluorescence, UV, NMR, TOC | 59 |
| 3M C18 SPE DISK: ODS | Seawater (2) | Up to 70 | UF coupled to SPE | FT-IR, DT-MS | 60 |
| Nanotubes: pristine MWCNTs Filtrsorb 400: activated carbon | Freshwater (3–9) | Up to 96 | Comparison of adsorbents (RO, MWCNTs, GAC) | TOC, FT-IR | 106 |
| PPL BOND ELUT: functionalised PS-DVB; ENV BOND ELUT: PS-DVB; C18 BOND ELUT: ODS; C8 BOND ELUT: C8-functionalised silica adsorbent; C18–OH BOND ELUT: monofunctional ODS | Seawater (2) | Up to 65 | Comparison of adsorbents | NMR | 57 |
| 3M C18 SPE DISK: ODS | Freshwater (2) | Up to 54 | — | ESI-MS, DT-MS, SEC-UV | 78 |
| Nanotubes: pristine MWCNTs; AG-MP5: anion exchange resin; AG1-X8: cation exchange resin | Seawater (1) adsorption, (10) desorption | Up to 81 | Adsorbents in series (cation, anion exchange resins + MWCNTs) | TOC, NDIR, UV | 107 |
| 3M C18 SPE DISK: ODS | Freshwater (2) | Up to 69 | Methods comparison (UF and SPE) | UV, TOC | 77 |
| SPME: Supelco PDMS (polydimethylsiloxane)-based adsorbent; Supelco PDMS-DVB (divinylbenzene)-based adsorbent; Supelco PDMS-DVB-based adsorbent | Aldrich humic acid mixture (6.8–7.1) | 95 | SPME | TOC, fluorescence, MS | 110 |
| Amberlite XAD-8™: MMA; C18 BOND ELUT: ODS; PPL BOND ELUT: functionalised PS-DVB; DEAE: cellulose-based adsorbent | Freshwater (2) | Up to 82 | Comparison of adsorbents | NMR, MS | 61 |
| PPL BOND ELUT: functionalised PS-DVB; HYPERCARB: activated carbon; LC column: Agilent Zorbax Eclipse XBD C18 | Freshwater seawater (2) | Up to 78 | SPE coupled to LC comparison of adsorbents | TOC, UV, MS | 102 and 309 |
Combined techniques for DOM isolation and desalting DOM,70–74 such as reverse osmosis (RO) and electrodialysis, can improve sample recovery (up to 95%), but are currently less commonly applied to DOM isolation than UF or SPE, likely due to the relative availability of the technique, but maybe also related to higher costs involved, and the need for more rigorous blank confirmations.61,62 For example, within recent studies, RO coupled to electrodialysis was found to be at least twice as expensive as SPE.62 In addition, there are also some reports that indicate the DOM extract obtained from reverse osmosis coupled to electrodialysis can contain high levels of inorganic matter.62,75
Clearly the above studies and observations point to some clear advantages and potential disadvantages of each approach (e.g. ease of use and cost of SPE, but with variable and limited recovery, compared to the availability and cost of RO, but which can provide excellent recovery). From the literature published there is certainly no obvious consensus as to the best approach to apply at this time, although it is clear that data generated from subsequent analysis and characterisation should be viewed with regard to the approach used and the inherent limitations thereof. The following sections present the applications of each extraction and isolation technique in individual detail.
UF typically involves higher sample flow rates, together with large surface area polysulfone or polyamide membranes, giving the possibility to extract large sample volumes relatively quickly, a considerable benefit of the technique (Table 2 and ESI Table S1†). However, MW fractions with sizes lower than the membrane cut-off are not retained onto the membrane, and membrane contamination and adsorption issues are occasionally encountered. Additionally, there is a need to carefully optimise operating parameters, and membrane conditioning procedures.30,31,56 Considerable variability in membrane performance and systems from different manufacturers has been observed, as well as between laboratories using the same UF systems.52,56,81,82
When UF is not combined with other extraction techniques, such as SPE, reported DOM recoveries have ranged from as low as 8% to 55% for marine samples, and up to 80% for freshwater DOM (Table 2).76,82,83 However, UF yield is reported to be tightly dependent upon salinity levels.56 Lower extraction efficiency is attributed to lower flocculation at higher salinity.56,77,78,84,85 It has also been observed that DOM recovery is also somewhat depth-dependent. Lower recoveries have been observed for deep water samples when compared to surface water equivalents.56,81,86,87 According to Skoog et al., this is related to the higher proportion of smaller molecules within deep water samples, which are not retained on the UF membrane. Conversely, surface water samples are richer in phytoplankton derived macromolecules.56,81,87
As mentioned above, potential problems associated with SPE include the contamination of isolated DOM, resulting from the leaching of material from the sorbent, (although this can be minimised through appropriate conditioning and wash procedures), together with any impact upon DOM arising from sample acidification, as it is not clear to what extent such treatments modify molecular structures and composition.30,78,88–90 Clearly, when using an SPE based extraction procedure, only those classes of compounds with affinity towards the selected sorbent will be isolated, which may translate to significantly lower recoveries compared to UF (Tables 2 and 3). Unless multiple SPE cartridges with complementary chemistry are used (e.g. combination of polar and apolar phases), it is difficult to extract the complete spectrum of compounds present in DOM. This presents a substantial hurdle to overcome when attempting to fully characterise this complex material. Despite these issues, SPE, particularly where automated (which is readily achievable), still represents perhaps the most practical option for DOM extraction, particularly in sample processing times and costs.62 SPE also provides the opportunity to introduce desired selectivity into the extraction procedure for more targeted studies. Together these advantages typically outweigh the above limitations and SPE remains a popular approach to DOM extraction, as demonstrated by the following methods and applications.
Non-ionic macroporous polymeric sorbents (e.g. XAD™) are typically formed from hydrophobic copolymers, displaying different extraction selectivity and capacity, reflecting their specific chemical and physical properties (i.e. surface area, porosity, % cross-linking etc.). The range of XAD resins reported within the literature for DOM isolation include XAD-2, XAD-4 and XAD-8 (Table 3). XAD-2 and XAD-4 have analogous chemical structure, both being poly(styrene-divinylbenzene) resins (PS-DVB), but with differing surface areas (330 m2 g−1 and 725 m2 g−1, respectively).91 XAD-8 has a similar surface area to XAD-2, but surface chemistry that is based upon a cross-linked poly(methylmetacrylate) (ESI Table S2† provides specific details on the physical and chemical nature of these and other sorbents used for the extraction of DOM).
The above XAD resins have been widely used in the past to extract DOM from natural waters and are reported to provide acceptable recoveries, together with the capacity to process large volumes of water (Table 3).31,59,67,79,92–95 When compared to material extracted using UF or alternative SPE sorbent, DOM obtained using XAD resins tends to show the lowest H/C ratios, and is characterised by a higher proportion of condensed aromatic moieties, typical of flavonoids and lignin-like materials.61 Extracted material is also reported to be relatively low in aliphatic and lipid-like moieties, which might lead to an underestimation of the hydrophobic portion of DOM.93
The use of XAD in DOM extraction involves thorough washing sequences with both organic solvents and aqueous solutions prior to use to reduce extensive sample contamination,30 often requiring multiple elution steps (considered a harsh extraction process). The exact retention mechanism exhibited by XAD resins has been discussed by Town et al., who proposed the potential for additional size-exclusion interactions,67 whilst other studies highlighted the existence of π–π interactions between aromatic compounds (i.e. lignin-like materials and humic substances) and aromatic structures on the resin surface.96,97 Alternative extraction phases to XAD resins are now more commonly used in DOM isolation, details of which are discussed below. However, as mentioned within Green et al., XAD resins still represent the most economically attractive technique in terms of equipment and extraction costs.62
Several alternative PS-DVB adsorbents for DOM recovery have been investigated by Roubeuf et al., (SUPELCLEAN ENVI-Chrom P) and more recently by Dittmar et al., (PPL BOND ELUT), with the latter sorbent described as a PS-DVB phase modified with a proprietary non-polar surface (Table 3).57,61,90 This particular sorbent exhibits a high surface area (600 m2 g−1) and offers significant retention of both non-polar and polar solutes, providing improved selectivity for the full range of compounds constituting the bulk of DOM (including CRAM). Such PS-DVB phases are also noted for their recovery of small molecules (<3 kDa), and recoveries of up to 62% have been reported (Table 3).57 Following these studies, SPE methods employing the above PS-DVB-based resins have seen widespread acceptance.
Although still requiring sample acidification to maximise recoveries, the sample obtained from these new PS-DVB phases has been deemed to be acceptably representative of the true DOM composition61 and according to Dittmar et al., the use of these PS-DVB-based resins allows for the isolation of molecules with polarity degrees ranging from highly polar to nonpolar.57 However, NMR spectra of DOM extracted using SPE with new PS-DVB-based sorbents indicate the extract is predominantly low polarity material, the bulk of which include aromatic groups, indicative of terrigenous origin. Additionally, when compared to other extraction techniques, relatively high CRAM and nitrogen contents have been reported, the latter an indication of higher recovery of solutes containing amino or amide groups, such as protein derived materials.61
Hydrophobic silica-based SPE sorbents are also applied in DOM extraction. The most widely used sorbents are well characterised C18-functionalised silica gels, typically applied to the extraction of non-polar to moderately polar compounds. The application of SPE using C18-functionalised silica sorbents dates back to the early 1980's, with studies such as those reported by Mills et al. (Table 3).98 Several years later, the same group compared recoveries and selectivity of alternative functionalised silica sorbents, such as: C2-, C18- and phenyl-bonded silica.99 In this work, cartridges were pre-rinsed with MeOH, 0.3 mM HCl, loaded with the sample, and eluted with MeOH and finally, deionised water. Relative composition of the isolated DOM samples was investigated using reversed-phase liquid chromatography (RP-LC). Phenyl-bonded silica gel was reported to show the highest recovery of the sorbents investigated (up to 27%), followed by C18 and C2-functionalised silica. More recently, Dittmar et al., also compared the efficiency of a number of C18-functionalised silica sorbents with PS-DVB based resins, including a non-endcapped C18-silica based sorbent (C18OH), which was reported to extensively contaminate the sample due to bleeding.57
Although highly variable, comparative studies such as that carried out by Dittmar et al.,57 have reported that C18-functionalised silica sorbents show similar, but slightly lower recoveries to those achievable using PS-DVB-based adsorbents, with NMR and HR-MS based characterisation suggesting that both types of sorbent generally extract analogous classes of compounds.57,100,101 However, points of difference include DOM from C18-functionalised silica seeming to exhibit a lower nitrogen content and higher H/C ratio, the latter indicative of strong retention of aliphatic compounds (i.e. lipids and terpenoids)61 or carbohydrates. PS-DVB resins exhibit a higher affinity towards compounds having aromatic and double or triple bonds.
PS-DVB, alkyl- and phenyl bonded silica are mainly designed for the extraction of hydrophobic and low polarity molecules. Ion-exchange based SPE extraction can be used for isolation of hydrophilic organic substances. Perminova et al., recently compared traditional DOM extraction methods and extraction based on the use of a diethylaminoethyl (DEAE) anion-exchange cellulose.61 In this work freshwater samples were loaded onto the DEAE sorbent and eluted with 0.1 M NaOH. Recoveries of up to 82% for DOC were reported (∼10–15% higher than the traditional approaches, namely C18-functionalised silica, PS-DVB and XAD-8™), however, the study found the DEAE-extracted DOM to be enriched in highly oxidised structures, such as polyhydroxyphenols, organic acids and carbohydrates.61 NMR data also showed a lower proportion of alkyl-chain protons and higher contributions from carbohydrate and aromatic protons, verifying that this DOM sample differs materially from DOM extracted using traditional sorbents. Based on these findings, the authors suggest the extracted material does not correspond to typical DOM compositional profile seen from the majority of former studies, and conclude by recommending the use of the SPE technique from Dittmar et al., based on the PS-DVB sorbent.57,61
Following on from the above comparative studies, Swenson et al., recently developed a novel SPE system based upon the use of two different kinds of extraction columns, which could be either applied coupled or in single mode.102 A PS-DVB-based stationary phase was coupled to a second cartridge containing an activated carbon phase, providing recoveries which were found to be higher than those obtained when a single extraction chemistry was used. The cartridge eluate was loaded directly onto a RP-LC analytical column operating in gradient mode (water/acetonitrile 0.1 M formic acid) with MS and/or UV-Vis detection.102
The use of multi-walled carbon nanotubes (MWCNTs) in SPE has been explored in the isolation of pollutants from aquatic streams, and in 2007 also for DOM extraction (Table 3).106 Su et al., studied the adsorption kinetics and thermodynamics of DOM onto this newly proposed material, achieving recoveries up to 95%. Prior to extraction, MWCNTs were thermally treated at 400 °C to remove amorphous carbon and adsorption experiments were conducted using 30 mg of adsorbent in 200 mL of DOM solution (pH range = 3 to 9). The solution was subsequently filtered to recover adsorbents, which were further reactivated through a N2 gas flow. This procedure was repeated ten times in order to maximise DOM recovery. DOM was found to be negatively charged across the solution pH range investigated, with the negative charge increasing with pH due to ionisation of carboxylic groups, which were found to be a prominent functional group together with phenolic groups and hydroxyl groups. DOM adsorption and desorption rates were found to be temperature dependent, with higher adsorption at lower temperatures and, conversely, greater desorption at higher temperatures. More recently, Sánchez-González et al., modified this procedure and applied their method to the isolation of DOM from seawater.107 In this case, 60 mg of sorbent were used for 250 mL of seawater, adjusted to pH 3. Desorption of DOM was carried out at pH 10, and the extract further characterised by means of size exclusion chromatography (SEC) (see Section 2.1.2). However, despite the reported high recoveries, the selectivity of MWCNTs for DOM as a whole still requires further clarification, particularly in comparison to previously discussed traditional SPE sorbents.
In a series of publications from Koprivnjak, Vetter and co-workers,70–72 the combination of RO and electrodialysis was reported to achieve enhanced recovery of DOM from both freshwater and seawater sources. The first demonstration of this approach was in 2006, applied to processed synthetic river water samples and obtaining extraction yields of up to 92%. Later, Vetter et al.,71 applied this technique to real seawater samples, and reported a recovery of organic carbon of up to 90%. However, large amounts of inorganic salts were still contained within the extracted sample, significantly higher than that existing in the extract reported by Dittmar et al.57
Koprivnjak et al.,72 reported 75% extraction efficiency for seawater derived DOM from a similar location. In this case, DOM was analysed by both NMR and Fourier Transform Ion Cyclotron Resonance Mass Spectrometry (FT-ICR-MS), which showed the sample to be comparable to the extracts obtained through UF. Koprivnjak et al., also compared their NMR spectra to those obtained by Hertkorn et al.,32 and underlined the presence of CRAM-like materials, together with differences in composition between non-coastal and coastal DOM (i.e. enrichment in terrestrially-derived molecules in the case of coastal DOM). Interestingly, within their review, Mopper et al.,31 suggested DOM extraction through the combination of RO and electrodiaysis is likely to provide a more representative material, and Koprivnjak et al.,72 did indeed observe additional peaks within their DOM NMR and FT-ICR-MS spectra, as compared to previously employed SPE based procedures.
More recently, in order to further confirm the more representative nature of DOM samples obtained through RO coupled with electrodialysis, Chen et al., analysed the isolated seawater DOM by means of ultrahigh resolution MS.75 Samples from two different locations (Atlantic and Pacific Ocean), each at three different depths, showed a significant number of common features (i.e. from 54 to 79% of the assigned molecular formulae), underlining inter- and intra-location analogies. The most significant differences were found within surface samples, characterised by higher H/C values. The authors related these findings to the degradation of aromatic compounds and the production of aliphatics and carbohydrates within surface waters. Furthermore, samples from the Pacific generally showed higher O/C values compared to those from the Atlantic, suggesting an enhanced degree of oxidation, which is possibly related to an enhanced microbial activity or remineralisation processes. The degree of intra-sample similarity suggests that a significant fraction of the extracted DOM is refractory in nature and many of the molecular formulae from these refractory moieties were also found within previously analysed freshwater samples.43,100,118 The study highlighted the representative nature of DOM obtained through RO with electrodialysis, a finding also confirmed upon calculation of the C/N ratio of the extracted samples, which was comparable to direct measurements obtained from the original seawater sample.75
The apparatus used by Lam and Simpson119 (see Fig. 2) consisted of an in-house constructed high-density polyethylene casing with pre-drilled holes containing a size-selective poly(vinylidene fluoride) (PVF) membrane and a DEAE functionalised exchange resin. The PVF membrane allowed the extraction of DOM with a MW lower than 1000 kDa, whereas the anion exchange resin was employed to concentrate negatively charged species (only suitable for freshwater systems). This extraction technique presents some clear advantages over UF and SPE procedures, such as the elimination of many potential sources of contamination arising from water sampling and associated sample handling/storage. There is also no need of sample pumping in passive systems and DOM can be concentrated from discrete depths at low cost.122 An obvious practical disadvantage of this technique is however sampling time. For example, Lam and Simpson reported excellent recoveries of between 72 and 89% from 10 ppm DOM solutions under laboratory conditions, but this was carried out over an extraction period of two weeks. In field experiments the authors deployed multiple samplers with a ratio of 250 mg of resin per 7 cm of membrane, over a similar two week period, enabling the isolation of an impressive 2.8 g of DOM.
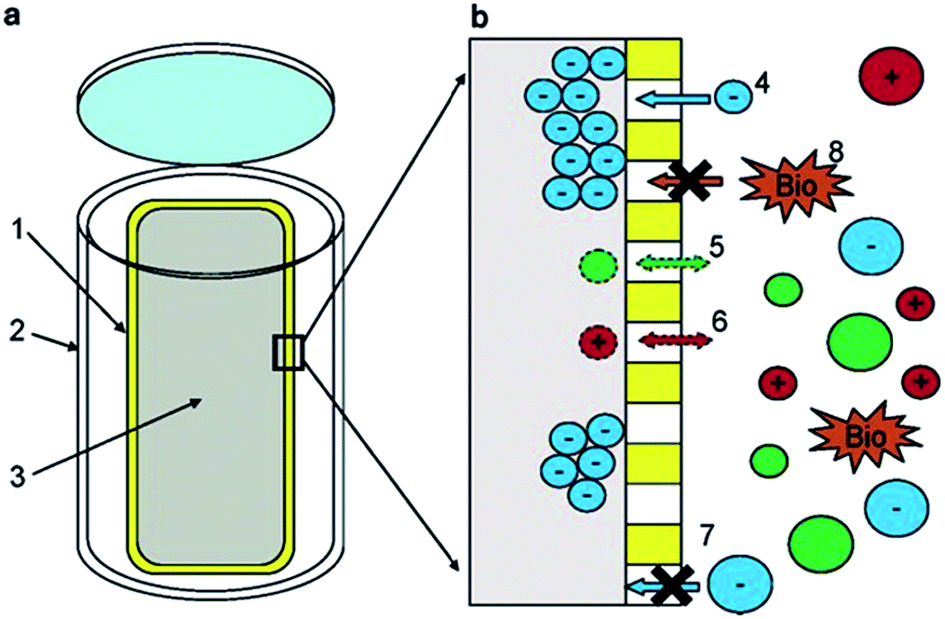 | ||
| Fig. 2 (a) Schematic showing the components of the passive sampler. (1) Poly(vinylidene fluoride) membrane. (2) High-density polyethylene casing. (3) Diethylaminoethyl-cellulose resin. (b) Region showing the resin/membrane/water interface. (4) Negatively charged DOM enters the membrane and is sorbed onto the resin, (5 + 6) neutral or positively charged DOM is not retained. (7 + 8) Large species cannot enter the membrane. Reproduced with permission from Lam et al.119 | ||
In a more recent study, McCaul et al., utilised similar passive samplers to those described above, deployed over a four week period to isolate and study the composition of lacustrine freshwater DOM.123 NMR spectra proved to be similar to those obtained from Lam et al.,119 showing the existence of representative classes of compounds such as: CRAM, MDLT, lignin-like materials, amino acids, proteins, peptides and carbohydrates. The same experiments also supported the presence of molecules typically derived from soil, plants and human activities (i.e. peptidoglycan, phenylalanine, lipoproteins and large polymeric carbohydrates).
In summary, although a promising approach, local conditions such as temperature, water movement, turbidity and biofouling could significantly affect the efficiency and selectivity of passive sampling. To help overcome these issues, reference compounds should be used to reduce and quantify the impact of such environmental parameters.124,125
2. Chromatography of dissolved organic matter
Typically following above mentioned isolation procedures, which aim to isolate and concentrate DOM, high-performance chromatographic techniques are mainly applied in an attempt to fractionate and separate the extracted DOM into its many different classes of compounds. To do so, different chromatographic methods have been applied, once again exploiting differences in compound polarity, shape, size, charge, volatility etc. The need for this additional simplification/fractionation step is quite clear, as discussed within the 2007 review of Mopper et al., who note the limitations of many analytical techniques when applied to direct DOM characterisation.31 Non-selective analytical methods only describe only bulk properties, or limited fractions of the total DOM pool, for example, total organic carbon (TOC) measurements, C:N ratios, or bulk fluorescence. Such approaches reduce DOM to an average theoretical material, with a characteristic fingerprint, which is often used for identification of the source, bulk transport and comparative studies of water bodies.31,126–129 For molecular level information, only MS and NMR (particularly HR-MS or multi-dimensional NMR) can begin to approach the level of selectivity required,32,33,36,80,101,130–134 although the complexity of the unfractionated material often results in extensive spectral overlap.135 Thus, the challenge currently sits in finding the right chromatographic approach to achieve DOM fractionation/separation prior to such HR-MS and NMR analyses.
2.1. Liquid chromatography
The following liquid chromatographic methods have all been applied to the fractionation and separation of DOM; RP-LC and normal phase liquid chromatography (NP-LC), SEC, hydrophilic interaction liquid chromatography (HILIC), ion exchange chromatography, silver ion chromatography, and most recently, high-performance counter-current chromatography (HPCCC). These various techniques have been applied in attempts to fractionate DOM into classes of compounds according to polarity (hydrophobicity/hydrophilicity), MW, charge, and degree of unsaturation (Tables 1 and 4). The following sections detail these approaches and applications thereof individually, followed by some summary and comparative observations.| Water source and isolation methoda | Column | Mobile phase | Detector(s)b | Ref. |
|---|---|---|---|---|
| a Abbreviations as in Scheme 1 and Tables 1–3. b LUM-FL: luminescence fluorescence, detector, TON: total organic nitrogen, ELSD: evaporative light scattering detection, ATR: attenuated total reflection, other abbreviations as in Scheme 1 and Tables 1–3. | ||||
| Reversed-phase liquid chromatography | ||||
| Seawater, SPE | Waters μBondapak C18 (3.9 × 300 mm, 10 μm) | Water/MeCN; ref. 43: water/MeCN H3PO4 (pH 3.2) | TOC, UV | 98 and 99 |
| Freshwater, filtration | LiChroCART (4.0 × 250 mm, 5 μm) | 50 mM phosphate buffer (pH 3.0), 1% dimethylformamide and 100% dimethylformamide | DAD, fluorescence | 143 |
| Seawater, SPE | Lichrosphere (4.0 × 250 mm, 5 μm) | 0.086% H3PO4 and MeOH/MeCN | DAD, TOC | 16 |
| Freshwater, SPE | C18 Supelcosil LC18 (4.6 × 150 mm, 5 μm) | Deuterated water/MeCN | DAD, NMR | 140 |
| Freshwater, filtration | C18 AQ 303, YMC (4.6 × 250 mm) | Water | TOC, MS | 205 |
| Seawater, SPE | Alltech Alltima C18 (2.1 × 150 mm, 5 μm) | Water/MeOH | DAD, TOC, MS | 22 |
| Seawater, SPE | C18 Phenomenex Synergi (4 × 250 mm, 4 μm) | Water/MeOH (pH 7) | Fluorescence, MS | 141 |
| Seawater, UF | RP-LC: Licrospher 100 RP 18 (4.5 × 250 mm, 5 μm) IEC: Dionex CarboPac-PA1 column (4 × 250 mm, 10 μm) | RP-LC: CH3COONa/MeOH (pH 6.8) IEC: 2 mM NaOH or 25 mM NaOH | TOC, fluorescence, PAD | 227 |
| Seawater, SPE | Waters Sunfire (2.1 × 150 mm, 3.5 μm | 0.7 mM phosphate buffer/MeCN | DAD, MS | 157 |
| Freshwater, SPE | C18 Prevail, Alltech (4.6 × 150 mm, 3 μm) | Water/MeOH | DAD, MS | 177 |
| Freshwater, filtration | Waters X-Bridge (4.6 × 150 mm, 3.5 μm) | Water/MeCN 0.1% formic acid | MS | 42 and 162 |
|
||||
| Size exclusion chromatography | ||||
| Freshwater, UF | Waters HPSEC | 2 mM phosphate buffer, 0.1 M NaCl (pH 6.8) | UV, TOC | 193 |
| Freshwater, UF | Protein Pak 125 (7.8 × 300 mm, 10 μm) | 20 mM phosphate buffer (pH 6.8) | UV | 189 |
| Freshwater, UF | Superdex 75 column (10 × 300 mm, 13 μm) | 25 mM phosphate buffer (pH 6.8) | CD, UV, TOC | 197 |
| Freshwater, activated carbon | TSK G3000SW (7.5 × 300 mm, 10 μm) | 10 mM sodium acetate buffer (pH 7) | UV, TOC | 200 |
| Freshwater, RO | TSK G3000SW (7.5 × 300 mm, 10 μm) | 20 mM phosphate buffer (pH 7) | UV, LUM-FL, TOC | 211 |
| Freshwater, RO, filtration | Protein Pak 125 (7.8 × 300 mm, 10 μm), TSK-50S (20 × 250 mm, 30 μm), Biogel P6 (5 × 900 mm, 90–180 μm) | Phosphate buffer (pH 6.8) | UV, TOC | 198 |
| Freshwater, seawater, UF | TSK-gel G3000 (7.8 × 300 mm, 5 μm) | 100 mM phosphate buffer (pH 7) | RI, UV, MS | 64 |
| Freshwater, RO, filtration | TSK-50S (2 × 250 mm, 30 μm) | Phosphate buffer (pH 6.8) | UV, fluorescence, TOC | 199 |
| Freshwater, filtration | TSK HW 40S (2 × 250 mm, 4 μm) | 28 mM phosphate buffer (pH 6.6) | UV, CD, TOC | 201 |
| Freshwater, filtration | PL-Aquagel-OH 30 (4.6 × 250 mm, 8 μm) | 10 mM carbonate buffer and MeOH | UV, TOC, MS | 204 |
| Freshwater, UF | Protein Pak 125 (7.8 × 300 mm, 10 μm) | 20 mM phosphate buffer (pH 6.8) | UV, TOC, NMR | 300 |
| Freshwater, UF, SPE | Waters protein Pak 125 (7.8 × 300 mm, 10 μm) | 20 mM phosphate buffer, 0.1 M NaCl | Fluorescence, UV, NMR, TOC | 59 |
| Freshwater, filtration | Waters ultra-hydrogel 250 (7.8 × 300 mm, 6 μm) | 2 mM phosphate buffer, 0.1 M NaCl (pH 6.8) | TOC, UV | 205 |
| Freshwater, UF, dialysis | BioSep-SEC-s3000 (21.2 × 600 mm, 40 μm) and TSK G3000SW (7.5 × 300 mm, 10 μm) | 10 mM sodium acetate (pH 7) | UV | 206 |
| Freshwater, filtration | Waters protein Pak 125 (7.8 × 300 mm, 10 μm) | 20 mM phosphate buffer (pH 6.85) | UV, fluorescence | 212 |
| Freshwater, RO, filtration | Ultra-hydrogel 250 and 120 (7.8 × 300 mm, 6 μm) | 30 mM ammonium and sodium chloride buffer (pH 11) | DAD, NMR | 132 |
| Freshwater, filtration | Tosoh TSK gel (7.8 × 300 mm, 5 μm) | 20 mM phosphate buffer (pH 6.8) | UV, TOC, NDIR | 208 |
| Freshwater, UF | Toyopearl HW 50S (20 × 250 mm, 45 μm) | Phosphate buffer (pH 6.85) | UV, TOC, TON | 217 |
| Seawater, MWCNTs | Superdex peptide 10/300 GL (10 × 300 mm, 13 μm) and TSK gel G2000SW (8 × 300 mm, 10 μm) | 5 mM ammonium sulphate and 5 mM diammonium hydrogen phosphate (pH 6.5) | UV, TOC, NDIR | 107 |
| Freshwater, SPE | PL-Aquagel-OH 30 (7.5 × 200 mm, 8 μm) | 10 mM carbonate buffer (pH 6.8) | DAD, TOC | 214 |
| Freshwater, filtration | Two in-line BioSep-SEC-s3000 (21.2 × 300 mm, 40 μm) | 10 mM sodium acetate (pH 7) | UV, fluorescence, TOC | 209 |
| Freshwater, UF | PL-Aquagel-OH 30 (4.6 × 250 mm, 8 μm) | 10 mM ammonium bicarbonate and MeOH | DAD, ATR, elemental analysis, TOC, FT-IR | 213 |
| Freshwater, cascade UF | TSK G2000SW Ultropac (7.5 × 300 mm, 10 μm) | 100 mM phosphate buffer (pH 7) | TOC, DAD | 76 |
| Freshwater, filtration | RP Kromasil (4.6 × 150 mm, 5 μm), Acclaim mixed-mode HILIC-1 (4.6 × 150 mm, 5 μm), PSS Suprema (8.0 × 150 mm, 10 μm) | RP-LC: 20% MeCN/water; mixed mode: 20 mM CH3COONH4 10% MeCN (pH 6.0); SEC: 20 mM NH4HCO3 11% MeCN (pH 8.0) | UV, fluorescence, ELSD | 210 |
|
||||
| Hydrophilic interaction chromatography | ||||
| Freshwater, RO, filtration | Phenomenex Luna (4.6 × 150 mm, 3 μm) | 100 mM deuterated ammonium acetate/MeCN | DAD, fluorescence, NMR | 133 |
| Freshwater, RO, filtration | Phenomenex Luna (4.6 × 150 mm, 3 μm), Phenomenex Kinetex (4.6 × 150 mm, 2.6 μm) | 100 mM deuterated ammonium acetate/MeCN | NMR | 134 |
|
||||
| Combined techniques | ||||
| Seawater, UF | Supelcogel Ag or Pb (7.8 × 300 mm, 8 μm) | Water | RI, NMR, MS | 241 |
| Seawater, UF | Supelcogel Ag (7.8 × 300 mm, 8 μm) | Water | RI, MS | 242 |
| Freshwater, SPE | HPCCC: 35 m PTFE tube, 0.8 mm id, total volume of 17.9 mL, external diameter 1.6 mm RP-LC: C18 waters Novapak (3.9 × 150 mm, 4 μm) | HPCCC: hexane/ethyl acetate (upper mobile phase), water/MeOH (lower stationary phase) RP-LC: water/MeCN | UV, MS | 246 |
| Freshwater, SPE | IEC: CarboPac-PA1 column (4 × 250 mm, 10 μm) RP-LC: C18 waters Novapak (3.9 × 150 mm, 4 μm) | IEC: 50–100 mM KOH RP-LC: water/MeOH 0.1% formic acid | PAD, MS | 230 |
Mills and Quinn were amongst the very first to use RP-LC (with UV detection) fractionation for DOM samples from an estuarine source in 1981.98 A water/MeCN mobile phase gradient was used with a 300 × 3.9 mm i.d. μBondapak C18 column. Although each chromatogram was dominated by several clusters of largely unresolved peaks, the largest of which eluted in the middle region of an applied MeCN gradient (suggesting intermediate polarity), each clearly showing specific features according to sampling location (see Fig. 3). Mills and co-workers later reported further application of this RP-LC method to estuarine DOM samples, following minor improvements, such as use of a buffered mobile phase (pH 3.2 with H3PO4).99 However, once again most of the detectable DOM components eluted within a similar gradient window as an unresolved ‘hump’, although large unretained peaks eluting at beginning of the chromatograms did indicate the presence of a significant fraction of highly polar organic material.
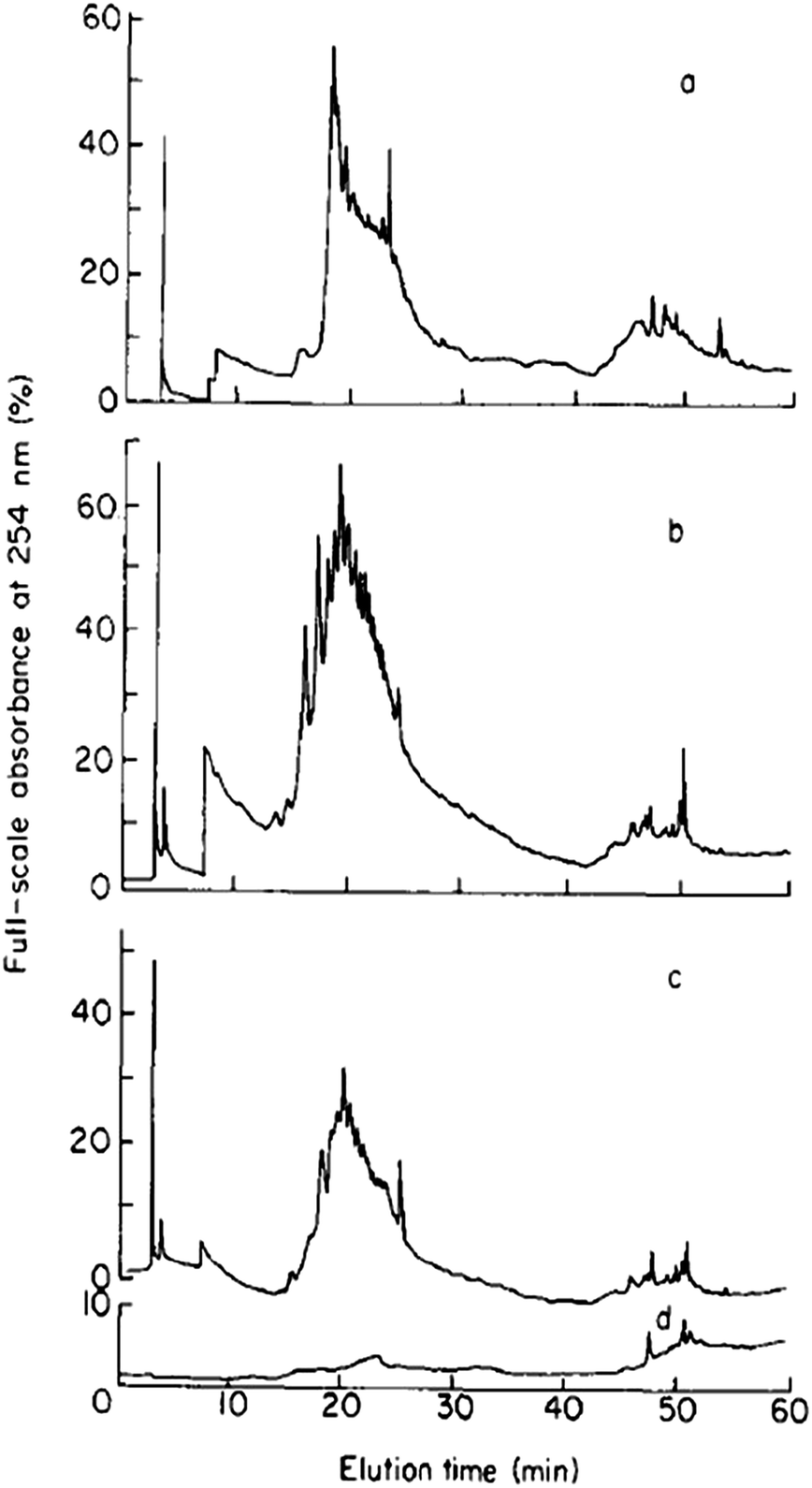 | ||
| Fig. 3 LC-UV chromatograms of DOM from different collection points (a–c), and (d) procedural blank. Reproduced with permission from Mills et al.98 | ||
Lignin-derived phenols are widely used to understand the transport of terrestrial organic matter and have also been analysed using RP-LC, on the basis of previously reported methods.16,136–138 Within one such study, terrestrially derived organic matter, in particular lignin, was oxidised by CuO and separated using a Lichrosphere 100 RP 18 (4 × 250 mm, 5 μm particle size) column and a mobile phase composed of phosphate buffer, MeOH and MeCN. Lignin-derived phenols were monitored through UV adsorption at 280 nm and identity confirmed by their absorbance spectra (230–340 nm). Together with the aid of carbon isotope analysis, this method underlined the presence of distinctive chemical patterns when analysing organic matter of marine origin and terrestrial origin, allowing for the comparison of samples from different collection points.
Parlanti et al., also used RP-LC with diode array detection (DAD), to compare the profiles of DOM from marine and freshwater sources (Table 4).139 Using a water–MeCN gradient, the authors were able to identify compositional differences (and similarities) between the two types of DOM sample, and were ultimately able to use the separation achieved to divide their DOM into multiple fractions according to polarity. These fractions were subsequently further separated by means of capillary zone electrophoresis (CZE), providing orthogonal selectivity to the RP-LC, with the authors suggesting CZE demonstrates considerable potential for DOM profiling and characterisation of DOM of varying origins (see Section 2.3).
In a similar study, Simpson et al., also investigated the use of RP-LC for DOM fractionation, here using a deuterated water–MeCN gradient, again with DAD, monitoring at 280 nm in order to detect compounds enriched in double bonds and aromatics (Table 4).140 The chromatograms recorded at this wavelength (for different freshwater sources of DOM) included large predominantly unresolved series of peaks, providing three fractions, and a separate more retained series of co-eluting peaks (fourth fraction). Each of these fractions was subsequently analysed by NMR. From the four RP-LC fractions obtained, a total of 150 NMR spectra were collected. The spectra from the early eluting fractions contained sharp aromatic peaks of relatively polar species (phenols and/or aromatic acids), which were eluted under almost purely aqueous conditions. The NMR spectra from the following fractions were dominated by broad signals, indicating an aggregation of co-eluting species. However, despite the broad profiles, differences could be identified between the spectra, indicating that the chromatography provided a certain degree of separation.
Koch et al., investigated the impact of pH (and the use of mobile phase buffers) upon the RP-LC separation of DOM, proposing a ‘bufferless’ pH-neutral water/MeOH gradient (Table 4).141 As MeOH can act as both proton acceptor and donor (whereas MeCN can only be a proton acceptor), MeOH can undergo polar or hydrogen bonding interactions with solutes, particularly when the pH of the mobile phase is neutral, so that any secondary interaction is prevented. Koch et al., thus found the absence of buffers and neutral pH approach resulted in more resolved peaks of the water soluble components (Fig. 4), whereas lower pH separations caused extensive co-elution. However, despite the partial success of this approach, the authors were clear to point out the necessity to further reduce the complexity of DOM samples prior to RP-LC and propose the use of a multi-dimensional chromatographic approach involving SEC.
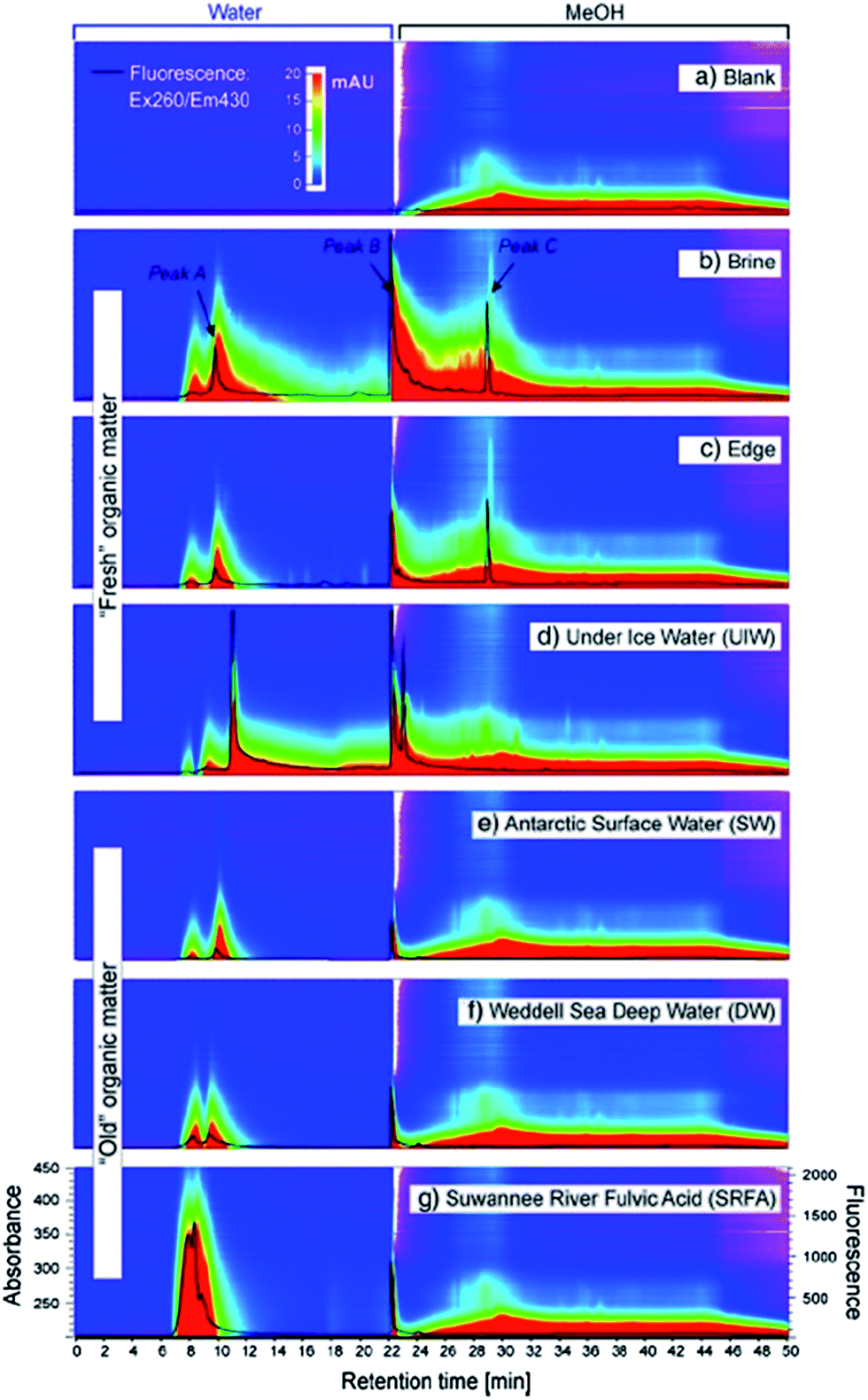 | ||
| Fig. 4 LC-diode array and fluorescence data (ex 260/em 430 nm) for (a) procedural blank and (b) six DOM samples. Reproduced with permission from Koch et al.141 | ||
Hutta and co-workers have extensively studied terrestrially derived organic matter (i.e. humic acids and lignin) and, based on their previous studies, which involved the use of a mobile phase gradient composed of a phosphate buffer and dimethylformamide, collected individual fractions of soil-derived humic acids from RP-LC with fluorescence detection. These were subsequently further separated by means of SEC (also with a phosphate buffer and dimethylformamide gradient and fluorescence detection).54,142–144 In both chromatographic steps dimethylformamide was chosen for its proven solvating power with regards to humic acids, polyelectrolytes and humic substances.142,143 This off-line 2D method provided increased resolution of certain compounds in the second dimension. However, a notable drawback of this procedure was the high boiling point of the mobile phase, which renders this method unsuitable for universal forms of detection such as MS, evaporative light scattering detection (ELSD) or charged aerosol detection (CAD).
Following collection of mass spectra, potential elemental formulae are assigned to the acquired monoisotopic mass of each molecular species, within the mass accuracy limits of the instrument used.44,131,154,155 Kendrick mass analysis plots and van Krevelen diagrams are commonly used in describing DOM composition and are a valuable aid in simplifying the enormous amount of data generated from these experiments.149,156,157 Kendrick mass defect highlights the presence of homologous series differing from each other by the number of CH2 groups and is usually plotted as function of nominal Kendrick mass. Within this representation, ions belonging to the same homologous series have the same Kendrick mass defect but different nominal Kendrick mass and are positioned along a horizontal line on the plot. This representation is often used in conjunction with van Krevelen diagrams, where H/C ratios of each identified molecule are plotted against the respective O/C ratios. These diagrams are useful in assessing the presence of various classes of compounds within DOM. However, it must be highlighted that different molecular formulae can be characterised by analogous H/C and O/C ratios and therefore be overlaid within such plots.154 By using these kind of plots, DOM from different sources can be readily compared, with considerably more detail than possible using simple UV or fluorescence based detection.43,75,149,158
More recent studies have begun to explore greater possibilities in MS detection for DOM characterisation. These include for example the use of tandem MS and hydrogen–deuterium exchange (H/D exchange) experiments.34,35,159–161 As most of the MS and MSn experiments are difficult to interpret, particularly identifying isobaric losses and the rearrangements that can occur during fragmentation, tools such as H/D exchange can help to distinguish functional groups such as hydroxyls from ethers or carbonyls.35,162 Additionally, due to the tendency of metal ions to form primarily even-m/z complexes within DOM, and in particular humic substances, Mg2+, Be2+, Cr3+ and Mn2+ have also been used to further simplify mass spectra.163–169 The resulting even m/z complexes stand out in the spectrum and can directly be characterised by molecular formulae assignments or tandem MS experiments.166,170–172
On the basis of previously developed HR-MS methods,42,156 Stenson et al., targeting humic substances within a Suwannee river fulvic acid standard,162 presented the separation of DOM isomers through RP-LC-HR-MS. Ions with identical formulae were found within different chromatographic fractions and analysed using the above H/D exchange protocol, providing for isotope differentiation. Structural isomers are different in the total number of exchangeable hydrogens and in the efficiency of each exchange. Spectra were obtained through ion molecule reaction, which avoids fragmentation during the ionisation process, rendering data interpretation more challenging due to the overlapping of fragmentation patterns.173 Spectra appear more resolved and less ambiguous, however ion molecule reaction is time consuming, requiring six minutes per scan. This means that only a small portion of sample can be processed. The investigated isomers not only had different retention times on the RP-LC chromatogram, but also reported different H/D exchange, which is evidence for the first isomeric fractionation of DOM.
In 2007, on the basis of previous experiments, Dittmar et al., applied RP-LC-MS to the mapping of terrestrially derived DOM along a river transect.22,174,175 RP-LC chromatograms showed an unresolved broad peak (mass range: 0.15 to 2 kDa), with no resolution of individual molecules, but demonstrating a peak maximum shifting towards increasing retention times for samples collected progressively further offshore. However, MS detection in this instance was able to further highlight how DOM also showed considerable variations due to photochemical modifications. Average MS spectra were used to ascertain that the estuary DOM displayed a bimodal mass distribution with an intensity-weighted average of 0.895 kDa, whereas 1.13 kDa was recorded in the case of terrigenous DOM. However, after irradiation, the latter more resembled the composition of estuary DOM and its intensity-weighted mass distribution decreased to 0.885 kDa, with a large fraction of UV-absorbing compounds not being detected after photodegradation.
In 2009, Reemtsma reviewed the issues encountered when coupling RP-LC to MS.176 Specifically, column overloading and signal to noise ratio issues were noted as limitations of the technique. As a solution to these problems, the author proposed the application of RP-LC fractionation followed by direct infusion to HR-MS, as already suggested by Koch et al.141 As previously mentioned, this work proposes the SEC pre-fractionation of DOM extracted using SPE according to Dittmar et al.57 The work underlines the complementarity of RP-LC and HR-MS, demonstrating that within each of the four fractions collected from RP-LC, approximately 400 to 900 different molecular formulae containing C, H and O were assigned. Single molecules were found to be fraction-specific, therefore allowing the technique to be usable in targeting potential biomarkers within DOM.
In a more recent study, Liu et al., used RP-LC with UV detection to obtain three to four fractions (according to the sample), which were first concentrated and subsequently injected into HR-MS for further characterisation.177 Within this work, only peaks with UV response at 254 nm were considered for collection, and MS and MS/MS analysis. MS spectra showed a peak distribution in the range of m/z 200–700, with peaks existing mainly at odd m/z and consisting of clusters of peaks at each nominal mass, which is consistent with earlier findings showing analogous m/z distributions.178,179 Minimally retained hydrophilic fractions typically included low MW compounds (<0.4 kDa), whereas most of the sample was characterised by hydrophobic components. This procedure reports the resolution of hundreds of compounds, however, as DOM was extracted through C18-functionalised silica SPE disks, the following chromatographic procedure represents a repetition of the extraction procedure, as an analogous stationary phase is used during RP-LC fractionation.22 For this reason, many authors have prescribed the direct analysis of SPE extracts (obtained from PS-DVB and C18-functionalised silica) via direct infusion HR-MS.43,152,155–157,177,180,181 Such a direct approach is less time consuming, can provide increased signal to noise ratios, and freedom from artefacts derived from the chromatographic procedure.182
However, in accepting the resolving power of MS detection, one has to also acknowledge potential biases originating from the ionisation source, which can be more efficient for certain classes of compounds over others, and the additional risk of in-source fragmentation.176,183 For example, ESI, which is the most popular ionisation source in DOM analysis, is particularly suited for ionic, high polarity compounds. Singly or multiply charged ions can be generated, and the number of charges retained by a particular analyte depends on factors such as molecular size, chemical composition, the solvent composition and the instrument parameters. In general, for molecules with mass lower than 2 kDa ESI generates singly, doubly, or, in some cases, triply charged ions, while for molecules with mass greater than 2 kDa, multiply charged ions are more common.22,75,118,162,182,184 Atmospheric pressure chemical ionisation (APCI) can also be found within DOM MS analysis, especially when attempting to target low polarity compounds. This technique generally provides singly charged ions: multiply charged species are not commonly observed as the ionisation process is more energetic if compared to ESI.159,185,186 Matrix-assisted laser desorption ionisation (MALDI) has also been used in DOM analysis but this soft ionisation technique mainly targets large molecules (up to 300 KDa) such as proteins and peptides, therefore not providing any information on the bulk of DOM. Thus currently there is no universal ionisation technique capable of unbiased ionisation of all of the classes of compounds within DOM. The ion source of choice commonly represents the best compromise in attempting to target the vast majority of DOM compounds. As already discussed by several authors, best approach is then to combine different HR-MS analysers, in order to complement the different kind of information that is delivered.39,187,188
2.1.3.1. Secondary interactions and choice of mobile phase. SEC has been widely used in the separation and fractionation of DOM and terrestrially-derived organic matter (i.e. humic and fulvic acids).191,192 Everett et al., used SEC to characterise freshwater DOM isolated by tangential flow UF (Table 4).193 The use of SEC on samples obtained using UF (1 kDa polysulfone membrane) proved the technique successfully isolated the >1 kDa fraction. However, this work also highlighted some of the limitations of SEC for DOM fractionation. Applying similar conditions to those proposed by Chin et al.,194 the SEC method used involved the addition of 0.1 M NaCl to the 2 mM phosphate buffer (pH 6.8) mobile phase to reduce secondary electrostatic interactions between the sample and the stationary phase. Chromatograms obtained under these conditions indicated several size fractions to be present within DOM samples, but these were very poorly resolved, presenting as a broad co-eluting peak. Interestingly, the authors did report that the presence of divalent cations within the DOM sample increased the observed MW distribution for DOM samples, which was lower following proton-exchange. This latter observation has obvious implications for the size fractionation of DOM following sample acidification.
Minor et al., employed SEC with a 100 mM phosphate buffer (pH 7) to analyse DOM samples extracted from UF (molecular weight cut-off: 1 KDa).64 Distinct variations were observed within apparent molecular size distributions from different samples, especially at high MW. High MW fractions were found to be rich in oligo- and polysaccharides containing aminosugars, deoxysugars, and methylated sugars, whereas the low MW portion was enriched in hexose containing oligosaccharides (Table 4). Schwede-Thomas et al., also used a NaCl containing mobile phase, similarly to Everett et al., however the phosphate buffer concentration was ten times higher.59,193 No size exclusion chromatograms were shown, however the authors observed MW distributions similar to those reported in previous works, and noted that terrestrially derived DOM possessed higher MW compared to their Antarctic counterparts.194,195
As underlined by Piccolo et al., high MW materials can sometimes be artefacts commonly observed within SEC separations of terrestrially-derived DOM.191,196 According to the authors, humic substances in solution result from the aggregation of heterogeneous moieties, which are held through hydrogen bonding and hydrophobic interactions. These can unpredictably interact with the stationary phase of the column in use, therefore rendering any measured MW distribution tightly dependent on the SEC column used. The authors underline that, due to the indefinite primary chemical structure of compounds such as humic substances, SEC can only provide approximate MW values, which resulted in the conclusion that SEC is more useful to compare changes in molecular sizes between different samples.
Pelekani et al., in their study comparing SEC with flow field – flow fractionation (FIFFF) for freshwater DOM size characterisation, also pointed out the significance of secondary solute–sorbent interactions in SEC of such samples.189 Using a series of carboxylated organic dyes as test solutes, significant evidence of both hydrophobic and electrostatic interactions were observed using a bonded silica gel SEC column, the latter of which were not eliminated through the use of a 0.1 M NaCl mobile phase. However, despite these limitations, reasonable agreement between the two independent size characterisation approaches for drinking water samples was achieved, providing validation of the technique for such applications.
Müller et al., compared two separate SEC columns for DOM fractionation (Superdex 75 HR10/30 and TSK HW-50 columns), each used with 25 mM phosphate buffer (pH 6.8), ionic strength 0.04 M, as mobile phase without the addition of NaCl (Table 4).197 The method provided a slightly improved separation of freshwater DOM, and enabled the collection of multiple fractions, which were then re-injected onto the SEC column. The re-injected fractions showed well defined Gaussian peaks of distinct elution volumes, which remained reproducible for periods of up to a week following fractionation. Both columns provided similar well defined fractions, which did support the hypothesis that molecular size was the dominant separation mechanism. However, collectively the peak area for the individual fractions was less than that recorded for the original sample, which suggested degree of irreversible adsorption of hydrophobic material.
Her et al., confirmed that significant ionic interactions occur in SEC when the ionic strength is low.198 At ionic strengths greater than 0.2, while such effects are suppressed, other secondary hydrophobic interactions remain. Aromatic species within DOM appear to be associated with most of the irreversible adsorption issues, with retention times shifts also observed. The columns evaluated within this study enabled the separation of species of size range 1–6 kDa (Biogel P6), 1–30 kDa (Protein Pak 125), and up to 5000 kDa (TSK 125). Given the uncertainty and variability of MW distributions within DOM, the most appropriate choice was found to be TSK 50S, as confirmed in a following publication.199 However, the type of stationary phase should also be considered. Biogel P6 is characterised by a polyacrylamide stationary phase, Protein PAK 125, by a silica-based stationary phase, and TSK-50S, by a hydroxylated organic stationary phase (Table 4). Both TSK 50S and Protein PAK 125 stationary phases are highly hydrophilic and therefore susceptible to hydrogen bonding interactions. This kind of secondary interaction can affect selectivity, causing hydrophilic compounds to be more retained, independently by their MW. On the other hand, a polyacrylamide stationary phase (Biogel P6) is more hydrophobic and for this reason, secondary effects from hydrogen bonding are less profound. The findings from Her et al., were also confirmed by Nissinen et al., who assessed that adsorption interactions and charge exclusion are altered by pH and ionic strength.200 Such observations led Her et al., to optimise their chromatographic method, and although peaks were not fully resolved in a subsequent study, DOM was separated into five fractions according to MW.199
The issue of secondary interactions has been reported in the majority of studies employing SEC to DOM characterisation (Table 4).201,202 According to Specht et al., secondary interactions take place regardless of whether the stationary phase is a polymer or silica based.203 Within this study, elution volumes obtained from two different columns were compared. The first column was a TSKHW50S, with a hydrophilic stationary phase obtained from the copolymerisation of ethylene glycol and methacrylate polymers, whereas the second a TSK G2000SW, with a bare silica stationary phase. Three categories of compounds were tested to understand the type of secondary interactions, namely amino acids, alcohols and carboxylic acids. Within these sets of experiments, performed using a phosphate buffer as the mobile phase (pH 6.8), both polymer and silica based columns were found to display hydrophobic interactions. Alcohols and monocarboxylic acids showed an increased elution volume which was proportional to the number of carbon atoms, whereas aromatic compounds were found to be strongly retained by both types of stationary phases.
Similar considerations were noted in the work of Reemtsma et al., who added MeOH to their SEC eluent (80/20 NH4HCO3/MeOH) to separate the fulvic and humic acid fractions of DOM (Table 4).204 Ammonium bicarbonate was used as the buffer, to decrease the secondary electrostatic interactions, here being sufficiently volatile, to facilitate direct coupling of the SEC column to ESI-MS detection.
Persson et al., compared MW distributions obtained through SEC-UV and RP-LC-ESI-MS. Lower MW molecules with exposed carboxylic groups were more readily ionised in MS, whereas, as previously mentioned by Her et al., higher MW compounds with greater specific absorbance in the UV (280 and 254 nm) appeared to be over-represented in SEC-UV.198,205 Further fractionation of DOM by using two preparative scale columns connected in series (and a NaCH3CO2 containing mobile phase), provided eight size-based portions of DOM.206,207 Pyrolysis-GC-MS analysis of the so-acquired fractions isolated single compounds. In a recent study by Woods et al., the coupling of SEC to NMR was reported (using an 0.1 M NaCl and 0.03 M NH4Cl mobile phase, pH 11) (Table 4).47,132 Two 7.8 × 300 mm columns (size exclusion limits = 1–80 kDa for the first column and 0.5 to 10 kDa for the second) were used in series in order to obtain three fractions of DOM according to size, prior to characterisation using NMR. The first fraction was enriched in carbohydrate and aromatic-like structures, whilst the second was representative of CRAM, and the third of MDLT. Even though the chromatography in this case could be improved, for the first time the authors demonstrated the partial separation of CRAM and MDLT. This was also the first SEC method reported applying a highly basic mobile phase to avoid any sample protonation. Due to the aforementioned issues regarding secondary interactions between sample and stationary phase, SEC is here only used as a means to size-fractionate DOM. Concerns regarding accuracy of any MW prediction meant no specific conclusions on DOM molecular weights were drawn.
Kawasaki et al., also used a phosphate buffer mobile phase (pH 6.8), with an OH-functionalised stationary phase (Table 4).208 The method used a smaller particle size (5 μm) column with a reduced injection volume (100 μL, representing a 20-fold decrease if compared to the study from Her et al.199). The optimised separation provided the fractionation of DOM within 35 minutes, and the authors reported higher sensitivities compared to previously reported methods.
On the basis of the methodology reported within Peuravuori et al., Romera-Castillo et al., further explored the fractionation of DOM and its variations according to pH.206,209 This study again confirmed the presence of supramolecular structures characterised by assemblies of small molecules with analogous fluorescence properties. After obtaining eight SEC fractions from DOM, fluorescence studies showed most of the molecules along a MW continuum, indicating similar nature, wide size distribution and a maximum fluorescence signal within the 0.18 to 2 kDa range.
To investigate the effects of ionic strength (buffer concentration) and pH of the mobile phase, Sanchez-Gonzalez et al.,107 investigated an ammonium sulphate/ammonium dihydrogenphosphate buffer (pH = 6.5) at increasing concentrations (5.0, 25, 50 and 100 mM). Improved fractionation of DOM was obtained at lower buffer concentrations (25 mM), while when higher buffer concentrations were used, the compounds appeared to be more retained, probably due to increased hydrophobic interactions. However, when different pH was tested, within the range 6.0 to 7.5, DOM fractionation was not dramatically affected (Fig. 5).
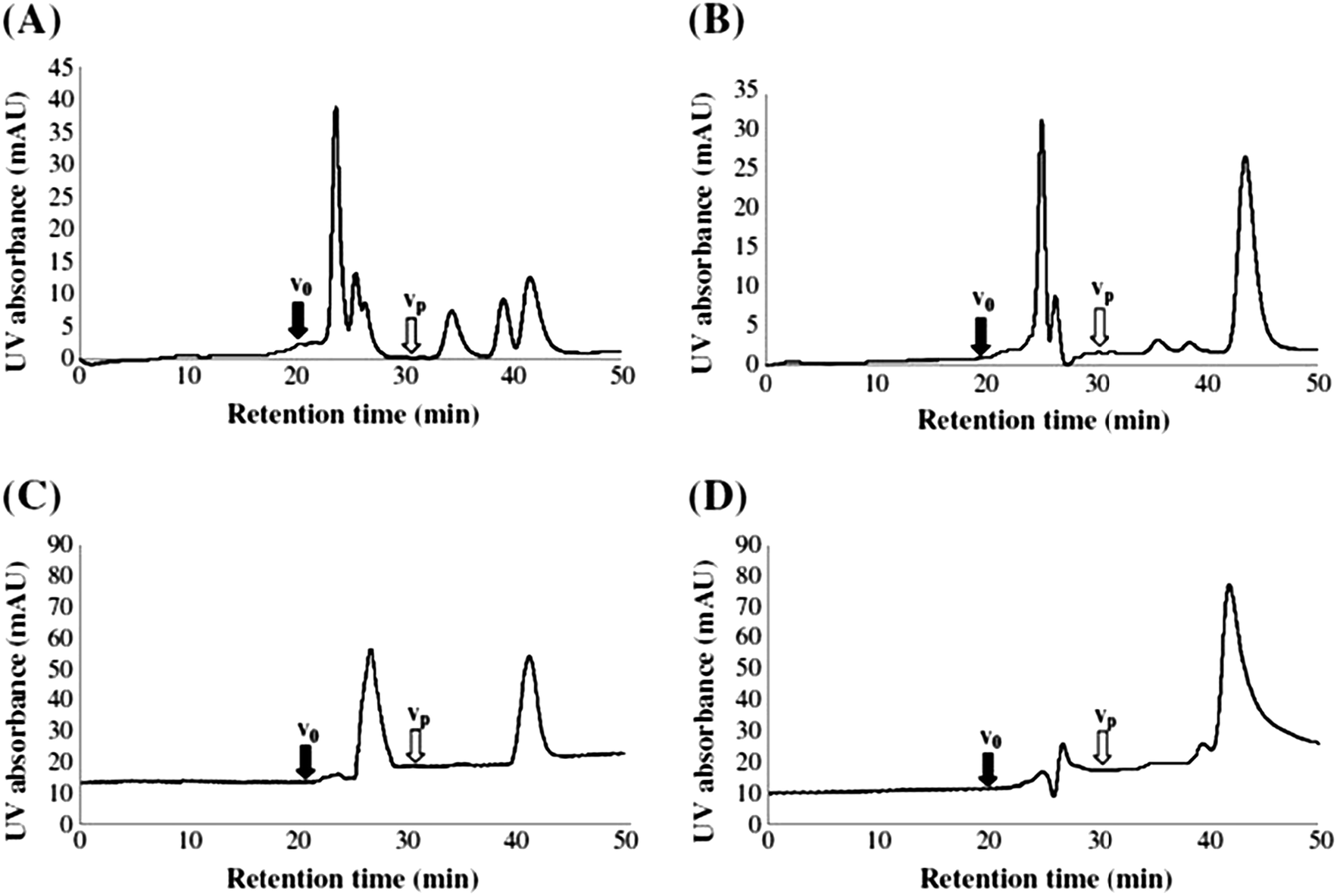 | ||
| Fig. 5 SEC-UV chromatograms for marine DOM eluted with alkaline methanol (pH 10) using ammonium sulphate/ammonium dihydrogenphosphate (pH 6.5 at the following ratios: 5.0 mM/5.0 mM (A), 25 mM/25 mM (B), 50 mM/50 mM (C), and 100 mM/100 mM (D)). V0 and Vp are the void volume and the permeation volume. Reproduced with permission from Sanchez-Gonzalez et al.107 | ||
In 2012, two separate LC × LC approaches were explored by Duarte et al., providing new information on MW distributions of humic and fulvic acids from the International Humic Substances Society.210 Within the first method, a C18-functionalised silica column (4.6 × 100 mm, 5 μm particle size) was used in isocratic mode (20% MeCN in water), prior to a second dimension SEC separation (polyhydroxymethacrylate copolymer stationary phase, 8 × 250 mm 10 μm particle size), also in isocratic mode (11% MeCN in 20 mM NH4HCO3, pH 8.0). In the second approach, the first dimension comprised an alkyl diol functionalised mixed mode HILIC column (4.6 × 100 mm, 5 μm particle size) operating in reversed-phase mode (10% MeCN in 20 mM CH3COONH4 at pH 6.0). As within the RP-LC × SEC method, SEC in isocratic mode (11% MeCN in 20 mM NH4HCO3, pH 8.0) was also used as second chromatographic dimension. Three detectors were used in both approaches: UV (254 nm), fluorescence (excitation: 240 nm, emission 450 nm) and ELSD. Both methods reported comparable results, with 2D chromatograms still showing fractions not completely resolved. However, those eluting at higher retention times within the second dimension seemed to be related to more hydrophobic moieties. The authors also underline the importance of method optimisation (i.e. mobile phase compatibility, modulation period and separation time), and found that, within SEC, MeCN contents higher than 20% provided poorer resolution and a move towards higher retention times.
2.1.3.2. The choice of SEC calibration standards. Correct calibration standards for MW determinations using SEC are critical. However, in the specific case of DOM, as a complex mixture of thousands of unknown molecules, it is clearly very challenging to select the appropriate standards. As already mentioned, the difficulty in determining precise MW distributions is also related to the type of stationary phase, as secondary interactions with the sample can occur. Therefore, Conte et al., point that the molecular weights determined by SEC should be regarded as relative to the system being used (i.e. type of sample and employed chromatographic conditions) rather than absolute values.191
Protein-based standards (up to approximately 80 kDa) were used in the study from Nissinen et al., whereas both proteins and polysaccharides were used in the work of Minor et al.64,200 However, each of these calibration standards only represent one of the many classes of compounds within DOM, and for this reason can be considered as non-representative of the whole organic mixture. When determining MW from different SEC DOM fractions, Minor et al., prepared two calibration curves, one obtained by using the protein-based standard and a second by using the polysaccharide-based standard. However, considerable variations were observed. For example, for the highest MW fraction, a MW of 660 kDa was estimated when using the calibration curve from the protein standard, as compared to 200 kDa in the case of the polysaccharide standard.64
Polystyrene and sulfonate standards from 1 to 35 kDa are the most widely used SEC calibrants.132,190,198,205,208–213 Within some studies, other side compounds such as glycerol, acetone, chlorobenzoic acid, polyethylene glycol, blue dextran and salicylic acid are added to extend the MW range.61,190,198,205,208 Although once again, the use of these kind of standards, given the variety of material within DOM, represents a compromise. Similar considerations can be applied to the work from Yan et al., where the selected calibrant was poly(ethylene glycol) (MW range: ∼0.1 kDa to ∼50 kDa),214 and although concluding an apparent DOM MW range from 3 to 16 kDa, also reported measurement errors ranging from ±10% to ±30%.
In order to overcome this issue, Peuravuori et al., used widespread classes of compounds with MW from 0.265 kDa to 169 kDa (pyridoxal-5-phosphate, a guaiacylglycerol-β-guaiacyl ether derivative, sucrose, sodium deoxycholate, sodium taurocholate, bierol, trypan blue, cyanocobalamin, tannic acid, polystyrene-sulfonates, polyethylene glycol, ribonuclease A, chymotrypsinogen A, ovalbumin, albumin and γ-globulin).206,207 These compounds resemble many classes of molecules present in DOM, however, within this extensive list of compounds, no terrestrially-derived compounds are present.
A number of studies have used humic and fulvic acid standards from the International Humic Substances Society with SEC.58,204,215–217 According to Huber et al. and Averett et al., nominal average MWs for these class of compounds are 0.711 and 1.066 kDa.58,217 However, due to the nature of humic and fulvic substances, which are themselves a very complex mixture of compounds, and the aforementioned secondary interactions occurring in SEC, the resulting MW estimations are only indicative. Despite this, the aromatic and polycarboxylated nature of humic and fulvic acids, which resemble some bulk properties of DOM, together with the standards proposed by Peuravuori et al., could be the most suitable and comprehensive model mixtures to aid in the estimation of DOM MW ranges.206,207
The first application of HILIC to the fractionation of DOM was reported by Woods et al., who collected fractions from their HILIC based separations for molecular characterisation using HR-NMR.47,133,134 In their initial studies, the group employed a diol functionalised silica column to generate up to 80 DOM fractions.133 Considerable co-elution between fractions was evident, however with greater retention, increasingly hydrophilic solutes were detected. Typical CRAM and MDLT-like components were eluted in decreasing polarity order along the entire chromatogram, demonstrating a wide diversity of chemical–physical properties within these classifications. Carbohydrates were found to elute towards the end of the chromatogram. Fig. 6 shows the HILIC separations of a freshwater DOM sample (Suwannee River) recorded using both UV absorbance DAD and fluorescence detection.
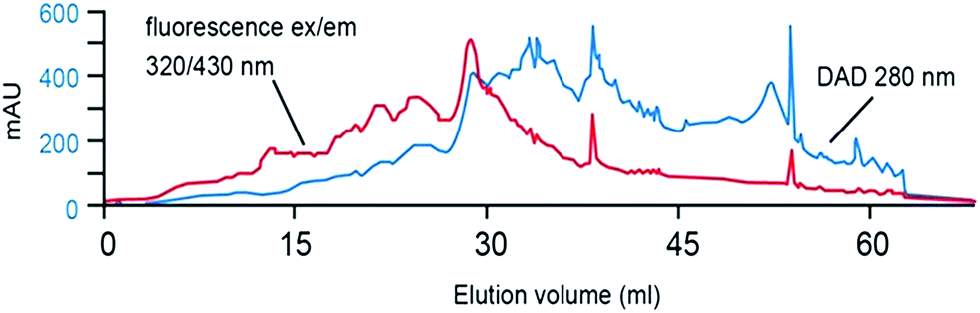 | ||
| Fig. 6 HILIC-UV separation for Suwannee River DOM. Reproduced with permission from Woods et al.133 | ||
More recently, in order to further improve chromatographic resolution, Woods et al., employed a two-dimensional chromatographic approach (HILIC × HILIC) coupled with NMR134 for the characterisation of fractionated freshwater DOM collected after isolation with ultrafiltration. The column employed as the first chromatographic dimension was the same as that used in previous mono-dimensional experiments,133 however, in the second dimension a normal-phase bare silica column was applied. Although not completely orthogonal in selectivity, some improvement in DOM fractionation appears to have been achieved (no two-dimensional chromatograms were shown), as less complex NMR spectra for each fraction were reported.
Kaiser et al., developed both RP-LC and IEC methods for the quantification of amino acids, amino- and neutral sugars in oceanic POM, high MW DOM (1–100 nm), and low MW DOM (<1 nm), obtained from varying depths.227 The developed IEC method used a CarboPac-PA1 anion-exchange column for the separation of amino- and neutral sugars under isocratic conditions (see Table 4). The study reported the concentrations of these small biomolecules fell sharply with depth, accounting for 55% of organic carbon in surface POM, but only 2% of organic carbon in low MW DOM in deep water, suggesting an upper ocean source and rapid microbial turnover.
Repeta and Aluwihare isolated monosaccharides from high MW DOM via acid hydrolysis, and desalting using a Biorex 5 anion exchange resin, with further fractionation of the collected neutral sugars using silver ion chromatography (see details below). These fractions were then further separated using two amino functionalised columns (Hamilton PXP-700) connected in series, for collection of individual sugar peaks for off-line compound-specific radiocarbon analysis.229
More recently, Sandron et al.,230 reported the use of IEC-PAD to investigate dissolved neutral sugars and their microbial conversion in both artificially prepared and naturally occurring freshwater and seawater DOM. Using a CarboPac-PA1 column and gradient elution with a KOH eluent, chromatograms for each sample, both natural and artificial, showed obvious similarities, notable a large retained composite peak eluting immediately before the well separated neutral sugars, several of which were readily detectable within the natural DOM samples (Fig. 7). The IEC based separation was used to generate fractions from the artificial DOM sample, prior to their further separation and analysis using RP-LC with HR-MS detection, as part of an off-line multi-dimensional chromatographic approach (Table 4). Fig. 7 shows the typical IEC chromatograms obtained for seawater DOM samples collected at 10 and 60 m depths.230
 | ||
| Fig. 7 IEC-PAD chromatograms obtained for (a) seawater and (b) freshwater DOM samples, overlaid with standard chromatograms for selected sugars. Reproduced with permission from Sandron et al.230 | ||
Specifically focussing on humic substances, Wu et al., reported a study comparing IMAC columns of differing metal form, including copper, nickel, cobalt and cadmium, for selective ligand retention (varying also eluent pH and ionic strength).234 The copper based method was reported to provide the greatest retention and humic substances binding capacity, which supports the common application of copper as the coordinating metal in most applications of IMAC in this area.
Silver ion or argentation chromatography, a close analogue of IMAC, is generally applied to the separation of unsaturated organic compounds, based upon the ability to form a charge-transfer type complex with immobilised silver ions. The unsaturated compound acts as an electron donor and the silver ion as an electron acceptor,235–240 with the stability of the complex increasing with the number of double bonds. Silver ion chromatography is commonly employed in the separation of apolar compounds such as lipid-like materials, and hexane-based mobile phases are employed, with the eluent strength commonly increased using MeCN.240 However, in DOM characterisation, silver ion chromatography has been applied by several groups for sample pre-fractionation in the study of methylated and neutral sugars.
Panagiotopoulos et al., used preparative silver ion chromatography as a fractionation method for methylated sugars in acid hydrolysed high MW DOM (seawater), prior to fractional analysis using GC-MS (Table 4).241,242 In this application, the positive charge on silver ions interacts with the partial negative charge on sugar hydroxyl groups, therefore enabling the retention of mono- and di-methylated sugars. Fractionation was carried out using a Supelcogel Ag column with a water mobile phase. Using the combined approach, up to 50 novel sugars were identified, and a trend observed, in which surface waters were enriched in mono- and di-methylated sugars, representing the 64% of the total methylated compounds, whereas deep water samples where richer in mono-methylated 6-deoxy sugars (42% of the total methylated compounds), being derived from predominantly bacteria sources.
As mentioned above, Repeta and Aluwihare isolated monosaccharides from high MW DOM via acid hydrolysis and desalination.229 The carbohydrate fraction was obtained using silver ion chromatography with refractive index (RI) detection, using two coupled sulfonated PS-DVB cation exchange columns in Ag+ form (0.8 cm I.D. × 30 cm L). Fig. 8A shows the resultant separation, which corresponds closely to that reported by Panagiotopoulos et al., for their similar high MW seawater derived DOM.241,242 As shown the selective retention of the carbohydrate fractions (eluting between 10–20 min) using silver ion chromatography is very clear.
 | ||
| Fig. 8 (a) Separation of neutral sugars from seawater DOM by IEC after acid hydrolysis and (b) by using a polymeric amino column before radiocarbon analyses. Reproduced with permission from Repeta et al.229 | ||
In their preliminary study, Sandron et al., recently reported the use of HPCCC in normal-phase mode in an attempt to fractionate DOM (Table 4).246 The developed separation provided five fractions which were further analysed by GC-MS or RP-LC with UV detection. In both cases the resulting chromatograms showed differences, supporting the fact that DOM was indeed fractionated into different classes of compounds. Although no HR-MS characterisation was reported, GC-MS fragmentation suggested an analogous molecular skeleton for the vast majority of the fractionated compounds. Complementary analysis via RP-LC with UV detection isolated a number of polar species, which were not detected by GC-MS.
From the above Sections detailing applications of liquid chromatographic techniques to the partial separation and/or fractionation of DOM, some summary points can be made. Both RP-LC and SEC have been applied extensively for such purposes, each providing an initial means of DOM fractionation, albeit based upon differing, and rather general selectivity. In both instances resolution is rather limited due to the complexity of the sample, and in many cases a secondary separation step (e.g. RP-LC fractionation followed by SEC separation, or vice versa) coupled to MS or HR-MS is applied. Clearly, the advantage of MS detection, especially HR-MS is that the technique and data obtained is complementary to the chromatographic separation. LC has the potential to separate isomers, reduces complexity and thus ion-suppression in the ESI source, and makes more of the DOM sample amenable to MS analysis. MS itself provides molecular formulas and confirms changes in composition between LC fractions and/or DOM samples.
Given the general selectivity of both RP-LC and SEC, the application of more specific modes of liquid chromatographic methods for targeted analysis, notably HILIC, IEC, IMAC and silver ion chromatography have been explored, often applied to pretreated or pre-fractionated DOM. With these methods the potential to better isolate specific classes of compounds (e.g. lipids, sugars etc.), and in some instances individual species exists.
2.2. Gas chromatography
Gas chromatography (GC) is only applicable to the separation of compounds that are volatile, or those which can be readily derivatised to volatile species. Prior to separation, compounds containing functional groups with active hydrogen atoms, such as –COOH, –OH, –NH, and –SH, may need to be protected as they tend to form intermolecular hydrogen bonds that can reduce volatility and interact adversely with many GC stationary phases. For complex mixtures such as DOM, with its diverse range of compound polarity, selection of an appropriate stationary phase is difficult, especially if a mono-dimensional GC approach is used. Another complicating issue in the GC analysis of DOM is that many compounds are thermally labile, meaning mode of injection, and injector and column temperature are important parameters to control.The degradation and derivatisation reactions employed in the GC analysis of DOM fall into three general categories, namely pyrolysis, alkylation, and silylation. Pyrolysis is essentially the cleavage of chemical bonds within large macromolecular structures into smaller and more volatile fragments by the application of heat. The limitation of this technique is the unintentional decomposition of thermally sensitive classes of molecules.247,248 Alkylation reactions replace active hydrogens from an organic acid or amine with an aliphatic group. This technique is used to transform carboxylic acids into esters, which are more volatile. A common reagent is tetramethylammonium hydroxide (TMAH), which allows the production of ethers, secondary amines and esters. Silylation replaces active hydrogens from acids, alcohols, thiols, amines, amides, enolisable ketones and aldehydes with a trimethylsilyl group, although there are also other silyl derivatives. Silylation reagents themselves (e.g. bis-trimethylsilyl trifluoroacetamide (BSTFA)) and silyl derivatives are unstable and must be protected from moisture.
The analysis of DOM by GC is either targeted to certain classes of molecules (i.e. lipids, lignin monomers), or non-targeted, in an attempt to provide a generic screening of the entire organic pool (Table 5). In the majority of published methods, the stationary phases employed have been relatively non-polar (based upon 5% phenyl/95% polydimethylsiloxane, e.g. DB5, VF5MS, RTX5MS, BPX5). More selective stationary phases have been generally avoided, due to the complicated range of chemical functionalities within DOM, which would see many compounds irreversibly adsorbed.
| Target compoundsa | Water source and isolation methodb | Sample treatmentc | Columnd | Temperature gradient (oC)e | Detector(s)f | Ref. |
|---|---|---|---|---|---|---|
| a Compounds targeted during the analysis. b Abbreviations as in Scheme 1 and Tables 1–3. c Derivatisation, pyrolysis or oxidation technique employed before analysis; CuO: copper(II) oxide; abbreviations as in Scheme 1. d Employed GC column. e Temperature gradient or isothermal applied. f AMS: microscale accelerator mass spectrometry, other abbreviations as in Scheme 1 and Tables 1–3. | ||||||
| DOM | Freshwater, SPE, UF | Pyrolysis | DB5 30 m 0.32 mm i.d. 0.25 mm film thickness | 40–250 | MS | 318 |
| Fatty acids, lignin | Freshwater, UF | TMAH | DB5 30 m or 60 m 0.32 mm i.d. 0.25 mm film thickness | 50–300 | MS | 257 |
| DOM | Freshwater, UF | TMAH | DB5 30 m 0.25 mm i.d. 0.25 mm film thickness | 60–280 | FID, MS | 254 |
| DOM | Freshwater, freeze drying | Pyrolysis | BPX 5 60 m 0.32 mm i.d. 1.0 μm film thickness | 36–300 | Elemental analysis, MS-IRMS | 260 |
| Terrigenous DOM (lignin) | Freshwater seawater, SPE, UF | CuO | DB5 30 m 0.32 mm i.d. 0.25 μm film thickness | 100–270 | FID, MS | 271 |
| Phenols | Freshwater, SPME | — | DB5.MS 30 m 0.32 mm i.d. 0.30 μm film thickness | 40–250 | TOC, MS | 109 |
| Sugars, lipids | Seawater, UF | BSTFA | DB5 30 m 0.32 mm i.d. 0.25 mm film thickness and DB5 30 m 0.25 mm i.d. 0.20 μm film thickness | 55–320 and 150–250 | AMS, FID, NMR | 261 |
| Lipids | Freshwater, RO, freeze-drying | TMAH | BPX 5 25 m 0.32 mm i.d. 0.25 μm film thickness | 150–280 | MS | 264 |
| DOM | Freshwater, SPE | TMAH | DB5 30 m 0.32 mm i.d. 1 μm film thickness | 60–280 | UV, MS, NMR | 265 |
| DOM | Freshwater, freeze drying | Pyrolysis | DB5 30 m 0.32 mm i.d. 0.2 μm film thickness | 35–280 | MS | 266 |
| DOM | Freshwater, freeze-drying | TMAH | RTX5MS 30 m 0.25 mm i.d. 0.1 μm film thickness | 40–310 | MS | 267 |
| DOM | Freshwater, SPE | TMAH | RTX5SilMS 30 m 0.25 mm i.d 0.5 mm film thickness | 50–300 | MS, NMR | 269 |
| Sugars, neutral lipids | Freshwater, UF | BSTFA, TMAH | DB5 30 m 0.25 mm i.d. 0.25 μm film thickness | 40–310 | Fluorescence, MS, NMR, TOC | 268 |
| DOM | Freshwater, SEC | TMAH, TMAAc | NB1701 50 m 0.32 mm i.d. 0.25 μm film thickness | 30–220 | MS | 206 |
| Sugars, lipids | Seawater, UF | NaBH4, acetylation, periodate over-oxidation, BSTFA | Supelco SP-2330 30 m 0.25 mm i.d. 0.2 μm film thickness and DB-XLB 60 m 0.25 mm i.d. 0.25 μm film thickness | 55–240 and 50–320 | FID, UV, NMR | 270 |
| Terrigenous DOM (lignin) | Freshwater, SPE | CuO, BSTFA | VF 5MS 30 m 0.25 mm i.d. 0.25 μm film thickness | 65–300 | Elemental analysis, MS-MS | 272 |
| DOM | Freshwater, oxidation to CO2 | Persulfate, heat | Poraplot Q 25 m 0.32 mm i.d. 5 μm film thickness | 60 (constant) | TOC, IR | 273 |
| Sugars | Seawater, UF | NaBH4, acetylation | DB5 30 m 0.25 mm i.d. 0.20 μm film thickness | 90–230 | MS | 242 |
| DOM | Freshwater, SPE | HPCCC pre-fractionation | EC-WAX 15 m, 0.53 mm i.d., 1.2 μm film thickness | 50–300 | UV, MS | 246 |
| Polycyclic aromatic hydrocarbons | Humic acid standard manually dissolved in water, SPME | — | HP-5MS 30 m 0.25 mm i.d. 0.25 μm film thickness | 60–310 | TOC, fluorescence, MS | 110 |
As mentioned in an early review by Aiken et al.,249 one of the first attempts to use GC in the analysis of DOM (freshwater) was reported by Stainton.250 This method reported a versatile yet simple extraction approach prior to GC analysis. Volatile species evolved from acidified water samples were collected via a gas stripping procedure with helium flow, the latter being used as carrier to deliver the sample to GC. The extraction efficiency of the method was highly dependent on the stripping time and on the nature of the sample, and applicable only to the highly volatile DOM fraction. Other than predictable co-elution issues, a limitation of the procedure described was the use of polypropylene (PPL) syringes during the gas stripping stage, as these can be a source of potential contamination.251,252
Due to the complexity of DOM and extensive co-elution, especially in the absence of sample derivatisation, Schulten et al., considered two approaches, namely pyrolysis-field ionisation MS and Curie-point pyrolysis GC-MS.253 A 30 m DB5 capillary column was used, characterised as a nonpolar stationary phase, targeting the separation of the mid to low polarity fraction of DOM. The aim of this study was to identify series of marker signals within freshwater DOM, which could allow inter-sample comparison. The obtained GC chromatogram showed fourteen prominent peaks and series of co-eluting compounds ranging from approximately m/z 200 to 500. Despite the authors highlighting the need for further method development (i.e. column selection, pyrolysis and MS conditions), classes of compounds such as benzenes (42 identified structures), phenols (26) and furans (35) were identified, which were further confirmed by following studies.254,255 Additionally, a wide range of ubiquitous substituted aromatic structures were found, which could not be identified. For this reason, Schulten et al., emphasise the need of complementary analysis such as isotope ratio measurements and HR-MS detection.
In a more targeted approach, Mannino et al., used GC-MS (mass range 0.05–0.6 kDa) to determine lignin phenols and lipids, following extraction using an organic solvent (CH2Cl2) and TMAH derivatisation (Table 5).256,257 The extraction technique used by Mannino et al., aimed to isolate the targeted classes of molecules, however, extraction of other DOM constituents, such as complex sterol-like materials, e.g. CRAM and terpenoids, was also evident, leading to substantial co-elution, particularly within the first and middle part of the chromatogram. However, using this method, the majority of lipids were extracted from river estuary samples, including fatty acids, with chain length typically ranging from 9 to 13 carbon atoms. Concentrations of lignin-like material were found to be higher in estuary regions than samples from other coastal regions, with terrestrially-derived DOM (i.e. humic and fulvic-like substances) also following an analogous trend. The study confirmed terrigenous DOM is enriched in lignin-like materials, whereas lipid-like materials, consistent with previous studies30 were found to have concentrations up to 1 μg mL−1.30,258,259
Kracht et al. applied pyrolysis to freeze-dried DOM.260 This study was the first to employ a combined form of detection involving elemental analysis and pyrolysis gas chromatography mass spectrometry-isotope ratio mass spectrometry (Py-GC/MS-IRMS), in order to correlate mass spectra to isotopic ratios and derive more comprehensive information on the origin of DOM (Table 5). Although using only one form of sample treatment, the authors actually proposed the treatment of the sample with different derivatisation techniques simultaneously. This approach could be used to detect other volatile compounds present in DOM, possibly converted as silyl derivatives, to compare their elution profile and detector responses to those obtained after thermal pyrolysis.
A limitation of the method developed by Kracht et al., relates to the extraction method employed. Although freeze-drying can provide a potentially uncontaminated extract (e.g. free from plastic-derived materials or artefacts), it is time consuming and requires additional sample desalting if seawater samples are processed. Freeze-drying as a process is also solute dependent, with every class of compound having different freeze-drying requirements, making optimisation difficult, leading to inconsistent dryness across the sample, reduced stability or rehydration.
Ohlenbusch et al., applied SPME with GC-MS to investigate the interaction between DOM and ten halogenated phenols.109 As with previous studies (Table 5), a DB5.MS column was used. This was chosen for its apolar and aromatic stationary phase (as targeted compounds were phenols). This study revealed the sorption of these compounds to DOM, which was directly proportional to the hydrophobicity of the phenol and inversely proportional to a pH increase. Furthermore, the authors were able to quantify the phenols by using selected ion monitoring mode when processing MS spectra collected in full scan mode.
Aluwihare et al., performed a targeted analysis on two different classes of compounds within DOM, lipids and monosaccharaides, which were separated and identified by GC, with flame ionisation detection (FID) and off-line NMR.261 Prior to GC-FID, DOM samples were liquid–liquid extracted with dichloromethane and derivatised using BSTFA to detect lipids, whereas acid hydrolysis was used for monosaccharides.262,263 In the case of carbohydrates, rhamnose, fucose, arabinose, xylose, mannose, galactose and glucose were identified. Unlike the studies from Mannino et al., free lipids were discarded and only ether- or ester-bound lipids targeted.257,259 It was found that hydrolysable lipids only represented the 2% of the total DOM. The presence of lipid and carbohydrate fractions within DOM was confirmed by means of 1H NMR, which also identified the presence of resonances corresponding to aromatic and acetate protons.
Lipids were also investigated by Jandl et al., both from seawater and freshwater DOM. The method comprised an extraction in CH2Cl2/acetone and TMAH derivatisation (Table 5).264 GC-MS data were compared to that available in databanks, confirming the presence of a C14:0 to C28:0n-alkyl fatty acid series. The highest concentration was observed by employing RO extraction on freshwater (river) samples (309.3 μg g−1), whereas in freeze-dried brown lake water the concentration was nearly halved (180.6 μg g−1). This finding not only further highlights the dependence of DOM upon its source, but also the different efficiencies from various extraction methods in use.
Weishaar et al., combined the information from 13C NMR, UV absorption at 254 nm and TMAH derivatised GC-MS, to focus upon the aromatic portion of DOM (Table 5).265 Within this study, both electron ionisation and chemical ionisation were used in order to comprehensively screen separated DOM. As already seen, the combination of on-line MS detection with off-line NMR spectra provided a more complete picture of the different classes of compounds within the DOM sample (i.e. proteins, ketones, chlorophyll pigments and aromatics).
Page et al., reported the treatment of a seawater sample with alum in order to remove color and turbidity prior to DOM extraction.266 The filtered material was then freeze-dried and characterised using pyrolysis GC-MS (Table 5), delivering semi-quantitative information on the components of DOM sensitive to this type of sample treatment. The alum-extracted samples were found to be rich in alkylbenzenes, alkylphenols and polycyclic hydrocarbons, whereas the fraction recalcitrant to alum treatment was characterised by the presence of polysaccharide-derived molecules. In the specific case of nitrogen containing compounds, alum treatment seemed not to affect the relative abundance of the detected compounds.
In a similar study, Frazier et al., were able to quantify the main compound classes discovered in the work of Page et al., (i.e. fatty acids, carbohydrates and lignin precursors) through TMAH derivatised GC-MS.267 The significance of this work arises from the potential to understand the variations these compounds can undergo within different water sources. For example, the chromatograms from four analysed samples showed analogous distributions for carbohydrate-derived compounds, whereas lignin-derived materials were found to be source-dependent and related to indigenous vegetation and local in-stream processes. Fatty acid methyl esters of microbial and plant origins were the most abundant aliphatic moieties. These were classified into low MW (number of carbons from 8 to 10 and no unsaturations) and high MW (number of carbons from 12 to 18 and no unsaturations). Their proportion showed differences in distribution depending upon the water source.
Multiple detection approaches were also employed by Maie et al.,268 and Templier et al.,269 who compared NMR data to that obtained using TMAH GC-MS (Table 5). Despite the possible contamination due to the fractionation method, the novelty of the Templier et al. study was based upon the combined use of different XAD™ resins to extract DOM, leading to the separation of two fractions with different polarity. This technique simplified the GC-MS chromatograms to an extent that, even if with low intensity, singly resolved peaks were detected (Fig. 9). The DOM sample was also characterised by the presence of large, late-eluting broad ‘humps’ of unresolved compounds. This unresolved portion of the chromatogram therefore needs to be separated by alternative chromatographic techniques, or via a multidimensional chromatography approach. NMR analysis was also improved by the initial DOM fractionation, and even though extensive spectral overlap was still evident, it was possible to recognise individual well defined classes of compounds.
 | ||
| Fig. 9 Pyrolysis GC-MS of (a) hydrophobic acid fraction and (b) transphilic acid fraction of freshwater DOM. Reproduced with permission from Templier et al.269 | ||
Quan et al., combined two different GC methods to investigate monosaccharides and lipids contained within DOM, and combined their findings with monodimensional NMR and UV spectroscopy.270 The method developed in 2002 by Aluwihare et al.,261 was employed in the determination of monosaccharides, whereas periodate oxidation was employed in the determination of lipids. After the oxidation reaction was complete, the lipids were extracted with CD2Cl2 and persylated by BSTFA. Even though the authors underlined the need for more reproducible and precise procedures, the periodate oxidation provided evidence for a carbohydrate fraction which was compositionally different from those analysed according to the method developed by Aluwihare et al. This fraction proved to be rich in both methyl and amino sugars, which seem to comprise 15% of the total carbohydrate content in the sample.
In an attempt to improve resolution, Peuravuori et al., employed a combined chromatographic approach, (LC and subsequently GC), in order to fractionate and then characterise DOM (Fig. 10).206 The DOM sample was firstly separated into eight fractions using SEC, according to decreasing MW, and then subsequently analysed using GC-MS using two alkylating reagents, namely TMAH, to reveal both esterified and free carboxylic acids, and tetramethylammonium acetate (TMAAc), to determine free carboxylic acids (Table 5). TMAH and TMAAc-treated DOM fractions obtained after SEC showed fraction to fraction carryover. However, up to 310 degradation products were detected, of which 185 were identified. These were classified in aromatics (mainly characterised by methyl derivatives of phenols, alkylphenols and phenolic acids) and aliphatics (mainly methyl esters of mono- and dicarboxylic acids). Other generated compounds were furans, cyclopentenones and nitrogen and sulfur-containing organic compounds.
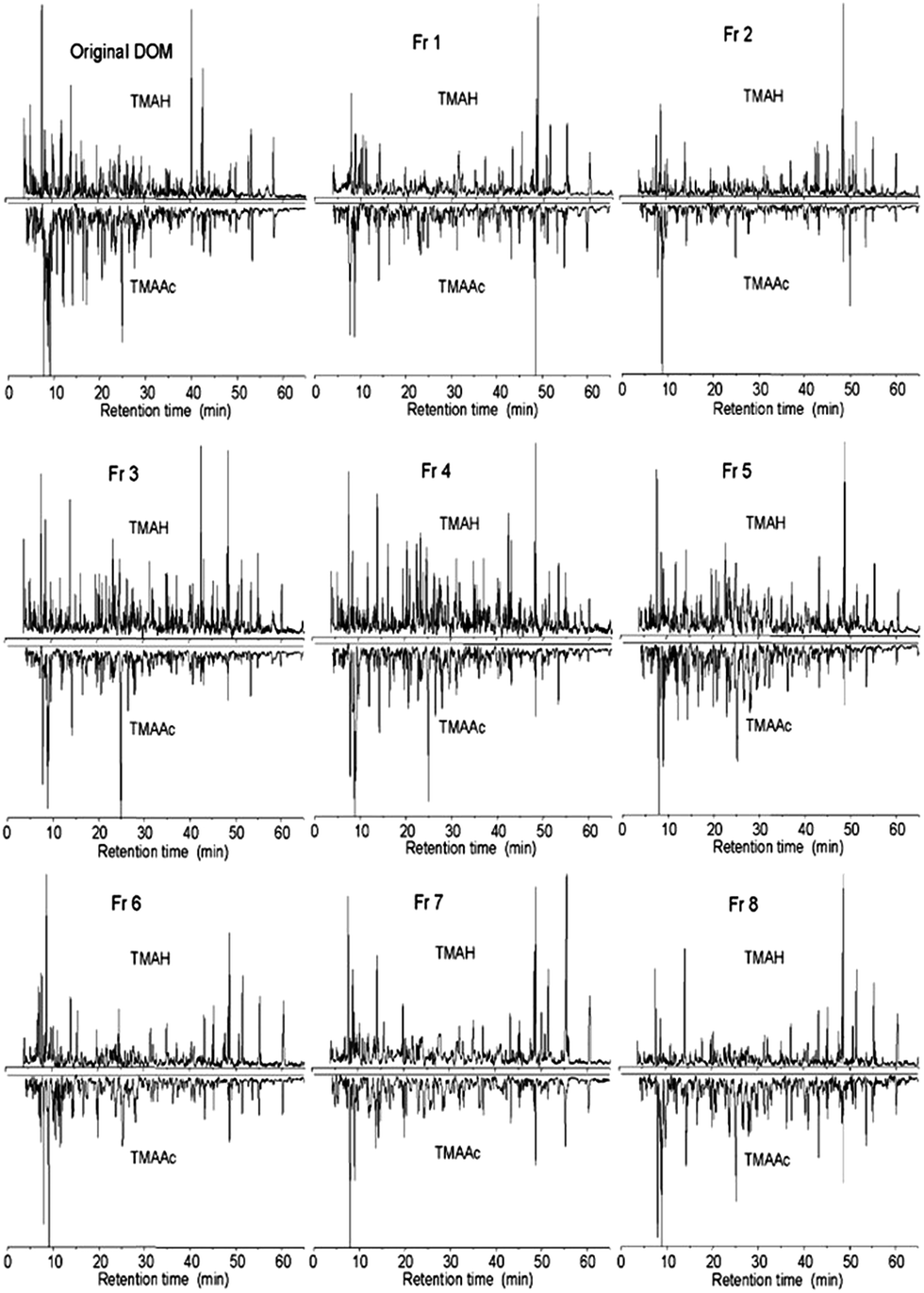 | ||
| Fig. 10 GC-MS chromatograms for TMAH- and TMAAc-treated freshwater DOM which was prefractionated through preparative SEC. Reproduced with permission from Peuravuori et al.206 | ||
Due to the importance of biologically derived compounds in marine ecosystems, a targeted analysis of crucial biomarkers was conducted by Louchoarn et al., who on the basis of previous experiments271 applied CuO oxidation with GC-MS/MS, with particular reference to lignin (Table 5).272 After oxidation, lignin was hydrolysed into its three building blocks, vanillyls, syringyls, and cinnamyls, which are readily identified by GC-MS and GC-MS/MS.
More recently, Lang et al., developed an innovative method for the isotopic analysis (δ13C) of organic samples by using a GasBench plumbing system. Within this approach, water soluble organic compounds were oxidised to CO2 using potassium persulfate, phosphoric acid and heat (Table 5).273 The developed gas was delivered through helium flow firstly to an injection valve and then to the GC column which separated CO2 from other interfering gases (i.e. N2O). The purified CO2 was then analysed by IRMS, with a limit of detection (LOD) of 1.2 μg of carbon. The authors suggest that this method can potentially be useful for determining the isotopic composition of LC-isolated fractions. However, a fundamental prerequisite would be a carbon-free or completely evaporated mobile phase. Another limitation underlined within this work is the possibility that the applied oxidation conditions could be not sufficient to convert refractory materials to CO2, limiting therefore its applicability.
2.3. Electrophoretic separation techniques
Electrophoretic techniques such as capillary electrophoresis (CE), isotachophoresis, isolelectric focusing, polyacrylamide gel electrophoresis (PAGE) and CZE, in combination with on-line and off-line detection methods such as MS, NMR, UV and fluorescence, have commonly been applied to the fractionation, and size and MW determination of many classes of compounds, including proteins, peptides, polymers (both natural and synthetic), and humic and fulvic substances.274,275 Linear relationships between electrophoretic mobility and MW had been demonstrated in the separation of humic substances, thus paving the way for the size and charge based fractionation of DOM.276One of the first applications to DOM characterisation using sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) in combination with Edman degradation and RP-LC, permitted the sequencing of proteins from oceanic waters.279 Within the electrophoretic separation, up to 30 proteins could be seen as unique bands. These were reported to have molecular masses from 14.3 to 66 kDa. Among these, porins were specifically identified. These outer membrane channel proteins of Gram-negative bacteria were found to have molecular masses ranging between 47 and 49 kDa.279 Within a following study, the same authors could also isolate further classes of proteins from oceanic waters, such as outer membrane protein A (OMP A) homologues,278 which are known to be resistant to enzymatic digestion.280,281 Here, proteins were separated and detected using SDS-PAGE in combination with western blotting or direct silver staining. Three major classes of proteins were isolated, namely porine homologues, glycoproteins and lectin-related proteins.
Gel filtration and SDS-PAGE were employed by Schulze et al., to separate proteins from the other organic molecules present within freshwater DOM.282 After silver staining, the gel was cut and subsequent tryptic digests separated and characterised using LC-MS/MS. The obtained spectra were searched against protein databases, and in most cases the sequences obtained were unique to a specific group of organisms. Up to 148 proteins were detected within the surface freshwater DOM, with 78% of them originating from bacteria. It was also observed that the types of proteins present was closely dependent on the season, depth and ecosystem type, as previously observed in a study by Crump et al., who applied denaturing gradient gel electrophoresis (DGGE) to monitor the seasonal variability in RNA samples from Arctic waters.283
Within a two dimensional approach by Yamada et al., SDS-PAGE and high resolution 2D electrophoresis were applied to the separation of proteins from seawater DOM.277 This technique resolved up to 412 protein spots from 10 different samples. The most prominent protein bands separated through SDS-PAGE were resolved within the second dimension, highlighting the presence of proteins with analogous molecular weights but different isoelectric points. In particular, two 34 and 39 kDa classes of glycoproteins were classified as isoforms, with the same amino-acid sequence, underlining a further presence of isomers in the DOM pool.32,33 The glycoforms of the 39 kDa protein were identified as low MW alkaline phosphatase, hydrolase enzymes belonging to the Pseudomonas group, a family of aerobic bacteria which are involved in the removal of phosphate groups from proteins or nucleic acids. Such enzymes play a key role in cellular metabolic pathways and can potentially be targeted as biomarkers to assess the MCP variations within different environmental conditions (i.e. pollution or seasonal change).278
In 2003, Schmitt-Kopplin et al., undertook a comparative study between free-flow electrophoresis (FFE) and CZE-ESI-MS for the separation of a freshwater DOM sample.284 Prior to this, separation conditions (i.e. pH buffer) were optimised using model compounds which can be found in DOM, such as benzene carboxylic acids. Further to this, for the same set of compounds, MS experiments were run in both positive and negative mode. The authors emphasise how different conditions and instrumental setup can affect analysis, causing for instance, the formation of adducts, multiply charged species and possible fragmentation issues. These phenomena are of high significance when trying to analyse a mixture of unknown compounds such as DOM. DOM separations (254 nm), obtained using an alkaline buffer, were characterised by a hump with similar m/z distribution. Lower m/z signals presented higher mobility, whereas higher m/z values were found at lower mobility. However, the authors point out that parameters such as size distribution and charge within DOM species is deeply affected by the separation conditions, therefore more experiments at different pH were proposed by the authors.
Due to the limitations identified within the above study, Vogt et al., employed multiple separation techniques, including CZE and capillary gel electrophoresis (CGE), together with SEC, all combined with the information from UV/Vis and FT-IR spectra, fluorescence emission spectra (FES), total luminescence spectra (TLS), electron spin resonance (ESR), MS, NMR and potentiometric pH titration.202 Five samples were processed using this array of analytical methods, with the results collectively highlighting clear differences according to location and seasonal changes. In particular, CZE and CGE were used in analogous conditions (i.e. sodium carbonate buffer at pH 9.3) to determine hydrodynamic radii within DOM components. As the variation in mobility from CZE to CGE is related to the molecular mass, the hydrodynamic radii could be calculated by using molecular mass distributions previously obtained when analysing polystyrene sulfonate standards. However, as discussed previously in relation to SEC, such standards poorly represent many classes of compounds within DOM, therefore, the calculated hydrodynamic radii have to be considered as indicative values.
CZE-ESI-MS has been employed by Hertkorn et al., in combination with CZE-UV (214 nm), NMR and HR-MS spectra, obtaining highly complementary data for seawater DOM collected at different depths.32,288 A 25 mM ammonium carbonate buffer (pH 9.4 and 11.4) was employed, and although extensive co-elution was also observed throughout the electropherograms (Fig. 11), major similarities in the resulting electropherograms were seen, allowing the authors to confirm, as already proved by NMR spectra, the absence of weakly acidic compounds (i.e. phenols). Within this paper CZE was directly hyphenated to MS, and no buffer removal or sample treatment was reported prior to entry into the electrospray chamber. The presence of the above mentioned alkaline buffer could potentially affect molecular weight distributions and in source sample fragmentation. However, CZE-MS chromatograms corresponded closely to those obtained through CZE-UV, with mass spectra deemed representative of the total DOM composition. The technique also enabled intra-sample comparison and showed that DOM collected at higher depths was characterised by a large fraction of highly charged aliphatic moieties. These compounds appeared to be consistent with CRAM, which were found to be more abundant within DOM collected at higher depths.
 | ||
| Fig. 11 (a) CZE-UV and (b) CE-ESI/MS electropherograms for surface and deep seawater DOM. Reproduced with permission from Hertkorn et al.32 | ||
2.4. Field-flow fractionation
Field flow fractionation (FFF) is a chromatographic technique that usually allows the fractionation of macromolecules according to their diffusion coefficient.289,290 This technique provides continuous molecular size distribution of macromolecules that can be detected off-line or via on-line coupling with various forms of detection (i.e. DAD, fluorescence). FFF is commonly used not only in the fractionation of colloidal organic matter,291–295 but also in the characterisation and determination of molecular size distribution of the chromophoric fraction of DOM (i.e. humic substances).295,296 Within early studies, FFF had only been coupled to absorbance detectors.294–296 However, Zanardi-Lamardo et al., on the basis of previous experiments, described the importance of multi-detector approaches, and also coupled FFF to a fluorescence detector.297–299 Once again the main issue with this technique is the use of polystyrene sulfonate standards as the calibrants, and additionally the surfactants commonly contained in the carrier solution. Similarly to SEC, polymeric materials share little similarity with the complex organic mixture that is DOM, therefore, erroneous MW estimations can be observed. Together with this, as for CE, the presence of surfactants can possibly induce denaturation of components within the sample, leading to a change in the tertiary structure of macromolecules. Such variation is dependent on the surfactant concentration and on the ionic strength of the carrier solution. The higher the ionic strength, the weaker the electrostatic interactions between surfactant and macromolecule.FFF was employed and complemented with solid-state NMR spectra in a study from Assemi et al., to characterise size and MW distributions of two freshwater DOM samples, which were separated into five fractions by UF according to their MW (lower than 0.5 kDa, from 0.5 to 3 kDa, from 3 to 10 kDa, from 10 to 30 kDa, and higher than 30 kDa).300 As the mobility in FFF is related to the particle size, usually the smaller the particles, the faster they elute from the channel. However, the fractograms obtained at 254 nm by using a deionised water carrier, show significant overlapping between certain fractions. This suggests that these fractions were not separated into discrete size ranges and/or the samples undergo secondary interactions, such as irreversible decomposition of large molecules into smaller units.69 This is further confirmed by the fact that when FFF (after calibration with a polystyrene sulfonate standard) was used to determine size and MW, these were found to be smaller than the nominal filter ranges. SEC was used to compare the MW distribution, and showed MW ranges analogous to those obtained through FFF in the case of only one of the two analysed samples.
Moon et al., were the first to evaluate the effects of ionic strength in FFF carrier solutions on the size determination of DOM, and to provide molecular sizes in terms of hydrodynamic effective size.301 Such approach was chosen to consider the influence of diffusion and convection flows during the separation and the interaction forces occurring between DOM and the membrane at the bottom of the FFF channel. To demonstrate the effect of the carrier solution on the separation, KCl and a detergent (FL-70) were used at different concentrations. However, substantial changes in the determination of DOM sizes with increasing ionic strength were not observed, although when FL-70 was used as carrier solution, DOM sizes were lower than those measured when using KCl (20 mM). This was explained by the fact that FL-70 is composed of anionic and nonionic species, allowing the solutes to be more dispersed and preventing aggregation and interactions between sample and the membrane surface. A higher concentration of FL-70 can therefore result in a more rapid elution of DOM, and in a consequent lower size determined by FFF.
Floge et al., used artificial seawater as carrier solution (salinity: 32, pH: 8.1) in a further FFF-UV study.293 The authors observed higher UV absorption in periods following phytoplankton blooms and the year-round presence of colloids (size higher than 18 kDa). Such findings further confirm the seasonal variability of DOM and that the colloidal species may have a refractory nature.
On the basis of previous experiments, Guéguen et al., also used a polystyrene sulfonate standard and a NaCl solution as a carrier, at ionic strengths analogous to natural waters.289,302 FFF-UV-DAD and excitation emission matrix (EEM) fluorescence were used to calculate the MW distribution of the chromophoric portion of DOM.303 Pre-fractionation or concentration methods such as UF or SPE were not used, therefore minimising the risk of contamination or additional fractionation. Despite the ubiquitous calibration issues, the mean MW distribution was found to range between 0.8 and 1.1 kDa, depending upon on the sampling location.
Analogous MW ranges (0.68–1.95 kDa) were also found by analysing the chromophoric fraction of DOM by asymmetrical flow field-flow fractionation (AF4) coupled to fluorescence parallel factor analysis (PARAFAC).303 AF4 was earlier introduced in the characterisation of DOC, coupled to both UV and DOC detection304 and has the advantage, if compared to symmetrical FFF, of a simpler channel construction and a transparent front plate, where the focusing band is visualised when a coloured analyte is injected.303 The analysed samples were fractionated, by using a 1 mM NaCl carrier solution, into five components, which showed humic-like fluorophores on fraction 1, 2 and 4, comprising the majority of the total fluorescence, and a protein-like fluorophore on fraction 5. The method could prove a stratification of such fluorophoric material, with surface samples having a higher total fluorescence, therefore a higher content in humic substances, if compared to deeper water samples.
3. Future directions and conclusions
It is hoped this review provides a comprehensive overview of the range and complexity of separation methods applied to this significant analytical challenge. Clearly, separation science remains central to greater understanding of this complex system, although the breadth of studies included within this review collectively highlight how no one approach individually is capable of providing the immense resolution required for molecular level separations, and this is likely to remain the case for the foreseeable future.As with most analytical problems, the first and most significant issue is collection of a representative sample. DOM provides the perfect demonstration of this principle. The difficulties in extracting uncontaminated and unbiased DOM are still considerable. This overriding issue is, in the authors' opinion, much overlooked in the vast majority of papers on DOM characterisation. As with subsequent separation techniques, it would appear multi-dimensional (multi-selective) approaches may provide a more comprehensive solution. How this is achieved practically, particularly for SPE, remains to be seen. Since the compounds constituting DOM are often at nanomolar or picomolar level, and given the complexity of the sample extraction procedures required, there is always a major risk in sample contamination, e.g. from storage containers, sample preparation (i.e. SPE, UF) and solvents used. This issue too is rarely commented upon and details of process blanks rare in most published studies. For this purpose, artificial seawater/freshwater should be employed and passed through all the extraction and separation procedures that the actual samples undergo. Using this procedure, it is possible to clearly identify and improve the extraction or chromatographic step where artefacts are generated.
DOM fractionation and subsequent separation appears to be a common approach within a great number of studies. Essentially this is off-line two dimensional chromatography, which attempts to provide some level of resolution prior to high-end detection techniques, such as HR-MS or 2D NMR. Woods et al., illustrated this very clearly by employing HILIC × HILIC separations of DOM, to deliver more resolved NMR spectra.134 Similar approaches using more orthogonal separation methods are likely to continue to emerge, including on-line multidimensional separation methods, both LC and GC based. However, chromatographically, we will only see peak capacities (resolution) of hundreds of peaks, a long way short of the tens (if not hundreds) of thousands of individual components thought to make up this complex substance. Thus the combination with HR-MS and 2D NMR will remain essential, providing the 3rd, 4th and 5th dimensions required for such molecular level resolution. In particular, HR-MS remains crucial to DOM characterisation, and the rapid development of such technology (including more universal ionisation techniques305–307) will ease, but not delete, the need for ever greater chromatographic resolution. For characterisation of the large number of isomers present in DOM, microgram-level NMR provides a solution, proving that MS and NMR spectra can and should be used to complement each other.
The above comments suggest that much more work remains to be done before obtaining a true understanding of the complexities of this abundant material.30 However, over the past decade this field has progressed rapidly, and the solid basis of understanding DOM and its role in the carbon cycle have been laid down by these pioneering studies.
Acknowledgements
This research was supported under Australian Research Council's Discovery Projects funding scheme (project number DP130101518). Robert Shellie is the recipient of an Australian Research Council Australian Research Fellowship (Project Number DP110104923). The authors' would also like to acknowledge the substantial and positive contributions and suggestions of the referees' in the preparation of this review.Notes and references
- F. Azam, N. Jiao and S. Sanders, The Microbial Carbon Pump in the Ocean, Science, 2011, 1–68 Search PubMed.
- C. Chen, J. Anaya, E. Chen, E. Farr and W. Chin, PLoS One, 2015, 10, 1–9 Search PubMed.
- P. Verdugo, A. L. Alldredge, F. Azam, D. L. Kirchman, U. Passow and P. H. Santschi, Mar. Chem., 2004, 92, 67–85 CrossRef CAS PubMed.
- W. C. Chin, M. V. Orellana and P. Verdugo, Nature, 1998, 391, 568–572 CrossRef CAS.
- E. Ortega-Retuerta, T. K. Frazer, C. M. Duarte, S. Ruiz-Halpern, A. Tovar-Sánchez, J. M. Arrieta and I. Rechea, Limnol. Oceanogr., 2009, 54, 1941–1950 CrossRef CAS.
- J. Arıstegui, C. M. Duarte, S. Agustı, M. Doval, X. A. Álvarez-Salgado and D. A. Hansell, Science, 2002, 298, 1967 CrossRef PubMed.
- J. A. Davis, Geochim. Cosmochim. Acta, 1984, 48, 679–691 CrossRef CAS.
- J. Reuter and E. Perdue, Geochim. Cosmochim. Acta, 1977, 41, 325–334 CrossRef CAS.
- P. A. Del Giorgio and C. M. Duarte, Nature, 2002, 420, 379–384 CrossRef CAS PubMed.
- C. Evans, D. Monteith and D. Cooper, Environ. Pollut., 2005, 137, 55–71 CrossRef CAS PubMed.
- T. Dittmar and J. Paeng, Nat. Geosci., 2009, 2, 175 CrossRef CAS PubMed.
- J. J. Cole, Y. T. Prairie, N. F. Caraco, W. H. McDowell, L. J. Tranvik, R. G. Striegl, C. M. Duarte, P. Kortelainen, J. A. Downing and J. J. Middelburg, Ecosystems, 2007, 10, 172–185 CrossRef.
- T. Dittmar and G. Kattner, Mar. Chem., 2003, 82, 115–123 CrossRef CAS.
- J. I. Hedges, G. Eglinton, P. G. Hatcher, D. L. Kirchman, C. Arnosti, S. Derenne, R. P. Evershed, I. Koegel-Knabner, J. W. de Leeuw, R. Littke, W. Michaelis and J. Rullkoetter, Org. Geochem., 2000, 31, 945–958 CrossRef CAS.
- T. Dittmar and R. J. Lara, Estuarine, Coastal Shelf Sci., 2001, 52, 249–259 CrossRef CAS.
- T. Dittmar, R. J. Lara and G. Kattner, Mar. Chem., 2001, 73, 253–271 CrossRef CAS.
- T. Dittmar and G. Kattner, Mar. Chem., 2003, 83, 103–120 CrossRef CAS.
- T. Dittmar, N. Hertkorn, G. Kattner and R. J. Lara, Global Biogeochem. Cycles, 2006, 20, 1–7 CrossRef.
- C. S. Hopkinson, I. Buffam, J. Hobbie, J. Vallino, M. Perdue, B. Eversmeyer, F. Prahl, J. Covert, R. Hodson and M. A. Moran, Biogeochemistry, 1998, 43, 211–234 CrossRef CAS.
- P. Kowalczuk, W. J. Cooper, M. J. Durako, A. E. Kahn, M. Gonsior and H. Young, Mar. Chem., 2010, 118, 22–36 CrossRef CAS PubMed.
- A. Stubbins, R. G. Spencer, H. Chen, P. G. Hatcher, K. Mopper, P. J. Hernes, V. L. Mwamba, A. M. Mangangu, J. N. Wabakanghanzi and J. Six, Limnol. Oceanogr., 2010, 55, 1467–1477 CAS.
- T. Dittmar, K. Whitehead, E. C. Minor and B. P. Koch, Mar. Chem., 2007, 107, 378–387 CrossRef CAS PubMed.
- E. Gacia and C. M. Duarte, Estuarine, Coastal Shelf Sci., 2001, 52, 505–514 CrossRef.
- D. A. Hansell, Annual Review of Marine Science, 2013, 5, 421–445 CrossRef PubMed.
- F. Guillemette and P. Del Giorgio, Environ. Microbiol., 2012, 1–12 Search PubMed.
- C. Romera-Castillo, H. Sarmento, X. A. Álvarez-Salgado, J. M. Gasol and C. Marrasé, Appl. Environ. Microbiol., 2011, 77, 7490–7498 CrossRef CAS PubMed.
- T. T. van Engeland, T. J. Bouma, E. P. Morris, F. G. Brun, G. Peralta, M. Lara, I. E. Hendriks, P. van Rijswijk, B. Veuger and K. Soetaert, Mar. Ecol.: Prog. Ser., 2013, 478, 87–100 CrossRef CAS.
- Y. Yi, J. Han, S. J. Birks, C. H. Borchers and J. J. Gibson, Sci. Total Environ., 2014, 518, 148–158 Search PubMed.
- F. Azam, Science, 1998, 280, 694–696 CrossRef CAS.
- D. A. Hansell and C. A. Carlson, Biogeochemistry of Marine Dissolved Organic Matter, Elsevier, 2002 Search PubMed.
- K. Mopper, A. Stubbins, J. D. Ritchie, H. M. Bialk and P. G. Hatcher, Chem. Rev., 2007, 107, 419–442 CrossRef CAS PubMed.
- N. Hertkorn, R. Benner, M. Frommberger, P. Schmitt-Kopplin, M. Witt, K. Kaiser, A. Kettrup and J. Hedges, Geochim. Cosmochim. Acta, 2006, 70, 2990–3010 CrossRef CAS PubMed.
- N. Hertkorn, M. Harir, B. P. Koch, B. Michalke and P. Schmitt-Kopplin, Biogeosciences, 2013, 10, 1583–1624 CAS.
- E. N. Capley, Ph.D thesis, University of South Alabama, 2011.
- E. N. Capley, J. D. Tipton, A. G. Marshall and A. C. Stenson, Anal. Chem., 2010, 82, 8194–8202 CrossRef CAS PubMed.
- F. Zhang, M. Harir, F. Moritz, J. Zhang, M. Witting, Y. Wu, P. Schmitt-Kopplin, A. Fekete, A. Gaspar and N. Hertkorn, Water Res., 2014, 57, 280–294 CrossRef CAS PubMed.
- H. A. Abdulla, R. L. Sleighter and P. G. Hatcher, Anal. Chem., 2013, 85, 3895–3902 CrossRef CAS PubMed.
- N. Hertkorn, M. Harir, B. P. Koch, B. Michalke and P. Schmitt-Kopplin, Biogeosciences Discussions, 2012, 9, 745–833 Search PubMed.
- J. D'Andrilli, T. Dittmar, B. P. Koch, J. M. Purcell, A. G. Marshall and W. T. Cooper, Rapid Commun. Mass Spectrom., 2010, 24, 643–650 CrossRef PubMed.
- R. L. Sleighter, G. A. McKee and P. G. Hatcher, Org. Geochem., 2009, 40, 119–125 CrossRef CAS PubMed.
- A. C. Stenson, Rapid Commun. Mass Spectrom., 2009, 23, 465 CrossRef CAS PubMed.
- A. C. Stenson, Environ. Sci. Technol., 2008, 42, 2060–2065 CrossRef CAS.
- R. L. Sleighter and P. G. Hatcher, Mar. Chem., 2008, 110, 140–152 CrossRef CAS PubMed.
- B. P. Koch, M. Witt, R. Engbrodt, T. Dittmar and G. Kattner, Geochim. Cosmochim. Acta, 2005, 69, 3299–3308 CrossRef CAS PubMed.
- G. St-Jean, Rapid Commun. Mass Spectrom., 2003, 17, 419–428 CrossRef CAS PubMed.
- A. Golchin, J. Oades, J. Skjemstad and P. Clarke, Soil Res., 1995, 33, 59–76 CrossRef.
- G. C. Woods, Ph.D thesis, University of Toronto, 2012.
- B. Lam, A. Baer, M. Alaee, B. Lefebvre, A. Moser, A. Williams and A. J. Simpson, Environ. Sci. Technol., 2007, 41, 8240–8247 CrossRef CAS.
- O. J. Lechtenfeld, N. Hertkorn, Y. Shen, M. Witt and R. Benner, Nat. Commun., 2015, 6, 1–8 Search PubMed.
- N. W. Davies, S. Sandron, P. N. Nesterenko, B. Paull, R. Wilson, P. Haddad, R. Shellie and A. Rojas, Environ. Sci.: Processes Impacts, 2015, 17, 495–496 CAS.
- B. Lam and A. J. Simpson, Analyst, 2008, 133, 263–269 RSC.
- R. Benner, J. D. Pakulski, M. McCarthy, J. I. Hedges and P. G. Hatcher, Science, 1992, 255, 1561–1564 CAS.
- B. G. Pautler, G. C. Woods, A. Dubnick, A. J. Simpson, M. J. Sharp, S. J. Fitzsimons and M. J. Simpson, Environ. Sci. Technol., 2012, 46, 3753–3761 CrossRef CAS PubMed.
- M. Hutta, R. Góra, R. Halko and M. Chalányová, J. Chromatogr. A, 2011, 1218, 8946–8957 CrossRef CAS PubMed.
- R. M. Duarte and A. C. Duarte, TrAC, Trends Anal. Chem., 2011, 30, 1659–1671 CrossRef CAS PubMed.
- E. C. Minor, M. M. Swenson, B. M. Mattson and A. R. Oyler, Environ. Sci.: Processes Impacts, 2014, 16, 2064–2079 Search PubMed.
- T. Dittmar, B. P. Koch, N. Hertkorn and G. Kattner, Limnol. Oceanogr.: Methods, 2008, 6, 230–235 CrossRef CAS.
- R. Averett, J. Leenheer, D. McKnight and K. Thorn, US Geological survey open file report, 1989, vol. 87, p. 557 Search PubMed.
- S. B. Schwede-Thomas, Y. P. Chin, K. J. Dria, P. Hatcher, E. Kaiser and B. Sulzberger, Aquat. Sci., 2005, 67, 61–71 CrossRef CAS.
- J.-P. Simjouw, E. C. Minor and K. Mopper, Mar. Chem., 2005, 96, 219–235 CrossRef CAS PubMed.
- I. V. Perminova, I. V. Dubinenkov, A. S. Kononikhin, A. I. Konstantinov, A. Y. Zherebker, M. M. Andzhushev, V. A. Lebedev, E. Bulygina, R. M. Holmes and Y. I. Kostyukevich, Environ. Sci. Technol., 2014, 48, 7461–7468 CrossRef CAS PubMed.
- N. W. Green, E. M. Perdue, G. R. Aiken, K. D. Butler, H. Chen, T. Dittmar, J. Niggemann and A. Stubbins, Mar. Chem., 2014, 161, 14–19 CrossRef CAS PubMed.
- V. I. Esteves, M. Otero, E. B. Santos and A. C. Duarte, Org. Geochem., 2007, 38, 957–966 CrossRef CAS PubMed.
- E. C. Minor, J. P. Simjouw, J. J. Boon, A. E. Kerkhoff and J. van der Horst, Mar. Chem., 2002, 78, 75–102 CrossRef CAS.
- R. Kerr and J. Quinn, Mar. Chem., 1980, 8, 217–229 CrossRef CAS.
- R. Kerr and J. Quinn, Deep-Sea Res., 1975, 22, 107–116 CAS.
- R. Town and K. Powell, Anal. Chim. Acta, 1993, 271, 195–202 CrossRef CAS.
- B. Little and J. Jacobus, Org. Geochem., 1985, 8, 27–33 CrossRef CAS.
- A. Zsolnay, Geoderma, 2003, 113, 187–209 CrossRef CAS.
- J. F. Koprivnjak, E. M. Perdue and P. H. Pfromm, Water Res., 2006, 40, 3385–3392 CrossRef CAS PubMed.
- T. A. Vetter, E. Perdue, E. Ingall, J. F. Koprivnjak and P. Pfromm, Sep. Purif. Technol., 2007, 56, 383–387 CrossRef CAS PubMed.
- J. F. Koprivnjak, P. H. Pfromm, E. Ingall, T. A. Vetter, P. Schmitt-Kopplin, N. Hertkorn, M. Frommberger, H. Knicker and E. M. Perdue, Geochim. Cosmochim. Acta, 2009, 73, 4215–4231 CrossRef CAS PubMed.
- J. Mao, X. Kong, K. Schmidt-Rohr, J. J. Pignatello and E. M. Perdue, Environ. Sci. Technol., 2012, 46, 5806–5814 CrossRef CAS PubMed.
- B. Gurtler, T. A. Vetter, E. Perdue, E. Ingall, J. F. Koprivnjak and P. H. Pfromm, J. Membr. Sci., 2008, 323, 328–336 CrossRef CAS PubMed.
- H. Chen, A. Stubbins, E. M. Perdue, N. W. Green, J. R. Helms, K. Mopper and P. G. Hatcher, Mar. Chem., 2014, 164, 48–59 CrossRef CAS PubMed.
- S. M. Ilina, O. Y. Drozdova, S. A. Lapitskiy, Y. V. Alekhin, V. V. Demin, Y. A. Zavgorodnyaya, L. S. Shirokova, J. Viers and O. S. Pokrovsky, Org. Geochem., 2014, 66, 14–24 CrossRef CAS PubMed.
- B. R. Kruger, B. J. Dalzell and E. C. Minor, Aquat. Sci., 2011, 73, 405–417 CrossRef CAS.
- B. J. Dalzell, E. C. Minor and K. M. Mopper, Org. Geochem., 2009, 40, 243–257 CrossRef CAS PubMed.
- A. Chow, R. Dahlgren and S. Gao, Aqua, 2005, 54, 475–507 CAS.
- H. A. N. Abdulla, E. C. Minor and P. G. Hatcher, Environ. Sci. Technol., 2010, 44, 8044–8049 CrossRef CAS PubMed.
- A. Skoog and R. Benner, Limnol. Oceanogr., 1997, 42, 1803–1813 CrossRef CAS.
- L. Guo and P. H. Santschi, Mar. Chem., 1996, 55, 113–127 CrossRef CAS.
- N. Ogura, Mar. Biol., 1974, 24, 305–312 CrossRef CAS.
- D. J. Repeta, T. M. Quan, L. I. Aluwiharea and A. Accardi, Geochim. Cosmochim. Acta, 2002, 66, 955–962 CrossRef CAS.
- L. I. Aluwihare, D. J. Repeta and R. F. Chen, Nature, 1997, 387, 166–169 CrossRef CAS PubMed.
- K. O. Buesseler, J. Bauer, R. Chen, T. Eglinton, O. Gustafsson, W. Landing, K. Mopper, S. Moran, P. Santschi and R. Vernon-Clark, Mar. Chem., 1996, 55, 1–31 CrossRef CAS.
- S. Opsahl and R. Benner, Limnol. Oceanogr., 1998, 43, 1297–1304 CrossRef CAS.
- M. L. Pace, I. Reche, J. J. Cole, A. Fernández-Barbero, I. P. Mazuecos and Y. T. Prairie, Biogeochemistry, 2012, 108, 109–118 CrossRef CAS.
- R. W. P. M. Laane, Mar. Chem., 1982, 11, 395–401 CrossRef CAS.
- V. Roubeuf, S. Mounier and J. Benaim, Org. Geochem., 2000, 31, 127–131 CrossRef CAS.
- G. R. Aiken, Humic Substances and their Role in the Environment, Wiley, 1988 Search PubMed.
- J. C. Knulst, R. C. Boerschke and S. Loemo, Environ. Sci. Technol., 1998, 32, 8–12 CrossRef CAS.
- R. J. Lara and D. Thomas, Mar. Chem., 1994, 47, 93–96 CrossRef CAS.
- J. A. Leenheer, Environ. Sci. Technol., 1981, 15, 578–587 CrossRef CAS PubMed.
- R. L. Malcolm and P. MacCarthy, Environ. Int., 1992, 18, 597–607 CrossRef CAS.
- V. Lepane, J. Chromatogr. A, 1999, 845, 329–335 CrossRef CAS.
- G. R. Aiken, E. Thurman, R. Malcolm and H. F. Walton, Anal. Chem., 1979, 51, 1799–1803 CrossRef CAS.
- G. L. Mills and J. G. Quinn, Mar. Chem., 1981, 10, 93–102 CrossRef CAS.
- G. L. Mills, E. McFadden and J. G. Quinn, Mar. Chem., 1987, 20, 313–325 CrossRef CAS.
- S. Kim, A. J. Simpson, E. B. Kujawinski, M. A. Freitas and P. G. Hatcher, Org. Geochem., 2003, 34, 1325–1335 CrossRef CAS.
- G. Morales-Cid, I. Gebefugi, B. Kanawati, M. Harir, N. Hertkorn, R. Rossello-Mora and P. Schmitt-Kopplin, Anal. Bioanal. Chem., 2009, 395, 797–807 CrossRef CAS PubMed.
- M. M. Swenson, A. R. Olyer and E. C. Minor, Limnol. Oceanogr.: Methods, 2014, 12, 713–728 CrossRef.
- T. H. Boyer and P. C. Singer, Water Res., 2005, 39, 1265–1276 CrossRef CAS PubMed.
- T. H. Boyer, P. C. Singer and G. R. Aiken, Environ. Sci. Technol., 2008, 42, 7431–7437 CrossRef CAS.
- T. H. Boyer and P. C. Singer, Environ. Sci. Technol., 2007, 42, 608–613 CrossRef.
- F. Su and C. Lu, J. Environ. Sci. Health, Part A: Toxic/Hazard. Subst. Environ. Eng., 2007, 42, 1543–1552 CrossRef CAS PubMed.
- J. Sánchez-González, N. García-Otero, A. Moreda-Piñeiro and P. Bermejo-Barrera, Microchem. J., 2012, 102, 75–82 CrossRef PubMed.
- Z. Mester, R. Sturgeon and J. Pawliszyn, Spectrochim. Acta, 2001, 56, 233–260 CrossRef.
- G. Ohlenbusch, M. U. Kumke and F. H. Frimmel, Sci. Total Environ., 2000, 253, 63–74 CrossRef CAS.
- C. de Perre, K. Le Ménach, F. Ibalot, E. Parlanti and H. Budzinski, Anal. Chim. Acta, 2014, 807, 51–60 CrossRef CAS PubMed.
- L. Sun, E. Perdue, J. Meyer and J. Weis, Limnol. Oceanogr., 1997, 42, 714–721 CrossRef CAS.
- L. Sun, E. M. Perdue and J. F. McCarthy, Water Res., 1995, 29, 1471–1477 CrossRef CAS.
- H. Ødegaard and S. Koottatep, Water Res., 1982, 16, 613–620 CrossRef.
- S. M. Serkiz and E. M. Perdue, Water Res., 1990, 24, 911–916 CrossRef CAS.
- T. A. Clair, J. R. Kramer, M. Sydor and D. Eaton, Water Res., 1991, 25, 1033–1037 CrossRef CAS.
- E. T. Gjessing, J. Alberts, A. Bruchet, P. K. Egeberg, E. Lydersen, L. B. McGown, J. J. Mobed, U. Münster, J. Pempkowiak and M. Perdue, Water Res., 1998, 32, 3108–3124 CrossRef CAS.
- I. C. Escobar, S. Hong and A. A. Randall, J. Membr. Sci., 2000, 175, 1–17 CrossRef CAS.
- A. C. Stenson, W. M. Landing, A. G. Marshall and W. T. Cooper, Anal. Chem., 2002, 74, 4397–4409 CrossRef CAS.
- B. Lam and A. J. Simpson, Anal. Chem., 2006, 78, 8194–8199 CrossRef CAS PubMed.
- B. Vrana, I. J. Allan, R. Greenwood, G. A. Mills, E. Dominiak, K. Svensson, J. Knutsson and G. Morrison, TrAC, Trends Anal. Chem., 2005, 24, 845–868 CrossRef CAS PubMed.
- P. Mayer, J. Tolls, J. L. Hermens and D. Mackay, Environ. Sci. Technol., 2003, 37, 184A–191A CrossRef.
- R. Greenwood, G. Mills and B. Vrana, Passive sampling techniques in environmental monitoring, Elsevier, 2007 Search PubMed.
- M. V. McCaul, D. Sutton, A. J. Simpson, A. Spence, D. J. McNally, B. W. Moran, A. Goel, B. O'Connor, K. Hart and B. P. Kelleher, Environ. Chem., 2011, 8, 146–154 CrossRef CAS.
- B. Zabiegała, A. Kot-Wasik, M. Urbanowicz and J. Namieśnik, Anal. Bioanal. Chem., 2010, 396, 273–296 CrossRef PubMed.
- A. Kot-Wasik, B. Zabiegała, M. Urbanowicz, E. Dominiak, A. Wasik and J. Namieśnik, Anal. Chim. Acta, 2007, 602, 141–163 CrossRef CAS PubMed.
- D. McKnight, E. W. Boyer, P. K. Westerhoff, P. T. Doran, T. Kulbe and D. T. Andersen, Limnol. Oceanogr., 2001, 46, 38–48 CrossRef CAS.
- G. M. Ferrari, M. D. Dowell, S. Grossi and C. Targa, Mar. Chem., 1996, 55, 299–316 CrossRef.
- P. Smart, B. Finlayson, W. Rylands and C. Ball, Water Res., 1976, 10, 805–811 CrossRef.
- S. A. Cumberland and A. Baker, Hydrol. Processes, 2007, 21, 2093–2099 CrossRef CAS PubMed.
- N. Hertkorn, C. Ruecker, M. Meringer, R. Gugisch, M. Frommberger, E. Perdue, M. Witt and P. Schmitt-Kopplin, Anal. Bioanal. Chem., 2007, 389, 1311–1327 CrossRef CAS PubMed.
- B. P. Koch, T. Dittmar, M. Witt and G. Kattner, Anal. Chem., 2007, 79, 1758–1763 CrossRef CAS PubMed.
- G. C. Woods, M. J. Simpson, B. P. Kelleher, M. McCaul, W. L. Kingery and A. J. Simpson, Environ. Sci. Technol., 2010, 44, 624–630 CrossRef CAS PubMed.
- G. C. Woods, M. J. Simpson, P. J. Koerner, A. Napoli and A. J. Simpson, Environ. Sci. Technol., 2011, 45, 3880–3886 CrossRef CAS PubMed.
- G. C. Woods, M. J. Simpson and A. J. Simpson, Water Res., 2012, 46, 3398–3408 CrossRef CAS PubMed.
- J. A. Leenheer, Ann. Environ. Sci., 2007, 1, 57–68 CAS.
- E. Kristensen, S. Bouillon, T. Dittmar and C. Marchand, Aquat. Bot., 2008, 89, 201–219 CrossRef CAS PubMed.
- J. R. M. Lobbes, H. P. Fitznar and G. Kattner, Anal. Chem., 1999, 71, 3008–3012 CrossRef CAS PubMed.
- S. Steinberg, M. Venkatesan and I. Kaplan, J. Chromatogr. A, 1984, 298, 427–434 CrossRef CAS.
- E. Parlanti, B. Morin and L. Vacher, Org. Geochem., 2002, 33, 221–236 CrossRef CAS.
- A. J. Simpson, L. H. Tseng, M. J. Simpson, M. Spraul, U. Braumann, W. L. Kingery, B. P. Kelleher and M. H. Hayes, Analyst, 2004, 129, 1216–1222 RSC.
- B. P. Koch, K.-U. Ludwichowski, G. Kattner, T. Dittmar and M. Witt, Mar. Chem., 2008, 111, 233–241 CrossRef CAS PubMed.
- R. Góra and M. Hutta, J. Chromatogr. A, 2005, 1084, 39–45 CrossRef PubMed.
- M. Hutta and R. Góra, J. Chromatogr. A, 2003, 1012, 67–79 CrossRef CAS.
- R. Góra, M. Hutta and P. Rohárik, J. Chromatogr. A, 2012, 1220, 44–49 CrossRef PubMed.
- J. Hedges, G. Eglinton, P. Hatcher, D. Kirchman, C. Arnosti, S. Derenne, R. Evershed, I. Kögel-Knabner, J. de Leeuw and R. Littke, Org. Geochem., 2000, 31, 945–958 CrossRef CAS.
- K. Longnecker and E. B. Kujawinski, Geochim. Cosmochim. Acta, 2011, 75, 2752–2761 CrossRef CAS PubMed.
- M. P. Bhatia, S. B. Das, K. Longnecker, M. A. Charette and E. B. Kujawinski, Geochim. Cosmochim. Acta, 2010, 74, 3768–3784 CrossRef CAS PubMed.
- J. D'Andrilli, J. P. Chanton, P. H. Glaser and W. T. Cooper, Org. Geochem., 2010, 41, 791–799 CrossRef PubMed.
- L. B. Tremblay, T. Dittmar, A. G. Marshall, W. J. Cooper and W. T. Cooper, Mar. Chem., 2007, 105, 15–29 CrossRef CAS PubMed.
- A. P. Bateman, S. A. Nizkorodov, J. Laskin and A. Laskin, Anal. Chem., 2010, 82, 8010–8016 CrossRef CAS PubMed.
- M. C. Kido Soule, K. Longnecker, S. J. Giovannoni and E. B. Kujawinski, Org. Geochem., 2010, 41, 725–733 CrossRef CAS PubMed.
- R. Flerus, B. P. Koch, P. Schmitt-Kopplin, M. Witt and G. Kattner, Mar. Chem., 2011, 124, 100–107 CrossRef CAS PubMed.
- C. E. Rostad and J. A. Leenheer, Anal. Chim. Acta, 2004, 523, 269–278 CrossRef CAS PubMed.
- T. Dittmar and B. P. Koch, Mar. Chem., 2006, 102, 208–217 CrossRef CAS PubMed.
- B. P. Koch and T. Dittmar, Rapid Commun. Mass Spectrom., 2006, 20, 926–932 CrossRef CAS PubMed.
- A. C. Stenson, A. G. Marshall and W. T. Cooper, Anal. Chem., 2003, 75, 1275–1284 CrossRef CAS.
- M. Gonsior, B. M. Peake, W. T. Cooper, D. C. Podgorski, J. D'Andrilli, T. Dittmar and W. J. Cooper, Mar. Chem., 2011, 123, 99–110 CrossRef CAS PubMed.
- M. Gonsior, B. M. Peake, W. T. Cooper, D. Podgorski, J. D'Andrilli and W. J. Cooper, Environ. Sci. Technol., 2009, 43, 698–703 CrossRef CAS.
- U. Klaus, T. Pfeifer and M. Spiteller, Environ. Sci. Technol., 2000, 34, 3514–3520 CrossRef CAS.
- C. McIntyre and C. McRae, Org. Geochem., 2005, 36, 543–553 CrossRef CAS PubMed.
- M. Witt, J. Fuchser and B. P. Koch, Anal. Chem., 2009, 81, 2688–2694 CrossRef CAS PubMed.
- A. C. Stenson, B. M. Ruddy and B. J. Bythell, Int. J. Mass Spectrom., 2014, 360, 45–53 CrossRef CAS PubMed.
- R. F. C. Mantoura, A. Dickson and J. P. Riley, Estuarine, Coastal Shelf Sci., 1978, 6, 387–408 CrossRef CAS.
- G. Castro, C. Padilha, J. Rocha, J. Valente, A. D. O. Florentino and P. Padilha, Ecletica Quim., 2005, 30, 45–51 CAS.
- M. Schnitzer and H. Kerndorff, Water, Air, Soil Pollut., 1981, 15, 97–108 CrossRef CAS.
- K. Wrobel, B. B. Sadi, K. Wrobel, J. R. Castillo and J. A. Caruso, Anal. Chem., 2003, 75, 761–767 CrossRef CAS.
- V. B. Di Marco and G. G. Bombi, Mass Spectrom. Rev., 2006, 25, 347–379 CrossRef CAS PubMed.
- F. Wu, R. Evans and P. Dillon, Anal. Chim. Acta, 2002, 464, 47–55 CrossRef CAS.
- F. Wu, R. Evans and P. Dillon, Environ. Sci. Technol., 2003, 37, 3687–3693 CrossRef CAS.
- F. Wu, D. Evans, P. Dillon and S. Schiff, J. Anal. At. Spectrom., 2004, 19, 979–983 RSC.
- A. C. Stenson, Rapid Commun. Mass Spectrom., 2009, 23, 465–476 CrossRef CAS PubMed.
- T. Riedel, H. Biester and T. Dittmar, Environ. Sci. Technol., 2012, 46, 4419–4426 CrossRef CAS PubMed.
- W. Lindinger, J. Hirber and H. Paretzke, Int. J. Mass Spectrom. Ion Processes, 1993, 129, 79–88 CrossRef CAS.
- S. P. Seitzinger, H. Hartnett, R. Lauck, M. Mazurek, T. Minegishi, G. Spyres and R. Styles, Limnol. Oceanogr., 2005, 50, 1–12 CrossRef CAS.
- S. P. Seitzinger, R. M. Styles, R. Lauck and M. A. Mazurek, Environ. Sci. Technol., 2003, 37, 131–137 CrossRef CAS.
- T. Reemtsma, J. Chromatogr. A, 2009, 1216, 3687–3701 CrossRef CAS PubMed.
- Z. Liu, R. L. Sleighter, J. Zhong and P. G. Hatcher, Estuarine, Coastal Shelf Sci., 2011, 92, 205–216 CrossRef CAS PubMed.
- E. B. Kujawinski, R. Del Vecchio, N. V. Blough, G. C. Klein and A. G. Marshall, Mar. Chem., 2004, 92, 23–37 CrossRef CAS PubMed.
- A. C. Stenson, A. G. Marshall and W. T. Cooper, Anal. Chem., 2003, 75, 1275–1284 CrossRef CAS.
- O. J. Lechtenfeld, G. Kattner, R. Flerus, S. L. McCallister, P. Schmitt-Kopplin and B. P. Koch, Geochim. Cosmochim. Acta, 2014, 126, 321–337 CrossRef CAS PubMed.
- P. E. Rossel, A. V. Vähätalo, M. Witt and T. Dittmar, Org. Geochem., 2013, 60, 62–71 CrossRef CAS PubMed.
- T. Riedel and T. Dittmar, Anal. Chem., 2014, 86, 8376–8382 CrossRef CAS PubMed.
- E. Hoffmann, Mass Spectrometry, Wiley Online Library, 1996 Search PubMed.
- T. B. Bittar, A. A. H. Vieira, A. Stubbins and K. Mopper, Limnol. Oceanogr., 2015, 60, 1172–1194 CrossRef PubMed.
- H. Li, P. A. Helm, G. Paterson and C. D. Metcalfe, Chemosphere, 2011, 83, 271–280 CrossRef CAS PubMed.
- V. Flotron, C. Delteil, Y. Padellec and V. Camel, Chemosphere, 2005, 59, 1427–1437 CrossRef CAS PubMed.
- S. Huclier-Markai, C. Landesman, H. Rogniaux, F. Monteau, A. Vinsot and B. Grambow, Rapid Commun. Mass Spectrom., 2010, 24, 191–202 CrossRef CAS PubMed.
- N. Hertkorn, M. Frommberger, M. Witt, B. P. Koch, P. Schmitt-Kopplin and E. M. Perdue, Anal. Chem., 2008, 80, 8908–8919 CrossRef CAS PubMed.
- G. C. Pelekani, G. Newcombe, V. L. Snoeyink, C. Hepplewhite, S. Assemi and R. Beckett, Environ. Sci. Technol., 1999, 33(33), 2807–2813 CrossRef.
- C. Landry and L. Trembay, Environ. Sci. Technol., 2012, 46, 1700–1707 CrossRef CAS PubMed.
- P. Conte and A. Piccolo, Chemosphere, 1999, 38, 517–528 CrossRef CAS.
- T. Vartiainen, A. Liimatainen and P. Kauranen, Sci. Total Environ., 1987, 62, 75–84 CrossRef CAS.
- C. R. Everett, G. R. Aiken and Y. P. Chin, Limnol. Oceanogr., 1999, 44, 1316–1322 CrossRef CAS.
- Y.-P. Chin, G. Aiken and E. O'Loughlin, Environ. Sci. Technol., 1994, 28, 1853–1858 CrossRef CAS PubMed.
- M. Meier, Y.-P. Chin and P. Maurice, Biogeochemistry, 2004, 67, 39–56 CrossRef CAS.
- A. Piccolo, S. Nardi and G. Concheri, Chemosphere, 1996, 33, 595–602 CrossRef CAS.
- M. B. Müller, D. Schmitt and F. H. Frimmel, Environ. Sci. Technol., 2000, 34, 4867–4872 CrossRef.
- N. Her, G. Amy, D. Foss, J. Cho, Y. Yoon and P. Kosenka, Environ. Sci. Technol., 2002, 36, 1069–1076 CrossRef CAS.
- N. Her, G. Amy, D. McKnight, J. Sohn and Y. Yoon, Water Res., 2003, 37, 4295–4303 CrossRef CAS.
- T. Nissinen, I. Miettinen, P. Martikainen and T. Vartiainen, Chemosphere, 2001, 45, 865–873 CrossRef CAS.
- T. Brinkmann, P. Hörsch, D. Sartorius and F. Frimmel, Environ. Sci. Technol., 2003, 37, 4190–4198 CrossRef CAS.
- R. D. Vogt, J. Akkanen, D. O. Andersen, R. Brüggemann, B. Chatterjee, E. Gjessing, J. V. Kukkonen, H. E. Larsen, J. Luster and A. Paul, Aquat. Sci., 2004, 66, 195–210 CrossRef CAS.
- C. Specht and F. H. Frimmel, Environ. Sci. Technol., 2000, 34, 2361–2366 CrossRef CAS.
- T. Reemtsma and A. These, Anal. Chem., 2003, 75, 1500–1507 CrossRef CAS.
- L. Persson, T. Alsberg, A. Ledin and G. Odham, Chemosphere, 2006, 64, 1093–1099 CrossRef CAS PubMed.
- J. Peuravuori and K. Pihlaja, Anal. Bioanal. Chem., 2007, 389, 475–491 CrossRef CAS PubMed.
- J. Peuravuori and K. Pihlaja, Environ. Sci. Technol., 2004, 38, 5958–5967 CrossRef CAS.
- N. Kawasaki, K. Matsushige, K. Komatsu, A. Kohzu, F. W. Nara, F. Ogishi, M. Yahata, H. Mikami, T. Goto and A. Imai, Water Res., 2011, 45, 6240–6248 CrossRef CAS PubMed.
- C. Romera-Castillo, M. Chen, Y. Yamashita and R. Jaffé, Water Res., 2014, 55, 40–51 CrossRef CAS PubMed.
- R. M. B. O. Duarte, A. C. Barros and A. C. Duarte, J. Chromatogr. A, 2012, 1249, 138–146 CrossRef CAS PubMed.
- J. J. Alberts, M. Takács and P. K. Egeberg, Org. Geochem., 2002, 33, 817–828 CrossRef CAS.
- J.-H. Park, Chemosphere, 2009, 77, 485–494 CrossRef CAS PubMed.
- C. Landry and L. Tremblay, Environ. Sci. Technol., 2012, 46, 1700–1707 CrossRef CAS PubMed.
- M. Yan, D. Wang, G. Korshin and Z. Cai, Chemosphere, 2012, 87, 879–885 CrossRef CAS PubMed.
- E. Perdue, J. Ritchie, K. Turekian and H. Holland, 5.10: Dissolved organic matter in freshwaters, Elsevier, 2003 Search PubMed.
- J. D. Ritchie and E. M. Perdue, Geochim. Cosmochim. Acta, 2003, 67, 85–96 CrossRef CAS.
- S. A. Huber, A. Balz, M. Abert and W. Pronk, Water Res., 2011, 45, 879–885 CrossRef CAS PubMed.
- P. Jandera, Anal. Chim. Acta, 2011, 692, 1–25 CrossRef CAS PubMed.
- Y. Guo and S. Gaiki, J. Chromatogr. A, 2011, 1218, 5920–5938 CrossRef CAS PubMed.
- P. Jandera, J. Sep. Sci., 2008, 31, 1421–1437 CrossRef CAS PubMed.
- A. J. Alpert, M. Shukla, A. K. Shukla, L. R. Zieske, S. W. Yuen, M. A. Ferguson, A. Mehlert, M. Pauly and R. Orlando, J. Chromatogr. A, 1994, 676, 191–202 CrossRef CAS.
- A. J. Alpert, J. Chromatogr. A, 1990, 499, 177–196 CrossRef CAS.
- K. Kaiser and R. Benner, Anal. Chem., 2000, 72, 2566–2572 CrossRef CAS.
- R. J. Wicks, M. A. Moran, L. J. Pittman and R. E. Hodson, Appl. Environ. Microbiol., 1991, 57, 3135–3143 CAS.
- K. Mopper, C. A. Schultz, L. Chevolot, C. Germain, R. Revuelta and R. Dawson, Environ. Sci. Technol., 1992, 26, 133–138 CrossRef CAS.
- N. H. Borch and D. L. Kirchman, Mar. Chem., 1997, 57, 85–95 CrossRef CAS.
- K. Kaiser and R. Benner, Mar. Chem., 2009, 113, 63–77 CrossRef CAS PubMed.
- A. Engel and N. Händel, Mar. Chem., 2011, 127, 180–191 CrossRef CAS PubMed.
- D. J. Repeta and L. I. Aluwihare, Limnol. Oceanogr., 2006, 51, 1045–1053 CrossRef CAS.
- S. Sandron, R. Wilson, R. Larragy, M. V. McCaul, P. N. Nesterenko, B. Kelleher and B. Paull, Anal. Methods, 2013, 6, 107–114 RSC.
- I. Paunovic, R. Schulin and B. Nowack, Eur. J. Soil Sci., 2008, 59, 198–207 CrossRef CAS PubMed.
- R. Prugue, Honors thesis, Florida State University, 2012.
- T. Midorikawa and E. Tanoue, Mar. Chem., 1996, 52, 157–171 CrossRef CAS.
- F. Wu, R. Evans and P. Dillon, Anal. Chim. Acta, 2002, 452, 85–93 CrossRef CAS.
- S. Momchilova and B. Nikolova-Damyanova, J. Sep. Sci., 2003, 26, 261–270 CrossRef CAS PubMed.
- L. Morris, J. Lipid Res., 1966, 7, 717–732 CAS.
- W. Christie, J. High Resolut. Chromatogr., 1987, 10, 148–150 CAS.
- W. W. Christie, Prog. Lipid Res., 1994, 33, 9–18 CrossRef CAS.
- G. Dobson, W. W. Christie and B. Nikolova-Damyanova, J. Chromatogr. B: Biomed. Sci. Appl., 1995, 671, 197–222 CrossRef CAS.
- B. Nikolova-Damyanova, Advances in lipid methodology-five, The Oily Press, 2003 Search PubMed.
- C. Panagiotopoulos, D. J. Repeta and C. G. Johnson, Org. Geochem., 2007, 38, 884–896 CrossRef CAS PubMed.
- C. Panagiotopoulos, D. J. Repeta, L. Mathieu, J.-F. Rontani and R. Sempéré, Mar. Chem., 2013, 154, 34–45 CrossRef CAS PubMed.
- A. Berthod, T. Maryutina, B. Spivakov, O. Shpigun and I. A. Sutherland, Pure Appl. Chem., 2009, 81, 355–387 CrossRef CAS.
- Y. Ito, J. Chromatogr. A, 2005, 1065, 145–168 CrossRef CAS PubMed.
- Y. Ito, J. Sandlin and W. G. Bowers, J. Chromatogr. A, 1982, 244, 247–258 CrossRef CAS.
- S. Sandron, P. N. Nesterenko, M. V. McCaul, B. Kelleher and B. Paull, J. Sep. Sci., 2014, 37, 135–142 CrossRef CAS PubMed.
- F. Gelin, J. W. de Leeuw, J. S. Sinninghe Damsté, S. Derenne, C. Largeau and P. Metzger, J. Anal. Appl. Pyrolysis, 1994, 28, 183–204 CrossRef CAS.
- J. A. Leenheer and J.-P. Croué, Environ. Sci. Technol., 2003, 37, 18A–26A CrossRef CAS.
- G. Aiken and J. Leenheer, Chem. Ecol., 1993, 8, 135–151 CrossRef CAS PubMed.
- M. P. Stainton, J. Fish. Res. Board Can., 1973, 30, 1441–1445 CrossRef CAS.
- G. A. Junk, H. J. Svec, R. D. Vick and M. J. Avery, Environ. Sci. Technol., 1974, 8, 1100–1106 CrossRef CAS.
- C. M. Curran and M. B. Tomson, Groundwater Monit. Rem., 1983, 3, 68–71 CrossRef CAS PubMed.
- H. R. Schulten, J. Anal. Appl. Pyrolysis, 1999, 49, 385–415 CrossRef CAS.
- J. W. de Leeuw, P. G. Hatcher, J. C. Del Río Andrade and J. D. H. van Heemst, Acta Hydrochim. Hydrobiol., 2000, 28, 69–76 CrossRef.
- C. Saiz-Jimenez and B. Hermosin, J. Anal. Appl. Pyrolysis, 1999, 49, 337–347 CrossRef CAS.
- A. Mannino and H. R. Harvey, Geochim. Cosmochim. Acta, 1999, 63, 2219–2235 CrossRef CAS.
- A. Mannino and H. R. Harvey, Org. Geochem., 2000, 31, 1611–1625 CrossRef CAS.
- K. A. Burns, J. K. Volkman, J. A. Cavanagh and D. Brinkman, Mar. Chem., 2003, 80, 103–128 CrossRef CAS.
- A. Mannino and H. R. Harvey, Geochim. Cosmochim. Acta, 1999, 63, 2219–2235 CrossRef CAS.
- O. Kracht and G. Gleixner, Org. Geochem., 2000, 31, 645–654 CrossRef CAS.
- L. I. Aluwihare, D. J. Repeta and R. F. Chen, Deep Sea Res., Part II, 2002, 49, 4421–4437 CrossRef CAS.
- W. S. York, A. G. Darvill, M. McNeil, T. T. Stevenson and P. Albersheim, Methods in Enzymology, Academic Press, 1986 Search PubMed.
- Z. Sidorczyk, U. Zahringer and E. T. Rietschel, Eur. J. Biochem., 1983, 137, 15–22 CrossRef CAS PubMed.
- G. Jandl, H. R. Schulten and P. Leinweber, J. Plant Nutr. Soil Sci., 2002, 165, 133–139 CrossRef CAS.
- J. L. Weishaar, G. R. Aiken, B. A. Bergamaschi, M. S. Fram, R. Fujii and K. Mopper, Environ. Sci. Technol., 2003, 37, 4702–4708 CrossRef CAS.
- D. W. Page, J. A. van Leeuwen, K. M. Spark and D. E. Mulcahy, J. Anal. Appl. Pyrolysis, 2003, 67, 247–262 CrossRef CAS.
- S. W. Frazier, K. O. Nowack, K. M. Goins, F. S. Cannon, L. A. Kaplan and P. G. Hatcher, J. Anal. Appl. Pyrolysis, 2003, 70, 99–128 CrossRef CAS.
- N. Maie, C. Yang, T. Miyoshi, K. Parish and R. Jaffé, Limnol. Oceanogr., 2005, 50, 23–35 CrossRef CAS.
- J. Templier, S. Derenne, J.-P. Croué and C. Largeau, Org. Geochem., 2005, 36, 1418–1442 CrossRef CAS PubMed.
- T. M. Quan and D. J. Repeta, Mar. Chem., 2007, 105, 183–193 CrossRef CAS PubMed.
- S. Opsahl, R. Benner and P. Louchouarn, Anal. Chem., 2000, 72, 2780–2787 CrossRef.
- P. Louchouarn, R. M. W. Amon, S. Duan, C. Pondell, S. M. Seward and N. White, Mar. Chem., 2010, 118, 85–97 CrossRef CAS PubMed.
- S. Lang, S. Bernasconi and G. Fruh-Green, Rapid Commun. Mass Spectrom., 2012, 26, 9–16 CrossRef CAS PubMed.
- G. Abbt-Braun, U. Lankes and F. H. Frimmel, Aquat. Sci., 2004, 66, 151–170 CrossRef CAS.
- P. P. Schmitt-Kopplin and J. Junkers, Iupac series on analytical and physical chemistry of environmental systems, Wiley, 2007 Search PubMed.
- M. de Nobili, G. Bragato and A. Mori, J. Chromatogr. A, 1999, 863, 195–204 CrossRef CAS.
- N. Yamada and E. Tanoue, Prog. Oceanogr., 2006, 69, 1–18 CrossRef PubMed.
- N. Yamada and E. Tanoue, Limnol. Oceanogr., 2003, 48, 1037–1048 CrossRef CAS.
- E. Tanoue, S. Nishiyama, M. Kamo and A. Tsugita, Geochim. Cosmochim. Acta, 1995, 59, 2643–2648 CrossRef CAS.
- E. C. A. H. J. Tomas Bergman, Anal. Biochem., 2001, 290, 74–82 CrossRef PubMed.
- J. M. J. Powell, J. N. Sutton, C. E. Del Castillo and A. T. Timperman, Mar. Chem., 2005, 95, 183–198 CrossRef PubMed.
- W. X. Schulze, G. Gleixner, K. Kaiser, G. Guggenberger, M. Mann and E.-D. Schulze, Oecologia, 2005, 142, 335–343 CrossRef PubMed.
- B. C. Crump, G. W. Kling, M. Bahr and J. E. Hobbie, Appl. Environ. Microbiol., 2003, 69, 2253–2268 CrossRef.
- P. Schmitt-Kopplin and A. Kettrup, Electrophoresis, 2003, 24, 3057–3066 CrossRef CAS PubMed.
- P. Schmitt-Kopplin and J. Junkers, J. Chromatogr. A, 2003, 998, 1–20 CrossRef CAS.
- G. Ping, P. Schmitt-Kopplin, N. Hertkorn, W. Zhang, Y. Zhang and A. Kettrup, Electrophoresis, 2003, 24, 958–969 CrossRef CAS PubMed.
- E. G. Davies and E. Ghabbour, Humic Substances: Nature's Most Versatile Materials, Taylor & Francis, 2004 Search PubMed.
- N. Hertkorn, H. Claus, P. Schmitt-Kopplin, E. M. Perdue and Z. Filip, Environ. Sci. Technol., 2002, 36, 4334–4345 CrossRef CAS.
- C. Guéguen and C. W. Cuss, J. Chromatogr. A, 2011, 1218, 4188–4198 CrossRef PubMed.
- J. C. Giddings, Science, 1993, 260, 1456–1465 CAS.
- B. Stolpe, L. Guo, A. M. Shiller and M. Hassellöv, Mar. Chem., 2010, 118, 119–128 CrossRef CAS PubMed.
- M. Benedetti, J. F. Ranville, M. Ponthieu and J. P. Pinheiro, Org. Geochem., 2002, 33, 269–279 CrossRef CAS.
- S. A. Floge and M. L. Wells, Limnol. Oceanogr., 2007, 52, 32–45 CrossRef CAS.
- B. Lyvén, M. Hassellöv, C. Haraldsson and D. Turner, Anal. Chim. Acta, 1997, 357, 187–196 CrossRef.
- R. Beckett and B. T. Hart, Environ. Part., 1993, 2, 165–205 Search PubMed.
- R. Beckett, Z. Jue and J. C. Giddings, Environ. Sci. Technol., 1987, 21, 289–295 CrossRef CAS PubMed.
- E. Zanardi-Lamardo, C. D. Clark, C. A. Moore and R. G. Zika, Environ. Sci. Technol., 2002, 36, 2806–2814 CrossRef CAS.
- E. Zanardi-Lamardo, C. D. Clark and R. G. Zika, Anal. Chim. Acta, 2001, 443, 171–181 CrossRef CAS.
- E. Zanardi-Lamardo, C. A. Moore and R. G. Zika, Mar. Chem., 2004, 89, 37–54 CrossRef CAS PubMed.
- S. Assemi, G. Newcombe, C. Hepplewhite and R. Beckett, Water Res., 2004, 38, 1467–1476 CrossRef CAS PubMed.
- J. J. Moon, S. H. Kim and J. Cho, Colloids Surf., A, 2006, 287, 232–236 CrossRef CAS PubMed.
- C. Cuss and C. Guéguen, Anal. Chim. Acta, 2012, 733, 98–102 CrossRef CAS PubMed.
- A. D. Pifer, D. R. Miskin, S. L. Cousins and J. L. Fairey, J. Chromatogr. A, 2011, 1218, 4167–4178 CrossRef CAS PubMed.
- T. N. Reszat and M. J. Hendry, Anal. Chem., 2005, 77, 4194–4200 CrossRef CAS PubMed.
- X. Ding and Y. Duan, Mass Spectrom. Rev., 2014, 34, 449–473 CrossRef PubMed.
- F. Hernández, M. Ibáñez, T. Portolés, M. I. Cervera, J. V. Sancho and F. J. López, J. Hazard. Mater., 2015, 282, 86–95 CrossRef PubMed.
- Y. Cho, A. Ahmed, A. Islam and S. Kim, Mass Spectrom. Rev., 2014, 34, 248–263 CrossRef PubMed.
- G. C. Woods, M. J. Simpson, B. G. Pautler, S. F. Lamoureux, M. J. Lafreniere and A. J. Simpson, Geochim. Cosmochim. Acta, 2011, 75, 7226–7241 CrossRef CAS PubMed.
- M. M. Swenson, Ph.D thesis, University of Minnesota Duluth, 2014.
- G. Aiken, D. McKnight, K. Thorn and E. Thurman, Org. Geochem., 1992, 18, 567–573 CrossRef CAS.
- C. Lu and F. Su, Sep. Purif. Technol., 2007, 58, 113–121 CrossRef CAS PubMed.
- S. Gur-Reznik, I. Katz and C. G. Dosoretz, Water Res., 2008, 42, 1595–1605 CrossRef CAS PubMed.
- P. Raimbault, W. Pouvesle, F. Diaz, N. Garcia and R. Sempéré, Mar. Chem., 1999, 66, 161–169 CrossRef CAS.
- G. V. Korshin, C.-W. Li and M. M. Benjamin, Water Res., 1997, 31, 1787–1795 CrossRef CAS.
- S. Liu, M. Lim, R. Fabris, C. W. Chow, M. Drikas, G. Korshin and R. Amal, Water Res., 2010, 44, 2525–2532 CrossRef CAS PubMed.
- L. Liu, C. Song, Z. Yan and F. Li, Chemosphere, 2009, 77, 15–21 CrossRef CAS PubMed.
- A. Bricaud, A. Morel and L. Prieur, Limnol. Oceanogr.: Methods, 1981, 26, 43–53 CrossRef CAS.
- H. R. Schulten and G. Gleixner, Water Res., 1999, 33, 2489–2498 CrossRef CAS.
- A. J. King, J. W. Readman and J. L. Zhou, Anal. Chim. Acta, 2004, 523, 259–267 CrossRef CAS PubMed.
- S. J. Goldberg, C. A. Carlson, M. Brzezinski, N. B. Nelson and D. A. Siegel, Geophys. Res. Lett., 2011, 38 CrossRef CAS.
- C. Panagiotopulos and R. Sempéré, Limnol. Oceanogr.: Methods, 2005, 3, 419–454 CrossRef.
- T. Midorikawa and E. Tanoue, Mar. Chem., 1998, 62, 219–239 CrossRef CAS.
- E. C. Minor, C. J. Steinbring, K. Longnecker and E. B. Kujawinski, Org. Geochem., 2012, 43, 1–11 CrossRef CAS PubMed.
- T. Brown and J. Rice, Anal. Chem., 2000, 72, 384–390 CrossRef CAS.
- L. M. de Souza, T. R. Cipriani, M. Iacomini, P. A. Gorin and G. L. Sassaki, J. Pharm. Biomed. Anal., 2008, 47, 59–67 CrossRef CAS PubMed.
- L. L. Zhang and Y. M. Lin, Molecules, 2008, 13, 2986–2997 CrossRef CAS PubMed.
- E. Kaiser, A. J. Simpson, K. J. Dria, B. Sulzberger and P. G. Hatcher, Environ. Sci. Technol., 2003, 37, 2929–2935 CrossRef CAS.
- C. Piccini, D. Conde, J. Pernthaler and R. Sommaruga, Photochem. Photobiol. Sci., 2009, 8, 1321–1328 CAS.
- U. Müller, M. Rätzsch, M. Schwanninger, M. Steiner and H. Zöbl, J. Photochem. Photobiol., B, 2003, 69, 97–105 CrossRef.
- S. Q. Lang, M. D. Lilley and J. I. Hedges, Mar. Chem., 2007, 103, 318–326 CrossRef CAS PubMed.
- O. J. Lechtenfeld, B. P. Koch, W. Geibert, K. U. Ludwichowski and G. Kattner, Anal. Chem., 2011, 83, 8968–8974 CrossRef CAS PubMed.
- D. B. Wagoner, R. F. Christman, G. Cauchon and R. Paulson, Environ. Sci. Technol., 1997, 31, 937–941 CrossRef CAS.
- D. R. Lovley and E. J. Phillips, Appl. Environ. Microbiol., 1986, 51, 683–689 CAS.
- C. A. Tumang, A. V. Krusche, R. L. Victoria and J. E. Richey, Acta Amazonica, 2009, 39, 397–404 CrossRef CAS.
- D. C. Johnson, D. Dobberpuhl, R. Roberts and P. Vandeberg, J. Chromatogr. A, 1993, 640, 79–96 CrossRef CAS.
- E. D. Dodds, M. R. McCoy, L. D. Rea and J. M. Kennish, Lipids, 2005, 40, 419–428 CrossRef CAS.
- X. Zhou and P. J. Wangersky, Mar. Chem., 1987, 20, 211–218 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Further details of the membrane-filtration approaches in the isolation of DOM (Table S1) and further details of the solid phase extraction approaches to the isolation of DOM (Table S2). See DOI: 10.1039/c5em00223k |
| This journal is © The Royal Society of Chemistry 2015 |