
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCu-catalyzed enantioselective decarboxylative cyanation via the synergistic merger of photocatalysis and electrochemistry†
Yin
Yuan
a,
Junfeng
Yang
 *ab and
Junliang
Zhang
*a
*ab and
Junliang
Zhang
*a
aDepartment of Chemistry, Fudan University, 2005 Songhu Road, Shanghai, 200438, P. R. China. E-mail: junliangzhang@fudan.edu.cn
bFudan Zhangjiang Institute, Shanghai 201203, P. R. China
First published on 7th December 2022
Abstract
The development of an efficient and straightforward method for decarboxylative coupling using common alkyl carboxylic acid is of great value. However, decarboxylative coupling with nucleophiles always needs stoichiometric chemical oxidants or substrate prefunctionalization. Herein, we report a protocol for Cu-catalyzed enantioselective decarboxylative cyanation via the merger of photocatalysis and electrochemistry. CeCl3 and Cu/BOX were used as co-catalysts to promote the decarboxylation and cyanation, and both catalysts were regenerated via anodic oxidation. This method establishes a proof of concept enantioselective transformation via photoelectrocatalysis. Studies by DFT calculations provided mechanistic insight on enantioselectivity control.
Introduction
Carboxylic acids are widely available and fundamental chemical feedstocks which are produced in huge amounts.1 Many alkyl carboxylic acids exist in nature as biomass and in a renewable form (e.g. amino acid, fatty acid, peptides and sugar acids).2 In this regard, the abundance of alkyl carboxylic acids makes it an attractive precursor in carbon–carbon or carbon-heteroatom bond-formation processes through decarboxylation.3–5 Much effort has been devoted to decarboxylative coupling methods, either by an ionic pathway3,5 or single electro transfer (SET) pathway conceptually (Scheme 1a).4 The ionic pathway is promoted thermally by transition metals3 or organocatalysis,5 with most substrates being limited to electron-deficient substrates that facilitate anionic decarboxylation. Instead of generating an inion intermediator, the SET pathway involves the single-electro oxidation process to give alkyl radicals, either through a Hunsdiecker type process6 or photoredox-catalytic approach,4a,7 which were subsequently intercepted by radical acceptors. This approach is particularly useful in enantioselective carbon–carbon bond-forming reactions with electrophilic organohalides via a redox-neutral strategy.7e,7f However, enantioselective decarboxylative carbon-heteroatom bond-formation reactions always require an oxidizing agent,8 or the prefunctionalization of the carboxylic acids9 to facilitate the decarboxylative generation of alkyl radical via single electron reduction of carboxylic acid derivatives, such as redox-active esters.4a,7a Seminal work by G. Liu and coworkers has demonstrated the enantioselective cyanation of redox-active esters by Ir photoredox catalysis and Cu catalysis.10 However, this protocol increases the synthetic effort and deteriorates the atom economy. In this context, enantioselective decarboxylative coupling with nucleophiles directly through simple alkyl carboxylic acids remains elusive (Scheme 1b).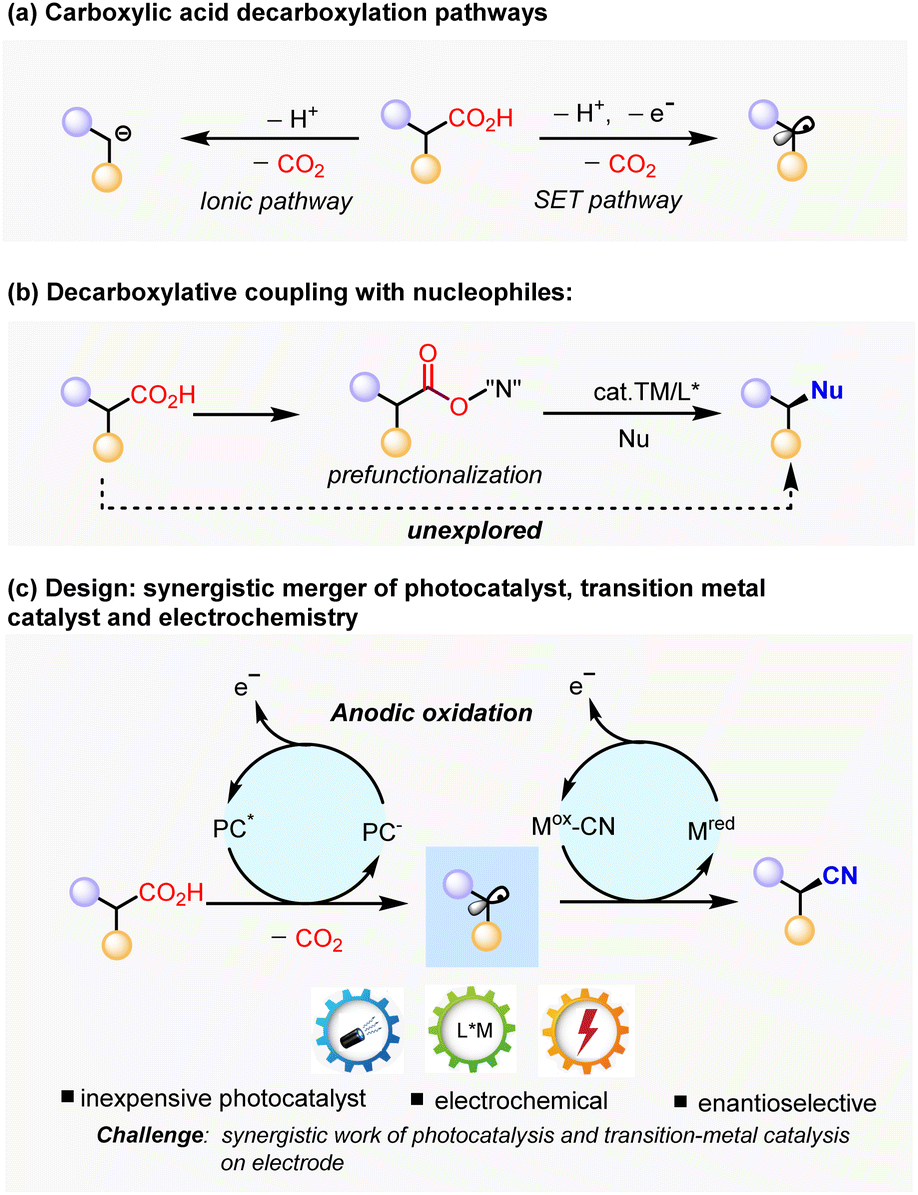 | ||
| Scheme 1 Background and project synopsis. | ||
Recently, electrochemistry has gained increasing attention as a sustainable approach for organic synthesis owing to its ability to generate highly reactive radical intermediates by SET on electrodes.11 In addition, the ability to control redox potential at the minimum level required for the desired redox transformation makes it an attractive approach of functionalizing complex synthetic intermediates. Therefore, we seek to use electrochemistry as a practical strategy to furnish the decarboxylation and regeneration of catalysis on electrodes. However, direct electrolysis of carboxylic acids (Kolbe electrolysis) suffers from poor selectivity, as the alkyl radical species tend to undergo dimerization or further oxidation to give alkyl cations, which would give a mixture of alkenes and racemic products.12 Inspired by the unique performance of CeCl3 as an efficient photocatalyst to promote decarboxylation13 and the good performance of Cu as a catalyst in radical coupling,14 as well as the similar redox potentials of CeCl3 and Cu(II) by anodic oxidization,15 we envision a synergistic approach to conduct enantioselective decarboxylative cyanation via the merger of photocatalysis and electrochemistry.16 If successful, the developed methodology would be highly valuable, because it avoids the usage of stoichiometric oxidatants, prefunctionalized substrates and noble photocatalysts. The main challenge in this scenario lies in the synergistic work of photochemistry, electrochemistry, and parallel generation of two catalytic species, in other words, the compatibility of photoredox-induced radical generation and electrochemically catalyst regeneration. Herein, we disclose copper catalyzed enantioselective decarboxylative cyanation via electrochemistry and photocatalysis (Scheme 1c).
Results and discussion
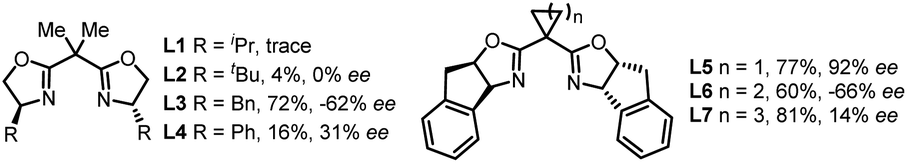
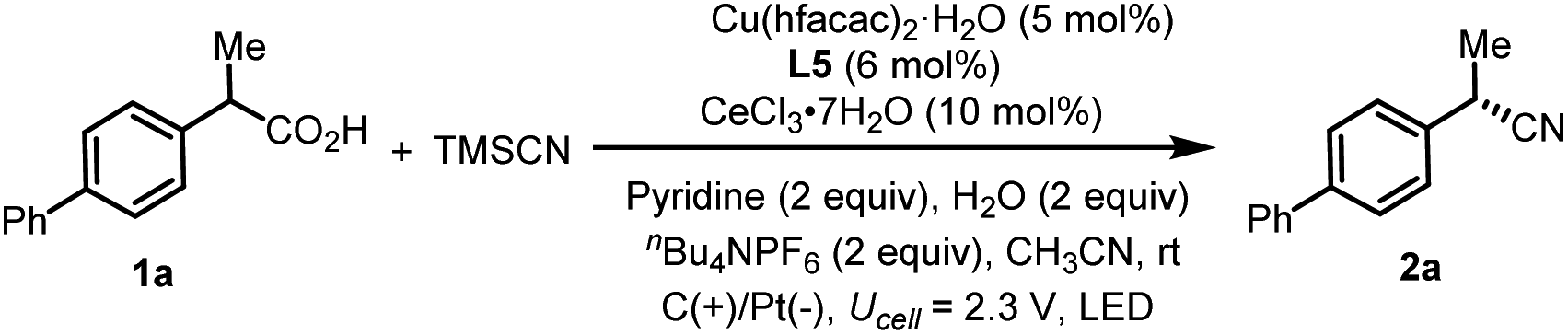
We commenced the enantioselective electrophotocatalytic decarboxylative cyanation using phenylpropanoic acids (1a) and TMSCN as starting materials (Table 1). A 465 nm blue LED was employed as the light source. Commercially inexpensive CeCl3·7H2O was found to be an efficient photocatalyst compared with other Ce sources (Table 1, entry 2). After extensive screening, the optimal reaction condition was established using 5 mol% of Cu(hfacac)2·H2O as a catalyst, 6 mol% of chiral bidentate BOX ligand (L5), 10 mol% of CeCl3·7H2O as a cocatalyst, 2.0 equiv. of nBu4NPF6 as supporting electrolyte, pyridine as the base and CH3CN as solvent in an undivided cell exposed to a 465 nm LED (40 W). Under the optimal conditions, the desired cyanation product 2a was isolated in 77% yield with 92% ee. A series of BOX ligands were examined, including those commonly used with the combination of Cu as Lewis acid or radical catalysis. L5 proved to be the best one, delivering the product with 92% ee. A DFT study disclosed that the non-covalent interaction between the indane structure of the ligand and the arene of the substrates is responsible for the high enantioselectivity (vide infra). The reaction is an overall oxidative transformation, which requires the proton as the electron scavenger. A batch of additives are examined as the electron scavenger and 2.0 equiv. of H2O proved to be beneficial for the yield and ee (Table 1, entry 7, see Table S4† for details). As for the electrode, replacing the Pt cathode with Ni (Table 1, entry 8) or other cathodes shows negative effects on the reaction efficiency. Moreover, further optimization revealed that the copper catalyst has a significant effect on the reactivity and Cu(hfacac)2·H2O gave the highest yield (Table 1, entry 10, see Table S5† for details). Conducting the reaction under air deteriorated the product formation (Table 1, entry 11). Control experiments indicated that light irradiation (Table 1, entry 12), electricity (Table 1, entry 13), the cerium catalyst (Table 1, entry 14) and Cu catalyst (Table 1, entry 15) were all critical for the reaction to proceed, as omission of any of these results in no desired decarboxylative cyanation product. Furthermore, lowering the Ce loading to 5 mol% results in decreased yield (Table 1, entry 15). This result highlights the importance of balancing the rates of the decarboxylation and cyanation process to ensure the optimal product selectivity.|
|
|||
|---|---|---|---|
| Entry | Deviation from the standard conditions | Yieldb [%] | eec [%] |
| a The reaction was performed using 0.2 mmol of 1a (0.05 M), 0.4 mmol TMSCN (0.1 M), Cu(hfacac)2·H2O (5 mol%), L5 (6 mol%), H2O (2 equiv.) nBu4NPF6 (0.4 mmol) and CH3CN (4 mL) in an undivided cell with a carbon felt anode and Pt cathode under constant cell potential conditions with irradiation for 6 h. b Yield was determined by GC using anisole as an internal standard. NR = no reaction. c Determined by HPLC using a chiral stationary phase. | |||
| 1 | None | 77 | 92 |
| 2 | Ce2SO4 (10 mol%) instead of CeCl3·7H2O | Trace | — |
| 3 | Cs2CO3 instead of pyridine | NR | — |
| 4 | Pyridine (50 mol%) was used | 36 | 92 |
| 5 | No pyridine | NR | — |
| 6 | DMF as solvent | Trace | — |
| 7 | No H2O | 73 | 90 |
| 8 | Ni foam (0.5 cm × 2 cm) was used as cathode | NR | — |
| 9 | U cell = 3.0 V | 58 | 89 |
| 10 | Cu(OTf)2 instead of Cu(hfacac)2·H2O | 45 | 90 |
| 11 | Under air | 43 | 91 |
| 12 | No LEDs | NR | — |
| 13 | No electricity | NR | — |
| 14 | No CeCl3·7H2O | Trace | — |
| 15 | No Cu | NR | — |
| 16 | CeCl3·7H2O (5 mol%) was used | 44 | 91 |
|
|||
With the optimized conditions in hand, we examined the scope of carboxylic acids in this reaction (Scheme 2). We were gratified to find that benzyl carboxylic acids featuring linear and branched aliphatic side chains were all suitable substrates for this reaction (2a–2d). Additionally, substituents such as terminal alkyne (2e), alkene (2f), cyanide (2g), and trifluoromethyl groups (2h) on the alkyl side chains were all tolerated. On the other hand, various functional groups of either electron-rich or -poor group on the phenyl rings of benzyl carboxylic acids were compatible under the current reaction conditions. In particular, functional groups that are susceptible to undergo oxidative degradation, such as electro-rich arenes (2m–2p) can be tolerated, delivering cyanation products in moderate to good yield with good enantioselectivity. Notably, halide groups, such as the chloride and bromide are well tolerated (2r–2t), which allows for further transformation by a cross-coupling reaction. Gratifyingly, commercially available racemic arylpropanoic acids, also known as nonsteroidal anti-inflammatory drugs, such as ibuprofen (1ab), naproxen (1ac), Ketoprofen (1ad) and flurbiprofen (1ae), were suitable substrates to deliver enantiomer enriched benzylic nitriles in good yields with good enantioselectivities (2ab–2ae). However, non-benzyl carboxylic acids are not competent substrates under the optimized conditions, probably due to the low stability of the radical intermediate.
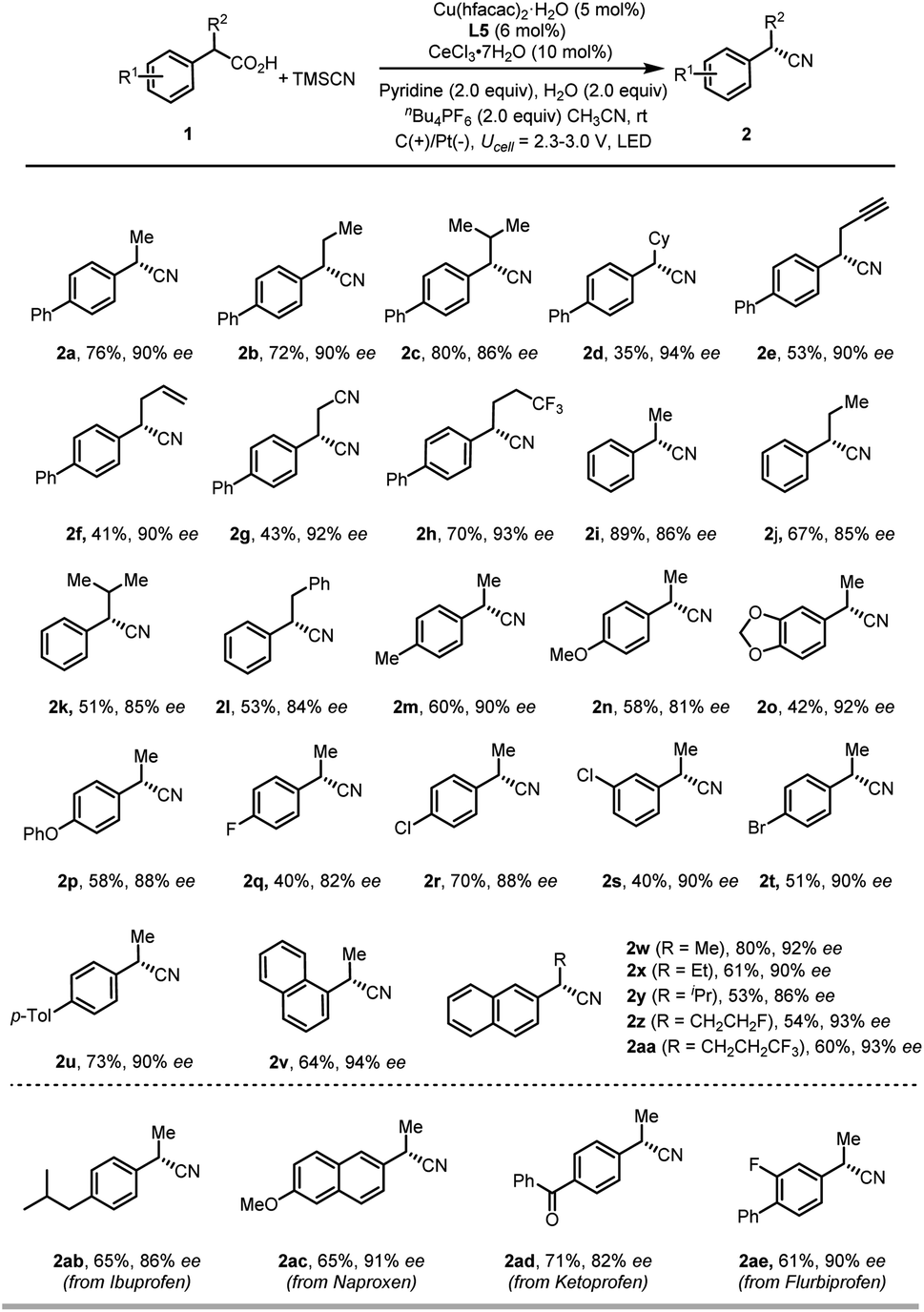 | ||
| Scheme 2 Scope of carboxylic acids. The reaction was performed on a 0.2 mmol scale under the conditions in Table 1, entry 1. | ||
To obtain experimental support for the mechanistic insight into this reaction, we conducted a few control experiments (Scheme 3). Firstly, when (S)-naproxen (ent-1ac) was used as the starting material, the same enantioselective product 2ac could be obtained (Scheme 3a). Meanwhile, no racemization of the starting material occurs under the reaction condition. This result revealed that the chiral ligand effectively controls the absolute configuration of the product, regardless of the stereochemistry of the electrophile, and that decarboxylation is essentially irreversible. Secondly, the reaction of radical clock substrate 3 under the standard conditions delivered a major ring-opening/cyanation product 4 in 32% yield with no detection of desired decarboxylative cyanation product (Scheme 3b). This result can serve as the direct evidence of the radical intermediate. Thirdly, when excessive water (6.0 equiv.) was added into the model reaction as the additive, we observed a high ratio of hydroxylation product with 18% ee (Scheme 3c), which is intriguing and provides the preliminary result for the promising enantioselective hydroxylation.
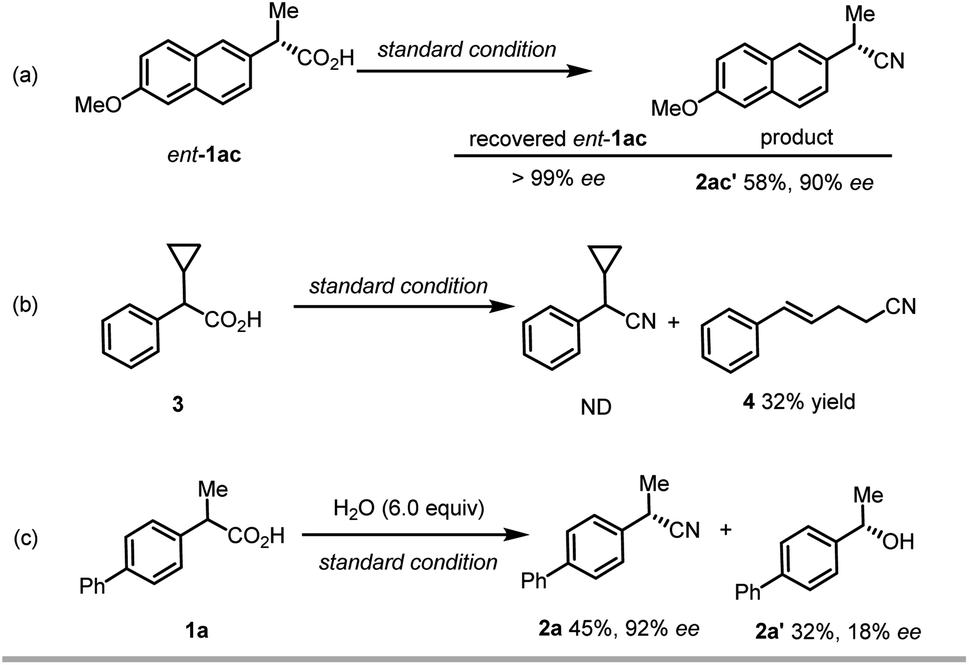 | ||
| Scheme 3 Mechanistic studies. | ||
Based on the above studies and related literature reports,10,15,17 a possible mechanism for the electrophotocatalytic decarboxylative cyanation was tentatively proposed (Scheme 4a). The reaction commences with the oxidation of Ce(III) to Ce(IV) (Ep/2red = 0.38 V vs. Ag/Ag+ in CH3CN) on the anode.13 The resulting Ce(IV) species coordinates with the carboxylic acid to give complex A, which then undergoes the photoinduced ligand-to-metal charge transfer (LMCT) to deliver the carboxylic radical B and the reduced Ce(III) species.18 The subsequent decarboxylation would generate the benzylic radical, which undergoes single-electron oxidative addition to the L*Cu(I)CN species, affording the alkyl-L*Cu(III)CN species. This event is followed by inner sphere reductive elimination to furnish the chiral cyanation product and regenerate the Cu(I) catalyst. Cu(I) species further undergoes anodic oxidation and combines with another CN− to give L*Cu(II)CN species. The possibility of the oxidation of Ce(III) by L*Cu(III)CN may be speculated. We measured the redox potentials of the different reaction species as shown in Scheme 4b using cyclic voltammetry (CV) data and found that the oxidation of the L*Cu(I)CN catalyst to the corresponding L*Cu(II)CN occurred at around 0.42 V (vs. Ag/Ag+). A visible drop of current is observed with the addition of CeCl3 (Scheme 4b), which may be attributed to the interaction between Ce(IV) and Cu(I). As a result, the oxidation of Cu(I) species by Ce(IV) cannot be excluded. It should also be noted that the complexation of Ce(III) with the carboxylic acid before the anode oxidation is highly possible based on the comparison of the CV data of CeCl3 and CeCl3 with 1a.
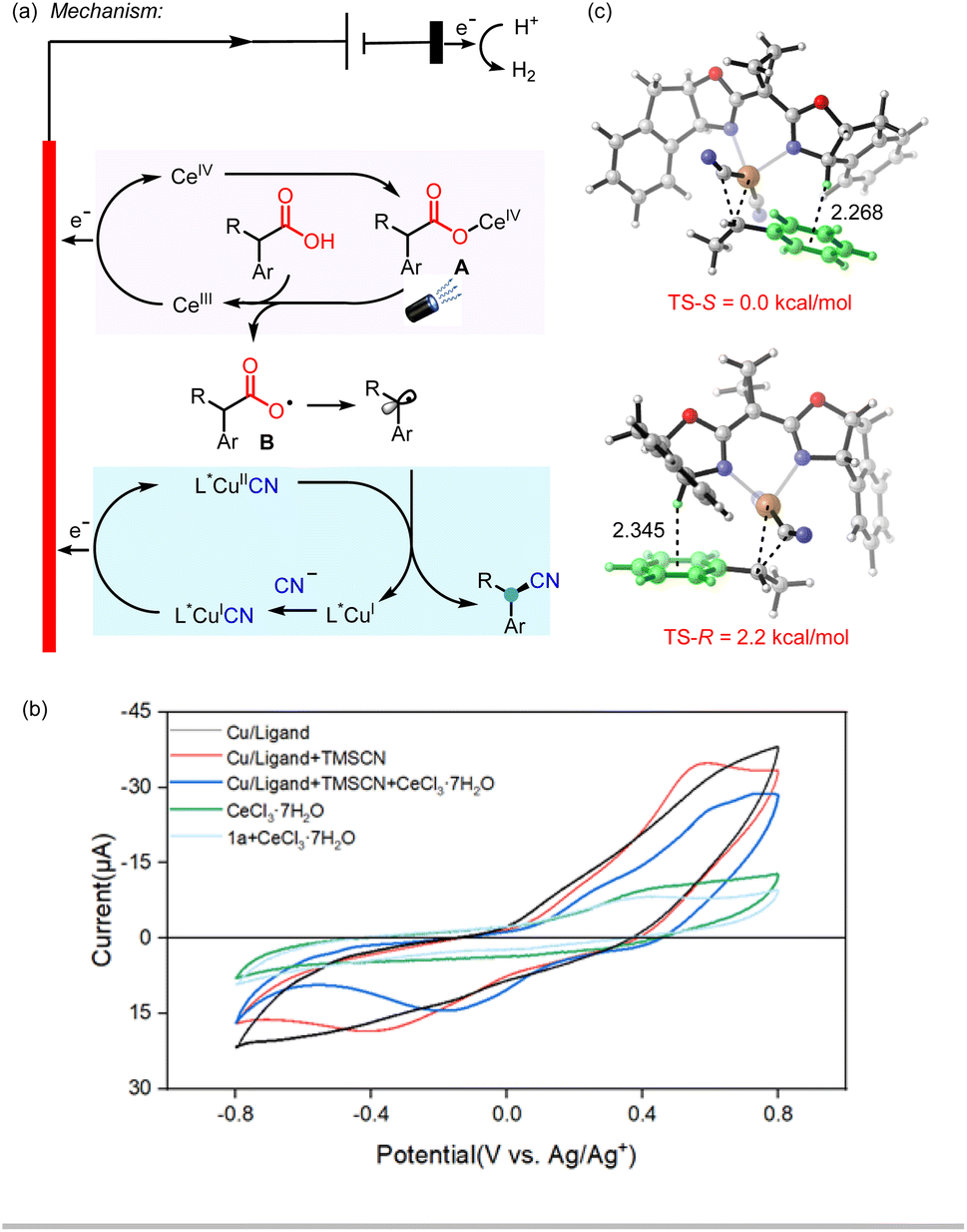 | ||
| Scheme 4 Proposed catalytic cycle, DFT study and cyclic voltammetry data. | ||
Even though the BOX ligand L5 has been extensively used in the Cu-catalyzed radical coupling reaction, the explanation of the stereo control of the ligand has not been well studied. A previous report has shown that the radical combination of a benzylic radical with L*Cu(III)CN is facile.17b Thus, we performed DFT calculations on the C–C reductive elimination to elucidate the role of the ligand in enantioselectivity. The calculations showed that the two transition states differ in free energy of activation by 2.2 kcal mol−1 favoring the S product, which is in good agreement with the experiments (Scheme 4c). In addition, the IGM analysis showed that C–H⋯π interactions between the benzylic proton of the ligand and the aryl group of the substrate plays a significant role in lowering the energy of the transition state (Scheme 4c, see the ESI† for details).15b This interaction could be further proved by our experimental observation that higher enantioselectivty could be obtained with a substrate containing more electro-rich aryl group. For example, substates with a para-phenyl group (1a) or naphthyl group (1v–1aa) afforded higher ee than the parent phenyl substrate (1i).
Conclusions
In summary, we have developed a Cu-catalyzed enantioselective decarboxylative cyanation reaction in a synergistic fashion with the merger of photocatalysis and electrochemistry, which allows the direct decarboxylation coupling of simple carboxylic acids.19 The current work demonstrates that this synergistic fashion could avoid the need for stoichiometric chemical oxidants or substrate prefunctionalization, in the meantime, enabling the reaction to operate under mild condition with high enantioselectivity and a reasonable range of functionalities. Further synthetic and mechanistic explorations of photoelectrocatalysis are currently underway.Data availability
All of the experimental and computational data is available and has been included in the ESI† of this publication.Author contributions
Y. Yuan conducted most of the reactions, experimental mechanistic studies and full characterization of the compounds. J. Yang conceptualized and directed the project, performed the DFT calculations and wrote the manuscript. All authors contributed to scientific discussions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the funding support of the National Key R&D Program of China (2021YFF0701600), NSFC (21901043, 21921003, and 22031004), STCSM (21ZR1445900) and Shanghai Municipal Education Commission (20212308).Notes and references
- A. J. J. Straathof, Chem. Rev., 2014, 114, 1871–1908 CrossRef CAS PubMed.
- E. Scott, F. Peter and J. Sanders, Appl. Microbiol. Biotechnol., 2007, 75, 751–762 CrossRef CAS PubMed.
- For recent reviews, see: (a) N. Rodríguez and L. J. Goossen, Chem. Soc. Rev., 2011, 40, 5030–5048 RSC; (b) J. D. Weaver, A. Recio iii, A. J. Grenning and J. A. Tunge, Chem. Rev., 2011, 111, 1846–1913 CrossRef CAS PubMed; (c) Y. Wei, P. Hu, M. Zhang and W. Su, Chem. Rev., 2017, 117, 8864–8907 CrossRef CAS PubMed; (d) P. J. Moon and R. J. Lundgren, ACS Catal., 2020, 10, 1742–1753 CrossRef CAS.
- For recent reviews, see: (a) J. Xuan, Z.-G. Zhang and W.-J. Xiao, Angew. Chem., Int. Ed., 2015, 54, 15632–15641 CrossRef CAS PubMed; (b) Y. Jin and H. Fu, Asian J. Org. Chem., 2017, 6, 368–385 CrossRef CAS; (c) Y. Li, L. Ge, M. T. Muhammad and H. Bao, Synthesis, 2017, 49, 5263–5284 CrossRef CAS; (d) T. Patra and D. Maiti, Chem.–Eur. J., 2017, 23, 7382–7401 CrossRef CAS PubMed; (e) J. Schwarz and B. König, Green Chem., 2018, 20, 323–361 RSC.
- S. Nakamura, Org. Biomol. Chem., 2014, 12, 394–405 RSC.
- (a) X. Liu, Z. Wang, X. Cheng and C. Li, J. Am. Chem. Soc., 2012, 134, 14330–14333 CrossRef CAS PubMed; (b) Z. Wang, L. Zhu, F. Yin, Z. Su, Z. Li and C. Li, J. Am. Chem. Soc., 2012, 134, 4258–4263 CrossRef CAS PubMed; (c) F. Yin, Z. Wang, Z. Li and C. Li, J. Am. Chem. Soc., 2012, 134, 10401–10404 CrossRef CAS PubMed; (d) C. Liu, X. Wang, Z. Li, L. Cui and C. Li, J. Am. Chem. Soc., 2015, 137, 9820–9823 CrossRef CAS PubMed; (e) X. Tan, Z. Liu, H. Shen, P. Zhang, Z. Zhang and C. Li, J. Am. Chem. Soc., 2017, 139, 12430–12433 CrossRef CAS PubMed.
- For reviews and leading references, see: (a) S. Murarka, Adv. Synth. Catal., 2018, 360, 1735–1753 CrossRef CAS; (b) A. Noble, R. S. Mega, D. Pflästerer, E. L. Myers and V. K. Aggarwal, Angew. Chem., Int. Ed., 2018, 57, 2155–2159 CrossRef CAS PubMed; (c) C. Shu, R. S. Mega, B. J. Andreassen, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2018, 57, 15430–15434 CrossRef CAS PubMed; (d) R. S. Mega, V. K. Duong, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2020, 59, 4375–4379 CrossRef CAS PubMed; (e) Z. Zuo, H. Cong, W. Li, J. Choi, G. C. Fu and D. W. C. MacMillan, J. Am. Chem. Soc., 2016, 138, 1832–1835 CrossRef CAS PubMed; (f) C. Pezzetta, D. Bonifazi and R. W. M. Davidson, Org. Lett., 2019, 21, 8957–8961 CrossRef CAS PubMed.
- G. a. Zhang, P. Huang, Z. Li, J. Guo, Y. Pei, M. Li, W. Ding and J. Wu, Cell Rep. Phys. Sci., 2022, 3, 101104 CrossRef CAS.
- Z. Zeng, A. Feceu, N. Sivendran and L. J. Gooßen, Adv. Synth. Catal., 2021, 363, 2678–2722 CrossRef CAS.
- D. Wang, N. Zhu, P. Chen, Z. Lin and G. Liu, J. Am. Chem. Soc., 2017, 139, 15632–15635 CrossRef CAS PubMed.
- For recent reviews see: (a) A. Jutand, Chem. Rev., 2008, 108, 2300–2347 CrossRef CAS PubMed; (b) J.-i. Yoshida, K. Kataoka, R. Horcajada and A. Nagaki, Chem. Rev., 2008, 108, 2265–2299 CrossRef CAS PubMed; (c) R. Francke and R. D. Little, Chem. Soc. Rev., 2014, 43, 2492–2521 RSC; (d) M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed; (e) M. D. Karkas, Chem. Soc. Rev., 2018, 47, 5786–5865 RSC; (f) K. Liu, C. Song and A. Lei, Org. Biomol. Chem., 2018, 16, 2375–2387 RSC; (g) S. Möhle, M. Zirbes, E. Rodrigo, T. Gieshoff, A. Wiebe and S. R. Waldvogel, Angew. Chem., Int. Ed., 2018, 57, 6018–6041 CrossRef PubMed; (h) C. Sandford, M. A. Edwards, K. J. Klunder, D. P. Hickey, M. Li, K. Barman, M. S. Sigman, H. S. White and S. D. Minteer, Chem. Sci., 2019, 10, 6404–6422 RSC; (i) H. Wang, X. Gao, Z. Lv, T. Abdelilah and A. Lei, Chem. Rev., 2019, 119, 6769–6787 CrossRef CAS PubMed; (j) C. Kingston, M. D. Palkowitz, Y. Takahira, J. C. Vantourout, B. K. Peters, Y. Kawamata and P. S. Baran, Acc. Chem. Res., 2020, 53, 72–83 CrossRef CAS PubMed; (k) D. Pollok and S. R. Waldvogel, Chem. Sci., 2020, 11, 12386–12400 RSC; (l) C. Ma, P. Fang, Z.-R. Liu, S.-S. Xu, K. Xu, X. Cheng, A. Lei, H.-C. Xu, C. Zeng and T.-S. Mei, Sci. Bull., 2021, 66, 2412–2429 CrossRef CAS; (m) S.-H. Shi, Y. Liang and N. Jiao, Chem. Rev., 2021, 121, 485–505 CrossRef CAS PubMed; (n) C. Zhu, N. W. J. Ang, T. H. Meyer, Y. Qiu and L. Ackermann, ACS Cent. Sci., 2021, 7, 415–431 CrossRef CAS PubMed.
- (a) H. Kolbe, Ann. Chem. Pharm., 1848, 64, 339–341 CrossRef; (b) H. Kolbe, Justus Liebigs Annalen der Chemie, 1849, 69, 257–294 CrossRef.
- (a) V. R. Yatham, P. Bellotti and B. König, Chem. Commun., 2019, 55, 3489–3492 RSC; (b) L. McMurray, T. M. McGuire and R. L. Howells, Synthesis, 2020, 52, 1719–1737 CrossRef CAS.
- (a) N. Fu, G. S. Sauer, A. Saha, A. Loo and S. Lin, Science, 2017, 357, 575–579 CrossRef CAS PubMed; (b) F. Wang, P. Chen and G. Liu, Acc. Chem. Res., 2018, 51, 2036–2046 CrossRef CAS PubMed; (c) X. Y. Dong, Y. F. Zhang, C. L. Ma, Q. S. Gu, F. L. Wang, Z. L. Li, S. P. Jiang and X. Y. Liu, Nat. Chem., 2019, 11, 1158–1166 CrossRef CAS PubMed.
- (a) N. Fu, L. Song, J. Liu, Y. Shen, J. C. Siu and S. Lin, J. Am. Chem. Soc., 2019, 141, 14480–14485 CrossRef CAS PubMed; (b) L. Song, N. Fu, B. G. Ernst, W. H. Lee, M. O. Frederick, R. A. DiStasio Jr and S. Lin, Nat. Chem., 2020, 12, 747–754 CrossRef CAS PubMed.
- (a) H. Huang, Z. M. Strater, M. Rauch, J. Shee, T. J. Sisto, C. Nuckolls and T. H. Lambert, Angew. Chem., Int. Ed., 2019, 58, 13318–13322 CrossRef CAS PubMed; (b) F. Wang and S. S. Stahl, Angew. Chem., Int. Ed., 2019, 58, 6385–6390 CrossRef CAS PubMed; (c) J. P. Barham and B. Konig, Angew. Chem., Int. Ed., 2020, 59, 11732–11747 CrossRef CAS PubMed; (d) H. Huang, Z. M. Strater and T. H. Lambert, J. Am. Chem. Soc., 2020, 142, 1698–1703 CrossRef CAS PubMed; (e) H. Kim, H. Kim, T. H. Lambert and S. Lin, J. Am. Chem. Soc., 2020, 142, 2087–2092 CrossRef CAS PubMed; (f) X. L. Lai, X. M. Shu, J. Song and H. C. Xu, Angew. Chem., Int. Ed., 2020, 59, 10626–10632 CrossRef CAS PubMed; (g) L. Niu, C. Jiang, Y. Liang, D. Liu, F. Bu, R. Shi, H. Chen, A. D. Chowdhury and A. Lei, J. Am. Chem. Soc., 2020, 142, 17693–17702 CrossRef CAS PubMed; (h) P. Xu, P. Y. Chen and H. C. Xu, Angew. Chem., Int. Ed., 2020, 59, 14275–14280 CrossRef CAS PubMed; (i) W. Zhang, K. L. Carpenter and S. Lin, Angew. Chem., Int. Ed., 2020, 59, 409–417 CrossRef CAS PubMed.
- (a) D. Wang, F. Wang, P. Chen, Z. Lin and G. Liu, Angew. Chem., Int. Ed., 2017, 56, 2054–2058 CrossRef CAS PubMed; (b) W. Zhang, F. Wang, S. D. McCann, D. Wang, P. Chen, S. S. Stahl and G. Liu, Science, 2016, 353, 1014–1018 CrossRef CAS PubMed; (c) J. Li, Z. Zhang, L. Wu, W. Zhang, P. Chen, Z. Lin and G. Liu, Nature, 2019, 574, 516–521 CrossRef CAS PubMed; (d) G. Zhang, S. Zhou, L. Fu, P. Chen, Y. Li, J. Zou and G. Liu, Angew. Chem., Int. Ed., 2020, 59, 20439–20444 CrossRef CAS PubMed.
- (a) A. Hu, J. J. Guo, H. Pan and Z. Zuo, Science, 2018, 361, 668–672 CrossRef CAS PubMed; (b) R. Zhao and L. Shi, Org. Chem. Front., 2018, 5, 3018–3021 RSC; (c) S. Shirase, S. Tamaki, K. Shinohara, K. Hirosawa, H. Tsurugi, T. Satoh and K. Mashima, J. Am. Chem. Soc., 2020, 142, 5668–5675 CrossRef CAS PubMed.
- During peer review of the manuscript, a similar work was reported by Song and Xu, X.-L. Lai, M. Chen, Y. Wang, J. Song and H.-C. Xu, J. Am. Chem. Soc., 2022, 144, 20201–20206 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc05428k |
| This journal is © The Royal Society of Chemistry 2023 |