
Recent advances in C–S bond formation via C–H bond functionalization and decarboxylation
Chao
Shen
ab,
Pengfei
Zhang
*a,
Qiang
Sun
c,
Shiqiang
Bai
d,
T. S. Andy
Hor
*cd and
Xiaogang
Liu
*cd
aCollege of Material Chemistry and Chemical Engineering, Hangzhou Normal University, Hangzhou, 310036, China. E-mail: chxyzpf@hotmail.com
bCollege of Biology and Environmental Engineering, Zhejiang Shuren University, Hangzhou 310015, China
cDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, Singapore, 117543, Singapore. E-mail: chmandyh@nus.edu.sg; chmlx@nus.edu.sg
dInstitute of Materials Research and Engineering, Agency for Science and Technology and Research, 3 Research Link, Singapore, 117602, Singapore. E-mail: andyhor@imre.a-star.edu.sg; liux@imre.a-star.edu.sg
First published on 13th October 2014
Abstract
The development of mild and general methods for C–S bond formation has received significant attention because the C–S bond is indispensable in many important biological and pharmaceutical compounds. Early examples for the synthesis of C–S bonds are generally limited to the condensation reaction between a metal thiolate and an organic halide. Recent chemical approaches for C–S bond formation, based upon direct C–H bond functionalization and decarboxylative reactions, not only provide new insights into the mechanistic understanding of C–S coupling reactions but also allow the synthesis of sulfur-containing compounds from more effective synthetic routes with high atom economy. This review intends to explore recent advances in C–S bond formation via C–H functionalization and decarboxylation, and the growing opportunities they present to the construction of complex chemical scaffolds for applications encompassing natural product synthesis, synthetic methodology development, and functional materials as well as nanotechnology.
 Chao Shen | Chao Shen was born in Zhejiang Province, China in 1983. He earned his BS degree in 2006 from Hangzhou Normal University. He received his MS degree (2009) from Hangzhou Normal University under the supervision of Prof. Pengfei Zhang and completed his PhD (2012) at Zhejiang University under the supervision of Prof. Xinzhi Chen and Prof. Pengfei Zhang. Then he joined the College of Biology and Environmental Engineering at Zhejiang Shuren University. His current research interest focuses on developing a new methodology toward sulfur-containing molecules and sugar-based heterocycles. |
 Pengfei Zhang | Pengfei Zhang was born in Anhui, China. He obtained his Bachelor's degree (1986) from Huaibei Normal University, China, and completed his PhD degree (2001) at Zhejiang University under the supervision of Prof. Zhenchu Chen. Currently, he is a professor in the College of Science at Hangzhou Normal University. His current research effort focuses on developing green and efficient synthetic methodologies for coupling reaction, carbohydrate chemistry, enzymatic catalytic reaction, and chemical industry. |
 Qiang Sun | Qiang Sun was born in Shandong, China. He received his BS (2007) and MS (2010) degrees in Applied Chemistry from Qingdao Agricultural University, under the supervision of Prof. Jinsheng Shi. He is currently pursuing a PhD degree under the supervision of Prof. Xiaogang Liu in the Department of Chemistry at the National University of Singapore. His research interest focuses on materials synthesis and optical characterizations. |
 Shiqiang Bai | Shiqiang Bai was born in Shandong, China. He received his BS (2000) degree in Chemistry Education and MS (2003) degree in Inorganic Chemistry under the supervision of Prof. Enqing Gao from Qufu Normal University. He completed his PhD (2006) in Inorganic Chemistry at Peking University under the supervision of Prof. Chunhua Yan. He then worked as a research fellow in the group of Prof. Andy Hor at the National University of Singapore in 2006. He is now a research scientist of the Institute of Materials Research and Engineering, Agency for Science, Technology and Research. His research interest focuses on novel organic–inorganic hybrid materials for catalysis and photoluminescence applications. |
 T. S. Andy Hor | T. S. Andy Hor obtained his BSc (Hon) from Imperial College, DPhil from Oxford University and postdoctorate in Yale University. He is the Executive Director of IMRE (A*STAR) and Professor of Chemistry and Fellow of the Teaching Academy of the National University of Singapore and Concurrent Professor of Fudan University (China). He is Fellow of the Singapore National Academy of Science (SNAS), Fellow of the Royal Society of Chemistry (FRSC), President of the Singapore National Institute of Chemistry (SNIC), and President of the Federation of the Asian Chemical Societies (FACS). His research interests are heterometallic materials, organometallic catalysis and supramolecular self-assembly, and has published ∼340 ISI paper with ∼500 annual citations in recent years. |
 Xiaogang Liu | Xiaogang Liu was born in Jiangxi, China. He earned his BE degree in Chemical Engineering from Beijing Technology and Business University, China. He received his MS degree in Chemistry from East Carolina University and completed his PhD at Northwestern University. He then worked as a postdoctoral fellow at MIT for two years before joining the faculty of the National University of Singapore. He holds a joint appointment with the Institute of Materials Research and Engineering. His interests include luminescent materials synthesis, supramolecular chemistry, transition metal-based catalysis, and surface science for sensing, optoelectronic and biomedical applications. |
1. Introduction
Sulfur, also known as brimstone (burn stone), is a reactive nonmetal found in nature and all living organisms.1 It is relatively scarce in the Earth's crust (∼6%). However, it is the third most abundant mineral after calcium and phosphorus in the human body. Sulfur bonding plays an important role in mediating electron transfer reactions and maintaining the strength and shape of proteins.2 For example, the presence of sulfur moieties is required for covalent assembly of naturally occurring polyketides and nonribosomal peptides.3 Biological macromolecules, such as enzymes and transfer ribonucleic acids (tRNAs), also contain a large amount of sulfur centers required for controlling their biological activity.4 The widespread attention to sulfur-based compounds is mainly due to their potential as novel pharmaceutical, agricultural and chemical agents.5,6 Most notably among these are sulfonamides,7 which have been extensively used as antibacterials, anticonvulsants, hypoglycemics and human immunodeficiency virus (HIV) protease inhibitors.8 Moreover, organic sulfur compounds are essential in materials science, in which the sulfur constituent can have a profound effect on the physical, electronic, and surface properties of the resultant materials.9The construction of sulfur-containing compounds by chemical synthesis through simple C–S bond-forming reactions is of utmost importance in synthetic and catalytic research fields.10 Much of the work on C–S bond formation has been devoted to direct coupling of organic halides with thiols and the addition of thiols to unsaturated C–C bonds under free-radical or metal-catalyzed conditions.11 Approaches that have proven successful for making sulfur-containing chiral compounds with high optical purity include asymmetric sulfa-Michael addition reaction,12 allylic sulfonation,13 and Diels–Alder reaction.14 Despite their usefulness in forming C–S bonds, these methods have significant limitations, requiring either the implementation of metal–ligand combination or highly pre-functionalized precursors. An important task for synthetic chemists is, therefore, to simplify the process of the C–S bond-forming reaction and minimize the amount of waste formed. Indeed, the current understanding of the mechanisms that govern the C–S coupling is surprisingly limited when compared to the knowledge gained from C–C or C–N coupling.15
In this review, we attempt to bring the readers up to date in the rapidly expanding field of research on C–S bond formation. Specifically, we will cover recent advances in catalytic C–S coupling processes involving C–H functionalization and decarboxylative reactions (Scheme 1). An emphasis will be placed on examining the scope and limitation of these catalytic transformations, highlighting gaps in knowledge and research, and understanding the mechanisms underlying the C–S bond-forming reactions. We conclude with a discussion of the likely directions of future research in the field.
 | ||
| Scheme 1 Catalytic C–S bond-forming processes via decarboxylative and C–H functionalization reactions. | ||
2. C–S coupling via C–H bond functionalization
The capacity to activate ubiquitous, but often inert C–H bonds has far-reaching implications in hydrocarbon functionalization that requires the cleavage of at least one C–H bond. Generally, C–H bonds are difficult to cleave directly due to their thermodynamic stability (calc. 420 kcal mol−1 for primary C–H bonds). Historically, C–C bond formation in alkanes via C–H bond activation usually relied on radical chemistry. The selective functionalization of a C–H bond with a more versatile sulfur group is less common because of the lack of suitable synthetic reagents that can tolerate the presence of sulfur functional groups. To expand this area in organic synthesis, the challenge is to identify new transition metal complexes that can overcome the thermodynamic and kinetic barriers to C–H bond activation and perform such difficult transformation with high fidelity.2.1 C–H activation catalyzed by transition metals
The utilization of C–H activation through choice of transition metals to form carbon–heteroatom bonds constitutes a powerful tool in pharmaceutical and medicinal chemistry.16 The wide range of oxidation states in transition metals accounts for their ability to act as ideal catalysts for C–H activation. During the past decade, numerous critical reviews on transition metal-catalyzed formation of C–C bonds via C–H activation have appeared. Despite the promising aspects of C–H activation catalyzed by transition metals, its utility for C–S bond forming reactions became known only until recently.17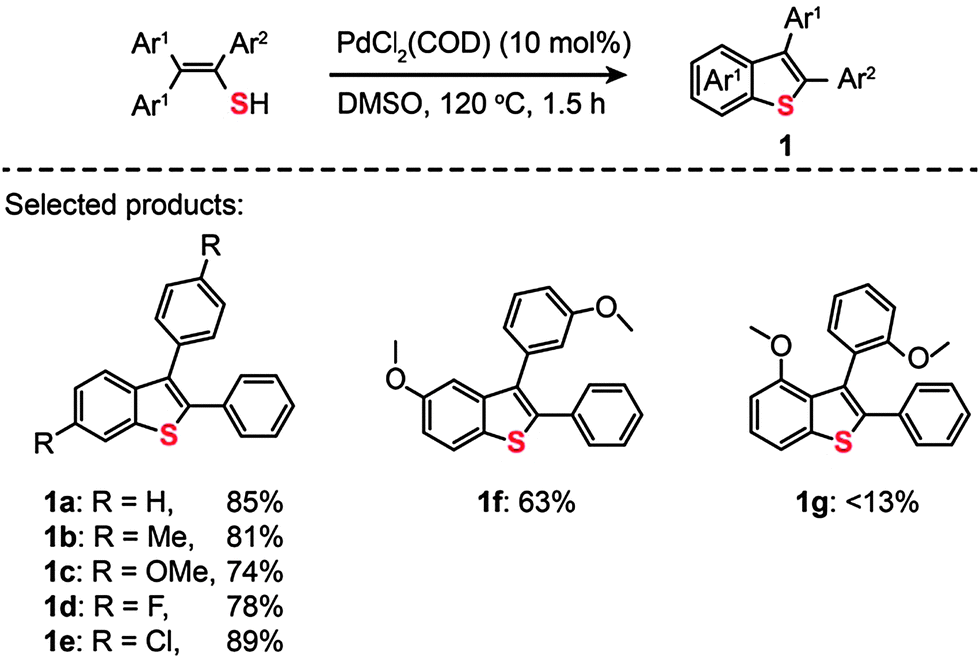 | ||
| Scheme 2 Inamoto's Pd(II)-catalyzed direct synthesis of benzo[b]thiophenes.18 | ||
 | ||
| Scheme 3 Inamoto's Pd(II)-catalyzed synthesis of 2-arylbenzothiazoles.19 | ||
In 2010, the same group designed new Pd(II)-based catalytic systems and succeeded in the cyclization of thiobenzanilides and thioureas to produce 2-aryl-benzothiazoles and 2-aminobenzothiazoles, respectively.20 Their protocol allowed molecular oxygen to be used as a reoxidant in combination with a Pd(II) catalyst. The use of molecular oxygen for aerobic oxidation in metal-catalyzed reactions is attractive from an environmental point of view. More recently, the authors took one step further and reported an intriguing system involving water as a reaction medium for Pd(II)-catalyzed cyclization of thiobenzanilides.21 Reactions proceeded under considerably mild conditions, providing a greener alternative to conventional methods for the synthesis of 2-arylbenzothiazoles. For a number of substrates, the addition of an amphiphilic surfactant greatly facilitates the cyclization reaction.
In a parallel investigation, Batey and Joyce developed a cocatalytic Pd(0)/Mn(IV) system toward the synthesis of 2-aminobenzothiazole (3) via an oxidative C–H functionalization/C–S bond formation (Scheme 4).22a The cocatalyst system includes Pd(PPh3)4 (3 mol%) and MnO2 (10 mol%) dispersed in CH3CN under an oxygen atmosphere. Using this synthetic strategy, various substituted N-arylthioureas could be converted to their corresponding products (3) in moderate to high yields under aerobic reaction conditions. To probe the mechanism of this reaction, the authors carried out a free-radical trap test. The reaction proceeded in the presence of galvinoxyl (30 mol%), precluding the existence of the radical pathway. The competing kinetic isotope effect (KIE) experiment also indicates that an electrophilic palladation mechanism does not hold.22b,c Based on these observations, the authors argued that the cyclization reaction occurs through C–H bond activation over a σ-bond metathesis, wherein proton abstraction is promoted by the Pd(II)–peroxo complex formed at the transition states (Scheme 4).
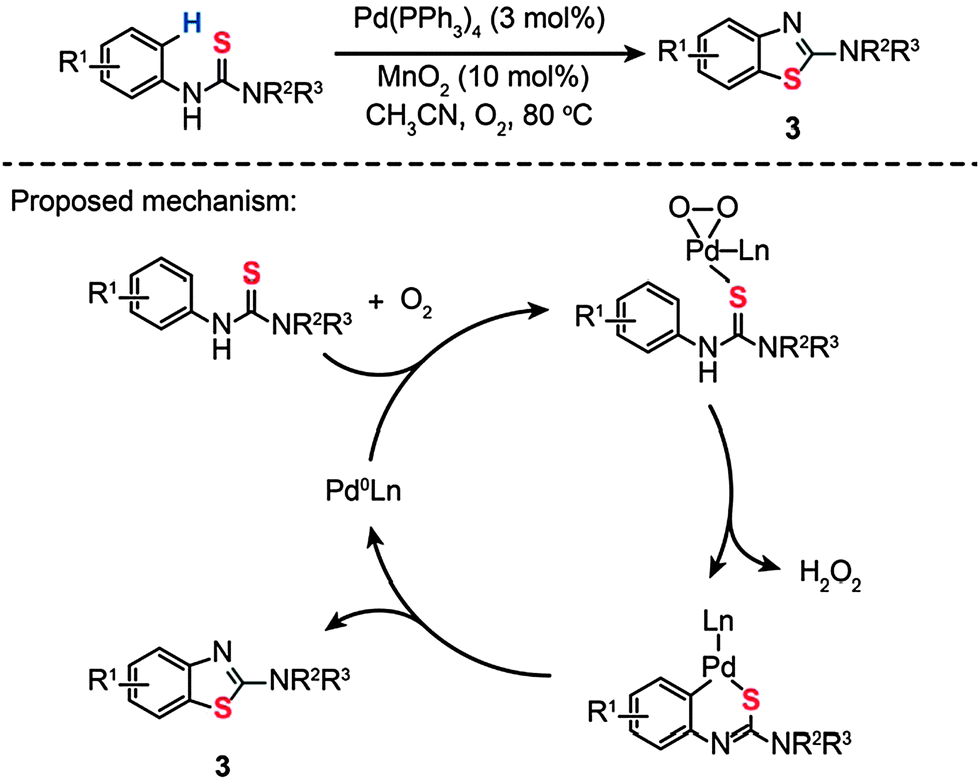 | ||
| Scheme 4 Batey's Pd(0)-catalyzed synthesis of 2-aminobenzothiazole.22a | ||
Sulfones constitute an interesting class of compounds that contain hexavalent sulfur atoms doubly bonded to two oxygen atoms and singly linked to two carbon atoms. Some of these compounds have found use in making plastics and as antibiotics to treat cancers. In 2009, the Dong group reported a Pd-catalyzed C–H bond activation/C–S cross-coupling with arylsulfonyl chlorides to produce sulfones (Scheme 5).23 Using a catalytic amount of PdCl2(CH3CN)2 (10 mol%), arylpyridine, arylpyrazole, and aryloxime ether substrates were successfully coupled to a stoichiometric amount of arylsulfonyl chloride to afford the corresponding diarylsulfone products (4a–f). The researchers found that the reaction conditions were tolerant of electron-donating (e.g., p-MeO) and electron-withdrawing (e.g., p-CF3) groups. However, aliphatic sulfonyl chlorides underwent decomposition under identical reaction conditions. Interestingly, competing C–H chlorination and arylation reactions could take place when the solvent, cocatalyst, and temperature were changed. The authors argued that the C–S bond-forming processes likely occurred through either a Pd(II)/Pd(0) or Pd(II)/Pd(IV) catalytic cycle.
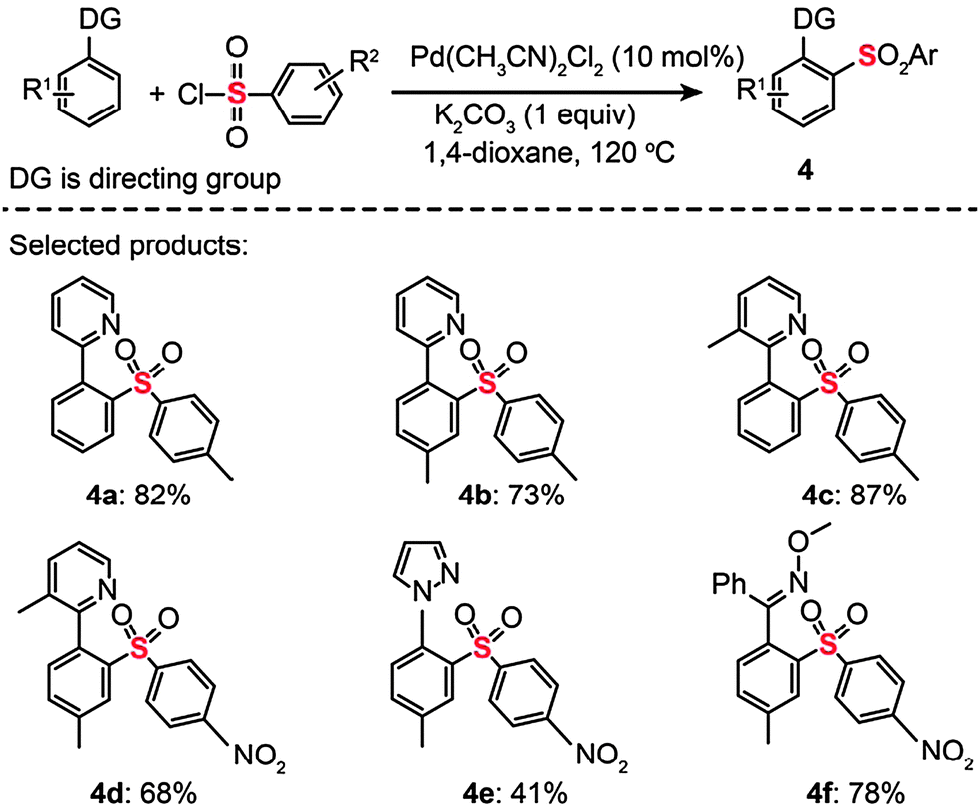 | ||
| Scheme 5 Dong's Pd(II)-catalyzed C–H bond sulfonylation.23 | ||
In 2010, Wu and co-workers reported the synthesis of 2-trifluoromethylbenzothiazoles shown in Scheme 6.24a These benzothiazoles (5a–e) were formed by the treatment of trifluoromethylimidoyl chlorides and sodium hydrosulfide hydrate via Pd-catalyzed C–H bond activation. Optimal reaction conditions for benzothiazole formation were found to be pre-heating of the imidoyl chloride substrate and sodium hydrosulfide hydrate for half an hour via a traditional cross-coupling reaction. Then the reaction was followed by addition of a PdCl2 catalyst (5 mol%) and heating of the resultant mixture at 110 °C for 3 h to complete the oxidative cross-coupling reaction. The reactions showed good tolerance to a number of functional groups such as the cyano, trifluoromethyl, and halogen groups. An attractive aspect of these Pd(II)-catalyzed reactions is the elimination of additives or oxidants typically required for this type of organic transformation. Recently, Liang and co-workers reported a similar double C–S bond formation reaction for the synthesis of benzothiazoles in which the C–S bond formation proceeded via both traditional cross-coupling and oxidative cross-coupling reactions.24b
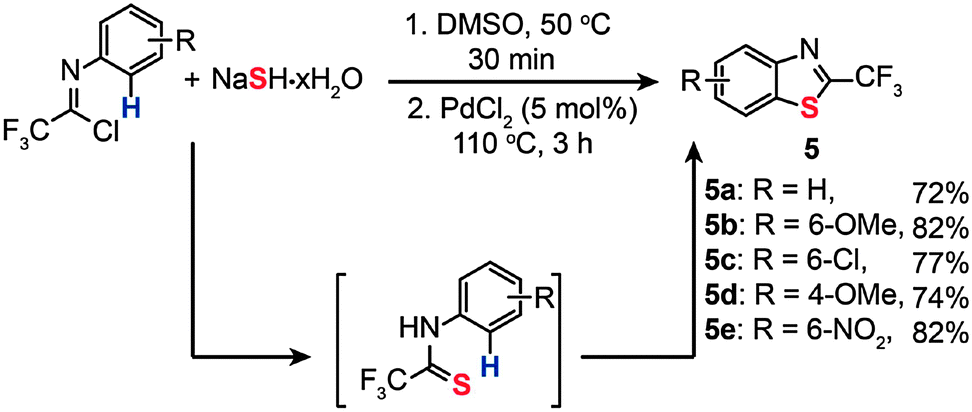 | ||
| Scheme 6 Wu's synthesis of various 2-trifuoromethylbenzothiazoles.24a | ||
Dibenzothiophene is an organosulfur compound harboring a thiophene skeleton fused to two benzene rings. It eluded synthetic attempts despite its importance as a model compound representative of thiophenic structures found in coal and oil. In 2011, Samanta and Antonchick devised a solution to this synthetic challenge by introducing an efficient double C–H activation method (Scheme 7).25 The authors found a way to synthesize dibenzothiophene (6) and its derivatives from benzyl phenyl sulfoxide precursors, which were catalyzed by PdCl2 (15 mol%) at 110 °C in the presence of AgOAc (2 equiv.) and p-fluoroiodobenzene (2 equiv.). The sulfoxides bearing electron-withdrawing or -donating groups were converted to the corresponding dibenzothiophene products in good yields. On the basis of their mechanistic studies, the authors postulated that Pd(II) acts as an electrophile and coordinates to the sulfur atom, resulting in the formation of a dinuclear species (Scheme 7). Subsequent oxidative addition of aryl iodide gives rise to a mononuclear complex of Pd(IV), which undergoes reductive elimination and C–H bond activation to yield a cyclic sulfoxide intermediate. In the presence of Pd(OAc)2 and AgOAc, the sulfoxide undertakes a Pummerer rearrangement to afford a mercaptoaldehyde.26 Concurrent with this Pummerer rearrangement, dibenzothiophene is formed by Pd(II)-catalyzed C–H activation via the formation of palladacycle intermediate H.
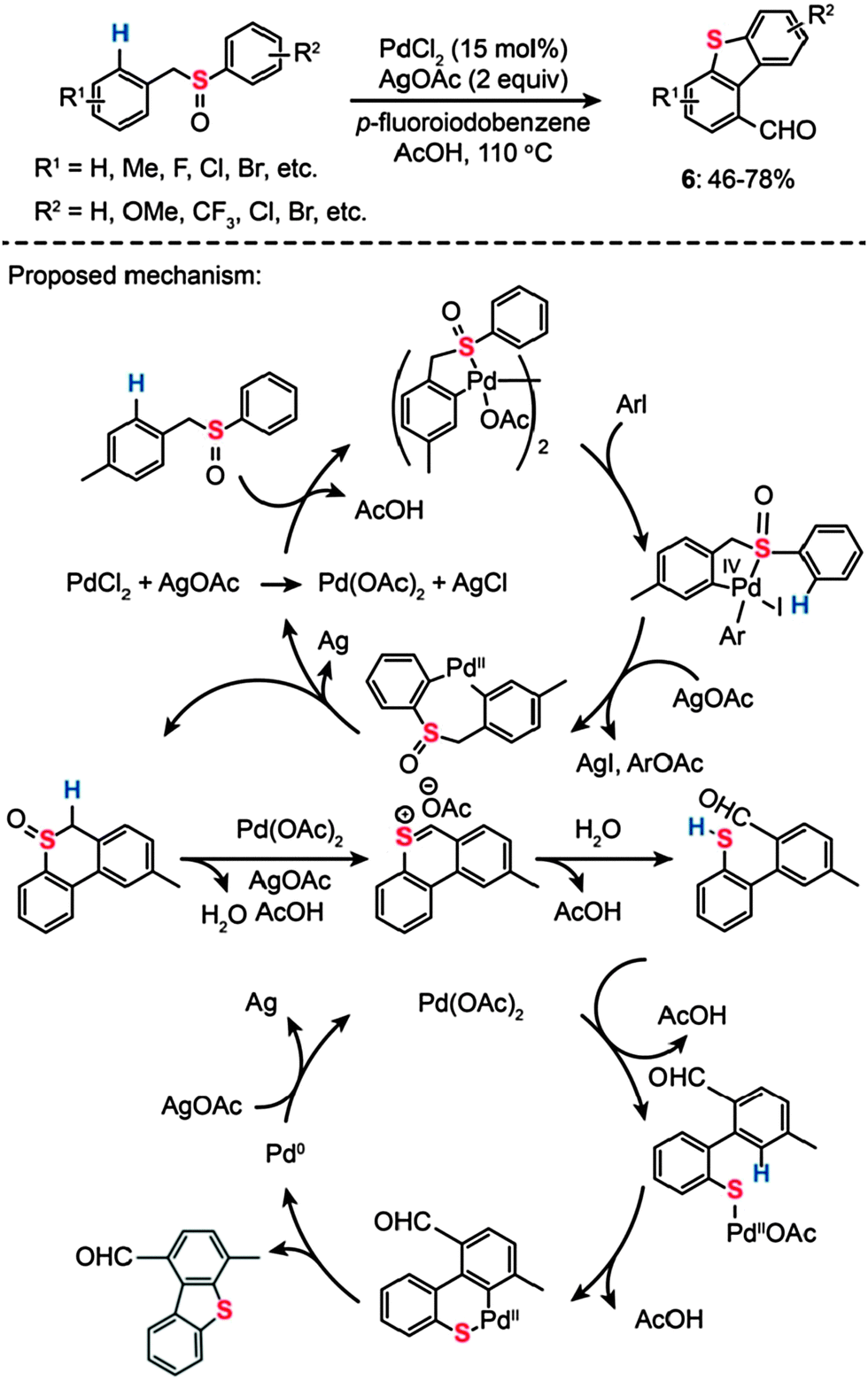 | ||
| Scheme 7 Antonchick's Pd(II)-catalyzed sulfoxide-directed synthesis of dibenzothiophenes.25 | ||
Recently, we reported a surprising discovery of synthesizing a wide range of sugar-functionalized benzothiazoles through Pd(II)-catalyzed, chemselective intramolecular C–S coupling of glycosyl thiourea substrates (Scheme 8).27 Organic compounds comprising sugar moieties have shown enormous potential for use as tumor markers and receptors, while benzothiazoles have found important therapeutic applications for treatment of diabetes and many forms of cancers. We reasoned that glycosyl benzothiazoles incorporating a combination of sugar and benzothiazole moieties would display synergistic therapeutic properties. After careful screening of metal catalysts, solvents and reaction temperatures, we found that a target glycosyl benzothiazole (7) can be prepared by choosing Pd(COD)Cl2 (10 mol%) and Bu4NBr (2 equiv.) heated at 100 °C in dimethyl sulfoxide (DMSO) after 12 h. Corroborated by theoretical calculations, our mechanistic investigations suggest that the coordination to the palladium by a pivaloyl carbonyl group and the presence of intramolecular hydrogen bonding play critical roles in dictating the efficiency and chemoselectivity of the process. The formation of a Pd(II)–arene complex shown in Scheme 8, promoted by Pd–S and Pd–O coordination, is believed to be the reaction pathway to sugar-based benzothiazole. Subsequent cyclopalladation of the Pd(II)–arene complex proceeds to form a cyclopalladated intermediate. Reductive elimination of this intermediate leads to the corresponding glycoconjugate, with concomitant generation of Pd(II) by the reoxidation of the Pd(0) species. To illustrate the principle of using the sugar-based benzothiazoles as biomarkers, we further assessed their antitumor activity and imaged the newly synthesized compounds in live mammalian cells.
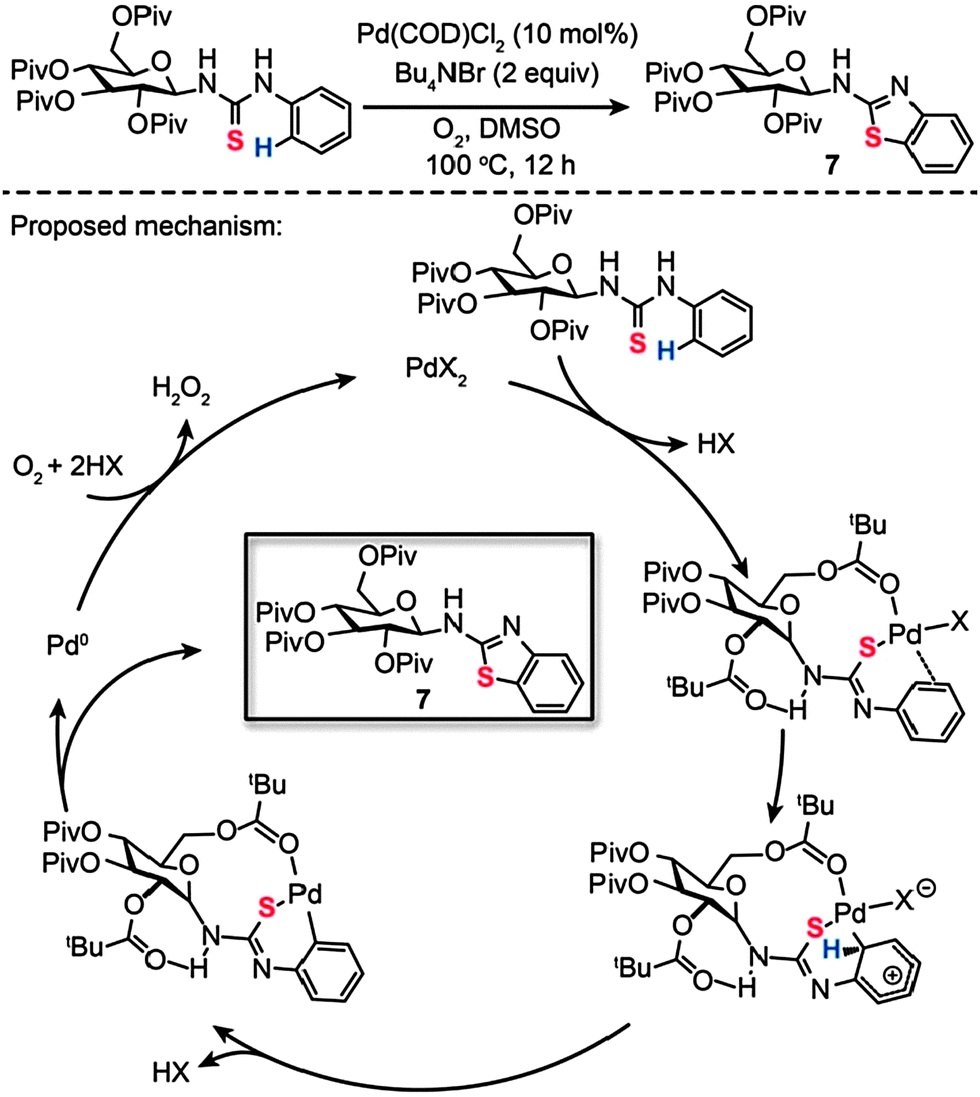 | ||
| Scheme 8 Zhang and Liu's Pd(II)-catalyzed intramolecular C–S bond formation for synthesis of sugar-based benzothiazoles.27 | ||
In 2012, Patel and co-workers found that regioselective formation of intramolecular C–S bonds (8 and 9) can take place during 2-aminobenzothiazole synthesis from 2-halo-substituted thioureas using either PdCl2 or CuI as the catalyst (Scheme 9).28 In most cases, the reactions for substrates with less reactive 2-halo groups proceeded via the C–H activation with the palladium in preference to the dehalogenation pathway. The specificity of the reactions appears to be controlled by the choice of the metal catalyst and the substituents on the aryl thioureas. Preliminary mechanistic studies suggested that the catalytic cycle is initiated by electrophilic attack of Pd(II) on the arene with subsequent transmetallation and reductive elimination to provide the desired benzothiazole products.
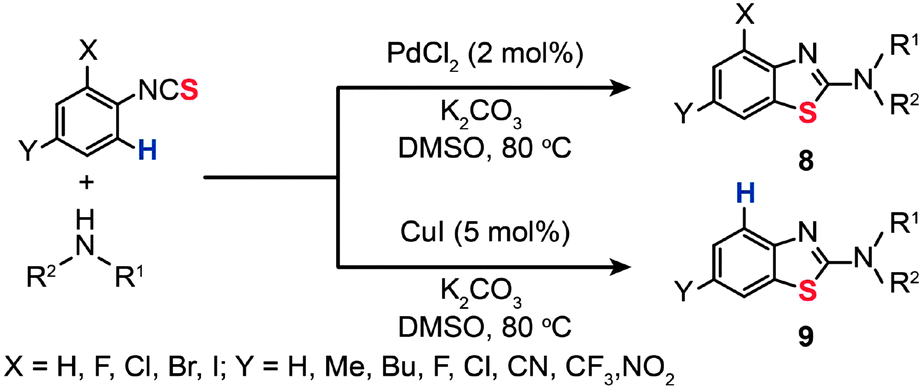 | ||
| Scheme 9 Patel's Pd/Cu-catalyzed synthesis of benzothiazoles.28 | ||
In 2011, Duan and co-workers reported the synthesis of dibenzothiophenes (10) and fulvenes through a catalytic ring-opening reaction of bromothiophenes with alkynes. The authors made use of Pd(II)-catalyzed C–H bond functionalization and the ring-rearrangement reaction of the bromothiophene to form a new C–S bond between the cleaved sulfur moiety and its neighboring phenyl group (Scheme 10).29 On the basis of related control experiments, the authors proposed that the catalytic cycle involves formation of a vinylic palladium intermediate, cycloaddition with an alkyne and reductive elimination of a six-membered palladacycle. More recently, the same group developed a new Pd(II)-catalyzed reaction of simple thiophenes with alkynes through domino C–H/C–S bond activation.30 Compared with their earlier work on dibenzothiophenes, no reactive α-C–Br bond was needed. During the course of the study, the authors found that the amount of the salt used had a significant impact on the efficiency and specificity of the reactions.
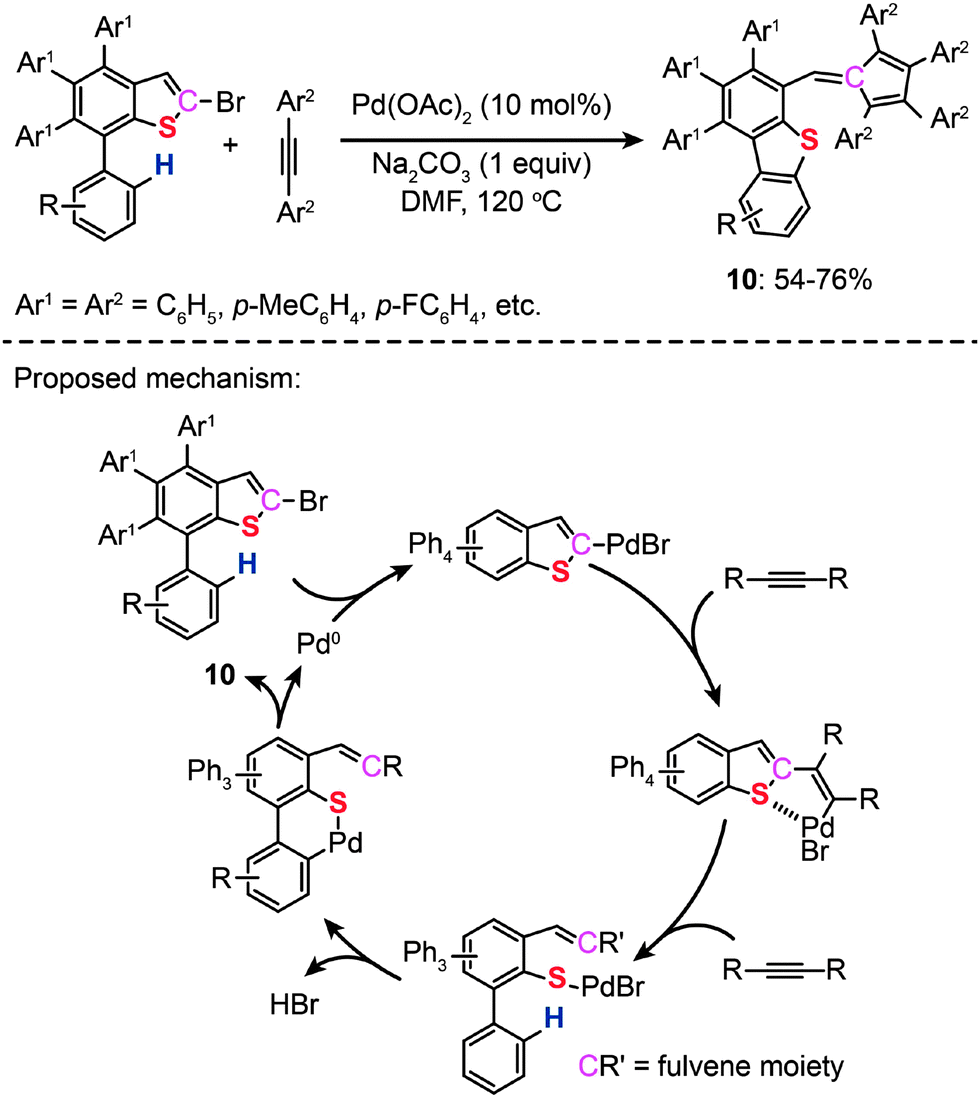 | ||
| Scheme 10 Duan's formation of bromobenzothiophenes with alkynes.29 | ||
More recently, Xu and Shen have shown that Pd(CH3CN)4(OTf)2 (10 mol%) is a more suitable homogeneous catalyst for highly selective intermolecular C–H monotrifluoromethylthiolation (11) in AcOH, also showing good tolerance to a variety of functional groups (Scheme 11).31 A kinetic isotope effect was observed by comparing the rate of an intramolecular reaction involving a C–H bond to that of the reaction involving a C–D bond, suggesting that the turnover limiting step of the catalytic trifluoromethylthiolation reaction did not involve C–H bond activation. On the basis of mechanistic investigations, the authors argued that the reaction was initiated by cyclopalladation of 2-pyridylbenzene with Pd(CH3CN)4(OTf)2, followed by oxidative addition of the N-SCF3 bond to form a trifluoromethylthiosubstituted Pd(III) or Pd(IV) intermediate. Concurrent with the yield of the desired product, the Pd(II) species was regenerated by reductive elimination (Scheme 11).
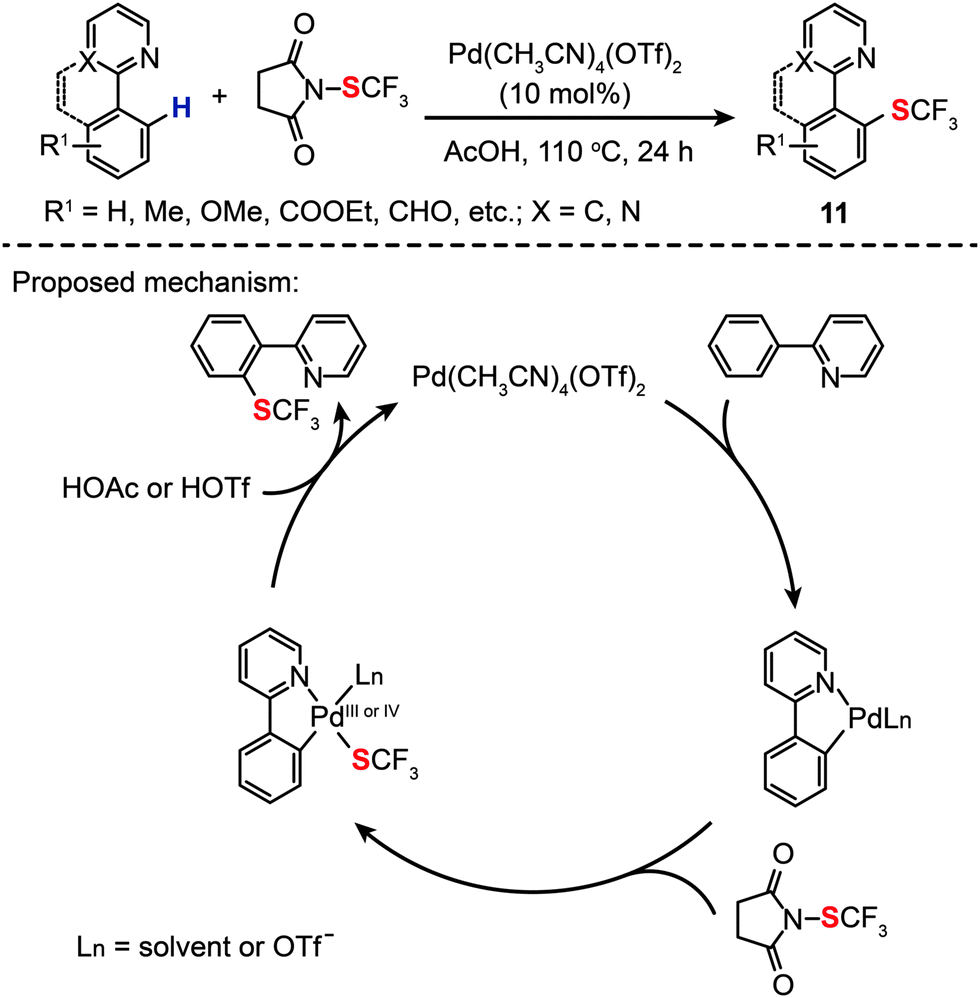 | ||
| Scheme 11 Shen's intramolecular C–H trifuoromethylthiolation of arenes.31 | ||
Recently, Nishihara and co-workers reported direct thiolation of aryl C–H bonds in arenes with disulfides or thiols to afford thiolated products (12a–i) (Scheme 12).32 The reaction proceeded with ortho-selectivity attributable to the directing groups such as 2-(3-methyl)pyridyl, 2-quinolyl, 2-pyrimidyl, and bidentate 8-aminoquinoline. This protocol tolerates a variety of functional groups and substrates under mild oxidative reaction conditions. A drawback of the protocol is the requirement for an inert atmosphere and a high reaction temperature (140 °C), which may limit its range of applications.
 | ||
| Scheme 12 Nishihara's direct thiolation of arenes with diphenyl disulfide.32 | ||
Palladium-catalyzed cross-coupling reactions of organic or organometallic reagents have emerged as a powerful tool for C–S bond construction via C–H functionalization. The emergence of this direct coupling method in synthesis arises from both the advantage of simplified reaction steps and the benefit of having low-level waste made up of non-halogenated compounds. Additionally, expensive phosphine ligands are not really a necessity – unlike in the case of conventional Pd-catalyzed C–S cross-couplings, whereupon the phosphine ligands are generally required for enhanced conversion efficiency. However, further development of Pd-catalyzed C–S couplings via C–H bond activation remains a challenge as one has to screen a vast combination of ligands, metals, and conditions in order to identify a particular set of parameters for the coupling reactions. Design of experiments involving combinatorial methods has to be developed for the discovery of highly efficient catalytic systems.
In 2006, Yu and co-workers demonstrated the cross-coupling of 2-phenylpyridine with thiophenol or dimethyl disulfide to yield thiolated products (13a and 13b) through Cu(OAc)2-catalyzed C–H functionalization (Scheme 13).35 The combined use of the soluble, inexpensive Cu(II) complex as the catalyst and O2 as a stoichiometric oxidant offers a practical advantage. This newly discovered reaction tolerates a variety of functional groups (such as alkene, alkoxy, and aldehyde), avoids the use of air-sensitive additives, and overcomes some of the limitations associated with Pd(0)-catalyzed analogues. No isotope effect was observed in an intramolecular competition experiment, suggesting a different reaction mechanism from Pd-catalyzed functionalization reactions typically governed by the isotope effect. The authors argued that a SET from the aryl ring to the pyridine-coordinated Cu(II) species leading to a cation-radical intermediate is the rate-limiting step. Their control reaction using a biphenyl group did not proceed under the identical conditions, suggesting the necessity of the pyridine–Cu(II) coordination for the SET process.
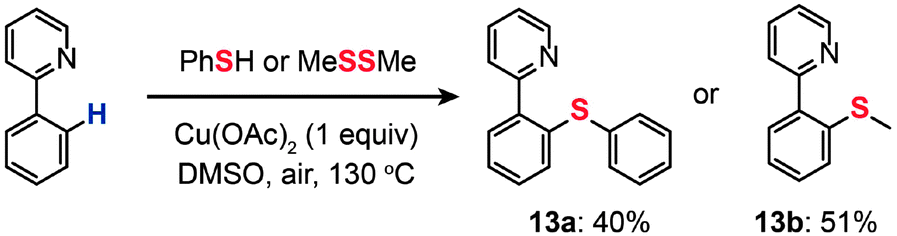 | ||
| Scheme 13 Yu's thioetherification of aryl C–H bonds.35 | ||
In 2009, Srogl and coworkers developed a Cu(I)-catalyzed intramolecular oxidation of iminodisulfides under an argon atmosphere. Under the optimal conditions, the starting iminodisulfide material was converted to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 2,3-dihydro-2-arylbenzothiazole (14a) and 2-arylbenzothiazole (14b) (Scheme 14, Path a).36a One year later, they presented an improved approach to synthesizing benzothiazoles (15) by the reaction of disulfide amines with aldehydes under relatively mild conditions (Scheme 14, Path b). The method builts upon a Cu-catalyzed activation strategy on a disulfide group, accompanied by C–H bond activation of its neighbouring imine moiety.36b The mechanism by which the disulfide amines and aldehydes promote the formation of benzothiazoles in the presence of Cu(I) has been extensively investigated. The authors introduced the concept of using disulfides as the oxidant for Cu(I) in the Cu-catalyzed organic transformation. Corroborated by the gas-phase data, these experimental results provide vital insight into the origin of the benzothiazole forming reaction. Similarly, the combination of o-iodoaniline with aldehydes and sulfur powder can afford benzothiazoles (16) in a simple one-pot procedure (Scheme 15).37
:1 mixture of 2,3-dihydro-2-arylbenzothiazole (14a) and 2-arylbenzothiazole (14b) (Scheme 14, Path a).36a One year later, they presented an improved approach to synthesizing benzothiazoles (15) by the reaction of disulfide amines with aldehydes under relatively mild conditions (Scheme 14, Path b). The method builts upon a Cu-catalyzed activation strategy on a disulfide group, accompanied by C–H bond activation of its neighbouring imine moiety.36b The mechanism by which the disulfide amines and aldehydes promote the formation of benzothiazoles in the presence of Cu(I) has been extensively investigated. The authors introduced the concept of using disulfides as the oxidant for Cu(I) in the Cu-catalyzed organic transformation. Corroborated by the gas-phase data, these experimental results provide vital insight into the origin of the benzothiazole forming reaction. Similarly, the combination of o-iodoaniline with aldehydes and sulfur powder can afford benzothiazoles (16) in a simple one-pot procedure (Scheme 15).37
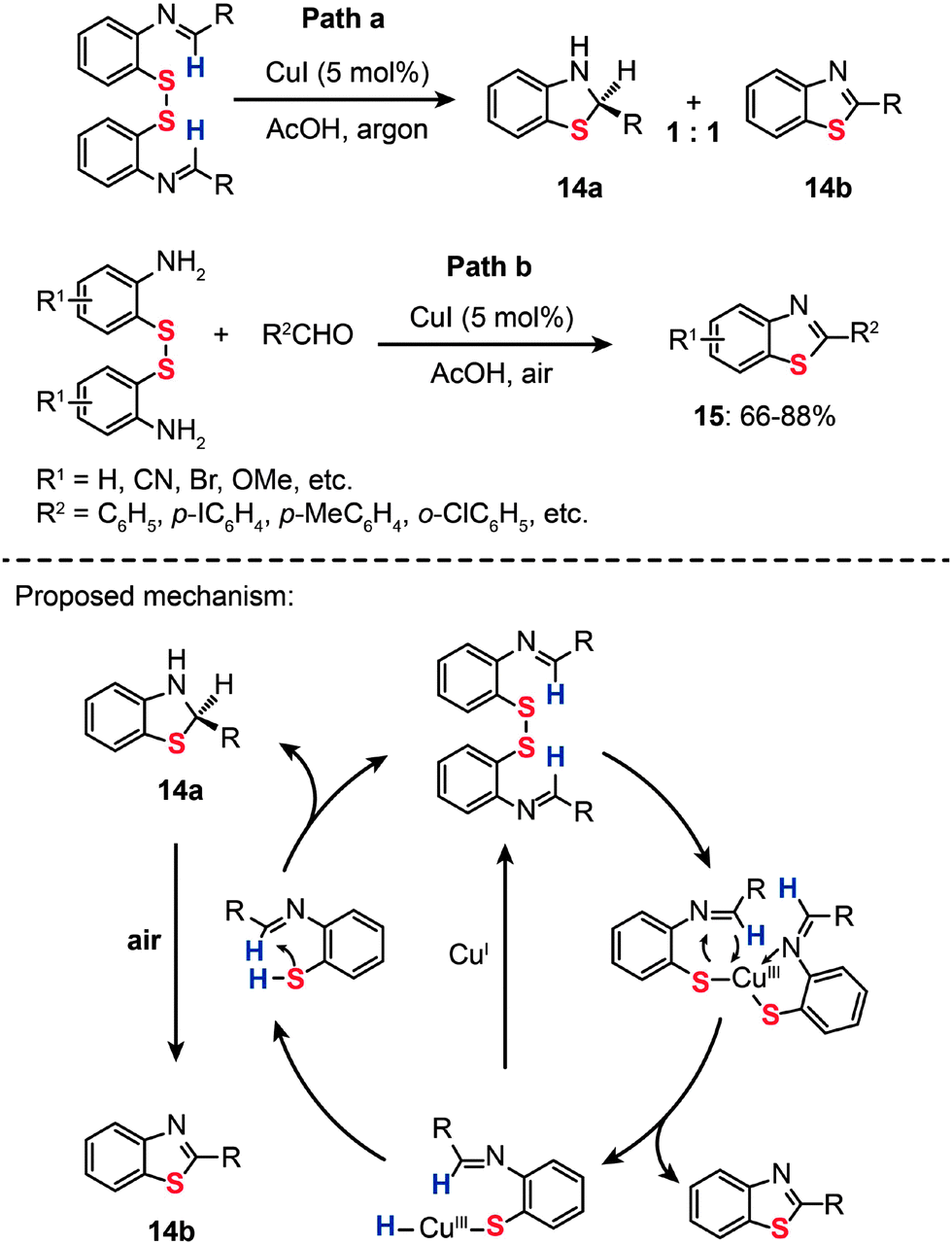 | ||
| Scheme 14 Srogl's Cu(I)-catalyzed transformation of imine disulfide.36 | ||
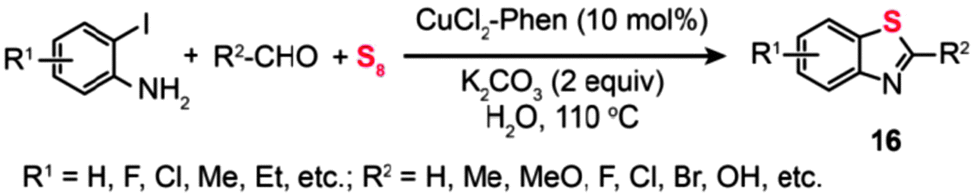 | ||
| Scheme 15 Zhou's Cu(II)-catalyzed three-component synthesis of substituted benzothiazoles.37 | ||
Sulfur-containing derivatives of 2-aminobenzothiazole have shown marked bacteriostatic and antitubercular activity. In 2013, Patel and co-workers described an efficient route to preparing 2-aminobenzothiazoles (17a–f) from aryl isothiocyanates by Cu(OTf)2-catalyzed intramolecular C–H functionalization/C–S bond formation (Scheme 16).38 In the presence of 10 mol% of Cu(OTf)2 in toluene and under 1 atm of O2 at 110 °C, both electron-poor and -rich substrates could be converted to the desired products.
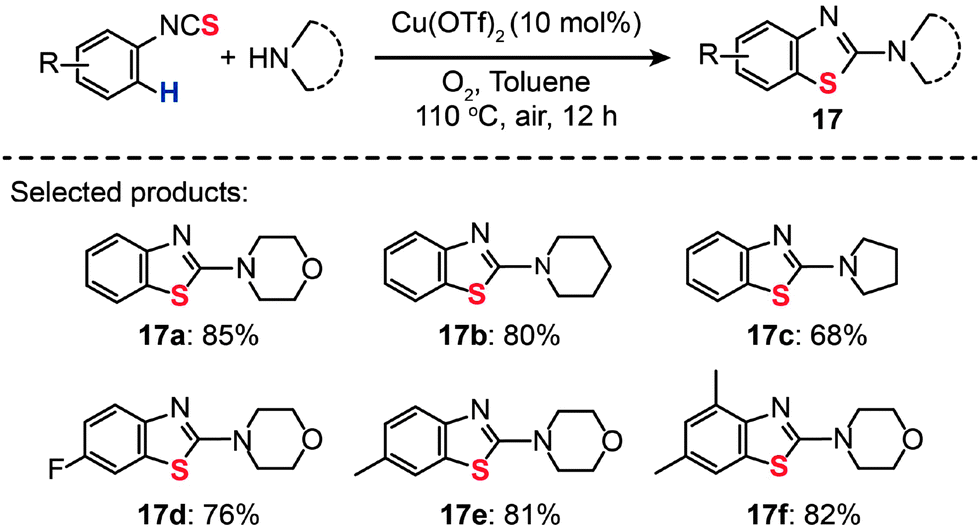 | ||
| Scheme 16 Patel's Cu(II)-catalyzed synthesis of 2-aminobenzothiazoles from aryl isothiocyanates.38 | ||
In 2009, Fukuzawa and co-workers performed the Cu(I)-catalyzed direct thiolation of benzoxazole with diaryl disulfides or aryl thiols in the presence of 10 mol% of CuI/Bpy, Cs2CO3, and 1 atm of O2 (Scheme 17).39 The cross-coupling reaction of benzoxazole with sulfides bearing electron-donating substituents afforded products (18) in good yields, while the reaction using sulfides with electron-deficient groups did not proceed.
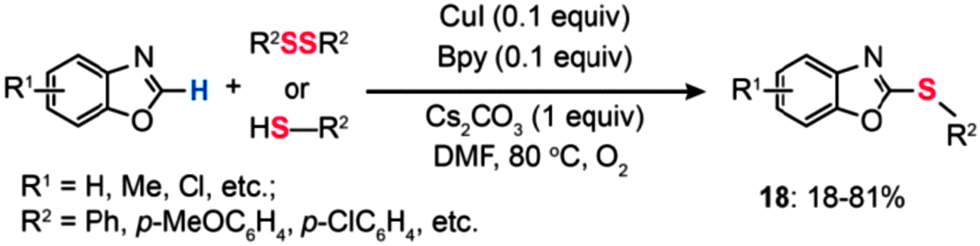 | ||
| Scheme 17 Fukuzawa's CuI/Bpy-mediated thiolation of benzoxazole with diphenyl disulfide or aryl thiols.39 | ||
Cheng and co-workers reported that a combination of CuI (20 mol%) and O2 catalyzes the thiolation of electron-rich arenes with diphenyl disulfides (Scheme 18).40 The thiolation reactions carried out at 120 °C in dimethylformamide (DMF) yielded the corresponding disulfides (19a–i) in 25–98% yields. The scope of this system seems to be limited to di- and trimethoxy-modified arenes.
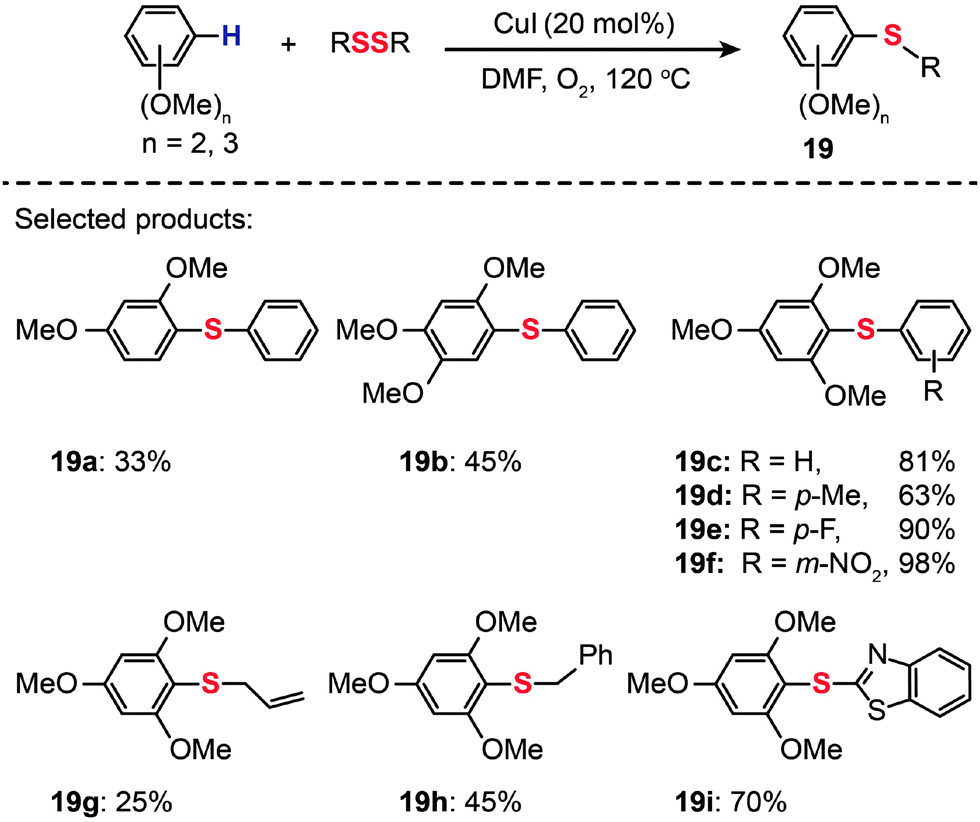 | ||
| Scheme 18 Cheng's CuI-catalyzed thiolation of di- or trimethoxybenzene C–H bonds.40 | ||
In 2011, Ranjit et al. developed a Cu(I)-catalyzed method for the direct thiolation of heteroarenes with aryl or alkyl thiols. The synthesis of a broad range of thiolated heteroarenes (20a–k) was carried out in the presence of stoichiometric CuI, Bpy, and Na2CO3 (Scheme 19).41 The heteroarenes under investigation included benzothiazole, benzimidazole, and indole substrates. Consistent with computational studies, the experimental results reveal that the first reactive intermediate in the form of a Cu–thiolate complex is responsible for the organic transformation. The computational studies also support a stepwise reaction mechanism involving a hydrogen-atom-abstraction pathway, which dominates over many other possible pathways, including β-hydride elimination, SET, hydrogen atom transfer, oxidative addition/reductive elimination, and σ-bond metathesis. Another example of direct thiolation of heteroarenes with thiols was reported by Zhou et al. through the use of a Cu(OAc)2-based catalyst.42 In their work, both aryl and aliphatic thiols furnished the thiolated products (21a–f) in moderate to good yields (Scheme 20). The reaction displayed exceptional compatibility with a wide range of heterocycles, including oxazole, thiazole, imidazole, and oxadiazole. Analogous to those proposed for similar Cu-catalyzed processes, a mechanism involving the generation of a Cu-thiolate species might be applicable to this thiolation procedure.
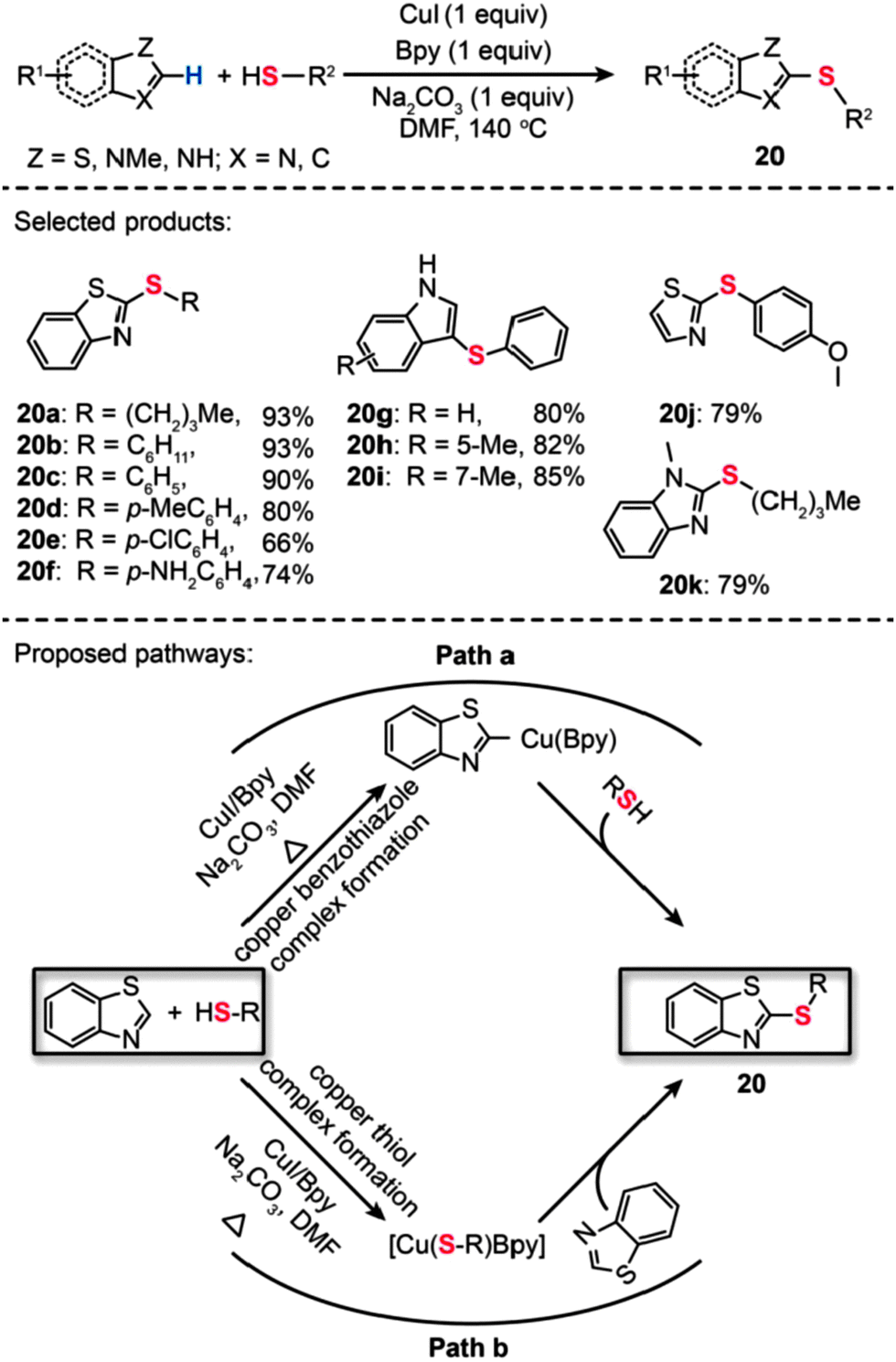 | ||
| Scheme 19 Liu's CuI/Bpy-mediated direct sulfurization of heteroarene with thiols.41 | ||
 | ||
| Scheme 20 Liu's Cu(II)-catalyzed direct thiolation of azoles with aliphatic thiols.42 | ||
For the regioselective synthesis of diaryl chalcogenides, direct C–H functionalization of arenes is plausible. Lee and co-workers developed a two-step tandem process to generate 3,5-disubstituted diaryl chalcogenides (22a–f) through Ir-catalyzed C–H borylation, followed by a CuCl-catalyzed C–S coupling reaction (Scheme 21).43 In contrast with monoarylthiolation, the bisarylthiolation process has proven to be more difficult. To confront this issue, Yu and co-workers in 2012 demonstrated a riveting Cu-catalyzed reaction of pentafluorobenzene with diaryl disulfides or aryl thiols through C–H bond and C–F bond activation (Scheme 22).44 The bisarylthiolation products (23) were obtained in high yields when CuBr (30 mol%) and tBuOLi (2 equiv.) were combined in DMSO at 60 °C under an oxygen atmosphere. The authors debated that the molecular oxygen is critical for the thiolation reaction, whereas the control performed under a nitrogen atmosphere afforded the products in low yields.
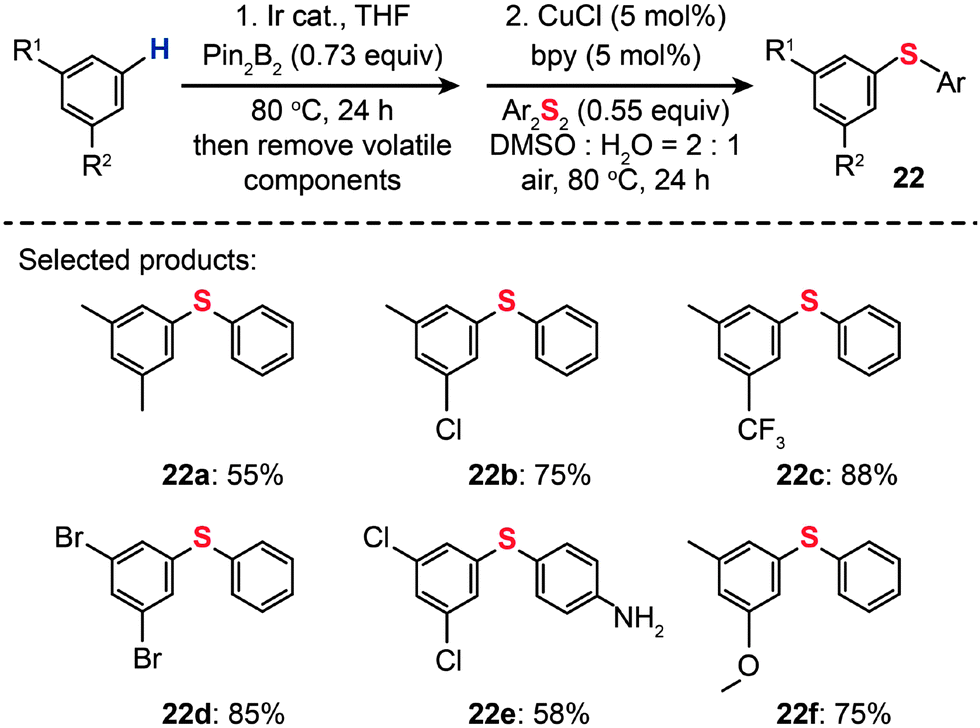 | ||
| Scheme 21 Lee's tandem C–H borylation and Cu-catalyzed C–S coupling reactions.43 | ||
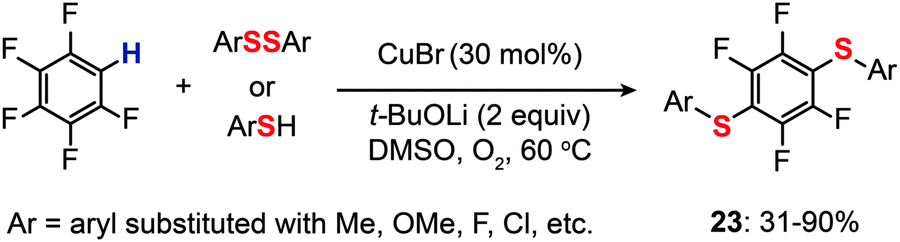 | ||
| Scheme 22 Yu's direct thiolation of pentafluorobenzene with diaryl disulfides.44 | ||
Transition metal ions are generally good electron-pair acceptors that readily bind electron-rich molecules and anions. Indeed, metal–ligand interaction can impart perturbation in the relevant frontier molecular orbitals, which potentially improves conditions for C–S coupling. Putting this idea into practice, Gao and co-workers developed transition metal-based Lewis acids as the catalysts for the synthesis of heteroaryl thioethers (24a–i and 25a–c) by direct C–H thiolation of heteroarenes (Scheme 23).45 The effective Lewis acids were found to be Ag(I), Ni(II), Au(I), Au(III), Cu(II), Zn(II), and Fe(II). The Lewis acid activation led to rate accelerations for the C–S coupling at relatively high catalyst loadings (0.2 equiv.).
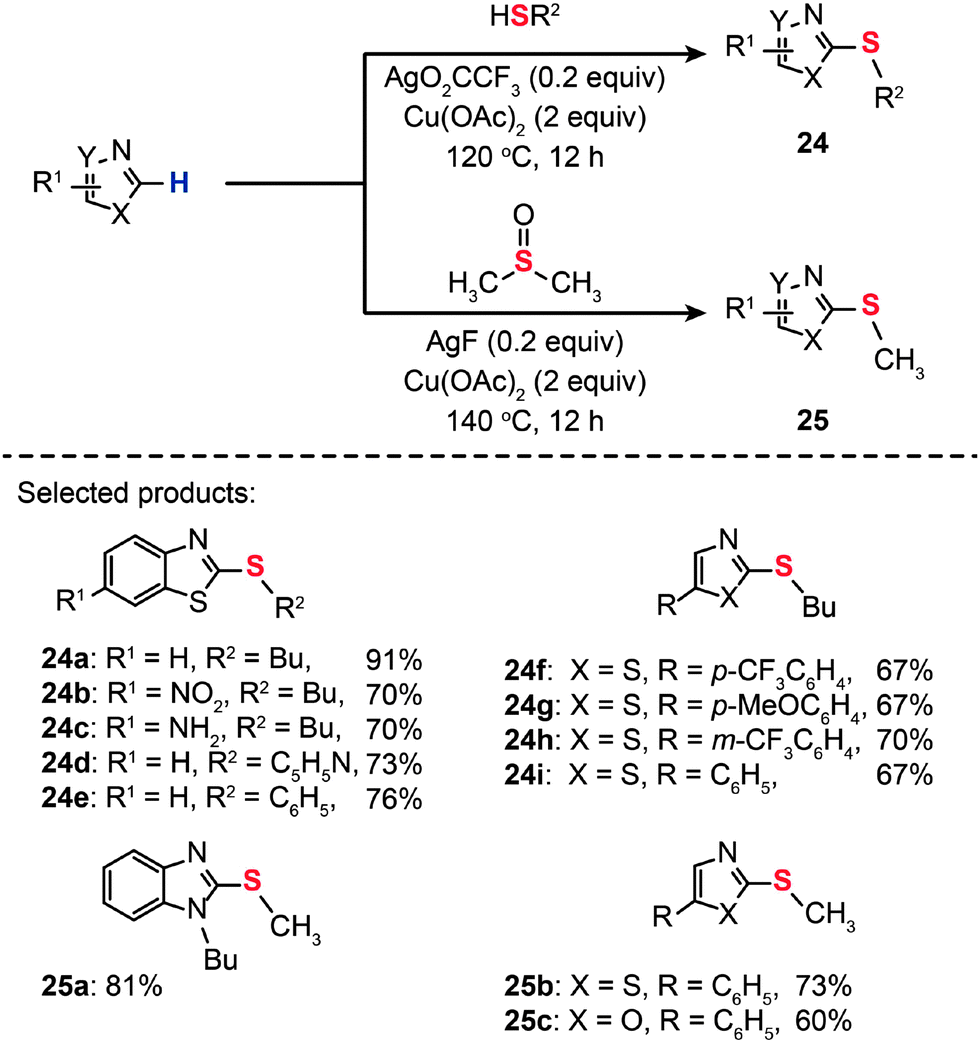 | ||
| Scheme 23 Xu's Lewis acid-catalyzed, Cu(II)-mediated synthesis of heteroaryl thioethers.45 | ||
The incorporation of a trifluoromethylthio group into arenes can result in a new class of chemical compounds that have found many important uses as agrochemical intermediates and antitumor drug candidates. Conventional methods typically rely on the condensation reaction between a metal thiolate and an aryl halides.46 Alternatively, aryl trifluoromethyl sulfides can be synthesized from aryl boronic acids under relatively harsh reaction conditions.47 In 2012, Daugulis et al. developed a method for an auxiliary-assisted sulfenylation of the β-C–H bonds in benzoic acid and benzylamine derivatives without needing extra steps to prefunctionalize precursors (Scheme 24).48 The optimal reaction employed catalytic or stoichiometric Cu(OAc)2, disulfide reagent, and DMSO solvent at elevated temperatures (90–110 °C). A broad range of arylamides bearing both electron-donating or -withdrawing groups were found to be suitable substrates for the synthesis of aryl trifluoromethyl sulfides (26a–l and 27) in moderate to good yields. Other effective sulfenylating reagents for C–H bond sulfenylation include aryl and alkyl disulfides. The authors further demonstrated the applicability of their method for Cu(II)-mediated sulfenylation of amine derivatives. When compared to the sulfenylation of carboxylic acid analogues, the sulfenylation of amine derivatives needs a stoichiometric amount of the Cu(II) catalyst and higher reaction temperatures (130 °C). It is reasonable to assume that this Cu(II)-catalyzed aerobic oxidative coupling should proceed via a mechanism similar to that of the reaction reported by Stahl and co-workers,49 however, details of the mechanism, such as on the reactive aryl Cu(III) species, are not clear at present.
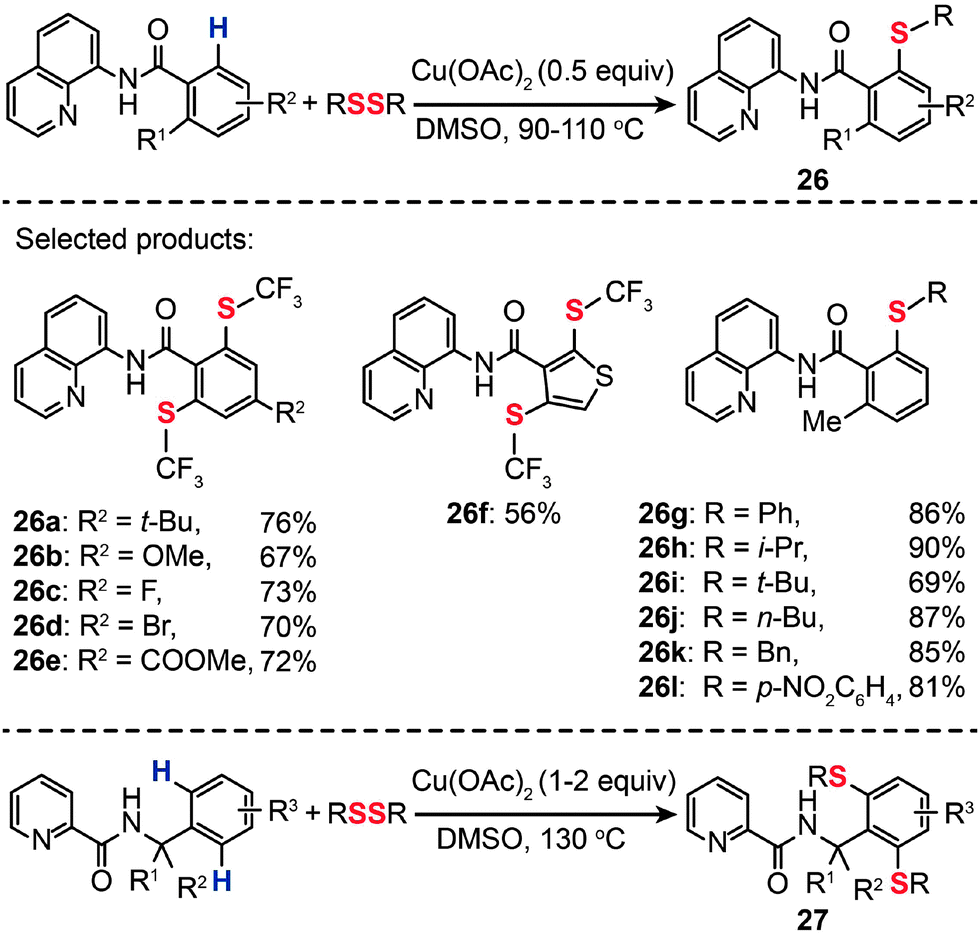 | ||
| Scheme 24 Daugulis's Cu(II)-mediated sulfenylation of carboxylic acid and amine derivatives.48 | ||
Recently, Alves and co-workers reported the synthesis of sulfenyl pyrroles by Cu(I)-catalyzed sulfenylation of pyrroles with organic disulfides or thiols (Scheme 25).50 The direct sulfenylation of pyrroles with organic disulfides was accomplished in the presence of 3 mol% of CuI in DMSO at 110 °C under an air atmosphere. By comparison, the sulfenylation of pyrroles by thiols succeeded only in the presence of CuI (5 mol%) under a nitrogen atmosphere. In most cases, the reaction products were dominated by 2-substituted sulfenyl pyrroles (28) in 65–93% yields obtained without the need for any ligands or additives. However, no product could be isolated when aliphatic disulfides were employed as the reactants.
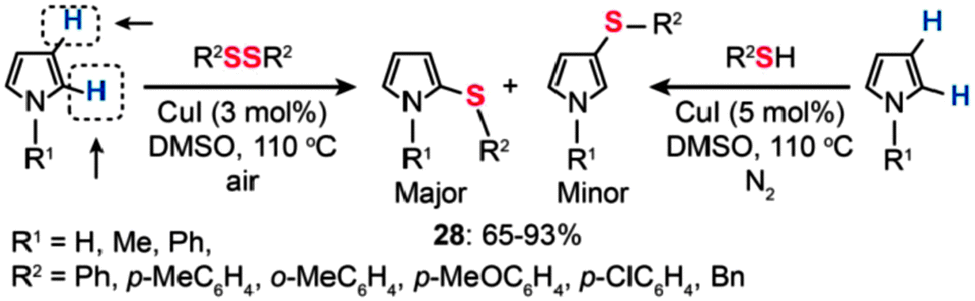 | ||
| Scheme 25 Alves's Cu(I)-catalyzed sulfenylation of pyrroles.50 | ||
In 2013, Wu and co-workers developed an efficient Cu(I)-catalyzed sulfonylation of quinoline N-oxides with aryl sulfonyl chlorides via C–H activation (Scheme 26).51 The best conversion efficiency was obtained for the reactions containing CuI (10 mol%) and K2CO3 (2 equiv.) in dichloroethane (DCE) at 100 °C. This approach provides a highly practical procedure for synthesizing 2-aryl sulfonyl quinolines (29) from commercially available and inexpensive aryl sulfonyl chlorides. Mechanistic investigations indicate that the reaction pathway involves the formation of a Cu(III) intermediate via oxidative addition (Scheme 26). A subsequent reductive elimination gives the sulfonylated product 29.
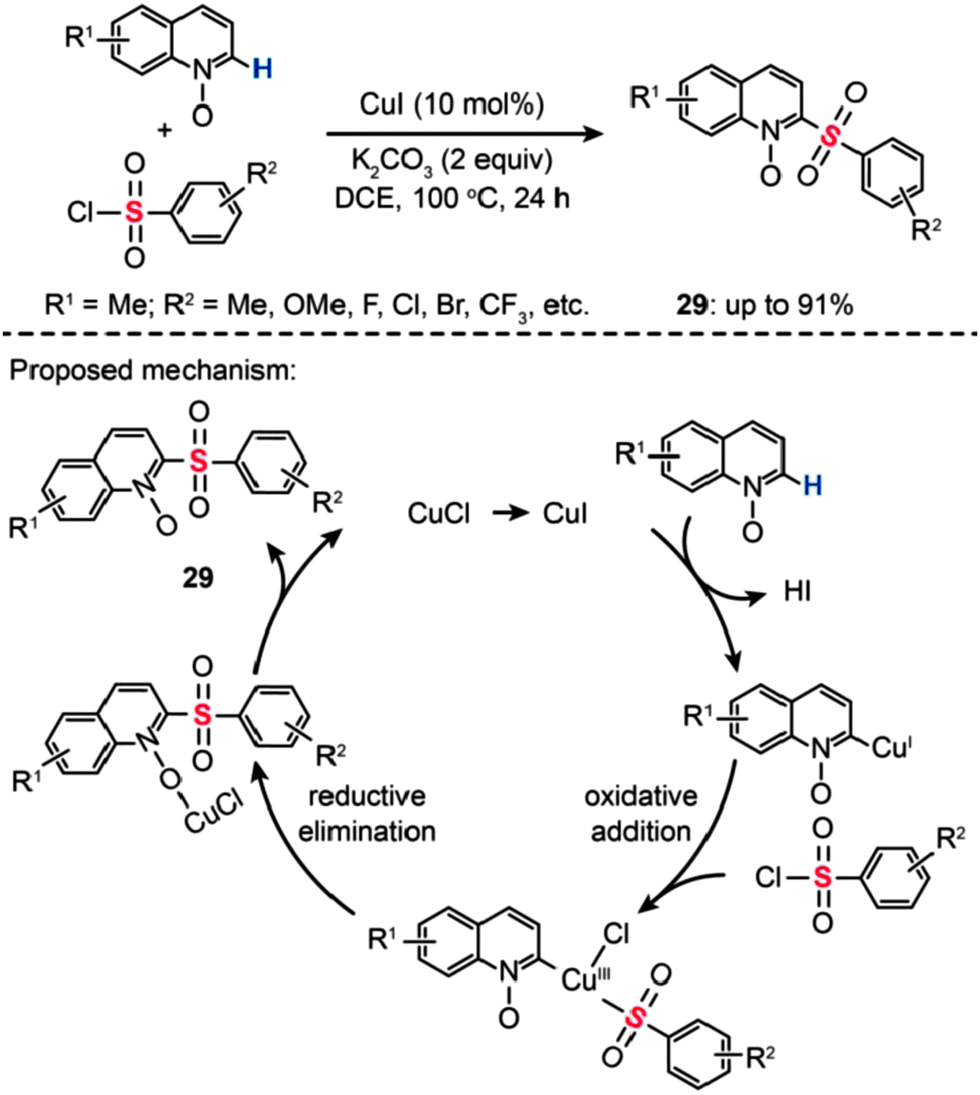 | ||
| Scheme 26 Wu's Cu(I)-mediated sulfonylation of quinoline N-oxides.51 | ||
On a fundamental level, the mechanisms of Cu(I)-catalyzed coupling reactions always involve three elementary steps: transmetalation with organometallic reagents, oxidative addition of an electrophile to Cu(I), and reductive elimination of the resulting organocopper(III) species. Cu-based catalysts are generally cheaper and more readily available than Pd-based catalysts, in addition to a host of other benefits. A Cu-based system can be a better replacement for the Pd-based counterpart when uncontrolled poisoning or deactivation of the latter by various mechanisms becomes a serious problem. For example, in the case of S8 or metal sulfides (e.g., Na2S, NaHS, H2S, Na2S4) selected as the source for sulfur, it turns out that the Cu-based catalysts are better at catalyzing the reactions in most cases.
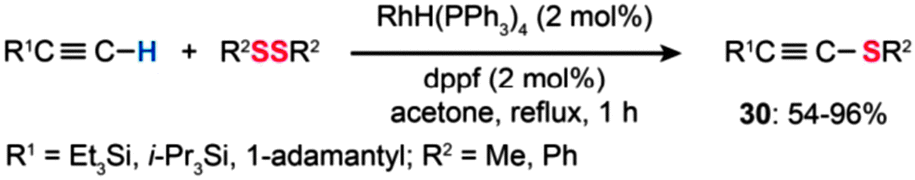 | ||
| Scheme 27 Yamaguchi's Rh-catalyzed alkylthiolation of 1-alkynes with disulfides.52 | ||
The usefulness of the RhH(PPh3)4 catalyst was also demonstrated by Yamaguchi and co-workers in the synthesis of thiolated heteroarenes (31), such as benzothiazoles (31a–e) and benzoxazoles (31f–j) (Scheme 28).54 When benzothiazole was reacted with α-(phenylthio)isobutyrophenone in refluxing chlorobenzene containing RhH(PPh3)4 (4 mol%) and dppe (8 mol%), 2-phenylthio-1,3-benzothiazole 31b was obtained in 92% yield. In stark contrast, the coupling reaction failed when attempts were made in the absence of either RhH(PPh3)4 or dppe. Notably, phenylthiolation of monocyclic heteroarenes, including 2-cyanothiophene (31k) and 1-methyl-1,2,3,4-tetrazole (31l), also provided the coupling products in good yields under the optimal reaction conditions.
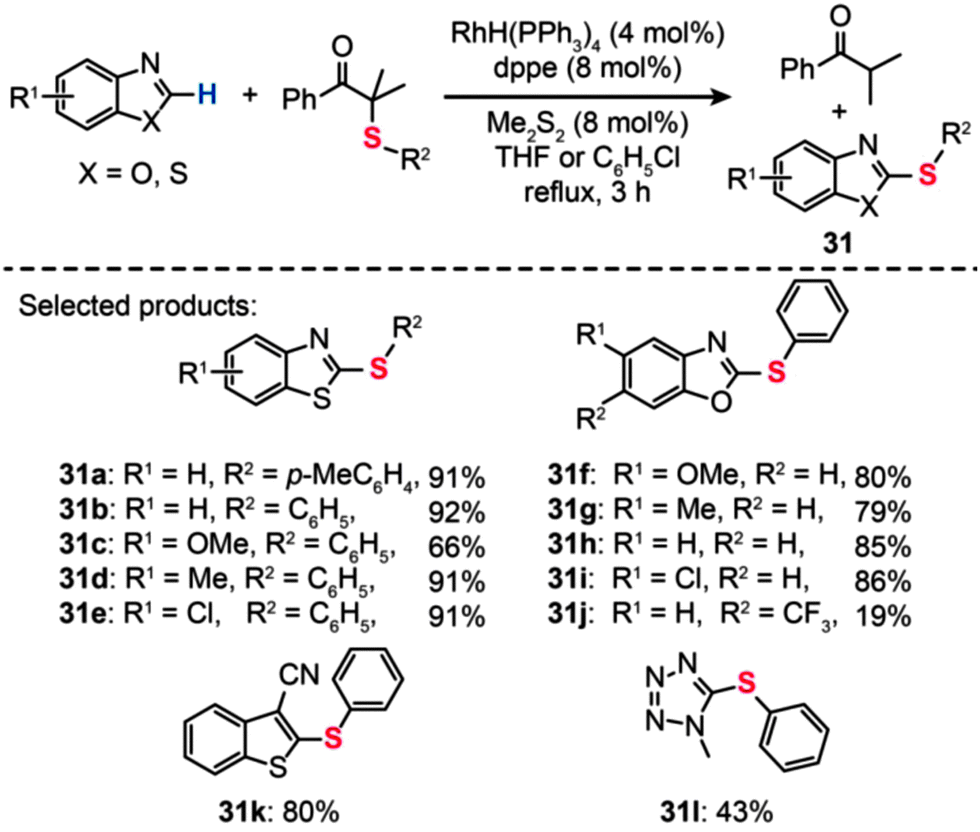 | ||
| Scheme 28 Yamaguchi's Rh-catalyzed phenylthiolation reaction of heteroaromatic compounds.54 | ||
Drawing inspiration from Daugulis's work on Cu(II)-catalyzed thiolation of arene C–H bonds,48 Li and co-workers recently reported the first example of Rh(III)-catalyzed direct C–H thioetherification using aryl and alkyl disulfides as thiolation reagents (Scheme 29).55 Under the optimal conditions ([RhCp*Cl2]2: 5 mol%, AgOTf: 20 mol%, Cu(OAc)2: 20 mol%, solvent: t-AmOH, temperature: 60 °C), a wide variety of aryl thioether products (32a–l) were prepared in good yields. In addition, the authors extended this principle to the selective mono- or dithiolation of arenes by controlling the reaction temperature. The broad substrate scope and high efficiency of the direct C–S coupling may allow the construction of sulfur-containing heterocycles not easily accessible by conventional cross-coupling reactions. A mechanism involving two different pathways was proposed by the authors. The [RhCp*Cl2]2 precursor first reacts with AgOTf to produce a Rh(III) complex. Then the Rh(III) complex reacts with 2-phenylpyridine to give an Ar–Rh(III) species which can undergo a nucleophilic-addition-type reaction with diphenyl disulfide to afford 32a (Path a). As an alternative reaction pathway (Path b), oxidative addition of the disulfide bond to Rh(III) gives rise to an intermediate Rh(V), followed by reductive elimination to afford the products of 32a and a Rh(III) species. Then the Rh(III) species can continue to react with 2-phenylpyridine to form a five-membered rhodacycle intermediate.
 | ||
| Scheme 29 Li's Rh-catalyzed ortho thiolation of arene C–H bonds.55 | ||
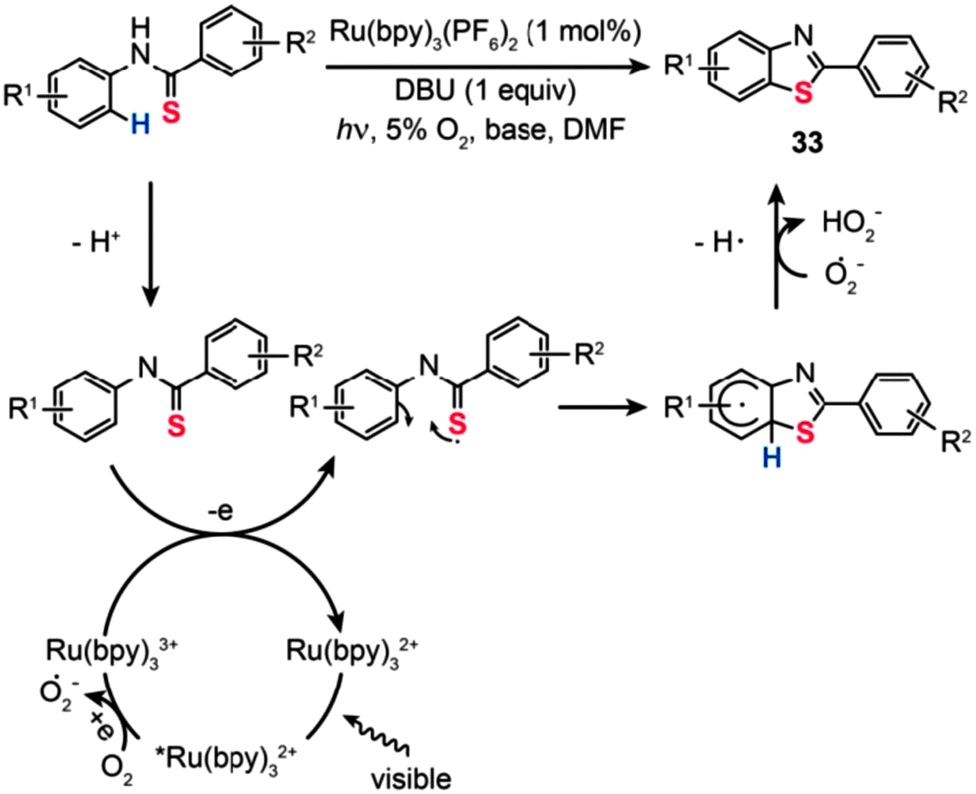 | ||
| Scheme 30 Li's aerobic visible-light photoredox synthesis of of 2-phenylbenzothiazoles.57 | ||
In 2011, Frost and co-workers developed a Ru(II)-catalyzed sulfonylation of 2-phenylpyridines that enables the synthesis of the meta-sulfonylation products (34) (Scheme 31).58 Moderate yields were obtained with the reactions carried out in CH3CN containing [Ru(p-cymene)Cl2]2 (2.5 mol%) and K2CO3 (2 equiv.). To unravel the regioselectivity at the meta position, some preliminary experiments with isotopically labeled 2-phenylpyridine 1-[D5] were carried out. No evidence of D/H exchange from adventitious water or solvent was observed. Based on the experimental data, the authors argued that the chelating pyridyl group facilitates the formation of a stable Ru–Caryl σ bond featuring a strong para-directing effect. Consistent with the hypothesis, treatment of an isolated cyclometalated Ru complex with p-toluenesulfonyl chloride (3 equiv.) under the standard reaction conditions afforded the meta-sulfonation product (35) quantitatively.
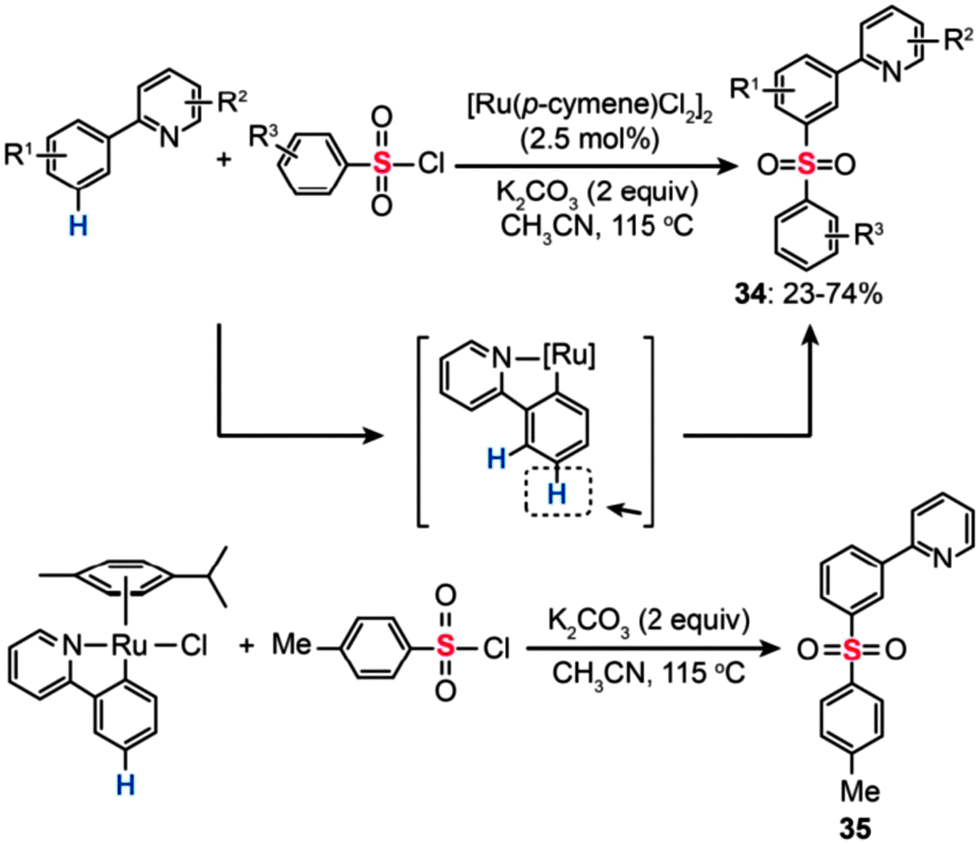 | ||
| Scheme 31 Frost's Ru(II)-catalyzed meta-sulfonylation of 2-phenylpyridine.58 | ||
Employing a similar concept with the Ru-based organometallic catalyst, Zheng and co-workers relied on photoredox reactions of N-methylindoles with arylsulfonyl chlorides to synthesize 1-methyl-3-(arylthio)-1H-indoles (36) in moderate yields.59 The exact reaction mechanism is still a matter of debate. Generally, the pivotal step in these reactions involves reductive quenching of Ru(bpy)32+ by N-methylindole to generate Ru(bpy)3+, followed by reduction of p-toluenesulfonyl chloride through electron transfer from Ru(bpy)3+. Although the detailed process for the conversion of TsCl to p-tolyl hypochlorothioite is unclear, partially reductive intermediate 4-methylbenzene-1-sulfinic chloride can be generated to react with N-methylindoles affording the product in moderate yield. After the conversion of the sulfonyl group to the sulfenyl moiety and the successive cross-coupling reaction with an indole substrate through C–H activation, 3-sulfenylated indoles can be obtained (Scheme 32). It is worth mentioning that this new reduction system has advantages over the conventional methods in its efficiency and easy operation. Additionally, one-pot and facile sulfenylation starting from non-volatile and non-stench aromatic sulfonyl chlorides is expected to be widely applicable for the synthesis of various useful organosulfur compounds.
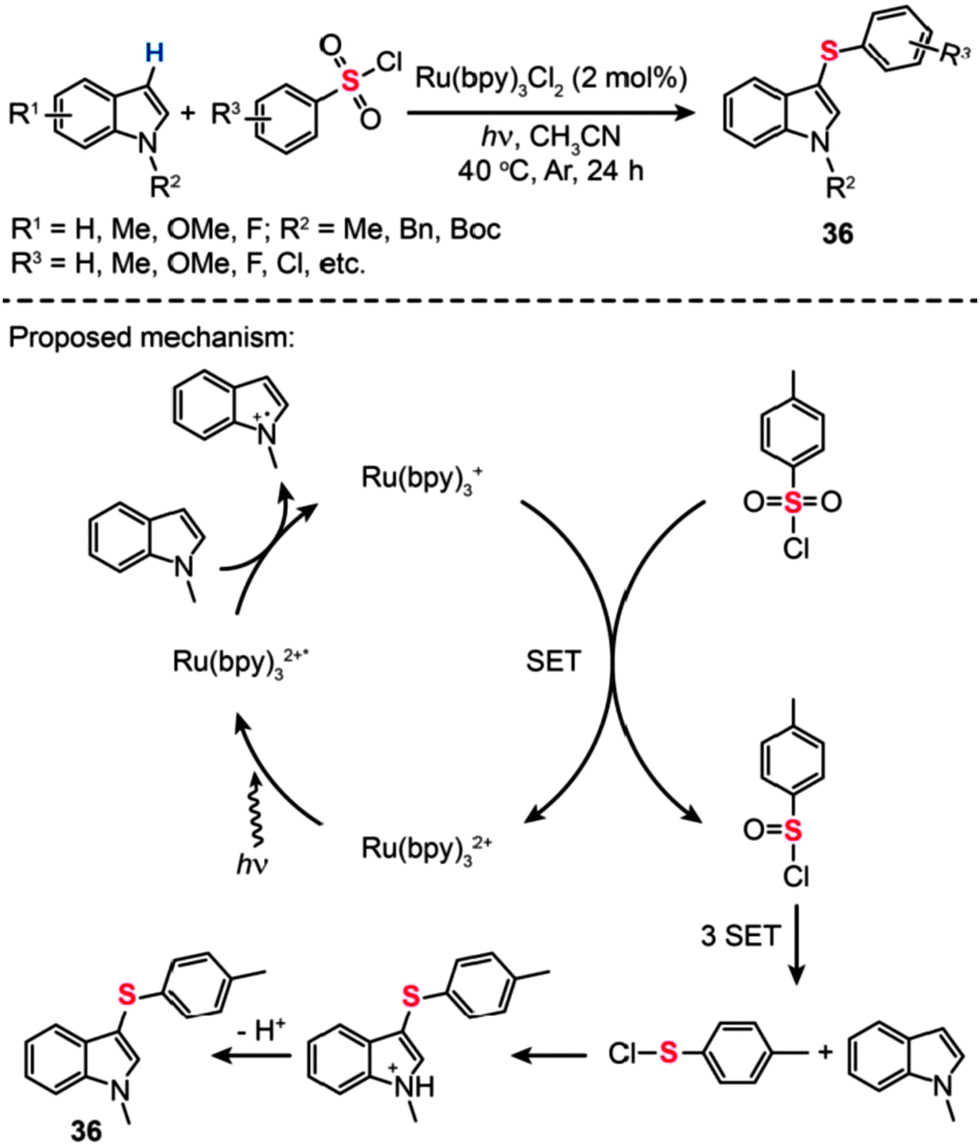 | ||
| Scheme 32 Zheng's visible light-induced 3-sulfenylation of N-methylindoles with arylsulfonyl chlorides.59 | ||
 | ||
| Scheme 33 Lei's Fe(III)-catalysed oxidative synthesis of 2-substituted benzothiazoles.61 | ||
In contrast to common catalysts derived from Pd, Cu and Rh metals, Fe-based catalysts are generally inexpensive, environmentally friendly, and easily accessible from commercial starting materials. Despite its advantages, an efficient Fe-based catalytic system for C–S cross-couplings via C–H bond activation is yet to be developed. A more detailed mechanistic understanding is essential for less efficient alkylboronic acids or esters to make them effective towards cross-coupling. At the same time, cost-effective catalysts are in high demand for successful application in industrial processes.
2.2 Direct C–H bond functionalization
In recent years, the development of transition-metal-free approaches appear to be particularly attractive.62 Many groups reported significant progress in this area including transition-metal-free protocols to construct C–C, C–N and C–O bonds.63 Importantly, the research groups of Yanagisawa, Shi and Lei have recently reported independently on the construction of biaryl compounds from unactivated aromatic rings by direct C–H bond activation with the aid of organocatalysts.64 Despite a significant achievement in the C–C bond formation by the sp2 C–H bond oxidative functionalization process,65 the carbon–heteroatom bond formation using a similar strategy receives less attention.66 Some of the examples are highlighted below.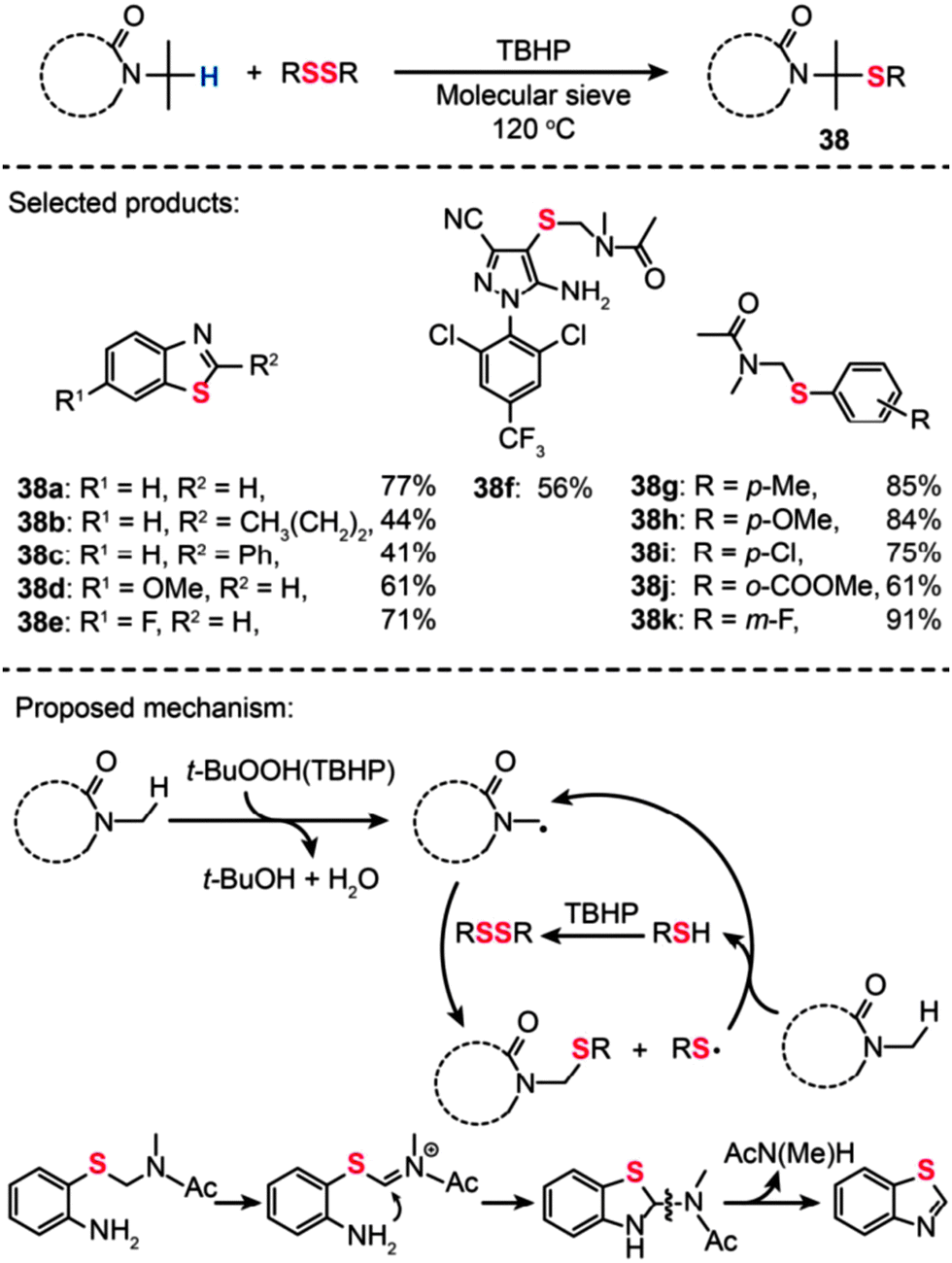
In 2014, Yuan and Xiang reported direct C–S bond formation via oxidative thiolation of commercially available ethers using di-tert-butyl peroxide (DTBP) as the oxidant (Scheme 35).68 A variety of substrates worked well to afford the target α-arylthio ethers (39a–f) smoothly. To probe the mechanism of this metal-catalyst-free reaction, a free-radical trap test was investigated. The authors found that the reaction did not proceed in the presence of TEMPO, providing evidence for the likely occurrence of a radical process in this reaction. On the basis of their mechanistic investigations, the authors proposed a mechanism that involves the formation of tert-butoxyl radicals from DTBP under heating conditions. The tert-butoxyl radical can abstract hydrogen atoms from the ether to generate an alkoxy radical intermediate, which can further react with ArSSAr to afford the product. The ArS˙ by-product can react with another alkoxy radical to furnish the target product.
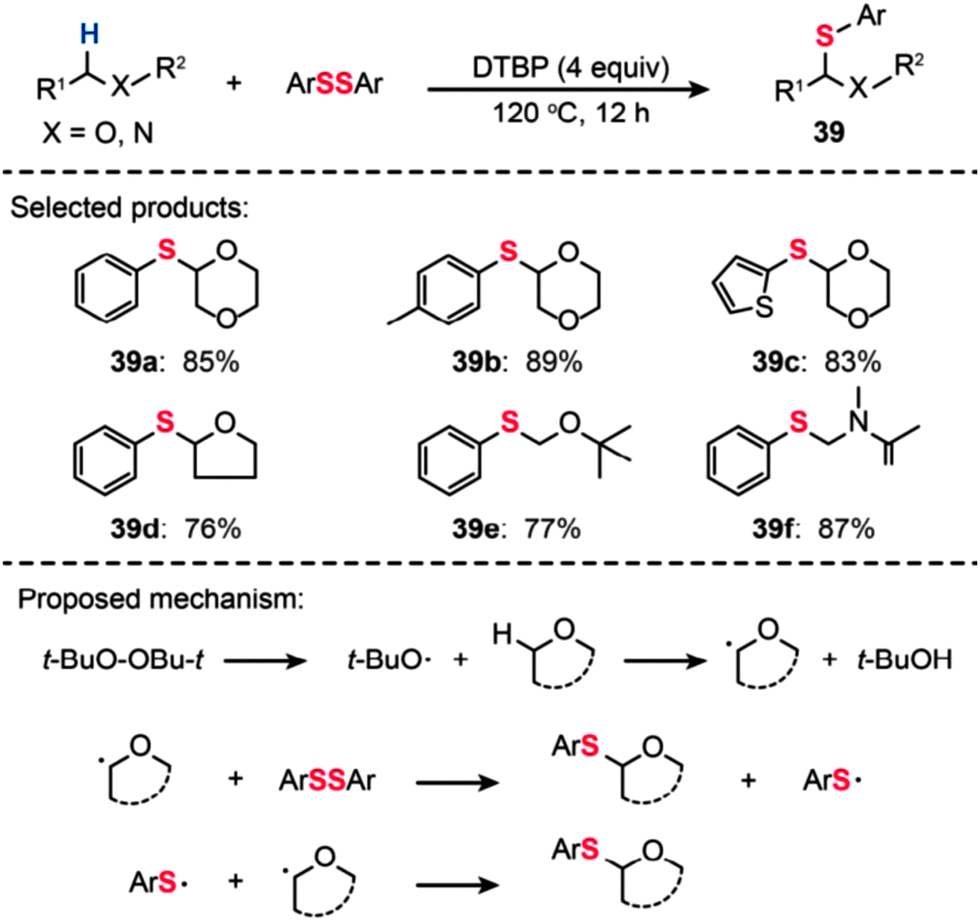 | ||
| Scheme 35 Yuan's DTBP-catalyzed thiolation of ethers and amides with diaryl disulfides.68 | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bond of DMSO results in the formation of a DMSO–HI adduct followed by protonation of the oxygen atom. Upon nucleophilic attack by I− on the iodine atom of the protonated DMSO–HI adduct, I2 can be regenerated along with the formation of water and dimethyl sulfide.
O double bond of DMSO results in the formation of a DMSO–HI adduct followed by protonation of the oxygen atom. Upon nucleophilic attack by I− on the iodine atom of the protonated DMSO–HI adduct, I2 can be regenerated along with the formation of water and dimethyl sulfide.
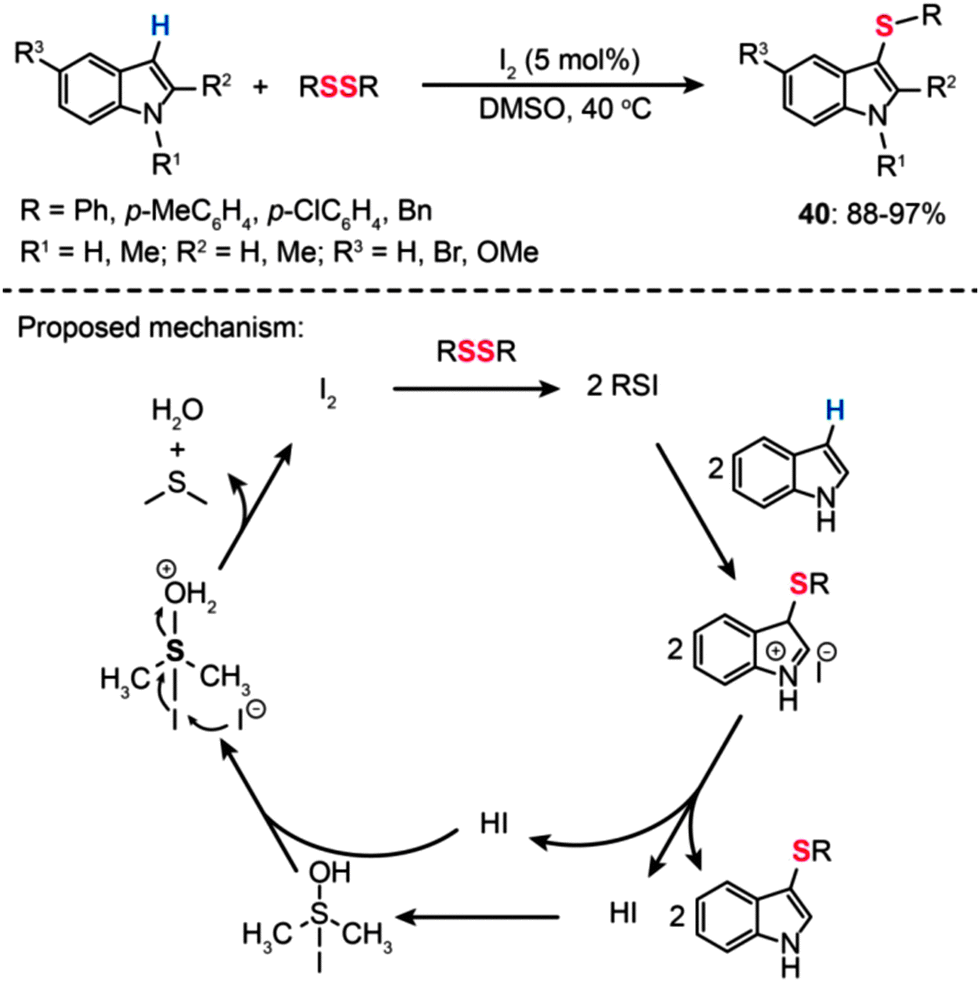 | ||
| Scheme 36 Wei's I2-catalyzed 3-sulfenylation of indoles with disulfides.69 | ||
The utilization of I2 as the oxidant for C–S bond formation was also demonstrated by Wu et al., who reported the discovery of an I2-promoted domino protocol for the conversion of aromatic ketones/unsaturated methyl ketones and o-aminobenzenethiols into the corresponding 2-acylbenzothiazoles (Scheme 37).70 Under the optimized conditions (1.5 equiv. of I2 at 100 °C in an aerobic environment), both electron-donating and electron-withdrawing groups at the ortho-, meta- or para-position of the phenyl group of aromatic ketones or unsaturated methyl ketones could afford the corresponding products (41a–i and 42a–e) with moderate to good yields. However, the aliphatic ketones such as acetone, cyclohexanone, and methylethylketone could not give the coupled products under the standard conditions. The same group also developed a multipathway-coupled domino strategy for preparing 2-acylbenzothiazoles from substrates in forms of arylethenes, arylacetylenes, 2-hydroxy-aryl ketones, and 1-arylethanol.71 Their protocol is presumed to follow four distinct reaction pathways. This provides a diverse synthetic approach to access 2-acylbenzothiazoles, which should be of great utility in developing organic methodologies for combinatorial chemistry. Additionally, the broad substrate scope and mild reaction conditions of this approach should make it a practical method for the synthesis of sulfur-containing heterocycles.
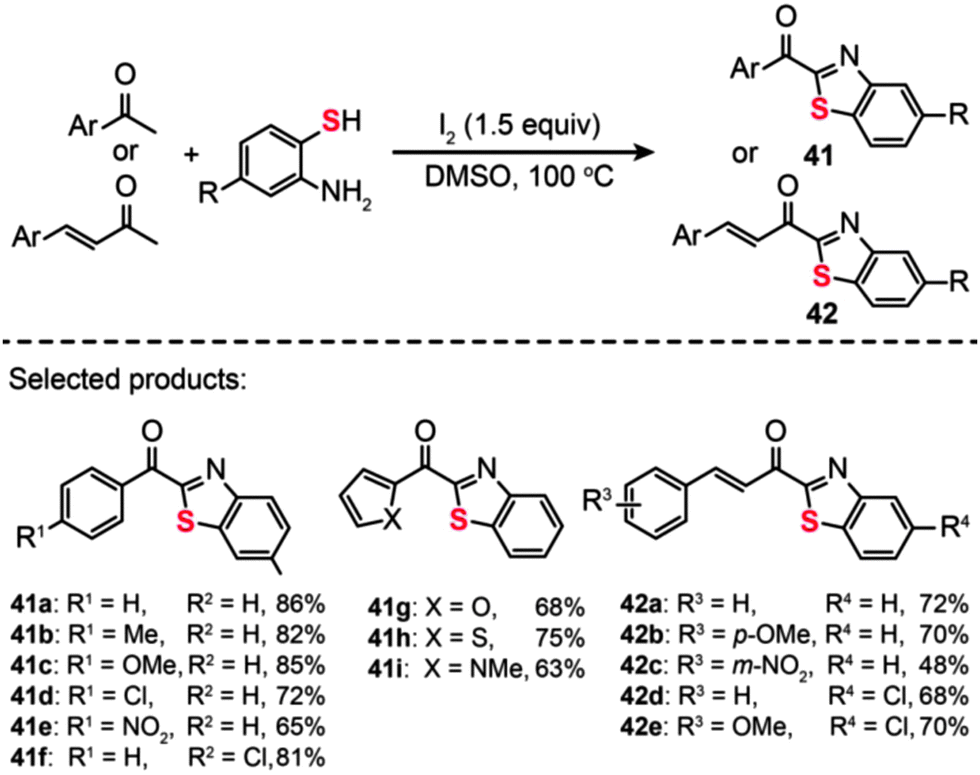 | ||
| Scheme 37 Wu's I2-promoted domino oxidative cyclization for one-pot synthesis of 2-acylbenzothiazoles.70 | ||
In the same year, Deng and co-workers developed a straightforward reaction, catalyzed by iodine in the presence of O2 under metal-free conditions, for the synthesis of 2-arylsulfanylphenols (43) using cyclohexanones as the source of phenol (Scheme 38).72 In this transformation, a catalytic amount of iodine was added to promote the oxidation process. The reaction of thiols with cyclohexanones proceeded smoothly, leading to the desired products 43 in moderate to good yields. Remarkably, the steric hindrance on the cyclohexanones drastically affects the efficiency of this reaction. Moreover, a wide range of substrates containing functional groups such as halo, hydroxyl, and ester groups were shown to be compatible with this transformation. The authors proposed a catalytic cycle involving the I2-mediated oxidation process (Scheme 38). A disulfide is first generated by the oxidation of an arylthiol in the presence of molecular oxygen. Subsequently, an electrophilic species ArSI is produced by the reaction of disulfide with I2, which can couple with a cyclohexanone to generate a 2-thio cyclohexanone intermediate and HI as the by-product. Dehydrogenation and tautomerization of the 2-thio cyclohexanone intermediate then affords the coupling product along with HI, which could be further oxidized by oxygen to give I2. However, the 2-arylsulfanylphenol product 43 obtained through such a mechanism still appears to be debatable as the oxidation of 2-thio cyclohexanones to 2-arylsulfanylphenols using I2 tends to be a daunting task.
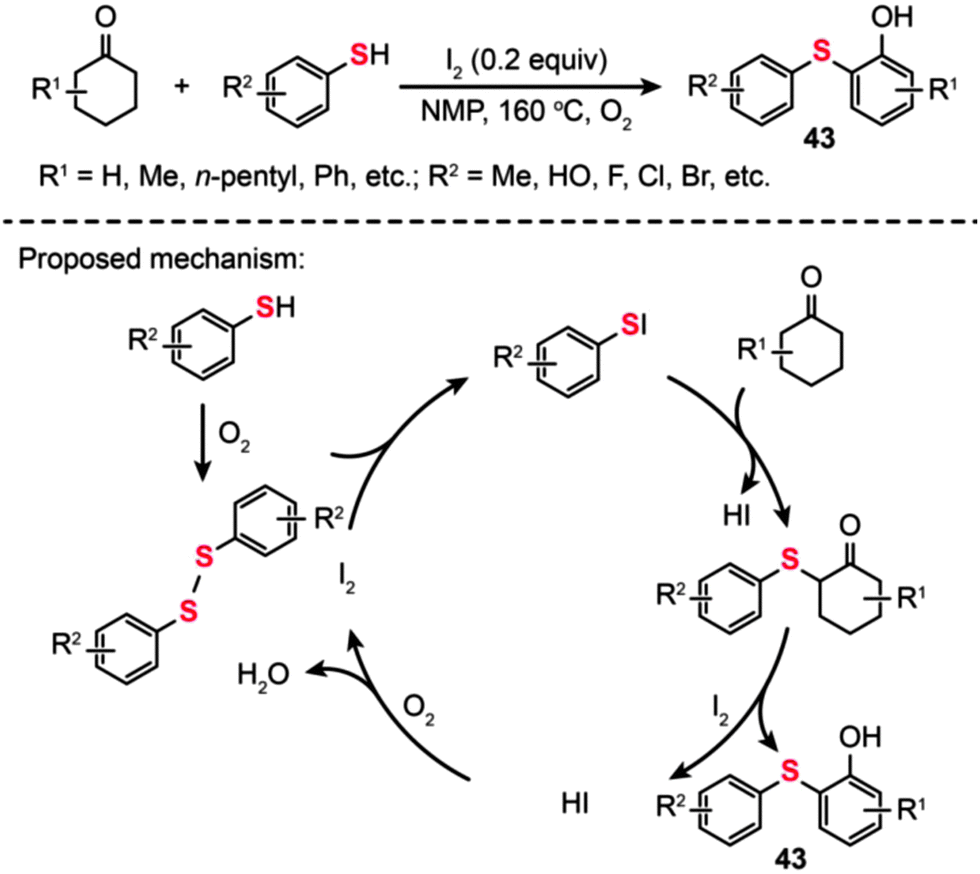 | ||
| Scheme 38 Deng's I2-catalyzed synthesis of 2-arylsulfanylphenols.72 | ||
Shortly after this report, Deng and co-workers took one step forward and demonstrated a simple and efficient method to prepare 3-arylthioindoles (44) from indole and sodium sulfinate precursors (Scheme 39).73 Their reactions were carried out in the presence of diethyl phosphite in anisole at 100 °C under an argon atmosphere. They could achieve a product yield as high as 93%. Mechanistic studies have revealed that 1,2-diphenyldisulfane is generated from sodium benzenesulfinate in the presence of I2 and diethyl phosphite, possibly via an S-phenyl phosphorothioate intermediate.74 This method affords an efficient alternative approach starting from easily accessible sodium sulfinates for the synthesis of heterodiaryl sulfides – synthetic intermediates widely used in the construction of biologically important compounds.
 | ||
| Scheme 39 Deng's I2-catalyzed sulfenylation of indoles with sodium sulfinates.73 | ||
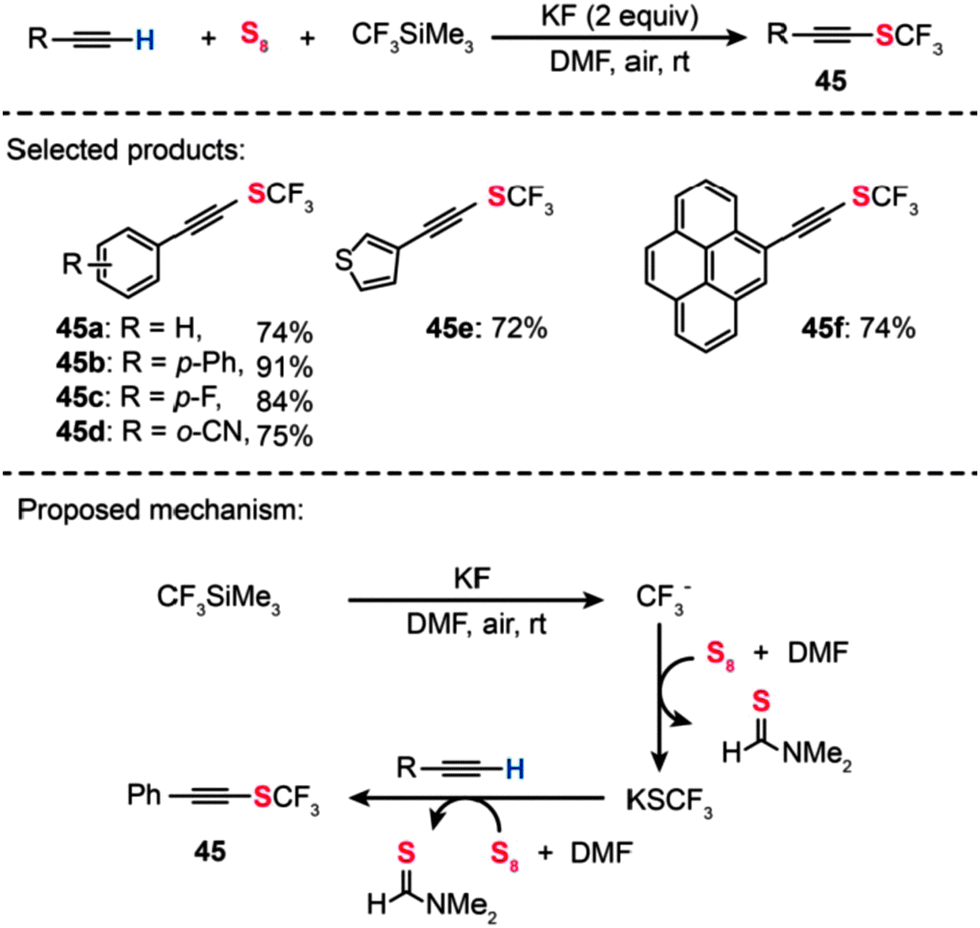 | ||
| Scheme 40 Qing's metal-free oxidative trifuoromethylthiolation of terminal alkynes.75 | ||
Bolm and co-workers described a transition metal-free procedure for the direct thiolation of 1,3,4-oxadiazole C–H bonds using diaryl disulfides (Scheme 41).76 A systematic screening of the reaction conditions with regard to catalyst, temperature, and solvent revealed that the use of Cs2CO3 (2 equiv.) in 1,4-dioxane at 130 °C was most effective for the synthesis of aryl sulfides (46). Notably, the authors observed that the cross-coupling reaction in acetonitrile gave a low yield and no product was isolated when using DMSO or DMF as the solvent. The methodology of direct thiolation was further extended to other substrates, including indoles, benzothiazoles, and N-phenylbenzimidazoles. In most cases, the reactions afforded the coupled products 46a–i in moderate to good yields. Presumably, the reaction involves initial heterocycle conversion to an anion species via deprotonation, followed by a nucleophilic reaction with the diaryl disulfide and a subsequent C–S bond-forming reaction.
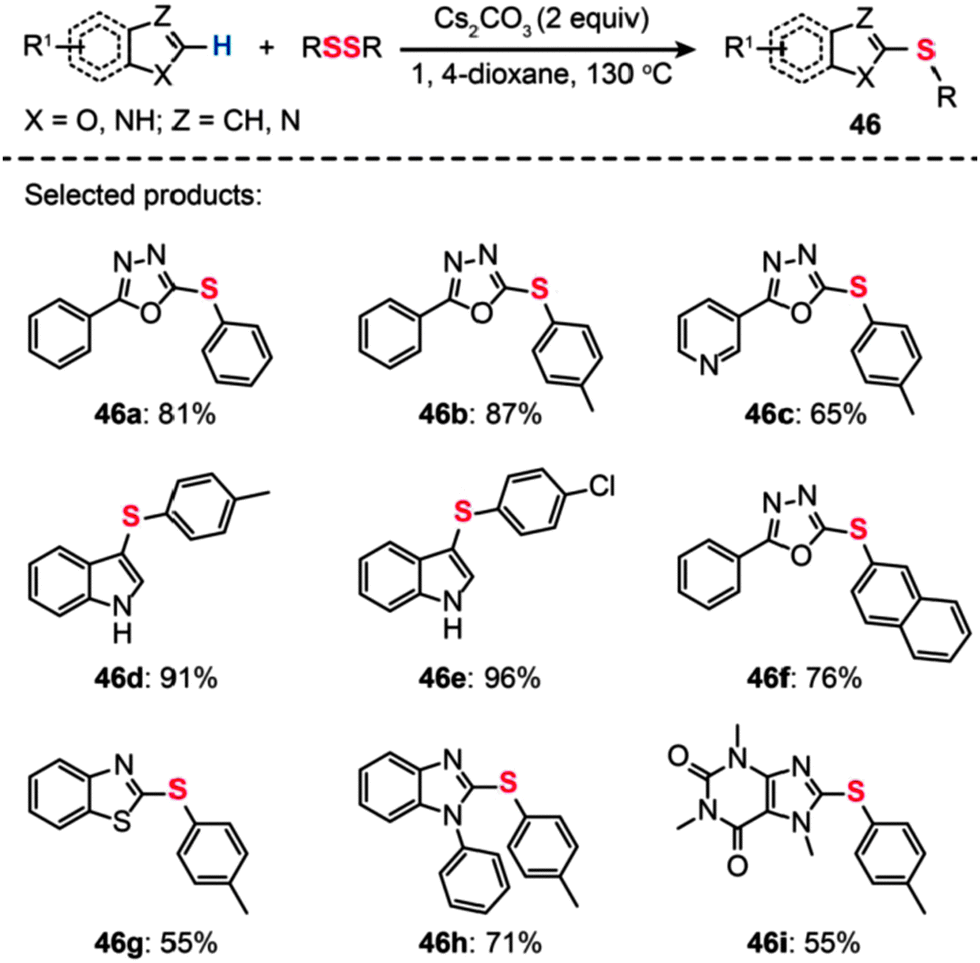 | ||
| Scheme 41 Bolm's transition metal-free direct thiolation of heteroarenes.76 | ||
In 2013, Shen and co-workers reported a new electrophilic hypervalent iodine reagent comprising a 1,2-benziodoxole moiety, which is effective for direct transfer of a trifluoromethylthio group (CF3S–) to carbonyl compounds.77 Various β-ketoesters derived from indanone, tetralone, or 1-benzosuberone reacted with the hypervalent iodine reagent in CH2Cl2 at room temperature to give the corresponding α-trifluoromethylthiolated derivatives (47a–i) in moderate to excellent yields when 4-dimethylaminopyridine (DMAP) was used as the base (Scheme 42). However, the above conditions failed completely when applied to reactions containing vinyl boronic acid and alkyne substrates. To overcome this limitation, the researchers used CuBr(SMe2) in 1,2-dichloroethane, combined with 2,2′-bipyridine as the ligand and K2CO3 as the base. Under the optimal conditions, a variety of electron-rich and electron-deficient terminal alkynes could be transformed into the corresponding alkynyl trifluoromethylsulfides 48 and trifluoromethylthiolated arenes 49 in good yields. A wide range of functional groups, including nitro, enolizable ketone, ester, fluoride, and bromide, were compatible with the reaction conditions. Aliphatic alkynes were also examined under the optimized conditions, furnishing alkynyl trifluoromethylthioethers in good to excellent yields.
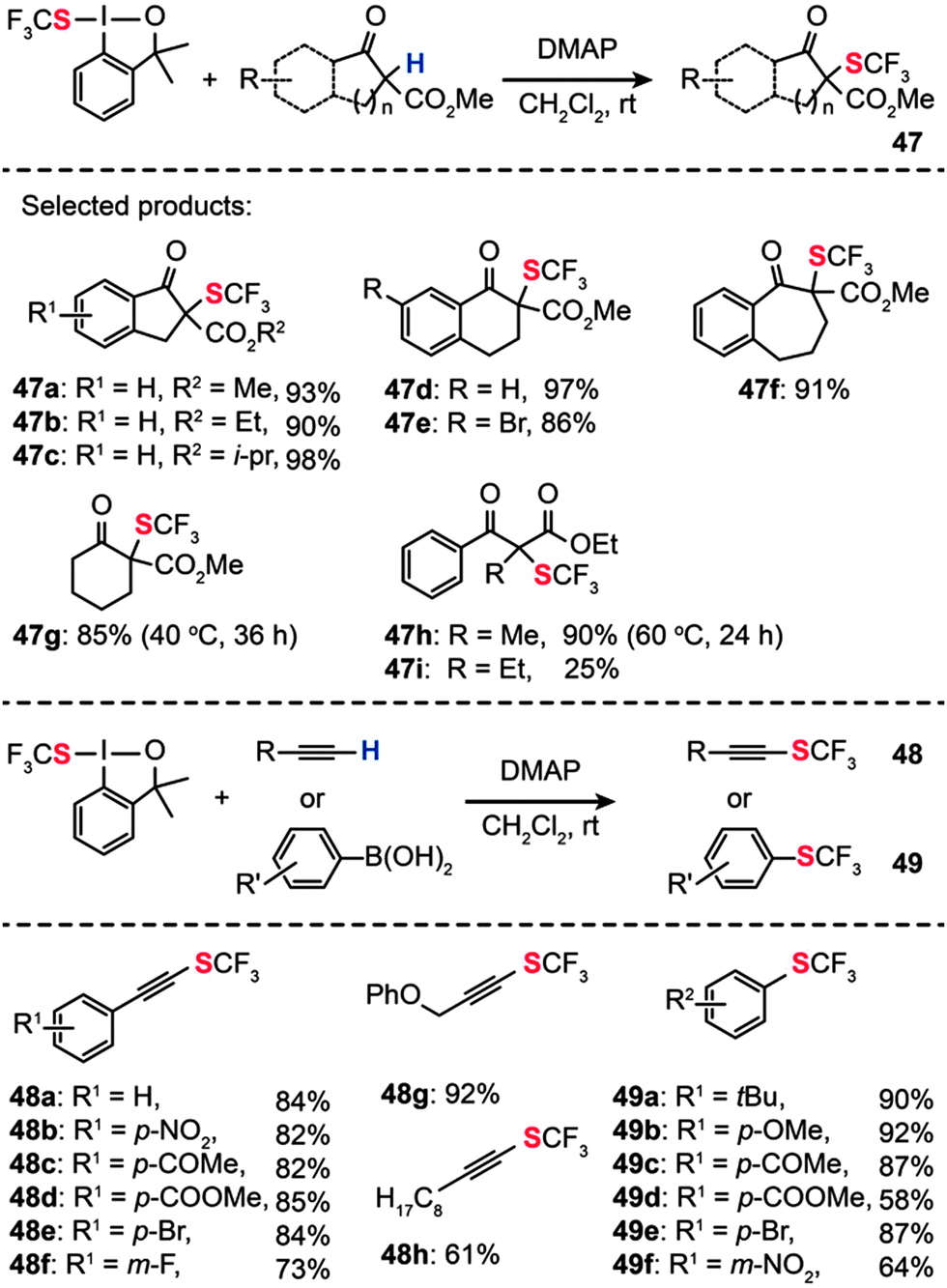 | ||
| Scheme 42 Shen's electrophilic α-trifluoromethylthiolation of β-ketoesters, aryl or vinyl boronic acids, and alkynes.77 | ||
Another important contribution was made by Zhang and co-workers, who recently developed a general strategy to achieve C–H sulfenylation of indoles by utilizing K2CO3 (Scheme 43).78 A range of indoles (50a–i) containing various substituents were obtained in excellent yields under the optimized conditions (50 mol% K2CO3 in DMSO at 100 °C). Furthermore, this reaction can be readily scaled up to produce multigram quantities of the product through a one-pot process without the need for stringent exclusion of air or moisture.
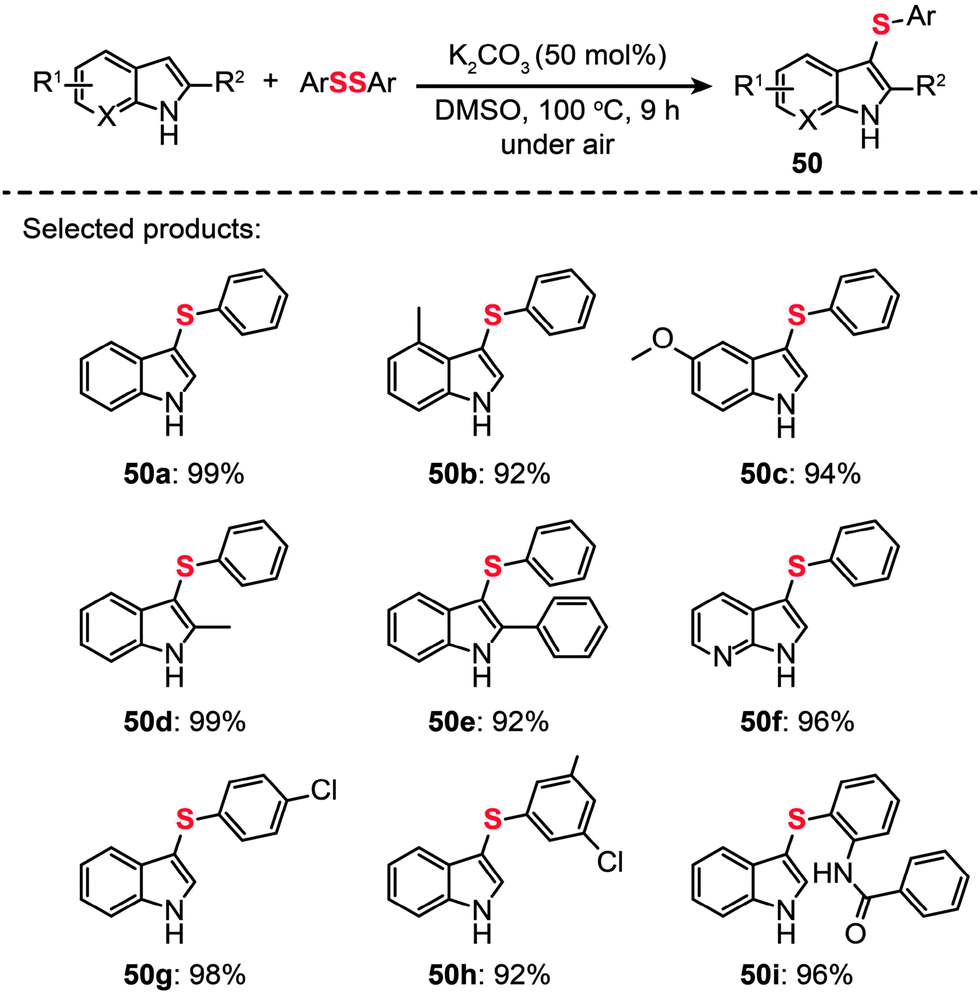 | ||
| Scheme 43 Zhang's K2CO3-promoted direct sulfenylation of indoles.78 | ||
In 2013, Kumar and co-workers reported a transition metal-free method for the synthesis of unsymmetrical diaryl chalcogenides (51) at room temperature starting from diaryl dichalcogenides and arenes under oxidative conditions by using potassium persulfate (K2S2O8) (Scheme 44).79 The optimized reaction conditions were determined in the presence of K2S2O8 as the oxidant and trifluoroacetic acid (TFA) as the solvent. Variously substituted arenes, including anisole, thioanisole, diphenyl ether, phenol, naphthol, di- and trimethoxy benzenes, xylene, mesitylene, N,N-dimethylaniline, and naphthalene, underwent C–S bond formation successfully, affording acceptable yields of the corresponding diaryl sulfides. The proposed reaction pathways for K2S2O8-mediated synthesis of unsymmetrical diaryl chalcogenides are shown in Scheme 44. The reaction is initiated by the reaction of the persulfate anion with a diphenyl disulfide to form a phenylchalcogenium ion intermediate. Electrophilic addition of the phenylchalcogenium ion with an electron-rich arene generates an arenium ion intermediate, which may give the diaryl sulfide product 51 upon removal of a proton from the arenium ion.
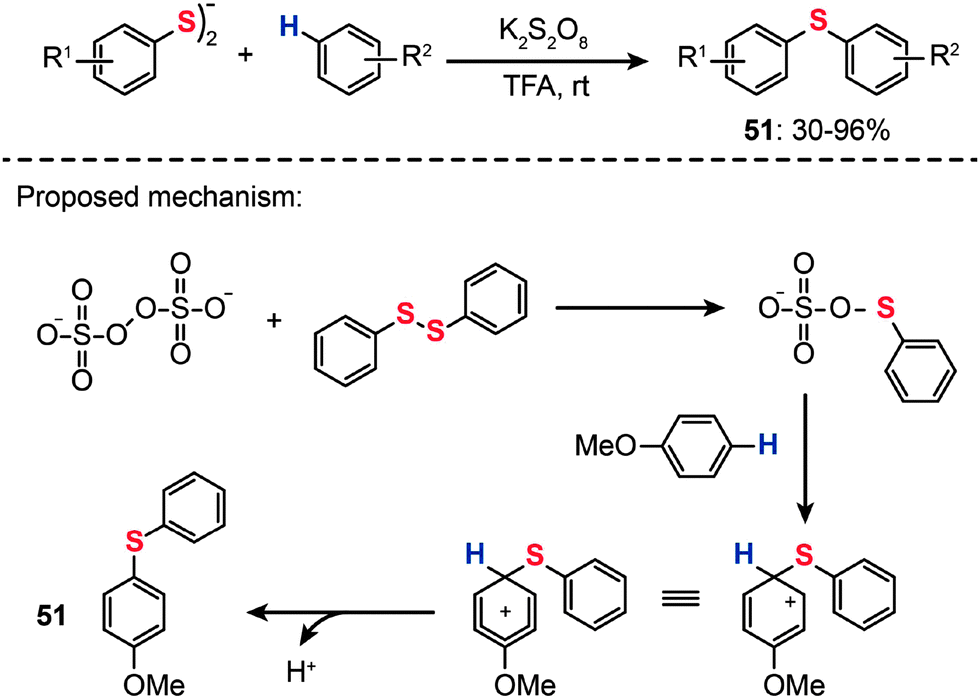 | ||
| Scheme 44 Kumar's transition-metal-free synthesis of diaryl sulfides.79 | ||
As an alternative sulfur source, arylsulfonyl chlorides have gained much attention in the light of their low cost, easy availability, and ease of operation. For example, You and co-workers developed a one-pot procedure for sulfenylation of arenes using arylsulfonyl chlorides in combination with triphenylphosphine (Scheme 45).80 This method represents a highly efficient preparation of diaryl thioethers (52a–f). It should be emphasized that the protocol for direct arene sulfenylation also works well for substituted indolizine and indole substrates. A plausible reaction mechanism proposed by the researchers involves the electrophilic attack of benzenesulfenyl chloride (PhSCl) by indolizine at the C3-position to form a di(hetero)aryl sulfide intermediate. The subsequent release of an acidic HCl gas leads to the generation of the corresponding coupling product.
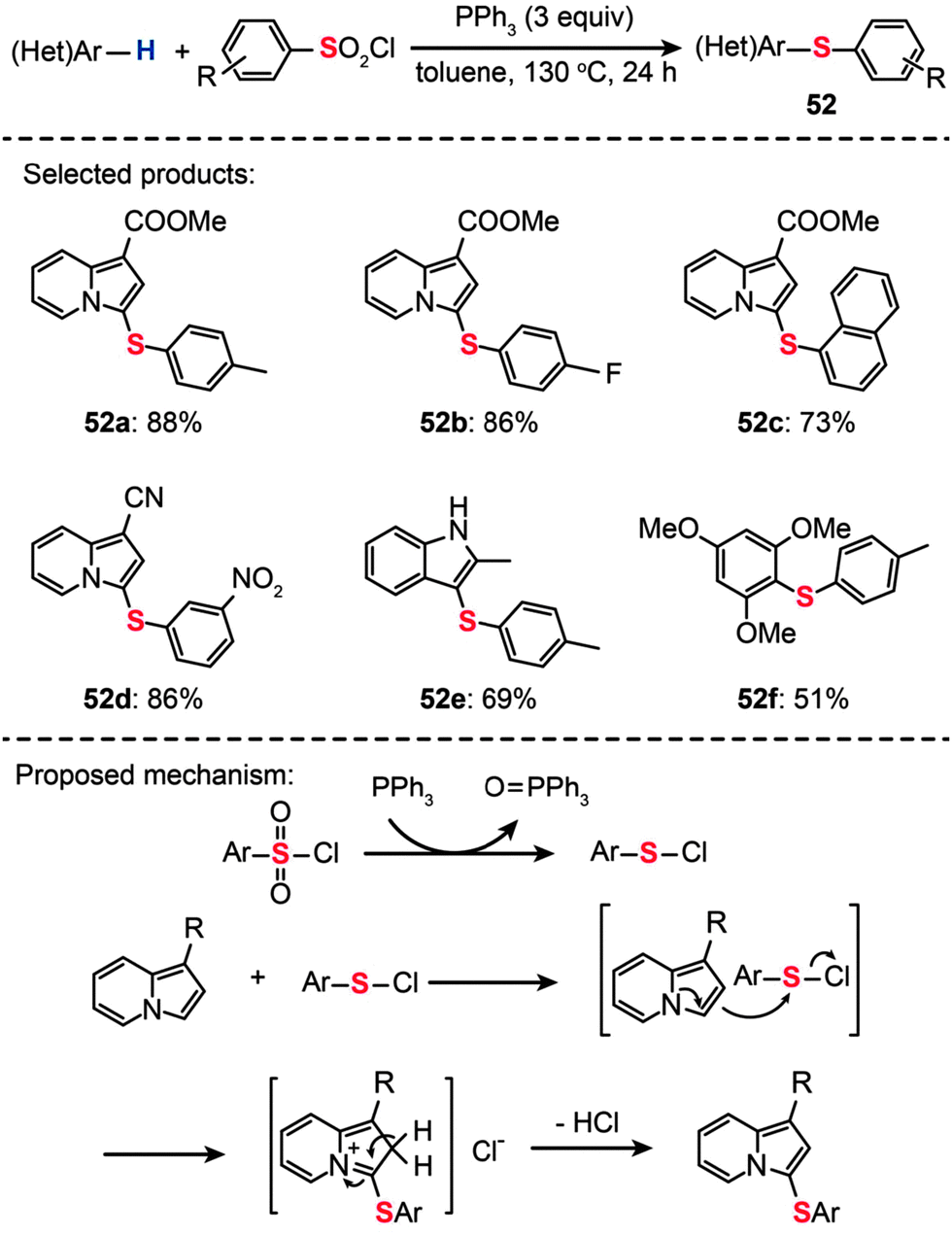 | ||
| Scheme 45 You's synthesis of di(hetero)aryl sulfides with sulfonyl chlorides.80 | ||
The use of molecular oxygen as the oxidant in the C–S bond formation via C–H functionalization deserves a special discussion as the molecular oxygen is safe for the environment. In 2012, Deng and co-workers developed the synthesis of 2-arylbenzothiazoles (53) by the treatment of 2-aminobenzenethiols with aryl ketones in the presence of molecular oxygen under metal- or iodine-free conditions (Scheme 46).81 Critically, the solvent used plays a key role in the reaction and the best yield is obtained in a mixture of DMSO–chlorobenzene. This protocol tolerates a variety of functional groups on the aminobenzenethiol, including methyl, methoxy, halo and nitro groups, giving their corresponding products in moderate to good yields. To unravel the underlying reaction mechanism, the authors carried out a 13C labeling experiment. Their results indicate that an imine intermediate is initially generated by the condensation of an amine with an aldehyde. Then, the enamine undergoes cyclization to form an isomer, followed by oxidation of the methyl group by molecular oxygen to generate an aldehyde intermediate and subsequent formation of the 2-arylbenzothiazole product 53 upon elimination of a proton and the CHO group.
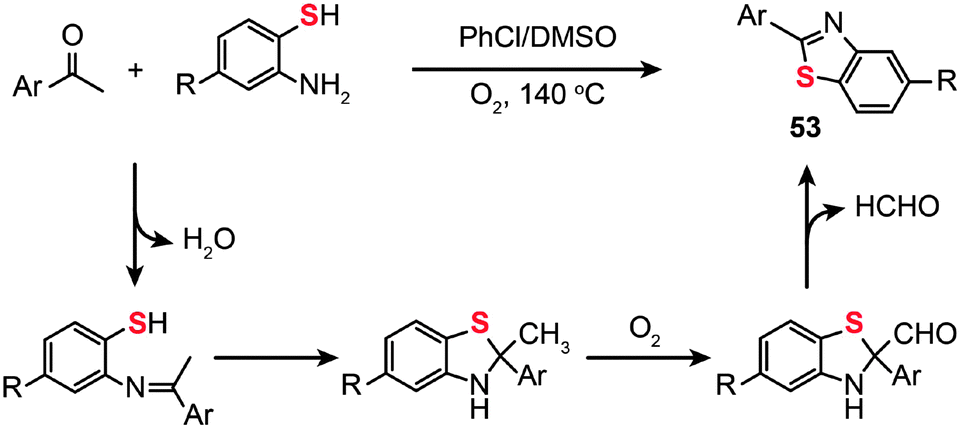 | ||
| Scheme 46 Deng's construction of 2-aryl benzothiazoles from aryl ketones and 2-aminobenzenethiols.81 | ||
It is quite evident that considerable progress has been made in transition-metal-free C–H functionalization. However, multifaceted challenges remain to be addressed in order to promote this methodology as a contender for highly efficient C–S bond formation. Research into efficient C–H functionalization of non-acidic sp3 C–H bonds is perhaps the most noticeable challenge for synthetic chemists in this field.
3. Decarboxylative C–S coupling
Recently, transition metal-catalyzed decarboxylative cross-coupling reactions have emerged as an attractive method to form C–C bonds starting from easily accessible carboxylic acids. These reactions are characterized by the cleavage of C–C bonds to carboxylative groups, followed by the formation of new C–C bonds at the site where the bond cleavage takes place.82 In 1968, Nilsson and Ullenius reported the first example of decarboxylative coupling by reacting 2-thenoic or 2-furoic acids with iodobenzene in the presence of a Cu(I) catalyst.83 Despite its potential utility, early examples of decarboxylative coupling had a limited impact on the synthesis of many types of organic molecules. It was not until the late 2000s that decarboxylative coupling became more prominent, as exemplified by the work of Myers84 and Goossen,85 in the field of metal-catalyzed cross-coupling.In contrast with C–H bond activation reactions, decarboxylative cross-coupling reactions through loss of CO2 generally do not need expensive organometallic reagents, while maintaining the advantage of regioselectivity offered by traditional cross-coupling reactions. However, these reactions are dominated by C–C and C–N bond-forming transformations.86 In 2009, Duan et al. reported the first direct decarboxylative coupling of ortho-substituted aryl carboxylic acids with thiols as an unprecedented synthetic entry to aryl sulfides (Scheme 47).87 Importantly, the direct decarboxylative C–S coupling provides an alternative access to aryl sulfides (54) without the need for halocarbon precursors. Optimized conditions for the reaction of 2-nitrobenzoic acid with 1-octanethiol (1.5 equiv.) were determined to be a catalytic amount of Pd(OAc)2 (5 mol%) and 1.5 equiv. of CuCO3·Cu(OH)2 in combination with KF (3 equiv.) in N-methylpyrrolidone (NMP) at 160 °C for 24 h. The products were obtained as mixtures of nitrobenzene and aminobenzene sulfides. Critically, primary aliphatic thiols were successfully transformed to aminobenzene sulfides in high yields, while cyclohexanethiol and aromatic thiols yielded nitrobenzene sulfides as major products. Notably, the use of disulfide precursors, which are advantageous over more reactive and odiferous thiols, also afforded the corresponding nitrobenzene sulfides in high yields. Despite the success of decarboxylative C–S coupling, these reactions suffer several limitations such as harsh reaction conditions, high temperature, strong bases, and often the need for toxic polar solvents. In addition, an electron-withdrawing group on the arenecarboxylic acid is required to afford good yields.
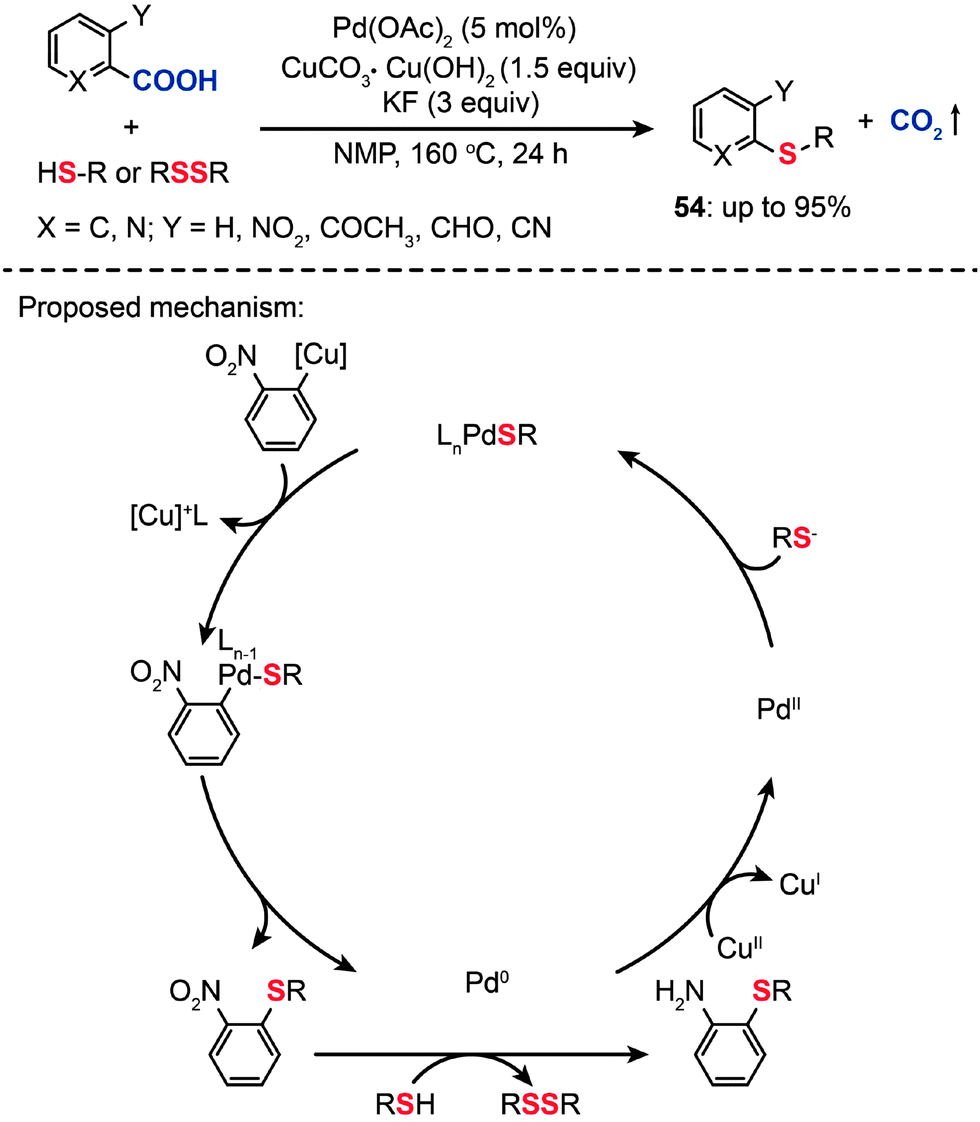 | ||
| Scheme 47 Liu's decarboxylative coupling of carboxylic acid with thiols or disulfides.87 | ||
The mechanism for the decarboxylative C–S coupling reaction is thought to be initiated by formation of an organometallic nucleophile upon decarboxylation, followed by the reaction with an electrophilic Pd(II) thiolate intermediate to form an aryl Pd(II) species (Scheme 47). Subsequent reductive elimination then generates the aryl sulfide coupling product that can undergo conversion to yield an aminobenzene sulfide. Oxidation of the reduced Pd(0) species by a Cu(II) catalyst regenerates the Pd(II) compound, thus supporting the continuation of the catalytic cycle for the palladium. On a separate note, it was discovered that a benzothiazole compound (55) can be alternatively obtained in moderate yield by reacting 2-nitrobenzoic acid with benzyl thiol under the standard decarboxylative cross-coupling conditions (Scheme 48).
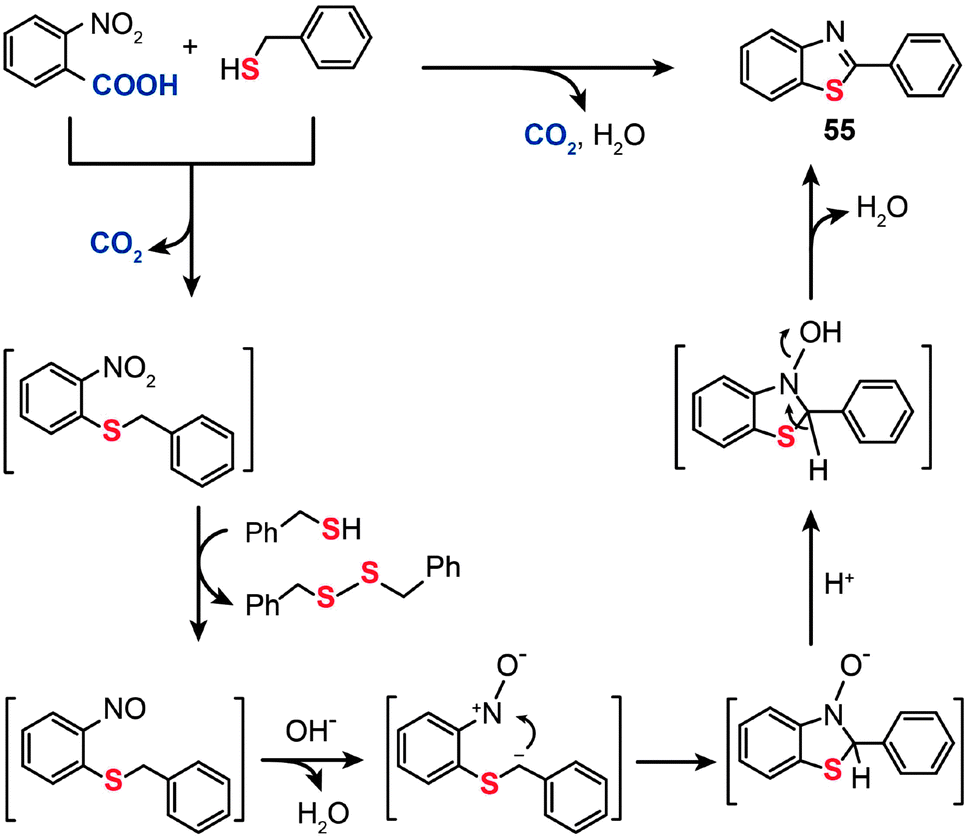 | ||
| Scheme 48 Liu's synthesis of 2-phenyl-benzothiazole by decarboxylative C–S cross-coupling.87 | ||
In a follow-up study, Ranjit et al. developed a versatile protocol, in which a CuI catalyst was used to initiate the decarboxylation of arylpropiolic acids, for the synthesis of vinyl sulfides (56) by the cross-coupling of the arylpropiolic acids with thiols (Scheme 49).88a The corresponding vinyl sulfides 56a–f were obtained in good to excellent yields with a high selectivity toward the Z-isomers. It is reasonable to assume that this Cu-catalyzed decarboxylative C–S cross-coupling should proceed via a mechanism similar to that of the reaction reported by the Zhang group.88b Notably, the formation of a cyclic alkene–carboxylate copper complex intermediate by the reductive C–S coupling is the key step that determines the stereoselectivity. This method not only expands our understanding of the decarboxylative reaction, but provides an entry to many intermediates and new pharmaceutically relevant compounds required for further investigations.
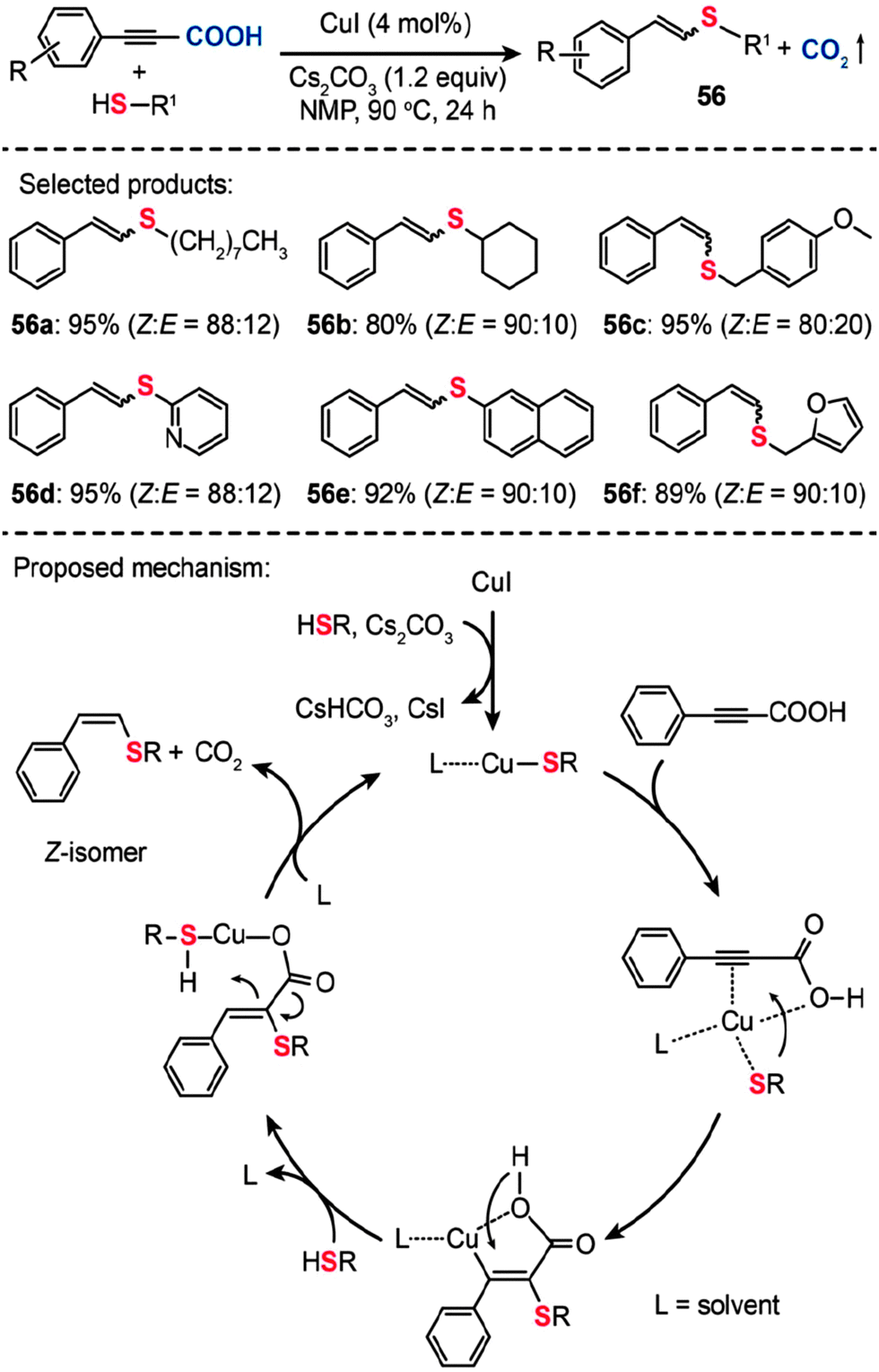 | ||
| Scheme 49 Liu and Zhang's Cu(I)-catalyzed decarboxylative C–S cross-coupling for synthesis of vinyl sulfides.88 | ||
In 2011, Becht and co-workers presented an efficient route to diaryl sulfides (57) from hindered electron-rich 2,6-disubstituted arenecarboxylic acids by decarboxylative C–S coupling (Scheme 50).89 Optimization reactions were carried out by treating 2,6-dimethoxybenzoic acid with diphenyl disulfide in conjunction with different catalysts, including PdCl2, Pd(OAc)2 and Pd(CF3CO2)2. The highest yield (75%) of the diaryl sulfide 57a was achieved in the presence of Pd(CF3CO2)2 (30 mol%), Ag2CO3 (2.2 equiv.), and a mixture of 1,4-dioxane–tetramethylene sulfoxide as the solvent. Furthermore, when PhSeSePh was subjected to catalysis under analogous conditions, the corresponding diaryl selenide product could be obtained in good yield. This reaction represents the first example of forming a C–Se bond from an arenecarboxylic acid.
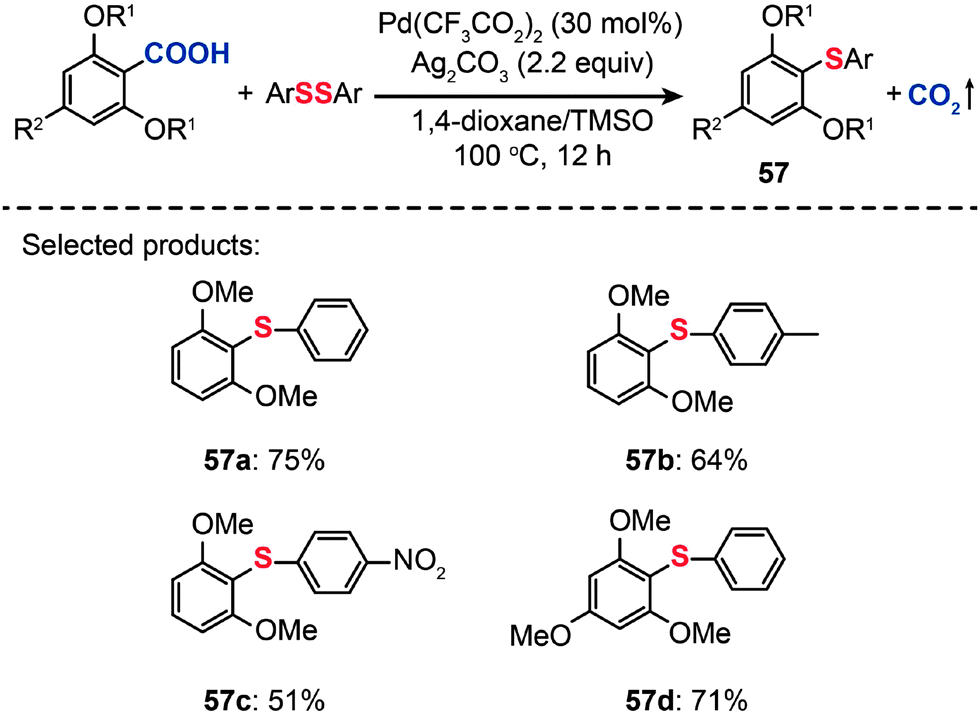 | ||
| Scheme 50 Becht's Pd-catalyzed decarboxylative cross-couplings of hindered 2,6-dialkoxybenzoic acids.89 | ||
As a result of the great benefits of decarboxylative coupling in chemical technology, considerable attempts have been undertaken to the development of suitable catalytic systems for decarboxylative C–S cross-coupling. Despite the efforts, these systems suffered from drastic conditions for complete cross-coupling and had intrinsic limitations. Best results were only obtained with ortho-substituted or heterocyclic carboxylic acids. A better mechanistic understanding of decarboxylative processes is essential for improving and optimizing process conditions, the catalysts and for making a broad range of aromatic or heteroaromatic acids more effective toward cross-coupling.
4. C–S coupling catalyzed by metal nanoparticles
The search for new catalysts that allow for simultaneous control over activity and selectivity is recognized as one of the major challenges of the chemical industry. Transition metal-based homogenous catalysts show remarkable activity and selectivity during chemical transformations, but often suffer from lack of recyclability and high cost of waste disposal. In contrast with homogeneous counterparts, nanoparticle-based catalysts provide the advantages of high surface area, excellent thermal stability, easy separation from reaction mixtures, and a high level of recyclability, but are generally less selective and active. To this end, the emergence of nanoparticle-based catalysts may enable the optimal control of chemical transformations via an atom-economical route.90–94 The nanoparticles can be recycled with negligible loss of selectivity and minimum loss of activity. Clever design and use of nanoparticle-based catalysts lead to highly efficient reactions, which are less energy demanding, less wasteful, and more cost effective.Indeed, highly selective and active nanoparticle-based catalysts have already been made, but mostly for C–C coupling reactions.90 By comparison, nanoparticle-catalyzed C–S bond-forming reactions were clearly underrepresented in the field of catalysis. In 2013, Zeni and co-workers led the charge in an exciting new field and reported the synthesis of aryl sulfides (58) by reacting substituted thiazoles and benzothiazoles with diphenyl disulfides in the presence of CuO nanoparticles (Scheme 51).95 The optimized conditions involve 20 mol% of CuO nanoparticles, K2CO3 (2 equiv.) as the base, DMF as the solvent, a temperature of 140 °C, and a reaction time of 24 h under an argon atmosphere. The nanoparticle-based coupling reaction is thought to proceed through a catalytic cycle involving ligand coordination of the nanoparticles by diphenyl dichalcogenides to give a thiol-stabilized nanoparticle intermediate. Metalation of the benzothiazole by the thiol-stablized nanoparticle followed by reductive elimination affords the aryl sulfide 58 to complete the catalytic cycle.
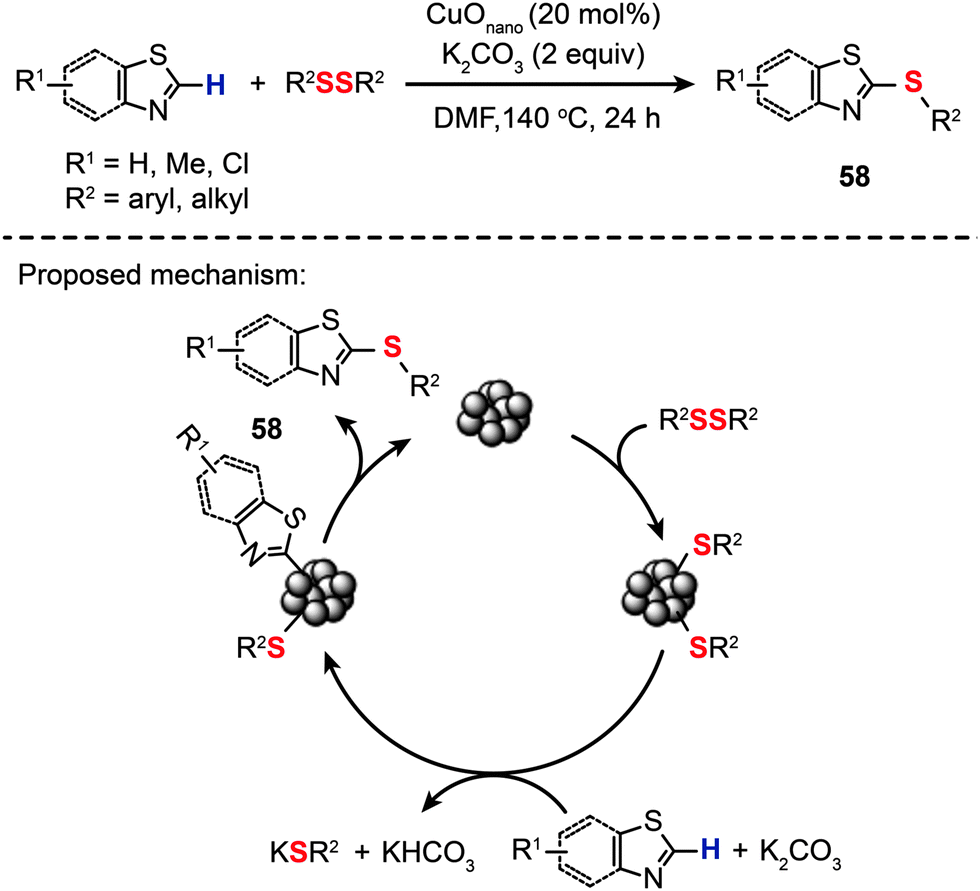 | ||
| Scheme 51 Zeni's CuO nanoparticle-catalyzed synthesis of 2-(organochalcogen)thiazoles.95 | ||
Although there are a few successful examples of C–H functionalization/C–S cross-coupling with metal or metal oxide nanoparticles, general catalytic systems with broad substrate scope are in high demand. The controversy on the homogeneous or heterogeneous nature of the cross-coupling reaction mechanism due to the issue of particle leaching remains to be addressed. Despite daunting synthetic challenges, it is likely that the next few years of catalytic research will advance this nanoparticle-based technology with much improved coupling efficiencies and selectivities while retaining maximum flexibility in waste recycling.
5. Conclusion
Over the past five years, extensive experimental work in metal-catalyzed cross-coupling reactions has resulted in significant advances for C–S bond formation via C–H functionalization and decarboxylation processes. In this regard, we have selected over 50 representative reactions as the key subjects of the review. Although these constitute only a small fraction of the state of the art, they amply illustrate the importance of this field to chemical synthesis and the science of constructing natural products and designed molecules. To further expand this area in organic synthesis, the challenge for synthetic chemists is to identify sustainable technologies for efficient C–S couplings under mild conditions and to unravel the underlying mechanisms that govern these organic transformations. To this end, we need to develop a range of new approaches that promote green chemistry for both economic and environmental benefits. Previously, there was skepticism on the profitability of sustainable technologies, but a number of examples of pharmaceutical synthesis have shown potential to greatly improve both environmental outcomes and profitability through catalyst innovation.Abbreviations
| Ar | Aryl group |
| Bn | Benzyl group |
| Boc | tert-Butoxycarbony |
| Bpy | 2,2′-Bipyridine |
| Bu | Butyl group |
| COD | 1,5-Cyclooctadiene |
| CF3 | Trifluoromethyl |
| CF3S– | Trifluoromethylthio group |
| Cp | Cyclopentadiene |
| DBU | 1,8-Diazabicycloundec-7-ene |
| DCE | 1,2-Dichloroethane |
| DMA | N,N-Dimethylacetamide |
| DMAP | 4-Dimethylaminopyridine |
| DMC | Dimethyl carbonate |
| DMF | Dimethylformamide |
| DMSO | Dimethyl sulfoxide |
| dppe | 1,2-Bis(diphenylphosphino)ethane |
| dppf | 1,10-Bis(diphenylphosphino) ferrocene |
| DTBP | Di-tert-butyl peroxide |
| EDG | Electron-donating group |
| Et | Ethyl group |
| EWG | Electron-withdrawing group |
| HIV | Human immunodeficiency virus |
| KIE | Kinetic isotope effect |
| Me | Methyl group |
| NMP | N-Methylpyrrolidone |
| OAc | Acetoxy group |
| OMe | Methoxy group |
| OTf | Trifluoromethanesulfonic acid |
| p-Cymene | 1-Methyl-4-(1-methylethyl)benzene |
| PH3 | Phosphine |
| PhSCl | Benzenesulfenyl chloride |
| Pin2B2 | Bis(pinacolato)diboron |
| Piv | Pivaloyl |
| PPh3 | Triphenylphosphine |
| Pr | Propyl group |
| SET | Single electron transfer |
| t-AmOH | tert-Amyl alcohol |
| TBHP | tert-Butyl hydroperoxide |
| TEMPO | 2,2,6,6-Tetramethylpiperidine-1-oxyl |
| TFA | Trifluoroacetic acid |
| THF | Tetrahydrofuran |
| TMSO | Tetramethylene sulfoxide |
| tRNA | Transfer ribonucleic acids |
Acknowledgements
C.S. acknowledges financial support from the National Natural Science Foundation of China (No. 21302171) and Science and Technology Plan of Zhejiang Province (No. 2014C31153). P.Z. acknowledges financial support from the National Natural Science Foundation of China (No. 21376058), Zhejiang Provincial Natural Science Foundation of China (No. LZ13B020001), and the Key-Sci-Tech Innovation Team of Zhejiang Province (No. 2010R50017). A.H. acknowledges the financial support from National University of Singapore, the Ministry of Education and the Agency for Science, Technology and Research (A*Star) of Singapore (No. R-143-000-426-305 and R143-000-426-331). X.L. acknowledges the National Research Foundation and the Economic Development Board (Singapore-Peking-Oxford Research Enterprise, COY-15-EWI-RCFSA/N197-1) and the Agency for Science, Technology and Research (A*STAR) for supporting this work. S.B. acknowledges the financial support from Institute of Materials Research Engineering (IMRE/12-1P0907).References
- M. Fontecave, S. Ollagnier-de-Choudens and E. Mulliez, Chem. Rev., 2003, 103, 2149–2166 CrossRef CAS PubMed.
- S. S. Mansy and J. A. Cowan, Acc. Chem. Res., 2004, 37, 719–725 CrossRef CAS PubMed.
- C. T. Walsh, Acc. Chem. Res., 2008, 41, 4–10 CrossRef CAS PubMed.
- N. S. Li, J. K. Frederiksen and J. A. Piccirilli, Acc. Chem. Res., 2011, 44, 1257–1269 CrossRef CAS PubMed.
- C. Hansch, P. G. Sammes and J. B. Taylor, Comprehensive medicinal chemistry; the rational design, mechanistic study & therapeutic application of chemical compounds, Pergamon Press, Oxford, New York, 1990, ch. 7.1, p. 2 Search PubMed.
- (a) A. Casini, J.-Y. Winum, J.-L. Montero, A. Scozzafava and C. T. Supuran, Bioorg. Med. Chem. Lett., 2003, 13, 837–840 CrossRef CAS; (b) M. Mellah, A. Voituriez and E. Schulz, Chem. Rev., 2007, 107, 5133–5209 CrossRef CAS PubMed.
- H. Haruki, M. G. Pedersen, K. I. Gorska, F. Pojer and K. Johnsson, Science, 2013, 340, 987–991 CrossRef CAS PubMed.
- (a) A. Natarajan, Y. Guo, F. Harbinski, Y.-H. Fan, H. Chen, L. Luus, J. Diercks, H. Aktas, M. Chorev and J. A. Halperin, J. Med. Chem., 2004, 47, 4979–4982 CrossRef CAS PubMed; (b) D. C. Cole, W. J. Lennox, S. Lombardi, J. W. Ellingboe, R. C. Bernotas, G. J. Tawa, H. Mazandarani, D. L. Smith, G. Zhang, J. Coupet and L. E. Schechter, J. Med. Chem., 2005, 48, 353–356 CrossRef CAS PubMed; (c) M. Banerjee, A. Poddar, G. Mitra, A. Surolia, T. Owa and B. Bhatta-charyya, J. Med. Chem., 2005, 48, 547–555 CrossRef CAS PubMed.
- A. R. Murphy and J. M. J. Fréchet, Chem. Rev., 2007, 107, 1066–1096 CrossRef CAS PubMed.
- (a) J. F. Hartwig, Nature, 2008, 314–322 CrossRef CAS PubMed; (b) Y.-M. Xi, B.-L. Dong, E. J. McClain, Q.-Y. Wang, T. L. Gregg, N. G. Akhmedov, J. L. Petersen and X.-D. Shi, Angew. Chem., Int. Ed., 2014, 53, 4657–4661 CrossRef CAS PubMed; (c) Q.-Q. Lu, J. Zhang, G.-L. Zhao, Y. Qi, H.-M. Wang and A. Lei, J. Am. Chem. Soc., 2013, 135, 11481–11484 CrossRef CAS PubMed; (d) Q. Lu, J. Zhang, F. L. Wei, Y. Qi, H. Wang, Z. Liu and A. Lei, Angew. Chem., Int. Ed., 2013, 52, 7156–7159 CrossRef CAS PubMed.
- E. Schaumann, Top. Curr. Chem., 2007, 274, 1–34 CrossRef CAS.
- D. Uraguchi, N. Kinoshita, D. Nakashima and T. Ooi, Chem. Sci., 2012, 3, 3161–3164 RSC.
- X. Wu, Y. Chen, M. Li, M. Zhou and S. K. Tian, J. Am. Chem. Soc., 2012, 134, 14694–14697 CrossRef CAS PubMed.
- A. Sakakura, H. Yamada and K. Ishihara, Org. Lett., 2012, 14, 2972–2975 CrossRef CAS PubMed.
- (a) C. Liu, H. Zhang, W. Shi and A. Lei, Chem. Rev., 2011, 111, 1780–1824 CrossRef CAS PubMed; (b) W. Shi, C. Liu and A. Lei, Chem. Soc. Rev., 2011, 40, 2761–2776 RSC; (c) M.-L. Louillat and F. W. Patureau, Chem. Soc. Rev., 2014, 43, 901–910 RSC.
- J. Magano and J. R. Dunetz, Chem. Rev., 2011, 111, 2177–2250 CrossRef CAS PubMed.
- S. R. Neufeldt and M. S. Sanford, Acc. Chem. Res., 2012, 45, 936–946 CrossRef CAS PubMed.
- K. Inamoto, Y. Arai, K. Hiroya and T. Doi, Chem. Commun., 2008, 5529–5531 RSC.
- K. Inamoto, C. Hasegawa, K. Hiroya and T. Doi, Org. Lett., 2008, 10, 5147–5150 CrossRef CAS PubMed.
- K. Inamoto, C. Hasegawa, J. Kawasaki, K. Hiroya and T. Doi, Adv. Synth. Catal., 2010, 352, 2643–2655 CrossRef CAS.
- K. Inamoto, K. Nozawa and Y. Kondo, Synlett, 2012, 1678–1682 CrossRef CAS PubMed.
- (a) L. L. Joyce and R. A. Batey, Org. Lett., 2009, 11, 2792–2795 CrossRef CAS PubMed; (b) D. Kalyani, N. R. Deprez, L. V. Desai and M. S. Sanford, J. Am. Chem. Soc., 2005, 127, 7330–7331 CrossRef CAS PubMed; (c) C. L. Campeau and K. Fagnou, Chem. Commun., 2006, 1253–1264 RSC.
- X. Zhao, E. Dimitrijeviće and V. M. Dong, J. Am. Chem. Soc., 2009, 131, 3466–3467 CrossRef CAS PubMed.
- (a) J. Zhu, Z. Chen, H. Xie, S. Li and Y. Wu, Org. Lett., 2010, 12, 2434–2436 CrossRef CAS PubMed; (b) X. Y. Zhang, W. L. Zeng, Y. Yang, H. Huang and Y. Liang, Org. Lett., 2014, 16, 876–879 CrossRef CAS PubMed.
- R. Samanta and A. P. Antonchick, Angew. Chem., Int. Ed., 2011, 50, 5217–5220 CrossRef CAS PubMed.
- L. H. S. Smith, S. C. Coote, H. F. Sneddon and D. J. Procter, Angew. Chem., Int. Ed., 2010, 49, 5832–5844 CrossRef CAS PubMed.
- C. Shen, H. Xia, H. Yan, X. Chen, S. Ranjit, X. Xie, D. Tan, R. Lee, Y. Yang, B. Xing, K.-W. Huang, P. Zhang and X. Liu, Chem. Sci., 2012, 3, 2388–2393 RSC.
- S. K. Sahoo, A. Banerjee, S. Chakraborty and B. K. Patel, ACS Catal., 2012, 2, 544–551 CrossRef CAS.
- H. Huang, J. Li, C. Lescop and Z. Duan, Org. Lett., 2011, 13, 5252–5255 CrossRef CAS PubMed.
- J. Li, H. Huang, W. Liang, Q. Gao and Z. Duan, Org. Lett., 2013, 15, 282–285 CrossRef CAS PubMed.
- C. F. Xu and Q. L. Shen, Org. Lett., 2014, 16, 2046–2049 CrossRef CAS PubMed.
- M. Iwasaki, M. Iyanaga, Y. Tsuchiya, Y. Nishimura, W.-J. Li, Z.-P. Li and Y. Nishihara, Chem. – Eur. J., 2014, 20, 2459–2462 CrossRef CAS PubMed.
- F. Monnier and M. Taillefer, Angew. Chem., Int. Ed., 2009, 48, 6954–6971 CrossRef CAS PubMed.
- C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3464–3484 RSC.
- X. Chen, X.-S. Hao, C. E. Goodhue and J.-Q. Yu, J. Am. Chem. Soc., 2006, 128, 6790–6791 CrossRef CAS PubMed.
- (a) J. Srogl, J. Hývl, A. Révész and D. Schröder, Chem. Commun., 2009, 3463–3465 RSC; (b) J. Hývl and J. Srogl, Eur. J. Org. Chem., 2010, 2849–2851 CrossRef.
- H. Deng, Z. Li, F. Ke and X. Zhou, Chem. – Eur. J., 2012, 18, 4840–4843 CrossRef CAS PubMed.
- A. Banerjee, S. Kumar Santra, S. K. Rout and B. K. Patel, Tetrahedron, 2013, 69, 9096–9104 CrossRef CAS PubMed.
- S. Fukuzawa, E. Shimizu, Y. Atsuumi, M. Haga and K. Ogata, Tetrahedron Lett., 2009, 50, 2374–2376 CrossRef CAS PubMed.
- S. Zhang, P. Qian, M. Zhang, M. Hu and J. Cheng, J. Org. Chem., 2010, 75, 6732–6735 CrossRef CAS PubMed.
- S. Ranjit, R. Lee, D. Heryadi, C. Shen, J. Wu, P. Zhang, K. Huang and X. Liu, J. Org. Chem., 2011, 76, 8999–9007 CrossRef CAS PubMed.
- A. Zhou, X. Liu, K. Yang, S. Zhao and Y. Liang, Org. Biomol. Chem., 2011, 9, 5456–5462 CAS.
- J. Cheng, C. Yi, T.-J. Liu and C. Lee, Chem. Commun., 2012, 48, 8440–8442 RSC.
- C. Yu, C. Zhang and X. Shi, Eur. J. Org. Chem., 2012, 1953–1959 CrossRef CAS.
- C. Dai, Z. Xu, F. Huang, Z. Yu and Y. Gao, J. Org. Chem., 2012, 77, 4414–4419 CrossRef CAS PubMed.
- C.-P. Zhang and D. A. Vicic, J. Am. Chem. Soc., 2012, 134, 183–185 CrossRef CAS PubMed.
- C. Chen, Y. Xie, L. Chu, R.-W. Wang, X. Zhang and F.-L. Qing, Angew. Chem., Int. Ed., 2012, 51, 2492–2495 CrossRef CAS PubMed.
- L. D. Tran, I. Popov and O. Daugulis, J. Am. Chem. Soc., 2012, 134, 18237–18240 CrossRef CAS PubMed.
- L. M. Huffman and S. S. Stahl, J. Am. Chem. Soc., 2008, 130, 9196–9197 CrossRef CAS PubMed.
- D. Alves, R. G. Lara, M. E. Contreira, C. S. Radatz, L. F. B. Duarte and G. Perin, Tetrahedron Lett., 2012, 53, 3364–3368 CrossRef CAS PubMed.
- Z. Wu, H. Song, X. Cui, C. Pi, W. Du and Y. Wu, Org. Lett., 2013, 15, 1270–1273 CrossRef CAS PubMed.
- M. Arisawa, K. Fujimoto, S. Morinaka and M. Yamaguchi, J. Am. Chem. Soc., 2005, 127, 12226–12227 CrossRef CAS PubMed.
- M. Arisawa, K. Suwa and M. Yamaguchi, Org. Lett., 2009, 11, 625–627 CrossRef CAS PubMed.
- M. Arisawa, F. Toriyama and M. Yamaguchi, Tetrahedron Lett., 2011, 52, 2344–2347 CrossRef CAS PubMed.
- Y.-X. Yang, W. Hou, L.-H. Qin, J.-J. Du, H.-J. Feng, B. Zhou and Y.-C. Li, Chem. – Eur. J., 2014, 20, 416–420 CrossRef CAS PubMed.
- J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- Y. Cheng, J. Yang, Y. Qu and P. Li, Org. Lett., 2012, 14, 98–101 CrossRef CAS PubMed.
- O. Saidi, J. Marae, A. E. W. Ledger, P. Liu, M. F. Mahon, G. Kociok-Köhn, M. K. Whittlesey and C. G. Frost, J. Am. Chem. Soc., 2011, 133, 19298–19301 CrossRef CAS PubMed.
- M. Chen, Z.-T. Huang and Q.-Y. Zheng, Chem. Commun., 2012, 48, 11686–11688 RSC.
- C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Rev., 2011, 111, 1293–1314 CrossRef CAS PubMed.
- H. Wang, L. Wang, J. Shang, X. Li, H. Wang, J. Gui and A. Lei, Chem. Commun., 2012, 48, 76–78 RSC.
- A. Studer and D. P. Curran, Angew. Chem., Int. Ed., 2011, 50, 5018–5022 CrossRef CAS PubMed.
- R. Cano, D. J. Ramón and M. Yus, J. Org. Chem., 2011, 76, 654–660 CrossRef CAS PubMed.
- Q. Xiao, L. Tian, R. Tan, Y. Xia, D. Qiu, Y. Zhang and J. Wang, Org. Lett., 2012, 14, 4230–4233 CrossRef CAS PubMed.
- S. Yanagisawa, K. Ueda, T. Taniguchi and K. Itami, Org. Lett., 2008, 10, 4673–4676 CrossRef CAS PubMed.
- (a) C. Sun, H. Li, D. Yu, M. Yu, X. Zhou, X. Lu, K. Huang, S. Zheng, B. Li and Z. Shi, Nat. Chem., 2010, 2, 1044–1049 CrossRef CAS PubMed; (b) W. Liu, H. Cao, H. Zhang, H. Zhang, K. Chung, C. He, H. Wang, F. Y. Kwong and A. Lei, J. Am. Chem. Soc., 2010, 132, 16737–16740 CrossRef CAS PubMed.
- R. Tang, Y. Xie, Y. Xie, J. Xiang and J. Li, Chem. Commun., 2011, 47, 12867–12869 RSC.
- S.-R. Guo, Y.-Q. Yuan and J.-N. Xiang, Org. Lett., 2014, 15, 4654–4657 CrossRef PubMed.
- W. Ge and Y. Wei, Green Chem., 2012, 14, 2066–2070 RSC.
- Y.-P. Zhu, M. Lian, F.-C. Jia, M.-C. Liu, J.-J. Yuan, Q.-H. Gao and A.-X. Wu, Chem. Commun., 2012, 48, 9086–9088 RSC.
- Y.-P. Zhu, F.-C. Jia, M.-C. Liu and A.-X. Wu, Org. Lett., 2014, 14, 4414–4417 CrossRef PubMed.
- Y.-F. Liao, P.-C. Jiang, S.-P. Chen, H.-R. Qi and G.-J. Deng, Green Chem., 2013, 15, 3302–3306 RSC.
- F.-H. Xiao, H. Xie, S.-W. Liu and G.-J. Deng, Adv. Synth. Catal., 2014, 356, 364–368 CrossRef CAS.
- H. W. Pinnick, M. A. Reynolds, R. T. McDonald Jr and W. D. Brewster, J. Org. Chem., 1980, 45, 930–932 CrossRef CAS.
- C. Chen, L.-L. Chu and F. Qing, J. Am. Chem. Soc., 2012, 134, 12454–12457 CrossRef CAS PubMed.
- L. Zou, J. Reball, J. Mottweiler and C. Bolm, Chem. Commun., 2012, 48, 11307–11309 RSC.
- X. Shao, X. Wang, T. Yang, L. Lu and Q. Shen, Angew. Chem., Int. Ed., 2013, 52, 3457–3460 CrossRef CAS PubMed.
- P. Sang, Z.-K. Chen, J.-W. Zou and Y.-H. Zhang, Green Chem., 2013, 15, 2096–2100 RSC.
- C. D. Prasad, S. J. Balkrishna, A. Kumar, B. S. Bhakuni, K. Shrimali, S. Biswas and S. Kumar, J. Org. Chem., 2013, 78, 1434–1443 CrossRef CAS PubMed.
- Q. Wu, D.-B. Zhao, X.-R. Qin, J.-B. Lan and J.-S. You, Chem. Commun., 2011, 47, 9188–9190 RSC.
- Y.-F. Liao, H.-R. Qi, S.-P. Chen, P.-C. Jiang, W. Zhou and G.-J. Deng, Org. Lett., 2012, 14, 6004–6007 CrossRef CAS PubMed.
- N. Rodríguez and L. J. Goossen, Chem. Soc. Rev., 2011, 40, 5030–5048 RSC.
- (a) M. Nilsson, Acta Chem. Scand., 1966, 20, 423–426 CrossRef CAS PubMed; (b) M. Nilsson and C. Ullenius, Acta Chem. Scand., 1968, 22, 1998–2002 CrossRef CAS PubMed.
- (a) A. G. Myers, D. Tanaka and M. R. Mannion, J. Am. Chem. Soc., 2002, 124, 11250–11251 CrossRef CAS PubMed; (b) D. Tanaka and A. G. Myers, Org. Lett., 2004, 6, 433–436 CrossRef CAS PubMed.
- L. J. Goossen, G. Deng and L. M. Levy, Science, 2006, 313, 662–664 CrossRef CAS PubMed.
- (a) W. Jia and N. Jiao, Org. Lett., 2010, 12, 2000–2003 CrossRef CAS PubMed; (b) P. Hu, Y. Shang and W. Su, Angew. Chem., Int. Ed., 2012, 51, 5945–5949 CrossRef CAS PubMed.
- Z. Duan, S. Ranjit, P. Zhang and X. Liu, Chem. – Eur. J., 2009, 15, 3666–3669 CrossRef CAS PubMed.
- (a) S. Ranjit, Z. Duan, P. Zhang and X. Liu, Org. Lett., 2010, 12, 4134–4136 CrossRef CAS PubMed; (b) S. N. Riduan, J. Y. Ying and Y. Zhang, Org. Lett., 2012, 14, 1780–1783 CrossRef CAS PubMed.
- J. M. Becht and C. L. Drian, J. Org. Chem., 2011, 76, 6327–6330 CrossRef CAS PubMed.
- A. Balanta, C. Godard and C. Claver, Chem. Soc. Rev., 2011, 40, 4973–4985 RSC.
- J. C. Garcia-Martinez, R. Lezutekong and R. M. Crooks, J. Am. Chem. Soc., 2005, 127, 5097–5103 CrossRef CAS PubMed.
- G. A. Somorjai and R. M. Rioux, Catal. Today, 2005, 100, 201–215 CrossRef CAS PubMed.
- C. Jiang, S. Ranjit, Z. Duan, Y. L. Zhong, K. P. Loh, C. Zhang and X. Liu, Nanoscale, 2009, 1, 391–394 RSC.
- L. T. Ball, G. C. Lloyd-Jones and C. A. Russell, Science, 2012, 337, 1644–1648 CrossRef CAS PubMed.
- A. R. Rosario, K. K. Casola, C. E. S. Oliveira and G. Zeni, Adv. Synth. Catal., 2013, 355, 2960–2966 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |