DOI:
10.1039/D4TA03317E
(Paper)
J. Mater. Chem. A, 2024,
12, 22539-22549
Stainless steel-derived nano-porous oxide: a cost-efficient, stable, and corrosion-resistant hydrogen evolution catalyst†
Received
13th May 2024
, Accepted 25th July 2024
First published on 26th July 2024
Abstract
Stainless steel (SS)-based electrocatalysts have attracted considerable attention in renewable energy research, emerging as viable replacements for precious noble metal-based catalysts owing to their cost-effectiveness and widespread availability. Further, SS's ability to function as an electrode material in alkaline water electrolyzers and its inherent corrosion resistance make it a key component of sustainable energy solutions. This study focuses on the fabrication of a self-ordered nanoporous oxide layer on SS through an anodization method, followed by an investigation into the electrocatalytic activity of the resulting films for hydrogen evolution reaction (HER) and corrosion properties. Anodization creates a highly organized nano-porous metal oxide structure featuring extensive surface areas that offer abundant active sites for electrochemical reactions via the oxide formation-dissolution mechanism. The nano-porous oxide film produced demonstrates exceptional HER activity, reaching a cathodic current density of 10 mA cm−2 with a minimal overpotential of 343 mV (versus the reversible hydrogen electrode (RHE)) in a strong alkaline medium. Moreover, the stability of the derived nano-porous oxide film remains unaffected during 50 hours of uninterrupted electrolysis, demonstrating remarkable operational resilience. Conversely, potentiodynamic polarization measurements reveal that the resulting films exhibit superior corrosion resistance in both NaCl and KOH electrolytes compared to bare SS. This enhanced resistance is attributed to the self-ordered nano-porous structure, which fosters the construction of a more uniform film with a more robust passive layer. The straightforwardness of the procedure and the widespread availability of the initial material make this an unexpectedly effective endeavour. The research demonstrated that anodized SS can achieve both corrosion resistance and catalytic activity for the HER, making this material system a viable candidate for alkaline water electrolyzer systems.
1. Introduction
Water electrolysis stands at the forefront of sustainable energy conversion technologies, offering a promising pathway towards producing clean and renewable hydrogen fuel. Among the various electrolysis methods, alkaline water electrolysis has emerged as a leading contender due to its simplicity, scalability, and compatibility with renewable energy sources.1–5 In alkaline water electrolyzers, water molecules are split into oxygen and hydrogen gases by the electrochemical oxygen evolution reaction (OER; 2OH− → ½O2 + H2O + 2e−) and hydrogen evolution reaction (HER; 2H2O + 2e− → H2 + 2OH−) under alkaline conditions. The efficiency and performance of alkaline water electrolysis systems are significantly influenced by the properties of the electrode materials employed, particularly their corrosion resistance and catalytic activity towards the HER.6–10 These two key properties play pivotal roles in determining the durability, efficiency, and economic viability of water electrolysis technologies. Corrosion resistance is paramount in alkaline water electrolysis systems due to the harsh chemical environment and high current densities involved in electrolysis operations. Electrodes are exposed to alkaline electrolytes, which can induce corrosive processes such as dissolution, pitting, and passivation. Corrosion not only compromises the structural integrity of the electrodes but also leads to loss of active surface area, reduced efficiency, and shortened lifespan of the electrolysis system.4,9 Therefore, electrode materials with high corrosion resistance are essential for ensuring the long-term durability and reliability of alkaline water electrolyzers. In an alkaline water electrolysis system, electrode surface interaction, reaction intermediate formation, and molecular hydrogen formation are crucial factors influencing catalytic performance and HER mechanisms.4,11–14 Electrodes with high catalytic activity towards the HER facilitate the rapid and efficient conversion of water into hydrogen gas, thereby improving the overall performance of the electrolysis system. Catalysts accelerate the hydrogen evolution process by limiting the activation energy barrier and enhancing adsorption and dissociation of water molecules on the electrode surface.11,15 Consequently, materials with superior HER catalytic activity enable higher electrolysis efficiency, lower overpotential, and increased hydrogen production rates. The synergy between corrosion resistance and HER catalytic activity is critical for optimizing the performance and longevity of alkaline water electrolysis systems.3,12,13,16 While corrosion-resistant materials protect electrodes from degradation, catalysts enhance hydrogen evolution efficiency, ultimately improving water electrolysis's overall energy conversion efficiency and cost-effectiveness. Therefore, the development of electrode materials with tailored properties to simultaneously address corrosion resistance and HER catalysis represents a promising approach towards advancing clean hydrogen production technologies.
Stainless steel (SS) has long been celebrated for its remarkable combination of mechanical strength, durability, and resistance to corrosion and also is a crucial material in alkaline water electrolyzers due to its cost-effectiveness, durability, and electrocatalytic properties.17–20 This alloy, primarily composed of iron (Fe), chromium (Cr), and other alloying elements, creates a passive oxide layer over the surface, which acts as a protective barrier against corrosive environments.18,21 However, despite its inherent corrosion resistance, SS can still be susceptible to certain aggressive conditions, particularly in applications where it is exposed to harsh chemical environments or high temperatures. To nullify this effect and further improve the performance of SS in demanding applications, researchers have turned to surface modification techniques such as anodization.21–25 Anodization involves the controlled electrochemical oxidation of the metal surface, leading to the formation of a thick and dense oxide layer with tailored properties. This process enhances the material's resistance to corrosion and introduces new functionalities, such as improved catalytic activity for electrochemical reactions.24–27 There has been growing interest in the anodization of SS for applications beyond traditional corrosion protection in recent years. One area of particular interest is the development of anodized SS electrodes with enhanced catalytic activity toward the electrochemical water splitting reactions in alkaline solutions, making it a practical choice for these electrolyzers.17,21–23,25 Despite recent advancements, there's still room for improvement in enhancing the HER activity, particularly in optimizing the anodization process and advancing binder-free electrodes. The anodization process typically involves an oxide layer formation on the SS surface, which can introduce surface defects like vacancies, dislocations, or edge sites.25,26,28 These defects can significantly influence electrocatalytic activity and the corrosion resistance properties of the material. Conversely, traditional electrodes often rely on binders to immobilize catalyst particles, which can hinder mass transport and create additional resistance, thereby limiting HER kinetics. However, binder-free electrodes offer a solution to these challenges by enabling direct contact between the catalyst and electrolyte, thus improving mass transport and enhancing electrochemical activity.29,30
By considering the importance of these key properties of SS and their interplay in water electrolysis systems, we develop a self-organized nano-porous oxide film on the surface of SS by an electrochemical anodization strategy using an ammonium fluoride and ethylene glycol-based solution. With this straightforward approach, SS surface morphology, chemical states, and electrochemical properties are tailored to create an electrochemically active nano-porous oxide film. This enhanced surface architecture facilitates the water molecules' adsorption and dissociation properties, thereby accelerating the kinetics of the HER and increasing the overall efficiency of electrochemical hydrogen production. Furthermore, the anodized oxide layer provides an additional barrier against corrosion, protecting the underlying SS substrate from degradation during prolonged electrochemical operation. This enhanced corrosion resistance is significant in alkaline electrolytes, where the HER typically occurs, as it ensures the long-term stability and durability of the electrode material.
2. Experimental
2.1 Materials
The anodization process was performed on stainless steel (SS 304 type) specimens containing 8–11 wt% Ni, 17–20 wt% Cr, and 2 wt% Mn, with the remainder being Fe. All chemicals and reagents were procured from Sigma-Aldrich and used without further purification. Before the anodization experiment, the specimens underwent pretreatment to remove surface impurities, including ultrasonic cleaning in ethanol and acetone for 10 minutes each, followed by a 10 minute rinse in water and drying with compressed air.
2.2 Anodization
Based on the method described in ref. 24, the anodization procedure consisted of two steps using a two-electrode setup (Fig. S1†) with a platinum gauze counter electrode and a stainless-steel working electrode. In the first step, the specimens were anodized in ethylene glycol with 10 vol% HClO4 at 40 V for 5 minutes, then washed with ethanol, deionized water, and air dried. The second step involved further anodization for 30 minutes at 65, 75, and 85 volts in an ethylene glycol solution containing 0.1 M NH4F. After rinsing with ethanol, the samples were oven-dried. Ice baths kept at approximately 5 °C were used throughout the anodization process. Following anodization, the specimens were thermally treated in the air at 450 °C for 1 hour, with a slow heating rate of 5 °C min−1 to prevent cracking of the nanoporous oxide layer. Finally, the specimens were gradually cooled to room temperature in the furnace.
3. Results and discussion
3.1 Structure, composition, and morphology of anodized SS
The surface oxidation (anodization), targeting the cost-effective SS 304 composed mainly of iron (Fe), chromium (Cr), and nickel (Ni), was carried out using a two-step procedure as outlined in Scheme 1. The anodization process of stainless steel with ethylene glycol and ammonium fluoride electrolyte involves a sequence of electrochemical reactions, complex formation, dissolution processes, and oxide growth mechanisms, ultimately resulting in the formation of a protective and functional anodic oxide layer on the surface of SS.
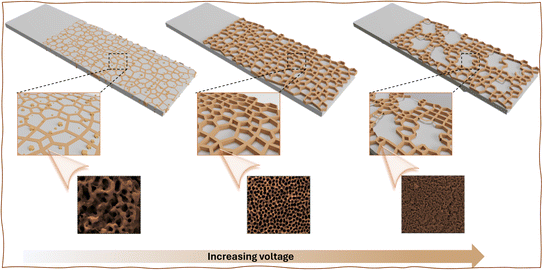 |
| | Scheme 1 Schematic illustrates an anodic oxidation process at different voltages, such as 65 V, 75 V, and 85 V, carried out on the cost-effective stainless steel (SS) 304, primarily containing iron (Fe), chromium (Cr), and nickel (Ni). Anodizing SS with ethylene glycol and ammonium fluoride electrolytes involves electrochemical reactions, complex formation processes, dissolution processes, and oxide growth mechanisms, forming a protective and functional nano-porous oxide layer. | |
A comprehensive theoretical hypothesis is presented in eqn (1)–(4) to understand the creation of oxide species from SS substrate. In first step, anodic oxidation occurs at SS, generating metal cations and electrons.24,31–33 Subsequently, in the second step, the ammonium fluoride in the electrolyte solution reacts with the metal cations (Mn+) to produce soluble metal fluoride complexes. These complexes aid in the dissolution of intermediate oxide layers formed during anodization, as observed in the third step. Meanwhile, in the fourth step, the applied voltage promotes the development of a dense and thick anodic oxide layer on the SS surface through electrochemical oxidation. In addition, gas evolution, specifically the release of hydrogen gas during anodization, leads to the creation of pores within the anodic oxide layer.34,35
| | | Mn+ + xNH4F → MFx + xNH4+ | (2) |
| | | MO + xNH4F → MFx + H2O + xNH4+ | (3) |
| | | MFx + yH2O → MxOy + zHF | (4) |
Moreover, Fluoride ions play two crucial roles in the oxide formation process. Firstly, they generate water-soluble [MFx]y− ions, which effectively hinder the formation of hydroxides.24,31,36 Secondly, they aid in the erosion of existing metal oxides, leading to the formation of a nanotubular layer rather than a dense one. Due to their ability to initiate chemical dissolution reactions and small ionic size, a small fraction of fluoride ions can be incorporated into the metal oxides.31,37,38 Upon annealing in air, fluoride ions are liberated from the metal oxide film surface as hydrogen fluoride (HF).38,39
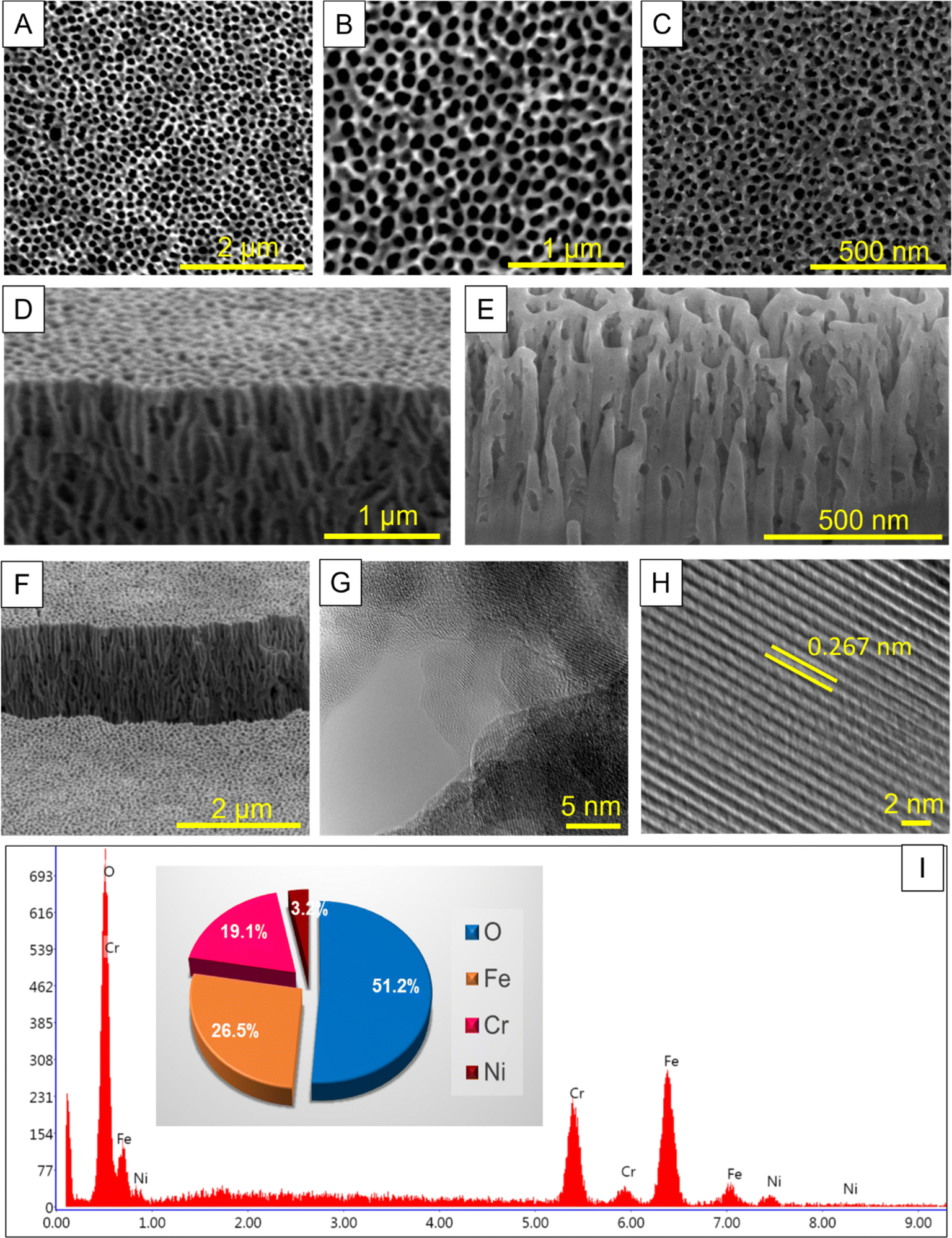
In addition, the voltage applied during anodic oxidation was significantly influenced by the formation of self-organized nanopore arrays, indicative of the consistency of pores on SS surfaces. Utilizing the observed influence, anodization was performed on SS samples at various operating voltages-specifically, 65, 75, and 85 V- for a duration of 30 minutes in an ethylene glycol electrolyte solution containing 0.1 mol per dm3 NH4F and 0.1 mol per dm3 H2O. The impact of varying anodization voltages on SS was assessed by analyzing surface morphology using scanning electron microscope (SEM) imaging. Fig. 1 displays the anodized SS's top surface, while Fig. S2† presents digital pictures depicting the films' macroscopic appearance. Moreover, SEM images provide comprehensive insights into the surface morphological characteristics. SEM imaging also confirmed the existence of self-organized nanopore arrays in all samples of anodized SS substrates. In addition, the SEM images display notable differences in the regularity of pores on the anodized SS films under an applied voltage of 75 V, contrasting with those untreated and anodized SS at 65, and 85 V (Fig. S3 and S5†). The SEM image of the sample's surface anodized at 75 V reveals a well-organized nanoporous oxide layer with a diameter ranging from 120 to 180 nm. However, the regularity of these highly ordered pores is notably disrupted by a further increase in voltage to 85 V (Fig. S5 and S6†). The voltage significantly affects the size and regularity of pores over the stainless steel's surface, which is concurrent with the literature.32,40 Previous investigations have proved that nanoporous arrays, including anodic aluminum oxide (AAO) and FeCrAl self-organizing porous structures, exhibit perfect self-ordered pore formations only when the potential does not exceed its breakdown threshold.32,41 Also, it can be observed that the interhole distance of the anodic porous oxide obtained during anodization depends on the anodization voltage, indicating a voltage-dependent formation of the nanohole arrays. The cross-sectional SEM image (Fig. 1D–F) authorized the presence of a nanoporous oxide layer comprising an ordered array of uniformly sized holes over the stainless steel's surface under the 75 V condition, exhibiting a curved and branched structure. Conversely, under the conditions of 65 V and 85 V, the cross-sectional SEM image revealed a disordered structure (Fig. S5†), indicating a significant influence of voltage on the formation of nanoporous arrays on the surface of SS. Based on the observed formation process of the ordered structure described above, it can be inferred that the anodic oxidation conducted in ethylene glycol-based electrolyte aligns more closely with the flow model proposed by Skeldon et al.42,43 In Fig. 1G and H, an HR-TEM image of the SS-75 V sample reveals distinct lattice fringes, with a lattice spacing of 0.267 nm corresponding to the (104) plane of the hematite phase, consistent with the previously reported work.24Fig. 1I displays the EDX spectra of the anodized SS sample at 75 V, while the inset of Fig. 1I illustrates the weight percentage of each element, accompanied by the corresponding EDX spectrum. The surfaces of the samples exhibit a significant enrichment in chromium (Cr) species and a considerable increase in oxygen (O) species following surface anodization compared to the bare SS (Fig. S4†). This augmentation of Cr and O species post-anodization aligns well with previous findings, indicating that the metal-oxide constitutes a major component of the nanoporous oxide layer.18,21,23,24 Furthermore, the well-ordered nano-array structures, resembling honeycomb-like patterns, possess high surface areas, and these structures would have been beneficial to improve the surface-anodized SS catalytic activity towards the HER and the bottom on the barrier layer (Cr-rich oxide layer; Fig. S7†) may impede the penetration of corrosive ions to the SS's surface atoms.
 |
| | Fig. 1 An overview of the morphology evolution of the as-prepared anodized SS-75 V sample as captured by SEM and TEM. (A–C) Top-view SEM images with low and high magnification. (D–F) Cross-sectional SEM images with different magnifications at an inclination (52°). (G and H) HR-TEM image of anodized SS-75 V sample. (I) EDX spectrum with the corresponding elements weight percentage. | |
Fig. 2A presents XRD results of both bare (untreated) SS and anodized SS samples. In all cases, three prominent peaks are observed at 43.85° (111), 51.06° (200), and 74.93° (220), which correspond to the austenite SS peaks (dot circle) originating from the SS304 substrate. These findings align well with previous studies and are consistent with the JCPDS reference card number 33-3097. Notably, the anodized samples (SS-65 V, SS-75 V, and SS-85 V) exhibited the presence of some low-intensity peaks with significant counts within the diffraction angle range of 20° to 40° (Fig. S8†). These peaks correspond to the Fe2O3 (hematite) phase of iron oxide (asterisk, JCPDS 33-0664). The XRD analysis reveals notable resemblances among the anodized samples, suggesting the absence of any crystallographic changes during the anodization process, regardless of the applied voltage variations. The consistent diffraction pattern observed across various anodized samples may be attributed to the relatively thin thickness of the nano-porous oxide film developed on the surface of SS. These thin films are challenging to detect using XRD due to its inherent limitation as a bulk characterization technique. Fig. 2B presents the Raman spectra obtained from the anodized SS samples, which were utilized to identify the chemical states of the samples. The spectra exhibit similarity and are characteristic of the α-Fe2O3 (hematite) phase, displaying peaks at approximately 228, 293, 416, 674, and 1331 cm−1. These observed peaks align closely with previously reported Raman data for the hematite phase.25,44–47 The peak at 228 cm−1 is attributed to the A1g mode, while those around 293 cm−1 and 416 cm−1 are assigned to the Eg modes. Additionally, the peak at approximately 674 cm−1 is linked to disorder effects and the existence of Fe2O3 nanocrystals, while the peak at around 1331 cm−1 is associated with hematite two-magnon scattering. It was further noted that the Raman spectrum of the anodized SS under voltages of 75 V and 85 V exhibits peaks characteristic of a magnetite (Fe3O4) phase, notably a strong band at 691 cm−1, alongside hematite peaks.25,44,47 This suggests that both samples contain a mixture of two phases. Specifically, in the case of 75 V anodization, the magnetite phase's composition appears slightly higher than the sample anodized at 85 V, as evidenced by the peak broadening in the respective Raman spectra. The coexistence of hematite and magnetite phases in the anodized SS sample may be attributed to the complex oxidation processes and the specific electrochemical conditions involved during the anodization process.48,49 The crystallographic structure and surface morphology of these iron oxide phases could be crucial in evaluating the electrocatalytic activity of the anodized SS towards the HER. Specific crystal facets and defects in these phases can promote hydrogen intermediate adsorption and enhance the overall catalytic efficiency.18,20
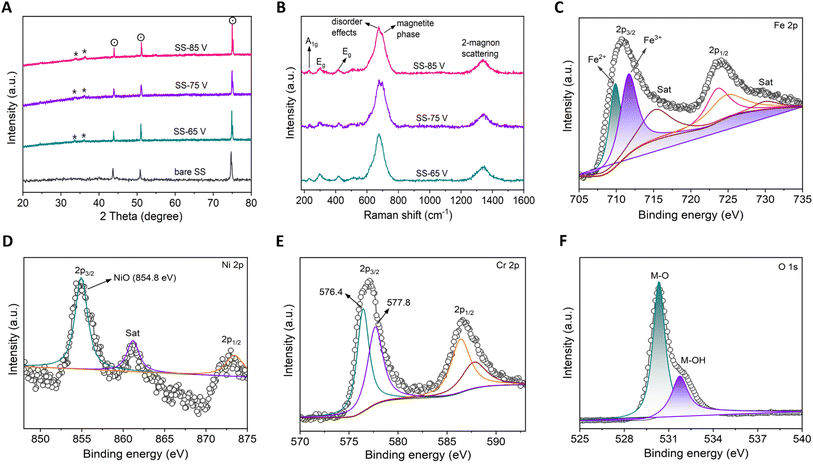 |
| | Fig. 2 Structural and spectroscopic characterization of the anodized SS nano-porous oxide layer. (A) X-ray diffractograms from the films. Identified phases are represented by symbols (dot circle: austenite SS peaks originating from the SS304 substrate; asterisk: hematite phase of iron oxide-Fe2O3). (B) Raman spectra of the anodized SS samples. The observed peaks are consistent with previously reported Raman data for hematite and magnetite phases.39,40 (C–F) XPS characterization of sample SS-75 V. (C) Fe 2p core levels – it shows Fe2+ and Fe3+ oxidation state. (D) Ni 2p core levels – Ni2+ oxidation state suggests NiO formation. (E) Cr 2p core levels – Cr is present in the form of CrIII. (F) O 1s core levels – this indicates mixed oxide states are present. a.u., arbitrary unit. | |
XPS was employed to conduct further investigations into the as-prepared material's surface chemical states and material constituents. The survey spectra (Fig. S9†) distinctly specified the existence of iron (Fe), nickel (Ni), chromium (Cr), and oxygen (O), implying the formation of nano-porous oxide thin films on the surface of the anodized SS. In the high-resolution Fe 2p spectrum of the SS-75 V anodized sample (Fig. 2C), peaks at 709.8 and 711.7 eV were observed, corresponding to Fe2+ and Fe3+.18,21,22,30 This observation suggests the coexistence of magnetite and hematite phases in SS-75 V, consistent with the findings from the Raman spectra. This confirmation indicates a reduction in the net oxidation state of Fe as the phase transition takes place, leading to the creation of oxygen vacancies. It is widely recognized that materials with self-doped oxygen vacancies, owing to the presence of vacant sites facilitating the transport of electrons or ions, may exhibit enhanced electron or ion conductivity compared to fully oxidized metal oxides.25 The presence of two peaks observed at 854.8 eV (2p3/2) and 873.2 eV (2p1/2) in the Ni 2p spectra (Fig. 2D) indicates the formation of NiO (Ni2+ oxidation state).21,24,50Fig. 2E shows a deconvolution of Cr 2p spectrum. In the surface-oxidized SS, Cr has undergone oxidation to a higher oxidation state, with the presence of Cr3+ (Cr2O3-576.4 eV; Cr(OH)3-577.8 eV) evident in the SS-75 V sample. This presence could manifest in the form of Cr2O3, Cr(III) hydroxide, or a combination of both forms.18,21,23,33 The O 1s spectra (Fig. 2F) exhibited two characteristic peaks at 530.4 eV and 531.8 eV, indicating the existence of passive metal oxides with mixed oxidation states on the surfaces of the anodized SS samples.22,24 The thorough and comparative XPS analysis has conclusively demonstrated the oxidation of crucial elements within SS, along with the notable observation of both iron oxide phases present on the surface of SS-75 V. Additionally, a pronounced presence of Ni2+ with FeCr oxides significantly influences the electrochemical behavior.
3.2 Electrocatalytic performance of anodized SS
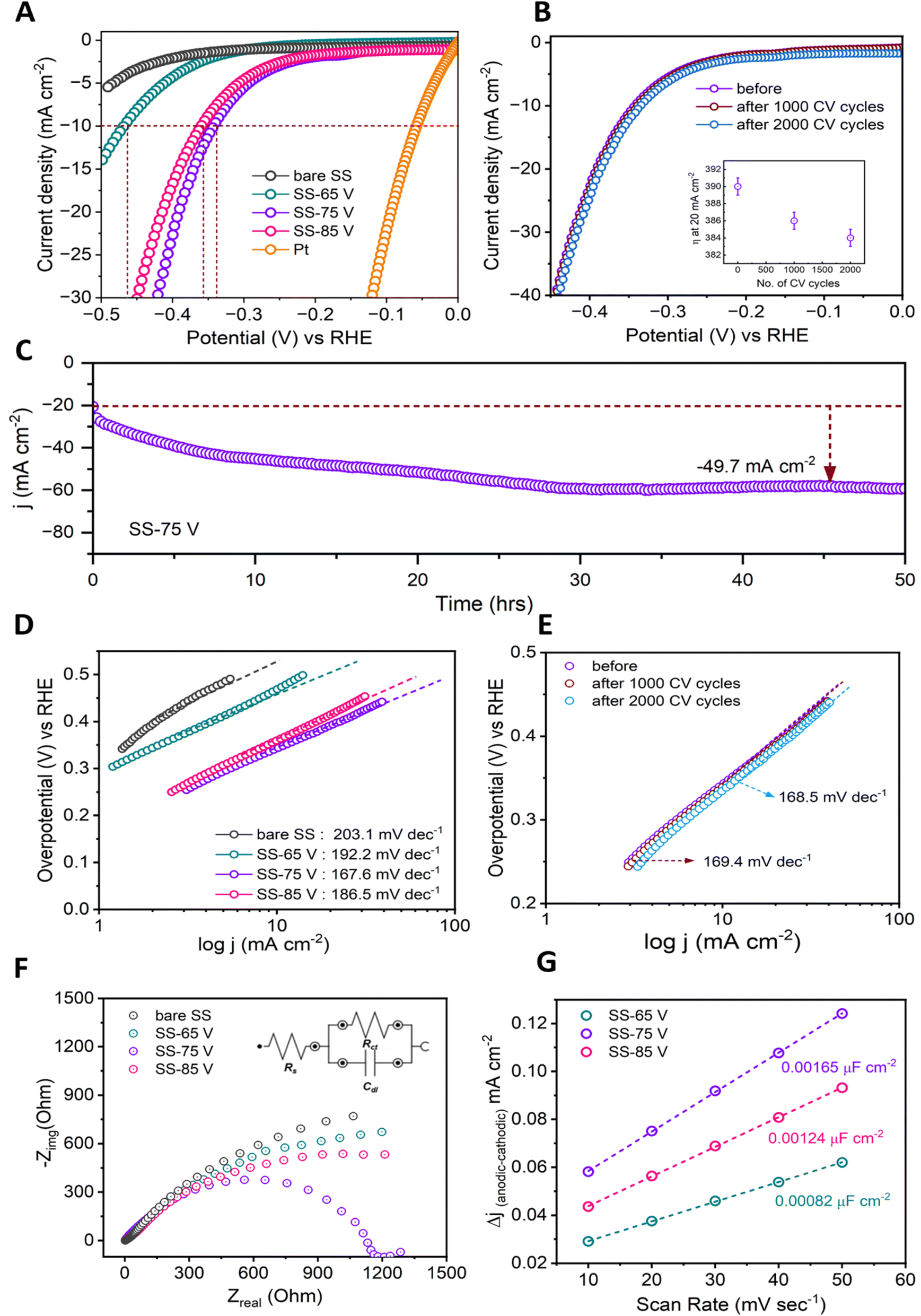
The HER performance of the prepared samples was evaluated in 1 M KOH electrolyte at room temperature using a standard three-electrode setup. Commercially available platinum (Pt) wire and untreated (bare) SS were also examined for comparison. Fig. 3A displays the linear sweep voltammetry (LSV) curves obtained on the reversible hydrogen electrode (RHE). The Pt wire exhibits outstanding activity, whereas bare SS demonstrates no HER activity within the investigated potential range. Surface-anodized SS electrodes exhibit significantly enhanced catalytic efficiency in HER compared to pristine SS electrode. The overpotential (η) needed to achieve a current density of 10 mA cm−2 serves as a valuable metric for assessing catalyst performance in a 12.3% efficiency solar water splitting device operating under 1 sun illumination.51 In this regard, the SS-75 V electrode demonstrates the highest catalytic activity, characterized by the lowest onset potential and requiring only a 343 mV overpotential to drive a current density of 10 mA cm−2. This value is notably lower than the overpotentials required by the anodized SS-65 V (468 mV) and SS-85 V (360 mV) electrodes to achieve a current density of 10 mA cm−2. Alongside assessing the HER activity, another crucial criterion is determining the long-term stability of the catalysts. The prolonged cycling performance of the SS-75 V electrode was investigated by conducting continuous cyclic voltammetry between −0.5 and 0.1 V versus RHE at a scan rate of 100 mV s−1. Fig. 3B presents the LSV curves obtained before and after 1000 and 2000 cycles, clearly illustrating the sustained electrochemical HER catalytic activity of the SS-75 V electrode. Interestingly, it was observed that the overpotential decreased from 390 mV to 386 mV after 1000 cycles and further to 384 mV after 2000 cycles at a current density of 20 mA cm−2. These findings highlight a significant improvement in the catalytic performance of the SS-75 V electrode, contrary to the typical trend of increasing overpotential with prolonged hydrogen reaction time. Furthermore, the enhanced catalytic performance, evidenced by the increase in current density, was corroborated through chronoamperometry (CA) measurements at −20 mA cm−2 (at constant potential −390 mV) over a duration of 50 hours, as shown in Fig. 4C. We also conducted a long-term durability assessment to confirm the practical availability of using CA on the SS-75 V electrode, operating at a high current density of 100 mA cm−2 under a constant potential condition for 24 hours (Fig. S10†). During the initial 4 hours, we observed a gradual increase in current density from 100 mA cm−2 to 172 mA cm−2. Following these 4 hours, the current density remained stable without any decline for the subsequent 20 hours. The underlying cause might be the self-activation of the electrode and the creation of metal hydroxides, leading to numerous active sites with extensive surface areas. The Tafel slope represents the electrocatalysts' intrinsic properties obtained through the rate-determining step of HER. The Tafel slopes, extracted from the polarization plots depicted in Fig. 4A and B, are illustrated in Fig. 4D and E. The Tafel slopes observed for both bare and anodized SS electrodes range from 167 to 203 mV dec−1, indicating adherence to the Volmer–Heyrovsky pathway in the HER. The widely accepted mechanism of the HER in alkaline media comprises three elementary reaction steps.52–55| | | M + H2O + e− = M–Hads + OH− | (5) |
| | | M–Hads + H2O + e− = M + H2 + OH− | (6) |
 |
| | Fig. 3 HER activity and stability of the as-developed binder-free nano-porous metal oxide electrodes. (A) Polarization curves after iR correction show the catalytic performance of the SS-75 V electrode compared to those of SS-65 V, SS-85 V, Pt wire, and bare SS in an alkaline solution of 1 M KOH. (B) Polarization curves of the SS-75 V electrode at its initial state and after 2000 CV cycles. The SS-75 V electrode exhibits sustained electrochemical HER catalytic activity initially and after 3000 cycles. (C) Long-term chronoamperometric (CA) stability measurement at 20 mA cm−2 (applied potential −390 mV) for the SS-75 V electrode. (D and E) The relevant Tafel plots were obtained from the corresponding polarization curves in (A and B). (F) Electrochemical impedance spectroscopy (EIS) was conducted to analyze the charge transfer resistance (Rct) of bare and anodized SS electrodes. The inset depicts a simplified Randles circuit model. (G) Calculation of double layer capacitance (Cdl): current density (Δj = ja − jc) plotted against scan rate. | |
 |
| | Fig. 4 Postcatalytic characterization of anodized SS-75 V electrode. (A) SEM image of after long-term stability test (magnification of 500 nm). (B) Raman spectra of SS-75 V electrode after CA testing. (C–F) Fe, Ni, Cr, and O valence state in SS-75 V electrode (after stability) examined by XPS analysis. (C) Fe 2p; (D) Ni 2p; (E) Cr 2p; (F) O 1s. | |
The Volmer step (5) involves breaking a water molecule and the adsorption of hydrogen onto a vacant site of the electrode/catalyst surface. Subsequently, hydrogen production occurs through either an electrochemical process (Heyrovsky step (6)) or a chemical process (Tafel step (7)). Several studies have indicated that, at low overpotentials, the HER mechanism entails the Volmer step followed by concurrent Heyrovsky and Tafel steps, while at higher overpotentials, the Tafel step becomes insignificant, and the reaction primarily follows the Volmer–Heyrovsky mechanism. In practical applications, a smaller Tafel slope leads to an enhanced HER rate with a steady increase in overpotential, thereby offering advantageous outcomes.
The EIS spectra were conducted during the HER activity to study the electrode kinetics (Fig. 4F). Additionally, a tedious R–C equivalent diagram was employed to analyse the kinetic disparities among these electrodes, as depicted in the inset of Fig. 4F. The smaller-semicircle observed in the low-frequency region of the Nyquist plot indicates a heightened HER process, correlating with reduced charge transfer resistance (Rct). Specifically, the charge-transfer resistance (Rct) observed for the SS-75 V electrode was notably lower compared to that of the SS-65 V and SS-85 V electrodes, suggesting a significantly enhanced electron transport capability for the hydrogen evolution reaction (HER) in the SS-75 V electrode. This trend aligns with the findings from the Tafel slopes and polarization curves. The robust stability and elevated HER activity of the developed binder-free electrode of SS-75 V, facilitated by the synergistic intervention of nanoporous oxide structure and active sites, may be assumed to occur by the following factors. The electrochemical active surface area (ESCA) of the anodized SS samples was estimated using the double-layer capacitance (Cdl) obtained from cyclic voltammetry (CV) curves. CV curves of the prepared electrodes at various scan rates in the 0.88–1.12 V vs. RHE are presented in Fig. S11.† The calculated Cdl values (Fig. 3G) were 0.00082, 0.00165, and 0.00124 μF cm−2 for SS-65 V, SS-75 V, and SS-85 V samples, respectively. Notably, SS-75 V demonstrated a significantly larger double-layer capacitance compared to the other two samples. This indicates that SS-75 V can expose more effective active sites due to its proportional relationship with surface area and material conductivity, thereby contributing to enhanced HER activity.
Further analysis of the SS-75 V sample was conducted using SEM, Raman, and XPS to determine any potential alterations to the valence state over the surface, phase change, and microstructure change after 50 hours of CA operation. Fig. 4A illustrates that the surface (nano-porous) morphology of the SS-75 V electrode remains unchanged even after continuous 50 h CA studies. Raman spectra (Fig. 4B) showed attenuation and broadening of peaks corresponding to the hematite and magnetite phases with the progression of the long-term stability test, and also, the Raman spectra data matched with the previously reported Raman data of Fe0.993+O(OH).44 This undoubtedly indicates that the crystallinity of the oxide layer decreased as metal hydroxide formed, which has a higher electrocatalytic activity.25,56 The Fe 2p, Ni 2p, and Cr 2p XPS spectra (Fig. 4C–E and S12†) were examined, with their corresponding peaks interpreted from Fig. 4C (Fe3+: 711.2 eV), Fig. 4D (Ni(OH)2: 855.4 eV), and Fig. 4E (Cr2O3: 576.3 eV; Cr(OH)3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :577.7 eV). These findings align with the characterization work on anodized SS samples previously reported by Kim et al.25Fig. 4F displays the O 1s spectrum, highlighting all component peaks and their respective contributions. The prominent band observed at a binding energy of 530.1 eV is because of the oxygen contribution from oxide ions (M–O), while the band at 531.6 eV is associated with oxygen from hydroxide (M–(OH)2).57,58 Additionally, the peaks at 532.9 eV are linked to water's physicochemical absorption.57,58 Together, these peaks indicate the construction of hydroxyls (OH−) and lattice oxygen (O2−) over the SS-75 V surface. Based on these findings, it is plausible to deduce that the formation of metal hydroxides and the preservation of highly organized nano-porous transition metal oxide structures with extensive surface areas occur. This facilitates the creation of multiple active sites for electrochemical reactions, which progressively multiply as cathodic current is applied for hydrogen evolution, resulting in the self-activation of the electrode and a gradual reduction in overpotential for HER.
:577.7 eV). These findings align with the characterization work on anodized SS samples previously reported by Kim et al.25Fig. 4F displays the O 1s spectrum, highlighting all component peaks and their respective contributions. The prominent band observed at a binding energy of 530.1 eV is because of the oxygen contribution from oxide ions (M–O), while the band at 531.6 eV is associated with oxygen from hydroxide (M–(OH)2).57,58 Additionally, the peaks at 532.9 eV are linked to water's physicochemical absorption.57,58 Together, these peaks indicate the construction of hydroxyls (OH−) and lattice oxygen (O2−) over the SS-75 V surface. Based on these findings, it is plausible to deduce that the formation of metal hydroxides and the preservation of highly organized nano-porous transition metal oxide structures with extensive surface areas occur. This facilitates the creation of multiple active sites for electrochemical reactions, which progressively multiply as cathodic current is applied for hydrogen evolution, resulting in the self-activation of the electrode and a gradual reduction in overpotential for HER.
3.3 Corrosion studies of surface-treated SS
A potentiodynamic polarization study was used to study the corrosion-resistant properties of anodized SS in 3.5% NaCl and 1 M KOH solutions. The anodic and cathodic Tafel curves of bare and anodized SS at different anodizing voltages in NaCl and KOH solutions are presented in Fig. 5, and corresponding polarization parameters derived from extrapolation methods are presented in Table 1. The anodic and cathodic Tafel curves display significant variations after anodization, likely due to differences in passive layer thickness, localized chemical gradients, or the number of free atoms or electrons available for oxidation. Careful observation of the polarization curves shows that corrosion potential (Ecorr) values of anodized SS shifted toward a more positive direction. In 3.5% NaCl solution, the anodization does not cause any significant shift in the corrosion potential (Ecorr); however, in 1 M KOH solution, the Tafel curves of anodized SS showed a prominent shift towards the positive direction. This observation indicates that anodization has a marked adverse effect on anodic dissolution reaction. The inspection of Table 1 reveals that the passive film could be more stable for prolonged as it shows the best corrosion resistance at SS-75 V sample. This is likely due to the highly protective and thick passive layer formed on the anodized SS surface. The enhanced chemical stability can be attributed to the absence of iron dissolution in the alkaline electrolyte. The passive layer is usually composed of a layer of Cr–Fe oxide on the inner side and a layer of Cr-hydroxide on the outer side. The lowest corrosion current density (Icorr) value was derived for SS-75 V of anodizing voltage in both electrolytes. The corrosion rate values of bare and anodized SS-85 V are comparable. This finding suggests that prolonged exposure to either neutral (3.5% NaCl) or basic (KOH) solution causes the diffusion of corrosive species such as chloride ions, hydroxide ions, and other active species and dissolves the surface protective passive layer.
 |
| | Fig. 5 Corrosion testing of anodized SS electrodes. (A and B) Potentiodynamic polarization curves for bare and anodized SS corrosion testing in (A) 3.5% NaCl and (B) 1 M KOH solution. | |
Table 1 Potentiodynamic polarization parameters for bare and anodized SS corrosion in 3.5% NaCl and 1 M KOH solution
| Parameter |
NaCl |
KOH |
| Bare SS |
SS-65 V |
SS-75 V |
SS-85 V |
Bare SS |
SS-65 V |
SS-75 V |
SS-85 V |
|
E
corr; (mV) |
−66.80 |
−15.50 |
−58.50 |
69.6 |
−212.0 |
−179.0 |
−343.0 |
−212.0 |
|
I
corr; (μA) |
16.10 |
12.90 |
0.915 |
4.930 |
139.0 |
65.70 |
6.680 |
102.0 |
|
β
a; (V per decade) |
104.2 × 10−3 |
82.40 × 10−3 |
311.1 × 10−3 |
486.0 × 10−3 |
1.233 |
987.6 × 10−3 |
97.90 × 10−3 |
2.052 |
|
β
c (V per decade) |
205.6 × 10−3 |
172.5 × 10−3 |
228.1 × 10−3 |
395.5 × 10−3 |
1.568 |
457.5 × 10−3 |
125.1 × 10−3 |
552.5 × 10−3 |
|
C
R (mpy) |
7.383 |
5.908 |
0.599 |
2.256 |
4.623 |
2.182 |
0.221 |
3.370 |
4. Conclusion
In summary, we have developed a meticulously structured nano-porous film on the surface of SS through a straightforward modification process of anodic oxidation. This resulted in the generation of catalytically active centers composed of Fe, Ni, and Cr, consequently leading to notable enhancements in electrocatalytic hydrogen evolution and corrosion resistance properties when operating in alkaline environments. Moreover, the SS-75 V electrode demonstrated consistent performance in the HER even after undergoing 2000 cycles of CV and 50 hours of chronoamperometric studies. This anodized SS electrode offer a synergistic combination of enhanced HER activity and superior corrosion resistance, making it an ideal candidate for use in alkaline water electrolyzers. Its ability to efficiently catalyse the HER while withstanding corrosion ensures the reliable and sustainable operation of electrolysis systems for hydrogen production. Furthermore, this methodology can be applied to modify the surfaces of various SS alloys, incorporating heteroatom doping such as sulphur, selenium, and phosphorus to enhance their overall catalytic activity.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Conflicts of interest
The authors have declared that they have no conflicts of interest.
Acknowledgements
This research was supported by the Emirates Nuclear Technology Center (ENTC) Research Grant at Khalifa University. The author thanks Mr Vasanth Rajendiran Jothi, Hanyang University, Seoul, for drawing a schematic diagram.
References
- J. C. Ehlers, A. A. Feidenhans'l, K. T. Therkildsen and G. O. Larrazábal, ACS Energy Lett., 2023, 8, 1502–1509 CrossRef CAS.
- M. David, C. Ocampo-Martínez and R. Sánchez-Peña, J. Energy Storage, 2019, 23, 392–403 CrossRef.
- A. S. Emam, M. O. Hamdan, B. A. Abu-Nabah and E. Elnajjar, Int. J. Hydrogen Energy, 2024, 64, 599–625 CrossRef CAS.
- H. Tüysüz, Acc. Chem. Res., 2024, 57, 558–567 Search PubMed.
- Y. Shen, S. He, Y. Zhuang, S. Huang, C. Meng, A. Yuan, W. Miao and H. Zhou, ACS Appl. Nano Mater., 2023, 6, 16873–16881 CrossRef CAS.
- L. Wan, Z. Xu, Q. Xu, M. Pang, D. Lin, J. Liu and B. Wang, Energy Environ. Sci., 2023, 16, 1384–1430 RSC.
- A. Parra-Puerto, K. L. Ng, K. Fahy, A. E. Goode, M. P. Ryan and A. Kucernak, ACS Catal., 2019, 9, 11515–11529 CrossRef CAS.
- X. Xu, Y. Lu, J. Shi, X. Hao, Z. Ma, K. Yang, T. Zhang, C. Li, D. Zhang, X. Huang and Y. He, Nat. Commun., 2023, 14, 7708 CrossRef CAS PubMed.
- N. Esfandiari, M. Aliofkhazraei, A. N. Colli, F. C. Walsh, S. Cherevko, L. A. Kibler, M. M. Elnagar, P. D. Lund, D. Zhang, S. Omanovic and J. Lee, Prog. Mater. Sci., 2024, 144, 101254 CrossRef CAS.
- J. Jiang, L. Zhu, Y. Sun, Y. Chen, H. Chen, S. Han and H. Lin, J. Power Sources, 2019, 426, 74–83 CrossRef CAS.
- A. H. Shah, Z. Zhang, C. Wan, S. Wang, A. Zhang, L. Wang, A. N. Alexandrova, Y. Huang and X. Duan, J. Am. Chem. Soc., 2024, 146, 9623–9630 CrossRef CAS PubMed.
- Z. Li, L. Sun, Y. Zhang, Y. Han, W. Zhuang, L. Tian and W. Tan, Coord. Chem. Rev., 2024, 510, 215837 CrossRef CAS.
- M. Zhou, Y. Zhao, Z. Liu, X. Zhao, E. Liu, L. Xiao, P. Yin, C. Dong, H. Liu, X. Du and J. Yang, Nano Res., 2024, 17, 4711–4719 CrossRef CAS.
- D. R. Kauffman, X. Deng, D. C. Sorescu, T.-D. Nguyen-Phan, C. Wang, C. M. Marin, E. Stavitski, I. Waluyo and A. Hunt, ACS Catal., 2019, 9, 5375–5382 CrossRef CAS.
- S. He, B. Chen, C. Meng, F. Shi, A. Yuan, W. Miao and H. Zhou, ACS Appl. Nano Mater., 2024, 7, 1138–1145 CrossRef CAS.
- Y. Zhang, B. Chen, Y. Qiao, Y. Duan, X. Qi, S. He, H. Zhou, J. Chen, A. Yuan and S. Zheng, J. Mater. Sci. Technol., 2024, 201, 157–165 CrossRef.
- S. Anantharaj, S. Chatterjee, K. C. Swaathini, T. S. Amarnath, E. Subhashini, D. K. Pattanayak and S. Kundu, ACS Sustain. Chem. Eng., 2018, 6, 2498–2509 CrossRef CAS.
- H. Schäfer, S. Sadaf, L. Walder, K. Kuepper, S. Dinklage, J. Wollschläger, L. Schneider, M. Steinhart, J. Hardege and D. Daum, Energy Environ. Sci., 2015, 8, 2685–2697 RSC.
- C. Dumas, A. Mollica, D. Féron, R. Basséguy, L. Etcheverry and A. Bergel, Electrochim. Acta, 2007, 53, 468–473 CrossRef CAS.
- X. Liu, B. You and Y. Sun, ACS Sustain. Chem. Eng., 2017, 5, 4778–4784 CrossRef CAS.
- H. Schäfer, S. M. Beladi-Mousavi, L. Walder, J. Wollschläger, O. Kuschel, S. Ichilmann, S. Sadaf, M. Steinhart, K. Küpper and L. Schneider, ACS Catal., 2015, 5, 2671–2680 CrossRef.
- S. Anantharaj, M. Venkatesh, A. S. Salunke, T. V. S. V. Simha, V. Prabu and S. Kundu, ACS Sustain. Chem. Eng., 2017, 5, 10072–10083 CrossRef CAS.
- A. K. Gomaa, B. S. Shaheen, G. E. Khedr, A. M. Mokhtar and N. K. Allam, Energy Fuels, 2022, 36, 7025–7034 CrossRef CAS.
- R. Bose, G. Suryaprakash Goud, M. I. Helal, I. Barsoum, S. O. Cho and A. Alfantazi, ACS Appl. Energy Mater., 2024, 7(10), 4402–4411 CrossRef CAS.
- M. Kim, J. Ha, N. Shin, Y.-T. Kim and J. Choi, Electrochim. Acta, 2020, 364, 137315 CrossRef CAS.
- F. Moureaux, P. Stevens, G. Toussaint and M. Chatenet, J. Power Sources, 2013, 229, 123–132 CrossRef CAS.
- J. Park, H. Yoo and J. Choi, J. Power Sources, 2019, 431, 25–30 CrossRef CAS.
- M. Lee, M. S. Jee, S. Y. Lee, M. K. Cho, J.-P. Ahn, H.-S. Oh, W. Kim, Y. J. Hwang and B. K. Min, ACS Appl. Mater. Interfaces, 2018, 10, 24499–24507 CrossRef CAS PubMed.
- X. Lu and C. Zhao, Nat. Commun., 2015, 6, 6616 CrossRef CAS PubMed.
- R. Bose, V. R. Jothi, K. Karuppasamy, A. Alfantazi and S. C. Yi, J. Mater. Chem. A, 2020, 8, 13795–13805 RSC.
- L. Suhadolnik, Ž. Marinko, M. Ponikvar-Svet, G. Tavčar, J. Kovač and M. Čeh, J. Phys. Chem. C, 2020, 124, 4073–4080 CrossRef CAS PubMed.
- K. Zhao, S. Wang, J. Lu, C. Ni, M. Wang and S. Wang, Surf. Coat. Technol., 2021, 425, 127707 CrossRef CAS.
- Y. Wang, G. Li, K. Wang and X. Chen, Appl. Surf. Sci., 2020, 505, 144497 CrossRef CAS.
- B. T. Lu, L. J. Qiao, J. L. Luo and K. W. Gao, Philos. Mag., 2011, 91, 208–228 CrossRef CAS.
- Q. Yang, J. G. Yu and J. L. Luo, J. Electrochem. Soc., 2003, 150, B389 CrossRef CAS.
- J. M. Macak, H. Tsuchiya, A. Ghicov, K. Yasuda, R. Hahn, S. Bauer and P. Schmuki, Curr. Opin. Solid State Mater. Sci., 2007, 11, 3–18 CrossRef CAS.
- K. Indira, U. K. Mudali, T. Nishimura and N. Rajendran, J. Bio- Tribo-Corros., 2015, 1, 28 CrossRef.
- J. Heo, S. Y. Lee, J. Lee, A. Alfantazi and S. O. Cho, Nanomaterials, 2021, 11(4), 838 CrossRef CAS PubMed.
- K. Kure, Y. Konno, E. Tsuji, P. Skeldon, G. E. Thompson and H. Habazaki, Electrochem. Commun., 2012, 21, 1–4 CrossRef CAS.
- H. Tsuchiya, T. Suzumura, Y. Terada and S. Fujimoto, Electrochim. Acta, 2012, 82, 333–338 CrossRef CAS.
- S. Z. Chu, K. Wada, S. Inoue, M. Isogai, Y. Katsuta and A. Yasumori, J. Electrochem. Soc., 2006, 153, B384 CrossRef CAS.
- P. Skeldon, G. E. Thompson, S. J. Garcia-Vergara, L. Iglesias-Rubianes and C. E. Blanco-Pinzon, Electrochem. Solid-State Lett., 2006, 9, B47 CrossRef CAS.
- S. J. Garcia-Vergara, P. Skeldon, G. E. Thompson and H. Habazaki, Electrochim. Acta, 2006, 52, 681–687 CrossRef CAS.
- D. L. A. de Faria, S. Venâncio Silva and M. T. de Oliveira, J. Raman Spectrosc., 1997, 28, 873–878 CrossRef CAS.
- D. Trpkov, M. Panjan, L. Kopanja and M. Tadić, Appl. Surf. Sci., 2018, 457, 427–438 CrossRef CAS.
- Y.-S. Hu, A. Kleiman-Shwarsctein, A. J. Forman, D. Hazen, J.-N. Park and E. W. McFarland, Chem. Mater., 2008, 20, 3803–3805 CrossRef CAS.
- A. M. Jubb and H. C. Allen, ACS Appl. Mater. Interfaces, 2010, 2, 2804–2812 CrossRef CAS.
- L. M. C. Pérez Pérez, J. Hernández Torres, L. Zamora Peredo, L. García González, M. García Hipólito, C. Falcony Guajardo, P. E. Cardoso Avila, O. Velázquez Camilo and A. Báez Rodríguez, ECS Trans., 2022, 106, 203 CrossRef.
- E. N'Dah, S. Tsipas, F. J. Bolívar, M. P. Hierro and F. J. Pérez, Oxid. Met., 2008, 69, 77–94 CrossRef.
- H. Zhong, J. Wang, F. Meng and X. Zhang, Angew. Chem., Int. Ed., 2016, 55, 9937–9941 CrossRef CAS PubMed.
- C. C. L. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef CAS PubMed.
- M. A. Gaikwad, V. V. Burungale, D. B. Malavekar, U. V. Ghorpade, U. P. Suryawanshi, S. Jang, X. Guo, S. W. Shin, J.-S. Ha, M. P. Suryawanshi and J. H. Kim, Adv. Energy Mater., 2024, 14, 2303730 CrossRef CAS.
- M. Ďurovič, J. Hnát and K. Bouzek, J. Power Sources, 2021, 493, 229708 CrossRef.
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC.
- J. O. M. Bockris and E. C. Potter, J. Chem. Phys., 1952, 20, 614–628 CrossRef CAS.
- K. Zhu, G. Zhu, J. Wang, J. Zhu, G. Sun, Y. Zhang, P. Li, Y. Zhu, W. Luo, Z. Zou and W. Huang, J. Mater. Chem. A, 2018, 6, 21360–21367 RSC.
- R. Bose, V. R. Jothi, D. B. Velusamy, P. Arunkumar and S. C. Yi, Part. Part. Syst. Charact., 2018, 35, 1800135 CrossRef.
- H. Zhou, F. Yu, Q. Zhu, J. Sun, F. Qin, L. Yu, J. Bao, Y. Yu, S. Chen and Z. Ren, Energy Environ. Sci., 2018, 11, 2858–2864 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Details of materials characterization, electrochemical experiments, digital images of the experimental setup, XRD and Raman data, SEM images of bare SS and anodized SS at 65, 85 and 90 V samples, and XPS survey spectrum of anodized samples. See DOI: https://doi.org/10.1039/d4ta03317e |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *ab,
Surya Prakash
Gajagouni
c,
Imad
Barsoum
c,
Sung Oh
Cho
*ab,
Surya Prakash
Gajagouni
c,
Imad
Barsoum
c,
Sung Oh
Cho