
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRole and dynamics of transition metal carbides in methane coupling†
Seraphine B. X. Y.
Zhang
a,
Quentin
Pessemesse
b,
Lukas
Lätsch
a,
Konstantin M.
Engel
a,
Wendelin J.
Stark
 a,
Alexander P.
van Bavel
c,
Andrew D.
Horton
c,
Pierre-Adrien
Payard
*b and
Christophe
Copéret
*a
a,
Alexander P.
van Bavel
c,
Andrew D.
Horton
c,
Pierre-Adrien
Payard
*b and
Christophe
Copéret
*a
aDepartment of Chemistry and Applied Biosciences, ETH Zurich, Vladimir-Prelog-Weg 1–5, 8093 Zürich, Switzerland. E-mail: ccoperet@ethz.ch
bUniversité de Lyon, Université Claude Bernard Lyon I, CNRS, INSA, CPE, UMR 5246, ICBMS, 1 rue Victor Grignard, 69622 Lyon, France. E-mail: pierre-adrien.payard@univ-lyon1.fr
cShell Global Solutions International B. V., Grasweg 31, 1031 HW Amsterdam, Netherlands
First published on 3rd May 2023
Abstract
Transition metal carbides have numerous applications and are known to excel in terms of hardness, thermal stability and conductivity. In particular, the Pt-like behavior of Mo and W carbides has led to the popularization of metal carbides in catalysis, ranging from electrochemically-driven reactions to thermal methane coupling. Herein, we show the active participation of carbidic carbon in the formation of C2 products during methane coupling at high temperature that is associated with the dynamics of Mo and W carbides. A detailed mechanistic study reveals that the catalyst performance of these metal carbides can be traced back to its carbon diffusivity and exchange capability upon interaction with methane (gas phase carbon). A stable C2 selectivity over time on stream for Mo carbide (Mo2C) can be rationalized by fast carbon diffusion dynamics, while W carbide (WC) shows loss of selectivity due to slow diffusion leading to surface carbon depletion. This finding showcases that the bulk carbidic carbon of the catalyst plays a crucial role and that the metal carbide is not only responsible for methyl radical formation. Overall, this study evidences the presence of a carbon equivalent to the Mars–Van Krevelen type mechanism for non-oxidative coupling of methane.
Introduction
Transition metal carbides1 are ubiquitous in catalysis today, particularly in the context of C–H activation.2–8,28 While molybdenum and tungsten are prominent metals in state-of-the-art catalysts for oxidative and non-oxidative coupling of methane,9,10 MoOx/ZSM-5 has become the gold standard for methane coupling to aromatics.9,11 For this system a characteristic induction period along with particle agglomeration is observed which is rationalized by a transformation of the molybdenum oxide to the corresponding oxycarbide or carbide species which is presumed to resemble the active species in methane coupling.12,13,27,29,30 Yet, the mechanism for non-oxidative coupling of methane is still a matter of debate. High reaction temperatures, i.e. in the range of 800–1100 °C, suggest the involvement of radical pathways in the gas phase and the catalyst activity is often ascribed to methyl radical formation which then couple to C2 products downstream of the catalyst bed.14,15,31 Previous reports addressing the involvement of carbidic carbon in metal carbide catalysts16,17,32 prompted us to investigate the role of carbon closer in bulk Mo and W carbides (Mo2C and WC) by applying a 13C-labelling strategy combined with several spectroscopic techniques (XPS, ss-NMR, pXRD) and metadynamics simulations. Mo and W carbides were tested as catalysts for the non-oxidative coupling of methane (NOCM). Both materials enable C2 product formation at 1100 °C with 21% C2 selectivity at 23% CH4 conversion for Mo2C (4.7% C2 yield) and 32% C2 selectivity at 10% CH4 conversion for WC (3.2% C2 yield). Furthermore, this study shows that carbidic carbon participates in C2 product formation and interacts with methane (gas phase carbon). Differences in methane coupling activity could be rationalized by investigating surface composition with XPS and carbon diffusion coefficients by metadynamics at the DFT level. Following the analysis, we concluded fast C exchange dynamics for Mo2C and slow C diffusion for WC. The incorporation of carbon originating from methane was evidenced by ss-NMR of the spent materials with carbidic carbon NMR signatures at 274 ppm and 307 ppm for Mo2C and WC, respectively. Hence, this study reveals the presence of a carbon equivalent to the Mars–Van Krevelen mechanism in non-oxidative coupling of methane, which is typically encountered in oxidation processes with certain metal oxides.18,19Results and discussion
Synthesis and activity of transition metal carbides
Mo2C and WC were prepared via carburization at 1000 °C, and their catalytic activity in NOCM was investigated (Fig. 1). They were characterized with pXRD, XPS and TEM. Detailed synthesis procedures, characterization and catalytic test conditions for Mo2C and WC can be found in the ESI.†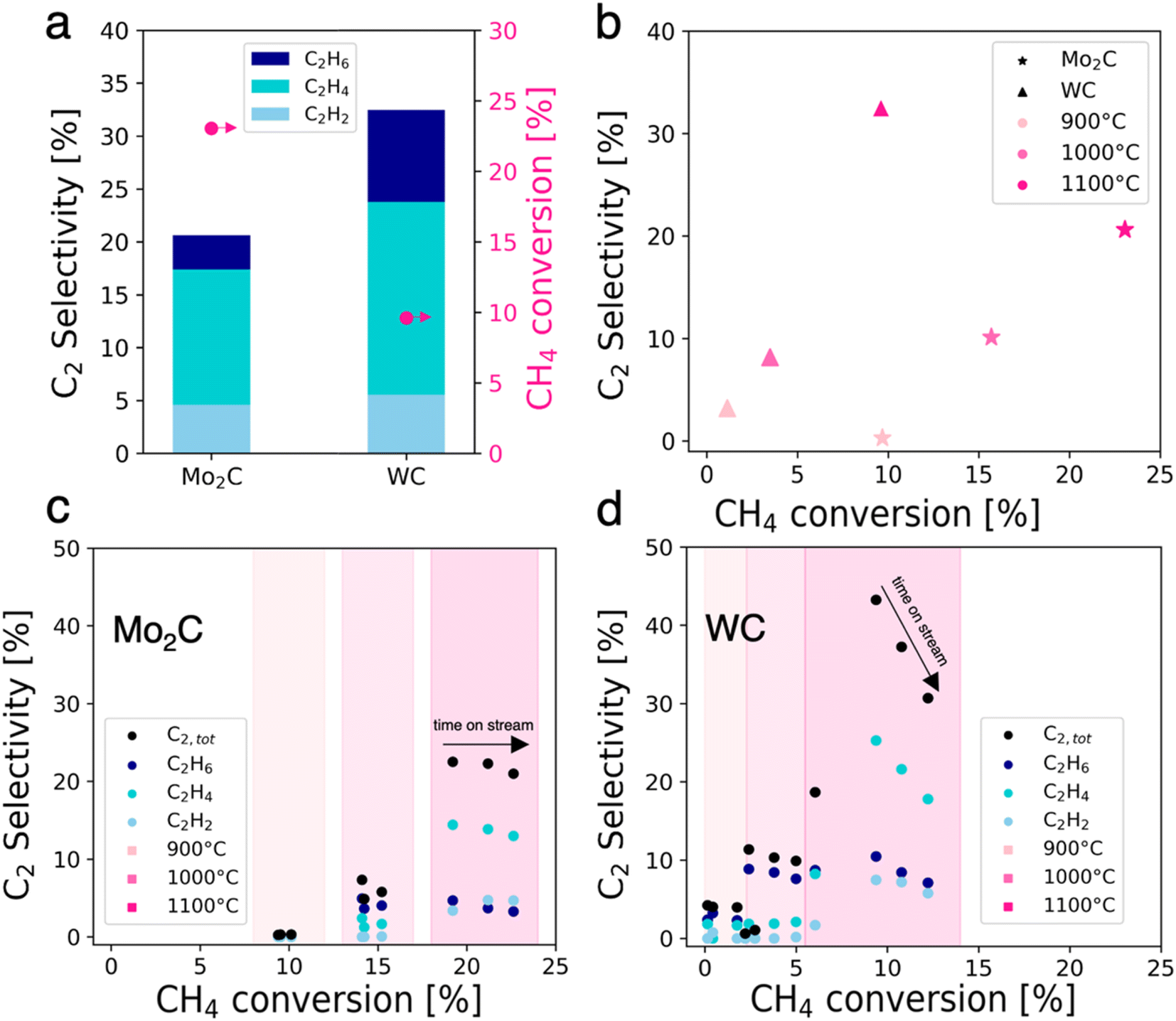 | ||
| Fig. 1 (a) Selectivity distribution for C2 species and CH4 conversion at 1100 °C with 10% CH4 in argon for Mo2C and WC (averaged over 1 h time on stream). (b) Effect of reaction temperatures on the C2 selectivity and CH4 conversion for the different catalysts. The color indicates the temperature and the form the catalyst. CH4 conversion vs. C2 selectivity at 900, 1000 and 1100 °C for C2 species over time on stream for (c) Mo2C and (d) WC; data points were taken every 15 min. | ||
Unless otherwise stated, all data sets are averages of 1-hour time on stream (TOS) at a given temperature. At 1100 °C, Mo2C and WC convert methaneto C2 products and coke with C2 yields of 4.7% for Mo2C and 3.2% for WC (Fig. 1a). For Mo2C, methane conversion increases with temperature: from 9.7% at 900 °C to values of up to 23.0% at 1100 °C with an average C2 selectivity of 20.6% which is stable over time on stream. In the case of WC, methane conversion reaches a maximum of 9.9% at 1100 °C with a C2 selectivity of 32.5%, while a loss of selectivity is observed over time on stream. Comparing both carbides, methane conversion is higher for Mo2C while the C2 selectivity is higher for WC. It is also noteworthy that at 900 °C WC already converts methane, albeit with a low C2 selectivity of 3.2% while no C2 products are detected for Mo2C. The best selectivity towards C2 products between the two catalysts was determined as 32.5% at 9.9% methane conversion for WC at 1100 °C, albeit the latter suffers from a fast deactivation.
In general, the highest conversion levels and C2 selectivities are obtained at high temperatures (Fig. 1b). Mo2C on the one hand exhibits stable selectivity at increasing conversion during time on stream at 1100 °C, forming coke and aromatics as major reaction byproducts (Fig. 1c). WC on the other hand experiences rapid loss of C2 selectivity over time on stream at 1100 °C along with increasing methane conversion (Fig. 1d).
Role of carbon in Mo and W carbides
The evolution of Mo2C and WC materials was furthermore investigated by comparing the materials before and after NOCM (3 h on stream at 900, 1000 and 1100 °C, 1 h at each temperature) using pXRD and XPS (Fig. 2). Based on pXRD, the bulk structures of Mo and W carbide remain unchanged for pristine and spent catalysts. XPS gives insight regarding differences in surface composition, namely carbon (surface) depletion for WC and structural retention for Mo2C. In the case of Mo2C, two components of the 3d5/2 peak are observed at binding energies of 228.6 and 229.4 eV associated with Mo2+ (Mo2C) and Mo4+ (MoC or MoO2), respectively.12,20,29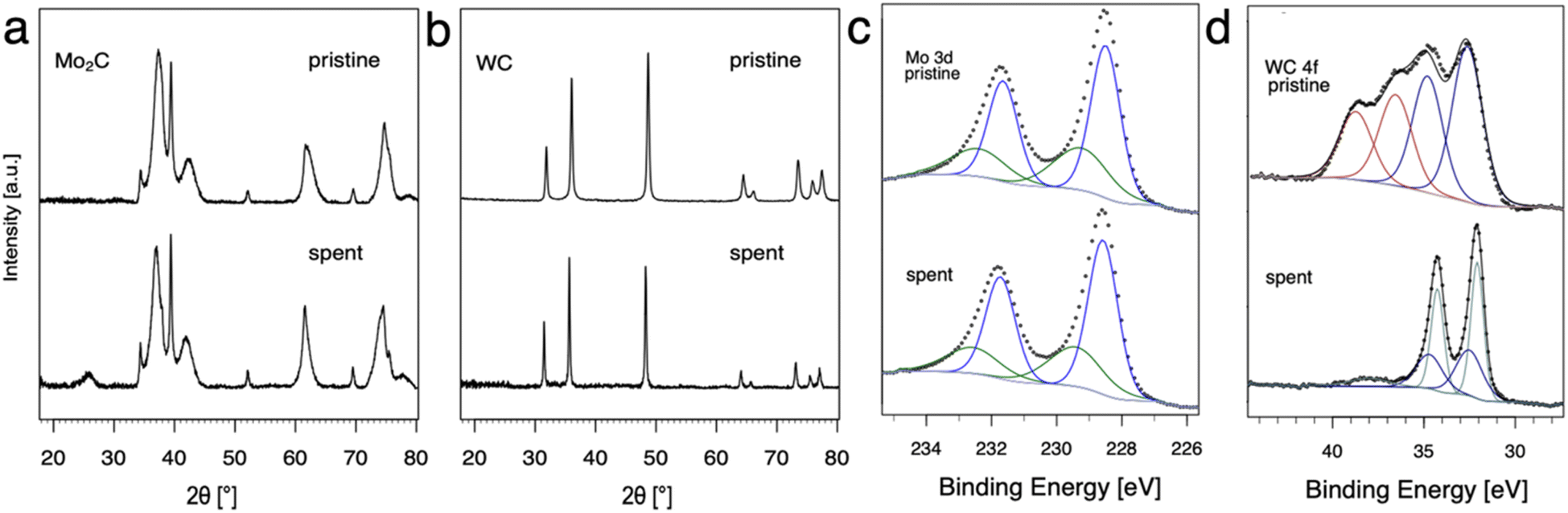 | ||
| Fig. 2 Structural transformations for pristine and spent catalyst materials are shown. pXRD for (a) Mo2C, (b) WC and XPS spectra of pristine and spent metal carbides: (c) Mo2C 3d, (d) WC 4f. The dots represent the recorded data and the solid lines show the results of a curve-fitting procedure. | ||
This finding remains unchanged considering pristine and spent catalyst surface. For WC, the XPS spectra (Fig. 3d) can be fitted as two multiplets that correspond to the W 4f7/2 and W 4f5/2 components which have a spin–orbit splitting of 2.2 eV. The WC (W4+) peaks were identified at 32.5 and 32.7 eV for the pristine and the spent catalyst, respectively. The W 4f7/2 peak at 36.6 eV was attributed to WO3. Pristine WC shows a mixture of WC (32.5 eV) and WO3 (36.6 eV) on the surface. While no WO3 was found in the spent WC, reduction to metallic W(0) with the 4f7/2 component at 32.1 eV was observed. For W(0) metal, a characteristic loss feature is reported, here it can be observed as a broad peak at 38 eV.21 Overall, XPS measurements show a change in composition for the surface from a mixture of WO3 and WC in the pristine catalyst to WC and W(0) in the spent catalyst hinting towards carbon depletion on the surface of WC with concomitant reduction to metallic W.
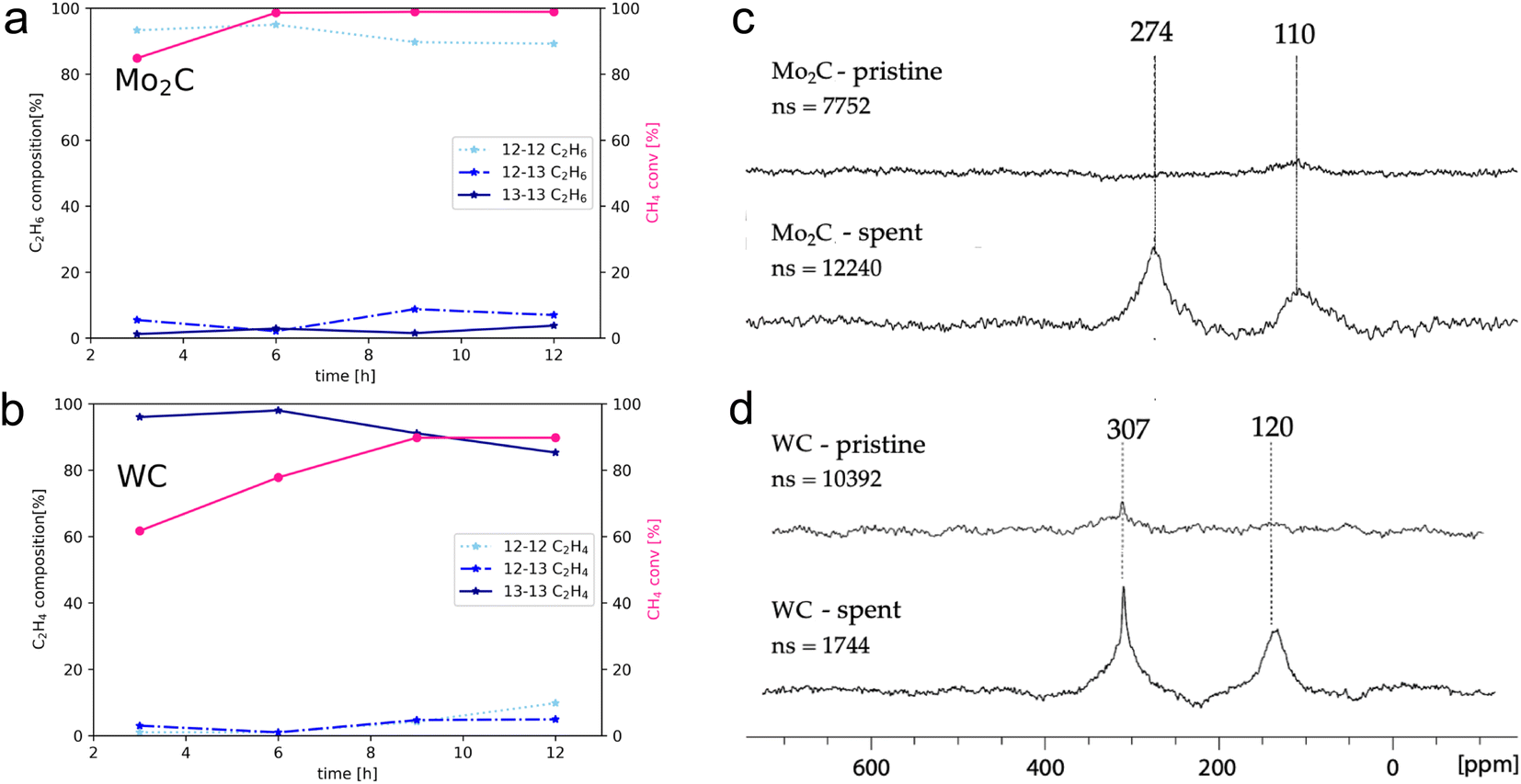 | ||
| Fig. 3 Left: Product distribution for dilabelled, monolabelled and non-labelled C2 products in case of Mo2C (a) and WC (b) as a function of contact time. Right: 13C MAS NMR characterization of pristine and spent (c) Mo2C and (d) WC. The catalyst was contacted with 200 mbar 13CH4 (at r.t.) in batch for 24 h at 1100 °C (molar ratio 7.2 WC: 1 13CH4 and 6.9 Mo2C: 1 13CH4). | ||
Intrigued by this finding, the reaction of 13CH4 with these materials was investigated in order to elucidate the role of surface C in transition metal carbides and to examine whether there is incorporation of the carbide surface C in the final products. 13CH4 labelling experiments were performed for Mo2C and WC in a batch reactor (see Fig. S10†). The reactor was filled with the respective metal carbide, 200 mbar of 13CH4 at r.t., heated to 1100 °C with a heating ramp of 400 °C h−1 and subsequent aliquots were taken after 3, 6, 9 and 12 h. Aliquots of the gas mixture are taken and analyzed with GCMS resulting in mass fragmentation patterns allowing to monitor the distribution of isotopomers among coupling products (non-, mono- and di-labelled C2 products) for ethane, ethylene and acetylene (see Table S2 and Fig. S7†).
The degree of carbidic 12C incorporation into C2 products was investigated by monitoring product evolution over the course of 12 h, with aliquots drawn every 3 h (Fig. 3a and b).
In the case of Mo2C, the isotopomer distribution is dominated by non-labelled C2 products, with ethane and ethylene as major gaseous products. The isotopomer distribution among ethane (Fig. 3a) is dominated by the non-labelled C2H6 (93%) after 3 hours, with mono- and di-labelled C2H6 being at 6% and 1%, respectively. The ratios of isotopomers change slightly with time; the amount of non-labelled C2H6 reaches 89% along with 7 and 4% of mono- and di-labelled isotopomers after 12 h. This finding suggests the incorporation of carbidic carbon in C2 products and C mobility in the Mo2C matrix. Considering that the surface and the bulk of Mo2C remains unchanged according to XPS and pXRD analysis, this labelling study further suggests fast C exchange with gas phase 13CH4 resulting in rapid reformation of the Mo2C and thus allows for maintaining a constant product selectivity. The exchanged carbidic C is replenished with 13C from the gas phase explaining the increase in labelled products over time.
For WC (Fig. 3b), ethylene is formed as major gaseous product under the same reaction conditions. However, in sharp contrast to Mo2C, the amount of di-labelled C2H4 isotopomer dominates (97%) for WC, with only a minor fraction of non- (1%) and mono-labelled (2%) C2H4 after 3 h. With increasing reaction time and conversion of 13CH4, the relative amount of 12C isotopomer increases with mono- and non-labelled isotopomers C2H4 reaching 14% and 9%, respectively after 12 h, indicating that WC can also act as a source of carbon, albeit much less efficiently than Mo2C. As discussed above, this process is accompanied by a reduction over time of WC to W(0) as evidenced by XPS (Fig. 2d), indicating a C depletion of the WC matrix. This change in catalyst state and the presence of metallic W offers a possible explanation for the loss of selectivity over time on stream with simultaneous increase in methane conversion. Considering the sharp contrast of reactivity patterns in these carbides (observed in the presence of labelled methane), one might pose the question whether Mo2C and WC can release carbon in the form of methane by interaction with H2 (10% H2 in Ar, 40 mL min−1, up to 1100 °C). As expected from their different carbon mobility (vide infra), methane evolution could only be observed for Mo2C (see Fig. S23†). In order to study 13C enrichment in the spent materials by exchange between the surface 12C of the carbide and the gas phase 13CH4, the materials were further examined by 13C MAS NMR (Fig. 3c and d). It should be noted that good quality spectra could only be obtained when diluting the sample with KBr. While hardly any signal is observed on pristine materials, in particular for Mo2C, intense signals are observed post-reaction, consistent with the exchange of carbon between the gas phase and the solid, thereby confirming enrichment of the spent catalyst with 13C. For both WC and Mo2C, two peaks are observed, corresponding to the carbidic carbon and sp2 carbon signals (graphitic carbon), the former appearing at 307 ppm for WC and at 274 ppm for Mo2C. The 13C shift of Mo2C agrees with previous reports,27 while the spectrum of WC has to date not been reported to the best of our knowledge.
One may note that the sp2 carbons have different chemical shifts for WC and Mo2C, indicating the presence of different types of carbons, possibly aromatic carbon deposit around 120 ppm vs. graphitic carbon around 110 ppm for WC and Mo2C, respectively.22,23 Summarizing these findings, the incorporation of surface/carbidic 12C from the carbide in the C2 products was shown for both Mo2C and WC, yet Mo2C exhibits a significantly higher C mobility of the carbide matrix than WC. The faster exchange/diffusion of carbidic carbon or the formation of methane from Mo2C are possible rationales for these contrasting isotopomer distributions.
Modelling diffusion of carbon in Mo2C and WC
In order to better understand the carbon exchange capability of molybdenum and tungsten carbides, the diffusion mechanism of carbon in the matrix at 1100 °C was explored using metadynamics at the DFT level (see the computational details for the method used).24,25,33–35 The diffusion of a single carbon atom through a periodic cell of the two most stable carbide steochiometries Mo2C and WC was investigated. The (001), (010) and (011) facets were considered to account for the anisotropy of the material; diffusion mechanisms and diffusion coefficients were similar in all cases so that only the (010) case will be discussed in the following, see ESI Section 7† for facets (001) and (011) and computational details. The cells consist of 6-atom layers with two fixed bottom layers. A potential bias was applied along selected collective variables, namely: the vertical position of the C atom (z), the C–C and C–M coordination numbers, Ncoord(C–C) and Ncoord(C–M) respectively, to accelerate diffusion. The free energy surfaces associated to z and Ncoord(C–C) were reconstructed by summing up the energy bias. The calculated free energy barrier (Fig. 4c, from z = 0.5 Å to z = 5.5 Å) to diffuse from the resting site to a higher lying site in Mo2C is ∼40 kcal mol−1, which corresponds to a diffusion coefficient of DMo2C1373K = 1.3 × 10−11 m2 s−1 at 1100 °C. This value suggests that carbon diffuses easily in the bulk at this temperature with an order of magnitude of the diffusion coefficient similar to the one obtained at ambient temperature for organic molecules in a liquid phase.26 The carbon diffusion in Mo2C takes place through migration of a C atom from a resting octahedral site (Oh) to vacant tetrahedral (Td) ones (Fig. 4a). As evidenced by the variation of the collective variables with time (Fig. 4b), two different tetrahedral sites are present in the crystal structure leading either to an isolated tetrahedral carbide (Ncoord(C–C) = 0, Fig. 4a, Td(C0)) or to the formation of a C2 moiety with an adjacent octahedral C neighbor (Ncoord(C–C) = 1, Fig. 4a, Td(C1)). In the latter, distortion of the crystal structure may lead to the formation of C2 defects with neighboring carbon sites, slowing down migration through these sites. As indicated by the free energy surface (FES, Fig. 4c, Ncoord(C–C) = 1), C2 defects are easily formed at this temperature and are likely to be involved in the subsequent formation of the coupling products. The contrasting behavior for WC reoccurs as the calculated diffusion of carbidic carbon in WC is significantly slower. The calculated free energy barrier for diffusion in WC (Fig. 4f between z = 0 Å and z = 4 Å) and the associated diffusion coefficient are ∼140 kcal mol−1 and DWC1373K = 7.5 × 10−30 m2 s−1, respectively.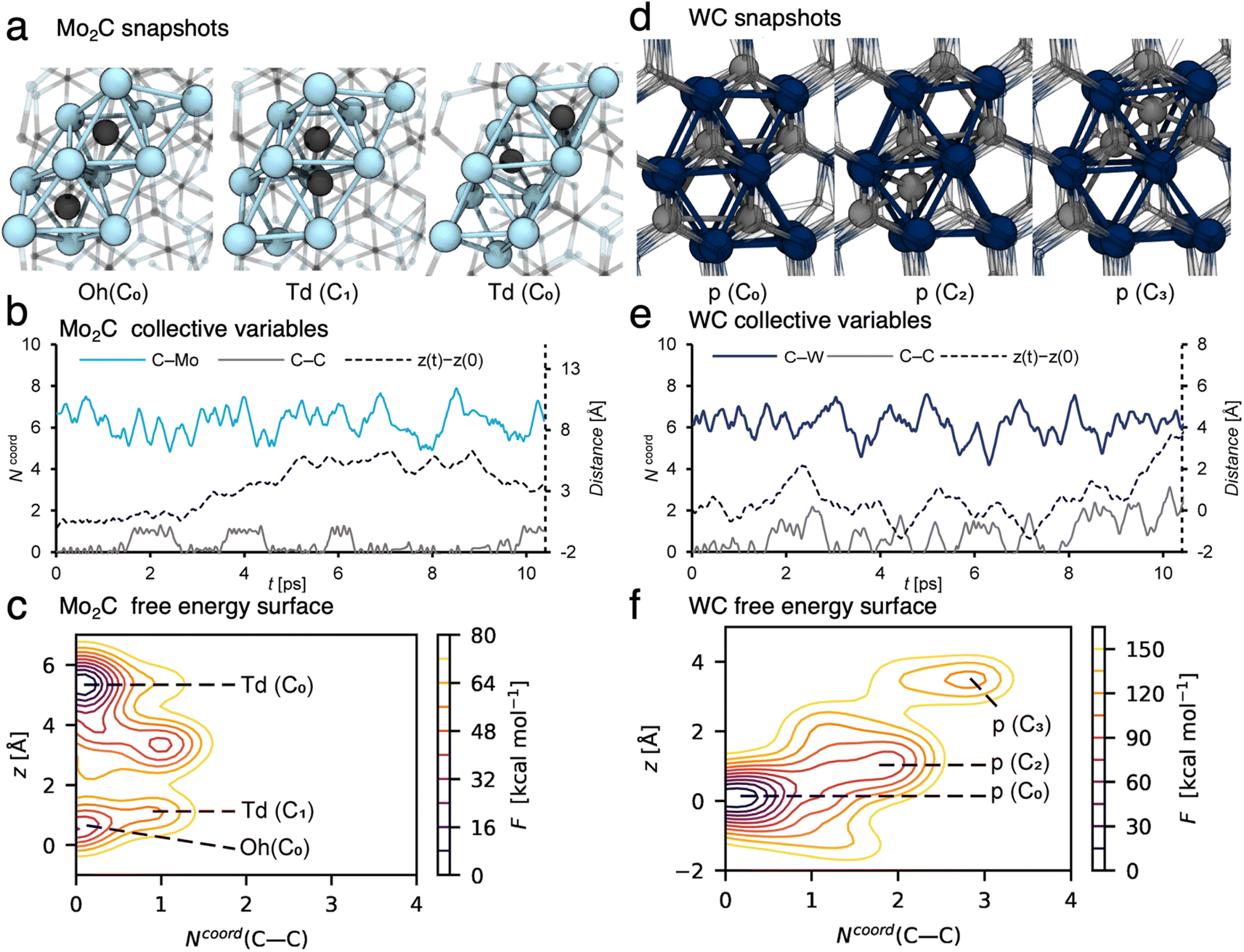 | ||
| Fig. 4 Successive sites involved in the diffusion of carbon towards the (010) facet in Mo2C ((a) octahedral Oh and tetrahedral Td sites with (C1) or without C–C interaction (C0)) and WC ((d) prismatic sites p with x C neighbors Cx). Biased collective variables as a function of the simulation time for the diffusion of carbon in Mo2C (b) and in WC (e). Free-energy surfaces as a function of the coordination number from C to C (Ncoord(C–C)) and distance along the z axis with respect to the initial position (z) for Mo2C (c) and WC (f). | ||
For WC, carbon has to move through surrounding unoccupied prismatic sites (Fig. 4d, p(C2) and p(C3)), and interacts with the carbon atoms occupying the neighboring prismatic sites (Fig. 4e, Ncoord(C–C) > 0), leading to the formation of transient Cx moieties (x = 3 and 4, Fig. 4e and f). The difference in calculated diffusion coefficients of around 20 orders of magnitude between Mo2C and WC is in agreement with the distribution of C2 products, which contain mainly non-labelled species originating from the 12C-bulk in the case of Mo2C, and mainly di-labelled species originating from the 13CH4 reactant gas in the case of WC. These results further confirm what was observed by XPS (vide supra). In WC, carbon diffusion is slow and carbon depletion is not compensated by incorporation of carbon from gas phase methane, thus leading to formation of metallic W(0). In the case of Mo2C, carbon easily diffuses through the bulk and is replenished rapidly at the surface as the spent catalyst's XPS signature resembles the pristine one.
Conclusions
In this study, the crucial role of carbidic carbon in the active catalytic materials was shown by (i) incorporation of carbidic carbon in C2 products, (ii) reduction of WC to W(0) leading to a loss of product selectivity, (iii) 13C exchange between gaseous methane and Mo/W carbide surfaces as evidenced by ss-NMR and (iv) computation of carbon diffusion coefficients in Mo and W carbides. Based on the incorporation of carbon from the bulk in the C2 products, a Mars–Van Krevelen type mechanism19 with participation of carbidic carbon of the metal carbide (instead of oxygen in a metal oxide) is most suited to rationalize our findings. This observation parallels what has been proposed in the Fischer-Tropsch process based on Fe carbide36 or for the methane aromatization on MoOx and WCx clusters on ZSM-5.17,37For Mo2C, fast carbon diffusion, as shown by labelling studies and computations, allows replenishment of the surface and a good catalytic performance (stable selectivity). In sharp contrast, under the same conditions, WC undergoes reduction of the surface species to W(0), consistent with the significantly lower fraction of carbidic carbon incorporated into the gaseous products and the slower calculated carbon diffusion, probably explaining the fast loss of C2 product selectivity.
For carbides with higher carbon diffusion coefficients like Mo2C, two carbidic carbons can recombine to form a C2 unit and desorb by combining with hydrogen atoms adsorbed on the surface; this would explain the absence of labelling of the hydrocarbon products at the initial stage, the labelling of the carbide without its reduction and the incorporation of 13C with increasing time.
With decreasing carbon mobility like for WC, the formation of products involves surface reactions, where C–H activation can form surface hydrocarbyls (alkyl, alkylidene and alkylidines) as previously proposed.38,39
CHx species on the surface can then recombine and desorb as C2 unit or can interact with a non-labelled surface C releasing a mono-labelled C2 product facilitated by Hads, as previously proposed.38,40,41 In the propagation of the catalytic process, the carbidic carbon on the surface can be regenerated by diffusion through the bulk or by interaction with methane in the gas phase. While above-described pathways might be more pronounced for certain metal carbides, it is most likely a combination thereof.
This study supports that a Mars–van Krevelen-like mechanism involving carbon atoms is at play, paralleling what is observed with oxygen and metal oxides in several oxidation reactions.16,27,32 While more insights in the mechanism of methane coupling are certainly needed to fully understand the product formation processes, this work shows that carbidic carbon and its dynamic behavior play a crucial role in methane coupling to higher hydrocarbon products; governing selectivity and conversion. It is further highlighted here that dynamics of metal carbides should be considered more broadly and integrated as design parameters in catalysis.
Data availability
All data are available upon request.Author contributions
S. B. X. Y. Z. performed all experiments and catalytic tests, measured pXRD, XPS, was involved in the development of the computational method and led the writing process. Q. P. performed metadynamics simulations under guidance of P.-A. P. L. L. measured NMR. K. E. synthesized WO3. All authors contributed to data interpretation and writing.Conflicts of interest
The authors declare no competing interests.Acknowledgements
We thank Shell Global Solution International B. V. for financial support. We thank Dennis Roelofszen (Shell) for consulting on technical details. Dr Nicolas Kaeffer (MPI CEC) and Dr Sourav Chatterjee (ETHZ) are acknowledged for fruitful discussions. Giovanni Cossu (ETHZ) is acknowledged for discussions and support regarding XPS. We thank the Mougel group (ETHZ) for access to XPS measurements. This work was carried out in part within the NCCR catalysis (financial support to the Stark group). P.-A. Payard and Q. Pessemesse are grateful to the Ecole Normale Supérieure de Lyon, Université Lyon 1 and the Region Auvergne Rhone Alpes for financial support and would like to thank Dr M.-E. L. Perrin (Univ Lyon) for precious advice and discussion. P.-A. P. and Q. P. are grateful to the CCIR of ICBMS, PSMN, GENCI-TGCC (Grants A0100812501 and A0120813435) for providing computational resources and technical support.Notes and references
- H. H. Hwu and J. G. Chen, Chem. Rev., 2005, 105, 185–212 CrossRef CAS PubMed.
- R. B. Levy and M. Boudart, Science, 1973, 181, 547–549 CrossRef CAS PubMed.
- Y. Deng, Y. Ge, M. Xu, Q. Yu, D. Xiao, S. Yao and D. Ma, Acc. Chem. Res., 2019, 52(12), 3372–3383 CrossRef CAS PubMed.
- B. Anasori, M. R. Lukatskaya and Y. Gogotsi, Nat. Rev. Mater., 2017, 2, 16098 CrossRef CAS.
- Z.-T. Xiong, L.-L. Chen, H.-B. Zhang, J.-L. Zeng and G.-D. Lin, Catal. Lett., 2001, 74, 227–232 CrossRef CAS.
- G. K. Dixit, M. Kumar, A. Katiyar, A. P. Jansen, A. P. van Bavel, R. Agrawal, P. M. Shenai and V. Srinivasan, Catal. Sci. Technol., 2021, 11, 7398–7411 RSC.
- C. Geng, T. Weiske, J. Li, S. Shaik and H. Schwarz, J. Am. Chem. Soc., 2019, 141, 599–610 CrossRef CAS PubMed.
- S. T. Hunt, M. Milina, A. C. Alba-Rubio, C. H. Hendon, J. A. Dumesic and Y. Roman-Leshkov, Science, 2016, 352, 974–978 CrossRef CAS PubMed.
- P. Schwach, X. Pan and X. Bao, Chem. Rev., 2017, 117, 8497–8520 CrossRef CAS PubMed.
- T. N. Nguyen, T. T. P. Nhat, K. Takimoto, A. Thakur, S. Nishimura, J. Ohyama, I. Miyazato, L. Takahashi, J. Fujima and K. Takahashi, ACS Catal., 2019, 10, 921–932 CrossRef.
- L. Wang, L. Tao, M. Xie, G. Xu, J. Huang and Y. Xu, Catal. Lett., 1993, 21, 35–41 CrossRef CAS.
- D. Wang, J. H. Lunsford and M. P. Rosynek, J. Catal., 1997, 169, 347–358 CrossRef CAS.
- W. Ding, S. Li, G. D. Meitzner and E. Iglesia, J. Phys. Chem. B, 2001, 105, 506–513 CrossRef CAS.
- T. V. Choudhary, E. Aksoylu and D. W. Goodman, Catal. Rev. - Sci. Eng., 2003, 45, 151–203 CrossRef CAS.
- A. Puente-Urbina, Z. Pan, V. Paunović, P. Šot, P. Hemberger and J. A. Bokhoven, Angew. Chem., Int. Ed., 2021, 60, 24002–24007 CrossRef CAS PubMed.
- C. Vogt, F. Meirer, M. Monai, E. Groeneveld, D. Ferri, R. A. van Santen, M. Nachtegaal, R. R. Unocic, A. I. Frenkel and B. M. Weckhuysen, Nat. Commun., 2021, 12, 1–10 CrossRef PubMed.
- I. Vollmer, B. Van Der Linden, S. Ould-Chikh, A. Aguilar-Tapia, I. Yarulina, E. Abou-Hamad, Y. G. Sneider, A. I. Olivos Suarez, J. L. Hazemann, F. Kapteijn and J. Gascon, Chem. Sci., 2018, 9, 4801–4807 RSC.
- C. Doornkamp and V. Ponec, J. Mol. Catal. A: Chem., 2000, 162, 19–32 CrossRef CAS.
- P. Mars and D. W. Van Krevelen, Chem. Eng. Sci., 1954, 3, 41–59 CrossRef CAS.
- F. Solymosi, J. Cserényi, A. Szöke, T. Bánsági and A. Oszkó, J. Catal., 1997, 165, 150–161 CrossRef CAS.
- C. Kalha, L. E. Ratcliff, J. G. Moreno, S. Mohr, M. Mantsinen, N. K. Fernando, P. K. Thakur, T.-L. Lee, H.-H. Tseng and T. S. Nunney, Phys. Rev. B, 2022, 105, 045129 CrossRef CAS.
- J. Yang, D. Ma, F. Deng, Q. Luo, M. Zhang, X. Bao and C. Ye, Chem. Commun., 2002, 8, 3046–3047 RSC.
- F. A. L. De Souza, A. R. Ambrozio, E. S. Souza, D. F. Cipriano, W. L. Scopel and J. C. C. Freitas, J. Phys. Chem. C, 2016, 120, 27707–27716 CrossRef CAS.
- A. Laio and M. Parrinello, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12562–12566 CrossRef CAS PubMed.
- G. Bussi and D. Branduardi, Rev. Comput. Chem., 2015, 28, 1–49 CAS.
- C. L. Yaws, in Transport Properties of Chemicals and Hydrocarbons, Elsevier, 2009, pp. 502–593 Search PubMed.
- I. Vollmer, I. Yarulina, F. Kapteijn and J. Gascon, ChemCatChem, 2019, 11, 39–52 CrossRef CAS.
- J. Gao, Y. Zheng, J.-M. Jehng, Y. Tang, I. E. Wachs and S. G. Podkolzin, Science, 2015, 348, 686–690 CrossRef CAS PubMed.
- K. Murugappan, E. M. Anderson, D. Teschner, T. E. Jones, K. Skorupska and Y. Román-Leshkov, Nat. Catal., 2018, 1, 960–967 CrossRef CAS.
- I. Vollmer, N. Kosinov, Á. Szécsényi, G. Li, I. Yarulina, E. Abou-Hamad, A. Gurinov, S. Ould-Chikh, A. Aguilar-Tapia, J.-L. Hazemann, E. Pidko, E. Hensen, F. Kapteijn and J. Gascon, J. Catal., 2019, 370, 321–331 CrossRef CAS.
- S. K. Kim, H. W. Kim, S. J. Han, S. W. Lee, J. Shin and Y. T. Kim, Commun. Chem., 2020, 3, 58 CrossRef CAS PubMed.
- N. K. Razdan and A. Bhan, J. Catal., 2020, 389, 667–676 CrossRef CAS.
- A. Barducci, G. Bussi and M. Parrinello, Phys. Rev. Lett., 2008, 100, 020603 CrossRef PubMed.
- A. Barducci, M. Bonomi and M. Parrinello, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2011, 1, 826–843 CrossRef CAS.
- O. Valsson, P. Tiwary and M. Parrinello, Annu. Rev. Phys. Chem., 2016, 67, 159–184 CrossRef CAS PubMed.
- V. V. Ordomsky, B. Legras, K. Cheng, S. Paul and A. Y. Khodakov, Catal. Sci. Technol., 2015, 5, 1433–1437 RSC.
- L. Q. Huang, Y. Z. Yuan, H. B. Zhang, Z. T. Xiong, J. L. Zeng and G. D. Lin, Stud. Surf. Sci. Catal., 2004, 147, 565–570 CrossRef CAS.
- T. Zhang, D. Holiharimanana, X. Yang and Q. Ge, J. Phys. Chem. C, 2020, 124(49), 26722–26729 CrossRef CAS.
- J. N. Carstens and A. T. Bell, J. Catal., 1996, 161, 423–429 CrossRef CAS.
- J. Eng, J. G. Chen, I. M. Abdelrehim and T. E. Madey, J. Phys. Chem. B, 1998, 102, 9687–9696 CrossRef CAS.
- M. Siaj, H. Oudghiri-Hassani, C. Maltais and P. H. McBreen, J. Phys. Chem. C, 2007, 111, 1725–1732 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc01054f |
| This journal is © The Royal Society of Chemistry 2023 |