A new environmentally friendly method for the Baeyer–Villiger oxidation of cyclobutanones catalyzed by thioureas using H2O2 as an oxidant
Niiha
Sasakura
,
Keiji
Nakano
,
Yoshiyasu
Ichikawa
and
Hiyoshizo
Kotsuki
*
Laboratory of Natural Products Chemistry, Faculty of Science, Kochi University, Akebono-cho, Kochi 780-8520, Japan. E-mail: kotsuki@kochi-u.ac.jp
First published on 28th May 2012
Abstract
A new environmentally friendly method for the Baeyer–Villiger oxidation of cyclobutanones has been developed. The reaction can be performed at room temperature by using thioureas as catalysts and H2O2 as the oxidant in toluene, and the desired γ-butyrolactone compounds are obtained in high yields.
Introduction
Since its discovery more than a century ago,1 Baeyer–Villiger oxidation has been used as a valuable tool in organic synthesis to transform cyclic or acyclic carbonyl compounds to the corresponding lactones or esters.2,3 A reaction of this type is usually performed with peracids or hydrogen peroxide as an oxidant. However, the former gives carboxylic acids as wasteful co-products. On the other hand, while the latter offers several advantages, i.e., cheap, clean, safe, and green (water is the sole by-product), it mostly requires the use of strong acids or metal-directed promoters due to its low activity.4Recently, considerable attention has been focused on the use of organocatalytic methods, mainly due to their environmentally friendly characteristics.5 Indeed, organocatalytic Baeyer–Villiger oxidation with hydrogen peroxide is considered to be one of the most challenging issues in this field.6,7
In our laboratory, we have been working to develop new methods for carbonyl group functionalization using hydrogen-bonding activation through thiourea-based organocatalysis.8,9 As an extension of these works, we expected that the Baeyer–Villiger oxidation of cyclic ketones with hydrogen peroxide might also be promoted by thioureas (Scheme 1).10 Herein we describe the realization of this expectation.
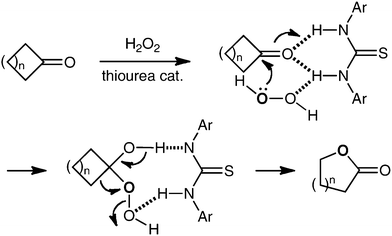 | ||
| Scheme 1 Thiourea-catalyzed Baeyer–Villiger oxidation. | ||
Results and discussion
First, we examined the reaction of 3-phenylcyclobutanone (1a) with aqueous or non-aqueous hydrogen peroxide in the presence of thiourea and related organocatalysts 2 (Chart 1) in toluene at room temperature as a model system. The results are summarized in Table 1.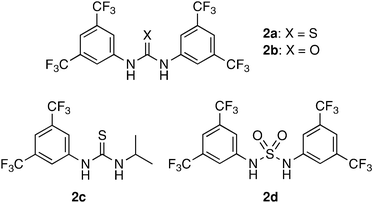 | ||
| Chart 1 Thiourea-related organocatalysts.11 | ||
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst(2) | Methoda | Time (h) | Yield(%)b |
| a Method A: 1a (0.2 mmol), 30% aq H2O2 (1.1 equiv) in toluene (1.0 mL) at r.t.; Method B: 1a (0.2 mmol), H2O2 in Et2O (1.1 equiv) in toluene (1.0 mL) at r.t.; Method C: 1a (0.2 mmol), H2O2 in Et2O (1.1 equiv) in CH2Cl2 (1.0 mL) at r.t. b Isolated yield. c 5 mol% of 2a was used. d 30 mol% of 2a was used. | ||||
| 1 | — | A | 30 | trace |
| 2 | 2a | A | 30 | 59 |
| 3 | 2a | B | 5 | 94 |
| 4c | 2a | B | 7 | 88 |
| 5d | 2a | B | 4 | 89 |
| 6 | 2b | B | 78 | 56 |
| 7 | 2c | B | 24 | 78 |
| 8 | 2d | B | 42 | 70 |
| 9 | 2a | C | 3 | 91 |

While no reaction was observed in the absence of a catalyst, we found that 2a could catalyze the desired reaction using 30% aqueous H2O2 as the oxidant, albeit in rather low yield after 30 h (Table 1, entries 1 and 2). After several experiments to optimize the conditions, we were delighted to find that the use of H2O2 in Et2O as the oxidant12 was most favorable for the present purpose.13 Thus, the treatment of 1a with 1.1 equiv of an ethereal solution of H2O2 (0.5∼0.6 M) in the presence of 10 mol% of 2a in toluene produced γ-butyrolactone 3a in 94% yield after 5 h at r.t. (Table 1, entry 3). With respect to the catalyst loading, we found that at least 10 mol% of the catalyst were necessary to achieve high yield, and a reduction to 5 mol% compromised the reactivity (Table 1, entries 3–5).14
Next, it became clear that there was a significant catalyst effect: urea homologue 2b and half-alkylated 2c were both less effective than 2a (Table 1, entries 6 and 7). Very similar observations were recognized in our previous study on Diels–Alder reactions.9 The fact that sulfamide 2d shows less catalytic activity means that the acidity of N–H is not proportional to the catalyst activity in this transformation (Table 1, entry 8).15 Finally, the use of CH2Cl2 as the solvent gave comparable results, and 3a was obtained in 91% yield after 3 h (Table 1, entry 9). Thus, the best conditions were as follows: H2O2 in Et2O (1.1 equiv) and catalyst 2a (10 mol%) in toluene at r.t.
We initially thought that the catalyst 2 can coordinate with cyclobutanones through hydrogen-bond formation. To confirm this speculation, 13C NMR experiments were performed (Fig. 1). A downfield shift of 0.2 ppm for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and an up-field shift of 0.019 and 0.01 ppm for CH2 and CHPh was observed when complexed with 2a, while 2b showed a rather weak interaction: a downfield shift of 0.076 ppm for CO and ±0.0 ppm for both CH2 and CHPh.
O and an up-field shift of 0.019 and 0.01 ppm for CH2 and CHPh was observed when complexed with 2a, while 2b showed a rather weak interaction: a downfield shift of 0.076 ppm for CO and ±0.0 ppm for both CH2 and CHPh.
 | ||
| Fig. 1 Chemical shift changes (Δδ) observed for the 13C NMR signals (125.8 MHz, CDCl3) of 1a complexed with 2a (30 mol%). | ||
With the optimized reaction conditions in hand, we then investigated the general scope of this chemistry by using various substituted cyclobutanones as substrates (Table 2).















As expected, a variety of aryl and alkyl group-substituted cyclobutanones smoothly underwent Baeyer–Villiger oxidation to give the corresponding γ-butyrolactones 3 in high yields (Table 2, entries 1–8). In all cases, the reaction follows the normal mode of migratory aptitude, but the norbornane-fused cyclobutanone 1i gave a regioisomeric mixture of 3i and 3j in a ratio of 87![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :13 (Table 2, entry 8). Unfortunately, we found that the present method was only successful for cyclobutanone substrates, and other cyclic ketones such as cyclopentanones and cyclohexanones gave only the corresponding H2O2 adducts (>95% conversion).16
:13 (Table 2, entry 8). Unfortunately, we found that the present method was only successful for cyclobutanone substrates, and other cyclic ketones such as cyclopentanones and cyclohexanones gave only the corresponding H2O2 adducts (>95% conversion).16
Finally, to demonstrate the synthetic value of this methodology, we briefly examined the possibility of extending it to asymmetric versions using chiral thiourea catalysts 2e–2h (Table 3).17 However, no remarkable results were obtained, probably due to the less efficient diastereoselective discrimination at the initial stage of the Criegee intermediate formation or the difficulty of controlling subsequent alkyl-group migration.
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst (2) | Time (h) | Yield (%)b | ee (%)c |
a
1a (0.2 mmol), H2O2 in Et2O (1.1 equiv), chiral cat 2 (10 mol%) in toluene (1.0 mL) at r.t.
b Isolated yield.
c Determined by chiral HPLC using Chiralpak AD. Based on our previous data, the absolute configuration of 3a was determined. See Ref. 18. 
|
||||
| 1 | 2e | 24 | 83 | 0 |
| 2 | 2f | 36 | 80 | 0 |
| 3 | 2g | 36 | 75 | 0 |
| 4 | 2h | 24 | 73 | 3 (R) |

Conclusions
In summary, we have developed a new efficient and significantly mild procedure for the Bayer-Villiger oxidation of cyclobutanones using H2O2 in Et2O as the oxidant and 2a as a catalyst. This method is of great value as a metal- and acid-free environment-friendly system.19 Although the extension of this reaction to asymmetric versions is still at an introductory stage, further studies to realize high levels of chiral induction are now in progress in our laboratory.Experimental section
General procedure
To a solution of cyclobutanones (0.2 mmol) and catalyst 2a (10 mg, 0.02 mmol) in toluene (1.0 mL) was added H2O2 in Et2O (0.5 M, 0.44 mL, 0.22 mmol) at r.t., and the mixture was stirred until the reaction was complete. The mixture was then diluted by the addition of Et2O and the excess of H2O2 was quenched by addition of aq Na2S2O3. The mixture was extracted with Et2O, and the extracts were washed with H2O and brine, dried (MgSO4), and concentrated. The crude product was purified by silica gel column chromatography (elution with hexane-AcOEt = 4:1) to afford the pure γ-lactones 3.
3-Phenyl-γ-butyrolactone (3a)20
Colorless solid, mp 45–46 °C (lit.20 40–41 °C); Rf 0.25 (hexane-AcOEt = 4:1); FTIR (KBr) ν 1765, 1496, 1456, 1422, 1355, 1164, 1011 cm−1; 1H NMR (500 MHz, CDCl3) δ 2.66–2.75 (1H, m), 2.91–2.99 (1H, m), 3.76–3.84 (1H, m), 4.26–4.32 (1H, m), 4.66–4.72 (1H, m), 7.23–7.40 (5H, m); 13C NMR (125.8 MHz, CDCl3) δ 35.7, 41.1, 74.0, 126.7 (×2), 127.7, 129.1 (×2), 139.3, 176.4.
3-(4-Methoxyphenyl)-γ-butyrolactone (3b)7b
Colorless solid, mp 68–70 °C (lit.7b 92–94 °C); Rf 0.18 (hexane-AcOEt = 4:1); FTIR (KBr) ν 1767, 1610, 1584, 1514, 1455, 1257, 1178, 1168, 1032, 1017 cm−1; 1H NMR (500 MHz, CDCl3) δ 2.64 (1H, dd, J = 17.5, 9.0 Hz), 2.90 (1H, dd, J = 17.5, 8.5 Hz), 3.74 (1H, quintet, J = 8.5 Hz), 3.81 (3H, s), 4.22 (1H, t, J = 8.5 Hz), 4.64 (1H, t, J = 8.5 Hz), 6.88–6.91 (2H, m), 7.14–7.16 (2H, m); 13C NMR (125.8 MHz, CDCl3) δ 35.9, 40.4, 55.3, 74.3, 114.4 (×2), 127.7 (×2), 131.1, 159.0, 176.5.
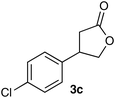
3-(4-Chlorophenyl)-γ-butyrolactone (3c)7b
Colorless solid, mp 51–52 °C (lit.7b 72–74 °C); Rf 0.20 (hexane-AcOEt = 4:1); FTIR (KBr) ν 1775, 1498, 1169, 1092, 1017 cm−1; 1H NMR (500 MHz, CDCl3) δ 2.63 (1H, dd, J = 17.5, 8.5 Hz), 2.94, (1H, dd, J = 17.5, 8.5 Hz), 3.77 (1H, quintet, J = 8.0 Hz), 4.24 (1H, dd, J = 9.5, 7.5 Hz), 4.66 (1H, dd, J = 9.0, 7.5 Hz), 7.17 (2H, d, J = 8.5 Hz), 7.33–7.35 (1H, m); 13C NMR (125.8 MHz, CDCl3) δ 35.6, 40.5, 73.8, 128.0 (×2), 129.3 (×2), 133.5, 137.9, 176.0.
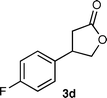
3-(4-Fluorophenyl)-γ-butyrolactone (3d)7b
Colorless solid, mp 63–65 °C (lit.7b 58–60 °C); Rf 0.15 (hexane-AcOEt = 4:1); FTIR (KBr) ν 1770, 1516, 1220, 1166, 1012 cm−1; 1H NMR (500 MHz, CDCl3) δ 2.64 (1H, dd, J = 17.5, 9.0 Hz), 2.93 (1H, dd, J = 17.5, 9.0 Hz), 3.79 (1H, quintet, J = 9.0 Hz), 4.24 (1H, dd, J = 9.0, 8.0 Hz), 4.66 (1H, dd, J = 9.0, 8.0 Hz), 7.04–7.08 (2H, m), 7.19–7.22 (2H, m); 13C NMR (125.8 MHz, CDCl3) δ 35.8, 40.4, 74.0, 115.9, 116.1, 128.2, 135.1, 161.1, 163.0, 176.1.
3-(2-Naphthyl)-γ-butyrolactone (3e)7b
Colorless solid, mp 118–119 °C (lit.7b 116–117 °C); Rf 0.21 (hexane-AcOEt = 4:1); FTIR (KBr) ν 1762, 1160, 1050, 1010 cm−1; 1H NMR (500 MHz, CDCl3) δ 2.79 (1H, dd, J = 17.5, 9.0 Hz), 3.00 (1H, dd, J = 17.5, 9.0 Hz), 3.95 (1H, quintet, J = 8.0 Hz), 4.37 (1H, dd, J = 9.5, 8.0 Hz), 4.74 (1H, dd, J = 9.5, 8.0 Hz), 7.33 (1H, dd, J = 8.5, 2.0 Hz), 7.47–7.52 (2H, m), 7.67 (1H, s), 7.80–7.84 (2H, m), 7.86 (1H, d, J = 9.0 Hz); 13C NMR (125.8 MHz, CDCl3) δ 35.7, 41.2, 73.9, 124.4, 125.5, 126.2, 126.6, 127.6(2), 127.6(6), 129.1, 132.6, 133.3, 136.6, 176.4.
4-Methyl-4-phenyl-γ-butyrolactone (3f)7b
Colorless solid, mp 45–48 °C; Rf 0.31 (hexane-AcOEt = 4:1); FTIR (KBr) ν 1776, 1497, 1381, 1281, 1174, 1021 cm−1; 1H NMR (500 MHz, CDCl3) δ 1.53 (3H, s), 2.69 (1H, d, JAB = 17.0 Hz), 2.93 (1H, d, JAB = 17.0 Hz), 4.42 (2H, AB q, JAB = 9.0 Hz), 7.17–7.19 (2H, m), 7.26–7.30 (1H, m), 7.36–7.39 (2H, m); 13C NMR (125.8 MHz, CDCl3) δ 28.1, 42.0, 44.1, 78.4, 125.1 (×2), 127.2, 129.0 (×2), 144.1, 176.3.
4-Dodecyl-γ-butyrolactone (3g)21
Colorless solid, mp 36–37 °C (lit.21 38–39 °C); Rf 0.31 (hexane–AcOEt = 4:1); FTIR (KBr) ν 1764, 1472, 1464, 1380, 1280, 1187, 1134 cm−1; 1H NMR (500 MHz, CDCl3) δ 0.88 (3H, t, J = 7.5 Hz), 1.26–1.39 (22H, m), 1.41–1.49 (1H, m), 1.56–1.63 (1H, m), 1.71–1.78 (1H, m), 1.82–1.90 (1H, m), 2.33 (1H, ddd, J = 12.8, 7.0, 6.5 Hz), 2.54 (2H, dd, J = 9.0, 7.5 Hz), 4.50 (1H, dq, J = 7.0, 6.5 Hz); 13C NMR (125.8 MHz, CDCl3) δ 14.1, 22.7, 25.2, 28.0, 28.9, 29.3(3), 29.3(5), 29.4(5), 29.5(2), 29.6 (×3), 31.9, 35.6, 81.2, 177.5.
Hexahydrobenzofuran-2-one (3h)22
Colorless oil; Rf 0.22 (hexane-AcOEt = 4:1); FTIR (neat) ν 1777, 1174 cm−1; 1H NMR (500 MHz, CDCl3) δ 1.22–1.31 (2H, m), 1.42–1.54 (2H, m), 1.61–1.75 (3H, m), 2.06–2.10 (1H, m), 2.25 (1H, dd, J = 17.0, 3.0 Hz), 2.36–2.42 (1H, m), 2.62 (1H, dd, J = 17.0, 7.0 Hz), 4.51 (1H, dt, J = 4.5, 4.0 Hz); 13C NMR (125.8 MHz, CDCl3) δ 19.8, 22.8, 27.1, 27.7, 34.8, 37.5, 79.1, 177.6.
Mixture of hexahydro-4,7-methanobenzofuran-2-one (3i) and hexahydro-4,7-methanoisobenzofuran-1-one (3j)23
Colorless oil; Rf 0.25 (hexane-AcOEt = 4:1); inseparable mixture of 3i : 3j = 87:13; FTIR (neat) ν 1772, 1360, 1174, 1026 cm−1.
Acknowledgements
This work was supported in part by the Yamada Science Foundation, and by a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) in Japan. One of the authors (N. S.) is grateful for a Sasakawa Scientific Research Grant from the Japan Science Society.References
- A. Baeyer and V. Villiger, Ber., 1899, 32, 3625 CrossRef.
- L. Kürti and B. Czakó, Strategic Applications of Named Reactions in Organic Synthesis, Elsevier, Amsterdam, 2005, pp 28–29 Search PubMed.
- (a) Reviews: G. R. Krow, Org. React., 1993, 43, 251 Search PubMed; (b) G. Strukul, Angew. Chem., Int. Ed., 1998, 37, 1198 CrossRef; (c) M. Renz and B. Meunier, Eur. J. Org. Chem., 1999, 737 CrossRef CAS.
- (a) Catalytic Oxidations with Hydrogen Peroxide as Oxidant, ed. G. Strukul, Kluwer Academic, Dordrecht, 1992 Search PubMed; (b) R. Noyori, M. Aoki and K. Sato, Chem. Commun., 2003, 1977 RSC; (c) J. Piera and J.-E. Bäckvall, Angew. Chem., Int. Ed., 2008, 47, 3506 CrossRef CAS; (d) R. A. Michelin, P. Sgarbossa, A. Scarso and G. Strukul, Coord. Chem. Rev., 2010, 254, 646 Search PubMed.
- Selected recent reviews: H. Pellissier, Tetrahedron, 2007, 63, 9267 Search PubMed; C. F. Barbas III, Angew. Chem., Int. Ed., 2008, 47, 42 CrossRef CAS; H. Kotsuki, H. Ikishima and A. Okuyama, Heterocycles, 2008, 75, 493 CrossRef; H. Kotsuki, H. Ikishima and A. Okuyama, Heterocycles, 2008, 75, 757 CAS; A. Dondoni and A. Massi, Angew. Chem., Int. Ed., 2008, 47, 4638 CrossRef CAS; P. Melchiorre, M. Marigo, A. Carlone and G. Bartoli, Angew. Chem., Int. Ed., 2008, 47, 6138 CrossRef; S. Bertelsen and K. A. Jørgensen, Chem. Soc. Rev., 2009, 38, 2178 CrossRef CAS; G. Valero, X. Companyó, N. Bravo, A.-N. Alba, A. Moyano and R. Rios, Synlett, 2010, 1883 RSC; A. Moyano and R. Rios, Chem. Rev., 2011, 111, 4703 CAS.
- (a) G.-J. ten Brink, I. W. C. E. Arends and R. A. Sheldon, Chem. Rev., 2004, 104, 4105 CrossRef; (b) T. Seiser, T. Saget, D. N. Tran and N. Cramer, Angew. Chem., Int. Ed., 2011, 50, 7740 CrossRef CAS.
- (a) Y. Imada, H. Iida, S.-I. Murahashi and T. Naota, Angew. Chem., Int. Ed., 2005, 44, 1704 CrossRef CAS; (b) S. Xu, Z. Wang, X. Zhang, X. Zhang and K. Ding, Angew. Chem., Int. Ed., 2008, 47, 2840 CrossRef CAS; (c) S. Xu, Z. Wang, Y. Li, X. Zhang, H. Wang and K. Ding, Chem.–Eur. J., 2010, 16, 3021 CrossRef CAS; (d) S. Xu, Z. Wang, X. Zhang and K. Ding, Eur. J. Org. Chem., 2011, 110 Search PubMed.
- (a) P. R. Schreiner, Chem. Soc. Rev., 2003, 32, 289 RSC; (b) Y. Takemoto, Org. Biomol. Chem., 2005, 3, 4299 RSC; (c) T. Akiyama, J. Itoh and K. Fuchibe, Adv. Synth. Catal., 2006, 348, 999 CrossRef CAS; (d) M. S. Taylor and E. N. Jacobsen, Angew. Chem., Int. Ed., 2006, 45, 1520 CrossRef CAS; (e) S. J. Connon, Chem.–Eur. J., 2006, 12, 5418 CrossRef; (f) A. G. Doyle and E. N. Jacobsen, Chem. Rev., 2007, 107, 5713 CrossRef CAS; (g) H. Miyabe and Y. Takemoto, Bull. Chem. Soc. Jpn., 2008, 81, 785 CrossRef CAS; (h) S. J. Connon, Chem. Commun., 2008, 2499 RSC; (i) S. J. Connon, Synlett, 2009, 354 CAS; (j) K. Etzenbach-Effers and A. Berkessel, Top. Curr. Chem., 2010, 291, 1 CrossRef CAS.
- (a) K. Mori, J. Maddaluno, K. Nakano, Y. Ichikawa and H. Kotsuki, Synlett, 2009, 2346 Search PubMed; (b) K. Mori, T. Yamauchi, J. Maddaluno, K. Nakano, Y. Ichikawa and H. Kotsuki, Synlett, 2011, 2080 Search PubMed.
- In related to this reaction scheme at the first step, Schreiner et al. reported the thiourea-catalyzed acetalization of aldehydes/ketones with alcohols: M. Kotke and P. R. Schreiner, Tetrahedron, 2006, 62, 434 Search PubMed.
- Preparation of these catalysts follows the literature protocol. 2a and 2b: H. Y. Kim and K. Oh, Org. Lett., 2011, 13, 1306. Search PubMed 2c: F. Z. Dorwald, J. B. Hansen, J. P. Mogensen, T. M. Tagmose, B. Pirotte, P. Lebrun, P. De Tullio, S. Boverie and J. Delarge, US Pat, 1997, 55193 Search PubMed. 2d: A. A. Rodriguez, H. Yoo, J. W. Ziller and K. J. Shea, Tetrahedron Lett., 2009, 50, 6830 Search PubMed.
- I. Saito, R. Nagata, K. Yuba and T. Matsuura, Tetrahedron Lett., 1983, 24, 1737 CrossRef CAS.
- The use of t-BuOOH in place of H2O2 was insufficient: the treatment of 1a (0.2 mmol) with t-BuOOH in toluene (13% solution, 1.1 equiv) at r.t. for 30 h gave 3a in 82% yield.
- We did not check the recyclability of the catalyst due to its some decomposition during the reaction.
- The pKa values of selected N–H bonds are reported by Yan et al.: J. Lao, X. Zhang, J. Wang, X. Li, M. Yan and H. Luo, Tetrahedron: Asymmetry, 2009, 20, 2818 Search PubMed
. - R. Criegee, Houben-Weyl, Vol. VIII, Georg Thieme Verlag, Stuttgart, 1968, pp 1–74 Search PubMed.
- Ref. 9b. 2f and 2g: A. Lattanzi, Synlett, 2007, 2106. Search PubMed 2h: M. Shi and X.-G. Liu, Org. Lett., 2008, 10, 1043 Search PubMed.
- T. Shinohara, S. Fujioka and H. Kotsuki, Heterocycles, 2001, 55, 237 Search PubMed.
- During the submission of this paper, a related work has been published: H.-J. Xu, F.-F. Zhu, Y.-Y. Shen, X. Wan and Y.-S. Feng, Tetrahedron, 2012, 68, 4145 Search PubMed.
- T. Satoh, Y. Kamide and S. Sugiyama, Tetrahedron, 2004, 60, 11805 Search PubMed.
- L. J. Gooβen, D. M. Ohlmann and M. Dierker, Green Chem., 2010, 12, 197 RSC.
- S. Fuchs, V. Berl and J.-P. Lepoittevin, Eur. J. Org. Chem., 2007, 1145 CrossRef CAS.
- M. Aoki and D. Seebach, Helv. Chim. Acta, 2001, 84, 187 CrossRef CAS.

| This journal is © The Royal Society of Chemistry 2012 |