Sonoporation of suspension cells with a single cavitation bubble in a microfluidic confinement
Séverine Le
Gac
a,
Ed
Zwaan
b,
Albert van den
Berg
*a and
Claus-Dieter
Ohl
bc
aBIOS The Lab-on-a-Chip group, MESA+ Institute for Nanotechnology, University of Twente, P.O. Box 217, 7500 AE Enschede, The Netherlands. E-mail: a.vandenberg@ewi.utwente.nl; Fax: +31-534893595; Tel: +31-534892691
bPhysics of Fluids, Faculty of Science and Technology, University of Twente, P.O. Box 217, 7500 AE Enschede, The Netherlands
cSchool of Physical and Mathematical Sciences, Division of Physics and Applied Physics, Nanyang Technological University, 6373616 Singapore
First published on 14th September 2007
Abstract
We report here the sonoporation of HL60 (human promyelocytic leukemia) suspension cells in a microfluidic confinement using a single laser-induced cavitation bubble. Cavitation bubbles can induce membrane poration of cells located in their close vicinity. Membrane integrity of suspension cells placed in a microfluidic chamber is probed through either the calcein release out of calcein-loaded cells or the uptake of trypan blue. Cells that are located farther away than four times Rmax (maximum bubble radius) from the cavitation bubble center remain fully unaffected, while cells closer than 0.75 Rmax become porated with a probability of >75%. These results enable us to define a distance of 0.75 Rmax as a critical interaction distance of the cavitation bubble with HL60 suspension cells. These experiments suggest that flow-induced poration of suspension cells is applicable in lab-on-a-chip systems, and this might be an interesting alternative to electroporation.
Introduction
Pore formation (poration) in cell membranes is a key-step for the introduction of foreign substances into cells, transfection of biomolecules, such as DNA fragments (vectors) or molecular delivery. It is widely used in various research areas, such as enzymology, stem cell research, pharmaceutics or food industry. A frequently used method is electroporation, which makes use of short electric field pulses to create pores in vitro in the cell membrane.1 Electroporation has been employed in microfluidic devices in order to enhance the control on the process and thereby its success.2–5 Yet, it implies the integration of microfabricated electrodes resulting in tedious fabrication and complex devices.Sonoporation is an alternative technique that uses acoustic-driven bubble pulsations to create pores in the membrane.6 Most of the previous sonoporation experiments have been performed in a bulk solution on a cell suspension, and using a population of ultrasound-excited microbubbles (contrast agents). A statistical analysis of the results enables one to extrapolate the potential effect of a single bubble to rupture a cell's membrane.
More recently, several groups made use of a single (or few) bubble(s) for sonoporation purposes to gain better insight and control on the phenomena. For instance, Prentice et al. used optical tweezers to trap a single microbubble excited with ultrasound.7 Both Okada et al.8 and van Wamel et al.9 positioned bubbles close to a cell layer with the help of buoyancy, before exposure to sound waves. Ohl et al.10 used shock waves to induce cavitation. Finally, a single bubble was created just above a monolayer of adherent cells for cell poration11 or lysis,12 using a laser pulse. All these recent studies showed successful cell poration/lysis, but were however limited to adherent cells immobilized on a substrate, since the bubble-induced flow without any confinement would result in the motion of suspension cells in the bulk.
Microfluidic systems provide a tool for cell confinement and for keeping track of cells over time. Munoz-Pinedo et al. reported on time-lapse imaging of apoptosis at the single cell level using a shallow microfluidic chamber.13 Valero et al. studied the fate of a small population of cells once they have been electroporated and transfected with a GFP fusion protein.3 Yang et al.14 and Eriksson et al.15 reported on the response of a single or a few cell(s) to environmental changes. Consequently, microfluidics is a promising tool for easily tracking cells after their exposure to a cavitation bubble. Moreover, this novel technique of bubble-induced poration does not require complicated microsystem fabrication, as is the case for electroporation, and it is also applicable to both adherent and suspension cells.
In this paper we report on the combination of sonoporation based on a single laser-induced bubble and microfluidic confinement to porate (single) (suspension) cells in a highly controlled way. We study here HL60 cells that are not immobilized on a substrate but are free to move in solution. The cells are introduced in a microchamber and submitted thereafter to a single bubble, which is created using a laser pulse. Loss of their membrane integrity is probed in two ways: by measuring either (i) release of previously loaded calcein from the cells or (ii) the uptake of trypan blue. After discussing the set-up and technique and their advantages, we describe here two series of experiments. The first one illustrates the poration of calcein-stained cells in a small 100–200 µm size microchamber, while the second aims at determining the interaction distance of the bubble with the cells in a larger (>400 µm) microchamber. To the best of our knowledge, this is the first report on cell sonoporation in microfluidic confinement, as well as the first attempt to porate suspension cells by means of a single cavitation bubble.
Materials and methods
Experimental set-up
Cavitation bubbles are generated by using focused light from a frequency-doubled pulsed laser (Nd : YAG laser at 532 nm, 6 ns pulse duration, Solo PIV, New Wave, CA, USA). Fig. 1 represents the set-up used for creation of cavitation bubbles in a microfluidic system. A microsystem including a microchamber loaded with cells is placed on the stage of a microscope (Carl Zeiss Axiovert 40 CF, Goettingen, Germany) and is positioned planar to the image plane of the microscope objective using a 3-axis translation stage and 2-tilt-axis. The light is focused into the fluid at a precise location in the microsystem using a 10× objective (N.A. 0.25). We have measured (after having removed the microsystem) that the actual laser energy at the focal point was 10–100 µJ, minimum and maximum, used in the experiments. With these energies, we were able to generate bubbles with lifetimes between 10 and 30 µs. In general, the laser energy necessary to reach a certain bubble size depends on the geometry of the channel and the concentration of the light absorbing compound. An even further increase of the laser energy leads to the formation of a more stable gas bubble, which then dissolves slowly due to surface tension. A cooled CCD camera (Sensicam QE, PCO, Weilheim, Germany) records the bubble oscillations and cell displacement within a time interval of circa 10 µs. Illumination is achieved by the means of a strobe light emitting diode (Lumiled V Star, Philips, San Jose, CA, USA). Fluorescent pictures of the cells are taken before creation of the bubble and once the bubble oscillation is finished, by directing light with a switching mirror from a mercury lamp through appropriate filter block(s) onto the dye-loaded cells.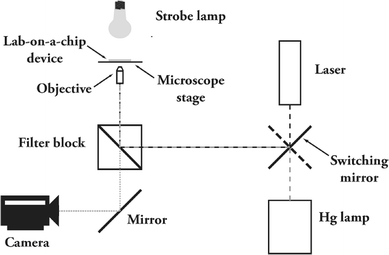 | ||
| Fig. 1 Set-up for creation of cavitation bubbles in a microfluidic system. The microfluidic system is placed on the stage of a microscope, equipped with a 10× objective. Bubbles are created by focusing short pulses of light generated by a green laser (532 nm). Illumination is achieved by using a strobe lamp with a light emitting diode and a camera records the bubble oscillation and motion of cells. For fluorescence measurements, a mercury lamp is used as well as appropriate filter blocks. A mirror is used for switching between the laser and the Hg lamp. | ||
Microsystem fabrication
Microchips are fabricated in an elastomeric material, PDMS (polydimethylsiloxane), using conventional molding techniques.16 In a first stage, silicon molds are fabricated in a clean-room environment using photolithography and dry-etching (Bosch process) techniques starting from a custom design. The height of the mold structure is chosen accordingly to the desired height for the microfluidic channels. After thorough cleaning (10 min in 10% HNO3) the surface of the etched silicon wafer is coated with vapor-phase FDHC (perfluoro-octadecyltrichlorosilane). A two-component kit (Sylgard 184, Dow Corning, Midland, Michigan, USA) is used for PDMS fabrication; it includes a pre-polymer of PDMS, as well as curing agent, which are mixed in a 10:1 weight ratio. The resulting mixture is degassed and poured on the mold, and curing is achieved for 1 h at 100 °C or 2 h at 60 °C. After curing, the PDMS layer is released from the mold, reservoirs punched using a blunt needle, and the PDMS cleaned (5 min in ethanol in an ultra-sonic bath) and carefully dried. Microchips are released and bonded one by one to individual microscope glass slides; for that purpose the surfaces to be bonded with each other are firstly activated using O2-plasma.Cell culture
Human promyelocytic leukemia HL60 cells are obtained from the German Collection of Microorganisms (Braunschweig, Germany). Tissue culture equipment is purchased from Greiner bio-one (Frickenhausen, Germany). HL60 cells are cultured in RPMI-1640 medium (Gibco, Invitrogen, Breda, The Netherlands), supplemented with 10% heat-inactivated and filter-sterilized fetal calf serum, 100 IU mL−1 penicillin, 100 mg mL−1 streptomycin, 2 mM L-glutamine and 250 µg mL−1 fungizone (RPMI+ medium). Supplements and antibiotics are purchased from Invitrogen (Breda, The Netherlands). Cell cultures are maintained at 37 °C in a humidified environment with 5% CO2. The medium is changed every 4–5 days. The cell concentration is assessed to be approximately 106 cells mL−1.Preparation of cell samples
Cell membrane poration is investigated by first staining the cells with calcein, a fluorescent dye, before they are introduced in the system. Therefore, the cells are resuspended in fresh medium with 1 µg mL−1 calcein AM (Molecular Probes, Invitrogen, Breda, The Netherlands). After 30–60 min incubation in the staining solution, the cells are washed twice with fresh medium and resuspended in a fresh medium supplemented with trypan blue (see below). The resulting solution is introduced into the microfluidic chambers.Microsystem preparation
Before cells are introduced into the microsystem the channels are prepared and coated to create a cell-friendly environment. The channels are first cleaned and sterilized with a 70% ethanol solution that is flushed and left in the system for about 5 min. Following this, the channels are rinsed with PBS buffer and filled with a coating solution. We use a BSA (bovine serum albumine, Sigma-Aldrich, Zwindrecht, The Netherlands) solution (3 mg mL−1) in PBS. The coating solution remains for approximately 2 h. Finally, the channels are rinsed with copious amounts of fresh medium before the cells are introduced. Note that the BSA coating makes the PDMS walls hydrophilic.For cavitation experiments, the solution is supplemented with a light-absorbing compound, trypan blue (Acros Organics, Geel, Belgium). Cells are consequently resuspended in fresh medium including trypan blue, before being introduced into the microsystem. The number of cells in the chamber is checked under a microscope before the experiment. When the number of cells is high enough, the flow is stopped by dropping equivalent volumes of liquid on the reservoirs before the microchip is placed on top of the microscope stage.
Results
Studying cell-bubble interactions
We have recently studied the fluid dynamics of cavitation bubbles within a microfluidic confinement using PDMS-based microsystems.17 A single cavitation bubble is nucleated at a precise location in the microfluidic network by means of a highly focused laser (see Fig. 1). The focused light of the laser pulse is absorbed by a suitable compound added to the solution and absorbing at 532 nm to cause rapid deposition of energy in a small volume of liquid leading to stress confinement.18 This creates a rapidly expanding vapor bubble with typical radii of 10–100 µm—the size of the bubble being adjusted via the laser energy—which in turn imparts an accelerating radial flow into the channel. These preliminary experiments showed that although the channel dimensions are small, fluid flow is inertia-dominated and Reynolds numbers (Re) reach about 500,17 as already suggested by Chen et al.19 Furthermore, we found that when a bubble collapses close to a boundary, e.g. a channel wall, a directed and fast jet flow is observed, resembling the jetting phenomenon obtained in 3D cavitation.20In the present paper, we go further and combine cavitation bubbles with biological cells in a microfluidic confinement to study the effect of the bubble on the cells. Bubble-induced flow has previously been reported to result both in cell reversible poration10,11 and lysis.12 A potential mechanism that has been suggested12 is that the bubble-induced flow exerts a shear stress on cells, and this leads to the rupture of their membrane. In a microfluidic system (height <40 µm), the bubble-induced flow is mainly confined in two dimensions: higher shear stress is expected, and this should lead in turn to a higher poration/lysis yield. Moreover, as stressed out in the introduction, sonoporation is limited to adherent cells in a conventional set-up, while confinement in a microfluidic chamber enables one to broaden its application to cells in suspension as well.
Human leukemia HL60 cells (suspension cells) are introduced into a microsystem and confined in a shallow microfluidic chamber with typical heights of 15–40 µm. Thereby, cells remain in the field of view13 and single cells can be tracked confocally during and after the cavitation event. Additionally, the microchamber is surrounded by grids to collect the cells therein (Fig. 2), and the effect of a single bubble can be studied on a small population of cells. Each microdevice also includes inlet and outlet channels upstream and downstream to the microchamber for the introduction of the solutions. For these experiments dealing with biological cells, the liquid is supplemented with trypan blue. This compound is chosen for two purposes in this series of experiments; it absorbs the laser energy (at 532 nm) and it can also be used as a probe for cell poration.2 The experiment is followed/monitored using both white light microscopy to study the bubble expansion and collapse and to track the cells, and in fluorescence mode to detect eventual cellular release of calcein (see below).
 | ||
| Fig. 2 PDMS-based microfluidic systems used for confined sonoporation experiments on suspension cells. (a) Photograph of such a microsystems having dimensions of 1.8 cm × 0.8 cm (one system includes two independent devices); (b) SEM photograph of a 100 µm diameter microchamber where cells are collected, scale bar denotes 25 µm. | ||
Probing poration of cells using calcein AM
In a first series of experiments we endeavor ourselves to study potential cell membrane poration using a single cavitation bubble created in the vicinity of cells in a microfluidic chamber. Cellular membrane poration can be probed either through the release of a compound out of the cells or the entry of a compound into the cell. One probe that is commonly used for this purpose is calcein, and depending whether calcein or calcein AM is used, either the entry or the release of a foreign substance can be assessed. In the former case, the solution is supplemented with calcein and its entry into the cells is studied,5,21 while in the latter case, cells are first loaded with calcein AM to become fluorescent and the release of calcein out of the cells is tracked.2 As studying the entry of calcein implies to work in a fluorescent medium, we have preferred to study the release of calcein out of the cells upon exposure to the bubble. Disruption of the membrane leads to calcein leakage into the medium and subsequently to both a decrease of the fluorescence in the cells and an increase of the fluorescent background signal in the surrounding liquid.Cells are placed in a small microchamber (100–200 µm diameter) and exposed to a single bubble expanding to a maximum diameter of approx. 80 µm. The pictures on Fig. 3 illustrate the bubble dynamics and the motion of the cells upon exposure to the bubble in a 160 µm diameter microchamber. Fig. 3a shows the microchamber before creation of the bubble; it includes 19 viable cells as well as cell debris that can be seen on the outlet grids as dark (trypan blue stained) spots. Fig. 3b represents the bubble 8 µs after its generation; the bubble shows a deformation which is caused by a fast flow through it towards the outlet grids. This so-called jetting flow phenomenon22 is induced by the asymmetry in the radial in-flow leading to flow focusing23 at the side of the bubble farthest away from the grid. This jet flow continues even after the bubble has disappeared and leads to a net displacement of the suspension cells. This net displacement is revealed by inspecting the cell position after the flow has stopped, 60 s later (see Fig. 3c). The largest displacement is observed for the cells closest to the outlet grids, while the two cells labeled 1 and 2 are slightly (or not) transported by the oscillating bubble. On other aspects, no dead cells are found within a few minutes after the bubble oscillation in the chamber.
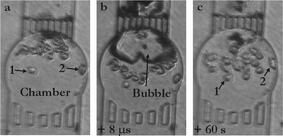 | ||
| Fig. 3 Exposure of HL60 cells to a cavitation bubble in a circular microchamber (diameter 160 µm; height 20 µm). (a) Chamber loaded with cells (19 viable HL60 cells); (b) single cavitation bubble already shrinking and jetting towards the outlet top grid (8 µs after its creation) causing motion of the cells; (c) 60 s after, most of the cells have undergone a net flow and been displaced towards the inlet bottom grid, while only two cells (numbered as 1 & 2) have hardly moved. | ||
Release of calcein out of the cells is depicted in Fig. 4; this figure represents the fluorescent pictures taken before and 85 s after the bubble oscillation (Fig. 4a & b, respectively). A 60% decrease in fluorescent intensity is observed for the set of cells closest to the bubble, while cells farther away (cells 1 & 2 in Fig. 4a & b) show a reduction of <3%. Moreover, dye leakage results in a marked increase in the fluorescence level in the surrounding liquid. Fig. 4c illustrates the variations of the fluorescence intensity before (white circles) and after (dark circles) creation of the cavitation bubble, as a function of the distance to the bubble center. This is done along a 250 µm line starting from the chamber (position ‘×’ in Fig. 4b) down to the microchannel on the other side of the inlet grids, as shown in the insert in Fig. 4c. The fluorescence intensity in the medium increases after the cavitation event and reaches the level observed prior to the cavitation event only far away and outside the chamber.
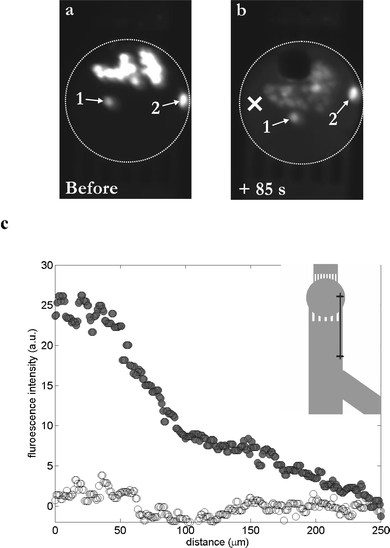 | ||
| Fig. 4 Probing the poration of HL60 cells upon their exposure to a single cavitation bubble in a microchamber. Cells are loaded with calcein AM and are fluorescent. (a) Fluorescent pattern before the creation of the bubble; viable cells are fluorescent. (b) Fluorescent pattern 85 s after exposure to a single cavitation bubble; almost all cells exhibit a lower fluorescent intensity, whereas the surrounding medium exhibits a light background fluorescence, indicating a leakage of dye out of the cells, and consequently poration of the cellular membrane. Two cells (numbered as 1 & 2) remain unaffected. (c) Graph representing the fluorescence level in the surrounding medium as a function of the distance from the center of the chamber (“×” symbol, see part (b)), before (white circles) and 85 s after (dark circles) the cavitation event; in insert: schematic of the 250 µm line along which the fluorescence intensity measurements have been done. | ||
In summary, this experiment demonstrates (i) the transient poration of suspension cells, causing a release of 60% of the dye into the surrounding medium, and (ii) the importance of the cell-bubble distance on the effect of the cavitation bubble on the cells. Nonetheless, the presence of structures in the microdevice changes the flow pattern with the creation of a jet that causes a net displacement of cells located close to the bubble center. This jet prevents the exclusive studying of the effect of a purely radial flow, that should only result in shear stress on cells and not cause any net displacement of the cells.
Determination of the interaction distance of the bubble with HL60 cells
A second set of experiments is carried out in larger microchambers to alleviate any jetting phenomena and to study the effect of a purely radial flow on cells. The chambers are of 400–1000 µm diameter and 35 µm height and the bubble is created far away from the chamber walls to avoid any jetting phenomena. In this case, the cells are not loaded with calcein but the effect of the bubble on cells is tested with trypan blue, whose concentration in the medium is increased to 1% wt. Our first idea was to study cell death as indicated by the cellular uptake of this viability probe. However, trypan blue can also enter cells upon viable poration, as reported by Khine et al.2 Therefore, trypan blue uptake reveals damage (either transient or permanent) of the cellular membrane. Fig. 5 represents the pictures corresponding to one of these experiments in a 600 µm diameter chamber with the cells before (Fig. 5a), during (Fig. 5b) and 47 s after (Fig. 5c) exposure to a 55 µm radius bubble. First of all, the cells in this experiment show no net displacement, as expected: this is consistent with a purely radial flow, where a fluid particle travels the same distance during the bubble expansion and collapse. On the contrary, for a jetting flow (see Fig. 3b), the radial symmetry is lost, which leads to a remnant (vortical) flow after the bubble collapse.17 Consequently, cells experience a net displacement which depends on their initial position to the bubble. Secondly, the cells (group 1) located close (approx. 100 µm) to the bubble at the beginning of the experiment become trypan blue positive after 50 s, as shown with their dark tinge. On the opposite, cells (group 2) located farther away (approx. 250 µm) remain unaffected. These data confirm the aforementioned observation on the importance of the bubble-to-cell distance on the outcome of the sonoporation experiment. If cells are located closer to the bubble they experience a higher shear stress so that they are porated and become trypan blue positive. On the contrary, when cells are farther away they feel reduced shear stress: they remain negative to the staining agent.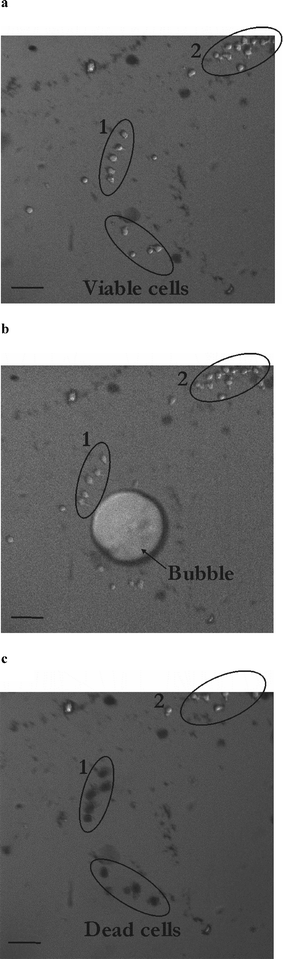 | ||
| Fig. 5 Determination of the interaction distance of a cavitation bubble with HL60 cells. Pictures of the microfluidic chamber (diameter 600 µm; height 35 µm) loaded with HL60 cells before (a), during (b) and 47 s after (c) exposure to a cavitation bubble. Black cells in (c) (group 1) have been affected by the cavitation bubble and are positive to trypan blue, whereas cells in group 2 remain viable. Scale bar denotes 50 µm. | ||
In order to get more statistical data on the effect of the bubble-cell distance, we have repeated this experiment 10 times using a new microsystem, each time filled with fresh HL60 cells. The distance of the cells to the bubble center is measured as a relative parameter d, defined as d = D/Rmax, where D is the initial distance of the cell to the bubble center and Rmax the maximum radius of the cavitation bubble. In total, 170 cells were exposed to a single cavitation bubble. Their distance to the bubble center lies in a range of d from 0 to 6. Fig. 6 represents the outcome of the experiment and the eventual uptake of trypan blue by cells after exposure to the bubble as a function of their relative distance d to the center of the bubble. The graph shows that HL60 cells located farther than d ∼ 4 are not positive at all to trypan blue, at least for the time duration of 50 s we study here. The probability of cell staining increases gradually for short distances, and we can define for this series of experiments a critical interaction distance value below which 75% of the HL60 cells are stained with trypan blue after the cavitation event, as dcrit ∼ 0.75. These data only apply to the system studied here, i.e. suspension HL60 cells and similar studies must be also carried out on other cell lines.
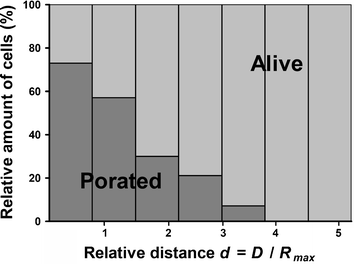 | ||
| Fig. 6 Determination of the interaction distance of the cavitation bubble with HL60 cells. Graph illustrating the poration of cells as a function of their relative distance d (d= D/Rmax) to the bubble center. Rmax stands for the bubble radius at its maximum size and D the distance between the cells and the bubble center. Data were collected for 170 cells through 10 experiments and the viability is averaged for bins that correspond to a 0.75 interval for d. The bins contain respectively 31 (0–0.75), 64 (0.75–1.5), 33 (1.5–2.25), 14 (2.25–3), 17 (3–3.75), 7 (3.75–4.5) and 4 (4.5–5.25) cells. | ||
Discussion
In this paper, we have demonstrated the benefit of a microfluidic confinement to carry out sonoporation experiments on suspension HL60 cells by means of a single cavitation bubble. Two series of experiments illustrate the importance of both the cell-bubble distance and the pattern of the flow induced by the bubble for the outcome of the experiment.Our second series of experiments have enabled us to determine the interaction distance of a cavitation bubble with HL60 cells. However, two effects can still be observed within this interaction distance dcrit, depending on the cell-bubble distance. Cells located closer to the bubble may undergo an irreversible poration (i.e. they die), while cells placed farther away would experience a reversible, and thus viable, poration. So to distinguish between these two cases, another system of probes is needed with two different compounds showing viable and permanent poration. Besides, trypan blue should be replaced by another light-absorbing compound that is large enough not to enter the cells upon poration. For further applications (see below), the entry of trypan blue should be avoided as this compound is poisonous to cells.
Other parameters may also affect the fate of the bubble and the outcome of the bubble oscillation. For instance, the height of the microfluidic chamber is worth studying, as it will influence the strength of the flow: the shallower the system, the stronger the flow for similar-sized cavitation bubbles. The experiments described in this paper have been carried out with systems of 20 or 35 µm height for the first and second series of experiments, respectively, but we have not tried to characterize yet the influence of this parameter on the results of the experiments.
Creating a single bubble using a laser pulse obviously results in a sudden and marked increase of the temperature, leading to the evaporation of the fluid at the laser focus. The potential detrimental effects of this short time duration temperature rise on the cell viability have not been assessed yet in these experiments. The rapid expansion of the bubble is expected to go along with an adiabatic cooling of its inside, thereby limiting the heated spot to the size of the laser focus. Here, as in other experiments with laser-induced cavitation bubbles, a mechanical and not a thermal effect seems to prevail.12,24 Yet, an experimental study of the temperature rise in the liquid is to be done. Similarly, cells absorb at the wavelength of the laser (532 nm) but the laser pulse is not targeted directly on the cells. Subsequently, little or no harm is expected for the cells from the use of laser pulses to generate bubbles in their vicinity.
Cell poration has been probed here by studying the release of a small fluorescent molecule (calcein) or the uptake of trypan blue upon exposure to a bubble, as this release phenomenon is easy to visualize. The same methodology is planned to be applied for similar applications as electroporation, for gene transfection or the delivery of other foreign substances (such as drugs or particles) into cells. Moreover, transfection experiments would help us to demonstrate the long-term viability and functionality of the porated cells, but in that case trypan blue should be avoided as the light-absorbing compound.
The current experiments have also been limited to HL60 suspension cells, as sonoporation of suspension cells has not been described yet. However, the herein described microfluidics-based methodology could be applied to other cell lines, other suspension cells or adherent cells. In the latter case, one can imagine of patterning adherent cells as circles with a diameter chosen, so as to be twice the optimal radius for cell poration. Cavitation bubbles would be created in the middle of the circles for simultaneous poration of every cell. Subsequently, the technique of sonoporation can easily be scaled up as the laser spot can be quickly steered at the desired locations, from one circle of cells to another one, thereby leading to high-yield poration (and for instance transfection) efficiency.
Cell lysis or inactivation is another promising application of this on-chip cell sonoporation.25 This may have many applications in the food industry, where microorganisms/bacteria are inactivated as a pasteurization method of fluid foods. Nonetheless, for this type of application, careful studies on temperature changes in the cell suspension are needed. A second application is cell lysis for analysis purposes. This is of great interest in a microfluidic format, as the cell content can be directly used and analyzed on-line on the chip. A major advantage of cavitation-based lysis is to “catch” the cell at a precise time as the lysis process is very fast; thereby it is possible to have a snapshot of a cell content at a precise time.26
Conclusions
We have consequently described a novel and very promising technique for cell poration that combines a single cavitation bubble and microfluidic confinement. This technique, which appears to be an alternative to electroporation, is very attractive as it can be easily applied in a microfluidic format without a complex fabrication process; the materials only need to be transparent to the laser pulse. Moreover, by carefully choosing the distance between the spot for bubble creation and the cells, the latter can be either killed or porated and transfected, but kept viable. Further work needs to be conducted on the temperature variations in the solution upon laser irradiation and bubble creation. Finally, another series of probes must be found to distinguish between viable and non viable poration within the interaction distance of the cavitation bubble with the cells, and trypan blue should be replaced by another compound.Acknowledgements
We thank Jiajie Li for his help in the fabrication of the silicon masters. C.D.O. is supported by N.W.O. (The Netherlands) through a VIDI grant.References
- E. Neumann, M. Schaeferridder, Y. Wang and P. H. Hofschneider, EMBO J., 1982, 1, 841–845 CAS.
- M. Khine, A. Lau, C. Ionescu-Zanetti, J. Seo and L. P. Lee, Lab Chip, 2005, 5, 38–43 RSC.
- A. Valero, J. N. Post, J. W. Van Nieuwkasteele, W. Kruijer, H. Andersson and A. Van den Berg, Proceedings of uTAS 2006 conference, 2006, p. 22 Search PubMed.
- M. B. Fox, D. C. Esveld, A. Valero, R. Luttge, H. C. Mastwijk, P. V. Bartels, A. van den Berg and R. M. Boom, Anal. Bioanal. Chem., 2006, 385, 474–485 CrossRef CAS.
- M. Khine, C. Ionescu-Zanetti, A. Blatz, L. P. Wang and L. P. Lee, Lab Chip, 2007, 7, 457–462 RSC.
- M. W. Miller, D. L. Miller and A. A. Brayman, Ultrasound Med. Biol., 1996, 22, 1131–1154 CrossRef CAS.
- P. Prentice, A. Cuschierp, K. Dholakia, M. Prausnitz and P. Campbell, Nature Physics, 2005, 1, 107–110 Search PubMed.
- K. Okada, N. Kudo, K. Niwa and K. Yamamoto, J. Med. Ultrason., 2005, 32, 3–11 Search PubMed.
- A. van Wamel, K. Kooiman, M. Harteveld, M. Emmer, F. J. ten Cate, M. Versluis and N. de Jong, J. Controlled Release, 2006, 112, 149–155 CrossRef CAS.
- C. D. Ohl, M. Arora, R. Ikink, N. de Jong, M. Versluis, M. Delius and D. Lohse, Biophys. J., 2006, 91, 4285–4295 CrossRef CAS.
- R. Dijkink, E. Nijhuis, S. Le Gac, A. van den Berg, I. Vermes and C.-D. Ohl, CAV 2006 conference, Wageningen, The Netherlands, 11–15, September 2006 Search PubMed.
- K. R. Rau, P. A. Quinto-Su, A. N. Hellman and V. Venugopalan, Biophys. J., 2006, 91, 317–329 CrossRef CAS.
- C. Munoz-Pinedo, D. R. Green and A. van den Berg, Lab Chip, 2005, 5, 628–633 RSC.
- M. S. Yang, C. W. Li and J. Yang, Anal. Chem., 2002, 74, 3991–4001 CrossRef CAS.
- E. Eriksson, J. Enger, B. Nordlander, N. Erjavec, K. Ramser, M. Goksor, S. Hohmann, T. Nystrom and D. Hanstorp, Lab Chip, 2007, 7, 71–76 RSC.
- C. S. Effenhauser, G. J. M. Bruin, A. Paulus and M. Ehrat, Anal. Chem., 1997, 69, 3451–3457 CrossRef CAS.
- E. Zwaan, S. Le Gac, K. Tsuji and C. D. Ohl, Phys. Rev. Lett., 2007, 98, 254501 CrossRef.
- G. Paltauf and P. E. Dyer, Chem. Rev., 2003, 103, 487–518 CrossRef CAS.
- Y. H. Chen, H. Y. Chu and I. Lin, Phys. Rev. Lett., 2006, 96, 034505 CrossRef.
- M. S. Plesset and A. Prosperetti, Annu. Rev. Fluid Mech., 1977, 9, 145–85 Search PubMed.
- L. Valat, S. Toutain, N. Courtois, F. Gaire, E. Decout, L. Pinck, M. C. Mauro and M. Burrus, Plant Sci., 2000, 155, 203–212 CrossRef CAS.
- J. R. Blake and D. C. Gibson, Annu. Rev. Fluid Mech, 1987, 19, 99–123 Search PubMed.
- B. W. Zeff, B. Kleber, J. Fineberg and D. P. Lathrop, Nature, 2000, 403, 401–404 CrossRef CAS.
- A. Vogel, S. Busch, K. Jungnickel and R. Birngruber, Lasers Surg. Med., 1994, 15, 32–43 CrossRef CAS.
- M. Fox, E. Esveld, R. Luttge and R. Boom, Lab Chip, 2005, 5, 943–948 RSC.
- C. E. Sims, G. D. Meredith, T. B. Krasieva, M. W. Berns, B. J. Tromberg and N. L. Allbritton, Anal. Chem., 1998, 70, 4570–4577 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2007 |