DOI:
10.1039/C3BM60161G
(Paper)
Biomater. Sci., 2013,
1, 1037-1045
Controllably degradable β-sheet nanofibers and gels from self-assembling depsipeptides†
Received
28th June 2013
, Accepted 25th July 2013
First published on 13th August 2013
Abstract
Self-assembled peptide materials have received considerable interest for a range of applications, including 3D cell culture, tissue engineering, and the delivery of cells and drugs. One challenge in applying such materials within these areas has been the extreme stability of β-sheet fibrillized peptides, which are resistant to proteolysis, degradation, and turnover in biological environments. In this study, we designed self-assembling depsipeptides containing ester bonds within the peptide backbone. Beta-sheet fibrillized nanofibers were formed in physiologic conditions, and two of these nanofiber-forming depsipeptides produced hydrogels that degraded controllably over the course of days-to-weeks via ester hydrolysis. With HPLC, TEM, and oscillating rheometry, we show that the rate of hydrolysis can be controlled in a straightforward manner by specifying the amino acid residues surrounding the ester bond. In 3D cell cultures, depsipeptide gels softened over the course of several days and permitted considerably more proliferation and spreading of C3H10T1/2 pluripotent stem cells than non-degradable analogs. This approach now provides a reliable and reproducible means to soften or clear β-sheet fibrillized peptide materials from biological environments.
Introduction
Since being introduced by Zhang in the 1990's, β-sheet fibrillizing peptides have received significant interest for a wide range of applications, including matrices for cell culture,1–6 tissue engineering,7–10 the delivery of cells and therapeutics,11–14 and vaccines.15–17 Self-assembled peptides are useful for such applications owing to their stimulus-sensitive assembly, their chemical definition, their generally good biocompatibility, and their ability to be directly appended with peptide ligands or epitopes.5,18,19 Collectively these properties enable self-assembled peptides to be used as modular scaffolds for cells or immunotherapies, where the content of many different functional components can be systematically and independently adjusted.6 Several other platforms that make use of β-rich fibril assembly have also been developed, including Fmoc-peptides,20–22 β-hairpins,13,23,24 peptide-amphiphiles,25–28 and multidomain peptides.29–31
In most of the applications listed above, the persistence, degradation, and turnover of the biomaterial are critical parameters. Matrix turnover in 3D culture is essential for permitting proliferation and morphogenic processes;32 controlled degradation has been a longstanding focus of tissue engineering approaches;33,34 and vaccine platforms can benefit from the predictable clearance of biomaterial components from a pharmacokinetic perspective. Despite these significant motivations, however, self-assembled peptide materials have largely remained resistant to degradation throughout their development. In early studies, Zhang and colleagues found that the peptide (AEAEAKAK)2 was highly resistant to proteolysis by enzymes such as α-chymotrypsin, trypsin, and papain,35 even though the peptide contained amino acid sequences expected to be cleaved by these enzymes. This extreme stability of β-sheet fibrils is also well known, both in amyloid biology and in the applied area of amyloid engineering.36,37 In engineered materials, several groups have addressed this problem by installing peptide sequences that can be cleaved by matrix metalloproteinases (MMPs). This has been accomplished in β-sheet fibrillizing peptides,38–40 β-hairpins,41 and peptide-amphiphiles.42 Although such an approach offers a much-needed route for degrading β-sheet fibrillar materials, it can be difficult to control or predict the expression of MMPs in specific culture conditions or in vivo applications.
As an alternative to MMP-sensitive materials, hydrolytically susceptible materials have received tremendous attention, owing to their relatively predictable degradation behavior, which is not dependent on controlling or knowing the concentration and activity of specific enzymes.34,43–45 The degradation of ester-containing polymers and materials is dependent on factors such as pH and the accessibility of the ester bond to water, which are more easily controlled in biological systems than enzyme action.46–49 Surprisingly, despite the common use of ester hydrolysis in biomaterials for tissue engineering, it has not, to our knowledge, been employed to soften β-sheet fibrillar peptide hydrogels over time. Instead, ester substitutions in β-sheet fibrillar peptides have largely been made to study hydrogen bonding patterns or to develop structure-breaking strategies.50–52 In most of these previous studies, ester substitution strongly disrupted fibrillization and gelation. For example, Gordon and Meredith found that depsipeptide analogs of Aβ16-20, in which two of the five amide bonds were switched to esters, were incapable of self-assembling.50 These depsipeptides also inhibited the aggregation of Aβ1-40 peptide, and even disassembled preformed Aβ1-40. Liskamp and colleagues, in studies of amylin (islet amyloid polypeptide, IAPP), found that one amide → ester substitution at a key residue was sufficient to dramatically postpone gelation and inhibit fibril formation.51 In separate studies, they also observed depsipeptides that were not capable of forming classical β-sheet fibrils, yet interestingly assembled into helical ribbon structures that were capable of producing hydrogels.52,53
Here, we report depsipeptides that are capable of self-assembling into β-sheet fibrillar materials and that also degrade via ester hydrolysis with rates controllable by the amino acid proximal to the ester bonds. Designs were based on the Q11 peptide sequence, which our group has studied extensively in the past, and which forms reproducible peptide nanofibers and gels useful for cell culture5,6,14 and vaccine platforms.15,17 Using TEM, HPLC, circular dichroism (CD), mass spectrometry, and oscillating rheometry, we monitored assembly and degradation in a variety of conditions, and we found that hydrolytic softening permitted the proliferation and spreading of pluripotent cells to a much greater degree than non-degrading analogs.
Results and discussion
Peptide design and synthesis
To engineer degradable self-assembling peptide nanofibers and hydrogels, we investigated a set of four depsipeptides derived from a self-assembling peptide previously reported by our group, Q11 (Ac-QQKFQ![[F with combining low line]](https://www.rsc.org/images/entities/char_0046_0332.gif) QFEQQ-Am).4 Depsipeptides were produced using Fmoc-based solid phase peptide synthesis, substituting the central phenylalanine (Phe6, underlined) with α-hydroxy acids, as described in the methods. Phe6 was chosen as the substitution site due to its central position; we hypothesized a substitution at this site would minimally disrupt β-sheet fibrillization, yet would still lead to fiber disassembly and degradation upon hydrolysis by generating fragments that would not aggregate. We further hypothesized that degradation rates could be controlled by adjusting the hydrophobicity and steric bulk of the side chain proximal to the ester bond, in an analogous fashion to biodegradable polyesters.54,55 Therefore, four depsipeptides were produced using four different α-hydroxy acids: glycolic acid, lactic acid, 2-hydroxycaproic acid, and 3-phenyllactic acid. These analogs of Gly, Ala, Leu, and Phe produced four Depsi-Q11s designated as Depsi-G, Depsi-A, Depsi-L and Depsi-F, respectively (Fig. 1), with Depsi-G having the smallest and least hydrophobic side chain, and Depsi-L and Depsi-F having the largest and most hydrophobic side chains. A control peptide was also synthesized with a Phe → Leu substitution at Phe6, but no ester linkage, and was designated L-Q11. Depsi-Q11s were synthesized with good yields and purified to >99% by reverse phase HPLC (about 45% yield crude, 20% yield pure). MALDI-TOF was used to confirm the depsipeptides’ identity (Table 1). All depsipeptides were soluble in water, with solubility limits between 10–30 mM, somewhat less than Q11 (60 mM).
QFEQQ-Am).4 Depsipeptides were produced using Fmoc-based solid phase peptide synthesis, substituting the central phenylalanine (Phe6, underlined) with α-hydroxy acids, as described in the methods. Phe6 was chosen as the substitution site due to its central position; we hypothesized a substitution at this site would minimally disrupt β-sheet fibrillization, yet would still lead to fiber disassembly and degradation upon hydrolysis by generating fragments that would not aggregate. We further hypothesized that degradation rates could be controlled by adjusting the hydrophobicity and steric bulk of the side chain proximal to the ester bond, in an analogous fashion to biodegradable polyesters.54,55 Therefore, four depsipeptides were produced using four different α-hydroxy acids: glycolic acid, lactic acid, 2-hydroxycaproic acid, and 3-phenyllactic acid. These analogs of Gly, Ala, Leu, and Phe produced four Depsi-Q11s designated as Depsi-G, Depsi-A, Depsi-L and Depsi-F, respectively (Fig. 1), with Depsi-G having the smallest and least hydrophobic side chain, and Depsi-L and Depsi-F having the largest and most hydrophobic side chains. A control peptide was also synthesized with a Phe → Leu substitution at Phe6, but no ester linkage, and was designated L-Q11. Depsi-Q11s were synthesized with good yields and purified to >99% by reverse phase HPLC (about 45% yield crude, 20% yield pure). MALDI-TOF was used to confirm the depsipeptides’ identity (Table 1). All depsipeptides were soluble in water, with solubility limits between 10–30 mM, somewhat less than Q11 (60 mM).
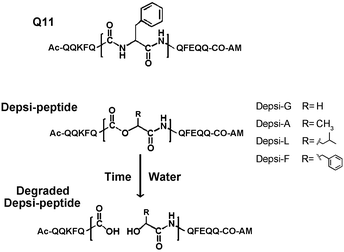 |
| | Fig. 1 Depsipeptide analogs of the self-assembling peptide Q11. The central Phe in Q11, shown expanded in the figure, was replaced with various α-hydroxy acids to generate Depsi-Q11s containing ester bonds. Over time, these peptides degraded by hydrolysis of the ester bond. | |
| Name |
Sequence |
[M + H]+m/z calc'd |
[M + H]+m/z found |
Solubility limit in water |
|
Ester bonds were produced between residues underlined in bold. m/z values were determined with MALDI-TOF mass spectrometry.
|
| Q11 |
QQKFQFQFEQQ |
1527.7 |
1528.3 |
60 mM |
| Depsi-G |
QQKF![[Q with combining low line]](https://www.rsc.org/images/entities/b_char_0051_0332.gif) ![[G with combining low line]](https://www.rsc.org/images/entities/b_char_0047_0332.gif) QFEQQ QFEQQ |
1438.5 |
1438.3 |
10 mM |
| Depsi-A |
QQKF![[A with combining low line]](https://www.rsc.org/images/entities/b_char_0041_0332.gif) QFEQQ QFEQQ |
1452.6 |
1451.8 |
15 mM |
| Depsi-L |
QQKF![[L with combining low line]](https://www.rsc.org/images/entities/b_char_004c_0332.gif) QFEQQ QFEQQ |
1494.7 |
1494.2 |
30 mM |
| L-Q11 |
QQKFQLQFEQQ |
1493.7 |
1492.9 |
60 mM |
| Depsi-F |
QQKF![[F with combining low line]](https://www.rsc.org/images/entities/b_char_0046_0332.gif) QFEQQ QFEQQ |
1528.7 |
1528.4 |
15 mM |
Initial characterization of fibrillization and degradation
We used transmission electron microscopy (TEM) to analyze the Depsi-Q11 peptides’ capacity to self-assemble into nanofibers, and for these fibers to degrade over time. Peptides and depsipeptides were dissolved in water at 4 mM, incubated overnight at room temperature (pH about 3), and subsequently diluted to 2 mM in PBS (see methods), during which no observable degradation occurred (see below). TEM was performed after 4 hours at final dilution, and at this time all four Depsi-Q11s formed clearly observable nanofibers (Fig. 2) that were morphologically similar to previously described Q11 nanofibers (ESI, Fig. S2†).4,15 Although some degradation may have occurred at this early time point (4 hours, as was later confirmed by HPLC, Fig. 3), many nanofibers were clearly visible for all groups. As an initial measure of degradation, these nanofibers were incubated at 37 °C for 7–14 days before imaging by TEM. By day 7, noticeably fewer fibers were observed in the Depsi-G and Depsi-A solutions (Fig. 2). Depsi-L fibers appeared to be generally shorter and thinner after 7 days, while Depsi-F fibers were not significantly different. Thus, the smaller and less hydrophobic side chains were correlated with a more rapid degradation of fibers. By 14 days, no intact fibers were observed for any Depsi-Q11s, indicating that all nanoscale assemblies were completely degraded.
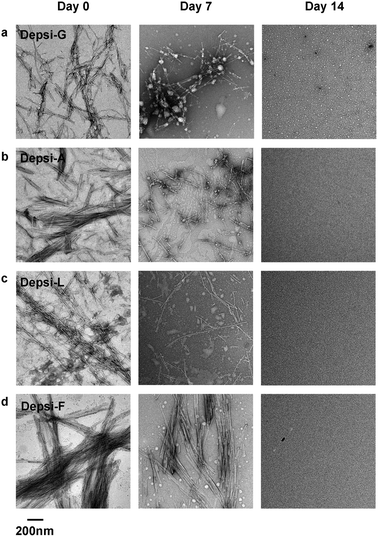 |
| | Fig. 2 In phosphate buffered saline, fibrillar structures were initially produced by Depsi-G, Depsi-A, Depsi-L, and Depsi-F (left column). After 14 days, all Depsi-Q11 assemblies had degraded, and no fibers were evident on TEM grids (right column). Compared to day 0, at intermediate time points (day 7) the remaining assembly state depended on the side chain: significantly smaller and fewer fibers were observed for Depsi-G and Depsi-A (a, b); numerous but shorter and thinner fibers were observed for Depsi-L (c); and numerous thick fibers were still observed for Depsi-F (d). | |
![Quantification of depsipeptide degradation by reverse-phase HPLC. On C18 columns with a water–acetonitrile gradient, Depsi-G eluted as a single peak at day 0 (a, peak 1) but as four peaks after 24 h of degradation at pH 6.5 (a, peaks 1–4, 2 mM peptide concentration). MALDI mass spectrometry confirmed that the peaks were the expected native and degraded species (peak 1, undegraded Depsi-G, m/z calc'd: 1438.5, found: 1438.3; peak 2, HO-GQFEQQ-Am [M + H+], m/z calc'd: 736.8, found: 736.7; peak 3, Ac-QQKFQ-COOH [M + H+], m/z calc'd: 720.8, found: 720.8; peak 4, aminoglutarimide derivative of peak 3, m/z calc'd: 701.8, found: 702.3). Degradation was tracked over time by the ratio of peak areas corresponding to the degraded versus total degraded and undegraded species (b–d). 15 mM Depsi-L gels (b) degraded more slowly than soluble nanofibers (c, d), in a temperature-dependent fashion. The amino acid placed C-terminally to the ester significantly influenced degradation rate, with more hydrophobic residues producing slower degradation (c, d). As expected, these rates were faster at pH 7.4 (d) than at pH 6.5 (c).](/image/article/2013/BM/c3bm60161g/c3bm60161g-f3.gif) |
| | Fig. 3 Quantification of depsipeptide degradation by reverse-phase HPLC. On C18 columns with a water–acetonitrile gradient, Depsi-G eluted as a single peak at day 0 (a, peak 1) but as four peaks after 24 h of degradation at pH 6.5 (a, peaks 1–4, 2 mM peptide concentration). MALDI mass spectrometry confirmed that the peaks were the expected native and degraded species (peak 1, undegraded Depsi-G, m/z calc'd: 1438.5, found: 1438.3; peak 2, HO-GQFEQQ-Am [M + H+], m/z calc'd: 736.8, found: 736.7; peak 3, Ac-QQKFQ-COOH [M + H+], m/z calc'd: 720.8, found: 720.8; peak 4, aminoglutarimide derivative of peak 3, m/z calc'd: 701.8, found: 702.3). Degradation was tracked over time by the ratio of peak areas corresponding to the degraded versus total degraded and undegraded species (b–d). 15 mM Depsi-L gels (b) degraded more slowly than soluble nanofibers (c, d), in a temperature-dependent fashion. The amino acid placed C-terminally to the ester significantly influenced degradation rate, with more hydrophobic residues producing slower degradation (c, d). As expected, these rates were faster at pH 7.4 (d) than at pH 6.5 (c). | |
Adjustment of degradation rate
Reverse-phase HPLC and MALDI-TOF mass spectrometry were next used to more closely measure the degradation of Depsi-Q11 variants at different concentrations, temperatures, and pH (Fig. 3). Stock peptide solutions were incubated overnight in water, then buffered and monitored over time. Immediately after buffering, Depsi-G was present as the un-degraded molecule, as it eluted as a single peak by HPLC (Fig. 3a, peak 1) and had a molecular mass corresponding to full molecule by MALDI (m/z [M + H]+ calc'd: 1438.5, found: 1438.3). After 24 h at pH 7.4, Depsi-G produced four peaks by HPLC, with the three new peaks appearing at shorter retention times (Fig. 3a). Peaks 2 and 3 were determined to be the two predicted hydrolytic degradation products (Peak 2, HO-GQFEQQ-Am [M + H]+m/z calc'd: 736.8, found: 736.7; Peak 3, Ac-QQKFQ-COOH, [M + H]+m/z calc'd: 720.8, found: 720.8). Peak 4 had a molecular mass 18 Da less than the N-terminal Ac-QQKFQ-COOH fragment, which we speculate could represent an aminoglutarimide at the C terminus.56,57 This derivative was also observed in other depsipeptides (not shown). Although we consider alkaline hydrolysis was most likely responsible for the degradation, another mechanism suggested by the product from peak 4 cannot be ruled out: the nitrogen in the side chain of the ester-forming glutamine could have attacked the carbonyl carbon in the ester bond to break the ester bond, thus leaving an aminoglutarimide-containing product. This mechanism could be further probed by substituting the glutamine with another similarly hydrophilic amino acid such as serine.
To measure and express the kinetics of degradation in various contexts, the areas under the three fragment HPLC peaks were added and then divided by the total area under all peptide peaks. First, Depsi-L was prepared at 15 mM in PBS, at which condition it formed a self-supporting hydrogel. Not unexpectedly, the degradation rate was greater at higher temperature; at 37 °C gelled Depsi-L was 50% degraded after 7–8 days, while at 25 °C less than 20% had degraded in the same time (Fig. 3b). Next, 2 mM solutions of all four Depsi-Q11 variants were prepared at two different pH conditions. Depsi-Q11s generally degraded more quickly at 37 °C in this less concentrated solution form compared to the gel form (Fig. 3c and d). In agreement with our hypothesis, Depsi-Q11s with increasingly hydrophobic and bulky side chains degraded progressively more slowly. Specifically, Depsi-G degraded the fastest, followed by Depsi-A, Depsi-L and Depsi-F. For all four variants, the degradation of the peptides in solution was faster at pH 7.4 than at pH 6.5, likely because the hydroxy group is the limiting reagent of basic ester hydrolysis in this pH range. In sum, changing the proximal side chain, pH, or temperature can vary the degradation rate of Depsi-Q11s.
Secondary structure influence of the ester bond
We next used circular dichroism spectroscopy (CD) to evaluate any influence of the ester bond on secondary structure, using Depsi-L, which was the most amenable to CD analysis. Depsi-L exhibited the best solubility and CD transparency in the buffers used, though strong CD absorbances at concentrations approaching 1 mM still prevented low wavelength analysis at these higher concentrations. After being incubated overnight in water at pH 3, then buffered and analyzed within 15 min to minimize any contribution from degraded peptides (see methods), Depsi-L adopted predominantly a random-coil structure at concentrations below 250 μM (Fig. 4a). However, it exhibited a clear conformational change at increased concentrations. At 1 mM, the trace suggested a β-sheet structure (Fig. 4a). For comparison, L-Q11 (Ac-QQKFQLQFEQQ-Am) displayed distinct β-sheet structure at all tested concentrations (Fig. 4b). Compared with the critical concentration of self-assembly for L-Q11 (tens of μM), this CD analysis indicated that the insertion of the ester bond hindered the peptide somewhat from assembling at low concentrations. The hindrance was, however, overcome by increasing the peptide concentration, and at least for Depsi-L, the peptide's secondary structure was likely to be predominantly β-sheet at the relevant working concentrations (2 mM for nanofibers and 15–30 mM for hydrogels).
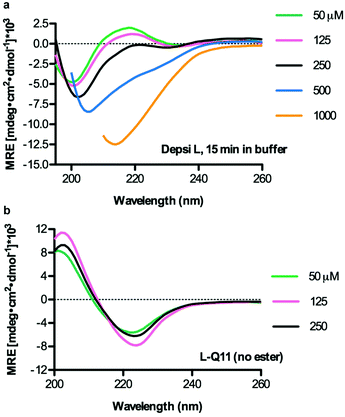 |
| | Fig. 4 Depsi-L adopted a random coil secondary structure at concentrations below 250 μM and assumed β-sheet character at higher concentrations (increasing β-sheet content at 500 μM, with predominant β-sheet structure at 1000 μM) (a). L-Q11 adopted a β-sheet secondary structure that was comparatively independent of concentration (b). Measurements were performed in 10 mM phosphate buffer containing 137 mM KF, pH 7.2–7.4. Data is shown for dynode voltage values below 500 V, reflecting adequate signal-to-noise ratios. | |
Mechanical properties of Depsi-Q11 gels
We employed rheometry to compare the mechanical properties of Depsi-Q11 gels with Q11 gels. We focused on Depsi-L and Depsi-F for this analysis, because the hydrogels formed by Depsi-G and Depsi-A were too fragile to be formed into cylindrical gels capable of being measured by parallel plate oscillating rheometry, even at their highest soluble concentrations. Surprisingly, Depsi-L formed substantially stiffer hydrogels than Q11, with storage moduli (G′) six times greater than Q11 hydrogels of the same concentration (30 mM, Fig. 5a). To probe whether this counterintuitive phenomenon was caused by the leucine side chain, the ester, or a combination of the two, we synthesized the all-amide peptide L-Q11 (Ac-QQKFQLQFEQQ-Am), which lacks the ester present in Depsi-L. L-Q11 at 30 mM formed a hydrogel with a G′ comparable to Q11, indicating that the leucine side chain did not by itself account for Depsi-L's high stiffness. To probe the role of the ester bond, we tested Depsi-F, which differs from Q11 only by the ester bond and from Depsi-L only by the single Leu → Phe substitution. Depsi-F also formed a hydrogel with a storage modulus comparable to Q11 (at 15 mM, the highest soluble concentration of Depsi-F, Fig. 5b). Taken together, it was a synergy between the ester bond and the leucine side chain that appeared to impart surprisingly high stiffness values to the Depsi-L hydrogels.
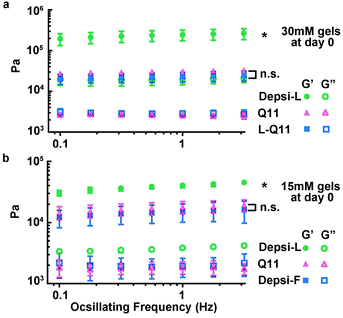 |
| | Fig. 5 Depsi-L produced hydrogels with storage moduli in excess of 100 kPa. At a concentration of 30 mM, the storage moduli of Depsi-L gels were about 6 times greater than Q11 (a). This stiffness arose from the combination of the ester substitution and the Phe → Leu substitution, as the amino acid substitution alone did not produce this effect (a, L-Q11 shown for comparison). Among the depsipeptides studied, only Depsi-L produced this unexpected stiffening; Depsi-F, shown in (b) for comparison, produced gels with moduli similar to Q11. *p < 0.05, ANOVA with Tukey post-hoc test calculated at 1 Hz. | |
We hypothesized that Depsi-Q11 hydrogels would soften over time as hydrolysis occurred. Indeed, the Depsi-L hydrogel's storage and loss moduli decreased significantly over time; by day 3, the G′ and G′′ of Depsi-L had decreased to levels comparable to newly formed Q11 hydrogels (Fig. 6a). The study was terminated after this time because the gels softened to a point where they detached from the substrate and became impossible to measure with rheometry. As a control, L-Q11 hydrogels were also tested; in contrast to the softening depsipeptide, they stiffened by day 3, as has been observed for Q11 and other self-assembling peptides.58 To visualize the process of Depsi-L hydrogel degradation, gels were stained with Congo red, a β-amyloid binding dye (Fig. 7). The 15 mM Depsi-L gel faded and disappeared over the course of 28 days, whereas Q11 hydrogels remained intact for over this time period (Fig. 7a,c,e).
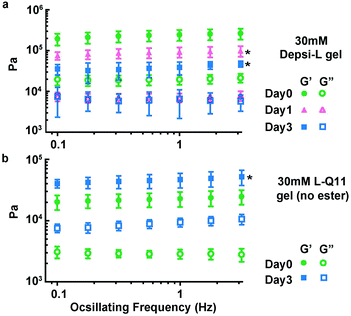 |
| | Fig. 6 Depsi-L gels softened over time as hydrolysis occurred (a), whereas control L-Q11 gels stiffened (b). *p < 0.05, compared to day 0; ANOVA with Tukey post-hoc test (a) or Student's t test (b), calculated at 1 Hz. | |
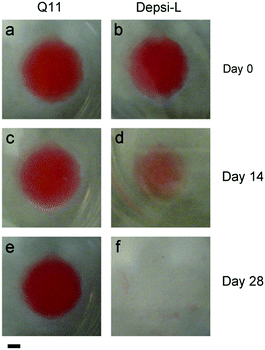 |
| | Fig. 7 Depsi-L formed β-sheet fibrillar hydrogels at 15 mM in phosphate buffered saline, similar to Q11 (gels stained with Congo red on day 0). Q11 gels showed no discernable degradation for up to 28 days (a, c, e), but Depsi-L gels degraded over this time period (b, d, f). Scale bar = 2 mm. Representative of 3 gels. | |
Encapsulation of mouse multipotent stem cells
Q11-based hydrogels have been utilized previously as 2D4–6 and 3D14 matrices for cell culture. We hypothesized that in comparison to Q11 and other fibrillized peptide materials, which are relatively resistant to remodeling and turnover,35–37 Depsi-Q11s would allow more cell invasion by degrading in the presence of encapsulated cells. Depsi-Q11 nanofibers were first found to be non-cytotoxic to C3H10T1/2 mouse pluripotent cells by MTS assay (Fig. S1†). C3H10T1/2 cells were next embedded into Depsi-L or Q11 hydrogels by quickly mixing cell suspensions with peptide solutions and pipetting the mixture into culture media, which effectively buffered the materials to physiological pH. This process resulted in suspended gels approximately 5 mm3 in size with entrapped cells (see methods). Cells encapsulated in Q11 and Depsi-L exhibited a similar survival rate at early timepoints but diverged in their behavior over the next few days (Fig. 8). Cells encapsulated in the Q11 gel were round and did not substantially proliferate over 5 days, whereas cells in the Depsi-L hydrogel exhibited a more spread morphology and proliferated (Fig. 8). Within 3D cell culture or tissue engineering, the degradation rate of a hydrogel should ideally be adjustable to meet the application's demands, ranging from a few weeks to several months.59,60 In the studies reported here, the integrity of the acellular Depsi-L hydrogel could be maintained for about 14 days (Fig. 7b,d). In principle, degradation was primarily a result of chemical hydrolysis, but we cannot rule out the possibility that cell-secreted esterases could also have contributed to degradation. Moreover, it is possible that differences in gel stiffness could account for some of the differences seen in cell behavior. To extend the durability of the gels, it may be possible to co-fibrillize Depsi-L with non-degradable Q11 peptide. In previous work we have shown that different variants of Q11 can be co-assembled into integrated nanofibers to combine more than one functionality.5,15 In this case we speculate that the degradation rate would scale with the depsipeptide/peptide ratio. In summary, we have developed self-assembling depsipeptides that form hydrogels capable of encapsulating cells, and have identified means for controlling the rate of degradation of this otherwise proteolysis-resistant class of materials. Although further optimization is required, we foresee that this technique could be broadly adopted for other common self-assembling peptide-based systems,9,20–31,61 to impart the property of tunable matrix degradation.
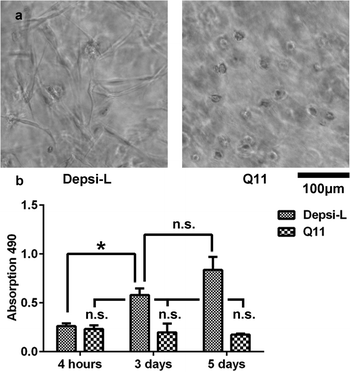 |
| | Fig. 8 C3H10T1/2 cells were encapsulated within Depsi-L and Q11 gels. By 72 hours, cells in Depsi-L gels were more highly spread than in Q11 gels (a, phase contrast). In Depsi-L gels, cells proliferated considerably, whereas little proliferation was observed in Q11 gels (b). *p < 0.05, ANOVA with Tukey's post-hoc test. | |
Conclusions
In this study, self-assembling depsipeptides were designed to produce hydrolytically degradable fibrillar hydrogels. Four Depsi-Q11 peptides were designed, and all retained the ability to fibrillize, while two formed hydrogels. The degradation rates of the four Depsi-Q11s were controlled by the choice of the side chain on the substituted residue and were also affected by environmental conditions such as peptide concentration, pH, and temperature. One variant, Depsi-L, formed a surprisingly stiff hydrogel, which softened and disassembled over time. C3H10T1/2 cells encapsulated in a Depsi-L gel exhibited spreading and significant proliferation, while no spreading or proliferation were observed from cells in a Q11 gel. This type of degradable self-assembling peptide can be readily applied in a variety of fields, including cell encapsulation/delivery or peptide-based vaccine adjuvants.15,17
Experimental
The peptides Q11 (Ac-QQKFQFQFEQQ-Am) and L-Q11 (Ac-QQKFQLQFEQQ-Am) were synthesized on a CS Bio 336X automated peptide synthesizer as previously reported, using standard Fmoc protocols.4 The Depsi-Q11s were similarly synthesized, except for the conjugation of the α-hydroxy acid and the adjacent amino acid, which was achieved by following the method demonstrated by Gordon and Meredith50 with slight modifications. Briefly, QFEQQ-resin (Rink Amide AM resin, NovaBiochem, 855004) was synthesized, N-terminally deprotected, removed from the synthesizer, and rinsed with dichloromethane (DCM, Fisher, D150). The chosen α-hydroxy acid, either glycolic acid (Sigma, 124737), lactic acid (Sigma, L1750), S-(−)-2-hydroxycaproic acid (Aldrich, 219827) or L-(−)-3-phenyllactic acid (Aldrich, 113069), was added to the resin in 3.2-fold molar excess. One equivalent (relative to the α-hydroxy acid) of hydroxybenzotriazole (HOBT, CS Bio, RS-H006), 1.14 equivalents of diisopropylcarbodiimide (DIC, Aldrich D125407), and 0.36 equivalents of N-ethylmorpholine (NEM, Fluka, 04499) were also added to the resin. We note that although NEM was used, it is not strictly necessary unless the peptides are synthesized using Boc chemistry. A 50%/50% (v/v) mixture of DCM and dimethylformamide (DMF, Fisher, BP1160) was used as the solvent. The reaction was stopped after 6 minutes by removing the reaction mixture and washing three times with DCM–DMF, to prevent a second conjugation of the α-hydroxy acid. The completion of the reaction was confirmed with a ninhydrin test. If the reaction did not reach completion, another 6-minute reaction was performed. The conjugation of the following amino acid, which generated the ester bond, was carried out in tetrahydrofuran (THF, Sigma, 401757). A 4-fold molar excess of amino acid was added to the resin. One equivalent (relative to the amino acid) of DIC, 0.55 equivalents of NEM, and 0.022 equivalents of 4-dimethylaminopyridine (DMAP, Aldrich, 522805) were added. The reaction was allowed to proceed for 3 hours, and then the resin was rinsed with DCM, collected, and transferred back to the synthesizer for the remainder of the synthesis. Depsi-Q11s were cleaved with a cocktail containing 95% trifluoroacetic acid (TFA, not anhydrous) and 5% triisopropylsilane (TIS, Acros 214920500), precipitated with diethyl ether, and purified with an Varian ProStar HPLC system, to obtain >99% purity before lyophilization. MALDI-TOF mass spectrometry (Applied Biosystems DE-Pro) was used to confirm peptide identity. Solubility was determined by dilution until the disappearance of gross turbidity.
Nanofiber and hydrogel preparation
Depsi-Q11s were dissolved in water (purified with a Millipore MilliQ system to resistivity >18.1 MΩ) at concentrations between 15–30 mM (for hydrogels) or at 4 mM (for nanofibers), sonicated (bath type, Branson model #3510) and incubated overnight at room temperature. These stock solutions of peptides and depsipeptides had pH values of about 3, and did not exhibit any substantial degradation overnight, as measured by HPLC, described below and in the results. To prepare nanofibers, one volume of 2× Dulbecco's phosphate buffered saline (PBS; Cellgro 20-030-CV) was added to the solution to produce 2 mM final peptide concentration in 1× PBS (0.1 g L−1 CaCl2; 0.2 g L−1 KCl; 0.2 g L−1 KH2PO4; 0.1 g L−1 MgCl2·6H2O; 8 g L−1 NaCl; 1.15 g L−1 Na2HPO4, anhydrous). For TEM, Depsi-Q11s were incubated additionally for 4 hours before analysis. To prepare hydrogels, peptide solutions were either mixed with one volume of 2× PBS (for HPLC), overlaid with an excess of PBS (for rheometry) or mixed with a 0.5 volume cell suspension (for microgel 3D cell culture), as described below.
Peptides and depsipeptides were dissolved in water at 4 mM, incubated overnight at room temperature, and subsequently diluted to 2 mM in PBS. Four hours later, these samples were then applied to a carbon-coated 400 mesh copper grid (FCF 400-Cu, EMS), negatively stained with 1% uranyl acetate, and analyzed with a FEI Tecnai F30 TEM. To monitor nanofiber degradation over time, Depsi-Q11s were incubated at their final dilutions in PBS for 7–14 days at 37 °C before TEM analysis.
HPLC characterization of degradation
Depsi-Q11 nanofibers (2 mM) and hydrogels (15 mM) were prepared as described above. After buffering with PBS, fibers and hydrogels were loaded on the HPLC immediately or were incubated at 25 °C (room temperature) or 37 °C for the indicated times before analysis. To render peptides monomeric for HPLC analysis, 20 μL of the peptide solution or 10 μL of hydrogel was mixed with 80 or 90 μL TFA, respectively. Samples were incubated for 10 min to allow fiber disassembly, and then were analyzed by HPLC using a C18 column (solvent A: water, solvent B: acetonitrile. Gradient: 15–35% in 40 min). The peptide quantity was determined by the absorption at 215 nm. Each data point in Fig. 3b–d represents the mean and standard deviation of 3 separately prepared samples.
Circular dichroism (CD)
Depsi-L and L-Q11 peptide solutions were prepared as described above, except that the 2× phosphate buffer was modified to contain 2 × 137 mM (274 mM) potassium fluoride instead of sodium chloride, to improve CD transparency. After 15 min, the peptide solutions were analyzed with an AVIV 202 Circular Dichroism Spectrometer in a 0.1 cm path length quartz cell. Triplicate scans at 25 °C were averaged.
A Malvern Kinexus Pro was used to measure the storage and loss moduli of hydrogels. After overnight incubation of peptide in water (30 mM or 15 mM), peptide solutions were cast on a glass microscope slide into an 8 mm circular template constructed from filter paper (Fisher, 12-550-143), as described previously.4,58 The peptide solution was then overlaid with PBS to induce gelation. After 2 hours, the PBS was removed, the upper plate was lowered, and the gels were analyzed using frequency sweeps (0.1 Hz to 3.162 Hz) with a constant strain of 0.1%. To measure the effect of degradation on the moduli of hydrogels, Depsi-L and L-Q11 hydrogels were submerged in PBS and incubated at 37 °C for 1 day or 3 days before analysis by rheometry. Each data point in Fig. 5 and 6 represents the mean and standard deviation of 3–5 separately prepared samples. The harmonic distortion of each data point was maintained at less than 10%.
Microscale visualization of degradation
To study the macroscopic degradation of Depsi-Q11 gels, 50 μM Congo red was incorporated into the aqueous peptide solution during the preparation of 15 mM Depsi-L and Q11 hydrogels. Peptide solutions were pipetted into multi-well plastic dishes, immersed in PBS at 37 °C, and photographed over 28 days.
3D cell culture
C3H10T1/2 cells were maintained with Eagle's Basal Medium supplemented with 10% FBS, 1× L-glutamine, 1 mM sodium pyruvate and 1× non-essential amino acids. For 3D culture, the cells were trypsinized and resuspended with 10% sucrose containing 180 mg mL−1 penicillin, 300 mg mL−1 streptomycin, and 0.75 mg mL−1 amphotericin B at a cell density of 6000 cells per μL. 10 μL of the cell suspension was then added to 20 μL of aqueous peptide solution (22.5 mM peptide) and was quickly mixed. Twenty microliters of mixture, containing 2000 cells per μL, was pipetted into culture media in 4–5 μL increments to induce gelation in the form of droplets. Encapsulated cells were imaged with a Zeiss Axioskop at day 3. An MTS assay was applied at 4 hours, 3 days and 5 days, as previously reported.4–6 Each data point represents the average and standard deviation of 3 wells of microgel culture, each containing 20 μL total volume of microgels.
Statistical analysis
Error bars represent standard deviations. For rheometry, G′ and G′′ measured at a frequency of 1 Hz were used to test for differences between groups. For rheometry and MTS assays, ANOVA with Tukey's post-hoc test was performed for comparisons between 3 or more groups, and a t-test was used for comparisons between 2 groups. The cutoff for statistical significance was p < 0.05.
Acknowledgements
This research was supported by the National Institutes of Health (NIBIB, 1R01EB009701; NCI, U54 CA151880), the National Science Foundation (CHE-0802286), and the Chicago Biomedical Consortium with support from the Searle Funds at The Chicago Community Trust. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH or the Chicago Biomedical Consortium. The authors have no conflicting financial interests. Circular dichroism was performed at the Biophysics Core Facility, and TEM was performed at the Electron Microscopy Core facility, both at the University of Chicago.
References
- M. E. Davis, P. C. Hsieh, T. Takahashi, Q. Song, S. Zhang, R. D. Kamm, A. J. Grodzinsky, P. Anversa and R. T. Lee, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8155–8160 CrossRef CAS.
- V. Jayawarna, S. M. Richardson, A. R. Hirst, N. W. Hodson, A. Saiani, J. E. Gough and R. V. Ulijn, Acta Biomater., 2009, 5, 934–943 CrossRef CAS.
- Y. Nagai, H. Yokoi, K. Kaihara and K. Naruse, Biomaterials, 2012, 33, 1044–1051 CrossRef CAS.
- J. P. Jung, J. L. Jones, S. A. Cronier and J. H. Collier, Biomaterials, 2008, 29, 2143–2151 CrossRef CAS.
- J. P. Jung, A. K. Nagaraj, E. K. Fox, J. S. Rudra, J. M. Devgun and J. H. Collier, Biomaterials, 2009, 30, 2400–2410 CrossRef CAS.
- J. P. Jung, J. V. Moyano and J. H. Collier, Integr. Biol., 2011, 3, 185–196 RSC.
- J. Kisiday, M. Jin, B. Kurz, H. Hung, C. Semino, S. Zhang and A. J. Grodzinsky, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 9996–10001 CrossRef CAS.
- M. E. Davis, J. P. M. Motion, D. A. Narmoneva, T. Takahashi, D. Hakuno, R. D. Kamm, S. G. Zhang and R. T. Lee, Circulation, 2005, 111, 442–450 CrossRef CAS.
- T. C. Holmes, S. de Lacalle, X. Su, G. S. Liu, A. Rich and S. G. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 6728–6733 CrossRef CAS.
- F. Gelain, S. Panseri, S. Antonini, C. Cunha, M. Donega, J. Lowery, F. Taraballi, G. Cerri, M. Montagna, F. Baldissera and A. Vescovi, ACS Nano, 2011, 5, 227–236 CrossRef CAS.
- M. Yolamanova, C. Meier, A. K. Shaytan, V. Vas, C. W. Bertoncini, F. Arnold, O. Zirafi, S. M. Usmani, J. A. Muller, D. Sauter, C. Goffinet, D. Palesch, P. Walther, N. R. Roan, H. Geiger, O. Lunov, T. Simmet, J. Bohne, H. Schrezenmeier, K. Schwarz, L. Standker, W. G. Forssmann, X. Salvatella, P. G. Khalatur, A. R. Khokhlov, T. P. J. Knowles, T. Weil, F. Kirchhoff and J. Munch, Nat. Nanotechnol., 2013, 8, 130–136 CrossRef CAS.
- N. Wiradharma, Y. W. Tong and Y. Y. Yang, Biomaterials, 2009, 30, 3100–3109 CrossRef CAS.
- L. Haines-Butterick, K. Rajagopal, M. Branco, D. Salick, R. Rughani, M. Pilarz, M. S. Lamm, D. J. Pochan and J. P. Schneider, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 7791–7796 CrossRef CAS.
- Y. F. Tian, J. M. Devgun and J. H. Collier, Soft Matter, 2011, 7, 6005–6011 RSC.
- J. S. Rudra, Y. F. Tian, J. P. Jung and J. H. Collier, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 622–627 CrossRef CAS.
- Z. H. Huang, L. Shi, J. W. Ma, Z. Y. Sun, H. Cai, Y. X. Chen, Y. F. Zhao and Y. M. Li, J. Am. Chem. Soc., 2012, 134, 8730–8733 CrossRef CAS.
- J. S. Rudra, T. Sun, K. C. Bird, M. D. Daniels, J. Z. Gasiorowski, A. S. Chong and J. H. Collier, ACS Nano, 2012, 6, 1557–1564 CrossRef CAS.
- E. Genove, C. Shen, S. G. Zhang and C. E. Semino, Biomaterials, 2005, 26, 3341–3351 CrossRef CAS.
- S. L. Gras, A. K. Tickler, A. M. Squires, G. L. Devlin, M. A. Horton, C. M. Dobson and C. E. MacPhee, Biomaterials, 2008, 29, 1553–1562 CrossRef CAS.
- V. Jayawarna, M. Ali, T. A. Jowitt, A. E. Miller, A. Saiani, J. E. Gough and R. V. Ulijn, Adv. Mater., 2006, 18, 611–614 CrossRef CAS.
- P. W. J. M. Frederix, R. V. Ulijn, N. T. Hunt and T. Tuttle, J. Phys. Chem. Lett., 2011, 2, 2380–2384 CrossRef CAS.
- A. M. Smith, R. J. Williams, C. Tang, P. Coppo, R. F. Collins, M. L. Turner, A. Saiani and R. V. Ulijn, Adv. Mater., 2008, 20, 37–41 CrossRef CAS.
- K. J. Nagy, M. C. Giano, A. Jin, D. J. Pochan and J. P. Schneider, J. Am. Chem. Soc., 2011, 133, 14975–14977 CrossRef CAS.
- C. Q. Yan, M. E. Mackay, K. Czymmek, R. P. Nagarkar, J. P. Schneider and D. J. Pochan, Langmuir, 2012, 28, 6076–6087 CrossRef CAS.
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Science, 2001, 294, 1684–1688 CrossRef CAS.
- G. A. Silva, C. Czeisler, K. L. Niece, E. Beniash, D. A. Harrington, J. A. Kessler and S. I. Stupp, Science, 2004, 303, 1352–1355 CrossRef CAS.
- T. D. Sargeant, M. O. Guler, S. M. Oppenheimer, A. Mata, R. L. Satcher, D. C. Dunand and S. I. Stupp, Biomaterials, 2008, 29, 161–171 CrossRef CAS.
- V. M. Tysseling-Mattiace, V. Sahni, K. L. Niece, D. Birch, C. Czeisler, M. G. Fehlings, S. I. Stupp and J. A. Kessler, J. Neurosci., 2008, 28, 3814–3823 CrossRef CAS.
- H. Dong, S. E. Paramonov, L. Aulisa, E. L. Bakota and J. D. Hartgerink, J. Am. Chem. Soc., 2007, 129, 12468–12472 CrossRef CAS.
- L. Aulisa, H. Dong and J. D. Hartgerink, Biomacromolecules, 2009, 10, 2694–2698 CrossRef CAS.
- E. L. Bakota, O. Sensoy, B. Ozgur, M. Sayar and J. D. Hartgerink, Biomacromolecules, 2013, 14, 1370–1378 CrossRef CAS.
- M. P. Lutolf and J. A. Hubbell, Nat. Biotechnol., 2005, 23, 47–55 CrossRef CAS.
- J. L. Drury and D. J. Mooney, Biomaterials, 2003, 24, 4337–4351 CrossRef CAS.
- S. F. Yang, K. F. Leong, Z. H. Du and C. K. Chua, Tissue Eng., 2001, 7, 679–689 CrossRef CAS.
- S. G. Zhang, T. Holmes, C. Lockshin and A. Rich, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 3334–3338 CrossRef CAS.
- O. S. Makin, E. Atkins, P. Sikorski, J. Johansson and L. C. Serpell, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 315–320 CrossRef CAS.
- K. E. Marshall and L. C. Serpell, Biochem. Soc. Trans., 2009, 37, 671–676 CrossRef CAS.
- Y. Chau, Y. Luo, A. C. Cheung, Y. Nagai, S. Zhang, J. B. Kobler, S. M. Zeitels and R. Langer, Biomaterials, 2008, 29, 1713–1719 CrossRef.
- K. M. Galler, L. Aulisa, K. R. Regan, R. N. D'Souza and J. D. Hartgerink, J. Am. Chem. Soc., 2010, 132, 3217–3223 CrossRef CAS.
- Y. Kumada, N. A. Hammond and S. Zhang, Soft Matter, 2010, 6, 5073 RSC.
- M. C. Giano, D. J. Pochan and J. P. Schneider, Biomaterials, 2011, 32, 6471–6477 CrossRef CAS.
- H. W. Jun, V. Yuwono, S. E. Paramonov and J. D. Hartgerink, Adv. Mater., 2005, 17, 2612–2617 CrossRef CAS.
- L. E. Freed, G. Vunjaknovakovic, R. J. Biron, D. B. Eagles, D. C. Lesnoy, S. K. Barlow and R. Langer, Biotechnology, 1994, 12, 689–693 CrossRef CAS.
- S. L. Ishaug, G. M. Crane, M. J. Miller, A. W. Yasko, M. J. Yaszemski and A. G. Mikos, J. Biomed. Mater. Res., 1997, 36, 17–28 CrossRef CAS.
- B. S. Kim and D. J. Mooney, Trends Biotechnol., 1998, 16, 224–230 CrossRef CAS.
- C. C. Chu, Ann. Surg., 1982, 195, 55–59 CrossRef CAS.
- D. M. Lynn, M. M. Amiji and R. Langer, Angew. Chem., Int. Ed., 2001, 40, 1707–1710 CrossRef CAS.
- E. Ron, T. Turek, E. Mathiowitz, M. Chasin, M. Hageman and R. Langer, Proc. Natl. Acad. Sci. U. S. A., 1993, 90, 4176–4180 CrossRef CAS.
- K. W. Leong, B. C. Brott and R. Langer, J. Biomed. Mater. Res., 1985, 19, 941–955 CrossRef CAS.
- D. J. Gordon and S. C. Meredith, Biochemistry, 2003, 42, 475–485 CrossRef CAS.
- D. T. S. Rijkers, J. W. M. Hoppener, G. Posthuma, C. J. M. Lips and R. M. J. Liskamp, Chem.–Eur. J., 2002, 8, 4285–4291 CrossRef CAS.
- R. C. Elgersma, T. Meijneke, G. Posthuma, D. T. S. Rijkers and R. M. J. Liskamp, Chem.–Eur. J., 2006, 12, 3714–3725 CrossRef CAS.
- R. C. Elgersma, G. Posthuma, D. T. S. Rijkers and R. M. J. Liskamp, J. Pept. Sci., 2007, 13, 709–716 CrossRef CAS.
- D. F. Detar and C. J. Tenpas, J. Am. Chem. Soc., 1976, 98, 7903–7908 CrossRef CAS.
- J. C. Middleton and A. J. Tipton, Biomaterials, 2000, 21, 2335–2346 CrossRef CAS.
- G. Amitai, B. Dassa and S. Pietrokovski, J. Biol. Chem., 2004, 279, 3121–3131 CrossRef CAS.
- K. V. Mills, J. S. Manning, A. M. Garcia and L. A. Wuerdeman, J. Biol. Chem., 2004, 279, 20685–20691 CrossRef CAS.
- J. Z. Gasiorowski and J. H. Collier, Biomacromolecules, 2011, 12, 3549–3558 CrossRef CAS.
- M. L. Cooper, J. F. Hansbrough, R. L. Spielvogel, R. Cohen, R. L. Bartel and G. Naughton, Biomaterials, 1991, 12, 243–248 CrossRef CAS.
- P. X. Ma and R. Langer, J. Biomed. Mater. Res., 1999, 44, 217–221 CrossRef CAS.
- D. M. Marini, W. Hwang, D. A. Lauffenburger, S. G. Zhang and R. D. Kamm, Nano Lett., 2002, 2, 295–299 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3bm60161g |
|
| This journal is © The Royal Society of Chemistry 2013 |
Click here to see how this site uses Cookies. View our privacy policy here.