
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Isoxazole to oxazole: a mild and unexpected transformation†
Raymond C. F.
Jones
*a,
Alexander
Chatterley
a,
Romain
Marty
a,
W. Martin
Owton
b and
Mark R. J.
Elsegood
a
aDepartment of Chemistry, Loughborough University, Leics. LE11 3TU, UK. E-mail: r.c.f.jones@lboro.ac.uk; Fax: +44 (0)1509 223926; Tel: +44 (0)1509 222557
bLilly UK, Erl Wood Manor, Windlesham, Surrey GU20 6PH, UK
First published on 26th November 2014
Abstract
3-Aryltetrahydrobenzisoxazoles prepared en route to the coleophomone natural products and analogues, were found to undergo a remarkable base-mediated rearrangement to 2-aryltetrahydrobenzoxazoles. The scope of this unprecedented, facile transformation was probed: a range of analogues was produced, a mechanism proposed, and an application demonstrated by synthesis of a known herbicidal compound.
As an extension of our interest in natural products containing the cyclic trione unit 1,1,2 we were attracted to the coleophomone natural products, exemplified by coleophomones A (2) and B (3), reported as being in equilibrium via an aldol process.3,4 This group of metabolites have enzyme inhibitory properties towards bacterial cell wall transglycosylase and human heart chymase.4,5
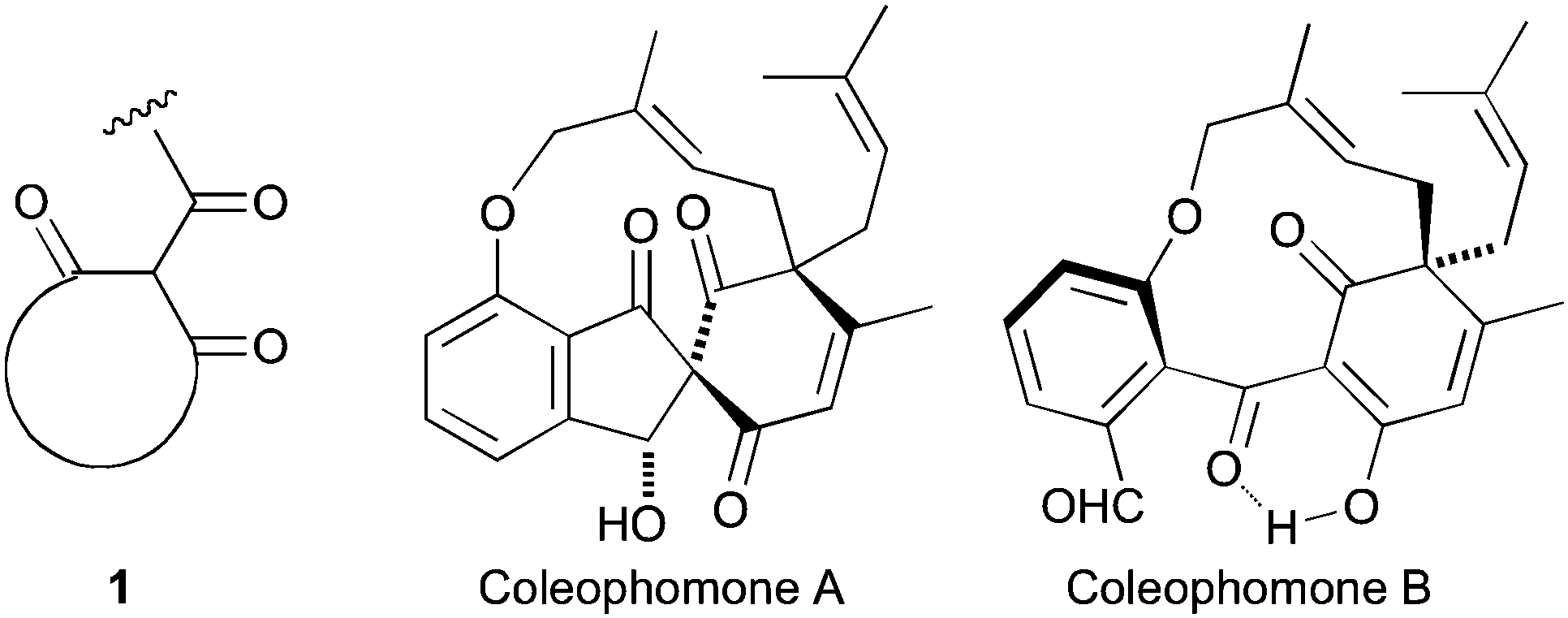
Applying our previously reported isoxazole masking strategy for the cyclic trione unit1,2 led us to propose the disconnection of Scheme 1, requiring 3-aryltetrahydrobenzisoxazole building blocks to access the natural products and (masked) analogues. Whilst manipulating one such arylbenzisoxazole, we observed a remarkable rearrangement to a 2-arylbenzoxazole. We report here our exploration of this unprecedented, facile transformation.
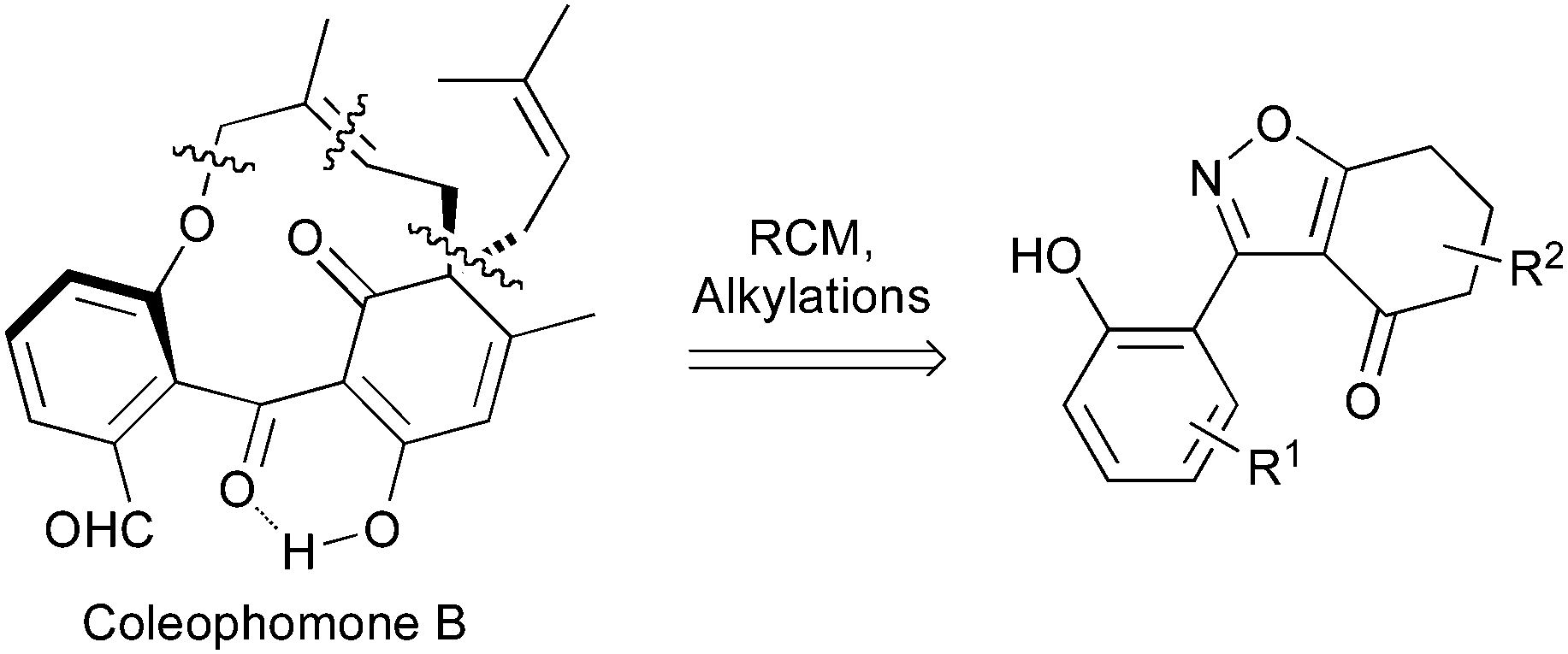 | ||
| Scheme 1 Strategic disconnection of coleophomones. | ||
A suitable set of 3-aryltetrahydrobenzisoxazoles 4 was prepared by 1,3-dipolar cycloaddition of aryl nitrile oxides [available from benzaldehyde oximes via C-chlorination (NCS, CHCl3 reflux) and 1,3-elimination] with cyclohexane-1,3-diones under basic conditions (Scheme 2).6
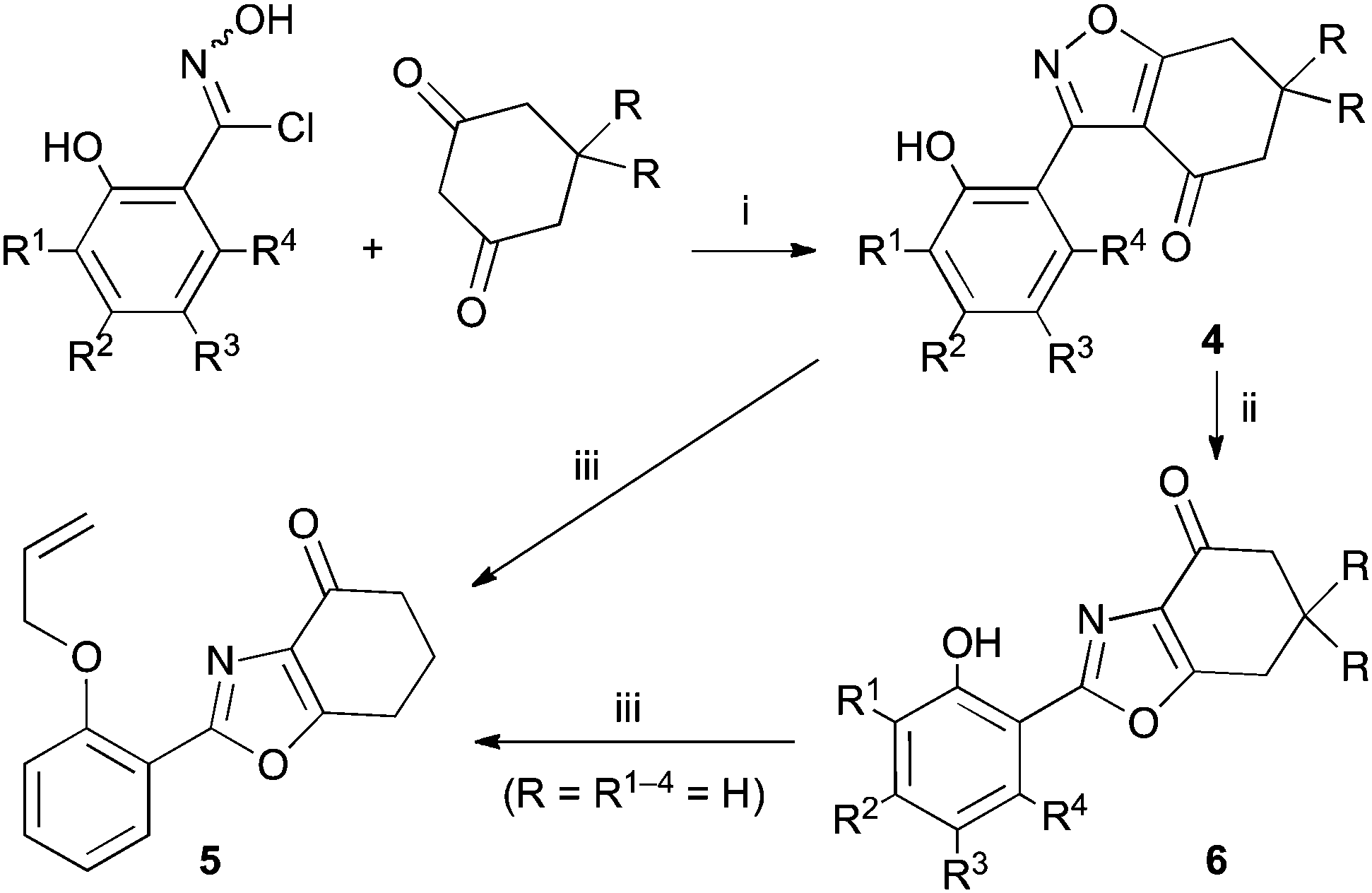 | ||
Scheme 2 Synthesis and rearrangement of 3-aryltetrahydrobenzisoxazoles 4. Reagents: (i), NaOi-Pr, i-PrOH; (ii), Cs2CO3, THF reflux; (iii), H2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCH2Br, Cs2CO3, THF reflux. CHCH2Br, Cs2CO3, THF reflux. | ||
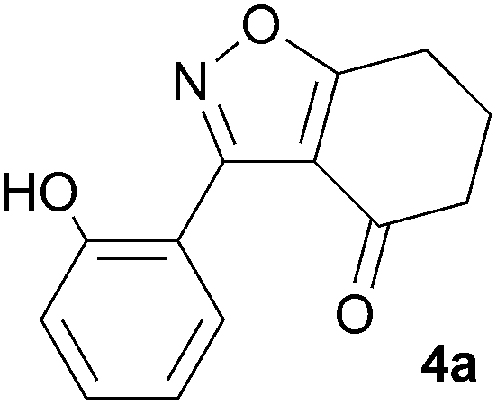
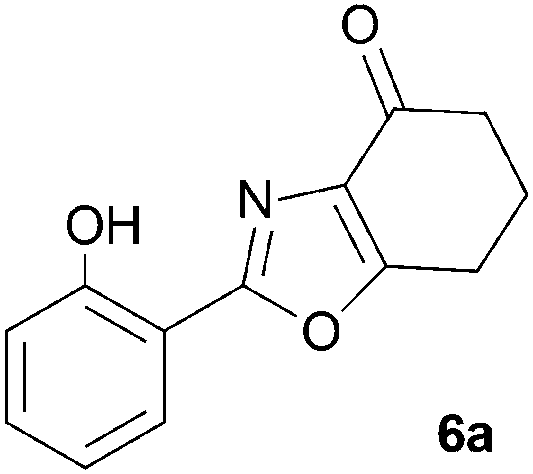
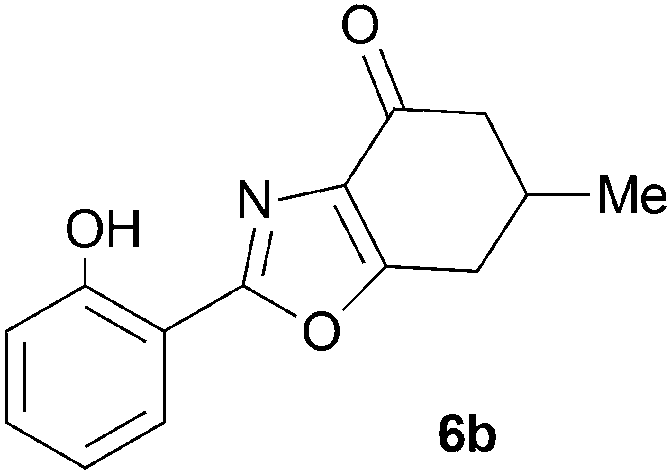
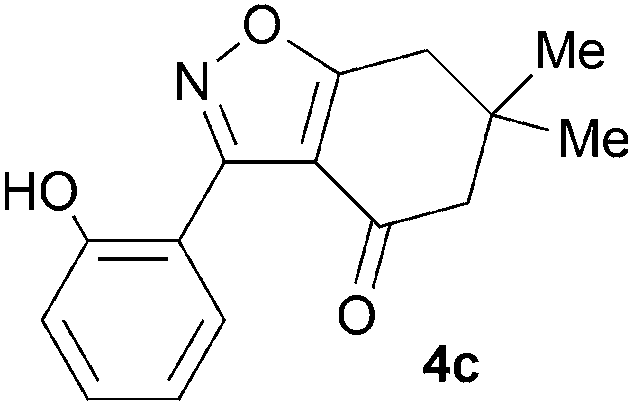
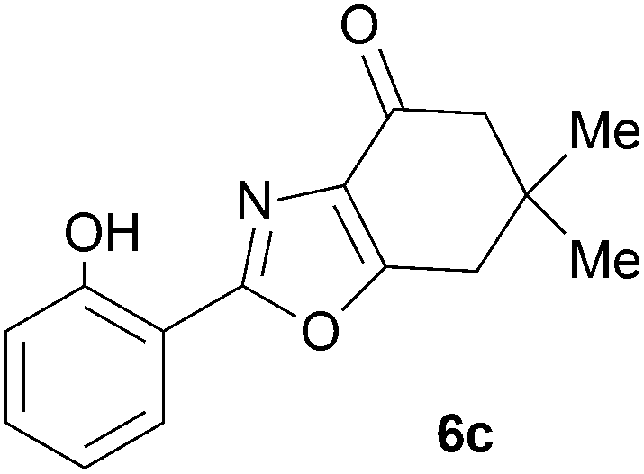
During attempts to complete O-allylation of 3-(2-hydroxyphenyl)benzisoxazole 4a (R = R1 = H) under standard basic conditions (Cs2CO3, THF reflux), we did not observe the expected product but instead isolated 2-(2-allyloxy)tetrahydrobenzoxazole 5. This rearrangement also took place in the absence of alkylating agent (Scheme 2); the phenolic product 6a (R = R1–4 = H) was stable to the basic conditions, and was successfully O-allylated to give ether 5 on addition of allyl bromide. We have verified the structures of both isoxazole 4a and dimethyl product oxazole 6b (R = Me, R1–4 = H; vide infra) through X-ray crystal structure determinations, Fig. 1a and b.‡
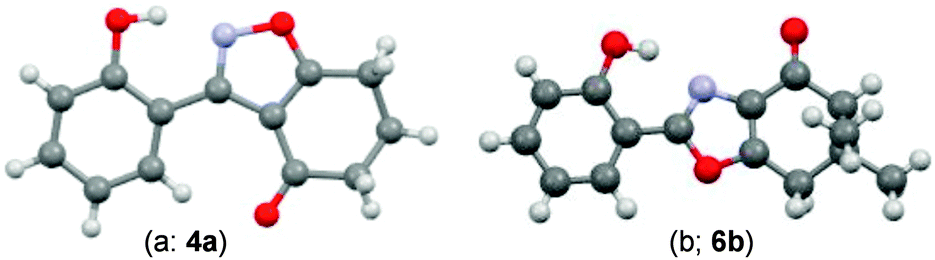 | ||
| Fig. 1 (a and b) X-Ray crystal structures of isoxazole 4a and oxazole 6b (O = red, N = blue, C = grey, H = light grey). | ||
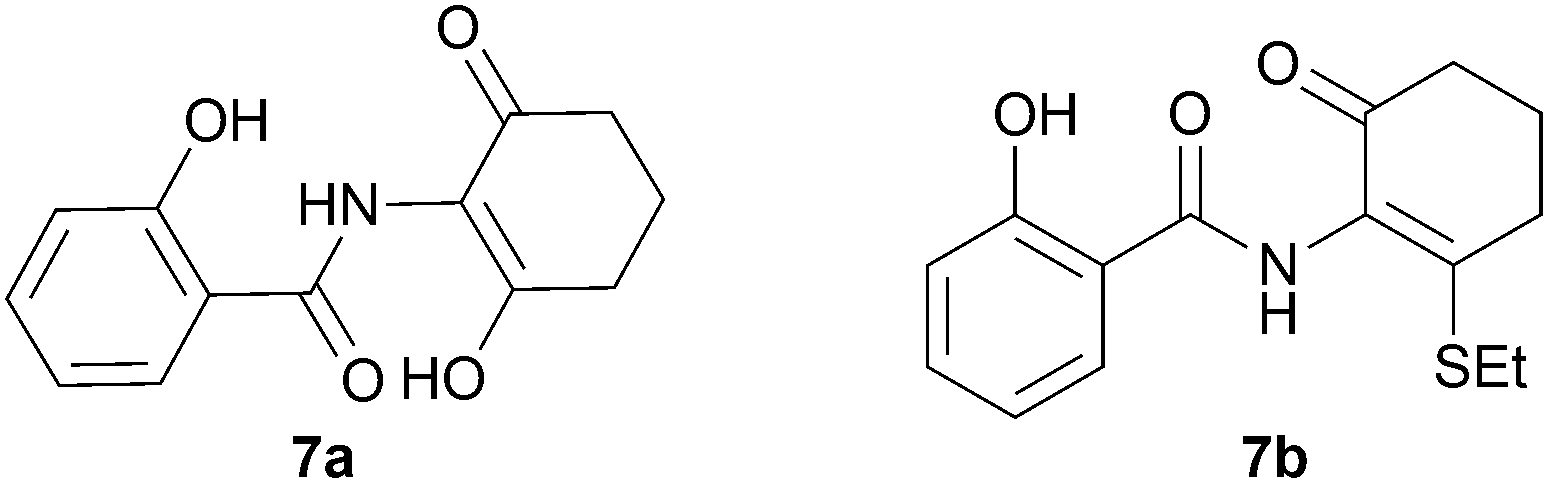
We further investigated the scope of the remarkable rearrangement of benzisoxazoles 4 to benzoxazoles 6. Using isoxazole 4a, rearrangement was found to occur in aprotic solvents with reaction time of 4 h under a range of basic conditions (Table 1) including carbonates, alkoxide and amidine, but failed with tertiary amines. In the presence of water or ethanethiol (entries 12, 13) the amide products 7a,b, respectively, of ring opening of the oxazole 6a were isolated; the constitutions of the amides were confirmed by X-ray crystal structures.7
| Entry | Base | Solvent | Yieldb (%) |
|---|---|---|---|
| a Isoxazole 1 (2.18 mmol), base (4.37 mmol), reaction time 4 h, solvent under reflux. b Isolated yields refer to 6a unless otherwise stated. | |||
| 1 | Cs2CO3 | THF | 87 |
| 2 | K2CO3 | THF | 84 |
| 3 | Na2CO3 | THF | 5 |
| 4 | Et3N | THF | 0 |
| 5 | DMAP | THF | 0 |
| 6 | DBU | THF | 83 |
| 7 | LDA | THF | 37 |
| 8 | Cs2CO3 | Toluene | 97 |
| 9 | NaOi-Pr | i-PrOH | 85 |
| 10 | Cs2CO3 | EtOH–H2O | 87 (for 7a) |
| 11 | None | H2O | 8 |
| 12 | Cs2CO3 | H2O | 91 (for 7a) |
| 13 | Cs2CO3 & EtSH | THF | 6 (for 7b) |
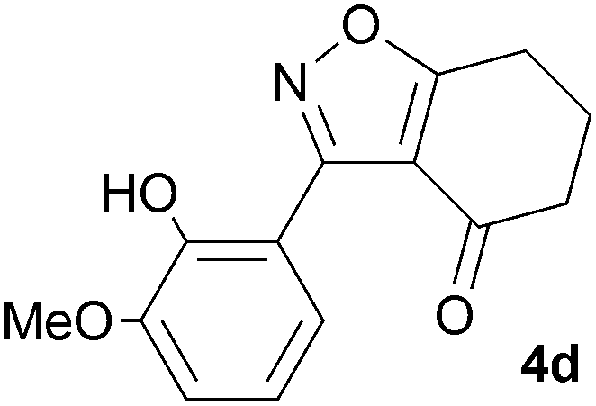
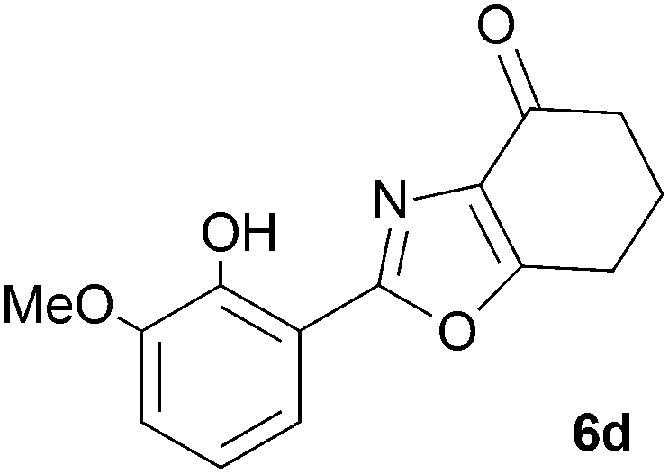
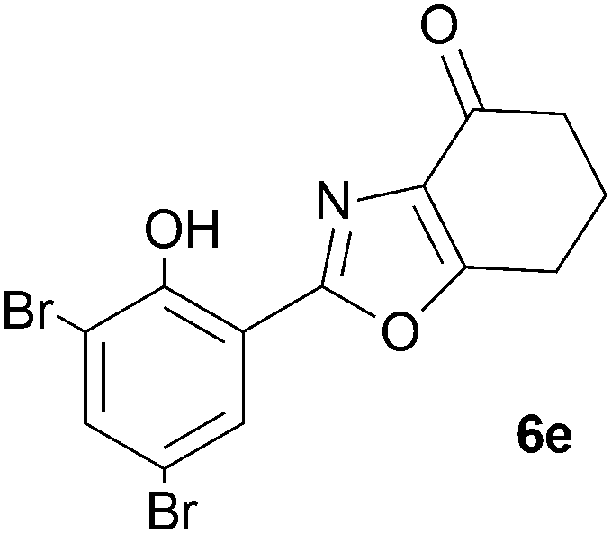
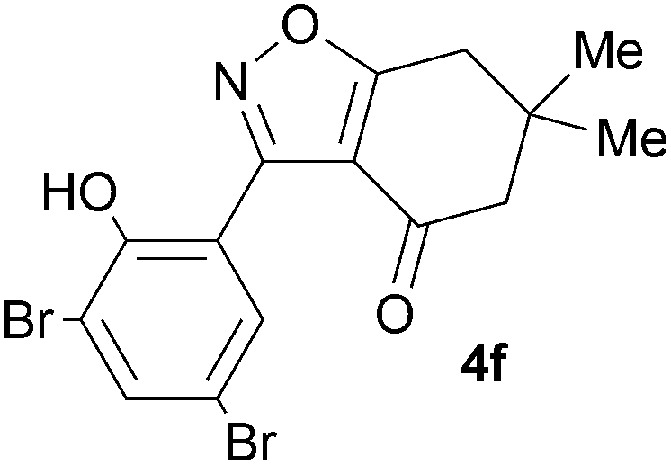
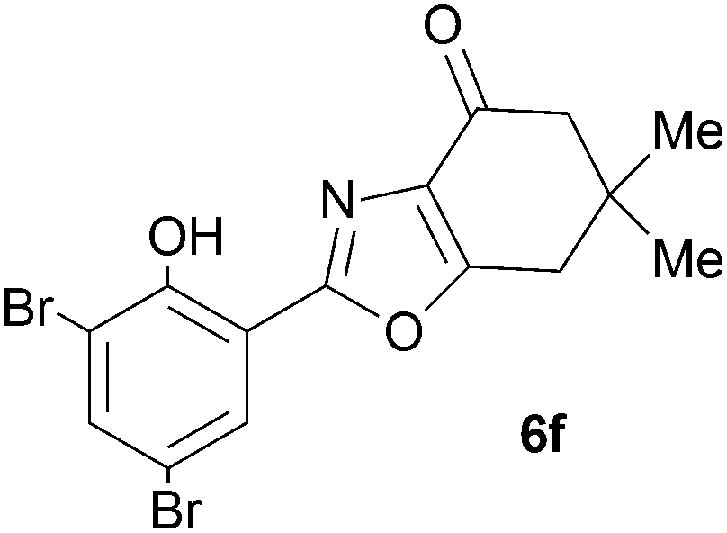
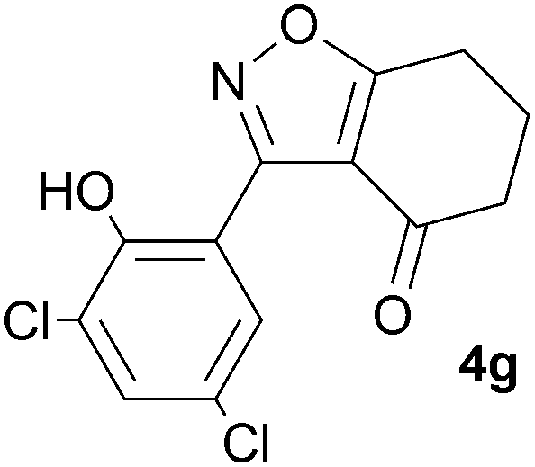
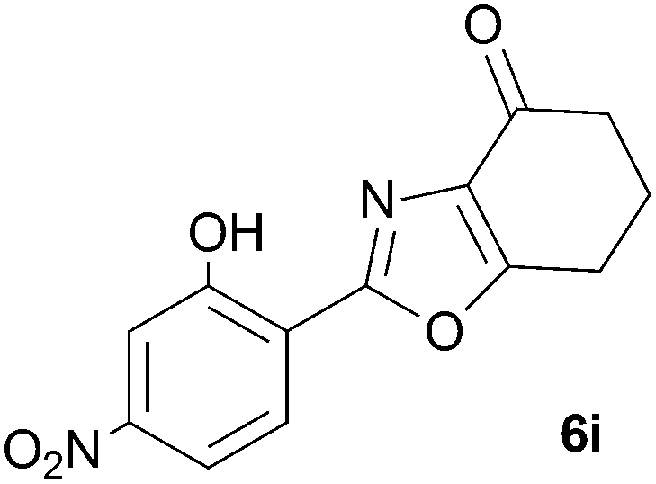
A range of 3-(2-hydroxyphenyl)tetrahydrobenzisoxazoles 4a–i, differently substituted in the aryl and the cyclohexane ring were shown to undergo rearrangement (Table 2) using the convenient Cs2CO3 conditions (THF reflux) to afford oxazoles 6a–i.§


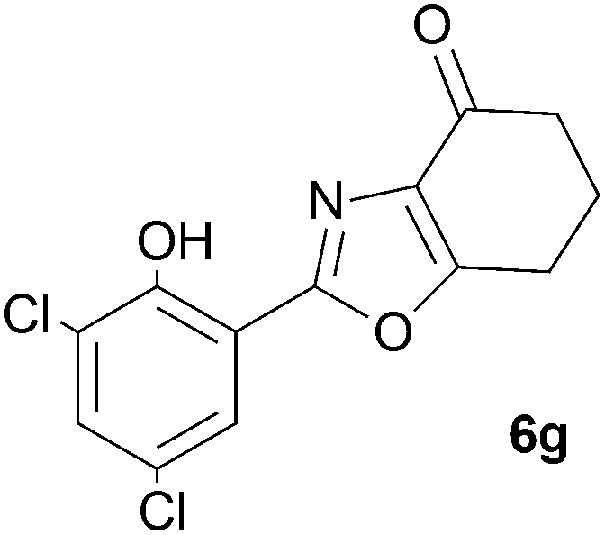
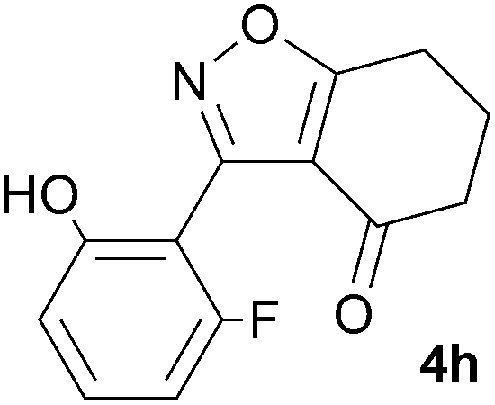
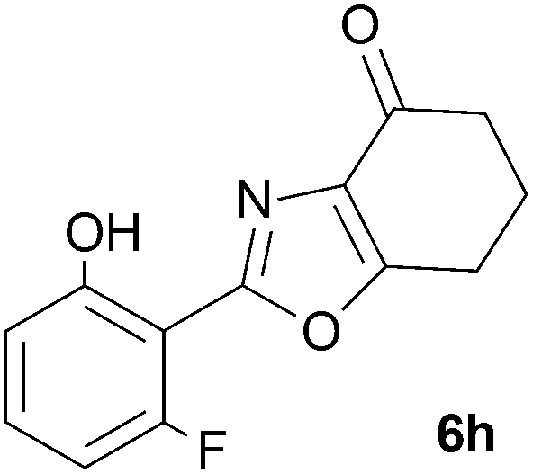
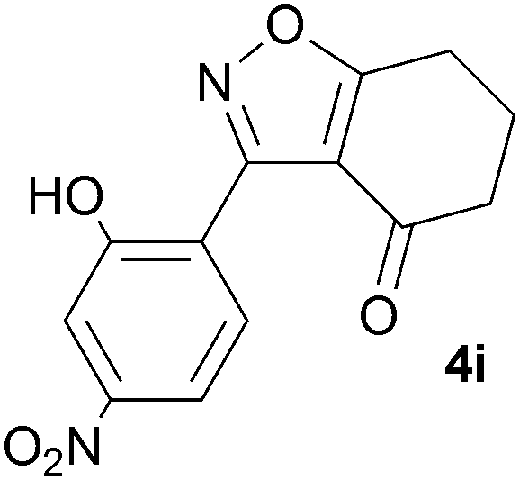
We propose the mechanism illustrated in Scheme 3 for the rearrangement. Until the oxazole structure was determined, we had supposed that a Boulton–Katritzky ring transposition8 (similar to that reported by Suzuki et al.9) was taking place, so we retain this as the initial step in this remarkable isoxazole-to-oxazole conversion.10 This can be followed by a Neber rearrangement11 to give an azirine, thus overall replacing the N–O bond of the isoxazole by an N–C bond. The azirine may be envisaged to be in equilibrium with a nitrile ylide12 stabilised at the formal negative end by the 1,3-dione system, and at the formal positive end by the electron-rich 2-hydroxyphenyl substituent. The 1,3-dipole finally collapses to the oxazole in a 6π electrocyclic ring closure.
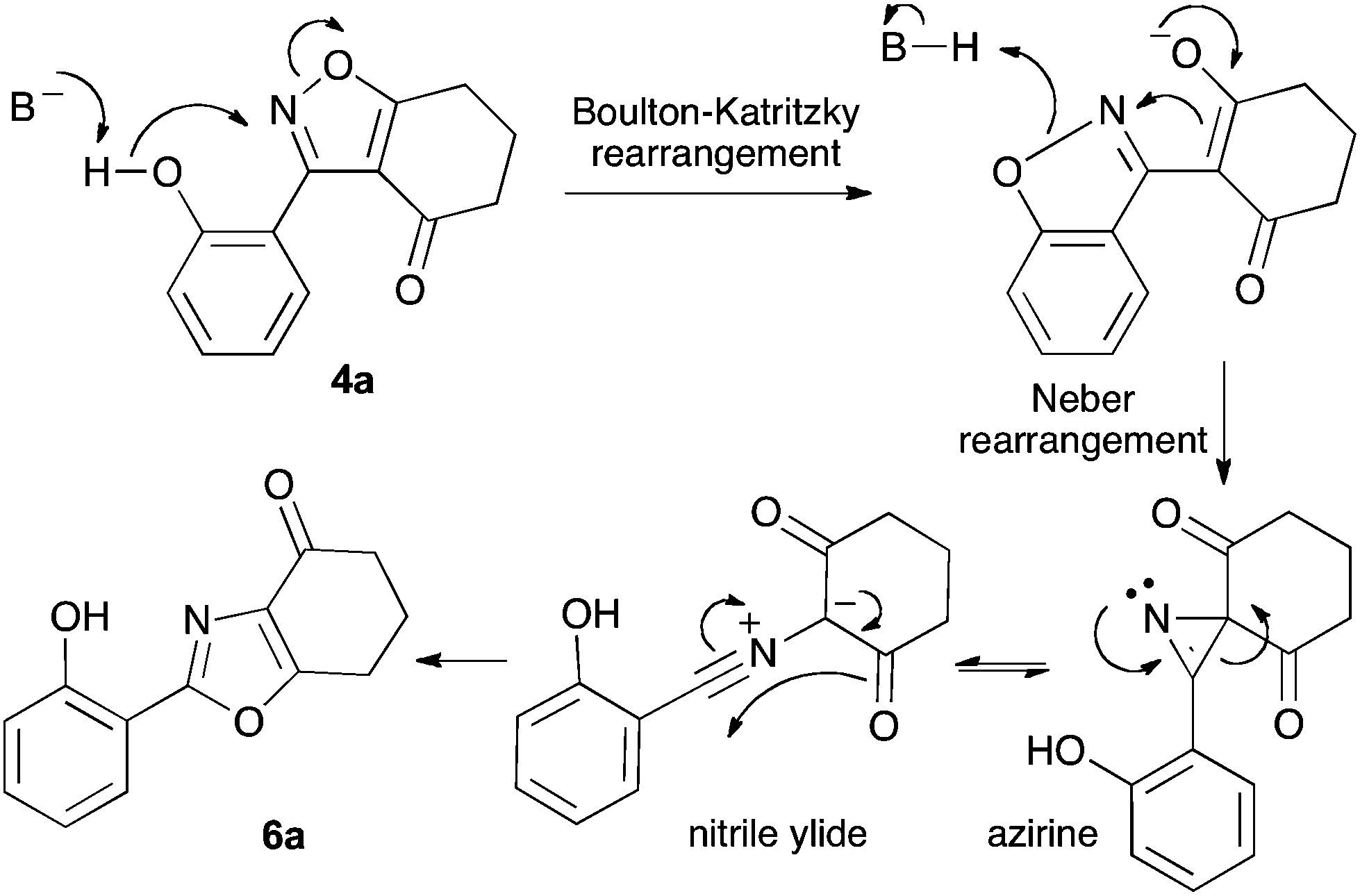 | ||
| Scheme 3 Proposed mechanism for isoxazole–oxazole rearrangement. | ||
Previous reports indicate that it is possible to form oxazoles from azirines, and also that an azirine can be generated from an isoxazole either thermally or photolytically.13,14 However, the energies required well exceed those of our reaction conditions and thus an alternative rationale was required. The Neber rearrangement is an alternative way of generating azirines given the appropriate leaving group.15 This mechanism implies that the base is catalytic, and this was supported by isolation of 6a (66%) from 4a using 0.1 mol equiv. of Cs2CO3 (THF reflux, 1.5 h). An intermediate with m/z identical to both the isoxazole and oxazole was observed by LC-MS during the rearrangements of 4a and 4c to 6a,c, respectively, and isolated by HPLC. We were not able to unambiguously identify the structure, but NMR studies indicate the cyclohexane portion to be symmetrical, supporting either the azirine or nitrile ylide formulation.16 An attempt to crystallise the dimethyl intermediate formed from 4c led merely to recovery of the oxazole 6c. The oxazole ring opening to form amides 7a,b is consistent with nucleophilic attack at C-5 of the oxazole.
To discount the possibility of the oxazoles being formed by retro-cycloaddition from the isoxazoles and recombination via a different connectivity, we have shown that treatment of a mixture of the two tetrahydrobenzisoxazoles 4c and 4i under the Cs2CO3–THF reflux conditions led only to the tetrahydrobenzoxazoles 6c and 6i predicted by the mechanism of Scheme 3, with no crossover products observed.
The tetrahydrobenzoxazoles prepared herein are closely related to a series of herbicides described in a patent by Ueda et al.17 Using benzoxazole 6a we have prepared an example 8 of this group by reaction with 2-chloropyrimidine (47%) (Scheme 4).
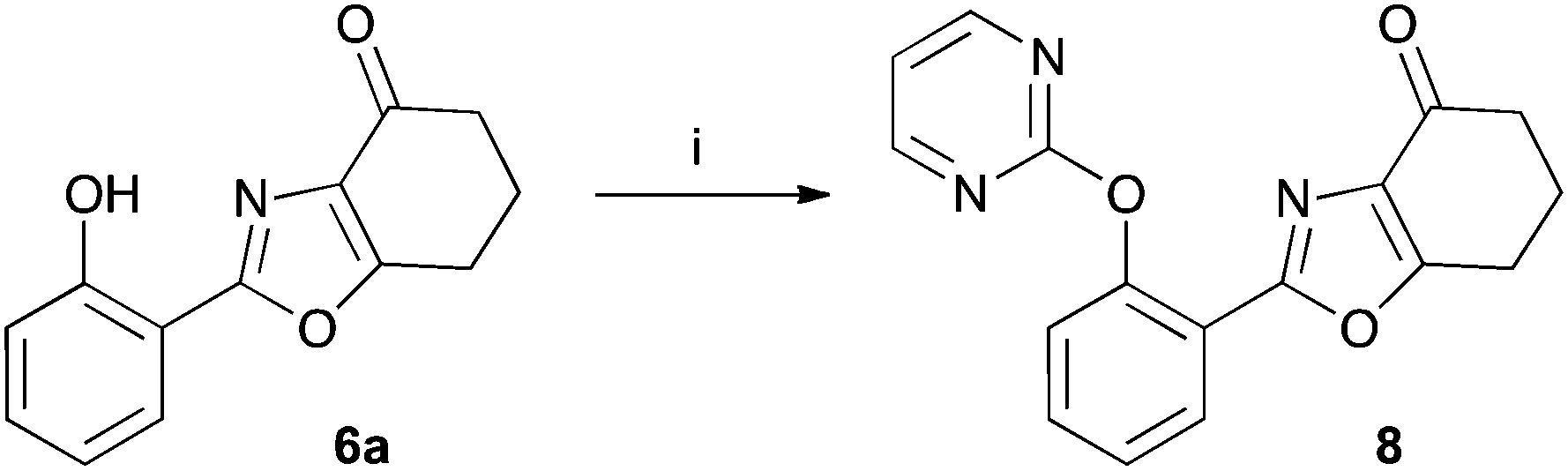 | ||
| Scheme 4 Reagents: (i), 2-chloropyrimidine, Cs2CO3, Cu, dry DMF, 1 day. | ||
In conclusion, we have discovered an unexpected, remarkably facile novel base-mediated rearrangement of tetrahydro-benzisoxazoles to tetrahydrobenzoxazoles, demonstrated the scope and probed the reaction mechanism of this surprising transformation. The synthetic utility of this rearrangement has been demonstrated by synthesis of a known bioactive compound.
The authors thank Loughborough University for a studentship (A. C.) and Lilly UK for financial support, and ENSIACET (Toulouse) for support for a work placement (R. M.).
Notes and references
- 3-Acyltetramic acids. For leading references, see: R. C. F. Jones and T. A. Pillainayagam, Synlett, 2004, 2815–2817 CrossRef CAS; R. C. F. Jones, C. C. M. Law and M. R. J. Elsegood, ARKIVOC, 2013, iii, 81–97 Search PubMed.
- 3-Acyl-4-hydroxypyridones. For leading references, see: R. C. F. Jones, A. K. Choudhury, J. N. Iley, M. E. Light, G. Loizou and T. A. Pillainayagam, Beilstein J. Org. Chem., 2012, 8, 308–312 CrossRef CAS PubMed; R. C. F. Jones, A. K. Choudhury, C. E. Dawson, C. Lumley and V. McKee, ARKIVOC, 2012, vii, 12–24 Search PubMed.
- For a total synthesis, see: K. C. Nicolaou, T. Montagnon, G. Vassilikogiannakis and C. J. N. Mathison, J. Am. Chem. Soc., 2005, 127, 8872–8888 CrossRef CAS PubMed.
- K. E. Wilson, N. N. Tsou, Z. Guan, C. L. Ruby, F. Pelaez, J. Gorrochategui, F. Vicente and H. R. Onishi, Tetrahedron Lett., 2000, 41, 8705–8709 CrossRef CAS.
- T. Kamigakinai, M. Nakashima and H. Tani, Japanese Pat., JP 10101666 A2, 1998 Search PubMed; T. Kamigaichi, M. Nakashima and H. Tani, Japanese Pat., JP 11158109 A2, 1999 Search PubMed.
- J. W. Bode, Y. Hachisu, T. Matsuura and K. Suzuki, Org. Lett., 2003, 5, 391–394 CrossRef CAS PubMed.
- Crystallographic data for amides 7a and 7b are deposited at the Cambridge Crystallographic Data Centre, CCDC 962974, 962975 respectively.
- For leading references, see: A. Palumbo Piccionello, S. Buscemi, N. Vivona and A. Pace, Org. Lett., 2010, 12, 3491–3493 CrossRef CAS PubMed; A. J. Boulton, A. R. Katritzky and A. M. Hamid, J. Chem. Soc. C, 1967, 2005–2007 RSC.
- J. W. Bode, H. Uesuka and K. Suzuki, Org. Lett., 2003, 5, 395–398 CrossRef CAS PubMed.
- Studies with the 3-phenyltetrahydrobenzisoxazole confirm the requirement for the phenolic substituent: unpublished observations.
- P. W. Neber and A. V. Friedolsheim, Justus Liebigs Ann. Chem., 1926, 449, 109–134 CrossRef CAS . For an application in synthesis, see: N. Garg, D. D. Caspi and B. M. Stoltz, J. Am. Chem. Soc., 2004, 126, 9552–9553 CrossRef PubMed.
- Cf. S. Lopes, C. M. Nunes, A. Gomez-Zavaglia and T. M. V. D. Pinho de Melo, Tetrahedron, 2011, 67, 7794–7804 CrossRef CAS.
- See, for example: I. J. Turchi and M. J. S. Dewar, Chem. Rev., 1974, 75, 389–437 CrossRef CAS; K. K. Zhigulev and M. A. Panina, Khim. Geterotsikl. Soedin., 1974, 4, 457–460 Search PubMed; M. Maeda and M. Kojima, J. Chem. Soc., Perkin Trans. 1, 1977, 239–247 RSC; S. M. Fonseca, H. D. Burrows, C. M. Nunes and T. M. V. D. Pinho de Melo, Chem. Phys. Lett., 2009, 474, 84–87 CrossRef; C. M. Nunes, I. Reva, T. M. V. D. Pinho de Melo, R. Fausto, T. Solomek and T. Bally, J. Am. Chem. Soc., 2011, 133, 18911–18923 CrossRef PubMed.
- For a base-catalysed isoxazole–oxazole transformation in which the oxazole ring atoms are not all derived from the isoxazole: G. Doleschall and P. Seres, J. Chem. Soc., Perkin Trans. 1, 1988, 1875–1879 RSC.
- A. Padwa, in Comprehensive Heterocyclic Chemistry III, ed. A. R. Katritzky, C. A. Ramsden, E. F. V. Scriven and R. J. K. Taylor, Elsevier, Oxford, 2008, vol. 1, pp. 94–95 Search PubMed.
- Selected spectral data for the intermediate in rearrangement of 4a to 6a: δH (500 MHz; CDCl3) 2.05–2.15 (m, 2H, CH2CH2CH2), 2.67 (4H, t, J = 6.4, 2 × CH2CO), 7.30–7.33 (1H, m, Ar–H), 7.52–7.60 (2H, m, 2 × Ar–H), 8.21 (1H, d, J = 8.2, Ar–H), 11.18 (s, 1H, OH).
- A. Ueda, Y. Miyazawa, Y. Hara, M. Koguchi, A. Takahashi and T. Kawana, US Pat., US 6268310 B1, 2001 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 962972–962975. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc07999j |
‡ Crystal data for 4a: C13H11NO3, M = 229.23, orthorhombic, Pca21, a = 15.024(3) Å, b = 21.464(4) Å, c = 13.309(3) Å, V = 4291.8(15) Å3, Z = 16, μ(Mo-Kα) = 0.102 mm−1, 36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 812 reflections measured, 8811 unique, Rint = 0.064, R1[for 5081 data with F2 > 2σ(F2)] = 0.056, wR2 (all data) = 0.168, absolute structure x = −0.4(19). Four molecules in asymmetric unit. For 6b: C15H15NO3, M = 257.28, orthorhombic, Pna21, a = 12.930(2) Å, b = 9.3159(15) Å, c = 21.663(4) Å, V = 2609.4(8) Å3, Z = 8, μ(Mo-Kα) = 0.09 mm−1, 25390 reflections measured, 6484 unique, Rint = 0.034, R1[for 5459 data with F2 > 2σ(F2)] = 0.035, wR2 (all data) = 0.088, absolute structure x = 0.2(4). Two molecules in asymmetric unit. CCDC 962972 and 962973. 812 reflections measured, 8811 unique, Rint = 0.064, R1[for 5081 data with F2 > 2σ(F2)] = 0.056, wR2 (all data) = 0.168, absolute structure x = −0.4(19). Four molecules in asymmetric unit. For 6b: C15H15NO3, M = 257.28, orthorhombic, Pna21, a = 12.930(2) Å, b = 9.3159(15) Å, c = 21.663(4) Å, V = 2609.4(8) Å3, Z = 8, μ(Mo-Kα) = 0.09 mm−1, 25390 reflections measured, 6484 unique, Rint = 0.034, R1[for 5459 data with F2 > 2σ(F2)] = 0.035, wR2 (all data) = 0.088, absolute structure x = 0.2(4). Two molecules in asymmetric unit. CCDC 962972 and 962973. |
| § Typical procedure for oxazole formation: 3-(2-hydroxyphenyl)-6,7-dihydrobenzo[d]isoxazol-4(5H)-one 4a (0.500 g, 2.18 mMol) and Cs2CO3 (1.42 g, 4.37 mMol) in dry THF (30.0 mL) was heated under reflux for 4 h. Hydrochloric acid (2 M; 5 mL) and CH2Cl2 (25 mL) were added after the reaction mixture had cooled to 20 °C. The mixture was separated and the combined organic layer washed with water (2 × 25 mL) and brine (25 mL). The organic layer was dried (MgSO4), filtered and evaporated to dryness under reduced pressure to yield 2-(2-hydroxyphenyl)-6,7-dihydrobenzo[d]oxazol-4(5H)-one 6a (0.435 g, 87%) as a beige solid, mp 202–204 °C (decomp.); νmax(CH2Cl2)/cm−1 3804, 1694; δH (400 MHz; CDCl3) 2.20–2.27 (2H, m, CH2), 2.57 (2H, t, J = 5.6, OxCH2), 3.00 (2H, t, J = 6.0, CH2CO), 6.86–6.90 (1H, m, Ar–CH), 7.10 (1H, dd, J = 0.8, 8.4, Ar–CH), 7.30–7.34 (1H, m, Ar–CH), 7.74 (1H, dd, J = 1.6, 8.0, Ar–CH) 10.58 (1H, br s, OH); δC (100 MHz; CDCl3) 22.2, 37.9 (CH2), 110.0 (C), 117.6, 119.5, 126.3, 133.2 (Ar–CH), 133.7, 157.7, 161.2, 163.0 (C), 190.7 (CO). HRMS: MH+ 230.0809; C13H11NO3 requires MH+ 229.0812. |
| This journal is © The Royal Society of Chemistry 2015 |