Recent advances in asymmetric organocatalytic conjugate addition of arenes and hetero-arenes
Pankaj
Chauhan
and
Swapandeep Singh
Chimni
*
Department of Chemistry, U.G.C. Centre of Advance Studies in Chemistry, Guru Nanak Dev University, Amritsar, 143005, India. Fax: (+)91-183-2258820; E-mail: sschimni@yahoo.com; sschimni.chem@gndu.ac.in
First published on 27th March 2012
Abstract
The asymmetric organocatalytic conjugate addition reaction of various nucleophiles to the unsaturated acceptor provides an important route for the synthesis of valuable chiral entities. Recently, aromatic and hetero-aromatic compounds have been successfully used as nucleophiles in the enantioselective conjugate addition reaction. This review provides an overview on the recent developments made in organocatalytic enantioselective conjugate addition of arenes and hetero-arenes to unsaturated acceptors.
Pankaj Chauhan (right) was born in 1984 at Bindal, a small village in Shimla District of Himachal Pradesh in India. He obtained his B.Sc. from Himachal Pradesh University in 2004. After completing his M.Sc. from Guru Nanak Dev University, Amritsar in 2007, he started his Ph.D. under the supervision of Dr Swapandeep Singh Chimni in the same University. His research interests include synthesis and application of chiral bifunctional organocatalysts for the development of new asymmetric transformation and green chemistry. |
 Swapandeep Singh Chimni (left) and Pankaj Chauhan (right) | Swapandeep Singh Chimni (left) was born in 1962 at Amritsar, India. He received his M.Sc. (Hons. Sch.) in Chemistry in 1985 and Ph.D. in 1991 from Guru Nanak Dev University, Amritsar. After two years as a lecturer at Regional Engineering College (now NIT) Jalandhar, he joined the Department of Chemistry, Guru Nanak Dev University as Lecturer in 1992. He is presently working as a Professor in the same department. He has a research experience of 21 years and published over 80 publications. He works in the area of synthetic organic chemistry with emphasis on asymmetric organocatalysis, biocatalysis and phase transfer catalysis as well as green chemistry. |
1. Introduction
The conjugate addition of various nucleophiles to electron-deficient alkenes is one of the most frequently used and widely studied carbon–carbon and carbon–heteroatom bond-forming reactions in organic synthesis. The catalytic asymmetric version of this reaction employing chiral catalysts has been extensively developed over the last few years. In the last decade, tremendous progress has been attained in asymmetric synthesis especially due to the emergence of asymmetric organocatalysis.1 Since the emergence of organocatalysis, the asymmetric addition of various nucleophiles to conjugate acceptor systems catalyzed by organocatalysts, has become one of the most significant C–C bond forming reactions not only for the standard collection of organic synthesis, but are also frequently used in the asymmetric synthesis of natural products and bioactive molecules.2 The popularity of the products and perfect atom economy of the process have made conjugate addition reactions very attractive in organic synthesis.Various anionic carbon species derived from nitroalkanes, dicarbonyls, ketoesters, ketoamides and hetero-atom nucleophiles, such as amines, thiols and alcohols add stereoselectively to the unsaturated acceptors such as α,β-unsaturated aldehydes, α,β-unsaturated ketones, unsaturated esters, nitroalkenes, etc. in the presence of chiral organocatalysts. In addition to the above mentioned nucleophiles, there exists a highly valuable class of nucleophiles, i.e. arenes and heteroarenes that have been reacted with unsaturated Michael acceptors in the presence of chiral organocatalysts to provide access to valuable chiral entities. This type of addition has also been visualized as a Michael-type Friedel–Crafts reaction, as this leads to direct alkylation of aromatic and heteroaromatic compounds and provides direct access to enantiopure aromatic compounds.
Many reviews have been published recently on asymmetric conjugate addition reactions.2 There are also some reviews on asymmetric organocatalytic aza-Michael3 and oxa-Michael4 reactions as well. Recently, some reviews have been published on the asymmetric organocatalytic Friedel–Crafts reaction,5 but none of them focused on the conjugate addition of aromatic and heteroaromatic compounds i.e. Michael-type Friedel–Crafts reaction. In this review we are focusing on the application of chiral organocatalysts for the conjugate addition of arenes and heteroarenes to the Michael acceptor.
Arene derivatives such as substituted anilines, phenols, naphthols and heteroarenes such as indole, dihydroindole and pyrrole derivatives have been successfully used as nucleophiles in asymmetric organocatalytic Friedel–Crafts reaction in order to obtain chiral arene derivatives (Fig. 1). On the other hand unsaturated electrophiles such as enals, enones, chalcones, nitroalkenes, β,γ-unsaturated α-ketoesters, α,α-dicyanoolefins have been activated by different organocatalysts by different modes of activation. Organocatalytic activation of enals and enones has been carried out by iminium catalysis.6 Activation of enals has been carried out by chiral amine organocatalysts, especially secondary amine catalysts, which form iminium ions with the enal, making the β-position in the enal labile to nucleophilic attack. Enones, excluding chalcones, have been activated by the reversible formation of an iminium ion with chiral primary amine organocatalysts. Unsaturated acceptors such as chalcones, nitroalkenes, β,γ-unsaturated-α-ketoesters, α,α-dicyanoolefins have been activated with hydrogen bonding or Brønsted acid organocatalysts such as thiourea, phosphoric acid, etc.7 For simplicity of presentation and understanding, this review has been classified according to the nature of Michael acceptor, which in turn is further classified on the basis of the mode of activation of Michael acceptor with different type of organocatalysts.
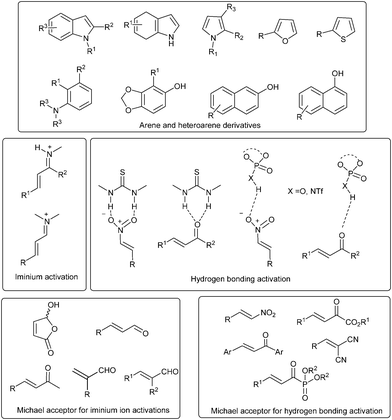 | ||
| Fig. 1 Arenes/heteroarenes, Michael acceptors and their mode of activation. | ||
2. Conjugate addition to α,β-unsaturated aldehydes
Organocatalytic activation of enals has been mostly carried out through iminium catalysis with secondary amine organocatalysts, while primary amine organocatalysts have been used for the activation of α-substituted enals.2.1. Secondary amine catalysis
In 2001, MacMillan's group reported the first organocatalytic enantioselective Michael-type Friedel–Crafts reaction of pyrrole (2) with enal (3) using the concept of LUMO-lowering activation of α,β-unsaturated aldehyde (3) via the reversible formation of iminium ion (A) with chiral imidazoldinone catalyst (1) to generate β-pyrrolyl carbonyls (4) (Scheme 1).8 The reaction proceeds well with a wide range of pyrroles (2) and enals (3) including electron-deficient unsaturated aldehydes, which do not readily participate in iminium ion formation, and the addition products (4) were obtained with good to high yield (68–90%) and high enantioselectivity (87–97% ee). The scaling up of the reaction to a level of 25 mmol did not alter the product yield and enantioselectivity. The catalyst (1) also controls the alkylation of both C–2 and C–5 of pyrrole with high enantioselectivity. When an excess of crotonaldehyde was used, the C2 isomer of disubstituted adduct (5) was obtained in 83% yield 98% ee and 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 dr. Unsymmetrical disubstituted pyrrole (6) was obtained in 72% yield over two steps with 99% ee and 90:10 dr when two different enals were used.
:10 dr. Unsymmetrical disubstituted pyrrole (6) was obtained in 72% yield over two steps with 99% ee and 90:10 dr when two different enals were used.
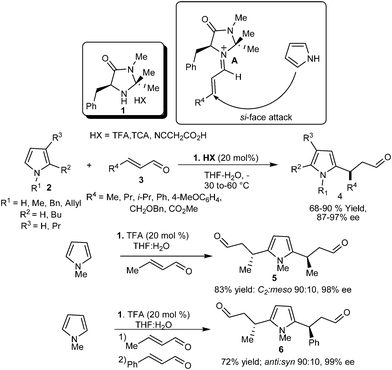 | ||
| Scheme 1 Enantioselective conjugate addition reaction of pyrroles with enals catalyzed by imidazolidinone (1). | ||
Houk and co-workers have explained the transition state structures for the chiral amine salt (1) catalyzed alkylation of N-methylpyrrole with enal, by using B3LYP/6-31G(d) density functional theory.9
A newly designed chiral imidazolidinone catalyst (7) efficiently catalyzes the conjugate addition of indole derivatives (8) with various enals (3) to provide access to 1,4-adducts (9) in good yield (74–90%) and good to high enantioselectivity (81–97% ee) (Scheme 2).10 Imidazolidinone (7) catalyzed enantioselective alkylation of 5-methoxy-2-methylindole with crotonaldehyde followed by oxidation of formyl moiety provides indolobutyric acid (10), a COX-2 inhibitor in 82% yield and 87% ee.
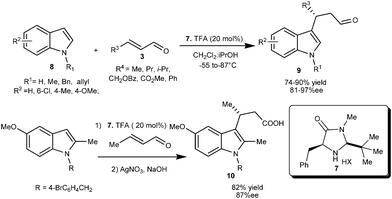 | ||
| Scheme 2 Enantioselective conjugate addition reaction of indoles with enals catalyzed by imidazolidinone (7). | ||
The flustramine ring structure was established by the conjugate addition of 3-alkylamino-substituted indole to enal, followed by domino cyclization of the corresponding 1,4-adducts in the presence of chiral imidazolidinone (Scheme 3).11 The TFA salt of imidazolidinones (7 or 11) catalyzes the enantioselective addition–cyclization of a wide range of tryptamines (12) and enals (3) to provide pyrroloindoline adducts (13) in good to excellent yield (66–99%) and high enantioselectivity (89–99% ee). This pyrroloindoline-forming reaction has been successfully used for the enantioselective synthesis of (−)-flustramine B (14). This amine-catalyzed sequence has been extended to the enantioselective construction of furanoindoline frameworks (16) in high levels of enantioinduction (93% ee). The catalytic cycle for the enantioselective addition–cyclization reaction involves the formation of an iminium ion intermediate (A) between enal and catalyst followed by the enantioselective conjugate addition from C3 of indole to the β-position of the iminium ion. The resulting intermediate (B) undergoes intramolecular cyclization to provide C, which on hydrolysis produces 13 and the catalyst gets regenerated for other catalytic cycle.
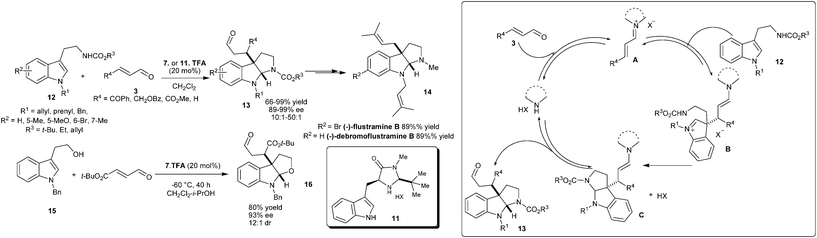 | ||
| Scheme 3 Enantioselective addition–cyclization reaction catalyzed by chiral imidazolidinones. | ||
The HCl salt of chiral imidazolidinone (7) catalyzes rthe enantioselective addition of substituted anilines (17) to α,β-unsaturated aldehydes (3) (Scheme 4).12 The conjugated products (18) with benzylic carbon stereocenter on aniline were obtained in good to high yield (66–97%) and high enantioselectivity (84–98% ee) from wide range of substituted anilines and unsaturated acceptors. In this transformation catalyst loadings as low as 1 mol% provide useful levels of enantioselectivity (88% ee). Kim and co-workers extended the iminium ion strategy of aniline alkylation with enal for the stereoselective synthesis of (+)-curcuphenol (21), a bioactive sesquiterpene phenol (Scheme 4).13 The initial step involves an imidazolidinone salt (19) catalyzed enantioselective conjugate addition of amino protected m-anisidine to crotonaldehyde which resulted in the chiral 1,4-adduct (20) in 90% yield and 90% ee. The corresponding adduct (20) was then converted into (+)-curcuphenol in six steps.
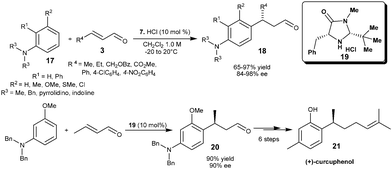 | ||
| Scheme 4 Enantioselective conjugate addition of substituted anilines to enals and stereoselective synthesis of (+)-curcuphenol. | ||
Instead of enal, unsaturated acetal (22) was used in imidazolidinone catalyzed intramolecular Michael addition of pyrroles, as aldehyde is readily generated in situ from acetal, which forms an iminium ion with imidazolidinone (1) (Scheme 5).14 Six- and seven-membered compounds (23) were synthesized in good yield (75–83%) and high enantioselectivity (87–96% ee), however the five-membered ring was not formed under the optimized reaction conditions. The intramolecular pyrrole addition strategy was extended for the total synthesis of natural alkaloids viz.(−)-rhazinal (26), (−)-rhazinilam (27), (−)-leuconolam (28) and (+)-epi-leuconolam (29) (Scheme 5).15 In the presence of a TFA salt of 1, the unsaturated aldehyde (24) undergoes an intramolecular conjugate addition reaction to form bicyclic aldehyde (25). The aldehyde (25) was then converted into (−)-rhazinilam and related natural products by subsequent transformations (Scheme 5).
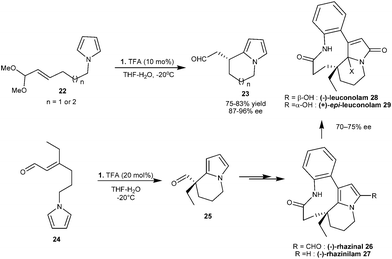 | ||
| Scheme 5 Intramolecular enantioselective conjugate addition of pyrrole and asymmetric synthesis of (-)-rhazinilam and related alkaloids. | ||
Imidazolidinone (7) catalyzes the conjugate addition of furans and thiophenes to variety of enals to provide adducts (31) in high enantioselectivity (93–98% ee) with high yield (74–95%) (Scheme 6).16
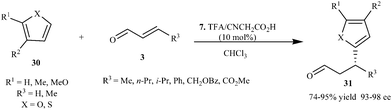 | ||
| Scheme 6 Enantioselective conjugate addition of furans and thiophenes to enals. | ||
MacMillan's research group developed the enantioselective organo-cascade catalysis, in which an imidazolidinone catalyst (32) imposed an orthogonal mode of substrate activation in the form of iminium (LUMO lowering) and enamine (HOMO rising) rising catalysis (Scheme 7).17 The organo-cascade sequence involves the formation of iminium ion (A) between enal and imidazolidinone which then stereoselectively accept various nucleophiles and rapid hydrolysis of the resulting iminium (C), the addition adduct (D) then enters a second catalytic cycle wherein the enamine (E) formation with the aldehyde would takes place, which then undergoes diastereoselective addition to the different electrophiles to generate 34via hydrolysis of iminium ion (F). Various arenes, enals and chlorinated quinone (33), tuned into an imidazolidinone (32) catalyzed cascade sequence providing α-chloro arene derivatives (34) in excellent enantioselectivity (99–>99% ee) and high diastereoselectivity (syn:anti 9:1–25:1) with significant liberty in the electronic and steric demand of the β-olefin. Various heteroaromatic nucleophiles such as furan, thiophenes, indoles, butenolides and tertiary amino lactones were found tolerable for primary catalytic cycle without any apparent impact on the secondary enamine cycle.
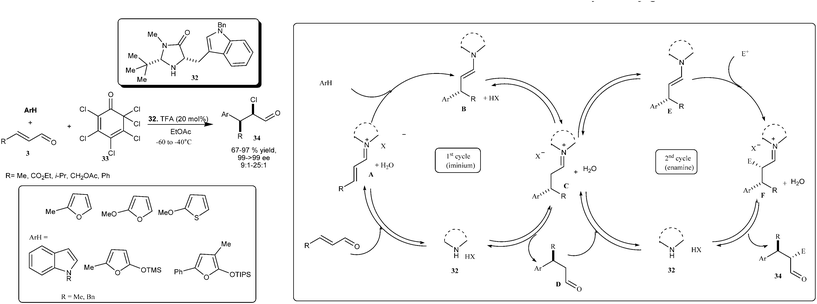 | ||
| Scheme 7 Asymmetric organo-cascade catalysis involving iminium and enamine catalysis. | ||
The imidazolidinone catalyst (35) efficiently catalyzes the 1,4-addition of 5-iodoindole to α,β-disubstituted α,β-unsaturated aldehyde (36) to provide conjugate adduct (37) in 75–84% yield and 84% ee. (Scheme 8)18 This strategy of indole alkylation with 36 was extended for the enantioselective synthesis of highly potent selective seretonin reuptake inhibitor-1 (39) via reductive amination of the aldehyde (37) to provide 38, which was then converted into 39.
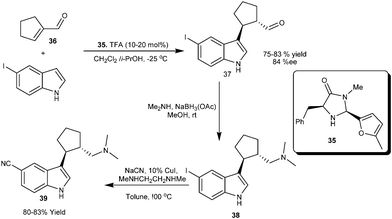 | ||
| Scheme 8 Asymmetric synthesis of seretonin reuptake inhibitor-1via imidazolidinone catalyzed conjugate addition of indole. | ||
Imidazolidinone (7) catalyzes the enantioselective intermolecular alkylation of various indolyl α,β-unsaturated aldehydes (40) (Scheme 9).19 The cyclized adducts (41) with good to high yield (48–95%) and high enantioselectivity (80–93% ee) were obtained from an intramolecular Michael type Friedel–Crafts reaction. This method is quite general as arene derivatives such as phenoxyl substituted α,β-unsaturated aldehyde (42) also undergoes enantioselective intramolecular alkylation to generate 43 with 88% yield and 90% ee.
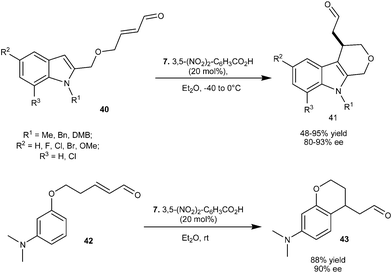 | ||
| Scheme 9 Enantioselective intramolecular alkylation of various indolyl α,β-unsaturated aldehydes and phenoxyl α,β-unsaturated aldehyde. | ||
Ying et al. reported the immobilization of imdazolidine-4-one on siliceous mesocellular foam (MCF) and polymer-coated MFC and demonstrated the application of these organocatalysts for conjugate addition of N-methylpyrrole to cinnamaldehyde and Diels–Alder cycloaddition (Scheme 10).20 The MCF-supported catalysts (44–47) have comparable reactivity to that of unsupported imidazolidinone, but their enantioselectivity and recyclability strongly depends on MCF surface modification.
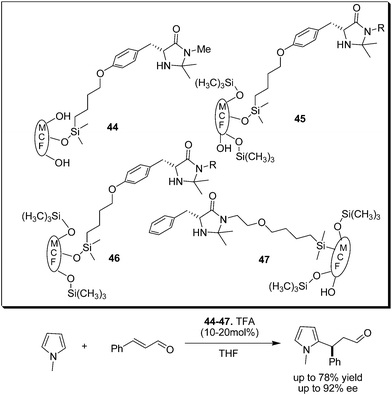 | ||
| Scheme 10 MCF supported imidazolidinone catalyzed enantioselective conjugate addition of pyrrole with cinnamaldehyde. | ||
A series of α-indolyl phosphonates (49) were synthesized in moderate to good yield (48–82%) and good to high enantioselectivity (73–96% ee) via organocatalytic asymmetric alkylation of indole derivatives (8) with dialkyl 3-oxoprop-1-enylphosphonates (48) using imidazolidinone catalyst (7) (Scheme 11).21 Imidazolidinone (7) also catalyzes the conjugate addition of anilines (17) with 48 to provide adducts (50) in good yield (75–90%) and good enantioselectivity (75–77% ee).
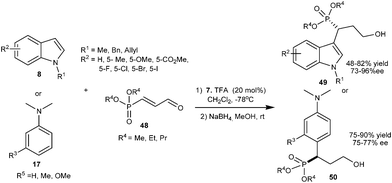 | ||
| Scheme 11 Enantioselective conjugate addition of indoles and N,N-disubstituted anilines to dialkyl 3-oxoprop-1-enylphosphonates. | ||
Chiral imidazolidinone organocatalyst (7) catalyzes the enantioselective conjugate addition of N-alkylated indoles to γ- hydroxy α,β-unsaturated aldehyde (51) to provide β-substituted γ-lactols (52) in moderate to high yield (51–98%) and good enantioselectivity (79–87% ee) (Scheme 12).22N-Alkylated pyrrole derivatives provided the corresponding γ-lactol (53) with 42–44% yield and 70–73% ee. The enantioselective conjugate addition reaction of electron-rich N,N-dialkylanilines (17) with γ-hydroxy-α,β-unsaturated aldehyde, provides γ-lactol (54) in 38–97% yield and 81% ee. Using protected tryptamine and tryptophol as a nucleophile for γ- hydroxy α,β-unsaturated aldehyde provides pyrroloindoline (55) and furanoindoline (56), respectively with poor to moderate levels of yield (25–56%) and poor enantioselectivity (3–8% ee).
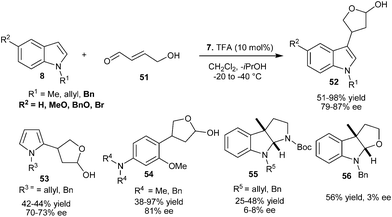 | ||
| Scheme 12 Enantioselective conjugate addition of arenes and heteroarenes to γ- hydroxy α,β-unsaturated aldehyde. | ||
Chiral imidazolidinone catalyst (32) catalyzes the enantioselective vinyl alkylation and Friedel–Crafts alkylation reaction of trifluoroborate salt with enals (Scheme 13).23 The nucleophilic BF3K salt forms a transition state which has extended enal scope without any restriction for the 1,4-addition. The HCl salt of 32 efficiently catalyzes the addition of potassium styryltrifluoroborate (57) to α,β-unsaturated aldehyde to provide the desired γ,δ-unsaturated aldehyde (59) with a high level of enantioselectivity (87–95% ee). Traditionally inert electron-deficient heteroaromatic compounds in iminium catalysis such as 2-formyl furan, benzofuran and N-Boc-indole became excellent nucleophiles when the BF3K moiety was incorporated into them and provide the corresponding 1,4-adducts (60) in excellent yield (69–94%) and high enantioselectivity (88–97% ee).
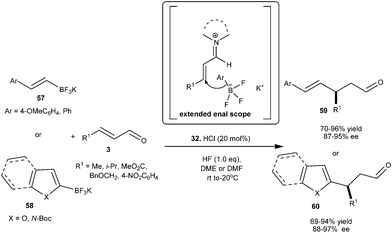 | ||
| Scheme 13 Enantioselective conjugate addition of trifluoroborate salt with enals. | ||
Wang's group reported Lewis base–Lewis base catalyzed asymmetric conjugate addition reaction of indole with α,β-unsaturated aldehydes (Scheme 14).24 Chiral diphenylprolinol silyl ether (61) in the presence of triethylamine acts as a bifunctional catalytic system, in which the chiral base activates the Michael acceptor by iminium ion formation and triethylamine activates the nucleophile by deprotonation or hydrogen bond interaction. This catalytic system efficiently catalyzes the conjugate addition of 1H-indole derivatives (8) substituted with electron releasing and electron withdrawing groups to different α,β-unsaturated aldehydes (3) to provide the chiral indole derivatives (9) in good to high yield (69–95%) and excellent enantioselectivity (92–98% ee). The failure of the reaction of N-methylindole with cinnamaldehyde was consistent with the fact that triethylamine was used to activate 1H-indole by deprotonation which was not possible with N-methyl indole.
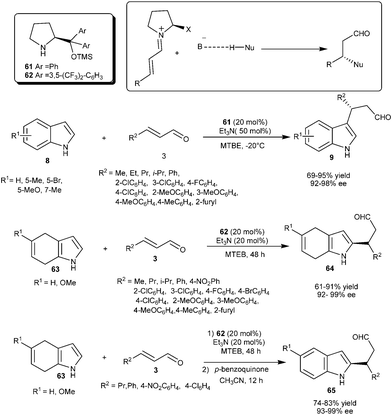 | ||
| Scheme 14 Lewis base–Lewis base catalyzed asymmetric conjugate addition of indoles and dihydroindoles to α,β-unsaturated aldehydes. | ||
Lewis base–Lewis base activation was successfully applied for the C–2 alkylation of 4,7-dihydroindole (63) with α,β-unsaturated aldehydes (3) (Scheme 14).25 The diarylprolinol silyl ether (62) in the presence of triethylamine catalyzes the enantioselective addition of dihydroindole (63) with α,β-unsaturated aldehydes (3). A diverse range α,β-unsaturated aldehydes react with 4,7-dihydroindole and its MeO derivative to provide the addition product (64) in good to high yield (61–91%) and excellent enantioselectivity (92–99% ee). The Lewis base–Lewis base catalytic system provides access to 2-substituted chiral indole derivatives (65) with good yield (74–83%) and high enantioselectivity (93–99% ee) via62 catalyzed enantioselective conjugate addition of 63 to 3, followed by subsequent oxidation of 1,4-adduct (64) with p-benzoquinone.
Bao and co-workers reported the enantioselective synthesis of 3-indolyl-3-arylpropanol (66) from indole and aromatic α,β-unsaturated aldehydes by diarylprolinol silyl ether (61) catalyzed conjugate addition reaction (Scheme 15).26 Diarylprolinol silyl ether (61) requires no acid or base additives in order to attain high efficiency in conjugate addition of various substituted indoles to aromatic α,β-unsaturated aldehydes to provide 1,4-adducts (66) in good yield (56–87%) and high enantioselectivity (86–95% ee). Alkyl α,β-unsaturated aldehydes such as crotonaldehyde was found to be unreactive under the optimized reaction conditions.
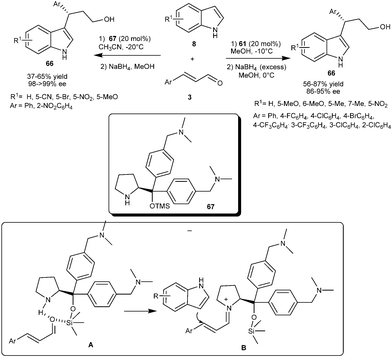 | ||
| Scheme 15 Enantioselective synthesis of 3-indolyl-3-aryllpropanols via diaryprolinol silyl ether catalyzed conjugate addition reaction. | ||
The multifunctional diarylprolinol catalyst (67) bearing two tertiary amine moieties on the aromatic ring proved an effective organocatalyst for enantioselective Michael-type addition of indoles (8) to aromatic enals (3) (Scheme 15).27 The catalyst (67) efficiently catalyzes the formation of chiral 3-indolyl-3-arylpropanol derivatives (66) in moderate yield (37–65%) and excellent enantioselectivity (98-> 99% ee) without the aid of acid or base additives. The experimental results and 29Si NMR analysis reveals that the silicon atom in 67 not only serves as a bulky group to strengthen steric repulsion, but also accelerates the iminium formation as silicon acts as a Lewis acid.
Enders's groups reported an organocatalytic one-pot multi-component process involving quadruple domino Friedel–Crafts-type–Michael–Michael–aldol condensation using acrolein (68), indoles (8) and nitroalkenes (69) as substrates (Scheme 16).28 Diphenylprolinol TMS-ether (61) catalyzes the straightforward and efficient entry to 3-(cyclohexenylmethyl)-indoles (70) bearing three vicinal stereogenic centers in moderate to good yield (23–84%) and excellent stereoselectivity (91:9–>95:5 dr, 94–>99% ee). A plausible catalytic cycle for the domino reaction involves activation of acrolein with the catalyst by formation of the vinylogous iminium ion (A), which undergoes an intermolecular Friedel–Crafts-type 1,4-addition reaction with indoles. The resulting enamine (B) subsequently undergoes an intermolecular Michael addition to nitroalkenes affording the intermediates (C). Subsequent hydrolysis of intermediate C leads to the formation of aldehydes (D), which enters into the second catalytic cycle and reacts with the iminium ion (A) to give an enamine intermediate (E). An intramolecular enamine-mediated aldol reaction resulted in the formation of intermediate (F), which undergoes dehydration and hydrolysis to afford 70, while the catalyst is regenerated for the next catalytic cycle.
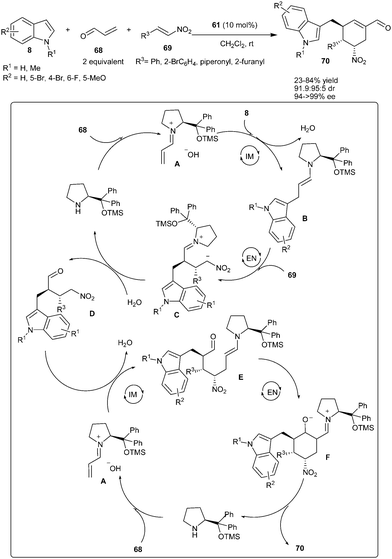 | ||
| Scheme 16 Organocatalytic one-pot multi-component quadruple domino Friedel–Crafts-type–Michael–Michael–aldol condensation. | ||
5-Hydroxyfuran-2(5H)-one (72) was used successfully as an electrophile in an organocatalyzed enantioselective conjugate addition of indoles in the presence of a prolinol catalyst (71) (Scheme 17).29 The catalyst (71) forms a zwitterionic iminium (A) with 72 and stereoselective nucleophilic addition of indole (8) to the iminium ion (A) results in the formation of adduct (B) which undergoes subsequent reduction and cyclization to afford chiral lactones (73). The methodology allows an efficient access to chiral lactones (73) in moderate to good yield (45–93%) and high enantioselectivity (80–96% ee) from various indole derivatives. The synthetic utility of 5-hydroxyfuran-2(5H)-one (72) was illustrated by a multicomponent reaction, involving sequential secondary amine (71) catalyzed enantioselective conjugate addition of indole followed by a Ugi 4-center 3-component reaction (U-4C-3CR). The corresponding chiral lactams (74) were obtained in good overall yield (59–82%) of two diastereomers and good enantioselectivity (78–88% ee).
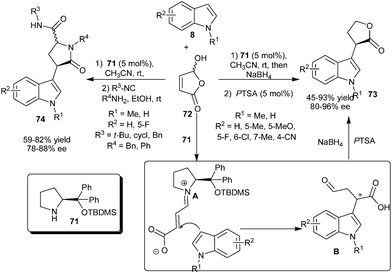 | ||
| Scheme 17 Enantioselective synthesis of chiral lactones and lactams from 5-hydroxyfuran-2(5H)-one. | ||
Wang and co-workers reported an enantioselective Michael-type Friedel–Crafts alkylation–cyclization cascade reaction of 1-naphthols (75) with α,β-unsaturated aldehydes (3) catalyzed by diphenylprolinol silyl ether (61) (Scheme 18).30 A secondary amine catalyst (61) in the presence of o-nitrobenzoic acid in wet toluene catalyzes the enantioselective alkylation–cyclization cascade reaction of 1-naphthols (75) with a wide range of α,β-unsaturated aldehydes (3) to provide addition–cyclization adducts (76) in good to high yield (63–93%) and high enantioselectivity (72–98% ee). The proposed mechanism for this addition–cyclization involves the formation of iminium ion (A) between α,β-unsaturated aldehydes and catalyst followed by the addition of 1-naphthol to the β-position of iminium ion to generate intermediate (B) which undergoes subsequent hydrolysis and half acetalization to afford the desired chromone (76).
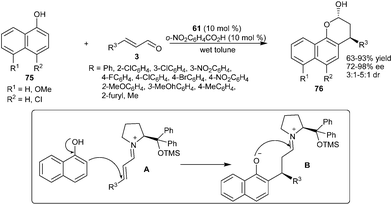 | ||
| Scheme 18 Enantioselective cascade reaction of 1-naphthols with α,β-unsaturated aldehydes. | ||
The amino acid derived diamine catalyst (77) catalyzes the enantioselective 1,4-conjugate addition of N-methylpyrrole (2) to cyclopent-1-ene carbaldehyde (36) (Scheme 19).31 The reaction parameters such as role of water, acid additives, as well as catalyst loading play an important role in stereoselectivity of this reaction. The addition of two equivalents of the strong acid HI to the diamine catalyst afforded 78, in 55% yield with the best diastereoselectivity (97:3 trans:cis) and 62% ee.
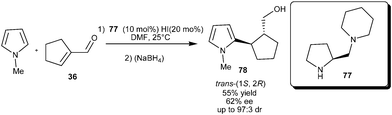 | ||
| Scheme 19 Enantioselective conjugate addition of N-methylpyrrole to cyclopent-1-ene carbaldehyde. | ||
Bonini and co-workers synthesized a series of azridin-2-yl-methanol organocatalysts and the utility of these organocatalyts were explored for the asymmetric conjugate addition reaction of N-alkylated nitrogen heterocycles with α,β-unsaturated aldehyde (Scheme 20).32 The pre-formed TFA salt of the 3-methyl-azridin-2-yl-methanols (79) efficiently catalyzes the conjugate addition of N-methylpyrrole as well as N-methylindole to enals to provide respective adducts (4 and 9) in good yield and enantioselectivity.
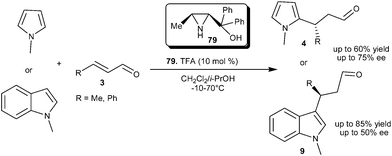 | ||
| Scheme 20 Azridin-2-yl-methanol catalyzed enantioselective conjugate addition. | ||
The N-terminal prolyl peptide catalyst (80) having a polyleucine tether was developed by Kudo's group for asymmetric conjugate addition of indoles to enals in aqueous media (Scheme 21).33 The importance of the hydrophobic polyleucine chain in the peptide catalyst was realized, since the catalyst without a polyleucine chain as well as proline TFA salt resulted in low catalytic efficiency. These results indicates that polyleucine moiety in the peptide provides a hydrophobic environment in aqueous media in which the reaction proceeds efficiently. The catalyst (80) efficiently catalyzes the addition of indoles (8) to enals providing products (66) in moderate to good yield (56–88%) and moderate to high enantioselectivity (52–94% ee). N-Methylpyrrole reacts well with enal in the presence of 80 to provide 81 in 77% ee. Recycling of the catalyst was carried out without much loss in enantioselectivity, though the yield decreased slightly.
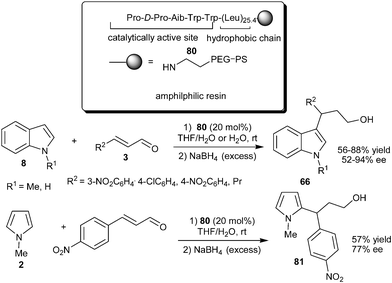 | ||
| Scheme 21 Enantioselective conjugate addition of indole and pyrrole catalyzed by 80. | ||
An N-isopropyl bipyrrolidine organocatalyst (82) was developed by Zhang's group for enantioselective addition of indole to enals (Scheme 22).34 Bipyrrolidine organocatalyst (82), in the presence of TfOH, facilitates the enantioselective conjugate addition reaction of indole derivatives with aliphatic enal derivatives to provide 3-alkylated indoles (9) in good to high yields (62–89%) and high levels of enantioselectivity (80–93% ee). The DFT calculations of the structure of iminium intermediate (A) reveals (i) the selective formation of (E)-iminium isomer to avoid nonbonding interactions between the olefin substrate and the other pyrrolidine group, (ii) the N-isopropyl group effectively shields the re-face of the iminium ion, leaving the si-face exposed to indole attack, and (iii) two benzyloxy groups on the catalyst further restricts the conformational flexibility for a positive effect on the enantioselectivity.
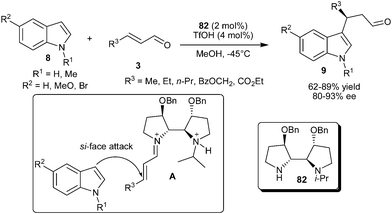
The camphor sulfonyl hydrazine organocatalyst (83), developed by Lee's group, catalyzes the enantioselective conjugate addition of N-alkylated indole with α,β-unsaturated aldehydes (Scheme 23).35 The secondary amine catalyst (83) in the presence of acid additive (TFA) catalyzes the enantioselective conjugate addition of N-benzylindole to the aliphatic and aromatic enals resulting in the formation of 1,4-adduct (66) in moderate to good yield (46–71%) and good enantioselectivity (81–88% ee).
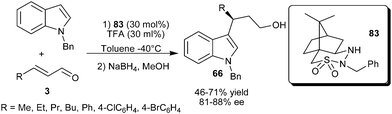 | ||
| Scheme 23 Camphor sulfonyl hydrazine catalyzed enantioselective conjugate addition of N-benzylindole to enals. | ||
2.2. Primary amine catalysis
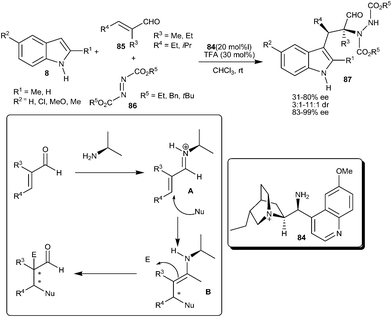
Melchiorre and co-workers reported the asymmetric organocatalytic cascade reaction of α-substituted α,β-unsaturated aldehydes indole and azo-dicarboxylate utilizing a Cinchona derived primary amine as catalyst (84) (Scheme 24).36 The TFA salt of 84 activates the α,α-disubstituted enals (85) towards an established iminium–enamine tandem sequence involving the Michael-type addition of the indole to iminium ion (A) followed by nucleophilic reaction of the resulting enamine (B) to the azo-compounds (86). This method provides the valuable precursors (87) of α-amino acids having vicinal tertiary and quaternary stereocenters, with an acceptable yield (31–80%) and high enantiomeric purity (83–99% ee and 3:1–11:1 dr). This protocol was extended to a cascade thio-amination reaction involving sulphur nucleophiles instead of indoles.
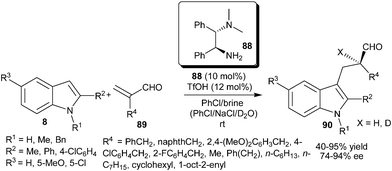
Very recently, Luo's group reported the enantioselective protonation of α-substituted acroleins (89) catalyzed by a primary amine organocatalyst (88) (Scheme 25).37 The primary amine catalyst (88) in the presence of an acid additive catalyzes the enantioselective protonation of 89via the initial conjugate addition of indole followed by enantioselective nucleophilic attack of the resulting enamine to a proton. The enantioselective deuteriation was also observed when the reaction was carried out in NaCl–D2O instead of brine. The corresponding chiral indole derivatives (90) were isolated in good yield (40–95%) and high enantioselectivity (74–94% ee) from a variety of indoles and unsaturated aldehydes.
 | ||
| Scheme 24 Cinchona derived primary amine catalyzed asymmetric organocatalytic cascade reaction. | ||
 | ||
| Scheme 25 Enantioselective protonation of α-substituted acroleins. | ||
3. Conjugate addition to α,β-unsaturated ketones
The organocatalytic enantioselective nucleophilic attack of arene derivatives to α,β-unsaturated ketones involves iminium and hydrogen bonding or Brønsted acid activation of enones.3.1. Secondary amine catalysis
Xiao and co-workers reported an organocatalytic 1,4-addition of indole to enones catalyzed by an equimolar amount of pyrrolidine and HClO4 (30 mol%) affording the corresponding β-indolyl ketones in good yield (60–92%).38 This idea of iminium activation of enones with secondary amine was extended to the enantioselective alkylation of indole with enone (92), using MacMillan's imidazolidinone catalyst (91), which provided a 1,4-adduct (93) in 52% yield and 28% ee (Scheme 26).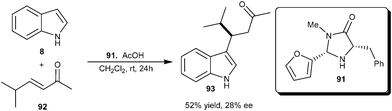 | ||
| Scheme 26 Enantioselective alkylation of indole with enone catalyzed by imidazolidinone. | ||
3.2. Primary amine catalysis
Chen's and Melchiorre's groups independently reported the enantioselective indole alkylation with α,β-unsaturated ketones by using a chiral primary amine catalyst derived from Cinchona alkaloids (Scheme 27).39,40 Chen and co-workers explained the reason for the limited success of the secondary amine catalyst for enantioselective conjugate addition to enones.39 The formation of an iminium ion (A) between enones and a secondary amine is less favoured than that of the primary amine due to steric reasons. Cinchona derived primary amine organocatalyst (94) in the presence of acid additive efficiently catalyzes the enantioselective addition of indole to an α,β-unsaturated ketone (92). The scope of this transformation was illustrated by the reaction between a variety of indoles and aliphatic, aromatic and heteroaromatic enone derivatives. The corresponding 1,4-adducts (93) were obtained in moderate to high yield (35–99%) and good enantioselectivity (56–89% ee). Melchiorre's group reported a novel catalytic system (95) bearing a chiral cation and chiral anion, which proved to be an efficient catalytic system for the iminium activation of enones.40 9-Amino(9-deoxy) epi-hydroqunine and N-Boc amino acid cooperatively catalyzed the enantioselective addition of indole derivatives to enones (92) to provide adducts (93) in high yield (56–99%) and enantioselectivity (62–96% ee). The catalytic salt of 9-amino Cinchona alkaloids acts as a bifunctional catalyst in which the 9-amino group activates the enone by forming an iminium ion and the quaternary ammonium salt activates and orients the indole through hydrogen bonding.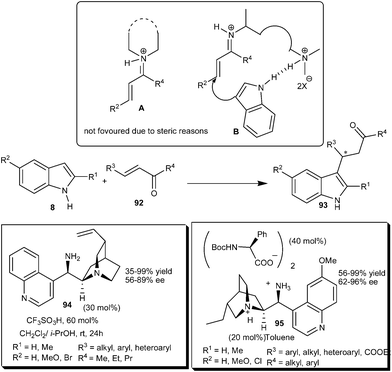 | ||
| Scheme 27 Cinchona alkaloid derived primary amine catalyzed enantioselective conjugate addition of indoles to enones. | ||
An amino acid derived primary amine–secondary amine organocatalyst (96) proved an efficient organocatalyst for the enantioselective alkylation of 4,7-dihydroindole (63) with enones (92) (Scheme 28).41 A leucine derived diamine catalyst (96) in the presence of TFA catalyzes the 1,4-addition of 4,7-dihydroindole (63) to a variety of enone derivatives (92) irrespective of steric and electronic factors and the desired products (97) were isolated in high optical purity (66–97% ee) and high yield (69–97%). Chalcone, a challenging substrate in iminium catalysis, also worked well as an acceptor in the presence of a primary amine catalyst and provided a good yield and enantioselectivity. The 2-functionalized indoles (98) were synthesized readily by oxidation of 2-substituted 4,7-dihydroindole adducts in good overall yield without any loss of enantioselectivity. In the transition state (99) the diamine catalyst acts as a bifunctional catalyst in which the primary amine forms the iminium ion and the secondary amine interacts with the N–H of the dihydroindole by hydrogen bonding and direct the addition of dihydroindole preferably on one enantioface of the double bond. This hydrogen bonding activation of the nucleophile can be rationalized by the fact that N-protected indole reacts sluggishly and a racemic product was obtained in only 10% yield.
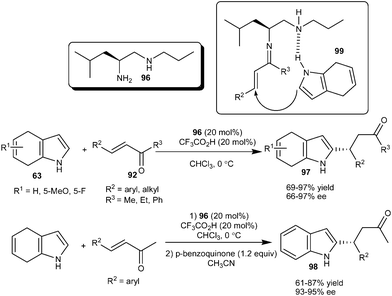 | ||
| Scheme 28 Leucine derived diamine catalyzed enantioselective conjugate addition of 4,7-dihydroindoles to enones. | ||
3.3. Brønsted acid catalysis
The catalytic amount of D-camphorsulfonic acid (D-CSA) (100) catalyzes the Michael-type addition of indole derivatives to chalcones (92) to provide 1,4-adducts (93) in good yield (75–96%) but poor enantioselectivity (0–37% ee) (Scheme 29).42 However, good conversion and improved enantioselectivity (13–58% ee) of the addition products (93) were attained using a BmimBr–CSA complex [1-butyl-3-methyl-1H-imidazolium bromide (BmimBr)]. The improved results in enantioselectivity and yield are attributed to the catalytic Lewis acid activation of the Brønsted acid.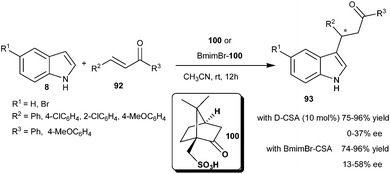
Chiral BINOL-derived phosphoric acid catalyzes the asymmetric Michael-type Friedel–Crafts alkylation of indoles with nonchelating α,β-unsaturated aromatic ketones (Scheme 30). The reactions of indoles (8) with chalcone derivatives (92) took place smoothly in the presence of 2 mol% of the catalyst (101) at room temperature affording chiral 3-substituted indole derivatives in moderate to good yield (63–92%) and moderate enantioselectivity (18–56% ee).43 The addition product of indole and cyclohexenone was isolated in 56% yield and 29% ee in the presence of 101. The phosphoric acid bearing 4-NO2C6H4 groups catalyzes the conjugate addition of indole to chalcones to provide Michael adducts in 44–98% yield and 41–54% ee in dichloromethane at room temperature.44 This transformation was later improved by Akiyama's group. Chiral phosphoric acid (102) catalyzes the conjugate addition reaction of indole derivatives with a wide variety of aryl and alkyl α,β-unsaturated aromatic ketones to provide access to the chiral indole derivatives (93) with improved yield (37–98%) and good to high enantioselectivity (58–92% ee).45 Phosphoric acid (103) catalyzes the addition of 4,7-dihydroindoles (63) to enone derivatives affording 2-substituted chiral indole derivatives (98) in good to high yield (51–90%) and good enantioselectivity (79–87% ee) with subsequent oxidation of dihydroindole adducts with 5 mol% of RuZrP in oxygen atmosphere. The transition state (104) for this transformation was illustrated as a ternary complex of indole, enone and catalyst, in which acidic proton of the catalyst activates the chalcone and oxygen of the catalyst activates the indole, in addition to activation the catalyst is also favourable for the reactants to provide a product with high enantioselectivity.
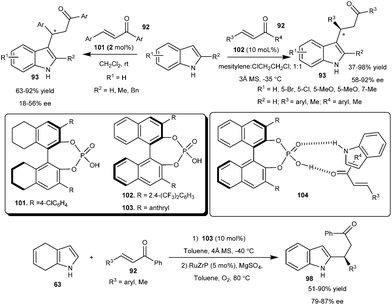 | ||
| Scheme 30 Chiral phosphoric acid catalyzed conjugate addition of indoles and 4,7-dihydroindoles to enones. | ||
4. Conjugate addition to nitroalkenes
4.1. Chiral thiourea catalysis
The first enantioselective organocatalytic Friedel–Crafts alkylation of indole with nitroalkene via 1,4-addition was developed by Ricci and co-workers (Scheme 31).46 The thiourea catalyst (105) efficiently catalyzes the C–3 alkylation of various indoles with nitroalkenes to produce optically active 3-indolylnitroalkane derivatives (106) in moderate to good yield (37–88%) and good enantioselectivity (71–89% ee). The thiourea catalysts with a protected –OH group and without the –OH group resulted in poor yield and low asymmetric induction. Also in the case of N-methylindole poor enantioselectivity was observed, which indicates that in the transition state (107), the thiourea catalyst exhibits a bifunctional mode of activation, in which two hydrogen atoms of the thiourea moiety activate the nitroalkene and the hydroxyl group of the catalyst interacts with the –NH of the indole via weak hydrogen bonding, thus directing the nucleophile on the one enantio-face of nitroalkene.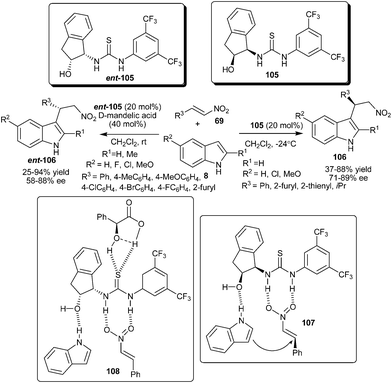 | ||
| Scheme 31 Enantioselective addition of indoles to nitroalkenes catalyzed by bifunctional thiourea catalysts. | ||
The addition of external Brønsted acid additives along with the thiourea catalyst (ent-105) in the asymmetric conjugate addition of indoles to nitroalkenes (69) results in improved yield (25–94%) and enantioselectivity (58–88% ee) of the desired adduct (ent-106) at room temperature (Scheme 31).47 This cooperative effect is presumptively attributed to an appropriate assembly between the Brønsted acid and thiourea moiety in the transition state (108), affording a more acidic and rigid catalytic complex.
The axially chiral bisarylthiourea organocatalysts (109) developed by Connon's group, catalyzes the asymmetric addition of N-methylindole to aryl, heteroaryl and alkyl substituted nitroalkenes (69) to provide 106 in good to excellent yield (54–98%) and low to moderate level of enantioselectivity (12–50% ee) (Scheme 32).48
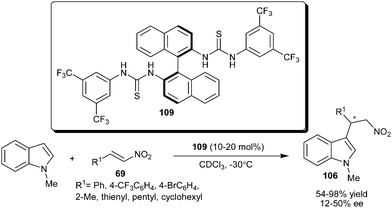 | ||
| Scheme 32 Chiral bisarylthiourea catalyzed conjugate addition of N-methyl indole with nitroalkenes. | ||
A Cinchona derived bifunctional thiourea–tertiary amine organocatalyst catalyzes the enantioselective conjugate addition of naphthol derivatives to variety of nitro-olefins (Scheme 33).49 The cinchonine derived thiourea catalyst (110) catalyzes the conjugate addition of 2-naphthol (113) to nitroalkenes to provide adducts (114) in good yield (69–83%) and high enantioselectivity (85–95% ee). The formation of 1,2-dihydronaphtho[2,1-b]furanyl-2-hydroxylamine derivatives (115) was observed with prolonged reaction time. These unpredicted dimeric adducts (115) were isolated in good yield (52–64%) and excellent enantioselectivity (>99.5% ee). The thiourea (112) catalyzes the alkylation of 1-naphthol (75) with nitroalkene to provide adduct (116) in 67% yield and 87% ee. The application of thiourea (111) catalyzed addition reaction of 2-naphthol with nitroalkene was extended for the asymmetric synthesis of indoline scaffold (118) of hypoxia selective prodrugs of DNA-alkylating agents related to the duocarmycin natural product.50
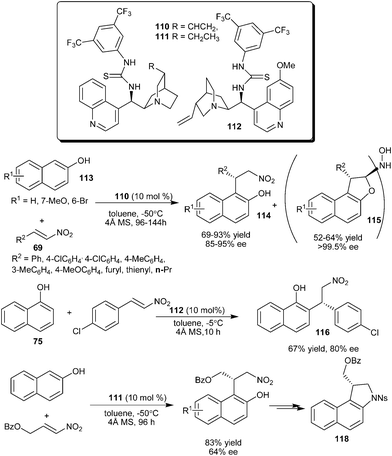 | ||
| Scheme 33 Cinchona derived thiourea-tertiary amine catalyzed enantioselective conjugate addition of naphthols to nitroolefins. | ||
The chiral thiourea–tertiary amine organocatalyst (110) derived from cinchonine was successfully employed to promote the enantioselective conjugate addition of sesamol derivatives (119) with various aromatic nitroolefins (Scheme 34).51 The corresponding products (120) were obtained in good yield (75–97%) and moderate to good enantioselectivity (59–90% ee).
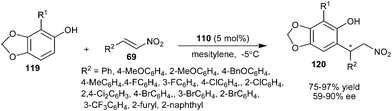 | ||
| Scheme 34 Enantioselective conjugate addition of sesamol derivatives to aromatic nitroolefins. | ||
Chiral C3-linked guanidine/bis-thiourea promotes a highly enantioselective 1,4-addition reaction of phenols with nitroalkenes (Scheme 35).52 The conformationally flexible guanidine–bisthiourea catalyst (121), catalyzes a temperature insensitive ortho-selective enantioselective alkylation of sesamol, 2-naphthol and 1-naphthol with nitroalkene derivatives to provide the desired products in good to excellent yield (66–99%) and high enantioselectivity (82–94% ee). Kinetic studies by using Eyring plots provide evidence that the differences in the activation entropies play a key role in the stereo-discrimination observed in this conjugate addition reaction catalyzed by 121. Stereoselective synthesis of N,N-bis(dihydrofuranyl)-hydroxyamines (115) from conjugate addition of sesamol and nitroolefins accomplished by means of cycle-specific catalysis with organocatalyst (121) and achiral base.53N,N-bis(Dihydrofuranyl)-hydroxyamines were synthesized in 51–99% yield, 90–99% ee and 6:1–>20:1 dr in a one-pot two-step approach involving enantioselective conjugate addition catalyzed by guanidine (121) followed by K2CO3 catalyzed hydroxylamine formation.
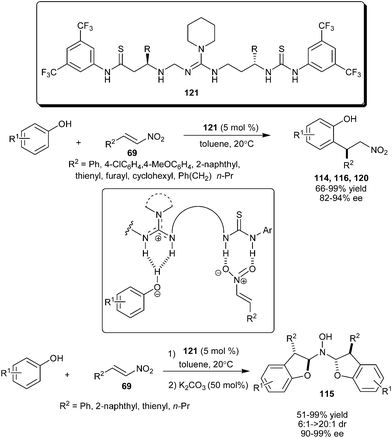 | ||
| Scheme 35 Chiral guanidine/bis-thiourea catalyzed enantioselective 1,4- conjugate addition reaction of phenols with nitroalkenes. | ||
4.2. Chiral phosphoric acid catalysis
The application of chiral phosphoric acid for enantioselective Michael-type Friedel–Crafts reaction of indole to nitroalkene was first explored by Akiyama's group (Scheme 36).54 This was the first report that demonstrated nitroalkene activation by a chiral phosphoric acid. Chiral phosphoric acid (122) in the presence of 3 Å molecular sieves catalyzes the Friedel–Crafts reaction of indole derivatives (8) with aromatic, heteroaromatic and aliphatic nitroalkenes (69) to provide enantioenriched 3-indolylnitroalkenes (106) in good to excellent yield (57–>99%) and high enantioselectivity (88–94% ee). The molecular sieves absorbs traces of water in the reaction medium, which facilitate better hydrogen bonding between catalyst and substrate. In the transition state (124), phosphoric acid acts as a bifunctional catalyst as it activates nitroalkene through its Brønsted acid part by hydrogen bonding and simultaneously activates indole through the oxygen of the catalyst by hydrogen bonding. This bifunctional mode of catalysis was supported by the observation in the reaction of N-methylindole with nitrostyrene, which results in considerable lowering of chemical yield (11%) and enantioselectivity (0% ee), due to the unavailability of N-H moiety for hydrogen bonding.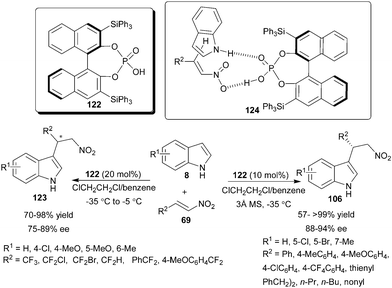 | ||
| Scheme 36 Enantioselective Michael type Friedel–Crafts reaction of indole with nitroalkene catalyzed with chiral phosphoric acid. | ||
The enantioselective Friedel–Crafts fluoroalkylation of indoles with fluoroalkylated nitroalkenes, catalyzed by chiral phosphoric acid has been developed recently (Scheme 36).55 With phosphoric acid (122) the fluoroalkylation of indole derivatives were carried out with high regioselectivity using tri-and di-fluoroalkyl nitroalkenes. The fluoroalkylated adducts (123) were obtained in good to excellent yield (70–98%) and good level of enantioselectivity (75–89% ee).
Chiral phosphoric acid (ent-103) catalyzes the enantioselective Friedel–Crafts reaction of 4,7-dihydroindoles (63) with nitroolefins (69) at low catalyst loading (Scheme 37).56 With slow addition of the nitroolefins by syringe pump, the reaction proceeds to completion in short reaction time with 0.5 mol% of catalyst (ent-103) and 4 Å molecular sieves as additive. 2-Substituted 4,7-dihydroindolylnitroalkanes (125) were isolated in excellent yield (93–98%) and low to high enantioselectivity (24–97% ee). Chiral 2-substituted indolylnitroalkanes (126) were isolated in high yield (85–91%) and enantioselectivity (88–95% ee) by subsequent oxidation of the conjugate adduct with p-benzoquinone in one pot.
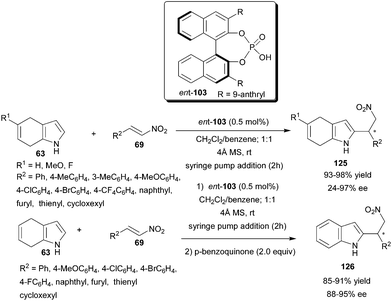 | ||
| Scheme 37 Chiral phosphoric acid catalyzed enantioselective conjugate addition of 4,7-dihydroindoles to nitroolefins. | ||
Density functional theory (DFT) studies on the chiral phosphoric acid-catalyzed Friedel–Crafts reaction between 4,7-dihydroindoles and nitroolefins reveals that higher reactivity of 4,7-dihydroindole in comparison to indole could be attributed to its higher HOMO energy as well as its more suitable trajectory to attack the nitroolefin in the transition state.57 The origin of the enantioselectivity of the chiral phosphoric acid catalyzed Friedel–Crafts reaction of 4,7-dihydroindole with nitroolefin was studied using complete models on the PBE1PBE/[6-311þG(d,p), 6-31G(d,p)] level, and shows that the enantioselectivity of the reaction is entirely controlled by the steric effect between the catalyst and the substrate in the case using (S)-phosphoric acid catalyst (ent-103), whereas in catalyst 122, bearing a silyl group, the enantioselectivity is determined by the solvent effect.
Chiral phosphoric acid (ent-103) efficiently catalyzes the enantioselective alkylation of pyrrole (2) with nitroalkenes to provide 2-substituted and 2,5-disubstituted pyrrole derivatives (127) in high yield (87–94%) and low to high enantioselectivity (11–94% ee) (Scheme 38).58 Chiral phosphoric acid acts as a bifunctional catalyst in a similar way as in the case of indole addition to nitroalkenes. The acidic proton and phosphoryl oxygen of the catalyst form the hydrogen bond with nitroolefin and pyrrole N–H, respectively. The proposed transition state was supported by the fact that the N–H of pyrrole is crucial for high enantioinduction as N-methyl pyrrole provides a product with only 6% ee with 65% yield.
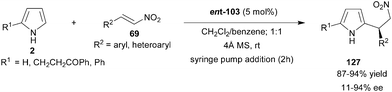 | ||
| Scheme 38 Chiral phosphoric acid catalyzed enantioselective conjugate addition of pyrrole derivatives to nitroolefins. | ||
4.3. Sulfonamide
Chiral bis-sulfonamide (128) catalyzes the enantioselective addition of indole to nitroolefins to provide products (106) in moderate to good yield (37–87%) and low to moderate level of enantioselectivity (13–63% ee) (Scheme 39).59 The addition product was converted into the optically active β-carbolines by a Pictet–Spengler reaction of the corresponding tryptamine without any loss in enantioselectivity.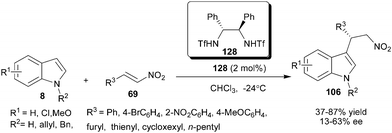 | ||
| Scheme 39 Chiral bis-sulfonamide catalyzed the enantioselective addition of indole to nitroolefins. | ||
Chiral bis-sulfonamide catalyst (ent-128) in the presence of acid additive catalyzes the double Michael addition–aromatization cascade of 2-propenylindoles (129) and nitroolefins (69) (Scheme 40).60 This strategy allows rapid and efficient access to diverse and structurally complex tetrahydrocarbazole derivatives (130) in moderate to good yield (42–86%) with good to excellent diastereoselectivity (80:20–99:1) and good enantioselectivity (82–92% ee). Plausible catalytic cycle involving double Michael addition–aromatization cascade sequence initiated by the chiral hydrogen bonding activation of nitroolefin with catalyst to generate an electrophilic complex (A), which could then enantioselectively intercept nucleophilic 2-propenylindole through a Friedel–Crafts-type Michael addition, resulting in a new active intermediate (B), which contains both iminium and nitronate components to accomplish the intramolecular Michael addition to form intermediate C. Subsequently, a rapid [1,3]-H migration regenerates the aromaticity of the indole ring, and therefore affords the corresponding tetrahydrocarbazole (130). The Friedel–Crafts alkylation product (131) would arise from the rearomatization of intermediate B.
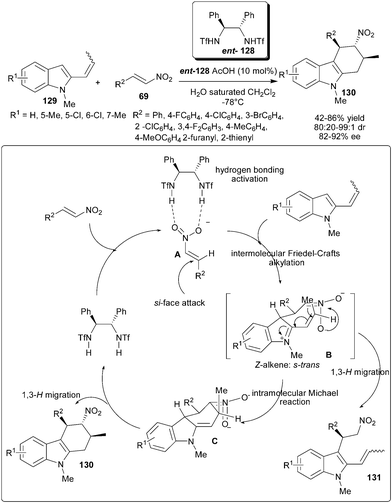 | ||
| Scheme 40 Double Michael addition–aromatization cascade of 2-propenylindoles and nitroolefins. | ||
4.4. Thioamide
Ganesh and Seidel mimicked the thiourea catalyst (105) to develop a new series of quinolinium thioamide organocatalysts (132) (Scheme 41).61 The thioamide organocatalyst (132) catalyzes the addition of the indole derivatives (8) to nitroalkenes (69) to provide addition products (106) in good to high yield (80–96%) and good to excellent enantioselectivity (90–98% ee). Compared with thiourea catalyst (105) the thioamide catalyst (132) is more efficient in terms of reaction rate and enantioselectivity. In the transition state (133), thioamide organocatalyst activates the nitroalkene with double hydrogen bonding and oxygen atom of the hydroxyl group atom activate and orient indole for si-face attack to provide an S-enantiomer of Michael adduct.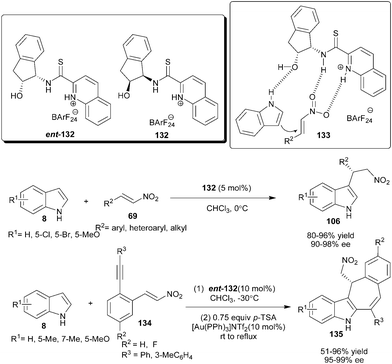 | ||
| Scheme 41 Enantioselective conjugate reaction catalyzed by quinolinium thioamide organocatalyst. | ||
The sequential combination of a quinolinium thioamide organocatalyst (ent-132) and gold catalyst provides an efficient one-pot access to tetracyclic indole derivatives (135) from the reaction of indole derivatives (8) with ortho-alkyne substituted nitroalkenes (134) (Scheme 41).62 The corresponding chiral heterocyclic adducts (135) were obtained in very good to high yield (51–96%) and excellent enantioselectivity (95–99% ee).
5. Conjugate addition to β,γ-unsaturated α-keto esters
Rueping's group reported the activation of an α,β-carbonyl compound with chiral Brønsted acid and developed the first organocatalytic asymmetric 1,2- and 1,4-addition of indole to β,γ-unsaturated α-keto esters (Scheme 42).63 The chiral N-triflylphosphoramides (136) catalyze the asymmetric synthesis of atropisomeric bisindoles by an unprecedented asymmetric 1,2-addition reaction of N-methylindole with β,γ-unsaturated α-keto ester (137) in up to 62% ee. Analogous to 136, the catalyst 139 provided predominantly bisindole (138), while 140 resulted in the formation of the addition product (141). This may be attributed to the steric properties of the catalyst. The 3,3′ Si–C bond is longer than the corresponding C–C bond in catalysts, and the spherically arranged phenyl groups on the silicon atom increase the steric demand at the catalytic center, resulting in better shielding of the carbonyl groups in the activation process and giving rise to the preferred regioselective addition of indole in the 4-position. With 5 mol% of 140 the 1,4-addition reaction of N-methylindole derivatives to β,γ-unsaturated α-keto esters (137) proceeds smoothly and the products (141) were isolated in an acceptable yield (43–88%) and high enantioselectivity (80–90% ee). A double Brønsted acid catalyzed reaction provides a convenient and direct access to new amino acid ester (142) via a Friedel–Crafts alkylation followed by a Hantzsch ester mediated reductive amination.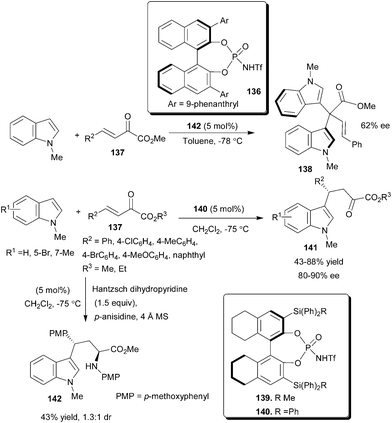 | ||
| Scheme 42 Organocatalytic asymmetric 1,2- and 1,4-addition of indole to β,γ-unsaturated α-keto esters. | ||
The chiral BINOL derived phosphoramide (143) with an attached phosphonium ion phase tag catalyzes the enantioselective conjugate addition reactions of indoles with enamides and β,γ-unsaturated α-ketoesters (137) (Scheme 43).64 The conjugate addition of indole to β,γ-unsaturated α-ketoesters provides 1,4-adducts (141) in high yield (89–98%) and moderate enantioselectivity (46–60% ee).
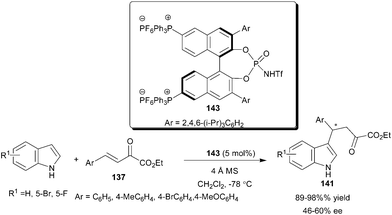 | ||
| Scheme 43 Phosphoramide (143) catalyzed enantioselective conjugate addition of indoles to β,γ-unsaturated α-ketoesters. | ||
Chiral N-triflylphosphoramide (144) catalyzes a highly enantioselective 1,4-addition reaction of 4,7-dihydroindoles (63) with β,γ-unsaturated α-keto esters (137), affording 2-substituted 4,7-dihydroindoles (145) in good to high yield (59–96%) and high enantioselectivity (91–98% ee) for a wide range of substrates (Scheme 44).65 Interestingly, when 5-methoxy-4,7-dihydroindole was used as the nucleophile, a hydrolyzed product (146) was obtained in 53% yield with 94% ee due to the hydrolysis of the enol methyl ether. The Friedel–Crafts alkylation together with a subsequent oxidation of the product (145) with p-benzoquinone led to a 2-alkylated indole derivative (147) in 58% yield and in 98% ee.
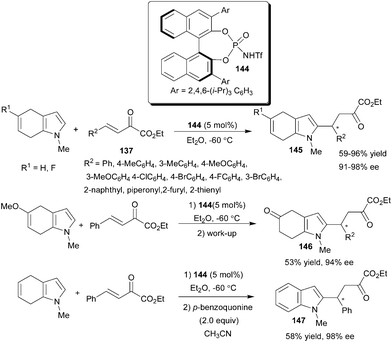 | ||
| Scheme 44 Chiral N-triflylphosphoramide (134) catalyzed 1,4-addition of 4,7-dihydroindoles to β,γ-unsaturated α-keto esters. | ||
The application of a new series of optically active 1,10-spirobiindane-7,7′-diol (SPINOL)-based phosphoric acids for enantioselective addition of indoles to aldimines and β,γ-unsaturated-α-ketoesters (137) was reported by Hu and co-workers (Scheme 45).66 Only 2 mol% of SPINOL-based phosphoric acid (148) catalyzes the regio and enantioselective 1,4-addition of indole derivatives to β,γ-unsaturated-α-ketoesters to provide desired adducts (141) in high yield (84–94%) and excellent enantioselectivity (91–99% ee).
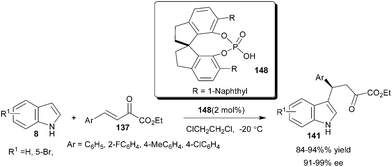 | ||
| Scheme 45 SPINOL-based phosphoric acids catalyzed enantioselective addition of indoles with β,γ-unsaturated-α-ketoesters. | ||
Takemoto's bifunctional thiourea (149) catalyzes the addition–cyclization reaction between 2-naphthols (113) and β,γ-unsaturated α-keto esters (137) (Scheme 46).67 The acid catalyzed dehydration of the corresponding adduct resulted in naphthopyran (150) derivatives in good yield (51–91%) and good enantioselectivity (57–90% ee) under mild conditions.
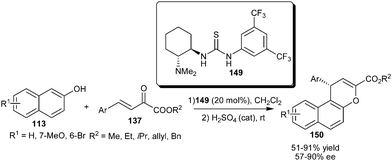 | ||
| Scheme 46 Takemoto's catalyst catalyzed addition–cyclization reaction between 2-naphthols and β,γ-unsaturated α-keto esters. | ||
Rosin-derived tertiary amine–thiourea organocatalysts catalyze the addition–cyclization reaction between naphthols and β,γ-unsaturated α-keto esters (Scheme 47).68 Rosin-derived tertiary amine–thiourea (151 or 152) catalyzes the formation of both enantiomers of potentially bioactive chiral functionalized chromones (153) in good yield (78–86%) and good to high enantioselectivity (78–96% ee) on reaction of 1-naphthols with aryl and heteroaryl β,γ-unsaturated α-keto esters. The organocatalyst (152) also catalyzes the addition–cyclization reaction of 2-naphthol with β,γ-unsaturated α-keto esters to provide chromones (154) in good yield (86–91%) and good enantioselectivity (86–90% ee).
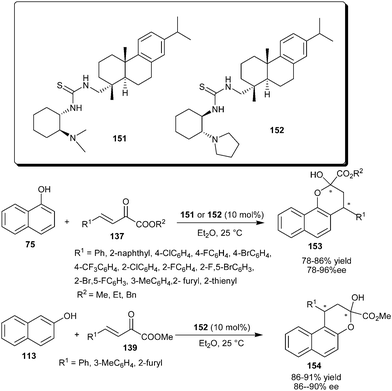 | ||
| Scheme 47 Rosin-derived tertiary amine–thiourea catalyzed addition–cyclization reaction of naphthols with β,γ-unsaturated α-keto esters. | ||
6. Conjugate addition to dicyanoolefins
The bifunctional thiourea tertiary amine organocatalyst (149) catalyzes the synthesis of naphthopyran derivatives via addition cyclization reaction of 2-naphthols (113) with α,α-dicyanoolefins (155) (Scheme 48).69 The reaction involves an initial Michael type Friedel–Crafts reaction of 2-naphthol with olefin followed by the cyclization reaction affording naphthopyrans (156) in moderate to high yield (38–99%) and moderate to good enantioselectivity (56–90% ee). A three component condensation reaction between 2-naphthol, aldehyde and malononitrile resulted in a cyclized adduct (156) but comparably lower enantioselectivity was observed compared with two component reaction.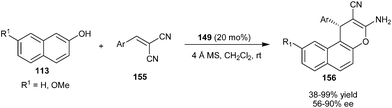 | ||
| Scheme 48 Enantioselective synthesis of naphthopyrans via addition cyclization reaction of 2-naphthol with α,α-dicyanoolefins. | ||
7. Conjugate addition to α,β-unsaturated acyl phosphonates
The thiourea catalyst (ent-105) catalyzes the conjugate addition reaction of indole derivatives to α,β-unsaturated acylphosphonates (157) to provide adducts (158) with subsequent DBU catalyzed nucleophilic displacement of phosphate ester with alcohols and amines (Scheme 49).70 The corresponding esters and amides were isolated in good to high yield (57–93%) with good enantioselectivity (72–90% ee).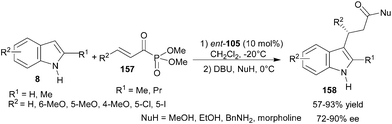 | ||
| Scheme 49 Enantioselective conjugate addition of indoles to α,β-unsaturated acylphosphonates. | ||
Conclusions
Since the first report on the organocatalyzed enantioselective conjugate addition of pyrrole to enals, over sixty significant examples of enantioselective conjugate addition of activated arenes and heteroarenes have been published in the last ten years. Remarkable levels of enantioselectivity have been achieved for electron-rich arenes such as indoles, pyrroles, naphthols etc. and electrophiles such as enals, enones, nitroolefins etc. The ready availability and stability of organocatalysts such as chiral imidazolidinones, prolinols, thioureas, BINOL-derived phosphoric acids, N-triflyl phosphoramides, and Cinchona alkaloid derivatives, make these enantioselective conjugate addition reactions more practical and operationally simple.Many enantioselective conjugate additions of arenes and heteroarenes require high catalyst loadings, which badly hamper their practical application. Therefore, there is a need to develop some new organocatalysts that have a higher turnover number. Although organocatalytic conjugate addition of arenes is a topic of great interest among synthetic organic chemists, the main challenge lies in the development of enantioselective addition of electron deficient arenes and heteroarenes. Further, the major focus of research remains confined to the Michael acceptors such as enals, enones and nitroalkenes. Other conjugated acceptor such as α,α-dicyanoolefins and α,β-unsaturated acylphosphonates are used very scarcely in the enantioselective addition of arenes and heteroarenes. Still, there is lots of room for the addition of arenes and heteroarenes to other Michael acceptors such as maleimide, α,β-unsaturated malonates, unsaturated sulfones, etc. There is also a wide scope for the development of organocatalytic cascade reactions using arenes and heteroarenes as one of the substrates.
We believe that designing some new organocatalysts having novel activation modes will make asymmetric conjugate addition reactions of arenes and heteroarenes fundamentally and practically important for the synthesis of valuable enantiopure compounds.
Acknowledgements
Our research work was supported by the research project sanctioned to S. S. C. by the University Grant Commission (UGC), India. Financial support from the Department of Science and Technology (DST), India under FIST program and UGC, India, under CAS-I is gratefully acknowledged.References
- (a) For selected reviews on asymmetric organocatalysis, see: P. I. Dalko and L. Moisan, Angew. Chem., Int. Ed., 2001, 40, 3726 CrossRef CAS; (b) P. I. Dalko and L. Moisan, Angew. Chem., Int. Ed., 2004, 43, 5138 CrossRef CAS; (c) A. Dondoni and A. Massi, Angew. Chem., Int. Ed., 2008, 47, 4638 CrossRef CAS.
- For selected reviews of asymmetric Michael additions, see: (a) O. M. Berner, L. Tedeschi and D. Enders, Eur. J. Org. Chem., 2002, 1877 CrossRef CAS; (b) R. Ballini, G. Bosica, D. Fiorini, A. Palmieri and M. Petrini, Chem. Rev., 2005, 105, 933 CrossRef CAS; (c) S. B. Tsogoeva, Eur. J. Org. Chem., 2007, 1701 CrossRef CAS; (d) D. Roca-Lopez, D. Sadaba, I. Delso, R. P. Herrera, T. Tejero and P. Merino, Tetrahedron: Asymmetry, 2010, 21, 2561 CrossRef CAS.
- D. Enders, C. Wang and J. X. Liebich, Chem.–Eur. J., 2009, 15, 11058 CrossRef CAS.
- (a) C. F. Nising and S. Brase, Chem. Soc. Rev., 2008, 37, 1218 RSC; (b) C. F. Nising and S. Brase, Chem. Soc. Rev., 2012, 41, 988 RSC.
- For selected reviews on organocatalytic asymmetric Friedel–Crafts reaction, see: (a) S.-L. You, Q. Cai and M. Zeng, Chem. Soc. Rev., 2009, 38, 2190 RSC; (b) V. Terrasson, R. M. de Figueiredo and J. M. Campagne, Eur. J. Org. Chem., 2010, 2635 CrossRef CAS; (c) M. Zen and S.-L. You, Synlett, 2010, 1289 Search PubMed.
- A. Erkkila, I. Majander and P. M. Pihko, Chem. Rev., 2007, 107, 5416 CrossRef.
- For selected reviews on hydrogen bond catalysis, see: (a) S. J. Connon, Chem.–Eur. J., 2006, 12, 5418 CrossRef; (b) S. J. Connon, Chem. Commun., 2008, 2499 RSC; (c) M. S. Taylor and E. N. Jacobsen, Angew. Chem., Int. Ed., 2006, 45, 1520 CrossRef CAS; (d) T. Akiyama, Chem. Rev., 2007, 107, 5744 CrossRef CAS; (e) P. Chauhan and S. S. Chimni, RSC Adv., 2012, 2, 737 RSC.
- N. A. Paras and D. W. C. MacMillan, J. Am. Chem. Soc., 2001, 123, 4370 CrossRef CAS.
- R. Gordillo, J. Carter and K. N. Houk, Adv. Synth. Catal., 2004, 346, 1175 CrossRef CAS.
- J. F. Austin and D. W. C. MacMillan, J. Am. Chem. Soc., 2002, 124, 1172 CrossRef CAS.
- J. F. Austin, S.-G. Kim, C. J. Sinz, W.-J. Xiao and D. W. C. MacMillan, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5482 CrossRef CAS.
- N. A. Paras and D. W. C. MacMillan, J. Am. Chem. Soc., 2002, 124, 7894 CrossRef CAS.
- S.-G. Kim, J. Kim and H. Jung, Tetrahedron Lett., 2005, 46, 2437 CrossRef CAS.
- M. G. Banwell, D. A. S. Beck and J. A. Smith, Org. Biomol. Chem., 2004, 2, 157 CAS.
- M. G. Banwell, D. A. S. Beck and A. C. Willis, Arkivoc, 2006,(iii), 163 CAS.
- S. P. Brown, Doctoral Dissertation, California Institute of Technology Pasadena, CA, 2005 Search PubMed.
- Y. Huang, A. M.Walji, C. H. Larsen and D. W. C. MacMillan, J. Am. Chem. Soc., 2005, 127, 15051 CrossRef CAS.
- D. J. Denhart, R. J. Mattson, J. L. Ditta and J. E. Macor, Tetrahedron Lett., 2004, 45, 3803 CrossRef CAS.
- C.-F. Li, H. Liu, J. Liao, Y.-J. Cao, X.-P. Liu and W.-J. Xiao, Org. Lett., 2007, 9, 1847 CrossRef CAS.
- Y. Zhang, L. Zhao, S. S. Lee and J. Y. Ying, Adv. Synth. Catal., 2006, 348, 2027 CrossRef CAS.
- Y.-C. Guo, D. -P. Li, Y.-L. Li, H.-M. Wang and W.-J. Xiao, Chirality, 2009, 21, 777 CrossRef CAS.
- S.-G. Kim, Bull. Korean Chem. Soc., 2009, 30, 2519 CrossRef CAS.
- S. Lee and D. W. C. MacMillan, J. Am. Chem. Soc., 2007, 129, 15438 CrossRef CAS.
- L. Hong, L. Wang, C. Chen, B. Zhang and R. Wang, Adv. Synth. Catal., 2009, 351, 772 CrossRef CAS.
- L. Hong, C. Liu, W. Sun, L. Wang, K. Wong and R. Wang, Org. Lett., 2009, 11, 2177 CrossRef CAS.
- Z.-J. Wang, J.-G. Yang, J. Jin, X. Lv and W. Bao, Synthesis, 2009, 3994 CAS.
- Z.-H. Shi, H. Sheng, K. -F. Yang, J.-X. Jiang, G. -Q. Lai, Y. Lu and L.-W. Xu, Eur. J. Org. Chem., 2011, 66 CrossRef.
- D. Enders, C. Wang, M. Mukanova and A. Greb, Chem. Commun., 2010, 46, 2447 RSC.
- E. Riguet, J. Org. Chem., 2011, 76, 8143 CrossRef CAS.
- L. hong, L. Wang, W. Sun, K. Wong and R. Wang, J. Org. Chem., 2009, 74, 6881 CrossRef CAS.
- P. Breistein, S. Karlsson and E. Hedenstrom, Tetrahedron: Asymmetry, 2006, 17, 107 CrossRef CAS.
- B. F. Bonini, E. Capito, M. Comes-Franchini, M. Fochi, A. Ricci and B. Zwanenburg, Tetrahedron: Asymmetry, 2006, 17, 3135 CrossRef CAS.
- K. Akagawa, T. Yamashita, S. Sakamoto and K. Kudo, Tetrahedron Lett., 2009, 50, 5602 CrossRef CAS.
- S. Jin, C. Li, Y. Ma, Y. Kan, Y. J. Zhang and W. Zhang, Org. Biomol. Chem., 2010, 8, 4011 CAS.
- T. Tian, B.-J. Pei, Q.-H. Li, H. He, L.-Y. Chen, X. Zhou, W.-H. Chan and A. W. M. Lee, Synlett, 2009, 2115 CAS.
- P. Galzerano, F. Pesciaioli, A. Mazzanti, G. Bartoli and P. Melchiorre, Angew. Chem., Int. Ed., 2009, 48, 7892 CrossRef CAS.
- N. Fu, L. Zhang, J. Li, S. Luo and J.-P. Cheng, Angew. Chem., Int. Ed., 2011, 50, 11451 CrossRef CAS.
- D.-P. Li, Y.-C. Guo, Y. Ding and W.-J. Xiao, Chem. Commun., 2006, 799 RSC.
- W. Chen, W. Du, L. Yue, R. Li, Y. Wu, L.-S. Ding and Y.-C. Chen, Org. Biomol. Chem., 2007, 5, 816 CAS.
- G. Bartoli, M. Bosco, A. Carlone, F. Pesciaioli, L. Sambri and P. Melchiorre, Org. Lett., 2007, 9, 1403 CrossRef CAS.
- L. Hong, W. Sun, C. Liu, L. Wang, K. Wong and R. Wang, Chem.–Eur. J., 2009, 15, 11105 CrossRef CAS.
- W. Zhou, L.-W. Xu, L. Li, L. Yang and C.-G. Xia, Eur. J. Org. Chem., 2006, 5225 CrossRef CAS.
- H.-Y. Tang, A. D. Lu, Z. -H. Zhou, G.-F. Zhao, L.-N. He and C.-C. Tang, Eur. J. Org. Chem., 2008, 1406 CrossRef CAS.
- A. Scettri, R. Villano and M. R. Acocella, Molecules, 2009, 14, 3030 CrossRef CAS.
- T. Sakamoto, J. Itoh, K. Mori and T. Akiyama, Org. Biomol. Chem., 2010, 8, 5448 CAS.
- R. P. Herrera, V. Sgarzani, L. Bernardi and A. Ricci, Angew. Chem., Int. Ed., 2005, 44, 6576 CrossRef CAS.
- E. Marqués-López, A. Alcaine, T. Tejero and R. P. Herrera, Eur. J. Org. Chem., 2011, 3700 CrossRef.
- E. M. Fleming, T. McCabe and S. J. Connon, Tetrahedron Lett., 2006, 47, 7037 CrossRef CAS.
- T.-Y. Liu, H.-L. Cui, Q. Chai, J. Long, B.-J. Li, Y. Wu, L.-S. Ding and Y.-C. Chen, Chem. Commun., 2007, 2228 RSC.
- D. M. Heinrich, J.-J. Youte, W. A. Denny and M. Tercel, Tetrahedron Lett., 2011, 52, 7000 CrossRef CAS.
- H. Zhang, Y.-H. Liao, W.-C. Yuan and X.-M. Zhang, Eur. J. Org. Chem., 2010, 3215 CrossRef CAS.
- Y. Sohtome, B. Shin, N. Horitsugi, R. Takagi, K. Noguchi and K. Nagasawa, Angew. Chem., Int. Ed., 2010, 49, 7299 CrossRef CAS.
- Y. Sohtome, T. Yamaguchi, B. Shin and K. Nagasawa, Chem. Lett., 2011, 40, 843 CrossRef CAS.
- J. Itoh, K. Fuchibe and T. Akiyama, Angew. Chem., Int. Ed., 2008, 47, 4016 CrossRef CAS.
- J.-H. Lin and J.-C. Xiao, Eur. J. Org. Chem., 2011, 4536 CrossRef CAS.
- Y.-F. Sheng, G.-Q. Li, Q. Kang, A.-J. Zhang and S.-L. You, Chem.–Eur. J., 2009, 15, 3351 CrossRef CAS.
- C. Zheng, Y.-F. Sheng, Y.-X. Li and S.-L. You, Tetrahedron, 2010, 66, 2875 CrossRef CAS.
- Y. -F. Sheng, Q. Gu, A.-J. Zhang and S-L. You, J. Org. Chem., 2009, 74, 6899 CrossRef CAS.
- W. Zhuang, R. G. Hazell and K. A. Jørgensen, Org. Biomol. Chem., 2005, 3, 2566 CAS.
- X.-F. Wang, J.-R. Chen, Y.-J. Cao, H.-G. Cheng and W.-J. Xiao, Org. Lett., 2010, 12, 1140 CrossRef CAS.
- M. Ganesh and D. Seidel, J. Am. Chem. Soc., 2008, 130, 16464 CrossRef CAS.
- C. C. J. Loh, J. Badorrek, G. Raabe and D. Enders, Chem.–Eur. J., 2011, 17, 13409 CrossRef CAS.
- M. Rueping, B. J. Nachtsheim, S. A. Moreth and M. Bolte, Angew. Chem., Int. Ed., 2008, 47, 593 CrossRef CAS.
- M. Zeng, Q. Kang, Q.-L. He and S.-L. You, Adv. Synth. Catal., 2008, 350, 2169 CrossRef CAS.
- J. Hermeke and P. H. Toy, Tetrahedron, 2011, 67, 4103 CrossRef CAS.
- C.-H. Xing, Y.-X. Liao, J. Ng and Q.-S. Hu, J. Org. Chem., 2011, 76, 4125 CrossRef CAS.
- X.-S. Wang, G.-S. Yang and G. Zhao, Tetrahedron: Asymmetry, 2008, 19, 709 CrossRef CAS.
- X. Jiang, L. Wu, Y. Xing, L. Wang, S. Wang, Z. Chen and R. Wang, Chem. Commun., 2012, 48, 446 RSC.
- X.-S. Wang, C.-W. Zheng, S.-L. Zhao, Z. Chai, G. Zhao and G.-S. Yang, Tetrahedron: Asymmetry, 2008, 19, 2699 CrossRef CAS.
- H. Jiang, M. W. Paixao, D. Monge and K. A. Jørgensen, J. Am. Chem. Soc., 2010, 132, 2775 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |