
K+ selectivity modulation in non-aqueous CO2 electroreduction on lead catalysts: from oxalic to tartaric acid production†
Eduardo
Arizono dos Reis
 ab,
Gelson T. S. T.
da Silva
c and
Caue
Ribeiro
*abc
ab,
Gelson T. S. T.
da Silva
c and
Caue
Ribeiro
*abc
aSão Carlos Institute of Chemistry, University of São Paulo (USP), São Carlos, São Paulo, Brazil
bNanotechnology National Laboratory for Agriculture (LNNA), Embrapa Instrumentation, São Carlos, São Paulo, Brazil. E-mail: caue.ribeiro@embrapa.com.br
cDepartment of Chemistry, Interdisciplinary Laboratory of Electrochemistry and Ceramics, Federal University of São Carlos, São Carlos, São Paulo, Brazil
First published on 21st May 2024
Abstract
Here, we show that the presence of potassium ions in the catholyte modulates the selectivity of a Pb plate electrode, leading to the formation of tartrate, a C4 compound, from CO2 reduction. A faradaic efficiency of 60% was achieved at −2.3 V (vs. Ag/Ag+) for tartrate using a proton exchange membrane and a high concentration of potassium-based supporting anolyte. The electrode microenvironment with a higher potassium concentration also inhibits cathode corrosion and deactivation. Remarkably, the electroreduction of CO2 changes the selectivity with the cationic availability in the anolyte. Higher FE to formic acid is observed with an increase in the proton concentration, and by increasing anolyte K+ availability, C–C coupled products (oxalate, C2, and tartrate, C4) are formed in the majority. Our results prove that controlling potassium ions and the proton concentration in the catholyte regulates the selectivity of the Pb plate electrode and can lead to the formation of a C2+ product from CO2 reduction.
1. Introduction
Implementing carbon-capture technology for CO2 directly from industrial waste gases can overcome the low benchmark atmosphere capture cost1,2 and use high purity CO2 emission to convert carbon dioxide into other molecules of industrial chemical interest.3–7 Among the various techniques for dioxide conversion, the electrochemical CO2 reduction reaction (CO2RR) is a growing approach8–10 due to the multiple pathways capable of producing all the top globally consumed carbonaceous products11 using a flexible suite of electricity-mediated reduction that can tune the selectivity of the value-added products obtained.8,11 One of the limitations of the CO2RR in aqueous media is driving the high-energy steps for activating CO2 reduction at a moderate potential for a specific product.12,13 Due to the high inertness of CO2 molecules associated with their low solubility in aqueous electrolytes (33 mM at 1 atm and 25 °C), the hydrogen evolution reaction (HER) is the main issue for achieving high current efficiencies.14 An effective way to circumvent the high reaction rates and suppress the HER is by engineering the reaction environment. Several strategies, such as ionic liquid electrolytes, gas phase reactions, and aprotic solvents, have been used.15–19 In the CO2RR, aprotic solvents can give more complex C2+ molecules as products (oxalic acid, glycolic acid, and glyoxylic acid), leading to new CO2-based polymers.20 Among the few electrocatalysts reported in the literature, lead-based electrodes are the most efficient catalyst to convert CO2 into C2+ products with higher faradaic efficiency,15,16,21–28 but their selectivity relies mainly on the electrolyte solvent19,22,29 and proton availability.22,30–32Using the non-aqueous electrolyte in a GDE flow cell configuration is far from an actual application.3,21 More basic studies are still necessary since old previous publications lack sufficient analysis, report misleading analytical data, and present conflicting reactivity.21,33 At the same time, microenvironment modulation has been found to be a critical factor in electrochemical reactions rather than just investigating new catalysts.34,35 The electrocatalyst microenvironment can play an essential role in the activity and selectivity of the reaction either by increasing the reactant at the interface or/and by the stabilization of the intermediates close to the electrode surface, favouring the formation of multielectron reduction products.36–40 In non-protic organic media, electrolyte ions, metal centre catalysts, solvent, and water content are the primary investigated interfacial microenvironment regulators for the CO2RR.19,22,32,41,42 Recently, high concentrations of local alkali ions have been demonstrated to inhibit proton diffusion and favour carbon–carbon (C–C) coupling reactions in aqueous electrolytes.43–47 Understanding the significance of alkali ions present in the electrolyte, regarding the solubility limit, and the ion migration effect on the selectivity of the CO2 reduction reaction is an unexplored field for non-protic electrolytes.
In this sense, we propose an unprecedented strategy to promote C2+ product formation in non-aqueous CO2RR, using a cation exchange membrane to regulate the reaction by the cation migration effect on flat Pb plate cathodes and tuning product selectivity by anolyte selection. We used electrochemical measurements to demonstrate that the anolyte pH influences the CO2 reduction activity and selectivity. Additionally, this work provides experimental evidence that proton availability and the migration of potassium ions through the membrane are selectivity regulators that can lead to the formation of C6 products from CO2 reduction.
2. Results and discussion
As a starting point, LSV measurements were made in the presence of CO2 and N2-saturated acetonitrile and 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) for each studied anolyte and are presented in Fig. 1a. Pb electrodes show higher activity for the CO2RR at a lower potential, with the higher pH anolyte exhibiting the start of the onset potential at −2.2 V (vs. Ag/Ag+), and no significant current density was observed for the electrode under N2-saturation. Moreover, at −2.5 V vs. Ag/Ag+, the alkaline anolyte (KOH 0.5 M) achieved three times higher current density than the near-neutral anolyte (KHCO3 0.5 M) and almost ten times the current density achieved for the acidic anolyte (H2SO4). Fig. 1b shows the stability of the Pb electrode with different electrolytes during CO2 reduction. From chronoamperometry curves collected during the reaction, it is possible to observe that the stability also depends on the anolyte type and pH. Over time, a current loss is observed in the acidic and near-neutral anolytes due to corrosion reactions at the electrode surface. | ||
| Fig. 1 (a) CO2RR polarization curve for a Pb plate electrode in dried acetonitrile 0.1 M TBAPF6 with H2SO4, KHCO3, and KOH anolytes and (b) chronoamperometric curve for the different anolytes on the Pb plate. | ||
Moreover, higher current and stability are achieved using the alkaline anolyte (KOH) since K+ ions, from the crossover, present near the Outer-Helmholtz plane, modulate the corrosion mechanism by electrostatic repulsion of H+, and it is associated with the carbon–carbon (C–C) coupling in the CO2RR since elevated potassium ion concentrations also stabilized the intermediates.44,47 Although the potassium ion can promote the precipitation of the CO2RR products and become a problem for some cell configurations,48 in this case, it was used as a strategy to favour the C–C coupling to lead to higher-value products achieving competitive current density in non-aqueous media compared with the literature.21,28,49 Electrochemical impedance spectroscopy spectra in Fig. 2a and b show that the reaction is accelerated with the availability of potassium ions in the anolyte. Low electron transfer resistance (see Table S2†) is observed in the following order: KOH < KHCO3 < H2SO4. With the increase in the pH value and the minor proton availability, K+ inhibits proton migration, and it is rapidly transported through the Nafion membrane to the catholyte solution,50 increasing the potassium ion concentration and favouring the promotion of the C–C coupling.
 | ||
| Fig. 2 Nyquist plots for the Pb plate under a CO2 atmosphere for the different anolytes (a) before and (b) after the cyclic voltammetry cleaning step. | ||
Furthermore, the EIS for the Pb plate before (Fig. 2a) and after (Fig. 2b) the cyclic voltammetry cleaning shows a decrease in the electron transfer resistance after the complete cleaning process for the alkaline anolyte. For the other anolytes (H2SO4 and KHCO3), an activity loss and a higher electron transfer resistance were observed, which can be associated with the intergranular corrosion (observed by SEM) that occurs right after the acidic cleaning and results in catalyst deactivation. The catalyst deactivation is also supported by the appearance of two charge-transfer resistances in series for KHCO3 and H2SO4 anolytes in the equivalent electrical circuit used for modelling the Pb electrode EIS, exhibited in Fig. S1.†
SEM images for each anolyte were obtained for the Pb electrode before and after the CO2RR to evaluate the cathodic corrosion behaviour (Fig. 3). Due to the high reaction overpotential, cathodic corrosion is observed for all the anolytes' pH, but harsh deterioration is observed for acid and neutral anolytes. A higher H+ concentration in the anolyte induces proton migration through the cation exchange membrane, leading to intergranular corrosion of the Pb plate with the reaction using the acidic and near-neutral anolyte, Fig. 3b and c, respectively. The electrode corrosion by intergranular corrosion causes activity loss for the CO2RR over time, as seen in the current decay on the chronoamperometry graph (see Fig. 1b). The intergranular attack was not observed for the alkaline anolyte (Fig. 3d), though K+ ions can promote the lead electrode's corrosion. Still, at higher magnification, restructuring of the flat Pb plate can be seen, similar to the Pb plate in the presence of a corrosion inhibitor.51 The restructuring can also be related to the corrosion and electrodeposition processes of the lead ions.52 The inductively coupled plasma-optical emission spectroscopy (ICP-OES) data presented in Table S1† showed that higher K+ ions modify the corrosion process instead of inhibiting corrosion. In the presence of H+ and low K+ ions, cathodic corrosion occurs by the intergranular attack, which leads to activity loss. However, with a higher K+ ion concentration, the anodic corrosion produces a porous structure that maintains high activity due to the metal etch pits formed on the surface. This phenomenon occurs due to the low stability of the dissolution intermediate in the non-protic medium,53 which causes the partial conversion of the ions to their metallic state to form agglomerated particle spots,54 as observed in the HRSEM images, Fig. 3. The formed porous structure can keep the catalyst's superficial active area even though a harsher metal dissolution chemical is used. These results suggest that the CO2RR intermediates can adsorb on the electrode surface in higher K+ ions due to the increase in surface porosity caused by the intense corrosion process and due to the potassium-stabilized effect of CO2 intermediates at the catalyst surface, enabling their conversion to various products.43,55 Even with the restructuring, only a minor current decay over time is seen, unlike the other anolyte pH that exhibits a significant current density decay over time. Post-reaction DRX analysis (Fig. 4) did not show the formation of different phases on the Pb plate surface even after the electrode corrosion for acid and near-neutral anolytes, showing no change in the bulk structure of the electrode.
 | ||
| Fig. 3 Scanning electron microscopy images for (a) cleaned Pb plate before the reaction and for the Pb plate after the reaction using (b) acidic anolyte (H2SO4), (c) near-neutral anolyte (KHCO3), and (d) alkaline anolyte (KOH). | ||
 | ||
| Fig. 4 XRD patterns of the Pb plate before and after CO2 reduction in acetonitrile 0.1 M TBAPF6 with different anolytes. | ||
The electrolysis using a K+ source anolyte (KHCO3 and KOH) produced a white precipitate due to cation migration through the Nafion membrane (Fig. 5), which was collected and washed in dry acetonitrile for analysis by FTIR and TGA. The FTIR spectrum (Fig. 6a) demonstrates that a mixture of compounds can be found in solid products. The CO2 reduction products in non-aqueous media have a considerably simple mechanism, as revised in ref. 15, 16 and 18, and can be identified using the FTIR spectrum combined with an analytical separation method, such as HPLC. Fig. 6a shows bands that can be assigned to the tartrate and oxalate ions signalled on the spectrum with the black and red arrows, respectively. The band at ∼3300 cm−1 can be assigned to the carboxylic acid in both compounds, and the band at 1585 cm−1 also confirms the presence of the products in the ion form due to the asymmetrical stretching of the carboxylate anions.56 The bands that correspond to the COO– stretching and bending vibration can be assigned to the tartrate56–58 (δCOO: 826 cm−1; 707 cm−1; and τCOO: 612 cm−1) and oxalate59–61 (νCOO: 1300 cm−1, βCOO: 771 cm−1, and WCOO: 520 cm−1). Additionally, no presence of the supporting electrolyte in the solid products and no characteristic band (∼1430 cm−1) for potassium carbonate are observed.62,63
 | ||
| Fig. 5 H-cell picture showing the precipitate formation on the catholyte side after the CO2RR in dry acetonitrile and KOH (0.5 M) as the anolyte. | ||
 | ||
| Fig. 6 (a) FTIR spectra for the precipitated CO2RR products and the supporting electrolyte; (b) TG and DTG analysis for the precipitated products. | ||
TGA/DTG analysis presented in Fig. 6b agrees with the FTIR spectrum. Three decomposition stages were verified from the TGA spectrum between 250 and 550 °C after water loss (>200 °C).64 These three primary decomposition stages are associated with the mixed products obtained from the CO2RR. The first and second stages (>350 °C) involve the decomposition of the tartrate ion into oxalate,65 corresponding to ∼20% weight loss due to the detachment of gaseous CO2 molecules from the tartrate crystal lattice.56 The third decomposition step involves the conversion of oxalate to carbonate with ∼7% weight loss between 450 and 550 °C, corresponding to the CO evolution.65,66 The mixture of the two solid products is due to the tartrate formation mechanism that occurs via the well-known process for converting CO2 to oxalate/oxalic acid by coupling two ·CO2− intermediates, followed by the dimerization of two oxalate molecules, summarized in Scheme 1 and described in Scheme 2a and b. With the main migration of H+ (H2SO4 anolyte), formic acid is the primary product of the CO2RR.67 However, with an increase in the K+ migration, more C–C coupled products are obtained, as represented in Scheme 1, and the tartrate formation reaction is presented in Scheme 2.
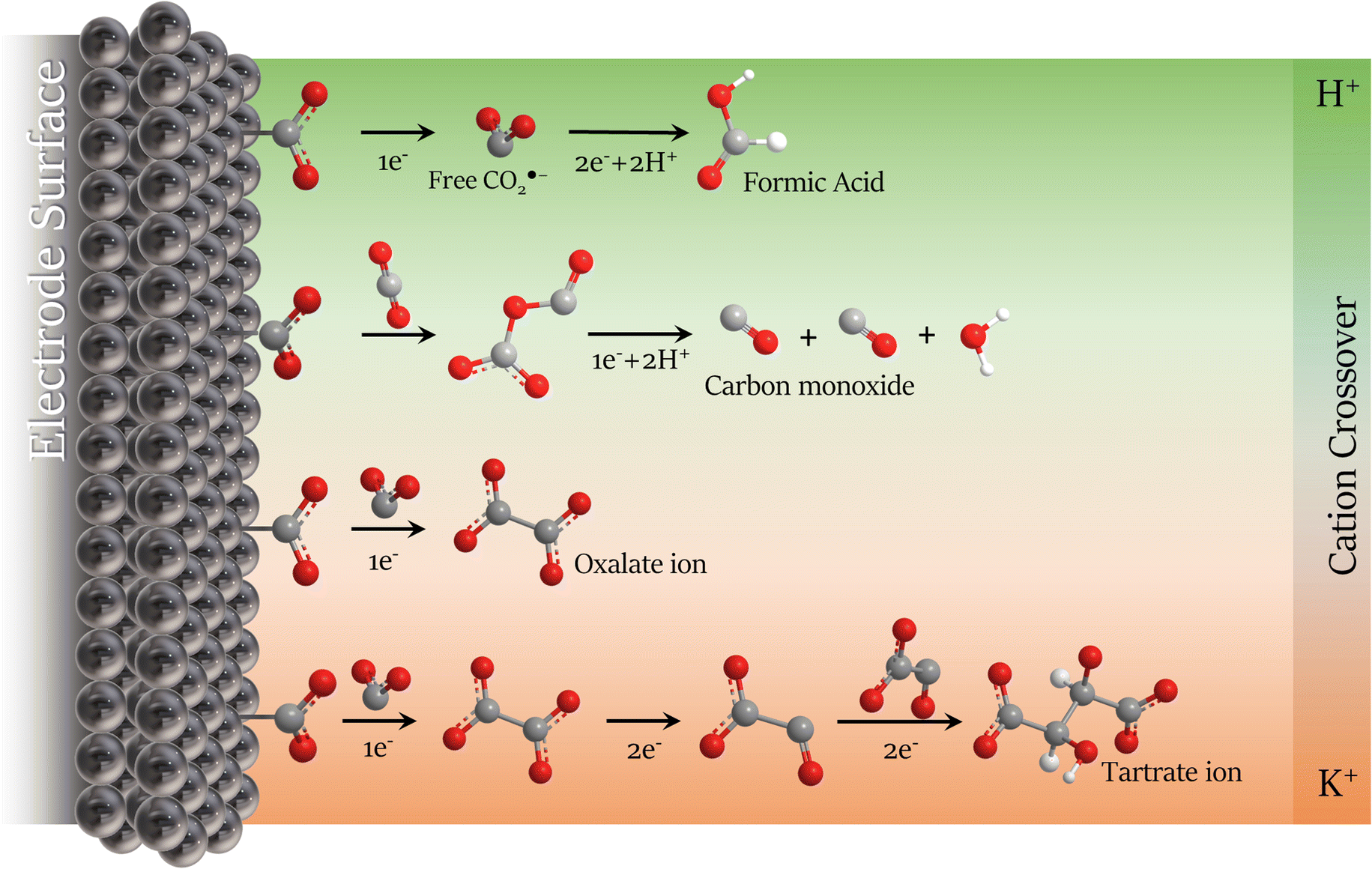 | ||
| Scheme 1 Schematic representation of CO2RR mechanism modulation in non-aqueous electrolyte with the cation migration preference from the anolyte on the Pb plate surface in dried acetonitrile. | ||
 | ||
| Scheme 2 The proposed tartrate mechanism formation is in high potassium ion availability media. | ||
The anolyte influence was verified by electrolysis at −2.3 V (vs. Ag/Ag+) for each anolyte, and an HPLC with a UV detector was used to quantify the products. Since the mobile phase for the HPLC analysis was acidic water (pH = 3), the CO2RR products were named by the protonated form. Also, it is essential to mention that experiments were carried out to evaluate the drying process in acetonitrile. In the collected post-reaction gas, only a trace of carbon monoxide from the CO2RR was detected in the CG, without the formation of hydrogen at −2.5 V vs. Ag/Ag+, showing the drying method's effectiveness. Fig. 7 exhibits the faradaic efficiency and the concentration of the detected products from the CO2RR. When potassium-based salts are used in the anolyte, higher faradaic efficiency is seen for the C2+ products. Besides, the presence of the K+ allows the formation of tartrate ions, which have been reported until now only by using an Ag-modified catalyst in the reduction reaction of glyoxylic acid18 and not as a product of CO2 reduction. To the best of our knowledge from the literature survey, Table S3,† this is reported for the first time for CO2RR with a Pb catalyst. Published papers usually use acidic or non-aqueous anolytes, achieving higher selectivity and faradaic efficiency for oxalate/oxalic acid (>80%).21,28,61,68 However, since the formation of tartrate depends on the viability of oxalate, the FE for carbonaceous products is expected to be lower. With the KHCO3 anolyte, the FE achieved by analyzing the cathodic electrolyte was 28%, 10%, and 37% for oxalic (OA), formic (FA), and tartaric acid (TA), respectively. The remaining faradaic efficiency was attributed to carbon monoxide, the only product detected in the gas phase. Higher faradaic efficiency was found using the KOH anolyte, 31% for OA, 16% for FA, and 53% for TA. The anolyte pH influences the amount of C2+ products on the catholyte side, as seen in Fig. 7b. Higher pH anolytes and low proton availability favour potassium migration in the crossover competition between the H+ and the K+. With the minimum migration of the H+, the reduction mechanism favours the C–C coupling,16,18 decreasing the energy barrier for C2+ products caused by the influence of potassium ions on the microenvironment of the electrode surface.34,43,69 These unprecedented results in non-aqueous electrolytes coincide with the recent reports for aqueous-based systems,44–46 where K+ ions can accelerate the carbon–carbon (C–C) coupling process, leading to more complex molecules from the CO2 reduction (Scheme 1).
 | ||
| Fig. 7 Anolyte influence on the (a) faradaic efficiency for CO2 reduction and on the (b) production rate for oxalic, formic, and tartaric acid at −2.2 V vs. Ag/Ag+. | ||
KOH was chosen as the anolyte to study the potential influence of CO2 conversion to tartrate since it showed the highest FE value for this product. The evaluated potentials were selected by LSV. The potentials studied were the potential right at the beginning of the onset, where the increase in the current density is observed (−2.2 V vs. Ag/Ag+), the potential at the end of the onset potential, where the current density growth is obvious (−2.3 V vs. Ag/Ag+), and the potential after the onset (−2.5 V vs. Ag/Ag+), see Fig. 1. Fig. 8a and b exhibit the faradaic efficiency and the concentration of the carbonaceous products at different potentials, respectively. In −2.2 V vs. Ag/Ag+, the most favoured reaction with more than 50% of FE was the formation of formic acid, even with the K+ ions. The amount of formic and oxalic acid produced was similar, but the rate of the tartaric acid was only 0.1 mM. At −2.5 V (vs. Ag/Ag+), more products were detected with a production rate of 0.17 μmol h−1 cm−2, 0.13 μmol h−1 cm−2, and 0.21 μmol h−1 cm−2 for oxalic, formic, and tartaric acid respectively. A loss in faradaic efficiency is observed due to the rapid formation and consumption of the ·CO2− intermediates that lead to the formation of gaseous CO (detected by GC). Fig. 8c and d show that when the anolyte concentration was improved, the electrolysis at the onset potential (−2.3 vs. Ag/Ag+) produced a significant amount of the C2+ products and a higher production rate was obtained (OA: 1.32 μmol h−1 cm−2 and TA: 1.49 μmol h−1 cm−2) with good faradaic efficiency, 35% and 60% for OA and TA, respectively. Compared with the literature, this is the first time tartaric acid was produced from the CO2RR using a Pb electrode in a non-aqueous medium (see Table S3†). Still, the tartaric acid formation achieved similar faradaic efficiency compared to recent reports.21,23,28
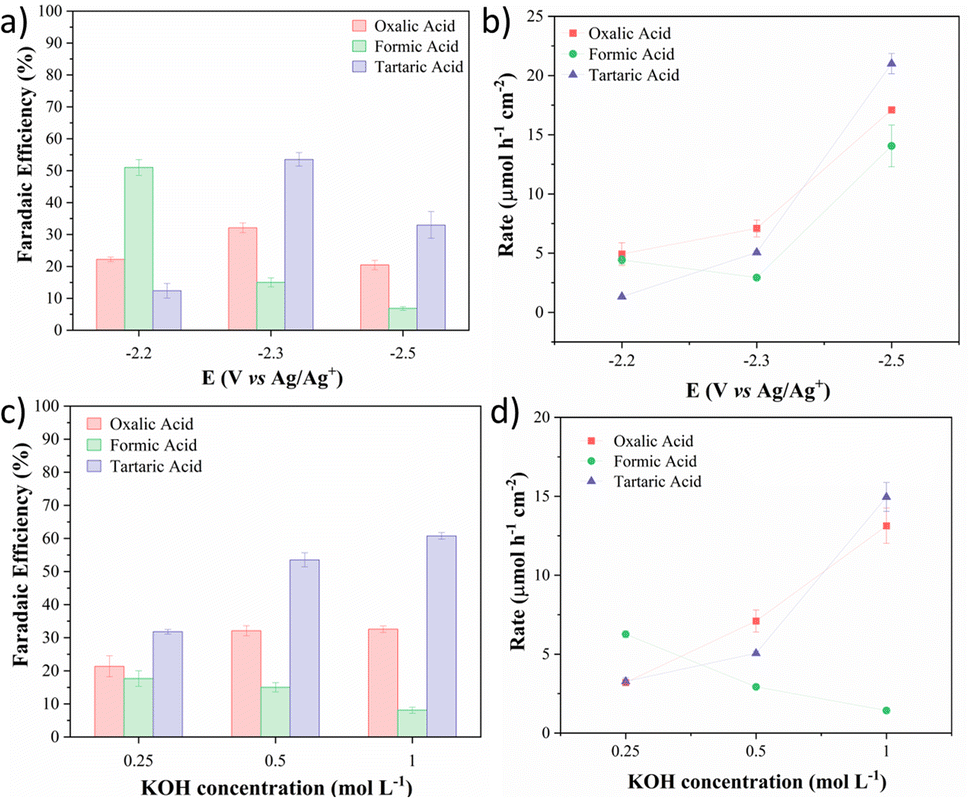 | ||
| Fig. 8 (a) FE of electrochemical CO2 reduction products and (b) production rate with applied potentials of −2.2, −2.3, and −2.5 V vs. Ag/Ag+ using KOH 0.1 M as the anolyte. (c) FE of CO2 reduction products with different anolyte potassium ion concentrations and the (d) production rate for oxalic, formic, and tartaric acids. | ||
Unlike the potential increase, the increase in the K+ concentration improved the production without losing energetic efficiency, emphasizing the potential of the potassium ion effect in non-aqueous media. All the studied parameters for the electrochemical CO2 reduction using aqueous anolytes are summarized in Table 1.
| Acids | Anolyte (0.5M) at −2.3 vs. Ag/Ag+ | KOH 5 M: potential (vs. Ag/Ag+) | KOH: anolyte concentration (M) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| H2SO4 | KHCO3 | KOH | −2.2 | −2.3 | −2.5 | 0.25 | 0.5 | 1.0 | |
| Faradaic efficiency (%) | |||||||||
| Formic | 88.5 | 7.3 | 15.3 | 51.1 | 15.3 | 6.8 | 17.5 | 15.3 | 8.14 |
| Oxalic | 9.6 | 27.6 | 32 | 22.4 | 32 | 20.4 | 21.4 | 32 | 32.7 |
| Tartaric | 2.7 | 37.6 | 53.6 | 12.7 | 53.6 | 32.9 | 31.9 | 53.6 | 60.8 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||||||||
| Production rate (μmol h−1 cm−2) | |||||||||
| Formic | 4.69 | 6.94 | 2.97 | 4.33 | 2.97 | 14.05 | 6.37 | 2.97 | 1.46 |
| Oxalic | 0.71 | 1.04 | 7.10 | 4.97 | 7.10 | 17.10 | 3.32 | 7.10 | 13.16 |
| Tartaric | 0.06 | 1.33 | 5.01 | 1.32 | 5.01 | 21.00 | 3.17 | 5.01 | 14.95 |
3. Conclusions
In summary, we showed a route to produce tartaric acid from the CO2RR in a non-aqueous medium by controlling the influence of the electrochemical configuration on modulating the faradaic efficiency and selectivity of the CO2RR in non-aqueous media. We synthesized tartaric acid/tartrate from gaseous CO2 using a commercial Pb plate as the cathode by modifying the anolyte to promote K+ migration. The anolyte and supporting electrolytes were essential to achieve higher value-aggregated products from CO2 waste. Tartaric acid production reached 60% faradaic efficiency with a higher KOH concentration at −2.3 V vs. Ag/Ag+, producing 1.5 μmol L−1 in 30 minutes of electrolysis. This multicarbon product formation is associated with the higher K+ crossover, favouring the C–C coupling of the intermediates in the CO2RR. In addition, the experimental results showed that the potassium ion has a more significant role in the coupling of intermediates than the increase in the reaction potential, and it is responsible for promoting the formation of C4+ products from CO2.The present work provided an innovative and promising approach to convert CO2 to multicarbon products, which are obtained in solid form (precipitated) and could be used strategically to design an easy and efficient separation process.
4. Experimental methods
4.1 Procedure for drying acetonitrile
Before the electrochemical experiments, the excess water in acetonitrile was removed using the methodology described by Williams and Lawton (2010).70 A 3 A° molecular sieve is pre-dried at 300 °C for 24 h immediately before use. After drying, the molecular sieve is added to the acetonitrile (HPLC grade) at 10% m/v and left for 24 hours in a nitrogen atmosphere. The dried acetonitrile was kept in a sealed vial with molecular sieves and stored for one week after the drying procedure.4.2 Ag/Ag+ reference electrode preparation and calibration
The silver–silver ion reference electrode was prepared according to Izutsu (2013).71 AgNO3 salt was dissolved (0.01 M) in dry acetonitrile with the tetrabutylammonium hexafluorophosphate (TBAPF6) supporting electrolyte (0.1 M). A cleaned silver wire was immersed in the freshly prepared solution using a glass shaft with a Teflon cap. The silver was cleaned by physical polishing with sandpaper and a chemical polishing step with 0.1 M HNO3 solution.The reference electrode was freshly prepared daily and calibrated using a ferrocene standard solution (5 mM ferrocene in 0.1 M TBAPF6 in CH3CN).
4.3 Electrochemical CO2 reduction setup
The experiments were carried out in an H-type cell configuration with a cation exchange membrane (Nafion 117) using a Pb plate (1.5 × 1 cm2), Pt mesh as working and counter electrodes, and an Ag/Ag+ (0.01 M AgNO3) reference electrode. The cell volume was 10 mL for the catholyte and 10 mL for the anolyte with magnetic stirring on the catholyte side. The catholyte used was 0.1 M TBAPF6 in dried acetonitrile. Before each reaction, the electrolyte was pre-saturated with CO2 for 30 minutes, and the investigated anolytes were H2SO4 (acid), KHCO3 (near neutral), and KOH (alkaline) for the potential −2.2, −2.3, and −2.5 V vs. Ag/Ag+.The Pb plate was previously cleaned by polishing it with wet sandpaper (2000 grits), followed by electrochemical cleaning by applying −1.8 V vs. Ag/AgCl (KCl 3 M) in an H2SO4 solution (0.1 M) for 500 seconds. Before each analysis, an additional cleaning step was made by cyclic voltammetry (−1 to −2.5 V vs. Ag/Ag+) until current stabilization (around eight cycles), see Fig. S2.† All the electrochemical measurements were made using a potentiostat/galvanostat Autolab (model PGSTAT30, Metrohm).
4.4 Characterization
The Pb plates were characterized using a Shimadzu XRD-6000 diffractometer with Cu-Kα radiation (λ = 1.5406 Å) and a scanning rate of 2° min−1 from 10 to 80° at room temperature. The morphological characterization of the electrodes was performed using a scanning electron microscope (SEM-JSM6510). The electrochemical characterization was performed in a potentiostat/galvanostat Autolab with an FRA 32M module in an H-type cell with a Nafion 117 membrane and Ag/Ag+ reference electrode and CO2 pre-saturated acetonitrile (0.1 M TBAPF6) as the catholyte. Thermogravimetric analysis was carried out on a TGAQ500 (TA instruments) analyzer. The samples were heated in a platinum pan from 25 to 800 °C with a ramp rate of 10 °C min−1 under N2 flux (40 mL min−1). ICP-OES analysis was performed on the catholyte after each reaction. The acetonitrile on the electrolyte was dried overnight using a water bath (80 °C), and the solid was dissolved in a 4 M H2SO4 solution.4.5 Product quantification
The total volume of the catholyte was collected, and the solid products were solubilized by adding water pH 3 (H2SO4) to the samples. The dilution was calculated for each experiment. The products were quantified using a high-performance liquid chromatography-HPLC (Shimadzu Prominence 20A liquid chromatograph LC-20AT equipped with a UV detector at 214 nm). An Agilent MetaCarb 87H (300 × 7.8 mm) column was used to separate the products at a temperature of 40 °C with an eluent flow rate of 0.6 mL min−1, water pH 3.0 (H2SO4) as the mobile phase, and a sample injection volume of 20 μL. The total run time was 22.00 minutes, and the retention times were 6, 7.2, and 12.5 min for oxalic acid, tartaric acid, and formic acid, respectively (see Fig. S3–S6†). Gaseous products were collected by coupling a Tedlar® gas sample bag (1 L) in the electrochemical cell to collect the post-reaction gases along with the CO2 flux. The collected gas was analyzed using argon as the carrier gas in a GC-Thermo gas chromatograph equipment with a carboxen 1010 plot capillary column.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to acknowledge the financial support provided by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior – Brasil (CAPES) – Finance Code 001. The authors are also grateful for the financial support of FAPESP (#2018/01258-5 and 2022/10255-5) and Shell Brazil and the strategic importance of the support given by ANP (the Brazilian National Oil, Natural Gas, and Biofuels Agency) under the R&D levy regulation.References
- Y. Abdullatif, A. Sodiq, N. Mir, Y. Bicer, T. Al-Ansari, M. H. El-Naas and A. I. Amhamed, RSC Adv., 2023, 13, 5687–5722 RSC.
- T. M. Gür, Prog. Energy Combust. Sci., 2022, 89, 100965 CrossRef.
- S. Sun, Y. Wang, X. Zhao, C. Zhang and C. Wu, Carbon Capture Sci. Technol., 2022, 5, 100078 CrossRef CAS.
- S. Sun, Y. Zhang, C. Li, Y. Wang, C. Zhang, X. Zhao, H. Sun and C. Wu, Sep. Purif. Technol., 2023, 308, 122956 CrossRef CAS.
- P. Wienchol, A. Szlęk and M. Ditaranto, Energy, 2020, 198, 117352 CrossRef CAS.
- L. Ou, S. Banerjee, H. Xu, A. M. Coleman, H. Cai, U. Lee, M. S. Wigmosta and T. R. Hawkins, J. Cleaner Prod., 2021, 321, 128779 CrossRef CAS.
- H. Choi, D.-K. Lee, M.-K. Han, G. Janani, S. Surendran, J. H. Kim, J. K. Kim, H. Cho and U. Sim, J. Electrochem. Soc., 2020, 167, 164503 CrossRef CAS.
- E. Ruiz-López, J. Gandara-Loe, F. Baena-Moreno, T. R. Reina and J. A. Odriozola, Renewable Sustainable Energy Rev., 2022, 161, 112329 CrossRef.
- F. Proietto, A. Galia and O. Scialdone, ChemElectroChem, 2021, 8, 2169–2179 CrossRef CAS.
- S. C. Jesudass, S. Surendran, G. Janani, T. H. Kim and U. Sim, Adv. Energy Mater., 2023, 13, 2301918 CrossRef CAS.
- Z. Huang, R. G. Grim, J. A. Schaidle and L. Tao, Energy Environ. Sci., 2021, 14, 3664–3678 RSC.
- F. Franco, C. Rettenmaier, H. S. Jeon and B. Roldan Cuenya, Chem. Soc. Rev., 2020, 49, 6884–6946 RSC.
- P. Saha, S. Amanullah and A. Dey, Acc. Chem. Res., 2022, 55, 134–144 CrossRef CAS.
- M. C. O. Monteiro, M. F. Philips, K. J. P. Schouten and M. T. M. Koper, Nat. Commun., 2021, 12(1), 1–7 CrossRef.
- Y. I. Hori, Electrochemical CO2 reduction on metal electrodes, Modern aspects of electrochemistry, 2008, pp. 89–189 Search PubMed.
- E. A. dos Reis, G. T. S. T. da Silva, E. I. Santiago and C. Ribeiro, Energy Technol., 2023, 11, 2201367 CrossRef CAS.
- J. Feng, S. Zeng, J. Feng, H. Dong and X. Zhang, Chin. J. Chem., 2018, 36, 961–970 CrossRef CAS.
- A. C. Garcia, C. Sánchez-Martínez, I. Bakker and E. Goetheer, ACS Sustain. Chem. Eng., 2020, 8, 10454–10460 CrossRef CAS.
- R. J. Gomes, C. Birch, M. M. Cencer, C. Li, S. B. Son, I. D. Bloom, R. S. Assary and C. V. Amanchukwu, J. Phys. Chem. C, 2022, 2022, 13595–13606 CrossRef.
- M. A. Murcia Valderrama, R. J. van Putten and G. J. M. Gruter, Eur. Polym. J., 2019, 119, 445–468 CrossRef CAS.
- V. Boor, J. E. B. M. Frijns, E. Perez-Gallent, E. Giling, A. T. Laitinen, E. L. V. Goetheer, L. J. P. van den Broeke, R. Kortlever, W. de Jong, O. A. Moultos, T. J. H. Vlugt and M. Ramdin, Ind. Eng. Chem. Res., 2022, 61(40), 14837–14846 CrossRef CAS PubMed.
- M. König, J. Vaes, E. Klemm and D. Pant, iScience, 2019, 19, 135–160 CrossRef.
- M. König, S. H. Lin, J. Vaes, D. Pant and E. Klemm, Faraday Discuss., 2021, 230, 360–374 RSC.
- Y. Tomita and Y. Hori, Stud. Surf. Sci. Catal., 1998, 114, 581–584 CrossRef CAS.
- W. Lv, R. Zhang, P. Gao, C. Gong and L. Lei, J. Solid State Electrochem., 2013, 17, 2789–2794 CrossRef CAS.
- B. Eneau-Innocent, D. Pasquier, F. Ropital, J. M. Léger and K. B. Kokoh, Appl. Catal., B, 2010, 98, 65–71 CrossRef CAS.
- E. Schuler, M. Demetriou, N. R. Shiju and G. J. M. Gruter, ChemSusChem, 2021, 14, 3636–3664 CrossRef CAS PubMed.
- Y. Cheng, P. Hou, H. Pan, H. Shi and P. Kang, Appl. Catal., B, 2020, 272, 118954 CrossRef CAS.
- B. J. Cook, G. N. Di Francesco, K. A. Abboud and L. J. Murray, J. Am. Chem. Soc., 2018, 140, 5696–5700 CrossRef CAS.
- A. Gennaro, A. A. Isse, M. G. Severin, E. Vianello, I. Bhugun and J. M. Savéant, J. Chem. Soc., Faraday Trans., 1996, 92, 3963–3968 RSC.
- C. Amatore and J. M. Savéant, J. Am. Chem. Soc., 1981, 103, 5021–5023 CrossRef CAS.
- M. M. Cencer, C. Li, G. Agarwal, R. J. Gomes Neto, C. V Amanchukwu and R. S. Assary, ACS Omega, 2022, 11, 43 Search PubMed.
- M. Marx, H. Frauendorf, A. Spannenberg, H. Neumann and M. Beller, JACS Au, 2022, 2, 731–744 CrossRef CAS PubMed.
- M. Schreier, P. Kenis, F. Che and A. S. Hall, ACS Energy Lett., 2023, 8, 3935–3940 CrossRef CAS.
- S. C. Jesudass, S. Surendran, J. Y. Kim, T. Y. An, G. Janani, T. H. Kim, J. K. Kim and U. Sim, Electrochem. Energy Rev., 2023, 6, 27 CrossRef CAS.
- S. Bin Dolmanan, A. Böhme, Z. Fan, A. J. King, A. Q. Fenwick, A. D. Handoko, W. R. Leow, A. Z. Weber, X. Ma, E. Khoo, H. A. Atwater and Y. Lum, J. Mater. Chem. A, 2023, 11, 13493–13501 RSC.
- Z. Xing, L. Hu, D. S. Ripatti, X. Hu and X. Feng, Nat. Commun., 2021, 12(1), 1–11 CrossRef PubMed.
- J. J. Lv, R. Yin, L. Zhou, J. Li, R. Kikas, T. Xu, Z. J. Wang, H. Jin, X. Wang and S. Wang, Angew. Chem., Int. Ed., 2022, 61, e202207252 CrossRef CAS PubMed.
- D. Wang, J. Mao, C. Zhang, J. Zhang, J. Li, Y. Zhang and Y. Zhu, eScience, 2023, 3, 100119 CrossRef.
- H. Wu, A. Singh-Morgan, K. Qi, Z. Zeng, V. Mougel and D. Voiry, ACS Catal., 2023, 13, 5375–5396 CrossRef CAS PubMed.
- T. C. Berto, L. Zhang, R. J. Hamers and J. F. Berry, ACS Catal., 2015, 5, 703–707 CrossRef CAS.
- M. Moura de Salles Pupo and R. Kortlever, ChemPhysChem, 2019, 20, 2926–2935 CrossRef CAS PubMed.
- S. Chandrashekar, H. P. I. van Montfort, D. Bohra, G. Filonenko, H. Geerlings, T. Burdyny and W. A. Smith, Nanoscale, 2022, 14, 14185–14190 RSC.
- X. Zi, Y. Zhou, L. Zhu, Q. Chen, Y. Tan, X. Wang, M. Sayed, E. Pensa, R. A. Geioushy, K. Liu, J. Fu, E. Cortés and M. Liu, Angew. Chem., Int. Ed., 2023, 62, e202309351 CrossRef CAS PubMed.
- J. Yu, M. Sun, J. Wang, Y. Wang, Y. Li, P. Lu, Y. Ma, J. Zhou, W. Chen, X. Zhou, C.-S. Lee, B. Huang and Z. Fan, Phys. Sci., 2023, 4(4), 101366 CAS.
- A. K. Surendran, G. L. Tripodi, E. Pluhařová, A. Y. Pereverzev, J. P. J. Bruekers, J. A. A. W. Elemans, E. J. Meijer and J. Roithová, Natural Sciences, 2023, 3, e20220019 CrossRef CAS.
- G. A. El-Nagar, F. Haun, S. Gupta, S. Stojkovikj and M. T. Mayer, Nat. Commun., 2023, 14(1), 1–10 Search PubMed.
- M. Sassenburg, M. Kelly, S. Subramanian, W. A. Smith and T. Burdyny, ACS Energy Lett., 2023, 8, 321–331 CrossRef CAS PubMed.
- M. König, S. H. Lin, J. Vaes, D. Pant and E. Klemm, Faraday Discuss., 2021, 230, 360–374 RSC.
- K. J. Chae, M. Choi, F. F. Ajayi, W. Park, I. S. Chang and I. S. Kim, Energy Fuels, 2008, 22, 169–176 CrossRef CAS.
- H. M. A. El-Lateef, A. R. El-Sayed, H. S. Mohran and H. A. S. Shilkamy, Int. J. Ind. Chem, 2019, 10, 31–47 CrossRef CAS.
- T. J. P. Hersbach and M. T. M. Koper, Curr. Opin. Electrochem., 2021, 26, 100653 CrossRef CAS.
- T. Wirtanen, T. Prenzel, J. P. Tessonnier and S. R. Waldvogel, Chem. Rev., 2021, 121, 10241–10270 CrossRef CAS PubMed.
- A. I. Yanson, P. Rodriguez, N. Garcia-Araez, R. V. Mom, F. D. Tichelaar and M. T. M. Koper, Angew. Chem., Int. Ed., 2011, 50, 6346–6350 CrossRef CAS PubMed.
- J. E. Huang, F. Li, A. Ozden, A. S. Rasouli, F. P. G. de Arquer, S. Liu, S. Zhang, M. Luo, X. Wang, Y. Lum, Y. Xu, K. Bertens, R. K. Miao, C. T. Dinh, D. Sinton and E. H. Sargent, Science, 2021, 372, 1074–1078 CrossRef CAS PubMed.
- T. S. Shyju, S. Anandhi and R. Gopalakrishnan, CrystEngComm, 2012, 14, 1387–1396 RSC.
- V. Mathivanan and M. Haris, Spectrochim. Acta, Part A, 2013, 102, 341–349 CrossRef CAS PubMed.
- G. P. Srivastava, S. Mohan and Y. S. Jain, J. Raman Spectrosc., 1982, 13, 25–29 CrossRef CAS.
- V. S. Tomar, H. D. Bist and D. P. Khandelwal, Appl. Spectrosc., 1970, 24(6), 598–601 CrossRef.
- M. C. Figueiredo, I. Ledezma-Yanez and M. T. M. Koper, ACS Catal., 2016, 6, 2382–2392 CrossRef CAS.
- S. Subramanian, K. R. Athira, M. A. Kulandainathan, S. S. Kumar and R. C. Barik, J. CO2 Util., 2020, 36, 105–115 CrossRef CAS.
- H. Du, C. T. Williams, A. D. Ebner and J. A. Ritter, Chem. Mater., 2010, 22, 3519–3526 CrossRef CAS.
- V. J. Bruckman and K. Wriessnig, Environ. Chem. Lett., 2013, 11, 65–70 CrossRef CAS PubMed.
- W. Liu, H. Wang, X. Gu, C. Quan and X. Dai, Anal. Methods, 2016, 8, 2845–2851 RSC.
- U. Tarpara, P. Vyas and M. J. Joshi, Int. J. Nanosci., 2015, 14(4), 1550013 CrossRef CAS.
- T. Yoshimori, Y. Asano, Y. Toriumi and T. Shiota, Talanta, 1978, 25, 603–605 CrossRef CAS PubMed.
- J. Hussain, H. Jónsson and E. Skúlason, ACS Catal., 2018, 8, 5240–5249 CrossRef CAS.
- A. V. Rudnev, U. E. Zhumaev, A. Kuzume, S. Vesztergom, J. Furrer, P. Broekmann and T. Wandlowski, Electrochim. Acta, 2016, 189, 38–44 CrossRef CAS.
- L. Gao, F. Bao, X. Tan, M. Li, Z. Shen, X. Chen, Z. Tang, W. Lai, Y. Lu, P. Huang, C. Ma, S. C. Smith, Z. Ye, Z. Hu and H. Huang, Energy Environ. Sci., 2023, 16, 285–294 RSC.
- D. B. G. Williams and M. Lawton, J. Org. Chem., 2010, 75, 8351–8354 CrossRef CAS PubMed.
- K. Izutsu, Reference Electrodes for Use in Nonaqueous Solutions, in Handbook of Reference Electrodes, ed. G. Inzelt, A. Lewenstam and F. Scholz, Springer, Berlin, Heidelberg, 2013, DOI:10.1007/978-3-642-36188-3_6.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta01172d |
| This journal is © The Royal Society of Chemistry 2024 |