
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Site-selective cleavage of peptides and proteins targeting aromatic amino acid residues
Ayan Bandyopadhyay
 ab and
Rajib Sarkar
*bc
ab and
Rajib Sarkar
*bc
aDepartment of Chemistry, Chapra Government College, Nadia-741123, West Bengal, India
bDepartment of Higher Education, Government of West Bengal, India. E-mail: rajibsarkar.org@gmail.com
cDepartment of Chemistry, Muragachha Government College, Nadia-741154, West Bengal, India
First published on 25th March 2025
Abstract
The site-selective cleavage of peptides and proteins at specific amino acid residues is an important strategy for the modification of biomolecules as it can potentially transmute the reactivity profile of the whole molecule. Moreover, precise cleavage of a specific amide bond in peptides and proteins has enormous applications in the domains of chemical biology, genetics, and protein engineering. Among the 20 proteinogenic amino acids, tryptophan (Trp, W), tyrosine (Tyr, Y), phenylalanine (Phe, F) and histidine (His, H) are classified as aromatic amino acids that maintain the function of protein folding through hydrophobic and π–π interactions. Thus, scissoring at a specific site of an aromatic amino acid may alter the structure and function of a peptide or protein. In the last 60–70 years, great success has been achieved in the development of methods for the aromatic amino acid (AAA)-selective cleavage of peptides and proteins. Generally, aromatic side chains are derivatized in the presence of specific reagents. Consequently, either the downstream or the upstream amide bond of the aromatic side chain is activated, and hydrolysis of the amide bond splits the peptide. Unfortunately, a systematic review covering this methodological development of the AAA-selective fission of peptide is lacking to date. Thus, in this review, we aim to showcase the up-to-date progress in the site-selective rupture of peptide bonds at aromatic amino acid residues with an emphasis on the postulated mechanisms, enabling future researchers to further drive progress in this research field.
 Ayan Bandyopadhyay | Ayan Bandyopadhyay obtained his M.Sc. in Chemistry in 2005 and received his Doctorate in 2013 in the area of asymmetric synthesis of biologically important natural products from the University of Kalyani, India. After completing his PhD, Dr Bandyopadhyay joined the same university as a CSIR-RA. He was awarded the D. S. Kothari Postdoctoral Fellowship and DST-Fast Track Young Scientist grant in 2015. Subsequently, he joined West Bengal Education Services as an Assistant Professor. His research interests include the development of asymmetric synthesis, and modification of biomolecules and natural products employing organocatalysts, C–H activation, and green chemistry. |
 Rajib Sarkar | Rajib Sarkar completed his Master of Science in Chemistry from Jadavpur University, Kolkata in 2012. After completing his PhD in Bioorganic and Supramolecular Chemistry from the Indian Institute of Science Education and Research, Kolkata in 2015, Dr Sarkar joined West Bengal Education Services as an Assistant Professor. He is a life-member of the Indian Chemical Society. His research group is engaged in developing smart protocols for the modification of biomolecules and natural products, employing organocatalysis and C–H activation. His research interest also includes supramolecular chemistry, studies of catalytic behavior of nanoparticles, and green chemistry. |
1. Introduction
Modification of a polypeptide in a specific amino acid residue can unlock a variety of chemical compounds with structural diversities and profound biological activities. Alteration of peptides and proteins can be achieved by installing new functional groups or through the bio-conjugation of small molecules into the core of amino acid residues and proteins,1–7 cyclization of polypeptides8–10 or cleaving long peptides or proteins.11–14 Among these modification techniques, the site-specific cleavage of peptide linkages is a noteworthy tool, which has exhibited promising applications in proteomics,15 site-selective derivatization,16,17 and design of novel therapeutics.18–22 Fission of peptide bonds produces new peptide fragments with a modified N- or C-terminus. Sometimes, some of the side chains of the residues are also modified during the cleaving process. Generally, these alterations metamorphose a molecule into a reactive intermediate, which can be further functionalized as drug leads and therapeutic agents.17,23 Moreover, the cleavage of the peptide bonds at specific residues followed by amino acid analysis or liquid chromatography with tandem mass spectrometry (LC-MS/MS) is a classical technique for the determination of the sequence of unknown peptides and proteins.24Amide bonds are one of the most fundamental and crucial chemical bonds in nature for the existence of life. They are very stable, exhibiting a half-life of approximately 350–600 per year for spontaneous hydrolysis at room temperature (RT) and neutral pH.25 As a result, conventional protocols for amide hydrolysis require harsh reaction conditions.26 However, nature can cleave peptides and proteins through various enzymatic processes under physiological conditions.
Aromatic amino acid (AAA) residues are some of the most important segments of proteins. Biomolecules that contain AAA, possess some unique properties and functions. Thus, the rupture of selective sites in the AAAs of peptides and proteins has long been a topic of interest for chemists and biologists. This particular research area emerged in the 1950s. The aromatic side chains of W-, Y-, F- and H-residues were targeted with chemoselective reagents, activating their derivatives at either the upstream or downstream amide bond. For example, oxidative halogenation of the side-chains of the W- and Y-residues can unlock the activation of the downstream amide bond. Alternatively, co-ordination of the imidazole side-chain of the H-residue with suitable metal ions can activate both the upstream and downstream amide bond depending upon the reaction conditions. Thus, the residue-specific and chemoselective scissoring of amide bonds became possible. Thereafter, numerous non-enzymatic protocols were developed gradually. Unfortunately, this research field was neglected for a long time. However, with the discovery of new proteins and the utilization of fragmented proteins as drug leads, recently this field has emerged again. Lots of protocols for the site-selective scissoring of peptides have been developed in the last two decades. An elegant book chapter was contributed by the Kanai group, which summarized the chemical approaches utilized in the site-selective cleavage of peptides and proteins over the last 10 years (2005–2015).27 Although not specifically focused on all AAAs, in 2023, the Brimble group published a review, in which a small section addressed the latest advances in the selective cleavage of peptides and proteins at the tyrosine sites only.28 The advancements in the site-selective cleavage of peptides at the AAA residues in recent years as well as their enormous application in the fields of drug design and proteomics necessitate an up-to-date summary of the current state of this field. We hope that this article will quench the thirst of young researchers.
2. Importance of site-selective cleavage of peptides at aromatic amino acid residues
As is known, among the 20 proteinogenic amino acids, the W-, Y-, F- and H-residues are categorized as AAA given that they each contain a side chain that is connected with an aromatic moiety. These residues are often acquired in helical and β-barrel membrane proteins and possess unique physicochemical properties. Their presence is very important for protein–protein interactions (PPI) in biological systems.29,30 The indolyl side chain in the W-residue contributes to PPI through π-interactions, hydrophobic interactions and hydrogen bonding.31 Thus, it has the potential to bind with a large number of residues simultaneously owing to its comparatively larger size than the other residues.32 Alternatively, the phenolic side chain of the Y-residue is amphiphilic in nature. Its hydroxyl group enhances the hydrophilic character by constructing hydrogen bonds, while its aromatic counterpart creates a balance by contributing hydrophobic interactions. In the context of PPI, the presence of the F-residue is indeed interesting. Despite its phenyl ring, its frequency in PPI is still one-third that of the Y-residue. The imidazole-tie knotted H-residue can interact with nonpolar and aromatic groups with its heteroaromatic moiety, while its heteroatoms can be involved in hydrogen bonding. Utilizing various protonated states of the imidazole segment, it can be engaged in salt-bridges with acidic groups of other biomolecules33 (Fig. 1). Actually, AAAs force the polypeptide chain to adopt a specific 3-dimensional structure through various covalent interactions such as hydrogen bonding, π–π stacking interactions, and hydrophobic interactions. Besides, they can induce interactions with specific reagents to. For example, W- and Y-residues react with N-bromosuccinimide (NBS) to afford the corresponding bromo-derivative that activates the downstream amide bond to cleave (Fig. 1). Similarly, H-residues can co-ordinate with selective metal ions to break selective amide bonds. Undoubtedly, AAAs play crucial roles and dictate the function of proteins in biological systems. Thus, scissoring at that specific site can snatch numerous inner information from the core of protein molecules.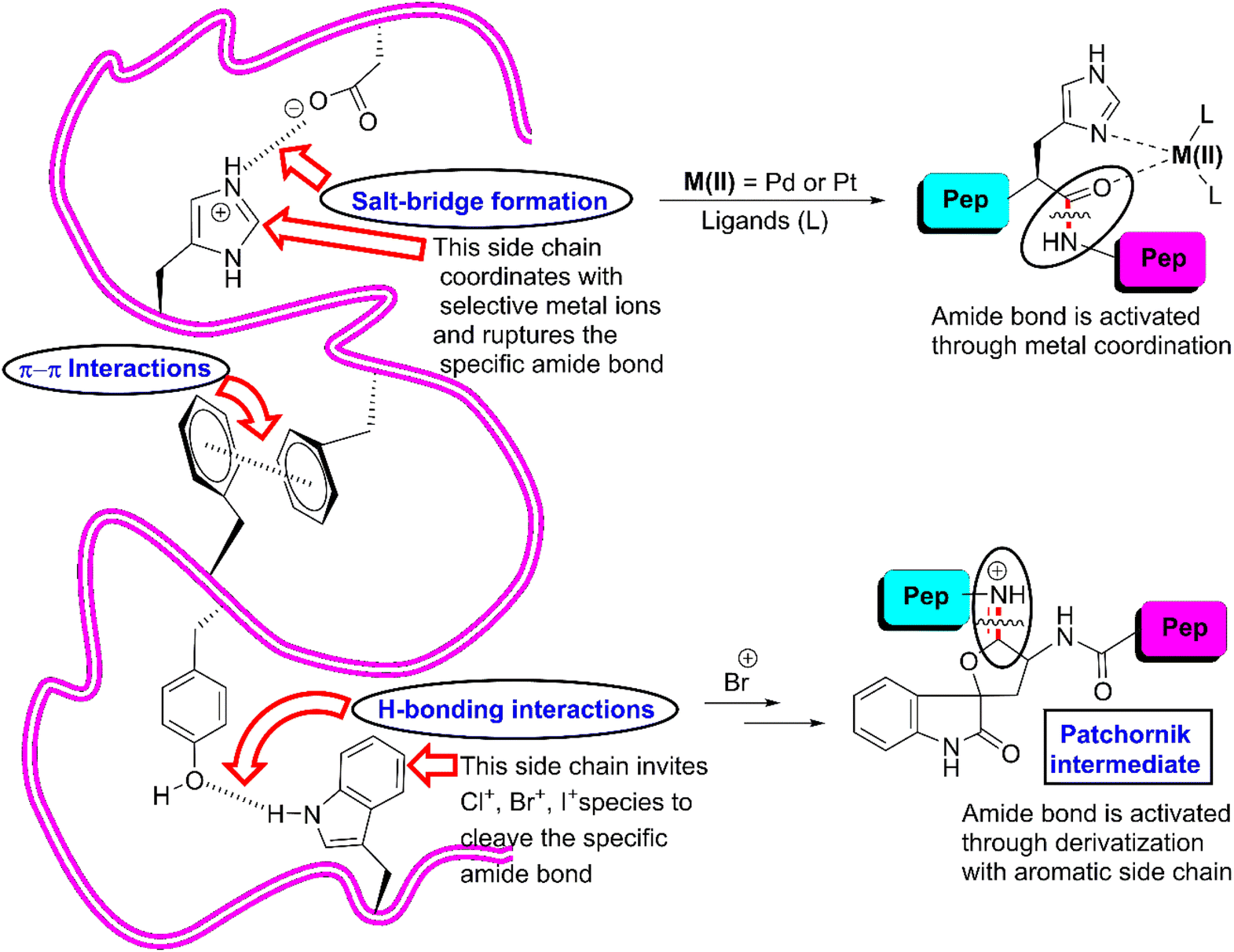 | ||
| Fig. 1 Key interactions exerted by AAAs that fix a polypeptide chain as well as activate amide bonds. | ||
3. Advances in site-selective cleavage of peptides at aromatic amino acid residue
3.1. Tryptophan
Tryptophan (Trp, W) is an essential amino acid, which is inserted into protein molecules by encoding the codon UGG. The W-residue is rare in proteins, but it plays significant structural or functional roles whenever it appears.34 Thus, the investigation of the specific function of W-residues in peptides and proteins is of growing interest to chemists as well as biologists. In this context, W-selective cleavage by means of various chemical, electrochemical, or chemoenzymatic techniques, followed by deep analysis may solve these purposes. The strategies that promote the W-selective fission of peptide bonds in peptides are showcased below. | ||
| Fig. 2 W-selective scissoring of peptides/proteins through chemical oxidation. [Reaction conditions: dNBS, aqueous acetate buffer (pH 4), RT, <1 min, 5–8%; eNBA, acidic buffer (pH 4), RT, 15–20 min, 20–40%, fNBS, 8 (M) aqueous urea, RT, 200 min, yield not reported; gBNPS-skatole/AcOH, RT, 20–30 min, 15%; hTBC/AcOH (pH 3), RT, 5–15 min, 50%; iNCS/50–80% acetic acid (pH 4–5), RT, 30 min, 35–40%; jNCS, 4.68 (M) urea, 27.5% acetic acid, RT, 30 min, 50%; kNaIO4, acetate buffer (pH 5), 0 °C, followed by acidic hydrolysis at RT, 92–94% at W-7 & 17–22% at W-14 of SWM; lOIBA, 80% acetic acid, RT, 30 min, 70–100%]. | ||
In 1964, Funatsu et al. introduced another mild oxidizing reagent, N-bromourea (prepared by adding NBS to aqueous urea), the reactivity of which is almost 200 times less than that of NBS (Fig. 2, path a).38 As a result, this reagent facilitates the cleavage of tryptophanyl peptide bonds without scissoring the tyrosinyl or histidinyl peptide bonds.
In 1970, the Omenn group reported the use of a sophisticated oxidizing agent, BNPS-skatole, a bromine adduct of 2-(2-nitrophenylsulfenyl)-3-methyl-3-indol, for the selective scissoring of the single tryptophan residue in the staphylococcal nuclease in 50–70% acetic acid (Fig. 2, path a).39 Later, the same reagent was utilized by Burnett et al. to split the A1 protein of bovine and human myelin.40 BNPS-skatole in 50% acetic acid was also applied by Prof. Angelo Fontana to scissor the W-selective sites of A1 encephalitogenic protein and horse heart cytochrome.41
Soon, the Patchornik group introduced another mild brominating reagent, 2,4,6-tribromo-4-methylcyclohexadieno (TBC), which can also cleave the tryptophanyl peptide bonds in protein in 65% acetic acid without scissoring the tyrosinyl or histidinyl peptide bonds (Fig. 2, path a).42 This reagent oxidizes methionine to methionine sulfoxide and cysteine to cysteic acid but never ruptures other peptide bonds.
In 1976, Shechter et al. cleaved the tryptophanyl peptide bonds utilizing 2 equiv. N-chlorosuccinimide (NCS) at pH 4–5 (Fig. 2, path b).43 Fortunately, all other peptide bonds are resistant to rupture by this reagent. Under these conditions, only the M- and C-residues are oxidized keeping the other amino acid residues intact. Later, horse heart cytochrome c was successfully fragmented by Lischwe et al. and Savige et al. by applying NCS/urea44 and DMSO/HX45 (X = Cl and Br) reagents, respectively (Fig. 2, path b). After investigating all these halogen-mediated reactions, it can be concluded that tryptophanyl peptide bond rupture occurs only when the peptide/protein attains the “Patchornik intermediate” and NCS in acetic acid is the most selective reagent for the cleavage of the tryptophanyl peptide bonds in proteins.
In 1967, M. Z. Atassi designed a protocol for the W-selective cleavage of sperm whale myoglobin (SWM) in acetate buffer at pH 5 (Fig. 2, path c).46 The protein was treated with sodium metaperiodate (NaIO4) at 0 °C for 7 h, which modifies the indole side chains of the W-residues into oxindole moieties that are structurally similar to the Patchornik intermediate. Upon stirring the oxidized apomyoglobin in 0.1 (N) HCI for 24 h at 25 °C, intact C-terminal peptide fragment 9 and modified N-terminal peptide segment 8 containing spiro-2-oxindole-lactone moieties were obtained. Sequencing of the fragmented peptides was accomplished, which revealed that the M- and Y-residues were modified during the reaction. The authors hoped that this procedure will be applicable to several biomolecules.
In 1977, Ozols et al. developed a methodology for the quantitative cleavage of the tryptophanyl peptide bonds in peptides and proteins with concentrated CNBr in the presence of anhydrous heptafluorobutyric acid (C3F7COOH).47 The investigators successfully applied their protocol for the quantitative W-selective cleavage of cytochrome b5. However, the selectivity of the reagent is hampered in the presence of M-residues.
In 1979, Mahoney and Hermodson claimed that o-iodosobenzoic acid (OIBA) is an effective and specific cleavage agent for tryptophanyl peptide linkages (Fig. 2, path-c).48 However, Wachter et al. proved that OIBA possesses lower selectivity than that reported by Mahoney and Hermodson because the selectivity of this reagent is disturbed in the presence of Y- and H-residues.49 Johnson et al.50 and the Fontana group51 confirmed the observation by Wachter et al. Later, Mahoney et al. improved their protocol by adding a scavenging agent of OIBA in the reaction mixture.52 Actually, excess OIBA cleaves the tyrosyl residues present in polypeptides. Thus, by adding p-cresol, the side reaction can be stopped. Truly, OIBA is a good oxidizing agent. It oxidizes the indol ring of the W-residue into an oxindole moiety, which approaches the Patchornik intermediate, and eventually cleave the tryptophanyl peptide bonds.
In 1966, Previero et al. ruptured C-terminal tryptophyl peptide bonds by passing a stream of ozone gas through a formic acid solution of tryptophyl peptide and resorcinol at 8–10 °C, followed by hydrolysis in strongly basic medium at 100 °C (Fig. 3).53 The reaction between the indole ring and ozone furnishes the N-formylkynurenine (NFK) intermediate 12 either through a dioxetane intermediate (10) or through 3-hydroperoxy intermediate (11). Upon treatment with NaHCO3–Na2CO3 at 100 °C, NFK ruptured into intact C-terminal peptide 9. However, the drawback of the protocol is that under these harsh conditions, denaturation of the protein can occur and other peptide bonds can also be cleaved. Thus, to avoid severe alkaline hydrolysis, Morishita et al. modified this protocol by treating NFK derivatives with hydrazine54 under much milder conditions to obtain N-terminal modified peptide segment 13 and C-terminal intact fragment 9 at RT. Fortunately, the modified protocol provided a better result for the cleavage of short peptides and W-bearing peptides of up to five residues were successfully cleaved at the specific site. However, both protocols were not tested for proteins or polypeptides.
 | ||
| Fig. 3 W-selective scissoring of peptides through NFK formation. | ||
Inspired by the coordinating ability of indole derivatives with metal ions in biological systems, the Kostic group developed an excellent protocol for the W-selective scissoring of peptides.55 When the Pd(II)-complex cis-[Pd(en)(sol)2]2+ was mixed with peptides bearing a W-residue in acetone, complex 14 was formed exclusively (Scheme 1). The formation of 14 was guided by the stereochemical preference of the six-membered ring (14) over seven-membered cyclic moieties (14A). In this complex, the indole moiety exists in its tautomeric form and the amide carbonyl nicely coordinates with the metal ion. This coordination converts the amide carbonyl to more electrophilic. As a result, upon hydrolysis with water, the C-terminal side of the W-residue was fragmented into 9 and 15 avoiding the N-terminal end. The yield of the reaction depended on the amino acid present next to the W-residue. Amino acids containing a bulky side chain reduced the yield of the reaction. In this reaction, the solvent choice is also a significant issue. Water drastically inhibits the tryptophan coordination with Pd(II), given that water itself can coordinate with the central metal ion. Besides, this methodology becomes vague if methionine and histidine residues are present in the peptide chain given that the coordinating ability of these amino acids with Pd(II) is greater than tryptophan. Later, the investigators investigated this protocol applying Pt(II)-complexes and obtained similar results.56
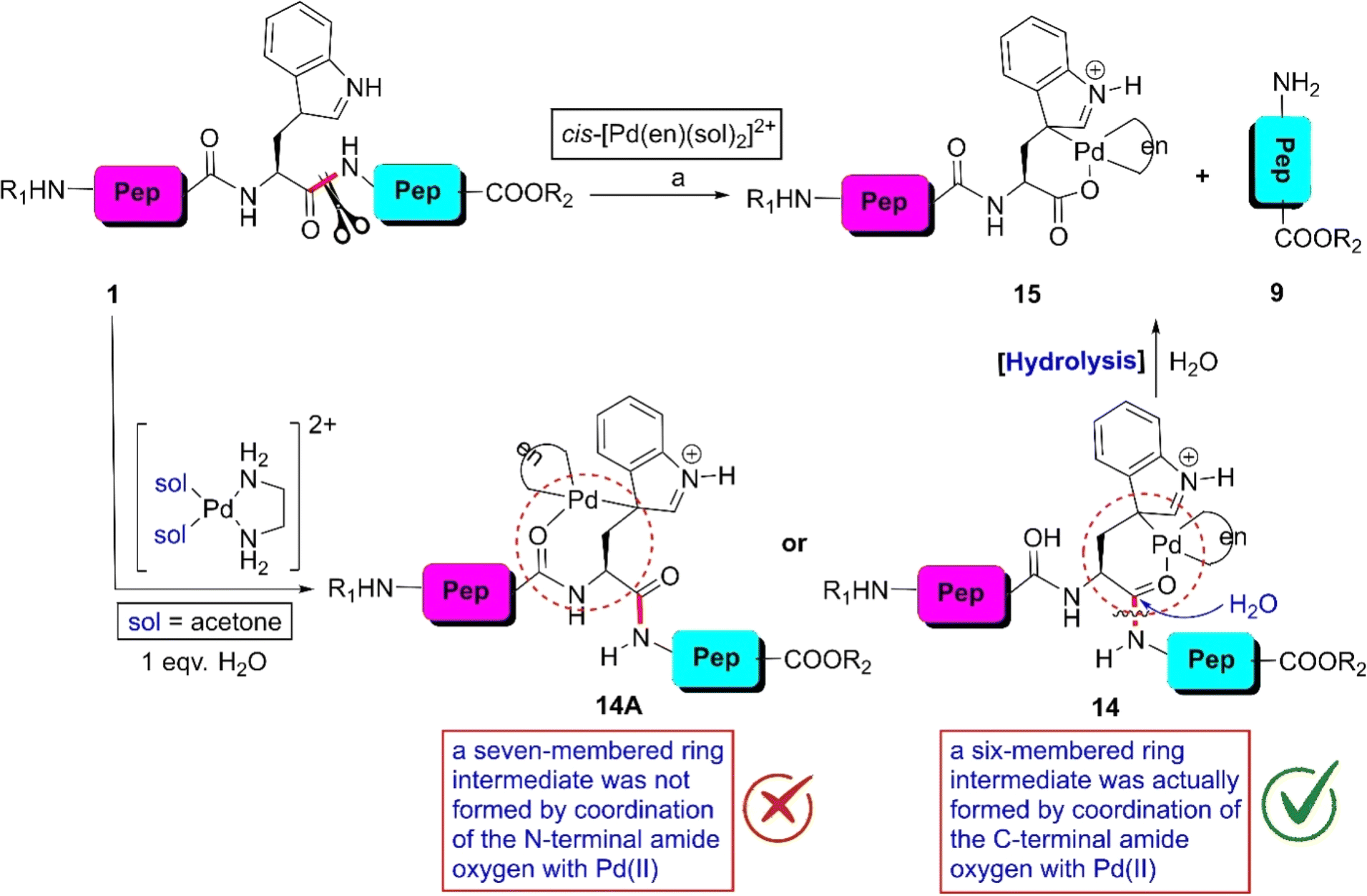 | ||
Scheme 1 W-selective scissoring of peptides through metal complexation. Reaction conditions: (a) peptide![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :metal complex = 1:1, acetone-d6, DClO4, 50 °C. (This experiment was performed in an NMR tube). :metal complex = 1:1, acetone-d6, DClO4, 50 °C. (This experiment was performed in an NMR tube). | ||
 | ||
| Scheme 2 W-selective cleavage of peptide bond via electrochemical approach. Reaction conditions: (a) divided cell setup, graphite-anode, Pt-cathode, +1.5 V potential, Britton–Robinson buffer (pH 3). | ||
In the case of electrochemical cleavage of W-bearing peptides, the reaction starts with the anodic oxidation of the indole moiety (1) in acetonitrile–water (1:1) solvent in the presence of 0.1% formic acid (pH 2.8) to generate oxindole intermediate 19 through several reactive species (Scheme 2). The reaction proceeded in a divided cell setup consisting of a graphite-anode and a Pt-cathode, applying a potential of +1.5 V. Next, 19 was transformed into the Patchornik intermediate (6) involving the downstream amide bond. Ultimately, activation of the amide bond furnished fragment 8 and fragment 9 through hydrolysis. It was observed that the efficiency of the reaction was the maximum at pH 3.
Several larger peptides and proteins of various sizes and compositions such as neuropeptide Y, insulin, RNase I, α-lactalbumin, apomyoglobin, and lysozyme were site-selectively cleaved as well as analyzed by Permentier and Bruins utilizing online electrochemistry-mass spectrometry (EC-MS) technology.64 The oligopeptides/proteins dissolved in water–acetonitrile–formic acid solutions (pH 2.8) were passed through a syringe pump at a flow rate of 2 μL min−1 connected with a Coulochem 5021 conditioning cell containing a porous graphite working electrode, a palladium auxiliary electrode, and a palladium reference electrode. The cell potential was varied from 0.8 to 1.2 V. The oxidized products analyzed by offline liquid chromatography-mass spectrometry (LC-MS) experiments ensured that W-selective peptide bond cleavage occurred successfully. Applying this protocol, a series of tripeptides consisting of W-residues (LWL, GWG, EWE, and KWK) were site-selectively cleaved as well as analyzed by the Bischoff group.65 Unlike the experiments reported by Permentier,64,66 these investigators scanned several adjacent amino acids with respect to the W-residue to verify the effect of size and charge on cleaving efficiency of the tripeptides. The effects of pH and electrolyte on the scissoring efficiency were also checked. Unstable reaction products were detected by online EC-MS, while isobaric oxidation products were separated by LC experiments. The LC-(MS/MS) technique accurately determined the yields of the reaction and product distribution. Based on this information, the authors proposed the reaction mechanism, as shown in Scheme 2. The advantage of this protocol is that the cleaving technique is fully instrumental. Thus, it has the potential for full automation. Moreover, the speed of the analysis is very rapid and the Y-selective peptide bond can also be ruptured by simply tuning the potential range. However, this protocol provides lower yields compared to enzymatic and chemical methods due to the competing side-chain oxidation reactions.
 | ||
| Scheme 3 W-selective cleavage of peptide bond via chemoenzymatic approach. Reaction conditions: (a) enzyme (LPO or HRP), KI, H2O2, acetate buffer (pH 5), RT, 10 min, 30–40%. | ||
In conclusion, in Section 3.1, the most commonly employed chemical protocol for the cleavage of the tryptophanyl peptide bond is chemical cleavage using OIBA due to its high yield and specificity compared to other methods.
3.2. Tyrosine
The tyrosine residue possesses a phenolic side chain consisting of some special features. Firstly, its aromatic ring is electron-rich, which promotes the tyrosine-selective sites of peptides to be ruptured by chemical approaches. Secondly, the redox-active behavior of the phenolic moiety provides an opportunity to scissor the tyrosyl-peptide bonds by employing sophisticated electrochemical technologies. Recently, an enzyme has been discovered that can lacerate the tyrosine-tagged sites by chemoenzymatic techniques. The strategies that initiated the Y-selective cleavage of peptide bonds in peptides are showcased.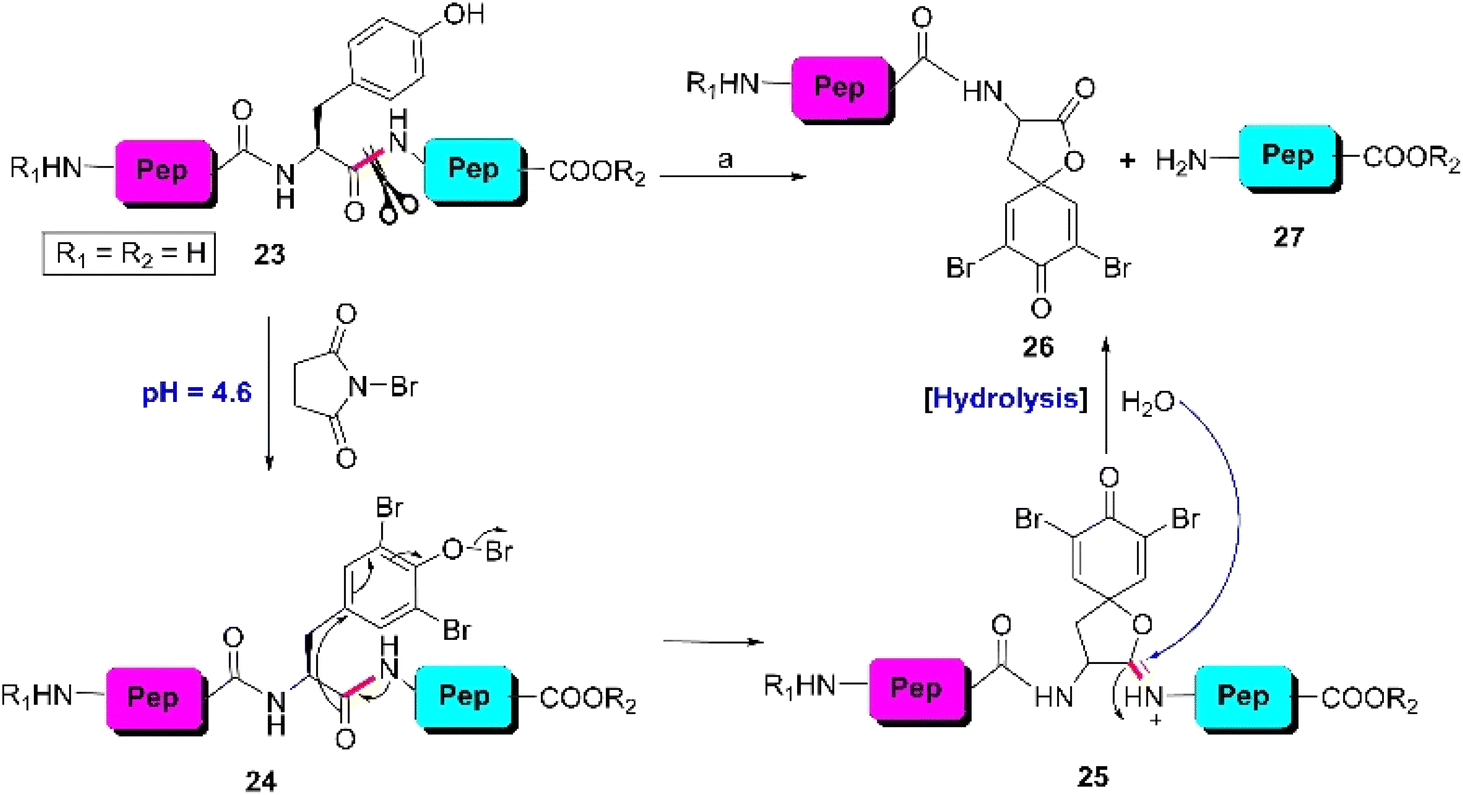 | ||
| Scheme 4 Y-selective cleavage of the peptide bond through oxidative bromination. Reaction conditions: (a) NBS, 20% acetonitrile–acetate buffer (pH 4.6), RT, 10–30 min, 40–50%. | ||
In 1969 Junek et al. replaced NBS with N-iodosuccinimide (NIS) and obtained similar results.73
In the mid-80's, Moriarty et al. observed that among the 20 proteinogenic amino acids, tyrosine selectively reacts with (diacetoxyiodo)benzene in the presence of alcoholic KOH to yield cyanomethylphenol (Scheme 5). Applying this reaction, the investigators tactfully cleaved a series of unprotected dipeptides (28) containing an N-terminal Y-residue under mild conditions.74 The reaction was initiated with the coordination of the hydroxyl group of the Y-residue with the iodine(III) center. Then, reductive elimination of phenyl iodide from intermediate 29 produced reactive species 4-methylenecyclohexadienone (30) and 31. Immediately, 30 transformed into 4-(methoxymethyl)phenol (32) in the presence of alkaline MeOH and 31 was converted into the corresponding C-terminal amino acid (34) through oxidation followed by immediate hydrolysis and decarboxylation (Scheme 5). However, due to the limited substrate scope for N-terminal tyrosyl peptides, no additional studies or applications of this reaction have been reported thus far.
 | ||
| Scheme 5 Y-selective cleavage of the peptide bond through oxidation using hypervalent iodine. Reaction conditions: (a) PhI(OAc)2, KOH–MeOH, 0–5 °C, 1.5 h, 60–80%. | ||
Recently, the Brimble group discovered that Dess–Martin periodinane (DMP) is an excellent reagent for the selective scission of the N-terminal amide bond of Y-residues in a mixture of DMSO and PBS buffer in a neutral medium (pH 7.0) at 40 °C.75 The rupture of the tyrosinyl peptide bond by DMP provided C-terminal peptide fragment 44 bearing an unusual hyperoxidized tyrosine moiety, 4,5,6,7-tetraoxo-1H-indole-2-carboxamide (TICA), together with unaltered N-terminal peptide fragment 45 (Scheme 6). Compared to the other Y-selective peptide bond cleavage protocols, it exhibited high selectivity for Y-residues together with high functional group tolerance of natural and diverse unnatural amino acids. During DMP-mediated oxidation, except for Cys, side chains of Trp, Ser, Thr, etc. remained unaffected. However, free peptide N-terminal amine impedes the cleavage of the tyrosinyl peptide bond. As a result, protection of N-terminal amine by the acetyl group becomes an unavoidable job before executing the DMP-mediated reaction. Applying this protocol, oligopeptides with up to 30 amino acid residues were modified easily. Besides, using DMP, naturally occurring cyclic peptides such as dichotomin J (isolated from Stellaria dichotoma76), szentiamide (isolated from Xenorhabdus szentirmaii77), and antifungal cyclic lipopeptide iturin A78 were successfully ruptured as well as linearized at the Y-selective sites. The linearized peptides were identified by the MS/MS technique. An interesting fact is that TICA is highly electrophilic in nature. Thus, DMP-modified peptides (44) can be further altered through selective bioconjugation and synthetic manipulation.
 | ||
| Scheme 6 Y-selective cleavage of the peptide bond through DMP-mediated oxidation. Reaction conditions: (a) DMP (excess), DMSO, phosphate buffer (pH 7), 40 °C, 2–3 h. | ||
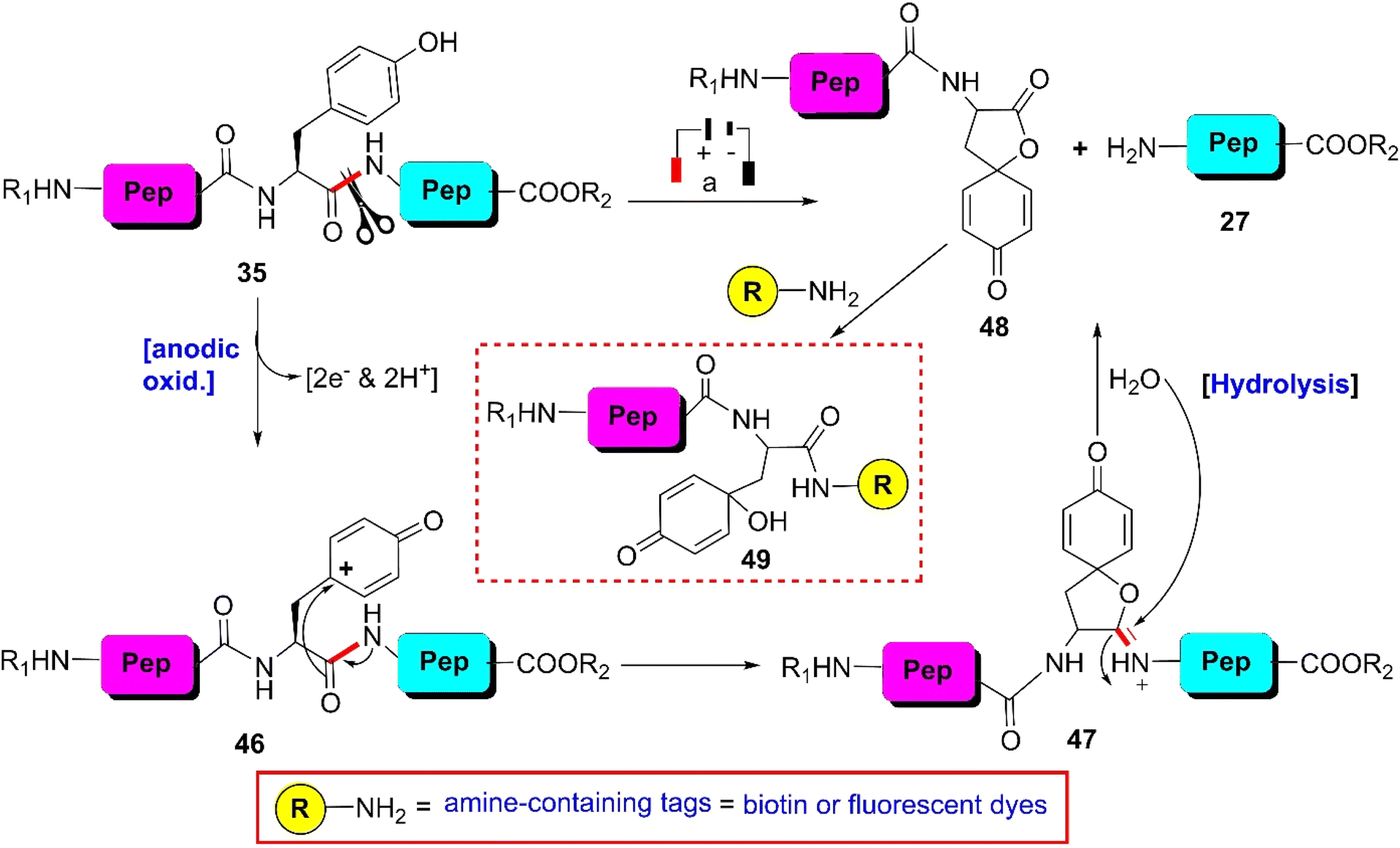 | ||
| Scheme 7 Y-selective cleavage of peptide bond via electrochemical approach. Reaction conditions: (a) porous graphite electrodes (vs. Pd reference electrode), constant potential (0.8 V or 1.0 V), acetonitrile–water–formic acid (pH 2.8), flow rate 2 μL min−1. | ||
A wide variety of substrates, ranging from tripeptides to proteins, have been successfully cleaved at the Y- and W-sites in electrochemical cells by just tuning the potential of the cell.64,65 The most remarkable feature of this cleavage method is its compatibility with MS and LC-MS analysis. The electrochemical cell can be directly coupled to a mass spectrometer, thereby allowing fast and real-time analysis of the complex protein digest without the need for extra sample preparation. However, the electrochemical oxidation of proteins and peptides suffered from inherent low yields given that a mixture of non-cleavable oxidation products was generated simultaneously during the cleavage. It is also interesting to note that the generated spirolactone moiety of cleaved N-terminal fragment 47 is susceptible to nucleophilic attack. Aminolysis of the spirolactone with several amine-containing tags, such as biotin and fluorescent dyes, would afford compound 49, enabling the selective labeling and enrichment of electrochemically cleaved peptides (Scheme 7).84
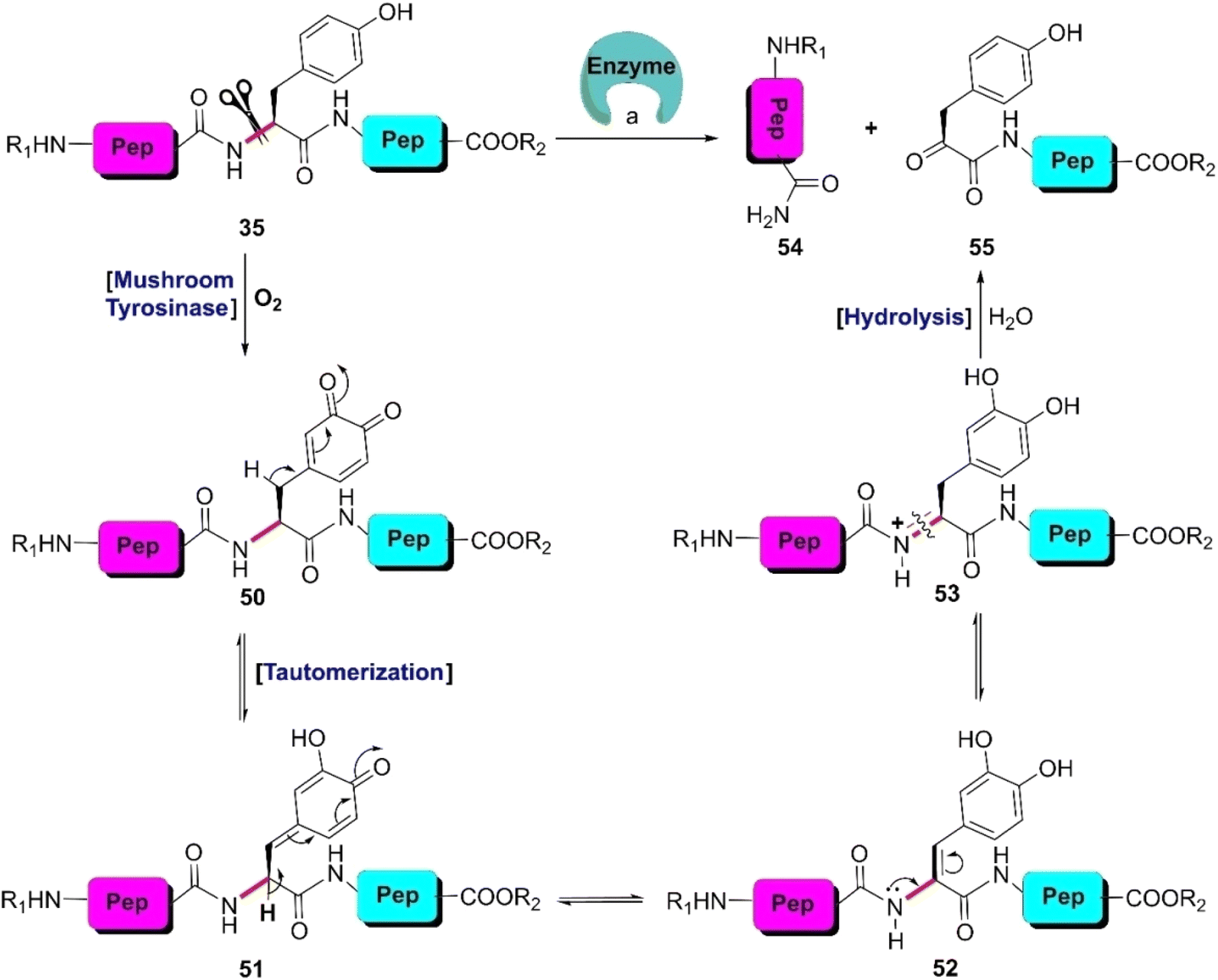 | ||
| Scheme 8 Y-selective cleavage of peptide bond via chemoenzymatic approach. Reaction conditions: (a) mushroom tyrosinase, phosphate buffer (pH 6.6), 37 °C. | ||
Besides mushroom tyrosinase, some enzymes present in peptides and proteins can selectively cleave engineered tyrosines such as phosphotyrosine, o-sulfated tyrosine, and 3-nitrotyrosine. In 2007, Knight et al. identified a subtilisin variant (E156R/P129G) that selectively cleaves phosphotyrosine peptides.86 Alternatively, Varadarajan et al. discovered two E. coli outer membrane protease (OmpT) variants that preferably rupture the sulfotyrosine residues in peptide substrates.87 Later, the same research group isolated a highly active variant of the E. coli endopeptidase OmpT that selectively hydrolyzes peptides after 3-nitrotyrosine.88
3.3. Phenylalanine
Phenylalanine (Phe or F) is an essential amino acid, which is introduced into protein molecules by encoding the codons UUU and UUC. It contains a benzyl side chain to introduce hydrophobicity into the molecule. F-residues play a crucial role in dictating the structure and folding of proteins. Furthermore, it is the precursor of many important biomolecules such as tyrosine, the neurotransmitter dopamine, and norepinephrine. Thus, F-selective cleavage by means of various chemical and chemoenzymatic techniques followed by analysis of the protein fragments may give in-depth information on proteins. Unfortunately, phenylalanine is a redox-inactive amino acid. Thus, there is no possibility for electrochemical cleavage of peptides at F-residues. Here, we describe the strategies that promoted the F-selective cleavage of peptide bonds in peptides and proteins. | ||
| Scheme 9 F-selective cleavage of peptide bond via the Birch reaction. Reaction conditions: (a) Li, CH3NH2, −70 °C, 2–3 h. (b) Br2/AcOH or NBS/AcOH. | ||
As an extension of their previous work,91 in 2014, Shimizu et al. accomplished the hydrazinolysis of several di- and tri-peptides in the presence of ammonium salt at 70 °C under microwave (MW) conditions.92 After the reaction, it was observed that the N-terminal peptide bond of the F-residue (62 or 63) was selectively fragmented to 64 in good to excellent yields without any epimerization of the α-position of the ruptured amino acids (Scheme 10). The authors assumed that the yield of the reaction and site-selectivity were directed by both steric and electronic factors.
 | ||
| Scheme 10 Y-selective cleavage of peptide bond via hydrazinolysis. Reaction conditions: (a) NH2NH2·H2O, NH4I, C2H5OH, and microwave heating at 70 °C until the release of the F-residue. | ||
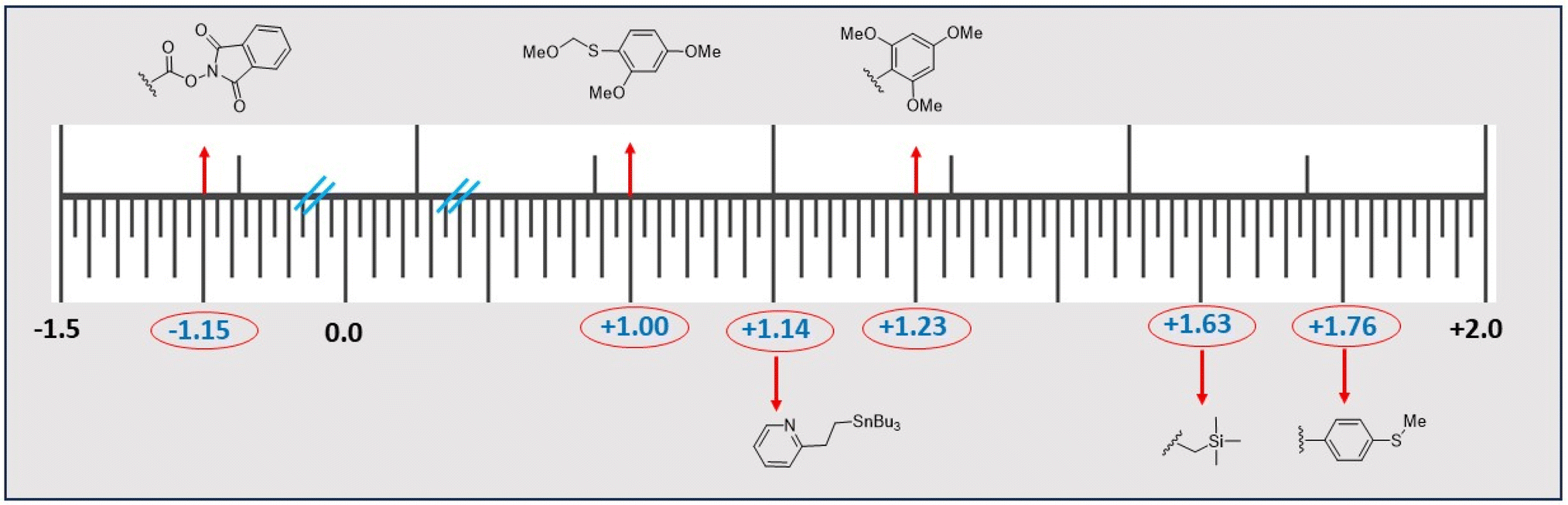 | ||
| Fig. 4 Popular electro-auxiliaries and their corresponding activation potentials (in voltage vs. Ag/AgCl). | ||
 | ||
| Scheme 11 F-selective cleavage of peptide bond via chemoenzymatic approach. Reaction conditions: (a) S24C:H64A, Tris–HCI (pH 8.0), dithiothreitol (DTT), dimethylsulfoxide (DMSO), phenylmethylsulfonyl fluoride (PMSF), 37 °C, 20 h. | ||
The investigators digested a ten-residue fragment (SYSMEHFRWG, 67) of human adrenocorticotropic hormone (ACTH) with S24C:H64A at 37 °C in the presence of Tris–HCI (pH 8.0), dithiothreitol (DTT), dimethyl sulfoxide (DMSO) and phenylmethylsulfonyl fluoride (PMSF) for 20 h and observed that the C-terminal peptide linkage of the F-residue was fragmented quantitatively to afford peptide fragments 68 and 69 (Scheme 11). Their further studies revealed that this bond rupturing is guided by the upstream H-residue present in the peptide. The same engineered enzyme was utilized to cleave the C-terminal peptide bond of a Y-residue present in the 20-residue fragment of the inhibin β chain.101
3.4. Histidine
Histidine (His or H) is an essential amino acid, which is inserted into protein molecules by encoding the codons CAU and CCA. It is an integral part of the catalytic mechanism of many enzymes. Histidine plays a crucial role in biological processes such as catalytic triads and proton shuttle mechanism. The imidazole moiety of the H-residue commonly serves as a ligand in metalloproteins. Naturally, cleavage at the H-selective site of peptides and proteins is very important. The protocols that enabled the H-selective cleavage of peptide bonds in peptides/proteins are detailed below.:10:19 v/v) split the tryptophyl and tyrosyl linkages only. Under these conditions, the histidyl residues were oxidized but not cleaved. After destroying the excess NBS and heating the reaction mixture, the histidyl peptides were fragmented.
 | ||
| Fig. 5 NBS-enabled cleavage of residue-selective peptide bonds. (X) Different substrates (Y) general mechanism. | ||
Utilizing the coordinating ability of H-residues (through the imidazolyl side chain) with selective metal ions, Mother Nature synthesizes several metalloproteins. Learning from this lesson in nature, in the 1990s, scientists accepted the challenge of synthesizing different metal complexes that can act as artificial peptidases. To the best of our knowledge, Prof. Kostic and colleagues were the first group of investigators who developed four Pd(II)-complexes (utilizing water, diethylenediamine, 1,5-dithiacyclooctane and its 3-hydroxy derivative as ligands) that could scissor the selective peptide bond of cytochrome c (75), a hemeprotein found in the inner membrane of the mitochondrion.103 The protein in a solution containing Pd(II)-complex was treated with a non-coordinating acid such as HBF4, HCIO4 or CF3COOH, causing it to exhibit the partially unfolded state and come closer to the Pd(II)-center to furnish complex 76. This aliquot (at pH 1.4) was incubated for 2 days at 40 °C and it was confirmed through various experiments that the peptide linkage between histidine 18–threonine 19 (H18–T19) was ruptured site-selectively to provide fragments 77 and 78 with RMM (approx.) of 2 kDa and 10 kDa respectively (Scheme 12). Cytochrome c consists of 104 residues, in which position-18, 26 and 33 are occupied by H-residues. However, the C-terminal peptide linkage of H18 (H18–T19) was fragmented selectively, avoiding the other H-connected peptide bonds. This site selectivity was achieved due to the presence of a cysteine residue (C17) just before H18. This C17 invites the metal complex to bind at that particular position and scissor the H18–T19 peptide linkage. In cytochrome c, there is another cysteine at position 14, after which there is an alanine residue (A15, bearing a non-coordinating methyl side chain). The C-residue has sufficient potential to co-ordinate with metal ions; however, in the sterically hindered protein environment, C14 alone cannot hold the metal ion. As a result, the peptide bond next to A15 is never fragmented. Thus, in the C17-directed fragmentation process, the imidazole side chain of H18 plays a vital role. To reveal the role of T19, in the fragmentation process, a model tripeptide (C–H–T) was selected and the threonine residue was replaced by alanine (to obtain tripeptide C–H–A) and glycine (to obtain tripeptide C–H–A) residues. It was observed that in all cases, the H–A and H–G bonds were split. However, in the presence of cis-[Pd(en)(H2O)2]2+, another model segment of cytochrome c C(Me)14–A15–Q16 (Q: glutamine), the C(Me)–A peptide bond was ruptured, and cleavage of the A–Q linkage was never observed. In this case, cysteine alone can hold the metal ion and rupture the scissile bond. This proves that in the fragmentation process, H19 does not play any significant role; rather, the coordinating ability of the C and H-residues is important.
 | ||
| Scheme 12 Site-selective cleavage of cytochrome C via Pd(II) complexation. Reaction conditions: (a) acidic promoter (HBF4, HCIO4 or CF3COOH), 40 °C, 2 days. | ||
Based on the experimental and theoretical results obtained by B3LYP104–106 density functional theory in the Gaussian98 program package using the 3-21G, 6-31G(d) and LanL2DZ basis sets, Zhu and co-workers proposed the mechanism of H18–T19 peptide bond scissoring in cytochrome c.107 During the cleavage process, Pd(II) coordinates with the carbonyl oxygen of H18, and simultaneously the hydrogen bond between the carbonyl oxygen and ancillary dimer water weakens as well as polarizes the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bond of the H-residue. As a result, the H18–T19 peptide bond ruptures site selectively.
O double bond of the H-residue. As a result, the H18–T19 peptide bond ruptures site selectively.
Generally, the cleavage of peptides and proteins guided by transition-metal complexes proceeds through stoichiometric reactions. However, in 1996, Kostic and Parac found that cis-[Pd(en)(H2O)2]2+ has potential to rupture the H-selective peptide bond of a series of di- and tri-peptides bearing a histidine residue in a catalytic manner.108 When a catalytic amount of cis-[Pd(en)(H2O)2]2+ was mixed with the peptide solution at pH 1.46, five NMR detectable complexes were formed spontaneously. Together with complexes 79, 80 and 81 (Fig. 6), another two complexes were constructed by the histidinyl peptides by holding two Pd(II) ions each. When the temperature of the reaction mixture reached 60 °C, the C-terminal amide bond of histidine was fragmented keeping the N-terminal amide bond intact. The rate of the reaction was pH dependent given that the attachment of the catalyst to the peptide was controlled by pH. It was observed that the maximum catalytic efficiency was achieved at pH 1.46. Additionally, the rate constant decreased with an increase in the steric bulk of the amino acid bonded with the carboxylic group of H-residue. Besides, intrapeptide hydrogen bonding through water molecules slowed down the rate of the reaction given that the hydrogen bonded water molecules veiled the catalyst, preventing it from coming closer to the scissile bond. Later, the same research group controlled the regioselective cleavage of H-bearing peptides by the choice of ligands in palladium(II) complexes. [Pd(H2O)4]2+ cleaves both the N- and C-terminal peptide bonds with respect to the H-residue, where [PdCl4]2− cleaves only the C-terminal peptide bond under the same reaction conditions (Fig. 6).109 The rate constant of the former reaction was 10 times greater than that of the latter. This reactivity difference is due to steric factors. The [Pd(H2O)4]2+ complex is smaller than [PdCl4]2−. Consequently, it can come closer to both the scissile bonds and both peptide linkages are fragmented.
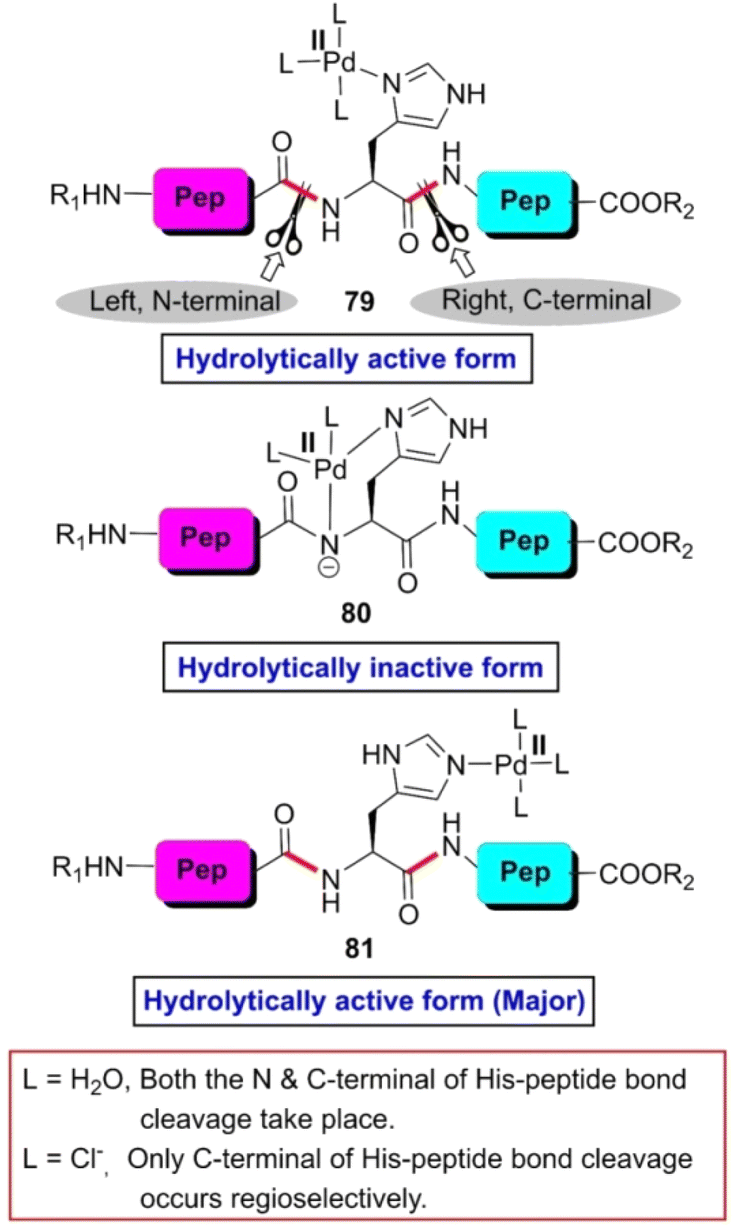 | ||
| Fig. 6 Modes of co-ordination of Pd(II)-complex with histidinyl peptide. Reaction conditions: (a) histidinyl peptide, cis-[Pd(en)(H2O)2]2+, HBF4 (promoter), pH 1.5–2 (0.1 M HCl or 70% HCOOH), 40 °C, 2 days, 60–80%. | ||
In 2010, Hong et al. could rapture the C-terminal peptide bond of the H-residue in a series of tripeptides applying a stoichiometric amount of Pt(II)-complexes (pH 2.65) at 60 °C.110 Histidinyl peptide 70 has the probability to construct complex-82 and complex 83 (Scheme 13). Between the two formations, 83 is stereochemically preferable and it is hydrolytically cleavable. As a result, the reaction goes through the formation of complex 83, and upon hydrolysis furnishes products 84 and 85. However, this protocol becomes useless if the tripeptides contain at least one cysteine or methionine residue. Sometimes, an excess amount of costly Pt(II)-complex is required, which reduces the utility of this protocol.
 | ||
| Scheme 13 H-selective cleavage of peptide bonds via metal complexation. Reaction conditions: (a) peptide: Pt(II)-complex = 1:1 or 1:2, H2O, pH 2.65, 60 °C. | ||
Recently, Rajkovic et al. designed and synthesized three dinuclear platinum(II) complexes by incorporating bidentate quinoxaline (qx), quinazoline (qz), phthalazine (phtz) ligands and investigated their catalytic behavior towards methioninylglycine and histidinylglycine at a pH of 2.0 < pH < 2.5 at 37 °C.111 Similar to platinum(II) complexes112,113 with pyrazine and pyridazine ligands, all three complexes coordinated with the methioninyl side chain of Ac–M–G and split the C-terminal amide bond. However, in the case of Ac–H–G, only the Pt(II) complex with the quinoxaline-bridging ligand could fragment the C-terminal peptide bond of the histidine residue to yield peptide fragments 85 and 89 (Scheme 14).
 | ||
| Scheme 14 H-selective cleavage of the peptide bond via metal complexation: role of ligands. Reaction conditions: (a) D2O, pH 2.0 < pH < 2.5, 37 °C. | ||
Applying [Pd(H2O)4]2+, cis-[Pd(en)(H2O)2]2+ and cis-[Pd(dtco-OH)(H2O)2]2+ complexes at pH 3.2, Zhu et al. could split myoglobin by digesting it for 24 h at 60 °C.114 SDS-PAGE electrophoresis and HPLC separation followed by ESIMS and MALDI-TOF experiments of the fragmented protein revealed that 13 selective sites were split, which were clustered around some of the histidine residues. However, most of the fragmented sites were ensured by methionine and arginine residues. Recently, in 2020, Jiao et al. improved this protocol by utilizing the binuclear [Pd2(μ-O–L–H)(μ-Cl)](ClO4)2 (L = 2,6-bis(N-2′-aminoethylaminomethyl)-p-cresol) complex, which cleaves the second upstream peptide bond from the H- and M-residues, avoiding C-orientated hydrolysis, although cysteine also can also serve as an excellent ligand.115 Based on theoretical results acquired from B3LYP70–72 density functional theory in the Gaussian09 program package using the Lanl2dy and 6-31g(d) basis sets, these investigators confirmed the process of hydrolysis. Synergically, one Pd(II) center coordinates with the selective site of Mb, and the other one binds with the amide bond and favours nucleophilic attack of the water on the peptide linkage. In the case of cysteine, two Pd(II)-centers construct a “closed” sulphur-bridge structure, which disfavors the approach of water towards the scissile bond.
4. Future prospect and conclusion
Based on the methodological development of the site-selective cleavage of AAAs in peptides/proteins over the last seven decades (1958–2024), there are tremendous possibilities of further innovation. Fig. 7 depicts the present status of this field in considering the research gaps and achievements achieved over time. According to this figure, it is crystal clear that the electrochemical technique has not been popularized to date for the residue-selective scissoring of peptides, although it has the capability of rupturing peptide bonds via clean and green reaction conditions. When a redox-active amino acid is inserted into a peptide or protein, the microenvironment of the amino acid is altered due to the change in the conformation of the protein. This conformational alteration process switches the redox potential of the amino acid side chains. This, by tuning the exact potential of this microenvironment, several selective sites of a protein can be fragmented in a single reaction vessel. Perhaps one day this technique will be beneficial for the instrumental improvement of peptide sequencing machines. | ||
| Fig. 7 Research achievement vs. research gap. | ||
In the last few decades, a fabulous technological improvement has been noticed in the engineering of diverse wild-type enzymes as well as chemoenzymatic processes. However, this has not yet employed in this research territory. Recently, the photochemistry-enabled fragmentation of peptides and proteins has become a catching topic of research. Applying photo-redox reaction, several complex proteins have been split site-selectively. Another review on this topic will be published in due course by our research group.
In most cases, peptide fragmentation reactions are low yielding and the introduction of a sufficient amount of protein in peptide sequencing is not possible. By employing flow-chemistry, this puzzling situation can be solved.
In conclusion, comprehensive methodological improvements in AAA-selective fragmentation have been exhibited over the last 60–70 years. However, there are still huge roadblocks to be overcome. At present, the number of species on Earth is nearly 8.7 million.118 Among them, 86% and 91% of species in the Earth and ocean, respectively, are yet to be discovered and analyzed. With their discoveries, chemists and biologists have to encounter new peptides/proteins with complex molecular segments. Analysis of these new proteins will require a huge technological improvement in this research arena. Moreover, the utility of fragmented proteins as drugs and therapeutic agents is increasing. Thus, more sophisticated techniques for the site-selective cleavage of proteins are required at the industrial level. We hope that young researchers will able to tackle all these challenges and this review will inspire the practitioners who are looking to enter this field.
Data availability
The data that support the findings of this study is already available in the literature.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Dr R. Sarkar and Dr A. Bandyopadhyay express their appreciation to the Department of Chemistry at Muragachha Government College and Chapra Government College for providing collaborative research facilities. The authors also thank the Department of Higher Education, Government of West Bengal, India, for their support. The authors also heartily acknowledge Dr D. Bose and Dr J. Bhattacharjee for their technical assistance.References
- C. D. Spicer and B. G. Davis, Nat. Commun., 2014, 5, 4740 CAS.
- J. N. deGruyter, L. R. Malins and P. S. Baran, Biochemistry, 2017, 56, 3863–3873 CAS.
- S. K. Kundu, A. Bandyopadhyay and R. Sarkar, Org. Biomol. Chem., 2025, 23, 1773–1793 RSC.
- R. Sarkar, K. Maji and D. Haldar, RSC Adv., 2015, 5, 59570–59575 RSC.
- R. Sarkar, M. Debnath, K. Maji and D. Haldar, RSC Adv., 2015, 5, 76257–76262 RSC.
- A. Bandyopadhyay, P. Biswas, S. K. Kundu and R. Sarkar, Org. Biomol. Chem., 2024, 22, 1085–1101 RSC.
- J. Chatterjee, A. Bandyopadhyay, M. Pattabiraman and R. Sarkar, Chem. Commun., 2024, 60, 8978–8996 RSC.
- C. White and A. Yudin, Nat. Chem., 2011, 3, 509–524 CrossRef CAS.
- C. H. Heather, Y. P. L. Louis and Yu-H. Tsai, Org. Biomol. Chem., 2021, 19, 3983–4001 Search PubMed.
- C. Bechtler and C. Lamers, RSC Med. Chem., 2021, 12, 1325–1351 RSC.
- T. F. Spande, B. Witkop, Y. Degani and A. Patchornik, Adv. Protein Chem., 1970, 24, 97–260 CrossRef CAS PubMed.
- K. K. Han, C. Richard and G. Biserte, Int. J. Biochem., 1983, 15, 875–884 CrossRef CAS.
- D. Hoyer, H. Cho and P. G. Schultz, J. Am. Chem. Soc., 1990, 112, 3249–3250 CAS.
- H. Hongjian, J. Wang, H. Wang, N. Zhou, D. Yang, R. G. Douglas and B. Xu, J. Am. Chem. Soc., 2018, 140, 1215–1218 CrossRef PubMed.
- R. L. Lundblad, Techniques in Protein Modification, CRC Press, Boca Raton, Florida, 1994 Search PubMed.
- H. E. Elashal, R. D. Cohen and M. Raj, Chem. Commun., 2016, 52, 9699–9702 RSC.
- H. E. Elashal, Y. E. Sim and M. Raj, Chem. Sci., 2017, 8, 117–123 RSC.
- T. Y. Lee and J. Suh, Chem. Soc. Rev., 2009, 38, 1949–1957 RSC.
- J. Thorner, S. D. Emr and J. N. Abelson, Methods Enzymol., 2000, 326, 613–617 Search PubMed.
- J. J. Thomas, R. Bakhtiar and G. Siuzdak, Acc. Chem. Res., 2000, 33, 179–187 CAS.
- T. Durek and C. F. W. Becker, Biomol. Eng., 2005, 22, 153–172 CAS.
- T. W. Muir, Annu. Rev. Biochem., 2003, 72, 249–289 CAS.
- T. Y. Lee and J. Suh, Chem. Soc. Rev., 2009, 38, 1949–1957 CAS.
- R. Aebersold and M. Mann, Nature, 2003, 422, 198–207 CAS.
- A. Radzicka and R. Wolfenden, J. Am. Chem. Soc., 1996, 118, 6105–6109 CAS.
- P. G. M. Wuts and T. W. Greene, Protective Groups in Organic Synthesis, Wiley, Hoboken, 4th edn, 2006, pp. 773–789 Search PubMed.
- J. Ni and M. Kanai, Top. Curr. Chem., 2016, 372, 103–124 CrossRef CAS PubMed.
- S. Zhang, L. M. D. L. Rodriguez, F. F. Li and M. A. Brimble, Chem. Sci., 2023, 14, 7782–7817 RSC.
- S. Burley and G. Petsko, Science, 1985, 229, 23–28 CrossRef CAS PubMed.
- A. K. Tewari and R. Dubey, Bioorg. Med. Chem., 2008, 16, 126–143 CrossRef CAS PubMed.
- I. S. Moreira, P. A. Fernandes and M. J. Ramos, Hot spots—a review of the protein–proteininterface determinant amino-acid residues, Proteins: Struct., Funct., Bioinf., 2007, 68, 803–812 CrossRef CAS PubMed.
- B. Ma and R. Nussinov, Curr. Top. Med. Chem., 2007, 7, 999–1005 CAS.
- R. Bhattacharyya, R. P. Saha, U. Samanta and P. Chakrabarti, J. Proteome Res., 2003, 2, 255–263 CAS.
- A. Fontana and C. Toniolo, Progress in the Chemistry of Organic Natural Products, Springer, Vienna, 1976, vol. 33, pp. 309–449 Search PubMed.
- A. Patchornik, W. B. Lawson and B. Witkop, J. Am. Chem. Soc., 1958, 80, 4747–4748 CrossRef CAS.
- A. Patchornik, W. B. Lawson, E. Gross and B. Witkop, J. Am. Chem. Soc., 1960, 82, 5923–5927 CrossRef CAS.
- L. K. Ramachandran and B. Witkop, J. Am. Chem. Soc., 1959, 81, 4028–4032 CrossRef CAS.
- M. Funatsu, N. M. Green and B. Witkop, J. Am. Chem. Soc., 1964, 86, 1846–1848 CrossRef CAS.
- G. S. Omenn, A. Fontana and C. B. Anfinsen, JBC, 1970, 245, 1895–1902 CrossRef CAS.
- P. R. Burnett and E. H. Eylar, JBC, 1971, 246, 3425–3430 CrossRef CAS.
- A. Fontana, Methods Enzymol., 1972, 25, 419–423 CAS.
- Y. Burstein and A. Patchornik, Biochemistry, 1972, 11, 4641–4650 CrossRef CAS PubMed.
- Y. Shechter, A. Patchornik and Y. Burstein, Biochemistry, 1976, 15, 5071–5075 CrossRef CAS PubMed.
- M. A. Lischwe and M. T. Sung, JBC, 1977, 252, 4976–4980 CrossRef CAS.
- W. E. Savige and A. Fontana, Methods Enzymol., 1977, 47, 459–469 CAS.
- M. Z. Atassi, Arch. Biochem. Biophys., 1967, 120, 56–59 CrossRef CAS PubMed.
- J. Ozols and C. Gerard, JBC, 1977, 252, 5986–5989 CrossRef CAS.
- W. C. Mahoney and M. A. Hermodson, Biochemistry, 1979, 18, 3810–3814 CrossRef CAS PubMed.
- E. Wachter, R. Werhrhn, Methods in Peptide and Protein Sequence Analysis, ed. C. Birr, Elsevier/North-Holland Biomedical Press, Amsterdam, 1980, pp. 323–328 Search PubMed.
- P. Johnson and V. B. Stockmal, Biochem. Biophys. Res. Commun., 1980, 94, 697–703 CrossRef CAS PubMed.
- A. Fontana and D. Dalzop, Biochemistry, 1981, 20, 6997–7004 CrossRef CAS PubMed.
- W. C. Mahoney, P. K. Smith and M. A. Hermodson, Biochemistry, 1981, 20, 443–448 CrossRef CAS PubMed.
- A. Previero, C. Previero and P. Jolles, Biochem. Biophys. Res. Commun., 1966, 22, 17–21 CrossRef CAS PubMed.
- M. Morishita and F. Sakiyama, BCSJ, 1970, 43, 524–530 CrossRef CAS.
- N. V. Kaminskaia, T. W. Johnson and N. M. Kostic, J. Am. Chem. Soc., 1999, 121, 8663–8664 CrossRef CAS.
- N. V. Kaminskaia and N. M. Kostic, Inorg. Chem., 2001, 40, 2368–2377 CrossRef CAS PubMed.
- P. Brunelle and A. Rauk, J. Phys. Chem. A, 2004, 108, 11032–11041 CrossRef CAS.
- A. Harriman, J. Phys. Chem., 1987, 91, 6102–6104 CrossRef CAS.
- P. C. Jocelyn, Eur. J. Biochem., 1967, 2, 327–331 CrossRef CAS PubMed.
- S. Navaratnam and B. J. Parsons, J. Chem. Soc., Faraday Trans., 1998, 94, 2577–2581 RSC.
- Sanaullah, G. S. Wilson and R. S. Glass, J. Inorg. Biochem., 1994, 55, 87–99 CrossRef CAS PubMed.
- S. M. MacDonald and S. G. Roscoe, Electrochim. Acta, 1997, 42, 1189–1200 CrossRef CAS.
- D. J. Walton, P. G. Richards, J. Heptinstall and B. Coles, Electrochim. Acta, 1997, 42, 2285–2294 CrossRef CAS.
- H. P. Permentier and A. P. Bruins, J. Am. Soc. Mass Spectrom., 2004, 15, 1707–1716 CrossRef CAS PubMed.
- J. Roeser, H. P. Permentier, A. P. Bruins and R. Bischoff, Anal. Chem., 2010, 82, 7556–7565 CrossRef CAS PubMed.
- H. P. Permentier, U. Jurva, B. Barroso and A. P. Bruins, Rapid Commun. Mass Spectrom., 2003, 17, 1585–1592 CrossRef CAS PubMed.
- N. M. Alexander, Biochem. Biophys. Res. Commun., 1973, 54, 614–621 CAS.
- N. M. Alexander, JBC, 1974, 249, 1946–1952 CrossRef CAS.
- G. L. Schmir, L. A. Cohen and B. Witkop, J. Am. Chem. Soc., 1959, 81, 2228–2233 CrossRef CAS.
- G. L. Schmir and L. A. Cohen, J. Am. Chem. Soc., 1961, 83, 723–728 CrossRef CAS.
- J. G. Wilson and L. A. Cohen, J. Am. Chem. Soc., 1963, 85, 560–564 CrossRef CAS.
- J. G. Wilson and L. A. Cohen, J. Am. Chem. Soc., 1963, 85, 564–567 CrossRef.
- H. Junek, K. L. Kirk and L. A. Cohen, Biochemistry, 1969, 8, 1844–1848 CrossRef CAS PubMed.
- R. M. Moriarty, M. Sultana and Y. Y. Ku, J. Chem. Soc. Chem. Commun., 1985, 14, 974–975 RSC.
- S. Zhang, L. M. D. L. Rodriguez, F. F. Li, R. Huang, I. K. H. Leung, P. W. R. Harris and M. A. Brimble, Chem. Sci., 2022, 13, 2753–2763 RSC.
- H. Morita, T. Iizuka, C. Y. Choo, K. L. Chan, H. Itokawa and K. Takeya, J. Nat. Prod., 2005, 68, 1686–1688 CrossRef CAS PubMed.
- B. Ohlendorf, S. Simon, J. Wiese and J. F. Imhoff, Nat. Prod. Commun., 2011, 6, 1247–1250 CrossRef CAS PubMed.
- A. Isogai, S. Takayama, S. Murakoshi and A. Suzuki, Tetrahedron Lett., 1982, 23, 3065–3068 CrossRef CAS.
- H. Iwasaki, L. A. Cohen and B. Witkop, J. Am. Chem. Soc., 1963, 85, 3701–3702 CrossRef CAS.
- H. Iwasaki and B. Witkop, J. Am. Chem. Soc., 1964, 86, 4698–4708 CrossRef CAS.
- L. Farber and L. A. Cohen, Biochemistry, 1966, 5, 1027 CrossRef CAS PubMed.
- S. Isoe and L. A. Cohen, Arch. Biochem. Biophys., 1968, 127, 522–527 CrossRef CAS PubMed.
- D. Alvarez-Dorta, C. Thobie-Gautier, M. Croyal, M. Bouzelha, M. Mevel, D. Deniaud, M. Boujtita and S. G. Gouin, J. Am. Chem. Soc., 2018, 140, 17120–17126 CrossRef CAS PubMed.
- T. Zhang, M. P. de Vries, H. P. Permentier and R. Bischoff, Anal. Chem., 2017, 89, 7123–7129 CrossRef CAS PubMed.
- M. J. Long and L. Hedstrom, ChemBioChem, 2012, 13, 1818–1825 CrossRef CAS PubMed.
- Z. A. Knight, J. L. Garrison, K. Chan, D. S. King and K. M. Shokat, J. Am. Chem. Soc., 2007, 129, 11672–11673 CrossRef CAS PubMed.
- N. Varadarajan, G. Georgiou and B. L. Iverson, Angew. Chem., Int. Ed., 2008, 47, 7861–7863 CrossRef CAS PubMed.
- N. Varadarajan, M. Pogson, G. Georgiou and B. L. Iverson, J. Am. Chem. Soc., 2009, 131, 18186–18190 CrossRef CAS PubMed.
- M. Wilchek and A. Patchornik, J. Am. Chem. Soc., 1962, 84, 4613–4614 CrossRef CAS.
- A. J. Birch, J. Chem. Soc., 1944, 66, 430–436 Search PubMed.
- Y. Shimizu, H. Morimoto, M. Zhang and T. Ohshima, Angew. Chem., Int. Ed., 2012, 51, 8564–8567 Search PubMed.
- Y. Shimizu, M. Noshita, Y. Mukai, H. Morimoto and T. Ohshima, Chem. Commun., 2014, 50, 12623–12625 CAS.
- V. Brabec and V. Mornstein, Biophys. Chem., 1980, 12, 159–165 CrossRef CAS PubMed.
- H. Sun, C. Martin, D. Kesselring, R. Keller and K. D. Moeller, J. Am. Chem. Soc., 2006, 128, 13761–13771 Search PubMed.
- H. Sun and K. D. Moeller, Org. Lett., 2002, 4, 1547–1550 CAS.
- J. I. Yoshida, M. Sugawara, M. Tatsumi and N. Kise, J. Org. Chem., 1998, 63, 5950–5961 CAS.
- J. I. Yoshida and M. Izawa, J. Am. Chem. Soc., 1997, 119, 9361–9365 CAS.
- T. Shoji, S. Kim, K. Yamamoto, T. Kawai, Y. Okada and K. Chiba, Org. Lett., 2014, 16, 6404–6407 CAS.
- X. Chen, X. Luo, X. Peng, J. Guo, J. Zai and P. Wang, Chem.–Eur. J., 2020, 26, 3226–3230 CAS.
- H. Wang, M. He, Y. Li, H. Zhang, D. Yang, M. Nagasaka, Z. Lv, Z. Guan, Y. Cao, F. Gong, Z. Zhou, J. Zhu, S. Samanta, A. D. Chowdhury and A. Lei, J. Am. Chem. Soc., 2021, 143, 3628–3637 CAS.
- P. Carter and J. A. Wells, Science, 1987, 237, 394–399 CAS.
- S. Shaltiel and A. Patchornik, J. Am. Chem. Soc., 1963, 85, 2799–2806 CAS.
- L. Zhu, L. Qin, T. N. Parac and N. M. Kostic, J. Am. Chem. Soc., 1994, 116, 5218–5224 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 564–5652 Search PubMed.
- X. J. Sun, L. Zhang, Y. Zhang, G. S. Yang, Z. J. Guo and L. G. Zhu, New J. Chem., 2003, 27, 818–822 RSC.
- T. N. Parac and N. M. Kostic, J. Am. Chem. Soc., 1996, 118, 51–58 CrossRef CAS.
- T. N. Parac, G. M. Ullmann and N. M. Kostic, J. Am. Chem. Soc., 1999, 121, 3127–3135 CrossRef CAS.
- J. Hong, Y. Jiao, W. He, Z. Guo, Z. Yu, J. Zhang and L. Zhu, Inorg. Chem., 2010, 49, 8148–8154 CrossRef CAS PubMed.
- S. Rajkovic, B. Warżajtis, M. D. Zivkovic, B. Đ. Glisic, U. Rychlewska and M. I. Djuran, Bioinorg. Chem. Appl., 2018, 2018 DOI:10.1155/2018/3294948.
- M. D. Zivković, S. Rajković, B. D. Glišić, N. S. Drašković and M. I. Djuran, Bioorg. Chem., 2017, 72, 190–198 CrossRef PubMed.
- S. Rajković, M. D. Živković, B. War˙azajtis, U. Rychlewska and M. I. Djuran, Polyhedron, 2016, 117, 367–376 CrossRef.
- L. Zhu, B. Ray and N. M. Kostic, JBIC, J. Biol. Inorg. Chem., 1998, 3, 383–391 Search PubMed.
- Y. Jiao, J. Hong, Y. Chen, Y. Zhang, Z. Guo, Z. Han and W. He, Dalton Trans., 2020, 49, 3164–3173 CAS.
- C. Tanner, S. Navaratnam and B. J. Parsons, Free Radical Med. Biol., 1998, 24, 671–678 Search PubMed.
- S. Navaratnam and B. J. Parsons, J. Chem. Soc., Faraday Trans., 1998, 94, 2577–2581 CAS.
- L. Sweetlove, Nature, 2011 DOI:10.1038/news.2011.498.
| This journal is © The Royal Society of Chemistry 2025 |