
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiscovery of a selective TC-PTP degrader for cancer immunotherapy†
Jinmin
Miao‡
a,
Jiajun
Dong‡
a,
Yiming
Miao‡
a,
Yunpeng
Bai
 a,
Zihan
Qu
b,
Brenson A.
Jassim
a,
Bo
Huang
b,
Quyen
Nguyen
b,
Yuan
Ma
a,
Allison A.
Murray
a,
Jinyue
Li
a,
Philip S.
Low
bcd and
Zhong-Yin
Zhang
*abcd
a,
Zihan
Qu
b,
Brenson A.
Jassim
a,
Bo
Huang
b,
Quyen
Nguyen
b,
Yuan
Ma
a,
Allison A.
Murray
a,
Jinyue
Li
a,
Philip S.
Low
bcd and
Zhong-Yin
Zhang
*abcd
aDepartment of Medicinal Chemistry and Molecular Pharmacology, Purdue University, West Lafayette, IN 47907, USA. E-mail: zhang-zy@purdue.edu
bDepartment of Chemistry, Purdue University, West Lafayette, IN 47907, USA
cInstitute for Cancer Research, Purdue University, West Lafayette, IN 47907, USA
dInstitute for Drug Discovery, Purdue University, West Lafayette, IN 47907, USA
First published on 24th October 2023
Abstract
T-cell protein tyrosine phosphatase (TC-PTP), encoded by PTPN2, has emerged as a promising target for cancer immunotherapy. TC-PTP deletion in B16 melanoma cells promotes tumor cell antigen presentation, while loss of TC-PTP in T-cells enhances T-cell receptor (TCR) signaling and stimulates cell proliferation and activation. Therefore, there is keen interest in developing TC-PTP inhibitors as novel immunotherapeutic agents. Through rational design and systematic screening, we discovered the first highly potent and selective TC-PTP PROTAC degrader, TP1L, which induces degradation of TC-PTP in multiple cell lines with low nanomolar DC50s and >110-fold selectivity over the closely related PTP1B. TP1L elevates the phosphorylation level of TC-PTP substrates including pSTAT1 and pJAK1, while pJAK2, the substrate of PTP1B, is unaffected by the TC-PTP degrader. TP1L also intensifies interferon gamma (IFN-γ) signaling and increases MHC-I expression. In Jurkat cells, TP1L activates TCR signaling through increased phosphorylation of LCK. Furthermore, in a CAR-T cell and KB tumor cell co-culture model, TP1L enhances CAR-T cell mediated tumor killing efficacy through activation of the CAR-T cells. Thus, we surmise that TP1L not only provides a unique opportunity for in-depth interrogation of TC-PTP biology but also serves as an excellent starting point for the development of novel immunotherapeutic agents targeting TC-PTP.
Introduction
The regulation of protein tyrosine phosphorylation, which is vital for controlling fundamental cellular processes, is managed by protein tyrosine kinases (PTKs) and protein tyrosine phosphatases (PTPs). As aberrant protein tyrosine phosphorylation is linked to various human diseases, PTKs and PTPs have emerged as compelling targets for drug discovery. While modifying PTKs for therapeutic purposes has proven successful,1 development of PTP-targeted therapies remain a largely underexplored area in drug discovery.2 T-cell protein tyrosine phosphatase (TC-PTP), encoded by PTPN2, is a non-receptor type PTP ubiquitously expressed in all tissues, with highest expression in hematopoietic cells.3 TC-PTP negatively regulates signaling of non-receptor tyrosine kinases including JAK1 and JAK3,4,5 receptor tyrosine kinases,6,7 transcription factors, such as STAT1,8 STAT3,9 and STAT5,5 and Src family kinases, including LCK.10 Recently, accumulated evidence has suggested that TC-PTP is an attractive target for cancer immunotherapy. By attenuating the JAK/STAT pathway, TC-PTP negatively regulates interferon gamma (IFN-γ) signaling and therefore restricts tumor cell recognition and elimination.11 An in vivo genetic screen of a mouse B16F10 transplantable tumor model showed that CRISPR/Cas9 mediated removal of TC-PTP from tumor cells increases the responsiveness of tumors to immunotherapy by amplifying the effects of IFN-γ on antigen presentation and growth inhibition.12 Furthermore, TC-PTP plays a fundamental role in T-cells by dampening T-cell receptor (TCR) signaling through dephosphorylation and inactivation of the Src family kinase LCK.13 TC-PTP also attenuates JAK/STAT1/5 signaling in response to cytokines, such as interferons (IFNs) and Interleukin-2 (IL-2), that are required for the activation, clonal expansion, and differentiation of T-cells.14 Genetic ablation of TC-PTP in T-cells promotes the activation, expansion, and survival of CD8+ T-cells and enhances the activation of CD8+ T-cells through amplified phosphorylation of LCK and STAT5.13,15 Moreover, TC-PTP deletion in chimeric antigen receptor (CAR)-T cells increased both CAR-T cell activation and tumor-homing in vivo.16 Collectively, the mounting evidence from these emerging observations necessitates the prompt discovery of potent inhibitors that specifically target TC-PTP for therapeutic applications.Over the past few decades, efforts to develop TC-PTP inhibitors have mainly focused on targeting the active site of the enzyme with small molecules that contain a phospho-tyrosine mimicking warhead.17–19 However, most of these inhibitors encounter limited specificity and bioavailability due to the highly conserved and positively charged nature of PTP active sites.20 Regardless of these challenges, Calico and AbbVie recently disclosed a series of 1,2,4-thiadiazolidinone based active-site-directed PTP1B/TC-PTP dual inhibitors with excellent potency and bioavailability, and the top compound ABBV-CLS-484 was advanced to a Phase I clinical trials for treatment of locally advanced or metastatic solid tumors.21
In a recent study, we employed a phosphonodifluoromethyl phenylalanine (F2PMP)-based PTP1B/TC-PTP dual competitive inhibitor to develop a potent dual PTP1B/TC-PTP degrader DU-14 which efficiently promotes antigen presentation by tumor cells and enhances TCR signaling.22 Despite these accomplishments, the prevalent similarity between the active sites of TC-PTP and PTP1B remains to be a challenge for the development of highly potent and selective TC-PTP inhibitors.23,24 Although simultaneously targeting of TC-PTP and PTP1B could benefit the effectiveness of immunotherapy treatments,25,26 it might also lead to considerable side effects as PTP1B is a major negative regulator of insulin and leptin signaling and a critical positive regulator of microglia activation and neuroinflammation.27–30 In addition, definitive roles for TC-PTP in tumorigenesis, antitumor immunity, and autoimmunity are still unclear.31 Consequently, highly specific small molecule TC-PTP inhibitors will serve as unique chemical tools to facilitate further biological studies and clinical translation of TC-PTP targeting.
Our lab previously reported an active-site-directed, potent, and cell permeable F2PMP-based TC-PTP inhibitor 3 (Fig. 1), which exhibits a Ki of 4.2 nM for TC-PTP and ∼7-fold selectivity over PTP1B.32 Given the aforementioned importance of target specificity, we sought to further augment the specificity of 3 towards TC-PTP by leveraging the proteolysis-targeting chimera (PROTAC) technology.33,34 PROTACs are dual-action compounds comprised of a ligand specific to the target protein, a linker, and an E3 ligase binding component that recruits the ubiquitin-proteasome system within cells to induce selective degradation of the target protein. The function of PROTACs requires the formation of a target protein – PROTAC – E3 ligase ternary complex, whereby specific protein–protein interactions between the E3 ubiquitin ligase and the target protein provides an opportunity to add another layer of target selectivity.35,36 The intriguing observation that PROTAC-mediated degradation of target proteins can surpass the binding selectivity of the corresponding ligand for its target protein motivated us to explore PROTAC molecules that can selectively degrade TC-PTP. Furthermore, in comparison to conventional inhibitors, the event-driven PROTAC degraders offer other distinct benefits, including the extended efficacy achieved through target elimination, and sub-stoichiometric concentrations needed for target engagement due to their catalytic mechanism of action. Herein, we report the development of a novel TC-PTP PROTAC – based on inhibitor 3 and using a Cereblon (CRBN) E3 ligase ligand – with excellent degradation efficacy and >110-fold selectivity over PTP1B, which is closely related to TC-PTP.
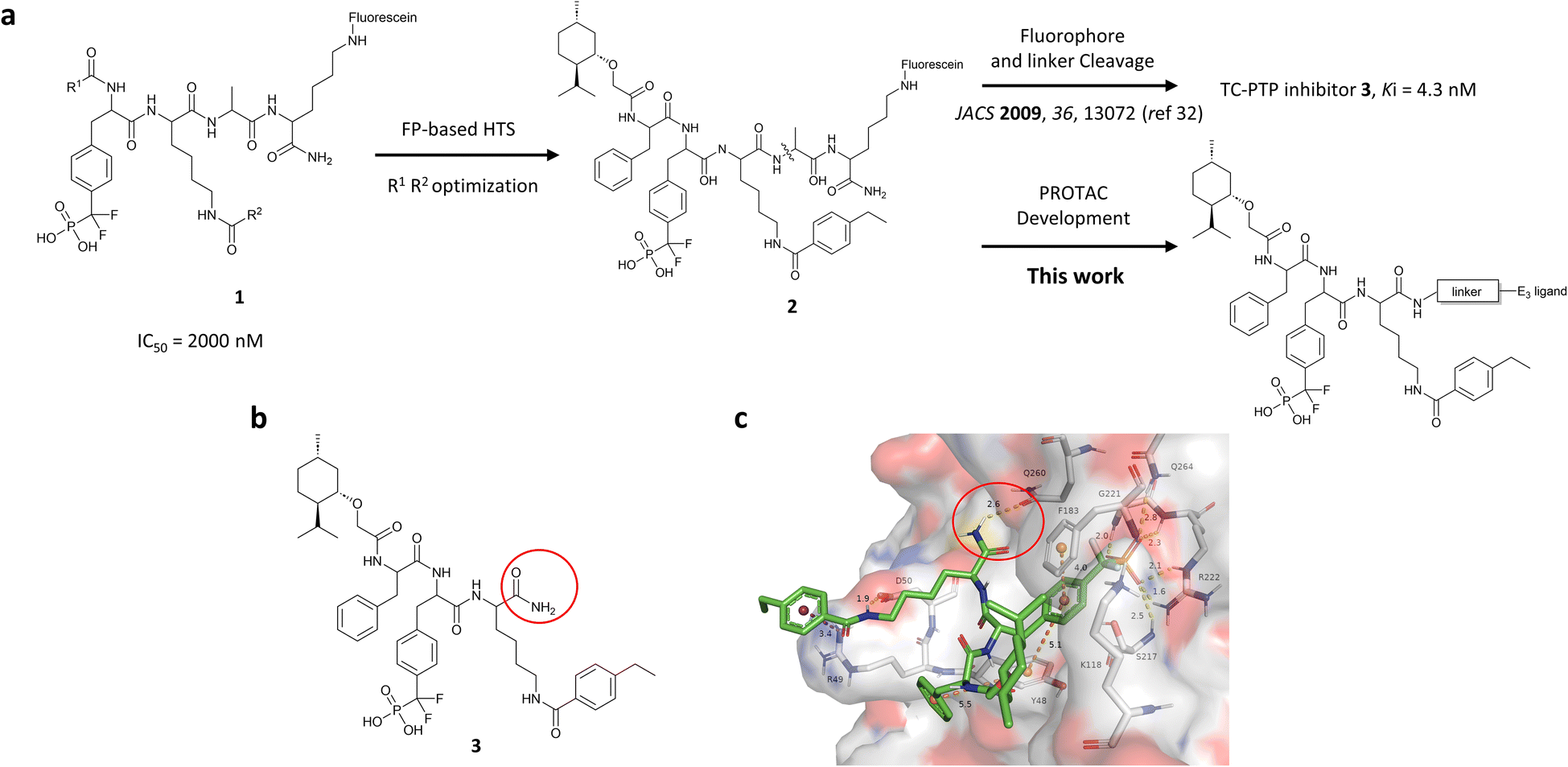 | ||
| Fig. 1 Design of TC-PTP PROTACs based on an active-site-directed TC-PTP inhibitor. (a) Development of TC-PTP inhibitor 3 and TC-PTP PROTACs. A fluorescein tagged TC-PTP probe 2 was previously utilized to develop a potent and selective inhibitor 3. The TC-PTP PROTACs were designed based on probe 2. (b) Structure of the selective TC-PTP active site inhibitor 3. (c) Docking pose of inhibitor 3 (green carbon stick) bound to TC-PTP (PDB 7F5O) in transparent surface representation with the solvent-exposing primary amide circled in red. Hydrogen bonds and ionic bonds are shown in yellow dashes, π–π stacking interactions are shown with orange dashes, and the cation–π interaction is shown with purple dashes. Distances for these interactions are labelled with their interaction distances (Å). | ||
Results and discussion
Design, synthesis, and evaluation of TC-PTP PROTAC degraders
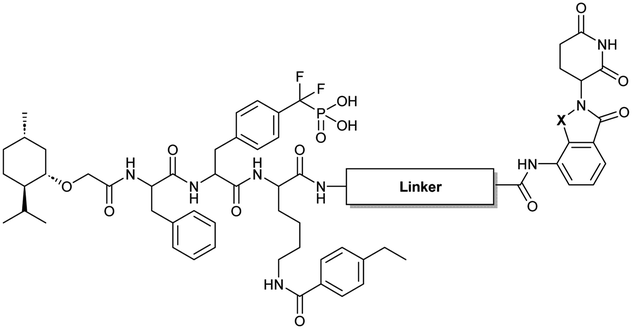
To obtain effective TC-PTP PROTACs, it is first necessary to identify an appropriate site on inhibitor 3 for linker instalment. Although no crystal structures of TC-PTP in complex with 3 are available to aid in the PROTAC design, the library screening strategy employed for the discovery of inhibitor 3 implicated a potential site for tethering the linker. In the previous work, a fluorescent-tagged probe 2 was synthesized to evaluate the binding affinity of 3 to TC-PTP by fluorescent polarization (FP) assay (Fig. 1a).32 The FP displacement results showed that ligand binding affinity to TC-PTP was not conspicuously reduced by the introduction of the fluorescein and linker, suggesting that the linker attachment on the amide moiety (Fig. 1b, circled in red) for PROTAC construction would be viable. To further ascertain the suitability of the primary amide in 3 as the TC-PTP PROTAC linker attachment site, we performed molecular docking experiments of 3 in complex with the TC-PTP catalytic domain. The top pose generated by our model is well-aligned with the SAR detailed in the previously reported discovery of inhibitor 3 (Fig. 1c). Notably, the scaffold's primary amide lies on the TC-PTP surface with the –NH2 of the amide pointing towards solvent, thus illustrating that this amide is a suitable vector for tethering of our PROTAC linker. Although the amide forms a hydrogen bond with Q260, this hydrogen bond is likely weak as it lies on the highly solvent-accessible protein surface, and a secondary amide moiety used for PROTAC linker attachment could still form this hydrogen bond, which minimizes a potential decrease in binding affinity. Therefore, we synthesized the TC-PTP ligand 16 with a free carboxylic acid as the linker tethering site (Scheme 1). First, fragment 6 was prepared from 4-ethylbenzoic acid and the protected lysine 4 through amide coupling and the appropriate protection/deprotection sequence. The tert-butyloxycarbonyl (Boc) protected F2PMP (10) was synthesized with the coupling reaction between the protected 4-iodophenylalanine 7 and the Cadmium reagent 8, followed by the deprotection of the carboxylic acid. Fragment 14 was prepared from methyl phenylalaninate 11 and (+)-menthoxyacetic acid 12via condensation and the subsequent hydrolysis of the methyl ester. Fragment 6 was then coupled with 10 to give the intermediate 15, followed by the amine deprotection and the subsequent coupling with fragment 14. The resulting intermediate was subjected to hydrogenation and hydrolysis to yield 16, which is ready for the construction of PROTACs.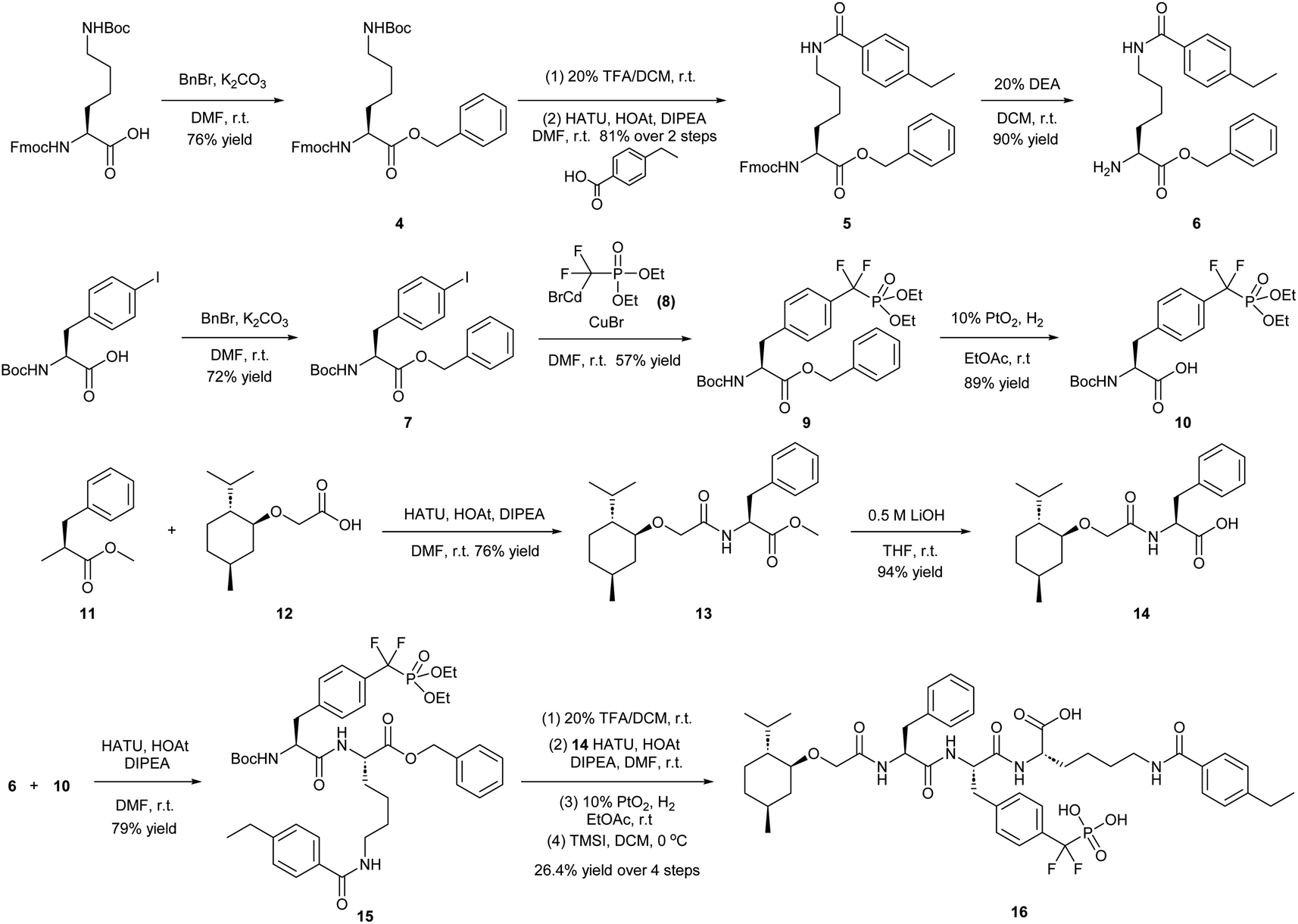 | ||
| Scheme 1 Synthetic route of TC-PTP ligand 16 for PROTAC construction. Abbreviations: DMF = N,N-dimethylformamide; r.t. = room temperature; TFA = trifluoroacetic acid; DCM = dichloromethane; HATU = hexafluorophosphate azabenzotriazole tetramethyl uranium; HOAt = 1-hydroxy-7-azabenzotriazole; DIPEA = N,N-diisopropylethylamine; DEA = diethylamine; THF = tetrahydrofuran; TMSI = trimethylsilyl iodide. | ||
The utilization of CRBN/cullin 4A E3 ligase complexes has proven to be successful in developing PROTAC degraders for various proteins.37 CRBN ligands, such as pomalidomide and lenalidomide, feature desirable physicochemical properties and were therefore employed in this study to develop TC-PTP degraders.38 To validate the linker tethering site and identify the optimal linker length, we synthesized an initial set of bivalent compounds with a linear alkyl chain linker of various lengths, using pomalidomide as the CRBN ligand (Table 1 and Fig. S1†). The in vitro biochemical IC50s of these PROTACs against PTP1B and TC-PTP were determined, and the data showed that they all inhibit TC-PTP with low nanomolar IC50s and ∼3–5-fold selectivity over PTP1B, indicating that the linker installment did not disturb the binding affinity and preference to the target protein (Table S1†). The compounds' ability to induce TC-PTP degradation was subsequently evaluated in HEK293 cells by western blot, and the dual TC-PTP/PTP1B PROTAC DU-14(ref. 22) was used as a positive control. Among this series of PROATC candidates, TC5P with an n-pentane linker presented the highest degradation potency for TC-PTP. After 16 hours of treatment, 2 μM of TC5P induced the depletion of 48% TC-PTP and only 8% PTP1B. The potency of TC-PTP degradation was significantly reduced when the linker length in TC5P was increased (TC6P) or decreased (TC4P) by one additional methylene. Compounds with even longer linkers exhibit negligible (TC7P) or no degradation (TC8P, TC9P). In addition, the parent inhibitor 3 did not induce any change on the protein level of TC-PTP in this assay. Hence, it was concluded that the n-pentane chain demonstrated the most optimal linker length for a TC-PTP PROTAC degrader based on inhibitor 3 and a CRBN-recruiting pomalidomide ligand.
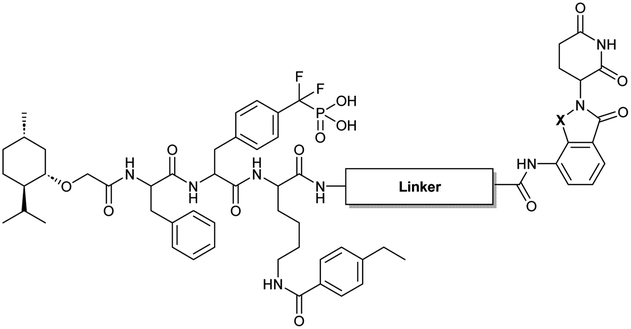
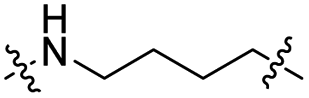
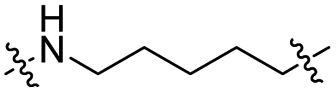
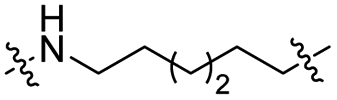
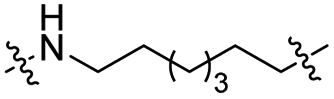
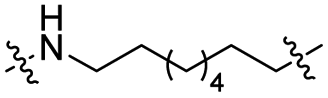
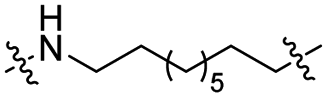
Lenalidomide, another CRBN binding ligand often used in PROTAC development, offers certain advantages over pomalidomide.38 The absence of one phthalimide carbonyl group in lenalidomide leads to reduced TPSA (total polar surface area), improved physicochemical properties, and increased metabolic and chemical stability. Therefore, to explore TC-PTP selective degraders with increased potency and bioavailability, we synthesized another set of PROTAC candidate compounds with a series of linear alkyl chain linkers, this time employing lenalidomide as the E3 ligand (Table 2 and Fig. 2). These compounds, and the pomalidomide based PROTACs TC5P and TC6P, were evaluated in HEK293 cells for their ability to degrade TC-PTP at 1 μM. To our delight, TC5L and TC6L showed improved potency on TC-PTP degradation compared to their pomalidomide counterparts, TC5P and TC6P, and nearly complete depletion of TC-PTP was achieved. However, degradation of PTP1B was also observed with these two compounds. Lenalidomide based PROTAC candidates with either short or long linkers did not show significant TC-PTP degradation under the same conditions (TC4L, TC7L, and TC8L). Apart from linker length, the chemical nature and flexibility of linkers are also known to influence the formation and stability of the target protein—PROTAC—E3 ligase ternary complex, and thus can alter the efficiency and selectivity of degradation.39 Therefore, we synthesized and evaluated a lenalidomide based PROTAC TP1L with an ethoxyethane linker instead of the alkyl chain in TC5L. The treatment of 1 μM TP1L resulted in an almost complete degradation of TC-PTP, while the PTP1B level remained unaffected. A PROTAC with a piperidine ring linker was also examined (TPiL), and interestingly, high degradative potency for both TC-PTP and PTP1B was observed with this molecule. Taken together, TP1L showed the optimal potency and selectivity for TC-PTP degradation and was therefore selected for thorough characterization.
|
|
||||
|---|---|---|---|---|
| Compound | Linker structure | X | PTP1B % degradation | TC-PTP % degradation |
| a HEK293 cells were treated with 1 μM PROTAC compounds for 16 hours. | ||||
| TC5P |
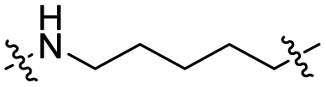
|
CO | 6% | 14% |
| TC6P |
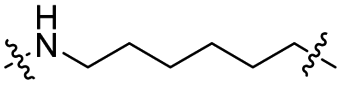
|
CO | 5% | 11% |
| TC4L |
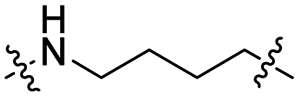
|
CH2 | <5% | <5% |
| TC5L |
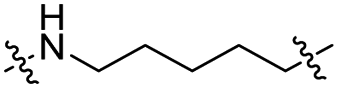
|
CH2 | 32% | 94% |
| TC6L |
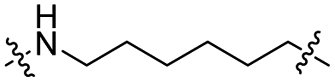
|
CH2 | 36% | 92% |
| TC7L |
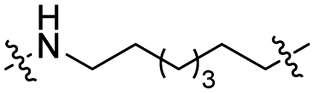
|
CH2 | <5% | <5% |
| TP8L |
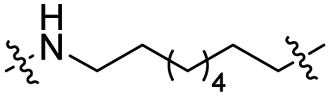
|
CH2 | <5% | <5% |
| TP1L |
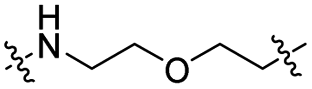
|
CH2 | <5% | >95% |
| TPiL |
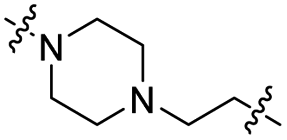
|
CH2 | 88% | >95% |
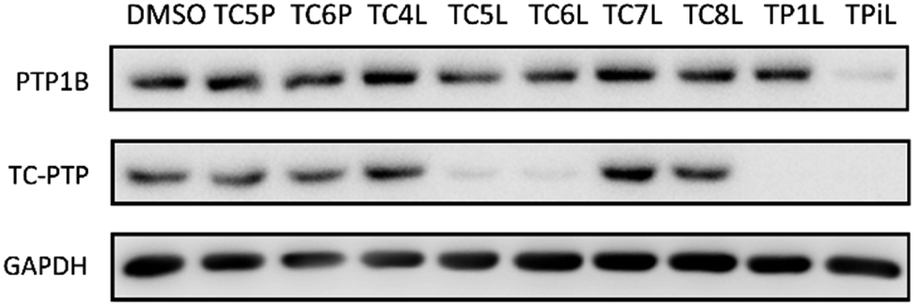 | ||
| Fig. 2 Degradation assay results of second-generation TC-PTP PROTACs. HEK293 cells were treated with DMSO or 1 μM indicated compounds for 16 hours. | ||
TP1L is a potent, selective, and bona fide TC-PTP PROTAC degrader
To evaluate TP1L as a genuine TC-PTP degrader, we first conducted degradation efficacy assessments across a broad compound concentration range in HEK293 cells (Fig. 3a). The DC50 (compound concentration needed to induce target protein degradation by 50%) for TP1L mediated TC-PTP degradation is 35.8 nM in HEK293 cells after 16 hours of treatment, while the level of PTP1B is not significantly altered at up to 4 μM TP1L concentration, demonstrating that this degrader is at least 110-fold selective for TC-PTP over PTP1B (Fig. 3b). The time course of TC-PTP and PTP1B degradation was then determined by treating HEK293 cells with 1 μM TP1L for 3, 6, 16, and 24 hours (Fig. 3c). Nearly complete depletion of TC-PTP was observed within 6 hours, and the suppression of TC-PTP protein level was sustained for at least 24 hours. It is noteworthy that the PTP1B protein level did not decrease during the entire period of TP1L treatment.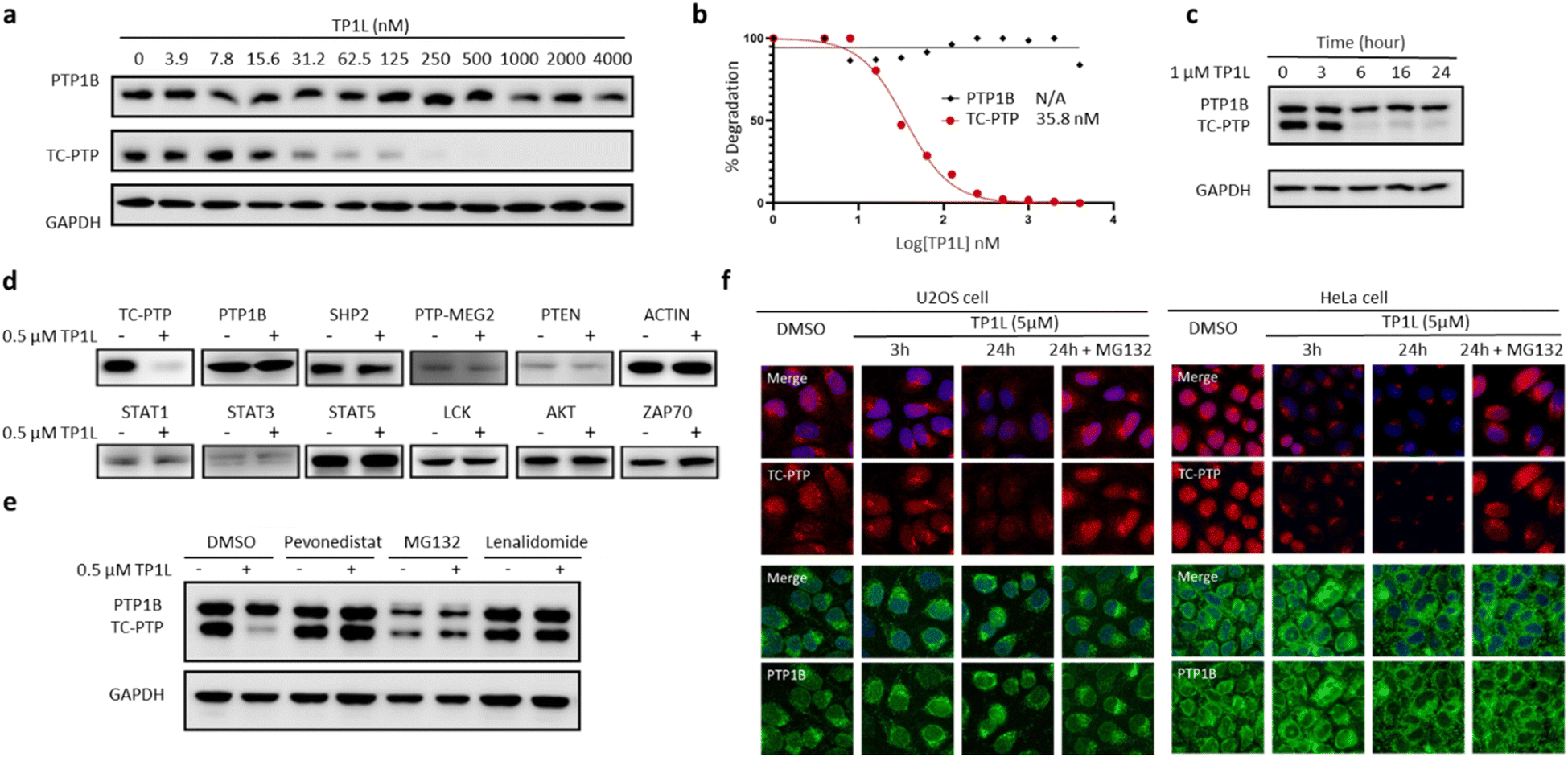 | ||
| Fig. 3 TP1L is a potent, selective, and bona fide TC-PTP PROTAC. (a and b) Immunoblots of cell lysates from HEK293 treated with TP1L at indicated concentrations for 16 hours showing dose-dependent selective degradation of TC-PTP with DC50 of 35.8 ± 1.4 nM. (c) Immunoblots of cell lysates from HEK293 treated with TP1L at 1 μM for indicated hours showing time-dependent degradation of TC-PTP. (d) TP1L selectively degrades TC-PTP in HEK293 cells. Treatment of 0.5 μM TP1L for 16 hours led to substantial degradation of TC-PTP, whereas none of the other PTPs and signaling proteins were affected. (e) Mechanistic investigation of TC-PTP degradation induced by TP1L in HEK293 cells. Cells were pre-treated with indicated concentration of Pevonedistat (prevents ubiquitination), MG132 (blocks proteasome activity), or Lenalidomide (prevents CRBN binding) followed by 6 h treatment with TP1L at 0.5 μM showing TP1L mediated TC-PTP degradation depends on the ubiquitination-proteasome pathway. (f) Immunofluorescences of TC-PTP (red) and PTP1B (green) with U2OS cells treated with DMSO, 5 μM TP1L for 3 and 24 hours showing TP1L degrades both nucleus and cytoplasm localized TC-PTP. 20 μM MG132 was used along with TP1L to block PTP1B and TC-PTP degradation in the control group. | ||
To investigate the binding affinity of TP1L to TC-PTP and selectivity towards other PTPs, we determined its enzymatic IC50s against a panel of 14 representative PTP family members, including receptor-like, nonreceptor-like, and dual specific PTPs (Table S2†). The results indicated that TP1L showed no significant inhibition towards other PTPs even at up to 10 μM TP1L concentration. Interestingly, the IC50 of TP1L for TC-PTP is only 6 times lower than that of PTP1B, indicating that TP1L's high degree of degradation preference to TC-PTP is likely due to the favorable propensity of the degrader to induce productive PTP-TP1L-CRBN ternary complex formation that is capable of catalyzing the subsequent ubiquitination of TC-PTP over PTP1B. Next, the selectivity of TP1L as a PROTAC degrader for TC-PTP over other PTP family members and different classes of enzymes was investigated (Fig. 3d). Western blots showed that treatment of HEK293 cells with 0.5 μM TP1L for 16 hours led to substantial degradation of TC-PTP, whereas none of the other PTPs including PTP1B, SHP2, PTP-MEG2, and PTEN were affected. In addition, under the same conditions, TP1L did not induce any appreciable degradation of other signaling proteins including STAT1, STAT3, STAT5, LCK, ACTIN, AKT, and ZAP70. Furthermore, no toxicity was observed when HEK293 cells were treated with TP1L at up to 20 μM concentration (Fig. S2†). Taken together, the above results validated TP1L as a potent and selective TC-PTP degrader.
The mechanism of action of TP1L in degrading TC-PTP was next investigated in the HEK293 cells by running a series of control experiments (Fig. 3e). Our data showed that TC-PTP degradation induced by TP1L can be effectively blocked by pretreatment with a NEDD8-activating enzyme (NAE) inhibitor Pevonedistat, a proteasome inhibitor MG132, or the CRBN ligand lenalidomide, together indicating that the degradation of TC-PTP by TP1L requires the recruitment of CRBN and is ubiquitination- and proteasome-dependent. In addition, the TP1L mediated TC-PTP degradation and proteasome dependency were verified by immunofluorescent imaging (Fig. 3f). In both U2OS and HeLa cells, TP1L selectively degrades TC-PTP in a time-dependent manner, and the proteasome inhibitor MG132 disrupted this process. This mechanistic data provides clear evidence that TP1L is a bona fide TC-PTP PROTAC.
TP1L amplifies IFN-γ signaling and promotes antigen presentation
TC-PTP negatively regulates the IFN-γ JAK/STAT signaling pathway by dephosphorylating JAK1 at residues Y1034/Y1035.5 In addition, TC-PTP can directly dephosphorylate STAT1 at Y701 in the nucleus to thoroughly suppress IFN-γ signaling.9 Accordingly, we next evaluated the ability of TP1L to modulate IFN-γ signaling in cells by measuring the phosphorylation levels of JAK1 and STAT1 upon the treatment of TP1L. As shown in Fig. 4a, the pJAK1 and pSTAT1 levels upon IFN-γ stimulation were enhanced by TP1L in a dose dependent manner, and their EC50s were determined to be 209 nM and 259 nM, respectively. Importantly, the phosphorylation level of JAK2, which is a substrate of PTP1B, was not affected by the presence of TP1L, demonstrating that the degrader is specific to TC-PTP. To further ascertain that TP1L boosts INF-γ signaling through degradation of TC-PTP, we utilized a TC-PTP knockout HEK293 cell line to run a control experiment (Fig. 4b). With the stimulation of IFN-γ and 500 nM treatment of TP1L, pJAK1 and pSTAT1 levels were significantly increased in wild-type HEK293 cells, whereas they were not changed in the TC-PTP knockout cells under the same conditions. As expected, pJAK2 and PTP1B levels in either cell line were not affected by TP1L. Furthermore, immunofluorescence microscopy also showed that the pSTAT1 signal in the nucleus is enhanced by treatment of TP1L in wild-type HEK293 cells, while no change was observed in TC-PTP knockout cells (Fig. 4c). These observations further substantiate the specificity of TP1L for TC-PTP. Moreover, in line with the increased activation of upstream IFN-γ signaling, we observed over 100% increase in the cell surface MHC-I expression in HEK293 cells induced by TP1L following IFN-γ stimulation, while the MHC-1 level in TC-PTP knockout cells was not affected (Fig. 4d and S3†). Collectively, we demonstrated that TP1L efficiently amplifies IFN-γ signaling through inhibition of TC-PTP catalyzed tyrosine dephosphorylation of JAK1 and STAT1, resulting in elevated cell surface MHC-I expression, which is crucial for efficient T-cell-mediated elimination of tumor cells.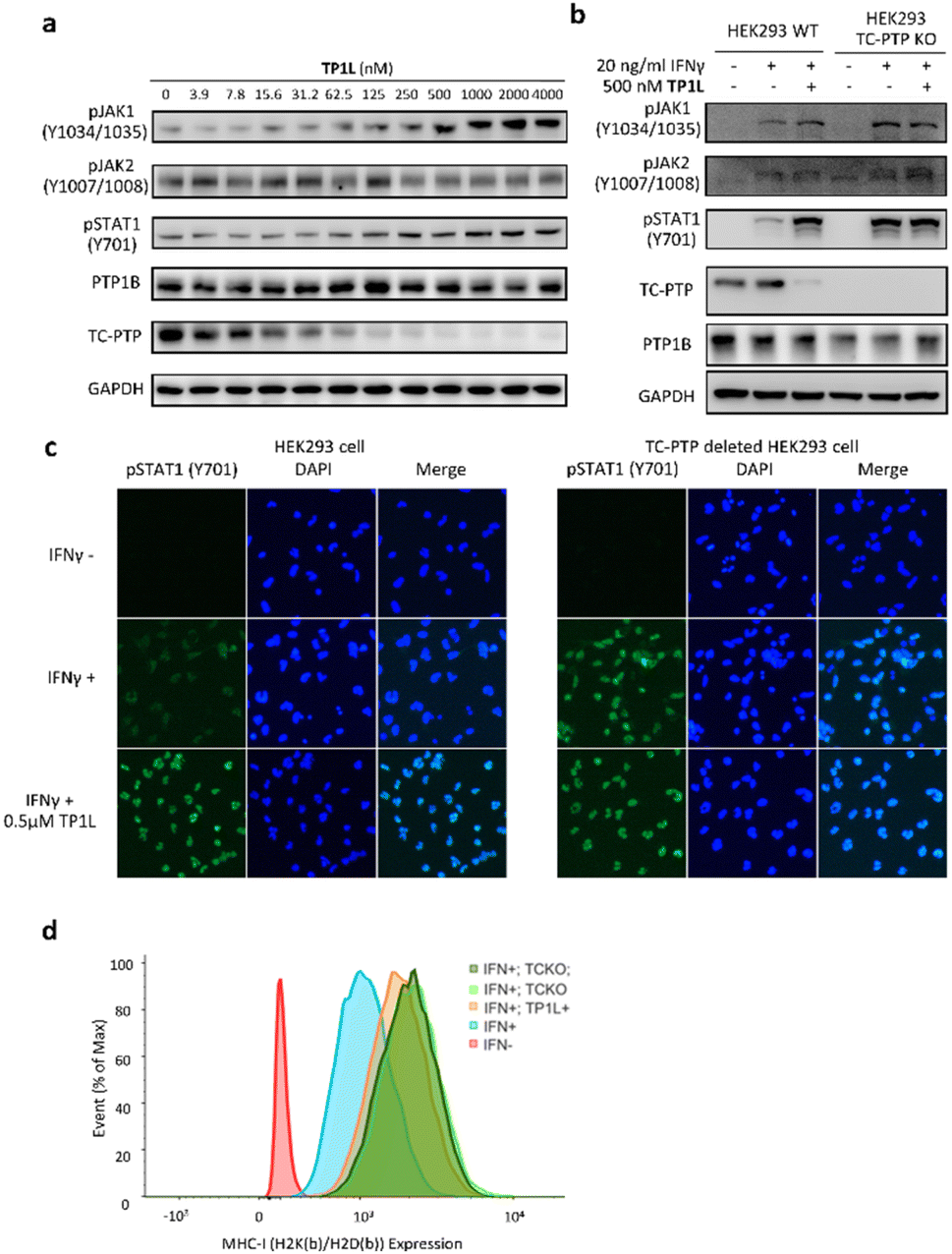 | ||
| Fig. 4 TP1L selectively enhances IFN-γ signaling and promotes antigen presentation in HEK293. (a) Immunoblots of whole cell lysates from HEK293 cells showed that pJAK1 and pSTAT1 levels were elevated by TP1L in a dose-dependent manner, along with TC-PTP degradation upon the treatment with TP1L for 16 hours and stimulation with 20 ng ml−1 human IFN-γ for 15 min. (b) Immunoblots of cell lysates from wild-type and TC-PTP deleted HEK293 cells that treated with 0.5 μM TP1L for 16 hours and stimulated with 20 ng ml−1 human IFN-γ for 15 min. Deletion of TC-PTP abolished the TP1L induced phosphorylation level increase of the TC-PTP substrate JAK1 and STAT1. (c) Immunofluorescence showing TP1L dramatically enhanced IFN-γ mediated STAT1 phosphorylation (green) and nucleus translocation through degradation of TC-PTP. Wild-type and TC-PTP deleted HEK293 cells were treated with DMSO or 0.5 μM TP1L for 16 hours and stimulated with 20 ng ml−1 IFN-γ for 30 minutes. (d) TP1L promotes MHC-1 expression in HEK293 cells. Wildtype and TC-PTP knockout HEK293 cells were treated with DMSO (blue and light green peak) or 500 nM TP1L (orange and dark green peak) for 16 hours for TC-PTP degradation then stimulated with 20 ng ml−1 mouse IFN-γ for 48 hours to induce MHC-I expression. Mouse MHC-I complex was stained with mouse H2K(b)/H2D(b) antibody and measured by flow cytometry. TCKO = TC-PTP knockout. | ||
TP1L promotes Jurkat T-cell and CAR-T cell activation, and augments tumor killing efficacy
The activation and clonal expansion of T-cells requires TCR signaling activation, which is mediated by a series of tyrosine phosphorylation events.40 TC-PTP suppresses TCR signaling by dephosphorylating and inactivating the Src family kinase LCK, which initiates TCR signaling upon recognition of the MHC-I complex, and deletion of TC-PTP in T-cells has been shown to significantly enhance anti-tumor immunity.13 Therefore, we assessed the impact of TP1L-induced TC-PTP degradation in T-cells. As we expected, anti-CD3 antibody stimulation of TP1L-treated Jurkat T-cells led to a dose-dependent decrease in the protein level of TC-PTP and a subsequent elevation in the phosphorylation level of LCK Y394 (Fig. 5a). It is noteworthy that the PTP1B protein level is unchanged under the same conditions, indicating that TP1L remains selective in Jurkat T-cells.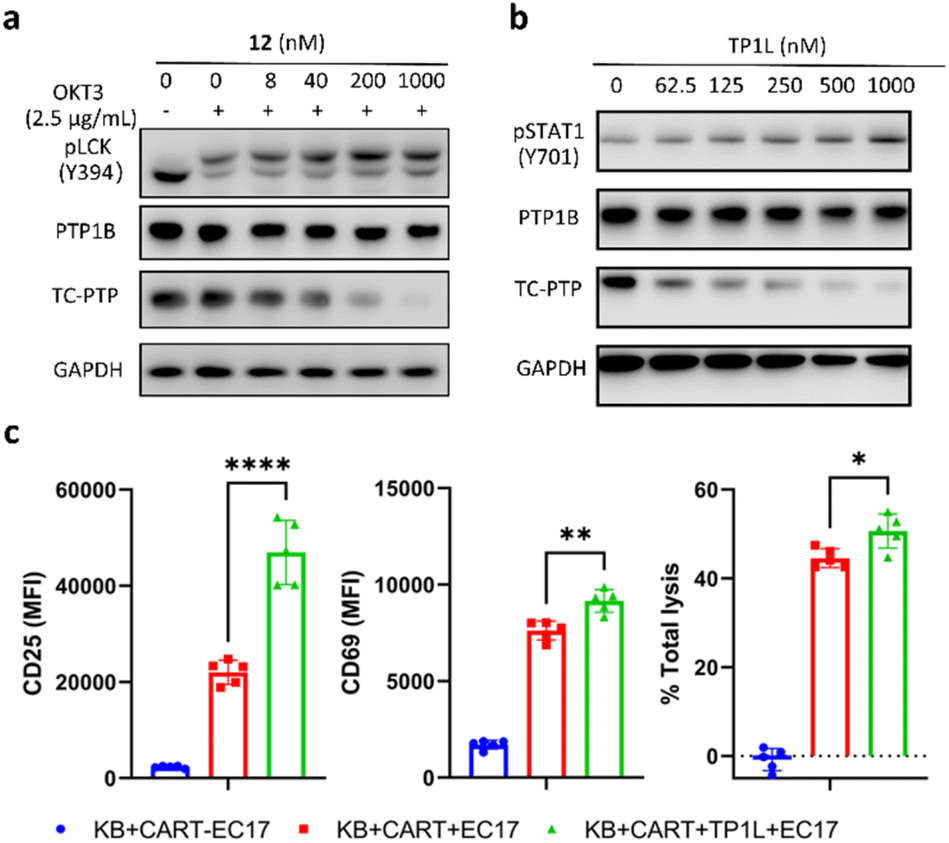 | ||
| Fig. 5 TP1L elevates pLCK in Jurkat T-cells and enhances CAR-T cells killing efficiency in a co-culture system. (a) Immunoblots of cell lysates showed that TP1L dose-dependently degrades TC-PTP and promotes phosphorylation of LCK in Jurkat T-cells. Cells were treated with TP1L at indicated concentrations for 16 hours and stimulated with 2.5 μg ml−1 anti-CD3 antibody for 10 min. (b) Immunoblots of cell lysates from KB cells treated with TP1L at indicated concentrations for 16 hours showing dose-dependent degradation of TC-PTP and elevation of pSTAT1. (c) Flow cytometry analysis of CD25 and CD69, and total lysis levels in cell lysates from KB and CAR-T cells with and without treatment of TP1L with the addition of bivalent molecule EC17. | ||
Previous studies have indicated that deletion of TC-PTP in T-cells promotes anti-tumor immunity and thus improves the efficacy of CAR-T cell therapy in solid tumors.16 To investigate the impact that degrading TC-PTP has on CAR-T cell therapy, we evaluate the effects of TP1L in an established epidermoid carcinoma KB tumor/CAR-T cell coculture system.41,42 The CAR-T cells were prepared as described in the previous reports (see ESI†).41,42 In this system, KB cells overexpress the folate receptor while the CAR-T-cells were engineered to recognize fluorescein. Once a FITC-folate bispecific adaptor small molecule (EC17) is added to the co-culture, the immunologic synapse will be formed in an EC17 dependent manner, therefore triggering CAR-T cell activation and subsequent tumor cell destruction. To first confirm whether TP1L mediates efficient TC-PTP degradation in KB cells, we treated KB cells with varying doses of TP1L for 16 hours and stimulated with human IFN-γ before harvesting the cells. Consistent with the previous results in HEK293 cells, TP1L treatment efficiently induced dose-dependent TC-PTP degradation and STAT1 phosphorylation, resulting in sensitization of KB cells to IFN-γ (Fig. 5b). To then evaluate the effect of TP1L mediated TC-PTP degradation on CAR-T cell activation, the early and late T-cell activation markers CD69 and CD25, respectively, were measured after co-culturing KB cells and CAR-T cells for 48 hours. As shown in Fig. 5c and S4L,† the presence of TP1L led to a 26% increase in the early activation marker CD69 and over 2-fold increase in the late activation marker CD25, thus indicating that TP1L-mediated TC-PTP degradation positively affects initial activation and significantly enhances persistence of CAR-T-cells. As a result of the enhanced CAR-T cell activation, the killing efficiency of the CAR-T cells were significantly boosted by the presence of TP1L as indicated by the total cell lysis level (Fig. 5c). Taken together, the results from the KB cell/CAR-T cell coculture system offer a promising application for selective TC-PTP degradation in cancer immunotherapy.
Conclusions
The remarkable clinical achievements of cancer immunotherapy indicate its potential to serve as the cornerstone for therapeutic treatments in a wide range of malignancies.43 Nonetheless, current approaches fail to attain consistent clinical responses in the majority of patients, highlighting the necessity for alternative immunotherapeutic strategies.44 Recent studies identified TC-PTP as a promising cancer immunotherapy target, as deletion of TC-PTP enhances tumor cell antigen presentation, T-cell and CAR-T cell functions, and in vivo antitumor immunity. However, due to the highly conserved and positively charged nature of PTP active sites, the development of inhibitors with sufficient selectivity and robust bioavailability has posed a persistent challenge for PTP-based drug discovery. In this study, we utilized PROTAC technology to convert an active-site-directed TC-PTP inhibitor into an eminently potent and selective TC-PTP degrader TP1L. This PROTAC compound specifically degrades TC-PTP in cells in an E3 ligase, ubiquitination, and proteasome dependent manner. TP1L amplifies IFN-γ signaling in cells through the degradation of TC-PTP and the subsequent elevation in pJAK1 and pSTAT1 levels, while JAK2, the substrate of the closely related PTP1B, is unaffected. In Jurkat T-cells, TP1L activates TCR signaling through the degradation of TC-PTP and thus the elevation of LCK phosphorylation. Notably, TP1L enhances the killing efficiency of CAR-T cells against KB tumor cells by boosting both the early and late T-cell activation. In conclusion, our study underscores the potential of targeting TC-PTP with a selective degrader as a novel therapeutic strategy in cancer immunotherapy. We anticipate that this unique chemical tool will enable in-depth investigations into the role of TC-PTP in various biological models and serve as a starting point for therapeutic development of novel cancer immunotherapeutic agents.Data availability
Supplemental figures and tables, further details of the experimental procedure, 1H and 13C NMR spectra, and original images of Western blots are available in the ESI.†Author contributions
J. M. and Z.-Y. Z. conceived the idea of the project. J. M., J. D., and Y. M. designed the experiments. J. M. and J. D. synthesized the PROTACs degraders. Y. M., J. D., Y. B., Z. Q., and A. A. M. performed evaluation and analysis of the degraders. B. A. J. conducted computational studies. Y. M., B. H., and J. L. performed the CAR-T cell experiments including data analysis. Z. Q. and Q. N. contributed to the preparation of key synthetic intermediates. P. S. L., and Z.-Y. Z. supervised the project. J. M. wrote the initial draft, and Z.-Y. Z. conducted reviewing and editing of the manuscript. All authors contributed to the finalization of the manuscript.Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
This work was supported in part by NIH RO1CA069202, NIH P30 CA023168, and the Robert C. and Charlotte Anderson Chair Endowment. The authors gratefully acknowledge the support of the Imaging Facility from the Institute for Cancer Research, and the Chemical Genomics Facility at the Institute for Drug Discovery.Notes and references
- M. M. Attwood, D. Fabbro, A. V. Sokolov, S. Knapp and H. B. Schiöth, Nat. Rev. Drug Discovery, 2021, 20, 839–861 CrossRef CAS PubMed
.
- A. D. Krabill and Z.-Y. Zhang, Biochem. Soc. Trans., 2021, 49, 1723–1734 CrossRef CAS PubMed
- B. Mosinger, U. Tillmann, H. Westphal and M. L. Tremblay, Proc. Natl. Acad. Sci. U.S.A., 1992, 89, 499–503 CrossRef CAS PubMed
- M. Kleppe, J. Soulier, V. Asnafi, N. Mentens, T. Hornakova, L. Knoops, S. Constantinescu, F. Sigaux, J. P. Meijerink, P. Vandenberghe, M. Tartaglia, R. Foa, E. Macintyre, T. Haferlach and J. Cools, Blood, 2011, 117, 7090–7098 CrossRef CAS PubMed
- P. D. Simoncic, A. Lee-Loy, D. L. Barber, M. L. Tremblay and C. J. McGlade, Curr. Biol., 2002, 12, 446–453 CrossRef CAS PubMed
- Y. Wang, S. Liu, T. Jia, Y. Feng, W. Zhang, X. Xu and D. Zhang, Front. Immunol., 2021, 12, 620333 CrossRef CAS PubMed
- D. Schmidt-Arras and F.-D. Böhmer, Trends Mol. Med., 2020, 26, 833–847 CrossRef CAS PubMed
- M. Kleppe, I. Lahortiga, T. El Chaar, K. De Keersmaecker, N. Mentens, C. Graux, K. Van Roosbroeck, A. A. Ferrando, A. W. Langerak, J. P. P. Meijerink, F. Sigaux, T. Haferlach, I. Wlodarska, P. Vandenberghe, J. Soulier and J. Cools, Nat. Genet., 2010, 42, 530–535 CrossRef CAS PubMed
- J. Ten Hoeve, M. De Jesus Ibarra-Sanchez, Y. Fu, W. Zhu, M. Tremblay, M. David and K. Shuai, Mol. Cell. Biol., 2002, 22, 5662–5668 CrossRef CAS PubMed
- C. Van Vliet, P. E. Bukczynska, M. A. Puryer, C. M. Sadek, B. J. Shields, M. L. Tremblay and T. Tiganis, Nat. Immunol., 2005, 6, 253–260 CrossRef CAS PubMed
- P. K. Goh, F. Wiede, M. N. Zeissig, K. L. Britt, S. Liang, T. Molloy, N. Goode, R. Xu, S. Loi, M. Muller, P. O. Humbert, C. McLean and T. Tiganis, Sci. Adv., 2022, 8, eabk3338 CrossRef CAS PubMed
- R. T. Manguso, H. W. Pope, M. D. Zimmer, F. D. Brown, K. B. Yates, B. C. Miller, N. B. Collins, K. Bi, M. W. LaFleur, V. R. Juneja, S. A. Weiss, J. Lo, D. E. Fisher, D. Miao, E. Van Allen, D. E. Root, A. H. Sharpe, J. G. Doench and W. N. Haining, Nature, 2017, 547, 413–418 CrossRef CAS PubMed
- F. Wiede, B. J. Shields, S. H. Chew, K. Kyparissoudis, C. van Vliet, S. Galic, M. L. Tremblay, S. M. Russell, D. I. Godfrey and T. Tiganis, J. Clin. Invest., 2011, 121, 4758–4774 CrossRef CAS
- M. W. LaFleur, T. H. Nguyen, M. A. Coxe, B. C. Miller, K. B. Yates, J. E. Gillis, D. R. Sen, E. F. Gaudiano, R. Al Abosy, G. J. Freeman, W. N. Haining and A. H. Sharpe, Nat. Immunol., 2019, 20, 1335–1347 CrossRef CAS PubMed
- M. Flosbach, S. G. Oberle, S. Scherer, J. Zecha, M. Von Hoesslin, F. Wiede, V. Chennupati, J. G. Cullen, M. List, J. K. Pauling, J. Baumbach, B. Kuster, T. Tiganis and D. Zehn, Cell Rep., 2020, 32, 107957 CrossRef CAS
- F. Wiede, K. Lu, X. Du, S. Liang, K. Hochheiser, G. T. Dodd, P. K. Goh, C. Kearney, D. Meyran, P. A. Beavis, M. A. Henderson, S. L. Park, J. Waithman, S. Zhang, Z. Zhang, J. Oliaro, T. Gebhardt, P. K. Darcy and T. Tiganis, EMBO J., 2020, 39, e103637 CrossRef CAS
- A. P. Combs, J. Med. Chem., 2010, 53, 2333–2344 CrossRef CAS
- R. He, Z. Yu, R. Zhang and Z. Zhang, Acta Pharmacol. Sin., 2014, 35, 1227–1246 CrossRef CAS
- Z.-Y. Zhang, Acc. Chem. Res., 2017, 50, 122–129 CrossRef CAS
- S. M. Stanford and N. Bottini, Trends Pharmacol. Sci., 2017, 38, 524–540 CrossRef CAS PubMed
- A. Iracheta-Vellve, H. Ebrahimi-Nik, T. R. Davis, K. E. Olander, S. Y. Kim, M. D. Yeary, J. C. Patti, I. C. Kohnle, C. K. Baumgartner, K. M. Hamel, K. A. McGuire, C. L. Chuong, Z. Xiong, E. P. Farney, J. M. Frost, M. Rees, A. Boghossian, M. Ronan, J. A. Roth, T. R. Golub, G. K. Griffin, C. Beauregard, P. R. Kym, K. B. Yates and R. T. Manguso, Cancer Res., 2022, 82, 606 CrossRef
- J. Dong, J. Miao, Y. Miao, Z. Qu, S. Zhang, P. Zhu, F. Wiede, B. A. Jassim, Y. Bai, Q. Nguyen, J. Lin, L. Chen, T. Tiganis, W. A. Tao and Z.-Y. Zhang, Angew. Chem., Int. Ed., 2023, e202303818 CAS
- J. N. Andersen, O. H. Mortensen, G. H. Peters, P. G. Drake, L. F. Iversen, O. H. Olsen, P. G. Jansen, H. S. Andersen, N. K. Tonks and N. P. H. Møller, Mol. Cell. Biol., 2001, 21, 7117–7136 CrossRef CAS PubMed
- T. Tiganis and A. M. Bennett, Biochem. J., 2007, 402, 1–15 CrossRef CAS PubMed
- K. M. Heinonen, A. Bourdeau, K. M. Doody and M. L. Tremblay, Proc. Natl. Acad. Sci. U.S.A., 2009, 106, 9368–9372 CrossRef CAS PubMed
- F. Wiede, K.-H. Lu, X. Du, M. N. Zeissig, R. Xu, P. K. Goh, C. E. Xirouchaki, S. J. Hogarth, S. Greatorex, K. Sek, R. J. Daly, P. A. Beavis, P. K. Darcy, N. K. Tonks and T. Tiganis, Cancer Discovery, 2022, 12, 752–773 CrossRef CAS PubMed
- A. Salmeen, J. N. Andersen, M. P. Myers, N. K. Tonks and D. Barford, Mol. Cell, 2000, 6, 1401–1412 CrossRef CAS
- Z.-Y. Zhang, G. T. Dodd and T. Tiganis, Trends Pharmacol. Sci., 2015, 36, 661–674 CrossRef CAS
- J. Olloquequi, A. Cano, E. Sanchez-López, M. Carrasco, E. Verdaguer, A. Fortuna, J. Folch, M. Bulló, C. Auladell, A. Camins and M. Ettcheto, Biomed. Pharmacother., 2022, 155, 113709 CrossRef CAS
- J. Pan, L. Zhou, C. Zhang, Q. Xu and Y. Sun, Signal Transduction Targeted Ther., 2022, 7, 177 CrossRef CAS
- S. M. Stanford and N. Bottini, Nat. Rev. Drug Discovery, 2023, 22, 273–294 CrossRef CAS
- S. Zhang, L. Chen, Y. Luo, A. Gunawan, D. S. Lawrence and Z.-Y. Zhang, J. Am. Chem. Soc., 2009, 131, 13072–13079 CrossRef CAS PubMed
- M. Békés, D. R. Langley and C. M. Crews, Nat. Rev. Drug Discovery, 2022, 21, 181–200 CrossRef PubMed
- K. Li and C. M. Crews, Chem. Soc. Rev., 2022, 51, 5214–5236 RSC
- R. G. Guenette, S. W. Yang, J. Min, B. Pei and P. R. Potts, Chem. Soc. Rev., 2022, 51, 5740–5756 RSC
- D. P. Bondeson, B. E. Smith, G. M. Burslem, A. D. Buhimschi, J. Hines, S. Jaime-Figueroa, J. Wang, B. D. Hamman, A. Ishchenko and C. M. Crews, Cell Chem. Biol., 2018, 25, 78–87.e5 CrossRef CAS PubMed
- M. Cieślak and M. Słowianek, Pharmaceutics, 2023, 15, 812 CrossRef PubMed
- A. Bricelj, C. Steinebach, R. Kuchta, M. Gütschow and I. Sosič, Front. Chem., 2021, 9, 707317 CrossRef CAS PubMed
- T. A. Bemis, J. J. La Clair and M. D. Burkart, J. Med. Chem., 2021, 64, 8042–8052 CrossRef CAS PubMed
- P. Castro-Sanchez, A. R. Teagle, S. Prade and R. Zamoyska, Front. Cell Dev. Biol., 2020, 8, 608747 CrossRef PubMed
- Y. G. Lee, H. Chu, Y. Lu, C. P. Leamon, M. Srinivasarao, K. S. Putt and P. S. Low, Nat. Commun., 2019, 10, 2681 CrossRef PubMed
- Y. G. Lee, I. Marks, M. Srinivasarao, A. K. Kanduluru, S. M. Mahalingam, X. Liu, H. Chu and P. S. Low, Cancer Res., 2019, 79, 387–396 CrossRef CAS PubMed
- A. D. Waldman, J. M. Fritz and M. J. Lenardo, Nat. Rev. Immunol., 2020, 20, 651–668 CrossRef CAS PubMed
- C. Peterson, N. Denlinger and Y. Yang, Cancers, 2022, 14, 3972 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Additional figures and data for degrader evaluation, material and methods, synthesis and characterization of compounds, and NMR spectra. See DOI: https://doi.org/10.1039/d3sc04541b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2023 |