
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBenzoyldiisopropylchlorosilane: a visible light photocleavable alcohol protecting group†
Florian
Lind
,
Kirill
Markelov
and
Armido
Studer
 *
*
Organisch-Chemisches Institut, Universität Münster, Corrensstrasse 40, 48149 Münster, Germany. E-mail: studer@uni-muenster.de
First published on 25th October 2023
Abstract
Silyl chlorides are highly valuable and popular reagents for the protection of alcohols. In this edge article we introduce a photocleavable alcohol protecting group on the basis of acyl silanes. To achieve this, acylchlorosilanes that represent a new class of acylsilanes were developed. They can be easily synthesized in a concise sequence of three steps in high overall yield. Alcohol silyl protection takes place under established mild conditions, akin to those associated with classical silicon-based protecting groups. The removal of the Si-group is achieved at room temperature through exposure to visible light (456 nm) in methanol. We demonstrate a broad spectrum of substrates with remarkable tolerance toward diverse functional groups, highlighting a substantial level of orthogonality with respect to other protecting groups. Furthermore, we showcase the robustness of this approach against various transformations.
Introduction
Protecting groups are widely established in organic chemistry and essential for the synthesis of complex molecules.1 Although their application is actually undesired,2 since they cause reduced step and atom economy, their usage remains essential, particularly in the total synthesis of complex natural products.3 Protecting groups offer a simple way to suppress undesired reactivity and to selectively manipulate specific functional groups within complex multifunctional compounds. Great efforts have thus been dedicated to the advancement of novel protecting groups. However, for utilization in organic synthesis, certain criteria must be satisfied.1 While employing protecting groups necessitates a minimum of two steps – protection and deprotection – it is of high importance that both these steps occur selectively, providing high yields, and without compromising the intended functionality or other functional groups. Additionally, the reagents for both steps should be easily accessible, and the protecting group should remain stable under diverse reaction conditions. Given the near-impossibility of satisfying all these criteria simultaneously for each synthesis problem, a wide array of protecting groups has been developed and established.Silyl ethers have gained widespread popularity as protecting groups for alcohols, primarily owing to the typically mild reaction conditions and high yields achievable through alcohol silylation using chlorosilanes (Scheme 1a).4 Their reactivity, readily modulated by the substituents on the silicon, coupled with their facile removal by fluoride anions, qualifies silyl ethers among the most valuable alcohol protecting groups.5 An exceptional silyl protective group is the sisyl group, tris(trimethylsilyl)silyl, which can be deprotected through photochemical means.5b
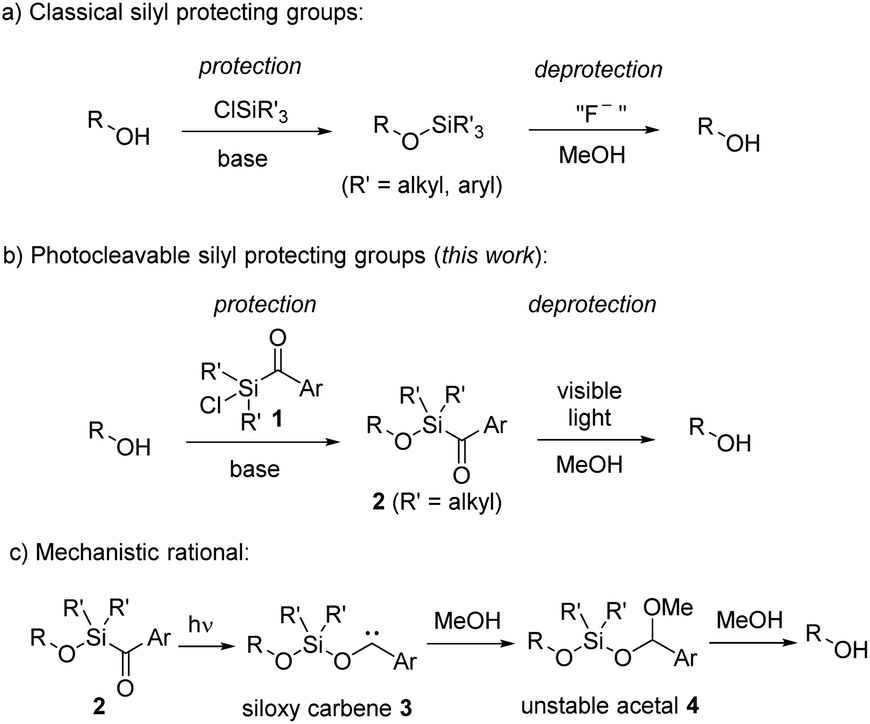 | ||
| Scheme 1 Alkyl/arylchlorosilanes as classical alcohol protecting groups and aryldialkylchlorosilanes as their photoactive congeners. | ||
Photocleavable groups belong to a special class of protecting groups,6 because they typically necessitate no external reagents, relying solely on light for the deprotection process. Utilizing light enables precise control over the spatial and temporal release of molecules. Along these lines, various photoactive protecting groups have been developed in the past; however, they are plagued by several drawbacks, including complex synthesis, multi-step protection procedures, and often requiring UV light for cleavage. Particularly, employing high-energy irradiation for deprotection introduces challenges to their application in synthesis, as only minimally functionalized molecules can endure such conditions. It is therefore highly desirable to use mild irradiation, such as visible light (>420 nm), for deprotection.
Herein we introduce aryldialkylchlorosilanes 1 as new photocleavable silicon-based alcohol protecting groups, by combining the advantages of chlorosilanes for protection and the ability of arylsilanes to decompose by irradiation with visible light under mild conditions for deprotection (Scheme 1b). Arylsilanes are known to form siloxy carbenes by irradiation with blue light. We assume that a similar photorearrangement is also occurring for alkoxyaroylsilanes 2 to give siloxy carbenes of type 3 (Scheme 1c). In the presence of MeOH, carbene insertion into the O–H bond should lead to unstable acetals 4,7 which upon solvolysis will eventually afford the deprotected free alcohols. We will demonstrate that protection and deprotection occur under mild conditions in high yields. In addition, we will show a broad spectrum of substrates with good tolerance toward diverse functional groups, and will highlight a substantial level of orthogonality with respect to other protecting groups.
Results and discussion
We commenced the investigations by developing an efficient synthesis for larger scale preparation of unprecedented aryldialkylchlorosilanes. Deprotonation of benzal bromide and subsequent reaction with commercial diisopropylchlorosilane provided the silane 5 that was further transformed to the benzoylsilane 6 by treatment with Ag2CO3 in DCM, as recently reported by us (Scheme 2).8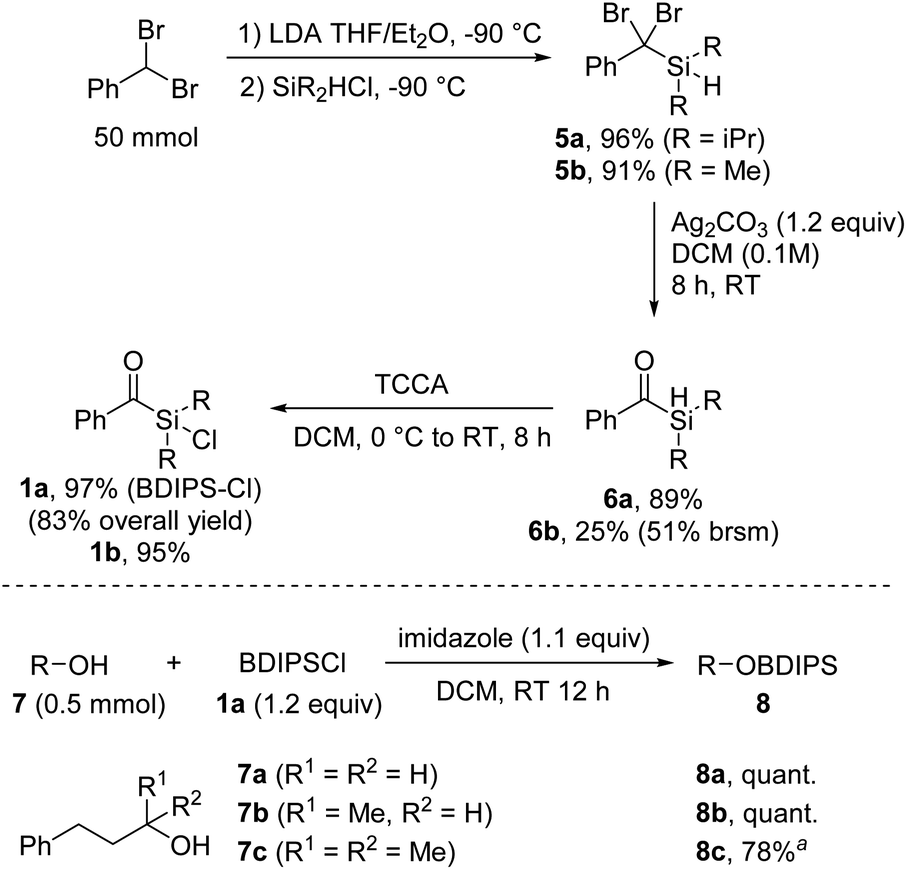 | ||
| Scheme 2 Synthesis of the chlorosilane 1a and alcohol protection. aReaction carried out using 1a (1.5 equiv.) and AgOTf (1.2 equiv.). | ||
The chlorination of the hydrosilane 6 was readily achieved with trichloroisocyanuric acid (TCCA) in DCM at room temperature to afford the targeted benzoyldisopropylsilylchloride (BDIPSCl, 1a) in near quantitative yield.9 The robustness of this sequence was demonstrated by the successful preparation of 1a on 50 mmol scale in a total yield of 83% over three steps, without the necessity of any column chromatography. Of note, benzal bromide is commercially available, but can alternatively be prepared on 200 mmol scale in quantitative yield within 4 hours (see the ESI†). An alternative but slightly less efficient synthesis route towards 1a starting from 2-phenyl-1,3-dithiane was also developed (see ESI†). Through the benzal bromide route, we additionally prepared benzoyldimethylsilylchloride (1b); however, the corresponding silylethers turned out to be unstable. Therefore, 1b was not further considered in this study.
We were pleased to find that the protection of primary and secondary alcohols 7a and 7b with the novel reagent 1a could be achieved with a typical silylation protocol4 by using imidazole as a base in DCM, and the protected alcohols 8a and 8b were obtained in quantitative yields. However, the standard protocol proved ineffective for tertiary alcohols, likely due to steric hindrances. Nonetheless, the challenges were overcome by in situ transformation of the acylchlorosilane 1a into the more reactive triflate silyl ester, achieved through the addition of silver triflate to the reaction mixture (8c, 78%).
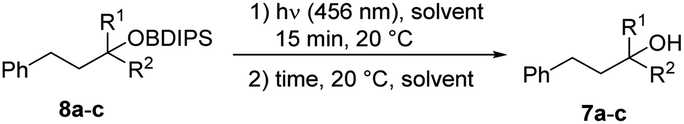
Next, the process of desilylation was examined utilizing silyl ethers 8a–c (Table 1). Deprotection was first studied in MeOH and it was found that irradiation of the BDIPS-protected primary alcohol 8a with a blue LED (456 nm) led to complete consumption of the starting silyl ether within 15 minutes. However, only a small amount of the alcohol 7a was identified, indicating that the further cleavage of acetal 4a (ref. 10) to 7a requires a longer reaction time. Indeed, after stirring for 4 hours at room temperature, full conversion to the alcohol 7a was achieved (Table 1, entry 1). In contrast, deprotection in water failed (Table 1, entry 2). Due to the substrate's insolubility in water, it became necessary to subject the substrate to a 2 hours irradiation process. This was done until the initial material was completely consumed. Despite subsequent stirring at room temperature, no formation of the desired alcohol occurred. The deprotection can also be accomplished in an acetone/methanol mixture (7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) upon stirring for 24 hours (Table 1, entry 3). Other solvents or solvent mixtures provided worse results (see ESI†). Deprotection of the sterically more demanding secondary silyl ether 8b in acetone/methanol was equally efficient, and 7b was obtained in a quantitative yield (Table 1, entry 4). For the tertiary silyl ether 8c, the acetone/MeOH protocol failed. Photo rearrangement occurred in short time, but solvolysis of the intermediate turned out to be inefficient under these conditions (Table 1, entry 5). However, in pure methanol, near quantitative deprotection was achieved for 8c (Table 1, entry 6). Further, we also determined the quantum yield for the reaction of 8a in acetone/MeOH (see ESI†). A quantum yield (Φ (456 nm)) of 0.209 was measured for that deprotection process.
:1) upon stirring for 24 hours (Table 1, entry 3). Other solvents or solvent mixtures provided worse results (see ESI†). Deprotection of the sterically more demanding secondary silyl ether 8b in acetone/methanol was equally efficient, and 7b was obtained in a quantitative yield (Table 1, entry 4). For the tertiary silyl ether 8c, the acetone/MeOH protocol failed. Photo rearrangement occurred in short time, but solvolysis of the intermediate turned out to be inefficient under these conditions (Table 1, entry 5). However, in pure methanol, near quantitative deprotection was achieved for 8c (Table 1, entry 6). Further, we also determined the quantum yield for the reaction of 8a in acetone/MeOH (see ESI†). A quantum yield (Φ (456 nm)) of 0.209 was measured for that deprotection process.
Stability study
Next, the stability of the selected model substrates under specific conditions was investigated by determining the half-life of the protected species. In pure methanol, the primary and secondary protected alcohols 8a and 8b showed partial desilylation over one day (Table 2, entry 1). In contrast, the tertiary silyl ether 8c exhibited a half-life of several days in MeOH. Notably, when subjected to an acetone/methanol mixture (7:1), the primary silyl ether 8a, which possessed the least steric hindrance, displayed a half-life of five days. As anticipated, 8b demonstrated greater stability under these conditions, with a half-life of 8 days. Additionally, the stability toward water was examined using aqueous acetone (10% water), revealing that the primary substrate 8a had a half-life of 2 days, whereas the secondary and tertiary counterparts exhibited significantly longer half-lives (6 days each, Table 2, entry 3). Lastly, we aimed to assess stability under both basic and acidic conditions following the methodology outlined by Davies, in order to make a comparative analysis of the BIDIPS protecting group against other well-established silyl protecting groups.11 The primary and secondary silyl ethers 8a and 8b decomposed within minutes under both basic and acidic conditions (Table 2, entries 3 and 4). A greatly improved stability was noted for the tertiary silyl ether 8c with half-life times of several hours (Table 2, entries 3 and 4).
Orthogonality study
We extended our research to explore the orthogonality of photochemical silyl ether deprotection in relation to the most commonly used alcohol protecting groups. For this purpose, we subjected the BDIPS-protected alcohol 8b, along with an equimolar quantity of a protected alcohol 7b-PG, to our photochemical deprotection conditions, and subsequently assessed the conversions of both substrates. The methyl ether (Table 3, entry 1), benzyl ether (Table 3, entry 2), p-methoxybenzyl ether (Table 3, entry 3), and methoxymethyl ether (Table 3, entry 4) showed no or very little reactivity, whereas 8b underwent complete deprotection. In contrast, the tetrahydropyranyl ether (THP) was not stable under the applied conditions (Table 3, entry 5).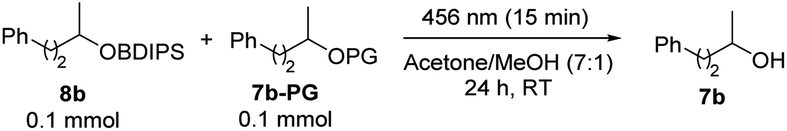
The TBDPS-protecting group showed an excellent degree of orthogonality (Table 3, entry 8), whereas the corresponding TBDMS-ether (Table 3, entry 6) and the TIPS-ether (Table 3, entry 7) underwent substantial desilylation. We assume that the desilylation is assisted by the silanol byproducts formed, since these TBDMS- and TIPS-ethers were perfectly stable in acetone/methanol (7:1) at room temperature. The three most prevalent ester protecting groups, namely acetyl (Table 3, entry 9), benzoyl (Table 3, entry 10), and pivaloyl (Table 3, entry 11) esters, were entirely compatible with the photomediated deprotection reaction. The most popular protecting group for photodeprotection, o-nitrobenzyl ether (oNBn), was also well tolerated under the deprotection conditions (Table 3, entry 12). The UV-spectrum showed that at 456 nm the o-nitrobenzyl ether does not absorb, explaining the perfect orthogonality of the two photoactive protecting groups (see ESI†).
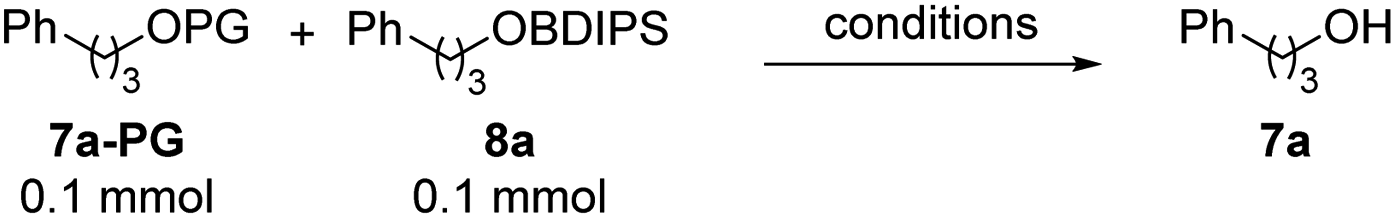
Then, orthogonality for deprotection of various protected alcohols 7a-PG in the presence of the BDIPS-ether 8a was investigated (Table 4). Deprotection of 7a-THP (Table 4, entry 1) and 7a-MOM (Table 4, entry 2) in the presence of 8a was achieved in very good orthogonality by using triethylsilyl triflate (TESOTf) and 2,2-bipyridine (Bipy).12 Deprotection of 7a-PMB occurred with excellent orthogonality with respect to 8a by applying a standard DDQ method (Table 4, entry 3).13 Pleasingly, photo orthogonal wave-length selective deprotection of 7a-oNBn was realized with near perfect chemoselectivity by irradiation with a 350 nm light source (Table 4, entry 4). 7a-TMS could be cleaved on silica with reasonable selectivity (Table 4, entry 5), while the bulkier 7a-TES showed a moderate chemoselectivity under these conditions (Table 4, entry 5). This also suggests that the steric protection provided by our photolabile silicon group is probably slightly greater than that offered by the triethylsilyl protecting group.
|
|
||||
|---|---|---|---|---|
| Entry | PG | Conditionsa | Conv. (7a-PG)b | Conv. (8a)b |
| a Reactions performed under argon. b Conversion determined by GC analysis. | ||||
| 1 | THP | TESOTf, Bipy, DCM, 0 °C, 6 h | 86% | 1% |
| 2 | MOM | TESOTf, Bipy, DCM, 0 °C, 2 h | 99% | 0% |
| 3 | PMB | DDQ, CH2Cl2/H2O, RT, 0.5 h | 99% | 0% |
| 4 | oNBn | 350 nm, RT, C6H6 | 99% | 2% |
| 5 | TMS | Silica, 40 °C, 6 h | 91% | 23% |
| 6 | TES | Silica, 50 °C, 8 h | 55% | 35% |
Scope
Studies were continued by investigating the compatibility of the BDIPS-protection and -deprotection procedures with various common functional groups (Scheme 3). We examined primary, secondary, and tertiary alcohols as substrates that all worked well (8a–d). Protection of 7e was achieved in a quantitative yield and photo deprotection of the corresponding silyl ether 8e was also realized quantitatively. The silylation of hydroxy esters 7f–h proceeded with high efficiency (87–99%). The deprotection of their silyl ethers 8f–h exhibited similar effectiveness. In the case of 8f, we increased the amount of methanol in the solvent mixture. Importantly, without any irradiation upon stirring in MeOH for 24 h, 96% of the silyl ether 8f were recovered. We achieved complete silylation of the lactone 7i, while a moderate yield was observed for the deprotection of 8i (54%).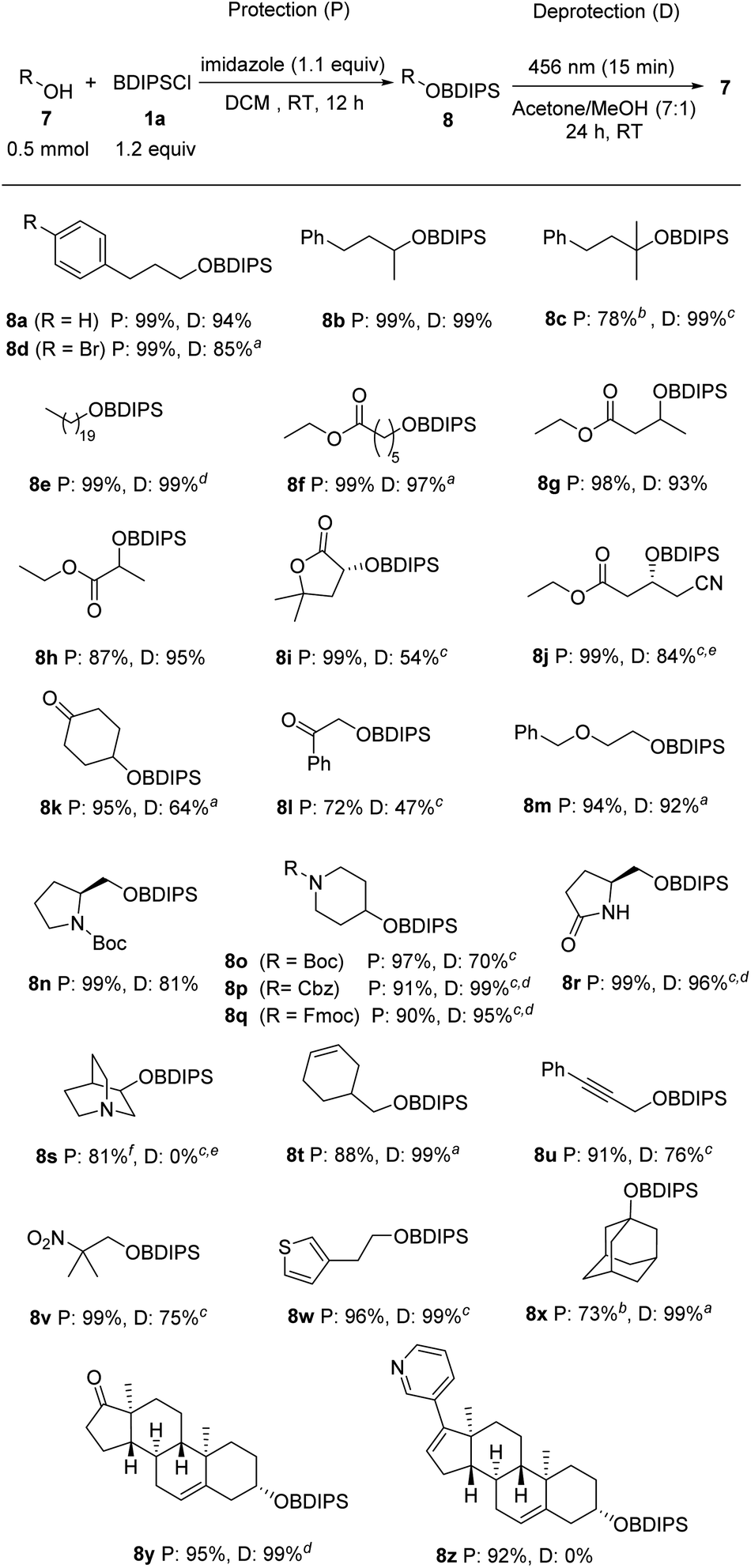 | ||
| Scheme 3 Scope. Yields for deprotection were determined by GC analysis. aReaction carried out in acetone/methanol (4:1). bReaction carried out using 1.5 equiv. of BDIPSCl and 1.2 equiv. of Ag(OTf). cReaction carried out in methanol. dIsolated yield. eReaction carried out under reflux. fReaction carried out in DMF. | ||
The cyanated hydroxyester 7j underwent efficient silylation (8j, 99%). However, attempts at desilylation at room temperature were unsuccessful. By elevating the temperature to reflux conditions for 24 hours, we facilitated the solvolysis of the intermediate, leading to the formation of the desired alcohol in 84% yield.
The ketone functionality demonstrates compatibility with silylation, as evidenced by the successful synthesis of 8k and 8l with yields ranging from 72% to 95%. The deprotection of these silyl ethers containing ketone groups proved to be less efficient, resulting in yields of 47% to 64%. Thus, the presence of the ketone moiety appears to enhance the stability of the intermediate. As anticipated, the benzyl ether functionality in 7m shows complete compatibility with the protection/deprotection processes. Similarly, the Boc-, Cbz-, and Fmoc-protected piperidinols 7o–q underwent highly efficient silylation with yields ranging from 90% to 97%. Subsequent deprotection using methanol yielded excellent results as well, ranging from 70% to 99%. Along with the carbamates, the lactam moiety is also compatible with the protection/deprotection sequence, as demonstrated for lactam 7r. Carbamate-protected secondary amines are well-tolerated within this context. However, it is important to note that tertiary amines are not compatible with the deprotection step, despite successful protection outcomes (as illustrated for 8s). This is in line with Brook's observations, where it was noted that the formed acetals remain stable under alkaline conditions.7 Additionally, the presence of alkenes and alkynes poses only small interference, as demonstrated by compounds 8t and 8u. Silylation of the hydroxy nitroalkane 7v occurred in a quantitative yield and also deprotection worked rather well in MeOH (75%). Notably, when placed in MeOH without any irradiation for a period of 24 hours, a recovery of 92% for compound 8v was observed. This underscores the essential nature of the photochemical process for the initial step. The silyl-protected hydroxyethylthiophene 8w was obtained with an excellent yield, and its deprotection was achieved quantitatively. The protection of 1-adamantanol (7x) was accomplished with a yield of 73%, and its corresponding silyl ether 8x underwent quantitative deprotection. Similarly, both the protection and deprotection of the steroidal alcohol 8y yielded excellent results. As for the free amine functionality, a pyridine moiety is not compatible with the deprotection step (see 8z), while initial silylation proceeded without any problems.
Of note, the reagent's dye-like property warrants attention from a practical point of view. The bright yellow color of aroylsilanes facilitates uncomplicated purification through column chromatography. The compound's visibility to the naked eye eliminates the necessity for TLC or test tubes, provided an adequate separation from potential byproducts has been achieved.
Additionally, the stability towards two typical transformations was documented (Scheme 4). Thus, reaction of 8k with a Wittig reagent led to the formation the desired product 8aa, while leaving the acylsilane untouched.14 This reaction also underlines the thermal stability of the protecting group. Hydration of double bonds was also tolerated and 8ac was obtained in a quantitative yield.15
 | ||
| Scheme 4 Compatibility of the BDIPS-ethers with two different chemical transformations. | ||
Desilylative benzoylation
Upon implementing the photo deprotection procedure on benzyl and allyl BDIPS-ethers, we observed an unexpected reactivity. When irradiating 8ae, we did not obtain the intended alcohol product; instead, we observed the quantitative formation of benzyl phenyl ketone 9a. This same outcome was reproduced in the absence of methanol in acetonitrile (Scheme 5). Primary and secondary benzylic BDIPS-ethers exhibited similar reactions under these conditions, yielding products 9b and 9c in high yields. Additionally, we observed a comparable desilylative rearrangement when attempting to deprotect the allylic alcohols 8ab and 8ah, resulting in the formation of α,β-unsaturated ketones 9d (91%) and 9e (70%). Notably, the initial ketone products also underwent a double bond isomerization in these two cases.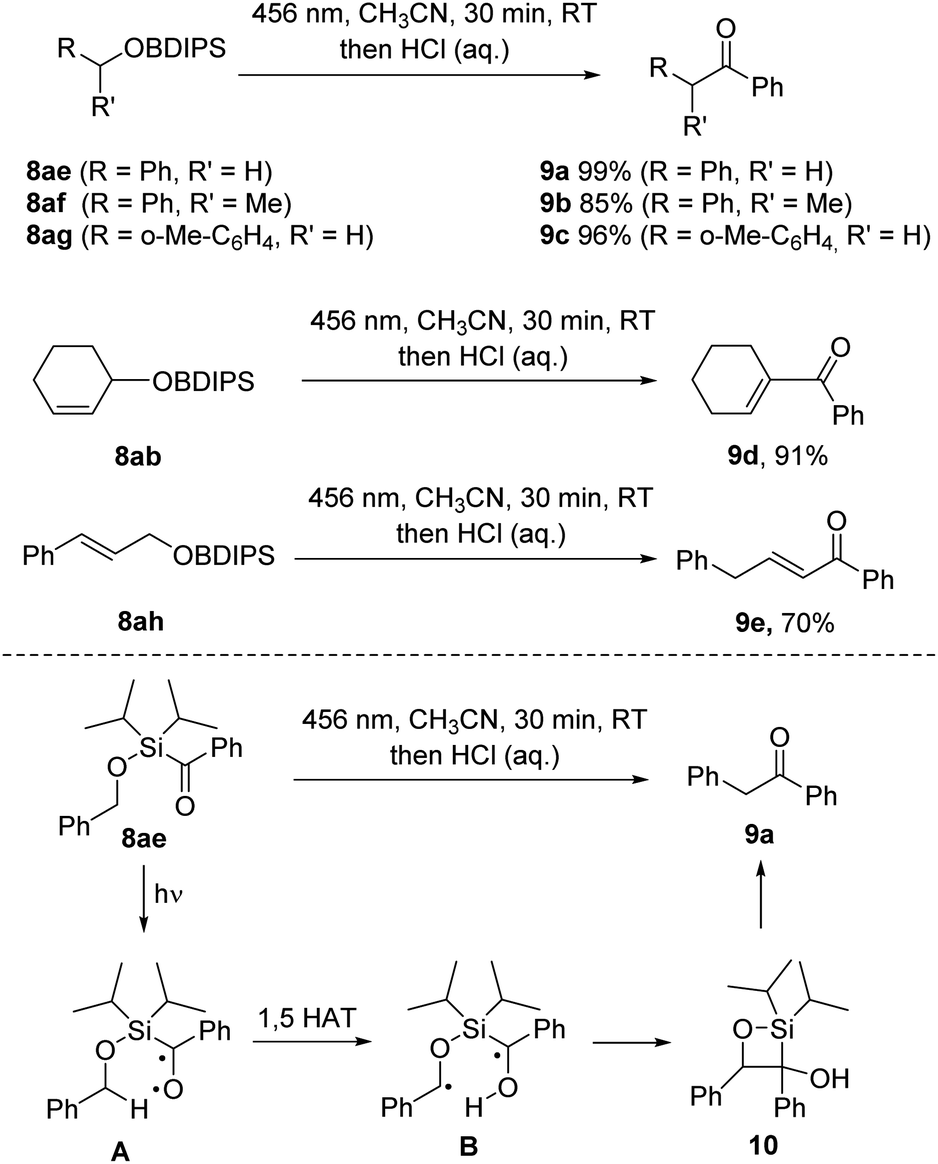 | ||
| Scheme 5 Desilylative rearrangement of benzyl and allyl BDIPS-ethers. | ||
Mechanistic NMR-studies using substrate 8ae (see ESI†) revealed the formation of both diastereomers (1:1) of silaoxetane 10 as unstable intermediates that further decomposed to the ketone 9a at room temperature in solution. Efforts to isolate compound 10 resulted in its complete conversion to 9a. We have outlined a plausible reaction mechanism in Scheme 5. Upon irradiation, the excited silyl ketone A is generated. Instead of undergoing silyl migration and forming a siloxy carbene, A undergoes a 1,5 hydrogen atom transfer (HAT) from the activated benzylic C–H bond, generating the diradical B, which then cyclizes via a Norrish type II process to yield silaoxetane 10. Subsequently, 10 further decomposes into ketone 9a. This observation is noteworthy since the formation of siloxycarbenes is typically strongly favoured for acylsilanes.16 Although Norrish type II reactions have been reported for alkanoylsilanes,17 to the best of our knowledge, they have not been previously documented for aroylsilanes.
Conclusion
In summary, benzoyldiisopropylchlorosilane (BDIPSCl) as a new visible light photocleavable silicon-based alcohol protecting group was introduced. BDIPSCl is readily prepared in large scale and can be stored for months under exclusion of moisture in the dark. Protection of primary, secondary and tertiary alcohols is easily achieved with common silylation protocols. To remove the Si-protecting group, a straightforward process involving exposure to visible light followed by stirring in MeOH or an acetone/MeOH solvent mixture at room temperature is employed. Notably, BDIPSCl exhibits high compatibility with various functional groups during both its formation and cleavage, demonstrating its versatility. Moreover, it displays a high degree of orthogonality in relation to other protecting groups. We are confident that BDIPSCl will significantly enhance the range of silicon-based protecting groups, by offering the option of a photochemical deprotection protocol.Data availability
The data that support the findings of this study are available in the ESI.†Author contributions
F. L. and K. M. conducted all experiments and characterized the novel compounds. F. L. and A. S. designed the experiments and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the University of Münster for supporting this work.Notes and references
- (a) T. W. Greene and P. G. M. Wuts, Protective Groups in Organic Synthesis, 3rd edn, J. Wiley, New York, 1999 CrossRef; (b) P. J. Kocieński, Protecting Groups, Thieme, Stuttgart, 1994 Search PubMed; (c) M. Schelhaas and H. Waldmann, Angew. Chem. Int. Ed., 1996, 35, 2056 ( Angew. Chem. , 1996 , 108 , 2192 ) CrossRef CAS.
- (a) P. Baran, T. Maimone and J. Richter, Nature, 2007, 446, 404 CrossRef CAS PubMed; (b) R. W. Hoffmann, Synthesis, 2006, 40, 3531 CrossRef.
- Selected examples: (a) R. W. Armstrong, J.-M. Beau, S. H. Cheon, W. J. Christ, H. Fujioka, W.-H. Ham, L. D. Hawkins, H. Jin, S. H. Kang, Y. Kishi, M. J. Martinelli, W. W. McWhorter Jr, M. Mizuno, M. Nakata, A. E. Stutz, F. X. Talamas, M. Taniguchi, J. A. Tino, K. Ueda, J. Uenishi, J. B. White and M. Yonag, J. Am. Chem. Soc., 1989, 111, 7530 CrossRef CAS; (b) T. M. Kamenecka and S. J. Danishefsky, Angew. Chem. Int. Ed., 1998, 37, 2995 ( Angew. Chem. , 1998 , 110 , 3164 ) CrossRef CAS.
- E. J. Corey and A. Venkateswarlu, J. Am. Chem. Soc., 1972, 94, 6190 CrossRef CAS.
- (a) T. D. Nelson and R. D. Crouch, Synthesis, 1996, 40, 1031 CrossRef; (b) M. A. Brook, C. Gottardo, S. Balduzzi and M. Mohamed, Tetrahedron Lett., 1997, 38, 6997 CrossRef CAS.
- (a) P. Klán, T. Šolomek, C. G. Bochet, A. Blanc, R. Givens, M. Rubina, V. Popik, A. Kostikov and J. Wirz, Chem. Rev., 2013, 113, 119 CrossRef PubMed; (b) R. Weinstain, T. Slanina, D. Kand and P. Klán, Chem. Rev., 2020, 120, 13135 CrossRef CAS PubMed; (c) C. G. Bochet, J. Chem. Soc., Perkin Trans., 2002, 1, 125 Search PubMed; (d) P. Wang, Asian J. Org. Chem., 2013, 2, 452 CrossRef CAS; (e) M. C. Pirrung, L. Fallon, J. Zhu and Y. R. Lee, J. Am. Chem. Soc., 2001, 123, 3638 CrossRef CAS PubMed.
- (a) G. Brook and J. M. Duff, J. Am. Chem. Soc., 1967, 89, 454–455 CrossRef; (b) C. Stuckhardt, M. Wissing and A. Studer, Angew. Chem. Int. Ed., 2021, 60, 18605–18611 ( Angew. Chem. , 2021 , 133 , 18753 ) CrossRef CAS PubMed.
- F. Lind and A. Studer, Chem. - Eur. J., 2023, 29, e202301120 CrossRef CAS PubMed.
- S. Lee, H. Lee and K. L. Tan, J. Am. Chem. Soc., 2013, 135, 18778 CrossRef CAS PubMed.
- Along with the acetal 4b, Ph(CH2)2CH(CH3)OSi(iPr)2OH was identified as major compound after irradiation (see ESI†).
- J. S. Davies, L. C. L. Higginbotham, E. J. Tremeer, C. Brown and R. C. Treadgold, J. Chem. Soc., Perkin Trans., 1992, 1, 3043 RSC.
- H. Fujioka, Y. Minamitsuji, O. Kubo, K. Senami and T. Maegawa, Tetrahedron, 2011, 67, 2949 CrossRef CAS.
- S. Hanessian, S. Marcotte, R. Machaalani and G. Huang, Org. Lett., 2003, 5, 4277 CrossRef CAS PubMed.
- S. P. Chavan, K. P. Pawar and S. Garai, RSC Adv., 2014, 4, 14468 RSC.
- R. Liffert, A. Linden and K. Gademann, J. Am. Chem. Soc., 2017, 139, 16096 CrossRef CAS PubMed.
- A. Alberti, G. Seconi, G. F. Pedulli and A. Degl'Innocenti, J. Organomet. Chem., 1983, 253, 291 CrossRef.
- (a) P. J. Wagner, Acc. Chem. Res., 1971, 4, 168 CrossRef CAS; (b) C. P. Casey and R. A. Boggs, J. Am. Chem. Soc., 1972, 94, 6457 CrossRef CAS; (c) M. E. Scheller and B. Frei, Helv. Chim. Acta, 1984, 67, 1734 CrossRef CAS; (d) M. E. Scheller and B. Frei, Helv. Chim. Acta, 1990, 73, 922 CrossRef CAS; (e) K. Ishida, H. Yamazaki, C. Hagiwara, M. Abe and H. Kusama, Chem. - Eur. J., 2020, 26, 1249 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc04975b |
| This journal is © The Royal Society of Chemistry 2023 |