
Silica-based nanocapsules: synthesis, structure control and biomedical applications
Yu
Zhang
a,
Benedict You Wei
Hsu
b,
Changliang
Ren
c,
Xu
Li
*c and
John
Wang
*ab
aDepartment of Materials Science & Engineering, National University of Singapore, Singapore. E-mail: msewangj@nus.edu.sg; msezy@nus.edu.sg
bNUS Graduate School for Integrative Sciences and Engineering, National University of Singapore, Singapore
cInstitute of Materials Research and Engineering, Agency for Science, Technology and Research (A*STAR), 3 Research Link, Singapore. E-mail: x-li@imre.a-star.edu.sg
First published on 13th October 2014
Abstract
Synergistically combining the merits of silica (e.g., mechanical robustness, biocompatibility and great versatility in surface functionalization) and capsular configurations (e.g., a large inner cavity, low density and favourable colloidal properties), silica-based nanocapsules (SNCs) with a size cutoff of ∼100 nm have gained growing interest in encapsulating bioactive molecules for bioimaging and controlled delivery applications. Within this context, this review provides a comprehensive overview of the synthetic strategies, structural control and biomedical applications of SNCs. Special emphasis is placed on size control at the nanoscale and material composition manipulation of each strategy and the newly emerging synthetic strategies. The applications of SNCs in bioimaging/diagnosis and drug delivery/therapy and the structure engineering that is critically important for the bio-performance of SNCs are also addressed in this review.
 Yu Zhang | Dr Yu Zhang received her Bachelor of Engineering degree in Polymer Science and Engineering and Master of Science degree in Materials Science from East China University of Science and Technology (ECUST) in 2000 and 2003, respectively. She received her PhD degree in Materials Science from National University of Singapore (NUS) in 2011. She is currently working in the group of Professor John Wang in the Department of Materials Science & Engineering at NUS on the development of bioactive silica-based nanocapsules. Her research interests include the synthesis of multifunctional silica-based nanocapsules for biomedical applications, and the synthesis and characterization of novel titania nanostructures via a block copolymer templating route and their applications in photocatalysis and dye-sensitized solar cells. |
 Benedict You Wei Hsu | Benedict You Wei Hsu received his Bachelor of Engineering degree in Materials Science and Engineering from NUS in 2011. Currently, he is pursuing his PhD degree in NUS under the supervision of Professor John Wang. His research interests mainly focus on the development of multifunctional nanomaterials for biomedical applications. |
 Changliang Ren | Dr Changliang Ren studied chemistry and received his BSc degree from Sichuan University in 2007. He received his PhD degree in chemistry from NUS in 2011 under the supervision of Dr Huaqiang Zeng. After 1 year of postdoctoral work with Prof. Young-Tae Chang at the department of chemistry, NUS, he joined Dr Xu Li's group as a research scientist at the Institute of Materials Research and Engineering (IMRE), Agency for Science, Technology and Research (A*STAR). His current research interests include the development of silica-based nanoparticles for biological applications including cell isolation, bioimaging and controlled drug delivery. |
 Xu Li | Dr Xu Li is a Senior Scientist at IMRE, A*STAR. He finished his PhD study in the Department of Chemistry, NUS, in 2001, and then joined IMRE. He has been an adjunct Associate Professor of the Department of Chemistry, NUS, since 2012. He has published more than 70 research papers and filed 15 international patents as a co-inventor. As a principle investigator, he is now leading a research team on hybrid and nano-structured material development for various applications, especially nanostructured particles for controlled drug delivery and bioimaging. |
 John Wang | Professor John Wang is Head of Department of Materials Science and Engineering, and Senior Faculty Member, NUS Graduate School for Integrative Sciences & Engineering (NGS), NUS. He has more than 30 years of experience in teaching and research of functional materials and materials chemistry. His current research focuses include: materials chemistry, biomaterials and nanohybrids for bioimaging and healthcare, and nanostructured materials for sustainable energy. He has published >300 papers in prestigious, top international refereed journals. He has been invited, on a regular basis, to give keynote/invited lectures at major international conferences/symposia/workshops. In 2003, he was elected Fellow of the Institute of Materials, Minerals and Mining (UK), which is the highest grade awarded to individuals who have achieved international excellence in materials, minerals or mining. |
1 Introduction
Capsules are defined as vesicular or “reservoir” systems in which hollow or soft material-filled cavities are surrounded by a shell of solid materials.1–3 With the fascinating structural features such as a large inner cavity, low density and favourable colloidal behaviour, capsules have been widely explored to modulate both chemical and physical characteristics of materials that enhance the overall stability in various environments when compared to their solid counterparts, as well as to increase the active area and pore volume for applications in catalysis, adsorption and energy storage.1–4 In addition, they are extremely useful to encapsulate various substances within their confined cavities so as to exhibit special optical, electrical, magnetic, chemical and biological properties.1–3 In particular, polymeric capsules of a wide variety of morphologies (e.g., micelles and vesicles) and compositions (e.g., polysaccharides, polypeptides, and amphiphilic fullerene derivatives), liposomes and inorganic hollow spheres have been demonstrated to be robust and tunable encapsulants for the immobilization of functional agents such as drugs, dyes, enzymes, genes, peptides, and other bioactive molecules.2,5–8 This has even driven the commercialization of some of the products (e.g., Doxil) in medicine and diagnosis.Silica capsules are ideal candidates as encapsulants for biomedical usage because they combine the merits of silica and capsular nanostructure. Silica is a non-toxic and biocompatible material.1 It is “generally recognized as safe” (GRAS) by the US Food and Drug Administration (FDA), as demonstrated by its common usage in the food additives and vitamin supplements. It also possesses the advantages of being low cost in production, mechanical and chemical stability, and optical transparency. The surface of silica is largely rich in hydroxyl groups, which renders it to be intrinsically hydrophilic and favourable for colloidal stability. Additionally, the silica surface can be easily modified by various functional moieties such as polymers and antibodies via well-established siloxane chemistry to gain multifunctionality.1,8
Since the pioneering work on templating synthesis of silica capsules from 1996 to 1998,9–12 a variety of synthesis paradigms have been employed to prepare varying types of silica capsules. They have dramatically expanded the range of silica capsules in sizes, shapes, and surface chemistry which has in turn catalysed new applications and fundamental research on silica capsules. In particular, silica-based nanocapsules (SNCs) on a truly nanoscale size (i.e., <100 nm) are gaining increasing attention, especially in the field of biomedical research. This is because the size cutoff of ∼100 nm offers considerable advantages such as reducing the reticuloendothelial system (RES) capture, prolonging blood circulation time, enhancing extravasation into targeted sites, increasing cellular uptake, and facilitating barrier penetration. A large number of SNCs have thus been developed in association with the advances of conventional templating synthesis and the emergence of new synthetic strategies (e.g., single micelle templating). The resulting SNCs have been extensively explored in various biomedical applications such as bioimaging/diagnosis and drug delivery/therapy. Within this context, this article reviews recent advances in the synthetic strategies, structural control of SNCs and their biomedical applications.
It is noted that although a number of reviews with regard to the synthesis of various silica nanostructures, such as spheres, core–shells, and mesoporous silica, and/or their applications have already been published,8,13,14 research on the synthesis and biomedical applications of SNCs with a hollow or hybrid capsular structure and in particular on a truly nanoscale size (i.e., <100 nm) has never been specifically and properly reviewed. To keep the review down to a manageable level, three dimensional porous silica (e.g. silica nanofoams), micro/meso/macroporous silica and silica nanotubes are not considered in this review.
In the first section of this review, we present a comprehensive overview of the synthetic strategies, which are categorized into cavity-generating (e.g., hard templating, soft templating and template-free) and shell-forming (e.g., stöber, reverse microemulsion and supramolecular-templated deposition). Special emphasis is placed on the key points of each strategy for a more precise control of the nanoscale size and material composition. Besides, the single-micelle templating approach is highlighted due to its effectiveness in fabricating SNCs. Next we address the structure control of SNCs in terms of the particle size, the level of shell porosity, cavity topology, cavity content, and surface functionality that are critical for their bio-performance. This is followed by the section that details a survey of SNCs as an excellent encapsulant to load a wide variety of bioactive agents for a broad range of biomedical applications. Finally, we conclude this review with some perspectives on future research and development of SNCs.
2 Design and synthetic strategies of silica-based nanocapsules
The development of SNCs generally involves three major steps, as illustrated in Fig. 1: (i) preparation of a nanosized template; (ii) formation of a silica-based shell on the template by depositing silica precursors (salts or alkoxides) onto the surface of the template; and followed by (iii) template removal through either calcination at high temperature or dissolution with a suitable solvent to generate a cavity inside the shell. In this review, the design and synthesis strategies of SNCs are categorized into cavity-generating and shell-forming respectively. More specifically, cavity-generating strategies include hard templating, soft templating, and the newly emerging template-free syntheses. In this regard, a wide panel of hard (i.e., rigid solid) templates such as polymer latex and inorganic nanoparticles (NPs) and soft (i.e., flexible solid, liquid, and gaseous) templates such as (micro)emulsion droplets, vesicles, micelles, polymer aggregates and gas bubbles have been employed to generate the cavity in SNCs. On the other hand, shell-forming strategies include stöber, reverse microemulsion, and supramolecular-templated deposition methods. In this section we present an overview of these synthetic strategies emphasizing on the aspects of size control at the nanoscale and silica composition control. | ||
| Fig. 1 Schematic illustration of the general steps for the formation of SNCs: (i) preparation of a nanosized template; (ii) deposition of a silica-based shell on the template; and (iii) template removal to generate a cavity inside the shell. | ||
2.1 Cavity-generating strategies
To date, both hard templating and soft templating have been widely explored for cavity generation in inorganic (silica and non-silica) hollow spheres. In general, when hard templates are employed, the templates must be eliminated eventually to leave behind a hollow shell. The shape and cavity size of the thus achieved hollow products are determined by the hard templates, which typically are monodisperse and have a well-defined morphology. However, removal of the templates through either thermal calcination or chemical etching may be uneconomical, time-consuming, and sometimes involves the utilization of toxic chemicals that can be harmful to the surrounding environment. As for the soft templating synthesis, although it is relatively easier to remove the templates, the as-prepared hollow products usually suffer from ill-defined morphologies and poor monodispersity due to the deformability and wide size distribution of the soft templates. Nonetheless, the recent emergence of single micelle templating, as well as the template-free approach, has been effective in synthesizing SNCs and thus will be highlighted in this section. | ||
| Fig. 2 Schematic illustration of the formation of SNCs by using functionalized PS templates. The scale bar in the TEM image is 100 nm. (Adapted from ref. 15 with permission from The Royal Society of Chemistry. Reprinted with permission from ref. 16. Copyright 2008 American Chemical Society.) | ||
In recent years, inorganic NPs have emerged as a new and promising class of hard templates for the fabrication of SNCs because they can be made very small (<100 nm) and highly monodisperse. Indeed, different inorganic NPs, including iron oxides (Fe2O3,20–22 and Fe3O423,24), CaCO3,25–30 PbS,31 and Au NPs,32,33 have been investigated for such purposes. These materials are selected because they can be dissolved in particular acids (e.g., hydrochloric acid,21–29 nitric acid,31,34 and oxalic acid20), alkalis (e.g., ammonia34), or an appropriate solvent (e.g., cyanide33) in which silica is not soluble. Thus, they allow for a selective dissolution of the templates without affecting the silica shell.
Microemulsion templating. Microemulsion is defined as an optically isotropic, thermodynamically stable and transparent (or translucent) system composed of two immiscible liquid phases (water and oil), a surfactant, and usually a co-surfactant. One liquid phase (i.e., the dispersed phase or droplet) is dispersed in the other (i.e., the continuous phase) to form droplet sizes in the order of 20–200 nm. A monolayer of surfactant molecules is self-assembled at the interface between the droplet and the continuous phase to facilitate the dispersion process by lowering the interfacial tension while the size, shape, and curvature of the droplets can be tuned by the surfactant type and structure. A microemulsion can be further classified as direct (oil-in-water, O/W), reverse (water-in-oil, W/O), or bicontinuous systems.
As to the microemulsion templating synthesis of hollow silica, the basic idea is to grow the silica shell exclusively at the interfacial region between the droplets and the continuous phase. In this regard, surfactants or co-surfactants should have good affinity for the silica precursors either through electrostatic interaction or hydrogen bonding. The liquid droplets, which act as the cavity-generating templates, can then be easily removed by gentle evaporation or dissolution in mild solvents such as water and ethanol after the shell formation.
Compared to hard templates, this method allows for easy template removal and incorporation of a wide range of functional molecules (hydrophilic and hydrophobic) due to the presence of both hydrophobic and hydrophilic domains. Hence, it is particularly attractive in drug delivery and pharmaceutical applications. However, it is rather challenging to obtain uniform droplets with a size smaller than 100 nm. This is because the droplets constantly move, collide and exchange with each other to cause droplet coalescence and Ostwald ripening, as a result, silica capsules derived from microemulsion templating are usually larger than 200 nm in size. They also exhibit broad size distributions or serious interparticle aggregation, which are great handicaps for their biological applications.
Over the years, several reports have described how both direct and reverse microemulsion systems are used for the preparation of monodisperse SNCs.35–37 As compared to direct microemulsion, reverse microemulsion demonstrates better capability to produce monodisperse SNCs with sizes in the sub-50 nm range. This is because the growth of silica is confined inside the water droplet. Thus, it does not interfere with the functioning of surfactant molecules, which are located at the interface of the W/O droplet, to prevent the SNCs from sticking together.35,37 Nevertheless, in most cases, the use of single-hydrocarbon chain surfactants (e.g., Triton X-100,35 cetyltrimethylammonium bromide (CTAB),36 and Tween-8037) alone is unable to reduce the interfacial tension sufficiently to form a stable microemulsion.35–37 One effective way to improve the system stability is to introduce a co-surfactant so as to reduce the interfacial tension and ensure sufficient flexibility of the interface. As a second surface-active species, the co-surfactant can be either another surfactant (e.g., lecithin37) or a medium sized aliphatic alcohol (e.g., hexanol).35,36 Another effective way to fortify the microemulsion droplets is to introduce a cross-linkable surfactant (e.g., octadecyltrimethoxysilane (C18TMS)37).
Vesicle templating. Amphiphilic surfactants or block copolymers with clearly distinct hydrophobic and hydrophilic groups are able to self-assemble into vesicles. Vesicles are hollow spheres with bilayer structured walls. The bilayers arise due to an inward orientation of the hydrophobic blocks and an outward protrusion of the hydrophilic blocks into the water-filled interior cavity as well as the external medium. With an inherent hollow structure and rich morphologies, vesicles are of particular interest as templates for the synthesis of SNCs. Various factors such as the type of surfactant, the components of surfactant mixtures, temperature, pH, or stirring rates have been delicately adjusted to determine the dimensions, morphology and stability of vesicles.38–40
Pinnavaia and co-workers were among the first who have successfully synthesized SNCs by using vesicle templating.12 In their study, vesicles were prepared from neutral gemini surfactants of CnH2n+1NH(CH2)2NH2 (n = 10, 12, 14) and the silica precursor of tetraethoxysilane (TEOS). Due to the penetration of TEOS into the vesicle layers to interact with the gemini through hydrogen bonding, as-prepared hollow silica exhibited ultrahigh thermal (1000 °C) and hydrothermal stabilities. However, the hollow silica was actually a mixture of single-walled hollow silica (SWHS) and multi-walled hollow silica (MWHS) with very wide size distributions that range from tens of nanometers to 1.4 micrometer. Nonetheless, in later work done by the same group,41 smaller SWHS (<40 nm) and MWHS (<100 nm) were successfully synthesized due to a proper balance of the hydrophilic and hydrophobic interactions during the assembly process of gemini surfactants.
Hubert et al. reported the first successful synthesis of SWHS by using a cationic surfactant of dioctadecyldimethylammonium bromide (DODAB).42 Alternatively, a catanionic surfactant, which is a cationic–anionic surfactant mixture, represents an interesting class of vesicle templates. Such a catanionic mixture initiates the formation of bilayer structures with high stiffness owing to the strong electrostatic interaction between the positively and negatively charged head groups.43 A catanionic mixture of cationic CTAB and anionic perfluorooctanoic acid (PFOA) has been employed to direct the formation of SWHS, MWHS, and MWHS with a mesoporous shell (MS-MWHS), respectively.43 In addition, non-ionic PEO-based triblock copolymers have also been applied to the formation of SWHS by delicately adjusting pH and temperature in salted buffer solutions.38
Compared to hydrogenated surfactants, partially fluorinated surfactants are more prone to forming stable vesicles because their fluorocarbon and hydrocarbon segments can increase the elasticity and flexibility of the bilayer respectively.39,44,45 Rankin's group first reported the use of 1H, 1H, 2H, and 2H-perfluorodecylpyridinium chloride (HFDePC) to synthesize SWHS with only a single layer of mesopore in the silica wall.39 Because of the stability of the fluorocarbon bilayers, they are used to direct the formation of organic–inorganic hybrid hollow silica from ethylene-bridged silanes. Ethane–silica of MSHS and MWHS has been synthesized using C3F7O(CFCF3CF2O)2CFCF3-CONH(CH2)3N+(C2H5)2CH3I− (FC4) and 1-(10-perfluorooctyldecyl)pyridinium bromide monohydrate (F8H10) respectively.44,45 This strategy is desirable because there is a rather uniform distribution of the organic ethylene-bridging groups within the silica framework. However, long-chain bridged silanes, such as octylene-bridged silane, were unsuccessful in achieving stable hollow particles. This is due to the flexibility of the walls which makes the particles susceptible to collapse upon drying. Additionally, the introduction of an amine group into the bridging chain could also prevent the formation of hollow structures, because the amine has either accelerated the hydrolysis process or increased the polarity of the bridging chain.44
Alternatively, Du et al. reported a successful synthesis of organic–inorganic hybrid hollow silica by using a home-designed amphiphilic diblock copolymer of PEO-block-poly(3-(trimethoxysilyl) propyl methacrylate) (PEO-PTMSPMA), which bears the reactive trimethoxysilane groups in one segment of the polymer to function as the silica precursor.46
Table 1 presents a summary of the SNCs synthesized by vesicle templating, along with their templating agents, morphologies, particle size and shell thickness. Hollow silica with a mesoporous shell is denoted as x-MS-SWHS or x-MS-MWHS, where x = d or h corresponding to disordered or hexagonal mesoporous shells, respectively.
| Templating agent | Morphology | Particle sizea (nm) | Shell thicknessa (nm) | Ref. | |
|---|---|---|---|---|---|
| a Determined by TEM. Only SNCs (including the ones coexistent with silica capsules larger than 100 nm) are considered. b Determined by DLS. c DDAB: didodecyldimethylammonium bromide; P123: poly(ethylene oxide)20-block-poly(propyl oxide)70-block-poly(ethylene oxide)20 (PEO20–PPO70–PEO20). | |||||
| Gemini surfactant | CnH2n+1NH(CH2)2NH2 | SWHS + MWHS | SWHS: 20–125; MWHS: 33–1400 | SWHS: 3; MWHS: 7–70 | 12 |
| CnH2n+1NH(CH2)mNH2 | SWHS | 18–37 | 2–3 | 41 | |
| MWHS | 18–100 | 8–40 | |||
| Catanionic surfactant | CTAB/PFOA | h-MS-MWHS | (100–300) × (50–150) | ∼25–50 | 43 |
| MWHS | 70–200 | ∼25–50 | |||
| SWHS | 60–120 | ∼10 | |||
| Cationic surfactant | DODAB | SWHS | 50–200 | 6–9b | 42 |
| DDABc | MWHS | 30–50 | 10 | 40 | |
| Nonionic surfactant | PEO20–PPO70–PEO20 (P123)c | SWHS | 50–150 | 5 | 38 |
| Partially fluorinated surfactant | HFDePC | d-MS-SWHS | ∼100 | 10–20 | 39 |
| FC4 + CTAB | h-MS-MWHS | 70–180 | 20 | 45 | |
| F8H10 | MWHS | 50–120 | 15–20 | 44 | |
| Reactive block copolymer | PEO-PTMSPMA | SWHS | 45.6 ± 12.1 | 16.9 ± 2.5 | 46 |
As summarized in Table 1, with only a few exceptions, a majority of the as-prepared silica have a wide distribution of sizes varying from nanometers to sub-micrometers and ill-defined morphologies. Hence, it is generally challenging to control the size of hollow silica to be fully below 100 nm through the vesicle templating approach. In most cases, SNCs are likely to coexist with hollow silica (>100 nm in size) in the same product, sometimes even constituting only a small fraction of the entirety. This can be ascribed to the characteristics of templating vesicles, which are broad in size span, as well as the bilayer shells, which are fragile and prone to collapse as conditions change.1
In addition, the resultant SWHS tends to possess a very thin wall in the range of 2–10 nm. Thickness of the wall can be increased if block copolymers of a large molecular weight are used as vesicle templates.46 On the other hand, for MWHS derived from multilamellar vesicles, the wall thickness is determined by the thickness of each layer and layer spacing.
Single micelle templating. Although the liquid characteristics of microemulsion droplets and the inherent hollow cavity of vesicles provide important advantages for the loading of drug and imaging contrast agents, they both suffer from wide size distribution, deformed morphology, and relatively poor stability. This would in turn prejudice their chance in biomedical applications. In this regard, polymeric micelles represent a promising class of templates for the fabrication of SNCs because of their remarkable capability for nanosize and monodispersity control. Indeed, the formation of both polymeric micelles and vesicles involves a similar supramolecular self-assembly process. Nonetheless, polymeric micelles are consistently smaller than vesicles because the former comprise of only a single hydrophilic outer layer that envelops the hydrophobic interior core.
Consequently, polymeric micelles are of particular interest as soft templates for the development of SNCs because their size is normally <50 nm, which is an ideal dimension for nanocarriers to extravasate into the leaky tumour tissues and achieve passive targeting. Additionally, numerous polymeric micelles employ PEO as the hydrophilic corona, thus ensuring an excellent aqueous solubility of the micelles and stealth protection against early recognition by RES. Furthermore, the hydrophobic compartment of the polymeric micelle allows the incorporation of various functional hydrophobic compounds, such as anti-cancer drugs and imaging contrast agents. As a result, there has been increasing interest in employing polymeric micelles as structure directing agents for the fabrication of SNCs, which are aimed for the development of novel intravenous injectable nanocarriers.
There are two general approaches that have been developed for the preparation of SNCs via single micelle templating.47,48 The first one involves synthesizing a designed block copolymer bearing specific functional groups. These functional groups are distributed along the corona of the polymeric micelles and serve as reservoirs for the adsorption of silica precursors, thereby confining the silica condensation in the corona segment of the micelles. For example, Koh and co-workers synthesized an amphiphilic block copolymer that is terminated with silanol (Si–OH) groups at the end of the hydrophilic blocks, namely poly(methyl methacrylate)-block-poly(poly(ethylene glycol) methyl ether monomethacrylate)-block-poly(poly(ethylene glycol) methyl ether monomethacrylate-random-methacryloxypropyltrimethoxysilane) (PMMA-PPEGMA-poly(PEGMA-r-MOPS).49 Upon the formation of polymeric micelles, the silanol groups are consequently located at the outermost region of micelles, which serve as reactive points for the formation of the silica shell, as illustrated in Fig. 3. It has been shown that hybrid SNCs with particle sizes of 25 and 42 nm were synthesized respectively by tuning the molecular weight of the block copolymers.
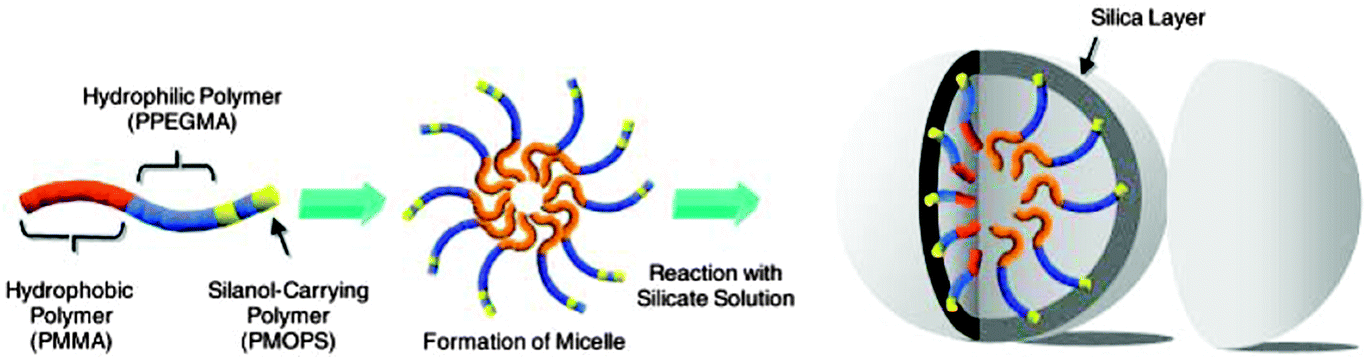 | ||
| Fig. 3 Schematic illustration of preparation of SNCs by using a new block copolymer functionalized with silanol groups. (Reprinted with permission from ref. 49. Copyright 2003 Wiley-VCH.) | ||
Similarly, Yuan and co-workers synthesized a cationic diblock copolymer of poly(2-(diisopropyl-amino)ethyl methacrylate)-block-2-(dimethylamino)ethyl methacrylate (PDPA-PDMA).50,51 The tertiary amine groups on the PDMA block could be partially (or fully) quaternized using methyl iodide. This resulted in the formation of polymeric micelles with PDPA blocks as the hydrophobic core and the cationic PDMA blocks as the micelle corona. The cationic PDMA blocks function not only as a water solubilisation and colloidal stabilization agent, but also as a physical scaffold for silica shell growth. Interestingly, the cationic PDMA corona catalyses the hydrolysis and condensation of tetramethoxysilane (TMOS) at neutral pH and room temperature, thereby eliminating the need to employ a catalyst such as an acid or a base.
Khanal and co-workers designed and prepared polymeric micelles with a core–shell–corona structure from tri-block copolymers of poly(styrene-block-2-vinyl pyridine-block-ethylene oxide) (PS-PVP-PEO) after protonating the PVP blocks in a low pH (<5) environment.52 The protonated PVP shell then acts as an acid catalyst site for the hydrolysis of the silica precursor (i.e., TMOS), as well as a reservoir for the adsorption of the hydrolysed TMOS via electrostatic interactions. The as-prepared SNCs were 20 and 30 nm in sizes, respectively.
Specifically designed block copolymers have been well established to prepare SNCs. The approach however may incur a rather complicated organic synthesis. Therefore, single micelle templating from commercially available block copolymers is highly desired. Good candidates for such a purpose are the PEO-based block copolymers, such as the Pluronic® (PEO–PPO–PEO) family, because PEO is known to have a strong affinity for the silanol groups of hydrolysed silica precursors through hydrogen bonding. In this regard, Huo and co-workers were the first to synthesize SNCs by using single micelle templating from PEO106–PPO70–PEO106 (F127) triblock copolymers.53 In their study, the synthesis was carried out under strong acidic conditions (pH < 1) so that the PEO blocks were protonated and could readily adsorb the hydrolysed silica precursors through electrostatic interactions. The highly acidic conditions, together with the use of a termination agent of diethoxydimethylsilane (DEDMS) with hydrophobicity and a slow hydrolysis rate, are effective in suppressing the condensation reaction between the silanol groups and thus preventing inter-particle cross-linking (i.e., aggregation). As a result, the silica shell is formed surrounding the core of the F127 micelles, leaving a part of the PEO blocks dangling on the as-prepared SNCs to serve as a steric stabilization agent, as illustrated in Fig. 4a (Route A).
 | ||
| Fig. 4 (a) Schematic illustration of the formation of hybrid SNCs by using F127 block copolymers as templates under strong acidic (Route A) or high temperature and pressure conditions (Route B). (b) TEM image of SNCs synthesized through Route B. (Reprinted with permission from ref. 54. Copyright 2008 American Chemical Society.) | ||
Alternatively, Liu and co-workers prepared organic–inorganic hybrid SNCs by depositing an organosilane of 1,2-bis(trimethoxysilyl)ethane (BTME) onto F127 micelles in sodium phosphate (NaH2PO4–Na2HPO4) buffer solution.48,54 This is followed by heating the polymeric micelles and silica precursors together in an autoclave vessel at a temperature range of 80–120 °C (Fig. 4a, Route B). The as-prepared ethane–silica hybrid SNCs feature microwindows (0.5–1.2 nm) in the shell, which allows for the diffusion of guest molecules into/out of the interior. In addition to the use of organosilanes to suppress the silanol cross-linking, the use of inorganic electrolytes (NaH2PO4–Na2HPO4) is also important. They are used to induce the micellization of F127 molecules at a lower concentration due to the salting-out effect. This thus generates a solution of “diluted” micelles favourable for suppression of aggregation of the micelle–silicate composites. In addition, the inorganic electrolytes can increase the ionic strength of the reaction solutions, and thus facilitate the self-assembly of the micelles and silica precursors. Other inorganic electrolytes, such as KCl, Na2SO4, K2SO4, and CH3COONa, have also been utilized for the same purpose of avoiding aggregation.47,48
By contrast, the present authors' group developed an interfacial templating condensation approach to synthesize well dispersed SNCs (Fig. 5).55 The synthesis was conducted in near-neutral pH aqueous solution and at room temperature, without the use of any inorganic electrolyte or organosilane. The basic idea is to confine the hydrolysis and condensation of the silica precursors to the core/corona interface of F127 micelles so that the free dangling PEO blocks can still provide steric stabilization to the SNCs. The resultant PEOlated SNCs exhibited excellent colloidal stability in both an aqueous environment and phosphate buffered solutions containing proteins (i.e., antifouling properties). The highly benign synthesis environment allows easy one-pot encapsulation of hydrophobic functional agents such as Fe3O4 nanocrystals,56,57 MnO NPs,58 quantum dots (QDs),59 fluorescent conjugated polymers60,61 and drugs,55 providing the material with magnetic resonance imaging (MRI), fluorescence imaging, and/or drug delivery capabilities.
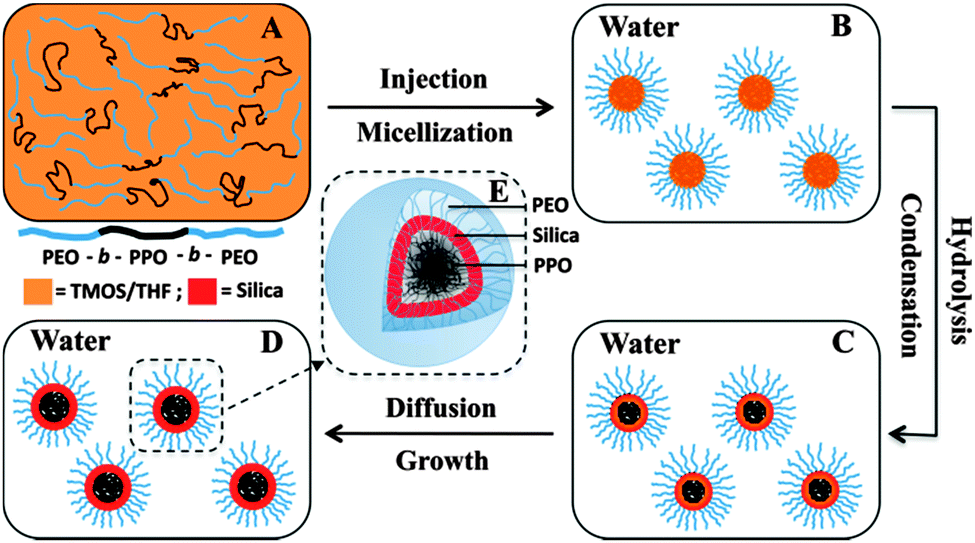 | ||
| Fig. 5 Schematic illustration of the formation of PEOlated SNCs through the interfacial templating condensation approach. (Reproduced from ref. 55 with permission from The Royal Society of Chemistry.) | ||
With the rapid development of the single micelle templating, it provides a versatile platform for the synthesis of SNCs of small size (<50 nm), a high degree of uniformity, and good colloidal stability in biological fluids. The PEOlation of the as-prepared SNCs helps to further slowdown the rate of clearance by inhibiting opsonization, thereby prolonging the blood circulation half-life of SNCs.
However, formation of second-order or higher-order aggregates cannot be avoided due to the cross-linking of silica. The loading capacities of SNCs are also limited by their small size. Hence, this may inevitably lead to faster drug release and less tumour-site specificity.
Polymer aggregate templating. Polymer aggregates derived from certain hydrophilic polymers such as poly(acryl acid) (PAA) and poly(vinyl pyrrolidone) (PVP) are another type of soft template.62–64 They are different from polymeric micelles and vesicles in that they consist of merely hydrophilic cross-linked or self-assembled polymer networks. A typical synthetic route of SNCs using PAA aggregates has been described by Yu et al.62 It involves three steps (Fig. 6): (1) formation of spherical PAA aggregates in a poor solvent (i.e., ethanol), (2) growth of silica on the PAA aggregates; this is driven by an interaction between the aminated carboxylic groups of PAA chains and the silanol groups of hydrolysed TEOS, and (3) subsequent removal of PAA by water washing. By simply adjusting the PAA concentration, this method provides a facile way to synthesize monodisperse hollow silica spheres with widely tunable diameters that range from 25 to 400 nm.
 | ||
| Fig. 6 (a) Schematic illustration of the formation of SNCs through PAA aggregate templating. (b) and (c) TEM images of SNCs synthesized through PAA aggregate templating. (Adapted with permission from ref. 62. Copyright 2008 American Chemical Society.) | ||
More complex systems such as hollow polymer aggregates are also available as soft templates. Jiang and co-workers reported the fabrication of pH-sensitive SNCs templated by hollow chitosan (CS)-PAA nanospheres,63 as shown in Fig. 7. The hollow CS-PAA templates are comprised of an outer shell of protonated and positively charged CS chains and an inner shell of CS-PAA polyelectrolyte complexes. The CS chains at the outer shell are deprotonated and act as nucleation sites for silica growth. After removing PAA by washing, CS–silica hybrid nanocapsules with double shells are achieved. The outside silica shell provides a good mechanical strength and high permeability, and the inside CS shell works as a pH-sensitive switch by swelling CS at pH = 4 and collapsing it at pH = 7.4 to open and close the pore channels in the shell, respectively. In addition, PVP aggregates are combined with dodecylamine (DDA) for the synthesis of MS-SNCs where DDA acts as a porogen in the shell.64
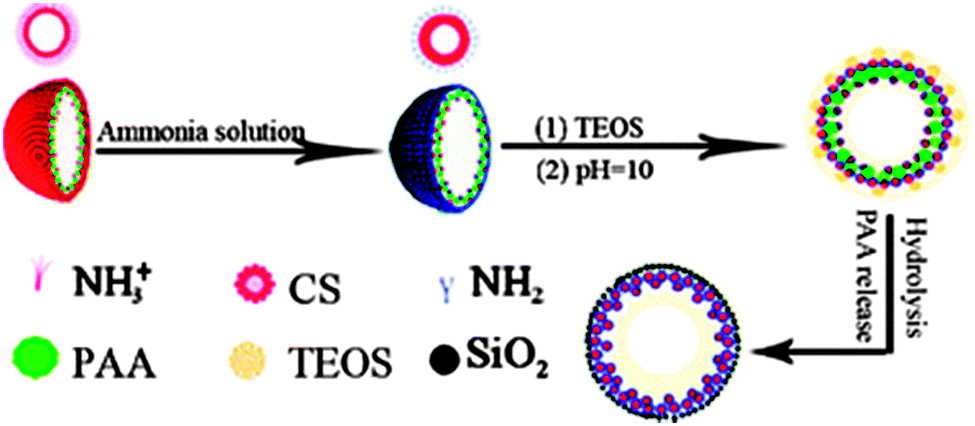 | ||
| Fig. 7 Schematic illustration of the formation of SNCs through hollow CS-PAA nanosphere templating. (Reproduced from ref. 63 with permission from The Royal Society of Chemistry.) | ||
Gas bubble templating. Another soft templating scheme for the preparation of SNCs is based on gas bubbles which can be generated from the blowing of gas or gas mixtures, in situ released gaseous products, or ultrasonic irradiation. Various types of gas bubbles (e.g., N2, O2, CO2, H2S, PH3, NH3, CH4, and air) have been applied as soft templates for the formation of hollow structures.1 With regard to the synthesis of SNCs, a surfactant-assisted sonochemical strategy has attracted much attention due to its characteristic rapid synthesis, no requirement of any cavity-directing reagent (except for the air bubbles generated from the ultrasonic irradiation), and a facile tuning of its particle size and shell structure.
In general, the process can be conceptualized as below. The sonochemistry arises from acoustic cavitation, which is a process of continuous formation, growth, and implosive collapse of bubbles within a liquid. Many tiny gas bubbles, which are generated from the collapse of the cavitational bubbles (∼200 μm in size), serve as soft templates for the formation of a hollow cavity. Surfactant molecules are enriched at the vapour–liquid interface of the tiny gas bubbles to stabilize the bubbles in a similar manner to normal liquid–liquid emulsions. They also induce a cooperative supramolecular-templated deposition of the surfactant and silica surrounding the bubbles, thus leading to the formation of a mesostructured shell. The SNCs prepared via the gas bubble templating strategy were reported to possess wide size distributions, in the range of 50 to 500 nm.65 The outer diameter and shell thickness of the SNCs could also be tuned by varying the sonochemical processing time, wherein more gas bubbles were available as templates with longer ultrasound irradiation times.
In summary, the quality of the soft template organization in terms of the size, size distribution, stability, and proper surface affinity to silica deposition is crucial to the synthesis of SNCs. In most cases, surfactants are indispensable because they either form the soft templates directly (in vesicle and single micelle templating), or stay at the interface to stabilize the templates (in microemulsion templating). This is of paramount importance for the synthesis of nanoscale structures because the system is usually unstable due to the high surface energy. Besides, the hydrophilic segments of surfactants can also facilitate the deposition of silica through electrostatic or hydrogen interactions.
Aerosol-assisted assembly strategy. The aerosol-assisted evaporation-induced self-assembly (EISA) approach was first reported by Brinker and co-workers in their work to synthesize mesoporous silica including MWHS.1,66 Briefly, it first develops a homogeneous dilute solution containing silica precursors, volatile solvents (typically ethanol, tetrahydrofuran, HCl, and H2O), catalysts, and surfactants. The solution is then nebulized as droplets before passing through a heated zone. Preferential evaporation of the volatile solvents causes the droplets to be enriched in silica and a surfactant with radial concentration gradients. Hence, the formation of a liquid-crystalline mesophase is initiated at the droplet surface and proceeds towards the droplet interior. In this way, MWHS is produced with a wide size distribution ranging from tens to hundreds of nanometers.1,66
In order to encapsulate functionalities into hollow silica, Tartaj and co-workers reported an aerosol pyrolysis method to incorporate γ-Fe2O3 into hollow silica.1,67 In the synthesis process, functional components (e.g., iron ammonium citrate) were dispersed into methanol solutions containing TEOS before nebulization. With the rapid evaporation of the methanol solvent, iron ammonium citrate and TEOS were successively precipitated and thermally decomposed at the surface of the droplet. As a result, hollow spheres of silica-coated γ-Fe2O3 with sizes of 50–250 nm were finally obtained. This strategy demonstrates that with the aerosol-assisted assembly approach, various moieties can be self-assembled or simply organized together within a confined droplet space. This approach is characteristic of a high production rate, cost effectiveness, and adjustable dimensions from 10 nm to a few tens of micrometers. However, it suffers from broad size distributions.
Structure difference-based selective etching strategy. There is a subtle structural difference between a silica layer, which is synthesized by the Stöber method with TEOS as the single silica precursor, and that achieved by a co-condensation of TEOS and a hydrophilic organosilane (e.g., aminopropyltrimethoxy silane (APTMS),68,69 or N-[3-(trimethoxysilyl)propyl]ethylenediamine (TSD)68,70). As the pure silica layer has a higher degree of condensation, it is more stable against etching than the latter which is made of organic–inorganic hybrid silica. This is the basic premise of the structure difference-based selective etching strategy, which is currently gaining more attention for the preparation of SNCs. For example, Haes et al. reported the synthesis of a three-layer sandwich structure of Au@APTMS/SiO2@SiO2 NPs wherein the faster ammonia etching of the middle APTMS/SiO2 layer has resulted in the formation of Au@SiO2 nanorattles.68,69 Likewise, Tang et al. synthesized SiO2@TSD/SiO2@SiO2 NPs, before using HF to selectively etch off the middle TSD/SiO2 layer to produce SiO2@SiO2 nanorattles (Fig. 8).68,70
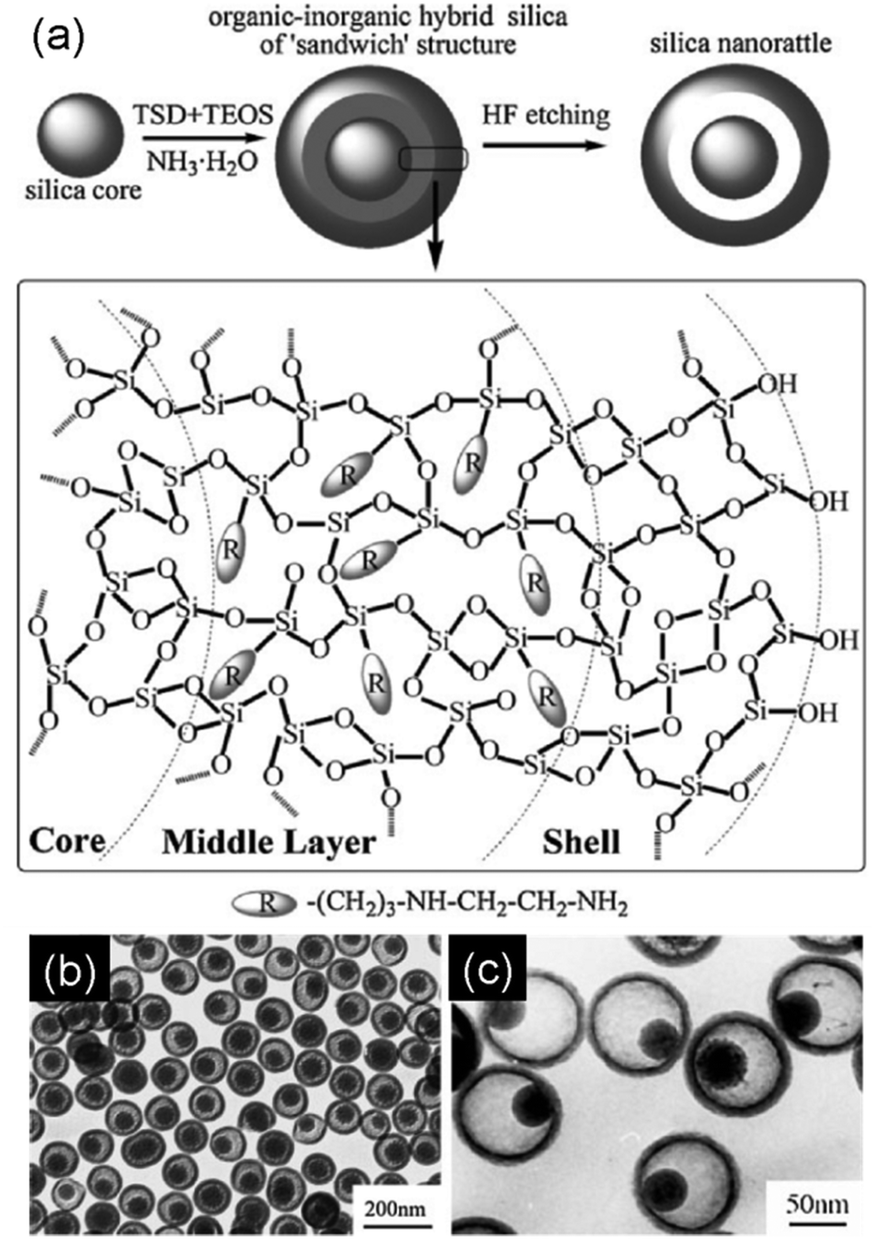 | ||
| Fig. 8 (a) Schematic representation of the synthesis of SiO2@SiO2 nanorattles through the structure difference-based selective etching strategy. (b) and (c) TEM images of silica nanorattles with different sizes and core–shell ratios. (Reprinted with permission from ref. 70. Copyright 2009 Wiley-VCH.) | ||
Meanwhile, it was found that the hydrophobic organosilanes were able to protect the silica shell against etchant attack. Yang and co-workers reported the deposition of hydrophobic organosilanes (e.g., BTME and 1,4-bis(triethoxysilyl)benzene) onto Fe3O4@SiO2 core–shell NPs to form a protective layer at the external surface of the silica shell.68,71 As a result, Fe3O4@SiO2 nanorattles were successfully produced because the inner silica was dissolved more rapidly than the external layer of silica. Similarly, Shi and co-workers synthesized SiO2@C18TMS–SiO2 core–shell NPs as templates.68,72 In this case, C18TMS is a silane coupling agent with a long hydrocarbon chain and can form hydrophobic domains within the silica shell. Consequently, it was observed that the SiO2 core had a significantly higher density of Si–OH and therefore a lower degree of condensation than the C18TMS/SiO2 shell. This causes the inner SiO2 core to be more prone to etching under alkaline conditions, thus favoring the formation of SNCs.
Dissolution re-deposition strategy. Lastly, a dissolution and re-deposition strategy was reported by Jang et al. in their synthesis of titania embedded SNCs.73,74 Unlike the other cavity-generating strategies, this method necessitates the presence of a silica/titania interface and sonication irradiation. Consequently, silica etching is first initiated at the core/shell interface of SiO2@TiO2 core/shell NPs when the NPs are subjected to sonication irradiation. Next, the etched silica and sometimes titania species are dissolved in the solution and diffused out of the NPs. However, due to the Ostwald ripening process, the dissolved silica is continuously re-deposited onto the outer surface of the NPs until the hollow nanostructures are eventually formed.
2.2 Shell-forming strategies
In order to grow smooth, uniform, and robust silica layers onto cavities, an attractive interaction between the cavity templates and the silicate oligomers is required.15 Functionalization/modification of the template surface with proper functional groups and/or electrostatic charges is sometimes necessary to ensure the compatibility between the template surface and shell material. Typically, stöber, reverse microemulsion, and supramolecular templating methods have been explored to grow silica shells. These methods are all based on a sol–gel process in which the hydrolysis of a silica precursor (e.g., silicon alkoxide, organosilane or silicate) is carried out. This is followed by the polycondensation of the silanol groups to afford the desired nanocapsules. The sol–gel reactions by using a silicon alkoxide as the precursor can be described as below:(i) hydrolysis
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Si–OR + H2O → Si–OH + ROH Si–OR + H2O → Si–OH + ROH | (1) |
(ii) condensation
| Si–OH + RO–Si → Si–O–Si + ROH | (2) |
| Si–OH + HO–Si → Si–O–Si + H2O | (3) |
The hydrolysis of silica precursors occurs by a nucleophilic attack of the oxygen in a water molecule on the silicon atom to produce silanol groups. Hydrolysis reaction can be catalysed by adding either an acid or a base into the system. Once silanol groups are formed, the condensation reaction will commence to produce the siloxane bond (Si–O–Si) and by-products of alcohol (ROH) or water. Sol–gel is a popular method to form silica because the hydrolysis and condensation of silica precursors in aqueous solution can be well controlled.
Stöber and reverse microemulsion methods are two popular sol–gel methods to synthesize silica shells. The Stöber synthesis involves the hydrolysis and condensation of a silica precursor, such as TEOS, catalysed by ammonia in an ethanol–water mixture. In this method, a uniform deposition of silica from hydrolysed TEOS onto a preformed core can be easily achieved to generate the core–shell nanostructures. On the other hand, in the reverse microemulsion method, nanometer-sized water droplets are dispersed in an organic phase and stabilized by surfactants. In the synthesis of core–shell nanostructures, the core materials must enter the interior water phase followed by ammonia-catalysed hydrolysis and condensation of silica precursors at the W/O interface or in the water phase.
Supramolecular-templated deposition is another shell-forming strategy that is exclusively employed to prepare mesoporous silica shells. It typically employs a cationic surfactant (e.g., CTAB) as the structure-directing agent; the strong electrostatic interaction between the positively charged surfactant molecules and the negatively charged silica ensures a cooperative deposition of the hydrolysed silica precursors, together with micelles as derived from the self-assembly of surfactant molecules, onto a preformed core. The micelles are eventually removed by pyrolysis or solvent extraction to form mesopores in the silica shell. This shell-forming strategy has been widely integrated with most of the cavity-generating strategies (as described in Section 2.1) except single-micelle templating for the preparation of MS-SNCs.
3 Structure control of silica-based nanocapsules
The structural features of SNCs and methods designed to control them have been under intensive investigation. In this section, we address their key structure features and control over their particle sizes, the level of shell porosity, cavity topology, cavity content and cargoes, and surface functionalities that are critically important for their bio-performance.3.1 Size control
Fig. 9 shows a statistic analysis of the particle size and distribution of silica capsules synthesized through various synthesis methods. It clearly demonstrates the strengths and weaknesses of each method in the control of the size and size distribution. In general, hard templating against inorganic NPs and single micelle templating are the most effective in controlling the particle size below 100 nm and attaining desired monodispersity. Although hard templating against polymer NPs can also generate monodisperse SNCs, it is more often used to prepare capsules larger than 100 nm in size. Microemulsion and emulsion (i.e., macroemulsion) templating can be employed to generate silica capsules that are smaller and larger than 100 nm, respectively. However, they tend to suffer from wide size distributions. Vesicle templating is susceptible to size non-uniformity to a certain extent as it often produces capsules ranging from tens of nanometers to micrometers. In general, polymer aggregate and gas bubble methods are less commonly applied to the synthesis of SNCs. In recent years, template-free strategies have attracted increasing attention for the development of SNCs because of their good size manipulation.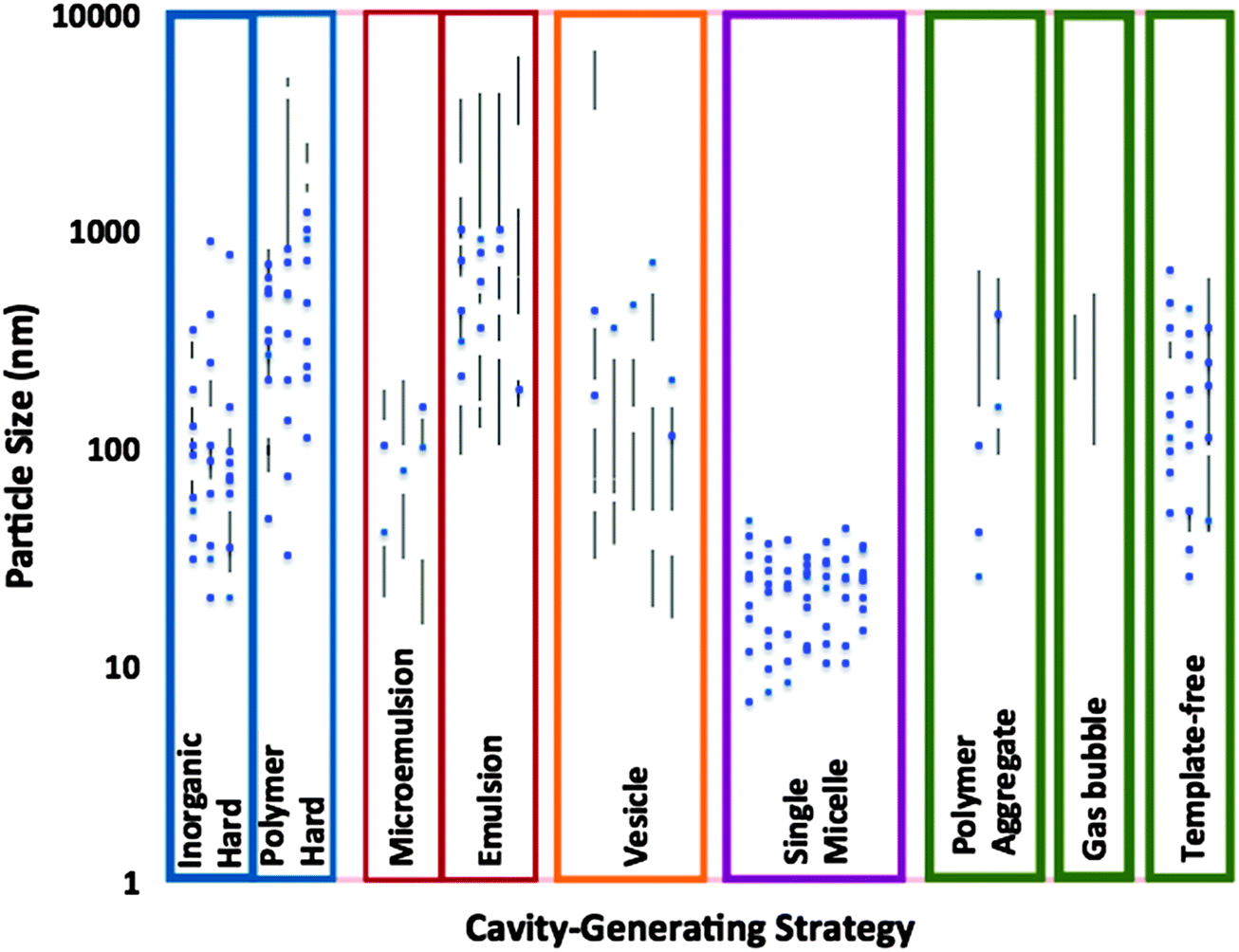 | ||
| Fig. 9 Particle size (up to 10 μM) and size distribution of silica capsules columned according to their synthetic strategies. The data are sourced from the publications on SNCs in 1996–2014. The size and size distributions are largely determined by TEM images. | ||
Nonetheless, it should be noted that Fig. 9 does not reflect the degree of particle aggregation which is otherwise significant for SNCs. This is because SNCs are very small and rich in silanol groups at the surface, thus rendering them more vulnerable to interparticle crosslinking. Such particle aggregation would hinder the use of SNCs for any application that requires a fine dispersion and function of discrete individual particles. Indeed, SNCs with small sizes and good colloidal dispersion are desirable for a prolonged blood circulation half-life. This is often a prerequisite for targeted drug delivery and sustained drug release in blood.
3.2 Porosity of silica shells
Another important attribute of SNCs is the density (or porosity) of the silica shell. This is determined by the presence of open channels/windows in the silica shell to provide pathways for the guest species to diffuse into/out of the hollow cavity. In this regard, silica-based shells can be broadly classified as purely silica shells, organic–silica hybrid shells, and supramolecular-templated mesoporous silica shells.Silica shells prepared via the sol–gel chemistry approach usually exhibit an amorphous nature and possess micropores within the shells. Nann et al. found that the pores within the shell can be roughly estimated to be of the sub-nanometer range.34 However, the permeability of these silica shells is not controllable, which strongly limits their applications. Hence, it is preferable to have a composite-type structure where the inorganic fraction yields mechanical strength while the organic fraction allows for controllable permeability by adjusting the hydrophobicity of the shell.53 This can be realized by the use of organosilanes that are composed of organic bridging groups.44,45,47,48,53 A good example is the synthesis of ethane-silica SNCs through the condensation of BTME around an inorganic-electrolyte-stabilized F127 micelle under mild buffer conditions (NaH2PO4–Na2HPO4, pH ∼ 7.0).48,54 The PEO group can penetrate into the organosilicate layer during the assembly process, thereby resulting in the formation of micropores in the shell of SNCs. The pore size distribution based on a Horvath–Kawazoe model clearly showed the presence of micropores with a diameter in the range of 0.5–1.2 nm in the shell. Other bridging groups, such as methylene (–CH2–), ethylene (–CH2CH2–), ethenylene (–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–), and phenylene (–C6H4–), have also been integrated in the silica framework.47 However, the limited size scale of microwindows in the organic–silica hybrid shell may still restrict certain applications, where relatively large window sizes are in demand for the adsorption and immobilization of large biomolecules.
CH–), and phenylene (–C6H4–), have also been integrated in the silica framework.47 However, the limited size scale of microwindows in the organic–silica hybrid shell may still restrict certain applications, where relatively large window sizes are in demand for the adsorption and immobilization of large biomolecules.
In order to enlarge the size of pores in silica shells, a supramolecular templating strategy was developed by using surfactants as the porogen for the mesopore formation in the silica shell. In this strategy, micelles derived from the self-assembly of surfactant molecules can direct the condensation of silica precursors surrounding the micelles through electrostatic interactions or hydrogen bonding. Such mesoporous silica shells with a tunable pore size, remarkable size homogeneity and a high level of porosity have attractive potential in controlled release bioapplications. A large and controllable level of porosity in the silica shell also allows it to house appreciable amounts of cargoes.
3.3 Cavity topology
As described in Section 2.1.2, the cavity configuration of SNCs can be of a single compartment or an “onion”-like structure with several concentric membranes. This is largely determined by their templates of unilamellar or multilamellar vesicles, respectively. Such morphology dissimilarity affects their biomedical performance in terms of loading capacity and release rate.In addition to the morphology variety, the cavity shape is another parameter that can be tailored. In general, SNCs take a spherical or near-spherical (probably due to the deformation of shells) shape in order to minimize the total surface free energy. The synthesis of non-spherical SNCs is much less established as compared to that of spherical SNCs. One difficulty lies in the poor availability of non-spherical templates especially for soft templates, such as micelles, vesicles and emulsion droplets, which in general prefer spherical shapes so that the interfacial energy can be minimized. Another difficulty is to deposit a uniform silica layer on surfaces with large variations in curvature.
Consequently, there have been only a few reports on the synthesis of non-spherical SNCs. In 2006, Brinker et al. reported the synthesis of MS-SNCs with a cubic cavity that is templated by in situ precipitated cubic NaCl crystals.75 Achieved by an aerosol-assisted assembly approach (Fig. 10a), the products still possessed a spherical external shape, which is largely determined by the spherical aerosol droplet. In 2008 Yu et al. observed the formation of unusual ellipsoidal-shaped MS-SNCs with a silkworm cocoon-like morphology through the vesicle templating of PFOA-CTAB co-templates (Fig. 10b).43 In 2011 Wang et al. synthesized MS-SNCs with both a cubic-shaped cavity and a cubic external shape (Fig. 10c and d).31 The synthesis strategy is based on the hard templating effect of cubic-shaped PbS NPs. In 2012, Crudden et al. synthesized short tube-shaped SNCs through the vesicle templating of catanionic surfactants (Fig. 10e and inset).76 Recently in 2014 Anker and co-workers achieved the synthesis of ellipsoidal MS-SNCs containing an iron nanocylinder core via hard templating against hematite nanospindles (Fig. 10f).20
 | ||
| Fig. 10 TEM images of non-spherical MS-SNCs. (Reprinted with permission from ref. 20, 75 and 76. Copyright 2014, 2006 and 2012 American Chemical Society, respectively. Reproduced from ref. 31 with permission from The Royal Society of Chemistry. Reprinted with permission from ref. 43. Copyright 2007 Wiley-VCH.) | ||
Rattle-type SNCs represent another unique nanostructure in which the solid NP(s) reside in a cavity but keep an interstitial space from the silica shell. The NPs are typically made from metals or oxides that offer SNCs corresponding functionalities. A comprehensive review on the synthetic strategies for preparing rattle-type hollow structures has been done by Lou et al.1 Here we will briefly focus on the strategies for the synthesis of rattle-type SNCs, as schematically illustrated in Fig. 11.
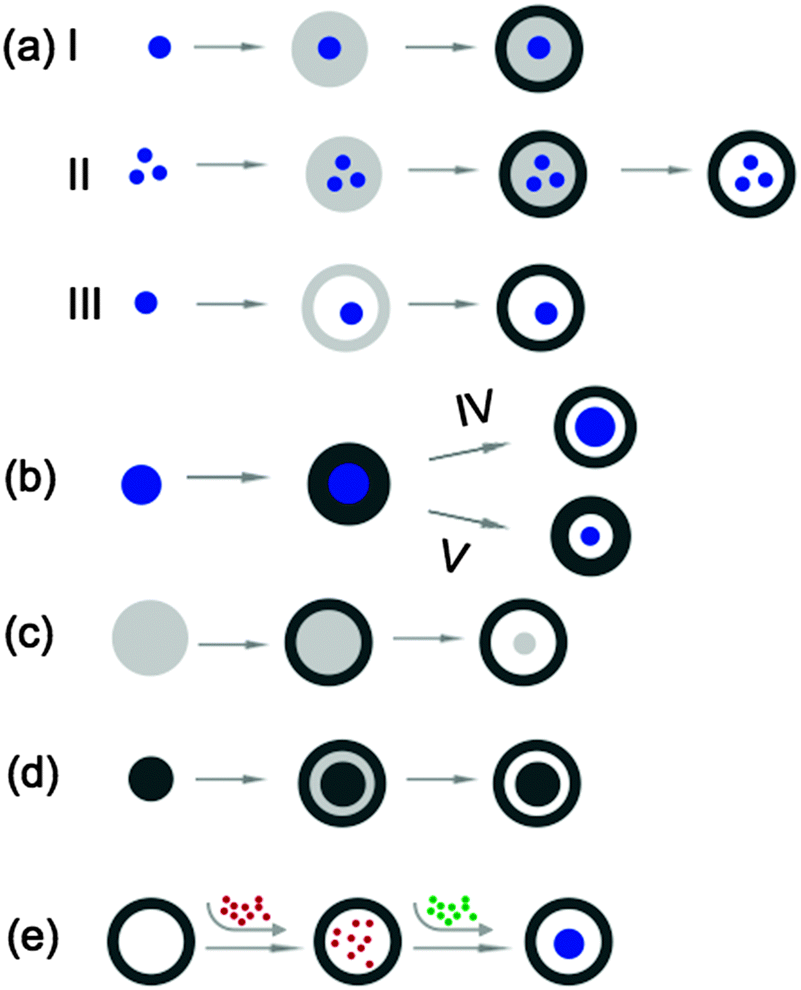 | ||
| Fig. 11 Schematic illustration of strategies for preparing rattle-type SNCs. | ||
Fig. 11a presents a bottom-up approach: formation of NPs (e.g., gold,32 Fe2O3,77 Fe3O4,55–57 MnO,58 and QDs59), encapsulation of single (Routes I and III) or multiple NPs (Route II) in a soft material, such as microemulsion droplets, micelles and vesicles, and followed by growth of an outer silica shell.55–59,77 The soft material-filled space or the hollow space after removing the soft material provides the nanorattles with the capability to immobilize other guest molecules. Fig. 11b shows another approach which also starts with the formation of NPs (e.g., gold,69 Fe2O3,20 and CdSe/ZnS34). The NPs are then coated with a silica layer to form core–shell nanostructures. The interstitial void is created by partially etching the silica shell from inside (Route IV) or the NP core (Route V).20,34,69 Sometimes, the yolk core and shell can be made of the same materials. Fig. 11c and d show two routes to form SiO2@SiO2 nanorattles.35,70 Their formation involves the structure-dependent selective etching mechanism, details of which have been described in Section 2.1.3. In addition, Fig. 11e illustrates a top-down approach, in which reactants (e.g., HAuCl4 and NaBH450) are sequentially introduced into the cavity of preformed SNCs. They react inside the cavity to form a yolk (e.g., Au) from within.
3.4 Cavity content and cargoes
The cavity of SNCs can be made “empty”, filled with solution, or may even consist of hydrophobic polymer chains, as seen in the case of single micelle templating, wherein the templates are maintained after silica shell formation.47,48,53,55–61,78 Interestingly, a broad variety of functional moieties have been encapsulated in the cavity of SNCs to offer bioimaging and controlled delivery capabilities. Table 2 presents an overview of the cargos that have been encapsulated in SNCs, along with their potential applications and the processes to synthesize such cargo-containing SNCs.| Cargo type | Cargo description | Templating strategy | Application | Ref. |
|---|---|---|---|---|
| a HPTSA: 8-hydroxypyrene-1,3,6-trisulfonic acid. b PN: N-phenyl-1-naphthylamine. c PA: polymer aggregate. d BP-PPV: poly{2-[2′-phenyl-4′,5′-di(3′′-methyl-butoxy)phenyl-1,4-phenylenevinylene]}. e C6PF: poly(9,9-dihexylfluorenyl-2,7-diyl). f MEH-PPV: poly[2-methoxy-5-(2-ethyl-hexyloxy)-1,4-phenylenevinylene]. g PDHFBT: poly(9,9-dihexylfluorenyl-2,7′-diylbithiophene). h LCH: lidocaine hydrochloride. i Gd-DTPA: diethylenetriaminepentaacetic acid gadolinium(III) dihydrogen salt hydrate. | ||||
| Organic dye | Coumarin 153 | Single micelle | Fluorescence; loading | 47 |
| Fast green FCF, HPTSA,a indigo, methylene blue, oil red O, orange II, orange OT, PNb | Single micelle | Loading | 53 | |
| Fluorescein, Nile red | Single micelle | Fluorescence cellular imaging | 53 | |
Hoechst 33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 342 342 |
PAc | Loading (2.01–3.3 wt%) & release | 64 | |
| Propidium iodide | PAc | Loading (3.22–4.82 wt%) & release | 64 | |
| Pyrene | Single micelle | Fluorescence | 53 | |
| Single micelle | Loading | 47 | ||
| Single micelle | Stability against dilution test | 53 and 55 | ||
| Sudan III | Single micelle | Loading | 47 and 53 | |
| Fluorescent conjugated polymer | BP-PPVd | Single micelle | Fluorescence cellular imaging | 55 |
| BP-PPVd, C6PF,e MEH-PPVf | Single micelle | Fluorescence | 55, 57 and 60 | |
| MEH-PPVf | Single micelle | Fluorescence cellular imaging | 57 and 60 | |
| Single micelle | In vivo fluorescence imaging | 61 | ||
| PDHFBTg | Single micelle | Fluorescence | 60 | |
| Drug | Bovine serum albumin (BSA) | Single micelle | Loading (170–585 mg g−1) | 48 |
| Cefradine | Hard | Loading & release | 27 | |
| Doxorubicin (DOX) | Hard | Loading (13.5 wt%20 and 18.4 wt%79) & release | 20, 31 and 79 | |
| PAc | Loading (4.2–8.9%) & release; fluorescence cellular imaging | 63 | ||
| Hard | In vivo anti-tumour growth assay | 31 | ||
| Ibuprofen (IBU) | Single micelle | Loading | 54 | |
| Microemulsion | Loading (13–18 wt%) & release | 80 | ||
| Indomethacin | Single micelle | Loading (8.1 wt%) & release | 55 | |
| Indomethacin, LCH,h procaine, vitamin B12 | Single micelle | Loading | 53 | |
| L-methionine | Template-free | Loading (1.29 g g−1) & release | 81 | |
| Nabumetone, naproxen, papaverine | Single micelle | Loading & release | 53 | |
| Inorganic NP | Au | Hard | Plasmon resonance | 32 |
| Template-free | Surface-enhanced Raman scattering | 69 | ||
| Vesicle | 77 | |||
| Single micelle | 50 | |||
| Fe | Hard | T 2-weighted MRI, r2 = 284.7 mM−1 s−1 | 20 | |
| Fe2O3 | Vesicle | 77 | ||
| Fe3O4 | Microemulsion | T 2-weighted MRI | 35 | |
| Single micelle | T 2-weighted MRI, r2 = 228,56 127.8,57 215.3,78 and 10.6–176.1 mM−1 s−1 (ref. 82) | 56, 57, 78 and 82 | ||
| Single micelle | In vivo MRI | 78 | ||
| Template-free | Superparamagnetism | 71 | ||
| MnO | Single micelle | T 1-weighted MRI, r1 = 1.17 mM−1 s−1 | 58 | |
| MnO (hollow) | Hard | T 1-weighted MRI, r1 = 0.14–0.283 and 0.99 mM−1 s−1 (ref. 84) | 83 and 84 | |
| Hard | In vivo MRI | 84 | ||
| Gd-DTPAi | Hard | T 1-weighted MRI, r1 = 8.6 mM−1 s−1 | 20 | |
| CdSe/CdS/ZnS | Single micelle | Fluorescence cellular imaging | 59 | |
| CdSe/ZnS | Microemulsion | Fluorescence | 80 | |
3.5 Surface functionalization
Extremely rich chemistry is available to functionalize the surface of SNCs, for example through co-condensation and post-synthetic grafting methods. In one-pot co-condensation, organosilanes with functional groups are added together with the silica precursors during the synthesis of SNCs.18,23,53,79,82,85 In this way, the functional groups are both entrapped into the silica framework and dangle on the capsule surface. In the post-synthetic grafting process, functional groups are conjugated to the exposed silica surface after the SNCs are formed.20,24,31,73,78,80,86 This technique allows conjugation of functional groups that are not stable during the SNC synthesis. Another widely employed method in the single micelle templating synthesis is to use templates with functional groups (e.g., PEO53,55–61 and carboxyl groups59,60) to functionalize the SNC surface.To date, a large variety of functional groups have been conjugated to the SNC surface; they can be broadly classified into four types. The first type is hydrocarbon groups including methyl,47,48,75,86 vinyl,47 propyl,47 phenyl,47 octyl,53 and other groups. They play several roles in the biomedical applications of SNCs, for example: (i) to increase the hydrophobicity of SNCs; (ii) to control the pore size of the silica shell; and (iii) to improve the particle dispersion by effectively preventing interparticle agglomeration. The second type of functional group includes amine,24,31,53,73,80 carboxyl,73 sulfonate,48 and thiol groups.18,78,82 They are employed to tune the surface charge of SNCs and provide sites for further anchoring of other bioactive groups such as dyes and ligands. The third type of surface functional group is bioactive molecules including fluorescein isothiocyanate (FITC),23,79,82,86 rhodamine B isothiocyanate (RITC),23,31,82,85 rhodamine B,78 and folic acid.59,60 Another important type of surface functional group is poly(ethylene glycol) (PEG).24,31,53,55–61,78 They form a dynamic “cloud” of hydrophilic and neutral chains over the surface of SNCs to prevent the nonspecific adsorption of proteins and enhance long blood circulation and colloidal stability under physiological conditions.
4 Functional silica-based nanocapsules
As described in Sections 3.4 and 3.5, SNCs can be designed for intrinsic multifunction by encapsulating imaging contrast agents, drugs, or multiple agents, within the cavity, in the silica shell and/or to the nanocapsule surface, either through “one-pot” synthesis or post-treatment, therefore making them potentially useful not only in cell imaging, but also in controlled drug delivery and targeted therapy. The following section will address the development of functional SNCs as bioimaging contrast agents, controlled drug delivery vehicles, and both.4.1 Silica-based nanocapsules for bioimaging
SNCs have been intensively pursued for bioimaging applications by incorporation of various types of fluorescent probes (e.g., organic dyes, conjugated polymers, and QDs) and magnetic nanocrystals (e.g., iron oxide, and manganese oxide) because of their high colloidal and chemical stability in the physiological media, protection for photosensitive compounds with the desired image contrast (a high signal-to-noise ratio), rich surface chemistry for avoiding nonspecific binding and enhancing target specificity, controllable nanoscale size with sufficiently long blood circulation, and transparency in the sense that they do not absorb much light in the near-infrared (NIR), visible, and ultraviolet regions or interfere with magnetic fields. In this section, we will briefly address the imaging capabilities of SNCs that are most commonly used for fluorescence imaging, MRI, or a combination of both modalities.Liu et al. reported the use of single micelle-templated SNCs for the delivery of Nile red to DU-145 human prostate cancer cells.53 It was shown that the SNCs may not be endocytosed by the cells; instead, the dye molecules were most likely first released from the nanocapsules, permeated the cell membrane, and then stayed in the cytoplasm of cells (Fig. 12a). In the work done by Yu et al.,86 FITC was covalently linked to the surface of single micelle-templated SNCs through conjugation with APTMS. These FITC-labelled SNCs were evaluated by HeLa cervical cancer cells to show that surface modification of SNCs with hydrophobic methyl groups can enhance the endocytosis properties (Fig. 12b). Jang et al. prepared various types of monodisperse titania–silica SNCs with diameters of 25, 50, 75, 100, and 125 nm and surface functional groups of amine, carboxylate, and methylene.73 In order to systematically investigate the size- and surface functionality-dependent cellular uptake, cytotoxicity, and innate immune response of SNCs in human breast cancer (SK-BR-3) and mouse alveolar macrophage (J774A.1) cells, FITC was conjugated to SNCs. The FITC-labelled SNCs were monitored to achieve a very high level of cellular uptake, cytotoxicity, and innate immune response when they are of 50 nm in size and surface functionalized with amine groups. Recently, Yu et al. synthesized ultrafine 10 nm-sized SNCs through single micelle templating and demonstrated an enhanced penetration ability of RITC-labelled SNCs in U87MG glioma spheroids, a three-dimensional solid tumour, as compared to that of conventional mesoporous silica NPs.85
 | ||
| Fig. 12 (a) Confocal fluorescence image of DU-145 human prostate cancer cells cultured with Nile red-labelled SNCs, showing red contrast inside the cell but outside the cell nuclei (stained in blue). The cell size ranges from 3 to 5 μm. (b) Merged fluorescence and bright-field image of HeLa cells cultured with FITC-labelled SNCs, showing that green contrast can even penetrate the membrane of the nucleus. (c)–(e) Confocal images of transgenic zebrafish larvae, illustrating the biodistribution and uptake of MEH-PPV-labelled SNCs (as indicated by red contrast) after 1 day post injection into the heart. (Reprinted with permission from ref. 53. Copyright 2006 American Chemical Society. Reproduced from ref. 61 and 86 with permission from The Royal Society of Chemistry.) | ||
In addition to the most commonly used organic dyes, fluorescent conjugated polymers have recently emerged as an alternatively promising class of fluorescent probes in cellular imaging. This is because they exhibit rather stable and bright emission, a high level of photostability, a low degree of toxicity and wide wavelength versatility. The present authors' group has reported the preparation of fluorescent SNCs by encapsulating a fluorescent conjugated polymer through the single micelle templating method.60 Four different fluorescent conjugated polymers, namely C6PF, BP-PPV, PDHFBT, and MEH-PPV, which emit blue, green, yellow, and red colours, respectively, were encapsulated in the SNCs. The in vitro studies by using BV-2 microglial cells and MDA-MB-231 breast cancer cells showed that the conjugated polymer-labelled SNCs were taken up and located at the cytoplasm of the cells.55,60 The MEH-PPV-labelled SNCs were further studied in vivo with transgenic zebrafish line TG(fli1:EGFP) as the vertebrate model.61 The biodistribution of fluorescent SNCs, which was examined in the whole larvae, 1 day post injection (Fig. 12c–e), clearly showed that the red contrast of MEH-PPV-labelled SNCs was predominantly detected close to the green fluorescent (GFP-positive) blood vessels. In vivo biocompatibility was also affirmed as the cellular internalization of these fluorescent SNCs did not interfere with larval development or affect vessel growth. Their biostability was verified by the presence of fluorescent-labelled cells even after 7 days since the initial exposure to the microinjected silica nanocarriers.
Alternatively, the present authors' group has recently encapsulated CdSe/CdS/ZnS QDs into single micelle-templated SNCs.59 The surface of SNCs was further conjugated with folate for target specificity. In vitro testing using MDA-MB-231 cells with overexpression of folate receptors demonstrated the efficacy of targeted delivery of QDs to the cancer cells.
T 1 magnetic resonance imaging. In recent years, the use of SNCs to encapsulate MRI T1 contrast agents (e.g., MnO NPs) has been demonstrated to enhance the ability of these “positive” contrast agents to exert a more hyperintense region and thus a higher r1 relaxivity. Silica itself has been recognized as a good candidate to coat T1 contrast agents because it is bio-inert and generally non-toxic. Coupled with the desired mechanical stability of silica, it ensures that the integrity of the nanocarrier can be maintained during the delivery of its payload in the bloodstream. Yet, it has been argued that the traditional MnO@SiO2 core–shell NPs may have a detrimental effect on the r1 relaxivity of the contrast agent. This is because a dense, non-porous silica coating, which is typically synthesized via the conventional Stöber method or the reverse microemulsion technique, can hinder the interactions between water molecules and the MnO NPs, thereby diminishing the r1 relaxivity. In this regard, adopting a nanocapsule structure has been demonstrated to be able to overcome some of these drawbacks that are associated with the core–shell morphology.
Single micelle-templated SNCs have been used to improve the water dispersibility, biocompatibility, and r1 relaxivity of MnO NPs. For example, the present authors' group has designed a hybrid coating to encapsulate MnO NPs.58 The coating layer that consists of a thin and porosified silica shell perforated by the PEO chains of F127 can allow for the rapid penetration of water molecules to the encapsulated MnO core. In addition, these SNCs exhibited a nanorattle structure, wherein the “empty” space surrounding the MnO core was believed to benefit the MRI T1 performance by facilitating the passage of water molecules to the surface Mn ions. The consequent enhancement of T1 contrast was further confirmed using a pre-clinical 7 T MRI scanner, wherein an r1 relaxivity of 1.17 mM−1 s−1 was measured when 15 nm-sized MnO NPs were encapsulated. This is indeed better than the r1 values reported for other 15 nm-sized MnO NPs that are encapsulated with commonly used surface coatings such as PEG-phospholipid or non-porous silica.
As a shell coating for MnO NPs, the use of mesoporous silica will also improve the accessibility of water molecules to the MnO core significantly due to the presence of mesopores in the silica shell. Hence, the water proton relaxation rate can be greatly increased by close proximity of the water molecules to Mn2+ ions on the surface of the MnO core. Moreover, by etching the MnO NPs with an acid solution or via the Kirkendall effect, a hollow MnO core can be produced for a higher surface-to-volume ratio. The higher Mn2+ concentration at the hollow inner surface will result in a higher r1 relaxivity of MnO NPs. Such effectiveness has been clearly demonstrated by several research groups.83,84 A key feature of these SNCs with a mesoporous shell and a hollow MnO core is that their interior surface still retains a thin MnO layer even after the nanoscale etching process.
T 2 magnetic resonance imaging. Development of T2 MRI contrast agents has been extensively studied and iron oxide nanocrystals with a high value of magnetization and a narrow size distribution are found to be excellent T2 MRI contrast agents. However, the application of iron oxide nanocrystals requires an appropriate coating on the surface to make them nontoxic and biocompatible. In this regard, SNCs endow the magnetic nanocrystals with not only good biocompatibility but also enhanced chemical and mechanical stability. The present authors' group has reported the fabrication of PEOlated SNCs with the encapsulation of hydrophobic Fe3O4 nanocrystal clusters.56 The products thus developed exhibited a high saturation magnetization (15.21 emu g−1). The T2-weighted properties were measured using a 1.5 T scanner. The r1 and r2 relaxivity values were reported to be 6.9 and 228 mM−1 s−1, respectively. The relaxivity ratio, r2/r1, was calculated to be 33; this is much higher than ferucarbotran (∼19) and other dextran-coated SPION (3–19).
Shi and co-workers have successfully synthesized another type of bi-functional SNC through the single micelle templating.78 Each SNC consists of multiple superparamagnetic nanocrystal cores, a rhodamine B-doped silica shell, a polymeric micelle of poly(ε-caprolactone)100-block-poly(acrylic acid)35, and PEG chains grafted onto the particle surface (PEGylated Fe3O4/dye@SNCs). T2-weighted MRI properties were measured using a 3 T scanner and the calculated r2 relaxivity value was 215.3 mM−1 s−1, which is higher than the non-clustered magnetite NP system. In view of the low cytotoxicity and excellent colloidal stability, the potential application of PEGylated Fe3O4/dye@SNCs as T2 contrast agents was further evaluated in vivo. For example, an in vivo test was performed by injecting PEGylated Fe3O4/dye@SNCs into tumour-bearing mice. After 40 minutes post-injection, significant accumulation of the SNCs was detected in the tumour area with a T2 signal intensity drop of 42% (Fig. 13). Thus, the passive targeting of PEGylated Fe3O4/dye@SNCs via the enhanced permeability and retention (EPR) effect was demonstrated. The accumulation was further confirmed by ex vivo Prussian blue staining of the tumour tissue from the sacrificed mouse at 5 hour post-injection.
 | ||
| Fig. 13 (a) In vivo T2-weighted MR images (upper) and colour mapped images (lower) of a tumour site before and 40 minutes after intravenous injection of PEGylated Fe3O4/dye@SNCs (the marked regions indicate the tumour site). (b) ex vivo Prussian blue staining images of tumour tissues which were selected from mice at 5 h post-injection of PEGylated Fe3O4/dye@SNCs. (Reproduced from ref. 78 with permission from The Royal Society of Chemistry.) | ||
Furthermore, the delivery of multifunctional SNCs in a targeted manner has been investigated by applying an external magnetic field.57 In the study, PEOlated Fe3O4/MEH-PPV@SNCs, which were synthesized via the single micelle templating approach, consist of the conjugated polymer MEH-PPV as the fluorescent emitter and SPIONs as the magnetic constituent, both of which are encapsulated inside the cavity of SNCs. The in vitro studies showed that the multifunctional SNCs can be guided to the HepG2 cancer cells by simply applying an external magnetic field. Therefore they demonstrate great potential for magnetic targeting biomedical applications (Fig. 14).
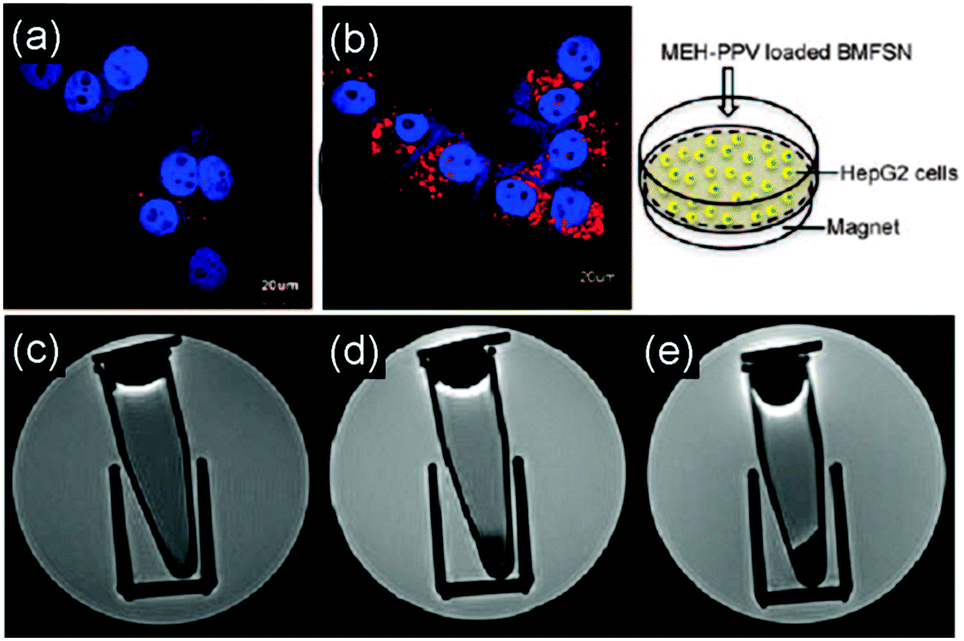 | ||
| Fig. 14 Confocal fluorescence images of HepG2 cells cultured with 120 μg mL−1 of PEOlated Fe3O4/MEH-PPV@SNCs without (a) and with (b) influence of an external magnetic field, showing a marked increase in the red emission of the cells cultured upon the application of an external magnetic field. (c)–(e) T2-weighted MR images of the HepG2 cells were acquired by using a spin-echo pulse sequence in a 7 T MR scanner: (c) control, (d) and (e) were incubated with 60 μg mL−1 of PEOlated Fe3O4/MEH-PPV@SNCs without and with the presence of an external magnetic field, respectively. A darker MR contrast was observed for the cells treated with the magnetic field relative to that of the cells cultured without the field. (Reprinted with permission from ref. 57. Copyright 2011 American Chemical Society.) | ||
MS-SNCs have been commonly used for dual MR and optical imaging applications. This is due to the good accessibility of water molecules through the mesoporous silica shell, thereby enabling them to increase the r1 relaxivity of encapsulated T1 MR contrast agents. Besides, the mesoporous silica shell can also be readily functionalized with various organic dyes for fluorescence imaging capabilities. In this regard, Chou et al. designed a bifunctional nanocomposite that is composed of a single hollow MnO NP encapsulated within an iridium (Ir)-functionalized mesoporous silica shell.83 Its versatility as a dual imaging probe was demonstrated because the Ir complex could yield a strong red phosphorescence even under in vitro conditions while the hollow magnetic core is responsible for the signal enhancement on T1-weighted MR images in vitro. Similarly, Hyeon and co-workers synthesized mesoporous silica-coated hollow manganese oxide NPs, with the fluorescent organic dye, RITC, conjugated to the silica surface using simple silane conjugation chemistry. Intracranial grafting of these nanoparticles enabled serial MR monitoring of cell transplants over a prolonged time period. Moreover, they could also be readily taken up by adipose-derived mesenchymal stem cells for cell labelling and cellular imaging in vitro.
4.2 Silica-based nanocapsules for drug delivery
Over the past couple of decades, cancer nanotherapeutics has rapidly progressed and many types of nanoscale drug carriers, such as liposomes, polymers (e.g., micelles, dendrimers, and polygels), and inorganic NPs (e.g., mesoporous silica and metallic NPs) have been developed, and some are now even available in the market. Within this context, the development of SNCs as controlled drug delivery vehicles has attracted much attention. They have been widely used to solubilize and carry drugs that exhibit relatively poor solubility, low stability, or undesirable pharmaceutical properties. Their small sizes and excellent colloidal and chemical stability allow them to circulate in the body for a long period of time. Hence, they are able to penetrate the leaky vasculature of tumours and accumulate in the affected area more easily.A range of drugs, both hydrophilic and hydrophobic, have been explored for encapsulation in SNCs. The most typical way is to soak SNCs in a drug solution so that the drug molecules are diffused into the SNC cavity.54,79 Alternatively, the drugs can also be facilely encapsulated in SNCs via a one-pot synthesis step.47 Specific drugs such as L-methionine were encapsulated within the spherical cavity at a high level of loading by repeated crystallization.81 However, the nanoscale size of SNCs can limit their physical space for drug encapsulation. Moreover, the use of SNCs (especially MS-SNCs) as drug carriers is susceptible to a rapid and near burst drug release from the nanostructure.27 Brinker et al. observed that L-methionine was released within 3 minutes in phosphate buffered saline because of the high aqueous solubility of the drug and fast diffusion rate of the small L-methionine molecules through mesoporous silica shell.81
To accomplish a better storage and sustained release of drugs, the pore openings in the silica shell of SNCs and the interactions between drugs and SNCs have to be manipulated, mostly through the surface modification of the SNCs. It was reported that the release rate of DOX from the PEG-modified SNCs significantly slowed down due to the reduced pore size by conjugating PEG molecules on the small pore walls.24 As a result, the sample exhibited near zero-order kinetics without initial burst release. It was also reported that the CH3-modified SNCs exhibited a decrease in pore size from 2.06 to 1.50 nm due to the occlusion of the pore channels with methyl groups.75 This greatly slowed down the release of NaCl (although it is not a drug) from several seconds to 80 hours. In addition, two types of interactions between drugs and SNCs, namely the electrostatic interaction and the hydrophobic interaction, have been explored to tune the loading capacity and release rate of SNCs. As has been reported, NH2-modified SNCs exhibited a higher loading capacity and a slower release rate of the drug of naproxen as compared to the unmodified SNCs. This is attributed to the electrostatic interaction between the carboxyl group of naproxen and the amino group of SNCs.53 Similarly, other carboxyl group-containing drugs (e.g., DOX and IBU) have been shown to exhibit a decreased release rate when encapsulated by the NH2-modified SNCs.24,80 Alternatively, increasing the condensation degree of silica or introducing organic groups to the silica matrix could increase the hydrophobicity of silica. As a result, there is a stronger hydrophobic interaction between the drugs and SNCs, thereby leading to a higher level of drug loading and a slower release rate.53 Yang et al. reported a shell-thickness-dependent drug release behaviour in MS-SNCs. The release amount and release rate of DOX were increased by a decrease in the shell thickness.79
In addition, the release behaviour can be controlled precisely by pH-responsive nanovalves.64 The concept is schematically illustrated in Fig. 15. Herein, the MS-SNCs contained anilinoalkane stalks (covalently bonded to the silica surface) and bulky nanovalves of α-CD (a cyclic molecule which encircles the stalks via non-covalent interactions). When the pH of the environment is changed, the bulky cyclic groups of α-CD will block or unblock the pores on the surface of MS-SNCs, respectively. For example, at pH ∼ 7, the α-CD rings are complexed with the stalks; this causes the bulky cyclic components to block the pores. Upon decreasing the pH environment, which occurs in several physiological and pathological processes such as endosome trafficking, tumour growth, inflammation, and myocardial ischemia, the aniline nitrogen atoms on the stalks are protonated. This lowers the binding affinity between α-CD and the stalks, thereby causing the α-CD to dissociate, unblock the pores and then release the encapsulated cargo molecules from the interior.
 | ||
| Fig. 15 Schematic illustration of a cross section of MS-SNC showing the worm-like pores connecting the interior to the surrounding medium. The stalks and the α-CD rings that control the pore openings are also shown. (Reprinted with permission from ref. 64. Copyright 2009 American Chemical Society.) | ||
4.3 Silica-based nanocapsules for nanotheranostics
Based on the functionalities briefly described above, SNCs represent an excellent scaffold for the facile loading of a wide variety of bioimaging and therapeutic moieties. As such, they are promising candidates for nanotheranostics for simultaneous imaging and therapy applications. This integrated system holds great promise because it could monitor the real time drug release and distribution, thus predicting and validating the efficacy of the treatment.To improve the performance of nanotheranostics, the surface of SNCs is modified by tethering protective ligands, such as PEG,24,31,53,55–61,78 to prolong the bioavailability of the nanotheranostics. Other modifications include conjugating targeting ligands, such as folic acid,59,60 to the SNC surface to trigger receptor-mediated endocytosis, thereby enhancing the biodistribution and uptake of the nanotheranostics in the targeting area. In addition, SNCs must retain their drug payload with “zero leakage” throughout the long circulation period before reaching the targeting site. The integrated system could then undergo a stimuli-responsive release of drugs at a satisfactory rate to ensure its bioavailability and pharmacological activity.
For example, Jiang and co-workers reported the use of CS–silica double shelled nanocapsules to load the anti-cancer drug of DOX for pH-sensitive nanotheranostics.63 The inner CS shell served as a pH-sensitive switch. Under the “off” status at pH = 7.4, CS was deprotonated and collapsed at the pore openings to restrict the release of DOX. Under the “on” status at pH = 4, the amino groups in CS were protonated. This caused the positively charged CS to swell and expose the pore openings so that the drug was released. Therefore, the release profile exhibited a pulse appearance while the drug release could be switched on and off by alternately changing the medium pH value (Fig. 16b). Fig. 16c showed the fluorescence image of DOX-loaded CS-silica nanocapsules incubated with LoVo cells as obtained in the green channel. It clearly showed that most of the DOX was inside the cells and even in the nucleus. The DOX-loaded CS-silica nanocapsules showed better cellular uptake than free DOX, probably due to the endocytosis mechanism, and thus a higher level of therapeutic efficiency (Fig. 16d).
 | ||
| Fig. 16 (a) TEM image of CS–silica double-shelled nanocapsules. (b) In vitro release profile of DOX-loaded CS–silica nanocapsules at 37 °C with pH values of the release medium alternately changed between 7.4 and 4.0. (c) Fluorescence image of DOX-loaded CS–silica nanocapsules incubated with LoVo cells for 4 hours. (d) In vitro cytotoxicity of free DOX and DOX-loaded CS–silica nanocapsules against LoVo cells. Inset is the cytotoxicity of empty CS–silica nanocapsules against LoVo cells. (Reproduced from ref. 63 with permission from The Royal Society of Chemistry.) | ||
In another study,31 multifunctional PEGylated MS-SNCs, which were labelled with RITC for fluorescence behaviour, were able to release DOX in tumour-bearing mice to demonstrate a more effective tumour growth inhibition efficacy (39.10%) than free DOX (22.33%).
Anker and co-workers recently reported a rattle-type SNC which can function as a dual MRI contrast agent, as well as a pH-responsive drug delivery system.20 The Fe/DOX/Gd-DTPA@SNC@AL/PLL NPs consist of an iron NP and an ellipsoidal silica shell that is loaded with DOX and the T1 contrast agent Gd-DTPA (Fig. 17). The surface of the ellipsoidal nanotheranostics was decorated with biocompatible poly-L-lysine (PLL) and sodium alginate (AL) to control the DOX release. The products thus derived demonstrated a very high saturation magnetization of 63.4 emu g−1. The T1-weighted and T2-weighted properties were studied with a 4.7 T MRI instrument. The r1 was measured as 8.6 mM−1 s−1, which is higher than that of the free Gd-DTPA and the r2 was measured as 285 mM−1 s−1, which is much higher than that of the FDA-approved iron oxide NP contrast agents. Furthermore, DOX was loaded into the cavity of the nanocapsules for controlled drug release. It was observed that the release of DOX was highly pH-sensitive as its release rate was significantly increased under acidic conditions. Cellular therapeutic efficiency, which was studied using MCF-7 breast cancer cells, also indicated that the NPs exhibited a higher cytotoxicity than free DOX.
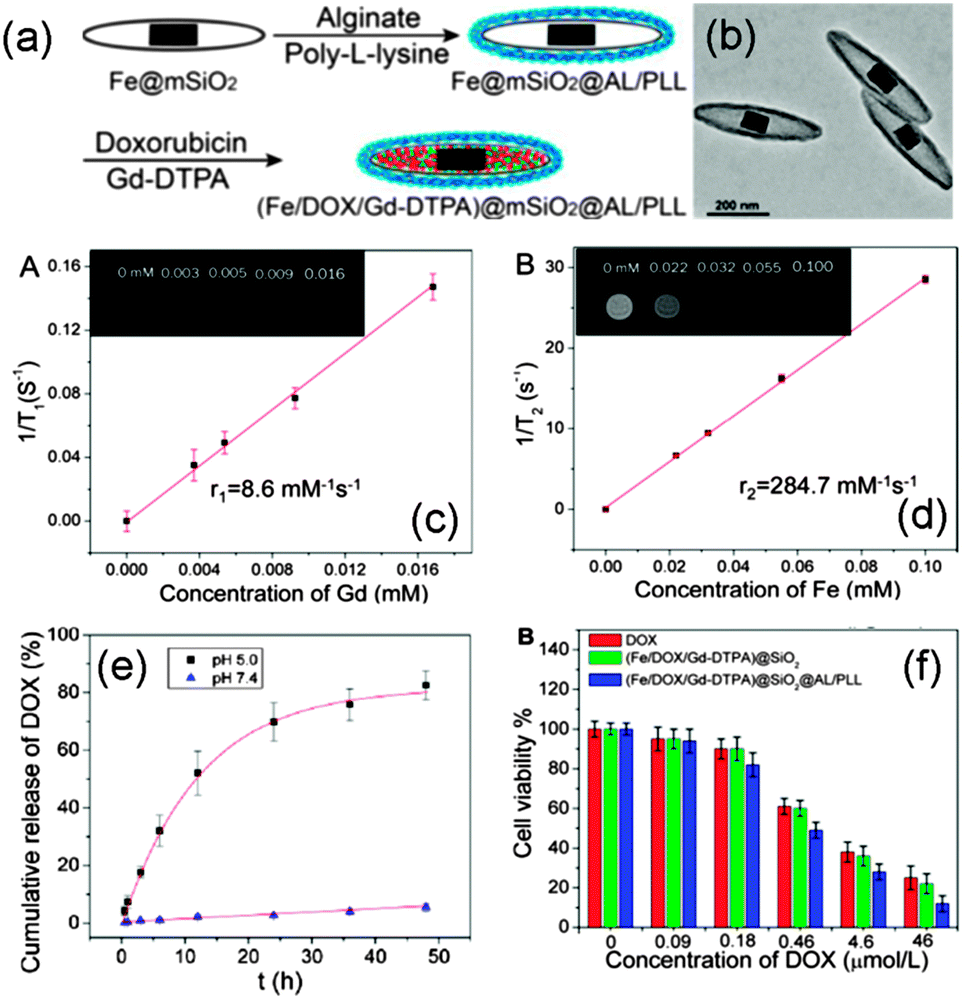 | ||
| Fig. 17 (a) Schematic illustration showing the synthesis route of Fe/DOX/Gd-DTPA@SiO2@AL/PLL. (b) TEM image of Fe@SNC@AL/PLL. (c) T1 and (d) T2-weighted images of magnetic at echo time of 4 ms. (e) pH-triggered release profile of DOX from Fe/DOX/Gd-DTPA)@SNC@AL/PLL. (f) Cell viability of MCF-7 cells after incubating with different drug formulations at different concentrations for 48 h. (Reprinted with permission from ref. 20. Copyright 2014 American Chemical Society.) | ||
The loading of SNCs with phthalocyanine photosensitizers has also been recently reported. Carriers of these therapeutic molecules were able to kill cancer cells (both in vitro and in vivo) by a combined photodynamic and photothermal effect.87
5 Conclusions and outlook
There has been intensive progress in syntheses, structure control, surface functionalization, and biomedical applications of SNCs, as described in this review. SNCs are promising platforms that can be engineered with varying structural configurations and numerous multifunctionalities for biomedical imaging and theranostic applications. Several synthetic strategies have been developed for SNCs, with varying degrees of success in terms of structure control and surface functionalization. Both in vitro and in vivo studies have demonstrated their potential as diagnostic and therapeutic agents. However, several areas still require considerable study before SNCs are fully established for the proposed applications. For example, SNCs are expected to be functionalized with more specific types of targeting ligands and designed to be more precisely responsive to the applied stimuli so that the cellular uptake and delivery process is more targetable and controllable. SNCs are also promising for gene therapy by conjugating gene binders such as nucleic acids to their outer surfaces. The in vivo biocompatibility, biodistribution, targeting efficiency, toxicity, biodegradability and clearance of SNCs also need to be further studied. The research focuses in this field will include design and synthesis of “smart” multifunctional SNC nanocarriers, in vivo performance evaluation using animal models and subsequent translational studies. Therefore, multi-disciplinary collaborations are critical among materials scientists, chemists and clinicians to enable the utilization of SNCs in clinics. Apart from the biomedical applications, SNCs also provide a unique platform for encapsulation of functional agents for heterogeneous catalysis, separation, optoelectronics, magnetic resonance and chemical sensing applications. They can serve as the solid templates for growth of various novel nanostructures. The synthetic strategies developed for SNCs can be extended to other organic and/or inorganic capsules.Acknowledgements
This work is supported by the BMRC-SERC Joint Program (grant No. 1121480002), and SERC (grant No. 1121202013), Agency for Science, Technology and Research (A*STAR), Singapore.Notes and references
- X. W. Lou, L. A. Archer and Z. Yang, Adv. Mater., 2008, 20, 3987–4019 CrossRef CAS.
- Y. Zhao and L. Jiang, Adv. Mater., 2009, 21, 3621–3638 CrossRef CAS.
- J. Hu, M. Chen, X. Fang and L. Wu, Chem. Soc. Rev., 2011, 40, 5472–5491 RSC.
- L. L. Zhang and X. S. Zhao, Chem. Soc. Rev., 2009, 38, 2520–2531 RSC.
- M. Antonietti and S. Förster, Adv. Mater., 2003, 15, 1323–1333 CrossRef CAS.
- H. J. Fan, U. Gösele and M. Zacharias, Small, 2007, 3, 1660–1671 CrossRef CAS PubMed.
- V. Sokolova and M. Epple, Angew. Chem., Int. Ed., 2008, 47, 1382–1395 CrossRef CAS PubMed.
- Z. Li, J. C. Barnes, A. Bosoy, J. F. Stoddart and J. I. Zink, Chem. Soc. Rev., 2012, 41, 2590–2605 RSC.
- P. T. Tanev and T. J. Pinnavaia, Science, 1996, 271, 1268–1269 Search PubMed.
- S. Schacht, Q. Huo, I. G. Voigt-Martin, G. D. Study and F. Schüth, Science, 1996, 273, 768–771 CAS.
- F. Caruso, R. A. Caruso and H. Möhwald, Science, 1998, 282, 1111–1114 CrossRef CAS.
- S. S. Kim, W. Zhang and T. J. Pinnavaia, Science, 1998, 282, 1302–1305 CrossRef CAS.
- J. L. Vivero-Escoto, R. C. Huxford-Phillips and W. Lin, Chem. Soc. Rev., 2012, 41, 2673–2685 RSC.
- A. Burns, H. Ow and U. Wiesner, Chem. Soc. Rev., 2006, 35, 1028–1042 RSC.
- J. J. L. M. Cornelissen, E. F. Connor, H.-C. Kim, V. Y. Lee, T. Magibitang, P. M. Rice, W. Volksen, L. K. Sundberg and R. D. Miller, Chem. Commun., 2003, 1010–1011 RSC.
- J. Yang, J. U. Lind and W. C. Trogler, Chem. Mater., 2008, 20, 2875–2877 CrossRef CAS.
- H. Blas, M. Save, P. Pasetto, C. Boissière, C. Sanchez and B. Charleux, Langmuir, 2008, 24, 13132–13137 CrossRef CAS PubMed.
- J. Yuan, D. Wan and Z. Yang, J. Phys. Chem. C, 2008, 112, 17156–17160 CAS.
- N. Kato, T. Ishii and S. Koumoto, Langmuir, 2010, 26, 14334–14344 CrossRef CAS PubMed.
- H. Chen, D. Sulejmanovic, T. Moore, D. C. Colvin, B. Qi, O. T. Mefford, J. C. Gore, F. Alexis, S.-J. Hwu and J. N. Anker, Chem. Mater., 2014, 26, 2105–2112 CrossRef CAS PubMed.
- D. K. Yi, S. S. Lee, G. C. Papaefthymiou and J. Y. Ying, Chem. Mater., 2006, 18, 614–619 CrossRef CAS.
- W. Zhao, M. Lang, Y. Li, L. Li and J. Shi, J. Mater. Chem., 2009, 19, 2778–2783 RSC.
- J. Kim, H. S. Kim, N. Lee, T. Kim, H. Kim, T. Yu, I. C. Song, W. K. Moon and T. Hyeon, Angew. Chem., Int. Ed., 2008, 47, 8438–8441 CrossRef CAS PubMed.
- J. Yang, J. Lee, J. Kang, K. Lee, J.-S. Suh, H.-G. Yoon, Y.-M. Huh and S. Haam, Langmuir, 2008, 24, 3417–3421 CrossRef CAS PubMed.
- Z.-Z. Li, L.-X. Wen, L. Shao and J.-F. Chen, J. Controlled Release, 2004, 98, 245–254 CrossRef CAS PubMed.
- J.-F. Chen, J.-X. Wang, R.-J. Liu, L. Shao and L.-X. Wen, Inorg. Chem. Commun., 2004, 7, 447–449 CrossRef CAS PubMed.
- J.-F. Chen, H.-M. Ding, J.-X. Wang and L. Shao, Biomaterials, 2004, 25, 723–727 CrossRef CAS.
- Z.-Z. Li, S.-A. Xu, L.-X. Wen, F. Liu, A.-Q. Liu, Q. Wang, H.-Y. Sun, W. Yu and J.-F. Chen, J. Controlled Release, 2006, 111, 81–88 CrossRef CAS PubMed.
- J. Wang, H. Ding, X. Tao and J. Chen, Nanotechnology, 2007, 18, 245705 CrossRef.
- R. V. Rivera-Virtudazo, M. Fuji, C. Takai and T. Shirai, Nanotechnology, 2012, 23, 485608 CrossRef CAS PubMed.
- T. Wang, F. Chai, Q. Fu, L. Zhang, H. Liu, L. Li, Y. Liao, Z. Su, C. Wang, B. Duan and D. Ren, J. Mater. Chem., 2011, 21, 5299–5306 RSC.
- S. Liu, Z. Zhang and M. Han, Adv. Mater., 2005, 17, 1862–1866 CrossRef CAS.
- Y. Liu, H. Miyoshi and M. Nakamura, Colloids Surf., B, 2007, 58, 180–187 CrossRef CAS PubMed.
- M. Darbandi, R. Thomann and T. Nann, Chem. Mater., 2007, 19, 1700–1703 CrossRef CAS.
- Y.-S. Lin, S.-H. Wu, C.-T. Tseng, Y. Hung, C. Chang and C.-Y. Mou, Chem. Commun., 2009, 3542–3544 RSC.
- S.-H. Wu, Y. Hung and C.-Y. Mou, Chem. Mater., 2013, 25, 352–364 CrossRef CAS.
- A. V. Jovanovic, R. S. Underhill, T. L. Bucholz and R. S. Duran, Chem. Mater., 2005, 17, 3375–3383 CrossRef CAS.
- H. Wang, Y. Wang, X. Zhou, L. Zhou, J. Tang, J. Lei and C. Yu, Adv. Funct. Mater., 2007, 17, 613–617 CrossRef CAS.
- B. Tan, H.-J. Lehmler, S. M. Vyas, B. L. Knutson and S. E. Rankin, Adv. Mater., 2005, 17, 2368–2371 CrossRef CAS.
- L. Zhang, P. Li, X. Liu, L. Du and E. Wang, Adv. Mater., 2007, 19, 4279–4283 CrossRef CAS.
- A. J. Karkamkar, S.-S. Kim, S. D. Mahanti and T. J. Pinnavaia, Adv. Funct. Mater., 2004, 14, 507–512 CrossRef CAS.
- D. H. W. Hubert, M. Jung, P. M. Frederik, P. H. H. Bomans, J. Meuldijk and A. L. German, Adv. Mater., 2000, 12, 1286–1290 CrossRef CAS.
- S. Yang, X. Zhou, P. Yuan, M. Yu, S. Xie, J. Zou, G. Q. Lu and C. Yu, Angew. Chem., Int. Ed., 2007, 46, 8579–8582 CrossRef CAS PubMed.
- B. Tan, S. M. Vyas, H.-J. Lehmler, B. L. Knutson and S. E. Rankin, Adv. Funct. Mater., 2007, 17, 2500–2508 CrossRef CAS.
- H. Djojoputro, X. F. Zhou, S. Z. Qiao, L. Z. Wang, C. Z. Yu and G. Q. Lu, J. Am. Chem. Soc., 2006, 128, 6320–6321 CrossRef CAS PubMed.
- J. Du, Y. Chen, Y. Zhang, C. C. Han, K. Fischer and M. Schmidt, J. Am. Chem. Soc., 2003, 125, 14710–14711 CrossRef CAS PubMed.
- M. Mandal and M. Kruk, Chem. Mater., 2012, 24, 123–132 CrossRef CAS , and references therein.
- X. Li, Y. Yang and Q. Yang, J. Mater. Chem. A, 2013, 1, 1525–1535 CAS , and references therein.
- K. Koh, K. Ohno, Y. Tsujii and T. Fukuda, Angew. Chem., Int. Ed., 2003, 42, 4194–4197 CrossRef CAS PubMed.
- J.-J. Yuan, O. O. Mykhaylyk, A. J. Ryan and S. P. Armes, J. Am. Chem. Soc., 2007, 129, 1717–1723 CrossRef CAS PubMed.
- Y. Li, J. Du and S. P. Armes, Macromol. Rapid Commun., 2009, 30, 464–468 CrossRef CAS PubMed.
- A. Khanal, Y. Inoue, M. Yada and K. Nakashima, J. Am. Chem. Soc., 2007, 129, 1534–1535 CrossRef CAS PubMed.
- Q. Huo, J. Liu, L.-Q. Wang, Y. Jiang, T. N. Lambert and E. Fang, J. Am. Chem. Soc., 2006, 128, 6447–6453 CrossRef CAS PubMed.
- J. Liu, Q. Yang, L. Zhang, H. Yang, J. Gao and C. Li, Chem. Mater., 2008, 20, 4268–4275 CAS.
- H. Tan, N. S. Liu, B. He, S. Y. Wong, Z.-K. Chen, X. Li and J. Wang, Chem. Commun., 2009, 6240–6242 RSC.
- H. Tan, J. M. Xue, B. Shuter, X. Li and J. Wang, Adv. Funct. Mater., 2010, 20, 722–731 CrossRef CAS.
- H. Tan, M. Wang, C.-T. Yang, S. Pant, K. K. Bhakoo, S. Y. Wong, Z.-K. Chen, X. Li and J. Wang, Chem. – Eur. J., 2011, 17, 6696–6706 CrossRef CAS PubMed.
- B. Y. W. Hsu, M. Wang, Y. Zhang, V. Vijayaragavan, S. Y. Wong, A. Y.-C. Chang, K. K. Bhakoo, X. Li and J. Wang, Nanoscale, 2014, 6, 293–299 RSC.
- Y. Zhang, M. Wang, Y.-G. Zheng, H. Tan, B. Y. W. Hsu, Z.-C. Yang, S. Y. Wong, A. Y.-C. Chang, M. Choolani, X. Li and J. Wang, Chem. Mater., 2013, 25, 2976–2985 CrossRef CAS.
- H. Tan, Y. Zhang, M. Wang, Z. Zhang, X. Zhang, A. M. Yong, S. Y. Wong, A. Y.-C. Chang, Z.-K. Chen, X. Li, M. Choolani and J. Wang, Biomaterials, 2012, 33, 237–246 CrossRef CAS PubMed.
- B. Y. W. Hsu, C. Teh, H. Tan, S. Y. Wong, Y. Zhang, V. Korzh, X. Li and J. Wang, RSC Adv., 2012, 2, 12392–12399 RSC.
- Y. Wan and S.-H. Yu, J. Phys. Chem. C, 2008, 112, 3641–3647 CAS.
- E. Yan, Y. Ding, C. Chen, R. Li, Y. Hu and X. Jiang, Chem. Commun., 2009, 2718–2720 RSC.
- L. Du, S. Liao, H. A. Khatib, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2009, 131, 15136–15142 CrossRef CAS PubMed.
- R. K. Rana, Y. Mastai and A. Gedanken, Adv. Mater., 2002, 14, 1414–1418 CrossRef CAS.
- Y. Lu, H. Fan, A. Stump, T. L. Ward, T. Rieker and C. J. Brinker, Nature, 1999, 398, 223–226 CrossRef CAS PubMed.
- P. Tartaj, T. González-Carreño and C. J. Serna, Adv. Mater., 2001, 13, 1620–1624 CrossRef CAS.
- X. Fang, X. Zhao, W. Fang, C. Chen and N. Zheng, Nanoscale, 2013, 5, 2205–2218 RSC , and references therein.
- M. Roca and A. J. Haes, J. Am. Chem. Soc., 2008, 130, 14273–14279 CrossRef CAS PubMed.
- D. Chen, L. Li, F. Tang and S. Qi, Adv. Mater., 2009, 21, 3804–3807 CrossRef CAS.
- Y. Yang, J. Liu, X. Li, X. Liu and Q. Yang, Chem. Mater., 2011, 23, 3676–3684 CrossRef CAS.
- Y. Chen, H. Chen, L. Guo, Q. He, F. Chen, J. Zhou, J. Feng and J. Shi, ACS Nano, 2010, 4, 529–539 CrossRef CAS PubMed.
- W.-K. Oh, S. Kim, M. Choi, C. Kim, Y. S. Jeong, B.-R. Cho, J.-S. Hahn and J. Jang, ACS Nano, 2010, 4, 5301–5313 CrossRef CAS PubMed.
- M. Choi, C. Kim, S. O. Jeon, K. S. Yook, J. Y. Lee and J. Jang, Chem. Commun., 2011, 47, 7092–7094 RSC.
- X. Jiang and C. J. Brinker, J. Am. Chem. Soc., 2006, 128, 4512–4513 CrossRef CAS PubMed.
- X. Wu and C. M. Crudden, Chem. Mater., 2012, 24, 3839–3846 CrossRef CAS.
- X.-J. Wu and D. Xu, J. Am. Chem. Soc., 2009, 131, 2774–2775 CrossRef CAS PubMed.
- D. Niu, X. Liu, Y. Li, Z. Ma, W. Dong, S. Chang, W. Zhao, J. Gu, S. Zhang and J. Shi, J. Mater. Chem., 2011, 21, 13825–13831 RSC.
- Y. Jiao, J. Guo, S. Shen, B. Chang, Y. Zhang, X. Jiang and W. Yang, J. Mater. Chem., 2012, 22, 17636–17643 RSC.
- J. Kim, J. E. Lee, J. Lee, J. H. Yu, B. C. Kim, K. An, Y. Hwang, C.-H. Shin, J.-G. Park, J. Kim and T. Hyeon, J. Am. Chem. Soc., 2006, 128, 688–689 CrossRef CAS PubMed.
- X. Jiang, T. L. Ward, Y.-S. Cheng, J. Liu and C. J. Brinker, Chem. Commun., 2010, 46, 3019–3021 RSC.
- D. Niu, Y. Li, Z. Ma, H. Diao, J. Gu, H. Chen, W. Zhao, M. Ruan, Y. Zhang and J. Shi, Adv. Funct. Mater., 2010, 20, 773–780 CrossRef CAS.
- Y.-K. Peng, C.-W. Lai, C.-L. Liu, H.-C. Chen, Y.-H. Hsiao, W.-L. Liu, K.-C. Tang, Y. Chi, J.-K. Hsiao, K.-E. Lim, H.-E. Liao, J.-J. Shyue and P.-T. Chou, ACS Nano, 2011, 5, 4177–4187 CrossRef CAS PubMed.
- T. Kim, E. Momin, J. Choi, K. Yuan, H. Zaidi, J. Kim, M. Park, N. Lee, M. T. McMahon, A. Quinones-Hinojosa, J. W. M. Bulte, T. Hyeon and A. A. Gilad, J. Am. Chem. Soc., 2011, 133, 2955–2961 CrossRef CAS PubMed.
- M. Yu, S. Karmakar, J. Yang, H. Zhang, Y. Yang, P. Thornb and C. Yu, Chem. Commun., 2014, 50, 1527–1529 RSC.
- J. Zhu, J. Tang, L. Zhao, X. Zhou, Y. Wang and C. Yu, Small, 2010, 6, 276–282 CrossRef CAS PubMed.
- J. Peng, L. Zhao, X. Zhu, Y. Sun, W. Feng, Y. Gao, L. Wang and F. Li, Biomaterials, 2013, 34, 7905–7912 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |