
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTandem electrocatalytic CO2 reduction with Fe-porphyrins and Cu nanocubes enhances ethylene production†
Min
Wang‡
a,
Vasilis
Nikolaou‡
 b,
Anna
Loiudice
ac,
Ian D.
Sharp
c,
Antoni
Llobet
*bd and
Raffaella
Buonsanti
*a
b,
Anna
Loiudice
ac,
Ian D.
Sharp
c,
Antoni
Llobet
*bd and
Raffaella
Buonsanti
*a
aLaboratory of Nanochemistry for Energy (LNCE), Institute of Chemical Sciences and Engineering (ISIC), École Polytechnique Fédérale de Lausanne, CH-1950 Sion, Switzerland. E-mail: raffaella.buonsanti@epfl.ch
bInstitute of Chemical Research of Catalonia (ICIQ), Barcelona Institute of Science and Technology (BIST), 43007, Tarragona, Spain. E-mail: allobet@iciq.cat
cWalter Schottky Institute and Physics Department, Technische Universität München, Am Coulombwall 4, 85748 Garching, Germany
dDepartament de Química, Universitat Autònoma de Barcelona (UAB), Cerdanyola del Vallès, 08193 Barcelona, Spain
First published on 18th October 2022
Abstract
Copper-based tandem schemes have emerged as promising strategies to promote the formation of multi-carbon products in the electrocatalytic CO2 reduction reaction. In such approaches, the CO-generating component of the tandem catalyst increases the local concentration of CO and thereby enhances the intrinsic carbon–carbon (C–C) coupling on copper. However, the optimal characteristics of the CO-generating catalyst for maximizing the C2 production are currently unknown. In this work, we developed tunable tandem catalysts comprising iron porphyrin (Fe-Por), as the CO-generating component, and Cu nanocubes (Cucub) to understand how the turnover frequency for CO (TOFCO) of the molecular catalysts impacts the C–C coupling on the Cu surface. First, we tuned the TOFCO of the Fe-Por by varying the number of orbitals involved in the π-system. Then, we coupled these molecular catalysts with the Cucub and assessed the current densities and faradaic efficiencies. We discovered that all of the designed Fe-Por boost ethylene production. The most efficient Cucub/Fe-Por tandem catalyst was the one including the Fe-Por with the highest TOFCO and exhibited a nearly 22-fold increase in the ethylene selectivity and 100 mV positive shift of the onset potential with respect to the pristine Cucub. These results reveal that coupling the TOFCO tunability of molecular catalysts with copper nanocatalysts opens up new possibilities towards the development of Cu-based catalysts with enhanced selectivity for multi-carbon product generation at low overpotential.
Introduction
The electrochemical CO2 reduction reaction (CO2RR) offers the opportunity to convert CO2 into fuels and chemical feedstocks, while mitigating anthropogenic CO2 emissions and storing renewable energy.1,2 Multi-carbon (C2+) compounds (e.g., ethylene, ethanol, propanol) are among the most attractive CO2RR products owing to their commercial value and high energy densities.1,2 However, optimizing the activity of CO2RR electrocatalysts towards one particular product remains a crucial challenge that is not trivial to address because multiple intermediates are involved in the CO2 to C2+ conversion pathway.3–5Cu-based materials have been widely employed as CO2RR catalysts due to their unique ability to facilitate the electroreduction of CO2 beyond two electron reduction products.5,6 Numerous studies on catalyst development and mechanistic investigations have demonstrated that the surface coverage of the adsorbed CO (indicated as *CO) regulates the activation barrier of the C–C coupling step and, thus, the formation of C2+ products.7–9 An attractive approach to realize this high *CO surface coverage is via tandem catalysis, wherein Cu is coupled with a CO-producing component to realize the sequential CO2-to-CO and CO-to-C2+ product conversion on complementary active sites that are coupled at the nanometer scale.10–21 However, the correlation between the intrinsic catalytic properties of the CO-generating component (e.g. turnover frequency, TOF, and overpotential) and its impact on the outcome of tandem catalysis (e.g. C–C coupling efficiency) is still critically missing. Thus, the fundamental design principles required to rationally promote C2+ production while decreasing the overpotential in tandem catalysis remain to be elucidated.
Bimetallic systems comprising Cu and another metal (Au, Ag, or Zn) as the CO-producing component have been extensively investigated as electrocatalysts in tandem CO2 reduction.15–21 One drawback of these catalysts is that composition (e.g. alloying) and morphology can change during operation.15–18 These changes affect both the physical and electronic structure, thus complicating the interpretation of the catalytic behavior and delaying the establishment of design principles required to further improve the catalytic activity of these systems.18–22 Additionally, tuning the intrinsic activity and efficiency of metals is difficult and requires their coupling with organic modifiers.23,24 Consequently, these systems are not ideal for guiding the understanding of the performance-determining factors in tandem catalysis.
Recent studies have suggested that CO-producing molecular catalysts are alternative candidates to metals in tandem schemes.25–27 Molecular components offer additional versatility compared to metals because their electronic and structural features can be tuned via synthetic modifications.28–31 While the examples are still limited, studies in the literature on tandem catalysts including CO-producing molecular catalysts and Cu are encouraging.25–27 For example, the addition of either an iron porphyrin or a cobalt phthalocyanine to sputtered Cu resulted in enhanced C2+ product yields.25,26 Specifically, the coupling of iron porphyrin with sputtered Cu resulted in increased faradaic efficiency for ethanol from 29% to 41% and in a shift the detection potential of this product from −0.84 to −0.82 V vs. RHE.25 The cobalt phthalocyanine/Cu catalyst enhanced the faradaic efficiency for C2+ products from 45% to 82% in a highly basic electrolyte.26 Instead, the combination of a cobalt phthalocyanine with a Zn–N–C catalyst promoted formation of methane with a total faradaic efficiency of 18%, a product not observed on the pristine Zn–N–C, again in a highly basic electrolyte.27 However, these studies have so far focused only on the integration of commercial molecular catalysts in tandem CO2RR, thus not profiting from the chemical tunability of these systems. Furthermore, they remain a few isolated examples each utilizing a different electrocatalyst and with testing performed under different conditions, which make any comparison among them difficult to make.
Herein, we exploit the intrinsic tunability of molecular catalysts (in terms of TOFCO and overpotential) to understand the impact of the CO-generating component on subsequent C–C coupling on Cu surfaces during tandem catalysis. Iron porphyrin (F-Por) molecular catalysts are selected as the CO-producing entity because of their high efficiency for CO2 to CO conversion at neutral pH when immobilized on conductive substrates.32–34 Furthermore, their versatile chemistry enables facile tuning of their redox behaviors via modification of the π-system.35–37 To this end, we synthesized three Fe-Por with different numbers of orbitals involved in the π-system. Colloidally dispersible Cu nanocubes (Cucub) were chosen as Cu catalysts. Their intrinsic selectivity towards C–C coupling, and ethylene production in particular, as well as the demonstrated possibility to integrate them in different electrochemical cell designs, justifies this choice.38–41 Furthermore, the fact that both Fe-Por and Cucub catalysts can be employed as inks is beneficial for catalyst processability, scalability, and device integration. By studying the designed series of Fe-Por molecules in tandem CO2RR, we investigate how their TOFCO impacts the C–C coupling on Cucub. Importantly, we discover that the most active Fe-Por generates an optimal local environment that boosts the intrinsic activity of Cucub towards ethylene while also increasing energy efficiency by anodically shifting the potential at which this product is detected.
Results and discussion
A series of Fe-Por complexes with a varying number of orbitals involved in the π-system of the porphyrin ligand were synthesized and tested as molecular catalysts for CO2RR (Fig. 1). These complexes are iron(III)-chlorido-tetrabicyclo[2.2.2]octadiene-tetraphenyl porphyrin (Fe-TbcTPP), iron(III)-chlorido-tetraphenyl porphyrin (Fe-TPP), and an iron(III)-chlorido-tetrabenzo-tetraphenyl porphyrin (Fe-TBTPP). The synthesis of Fe-TPP was carried out following a procedure reported in the literature,42 whereas Fe-TbcTPP and Fe-TBTPP were prepared according to the synthetic approach described in detail in the ESI and illustrated in Scheme S1†. In brief, the initial step corresponds to the synthesis of the free base porphyrin H2TbcTPPvia a “Lindsey condensation reaction”.43 Refluxing H2TbcTPP in a tetrahydrofuran (THF) solution containing FeBr2 and 2,6-lutidine generates the desired Fe-TbcTPP. The last step is a retro-Diels–Alder reaction achieved by heating the Fe-TbcTPP complex at 200 °C under vacuum to produce Fe-TBTPP. The 1H and 13C-NMR spectra of H2TbcTPP are presented in Fig. S1 and S2,† while the MALDI-TOF spectra of the iron-derivatives (Fe-TbcTPP and Fe-TBTPP) are provided in Fig. S3 and S4.† The aromatic delocalization effect in Fe-Por is revealed by optical absorption spectroscopy (Fig. 1 and Table S1†). According to expectation, as the number of π-orbitals contributing to the system increases, the respective Soret band red-shifts, following the peak wavelength (λmax) order λmax (Fe-TBTPP) > λmax (Fe-TPP) > λmax (Fe-TbcTPP).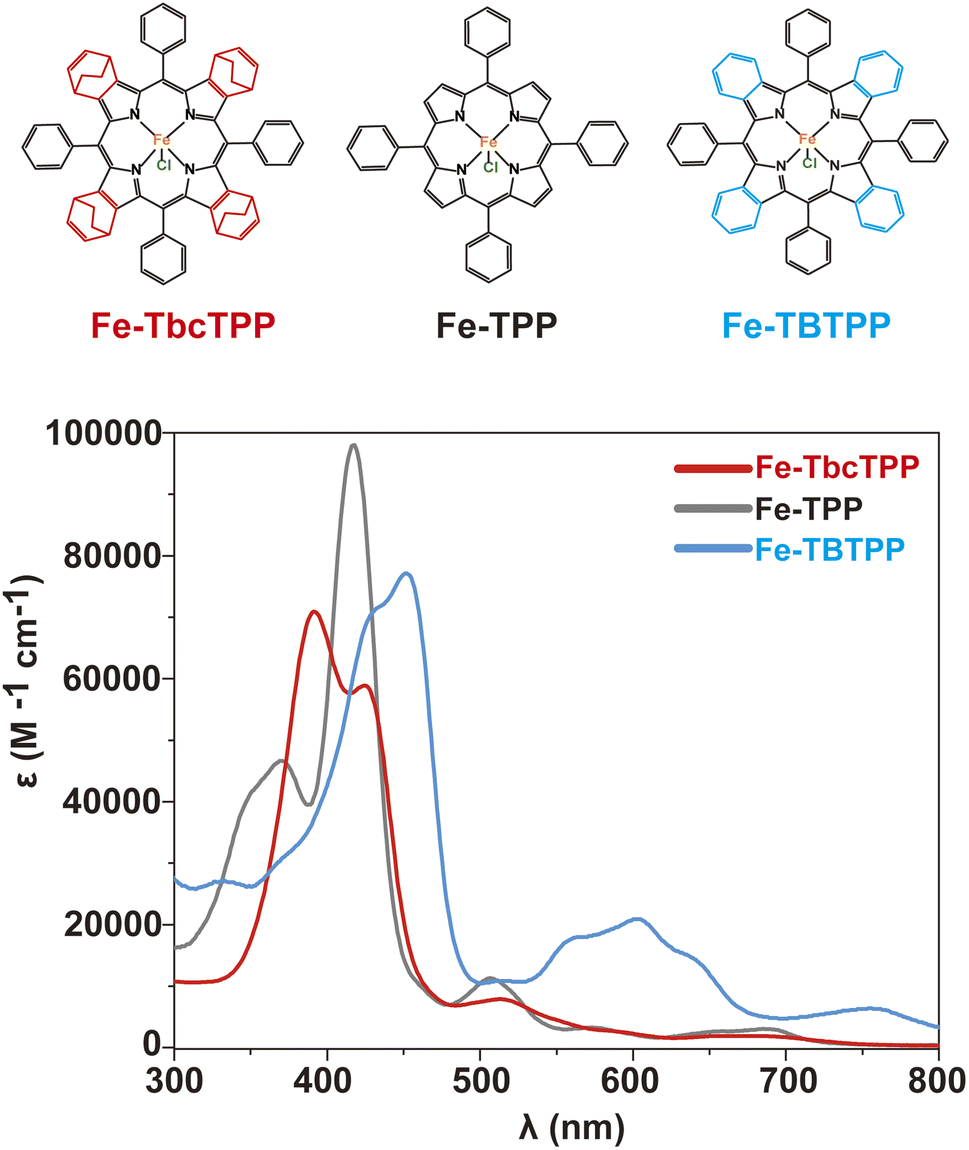 | ||
| Fig. 1 The Fe-Por molecules synthesized and investigated as molecular catalysts in this study (top) along with the UV-vis spectra of the same in THF (bottom). | ||
To understand the impact of the different porphyrin ligands on the electrocatalytic properties, we performed cyclic voltammetry (CV) experiments in homogeneous conditions (Fig. 2 and ESI† for details). A very similar behavior was observed for all Fe-Por, with each characterized by three redox processes that are assigned to FeIII/FeII, FeII/FeI, and FeI/Fe0, as depicted in Fig. 2 and Table S1.† The second and third reduction waves are associated with two consecutive one electron reductions that are mainly ligand-based.44,45 As expected, the different degree of π-delocalization of the Fe-Por results in a significant shift of the respective redox potentials. More specifically, the Fe-TbcTPP containing the more σ-donating and less π-accepting bicyclo[2.2.2]octadiene groups has the lowest potentials. On the other hand, the Fe-TBTPP containing benzo groups and phenyl substitution at the meso positions has more anodic values. The Fe-TPP with no additional functionalization onto the pyrrole moieties has redox potentials between those of Fe-TbcTPP and Fe-TBTPP.
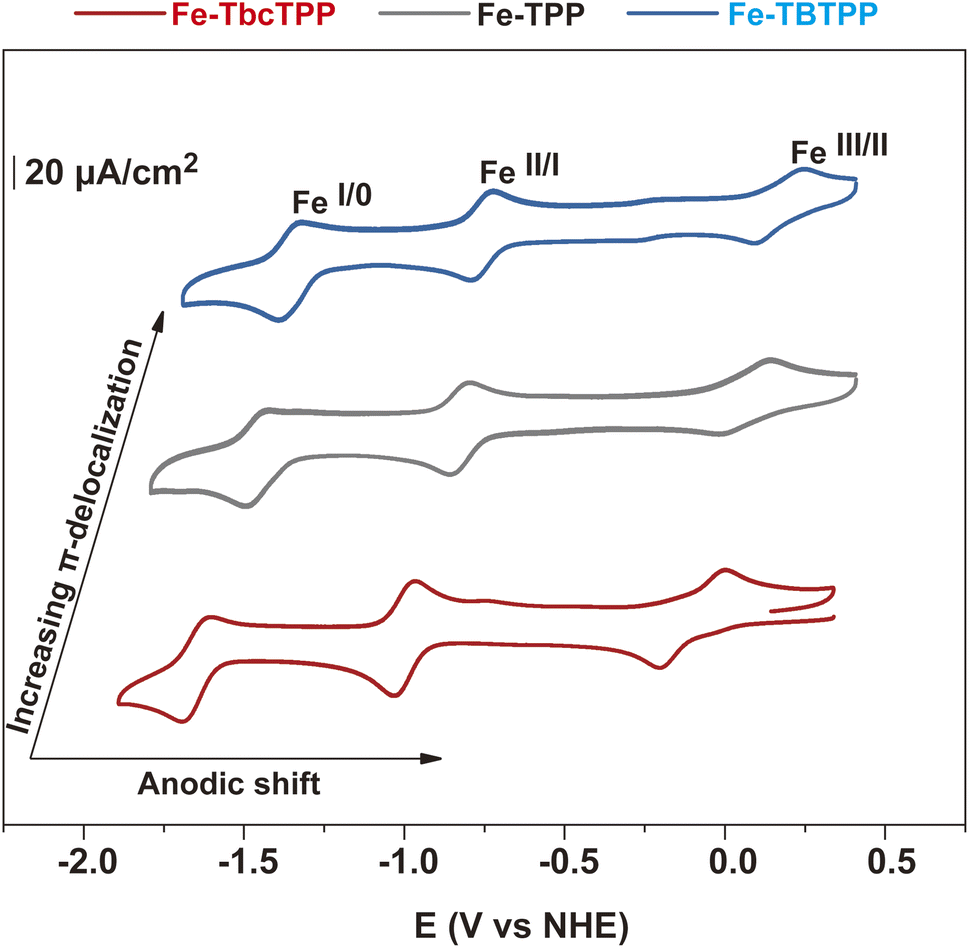 | ||
| Fig. 2 Cyclic voltammetry of 0.25 mM Fe-Por dissolved in DMF containing 0.1 M TBAPF6 as the supporting electrolyte, carried out at a scan rate of 100 mV s−1. Fe-TbcTPP (red), Fe-TPP (gray), and Fe-TBTPP (blue). | ||
We then investigated the CO2RR catalytic properties of the Fe-Por in an H-cell using CO2 saturated 0.1 M KHCO3 as the electrolyte (see ESI† for details). Results of these experiments are presented in Fig. 3. Linear sweep voltammetry (LSV) (Fig. 3a) reveals an anodic shift of the onset potential (i.e. the potential at which the current starts to be detectable from the background) for CO2 reduction from −0.57 V (Fe-TbcTPP) to −0.50 V (Fe-TPP) and to −0.40 V (Fe-TBTPP) vs. RHE (Reversible Hydrogen Electrode). This anodic shift follows the π-delocalization trend of the Fe-Por reported above.46–48 Furthermore, the faradaic efficiency for CO (FECO) is higher for Fe-TBTPP than for either Fe-TPP or Fe-TbcTPP across the entire potential range (Fig. 3b and S5†), with a maximum of 91% at −0.55 V vs. RHE. The TOFCO (see ESI† for details) follows the same trend, with TOFCO (Fe-TBTPP) > TOFCO (Fe-TPP) > TOFCO (Fe-TbcTPP) (Fig. 3c). Specifically, Fe-TBTPP exhibits the highest TOFCO (0.26 s−1) compared to Fe-TPP (0.11 s−1) and Fe-TbcTPP (0.02 s−1) at −0.95 V vs. RHE.
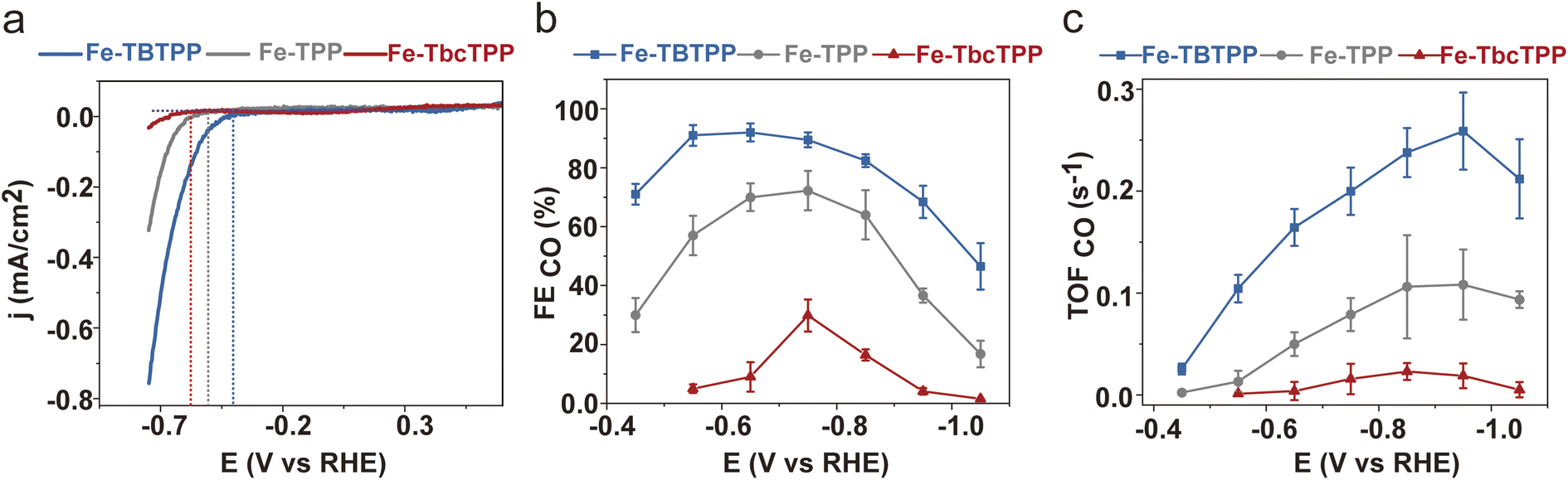 | ||
| Fig. 3 (a) LSV curves of the Fe-Por catalysts measured at a scan rate of 10 mV s−1, with the vertical dashed lines representing the onset potentials for each catalyst; (b) FECO and (c) TOFCO as a function of applied potential. These measurements were performed at Fe-Por loadings of 16 nmol cm−2 on carbon nanotubes deposited onto glassy carbon electrodes (S = 1.33 cm2) using an H-cell with CO2 saturated 0.1 M KHCO3 electrolyte. The carbon nanotubes were chosen as commonly used as a conductive support to homogeneously disperse the molecular catalysts via non-covalent interactions.32–34 The reported FE and TOF values are the average of three independent experiments with error bars indicating the standard deviations. | ||
Following the analysis of the Fe-Por, we prepared the Cucub/Fe-Por by mixing the Fe-Por and the Cucub (Fig. S6†) (see ESI† for details). The inks were then drop cast onto glassy carbon electrodes and the activities of the tandem catalysts were tested from −0.45 V to −1.05 V vs. RHE. Fig. 4 and S7† provide an overview of the obtained data. At low potentials (−0.45 V and −0.55 V vs. RHE), the addition of the Fe-Por to Cucub suppresses the hydrogen evolution reaction (HER) and enhances the production of CO and formate. However, no evidence of C–C coupling reactions is observed at these potentials (Fig. S7†). The HER suppression and CO2RR promotion induced by the Fe-Por become more pronounced at more negative potentials (−0.65 V to −1.05 V vs. RHE, Fig. 4a). Indeed, the enhancement of C2H4 faradaic efficiency from Cucub/Fe-Por relative to Cucub is clearly observed for all tandem-catalysts (Fig. 4b). Interestingly, C2H4 is produced at a potential as low as −0.65 V vs. RHE for the Cucub/Fe-TBTPP, which maintains the highest FEC2H4 across the entire potential range (Fig. 4b), reaching a FEC2H4 of 36% at −1.05 V vs. RHE. By comparison, the Cucub in the absence of Fe-Por only achieved a FEC2H4 of 19% at the same potential. At more negative potential, CH4 is favored over C2H4 (Fig. S8†). Thus, while Fe-TBTPP still promotes C–C coupling, the FEC2H4 decreases (∼25% at −1.15 V). The FEC2H4 was much lower also for Cusphere/Fe-TBTPP (∼15% at −1.05 V), which highlights the importance of the Cu morphology to maximize ethylene production (Fig. S9†).
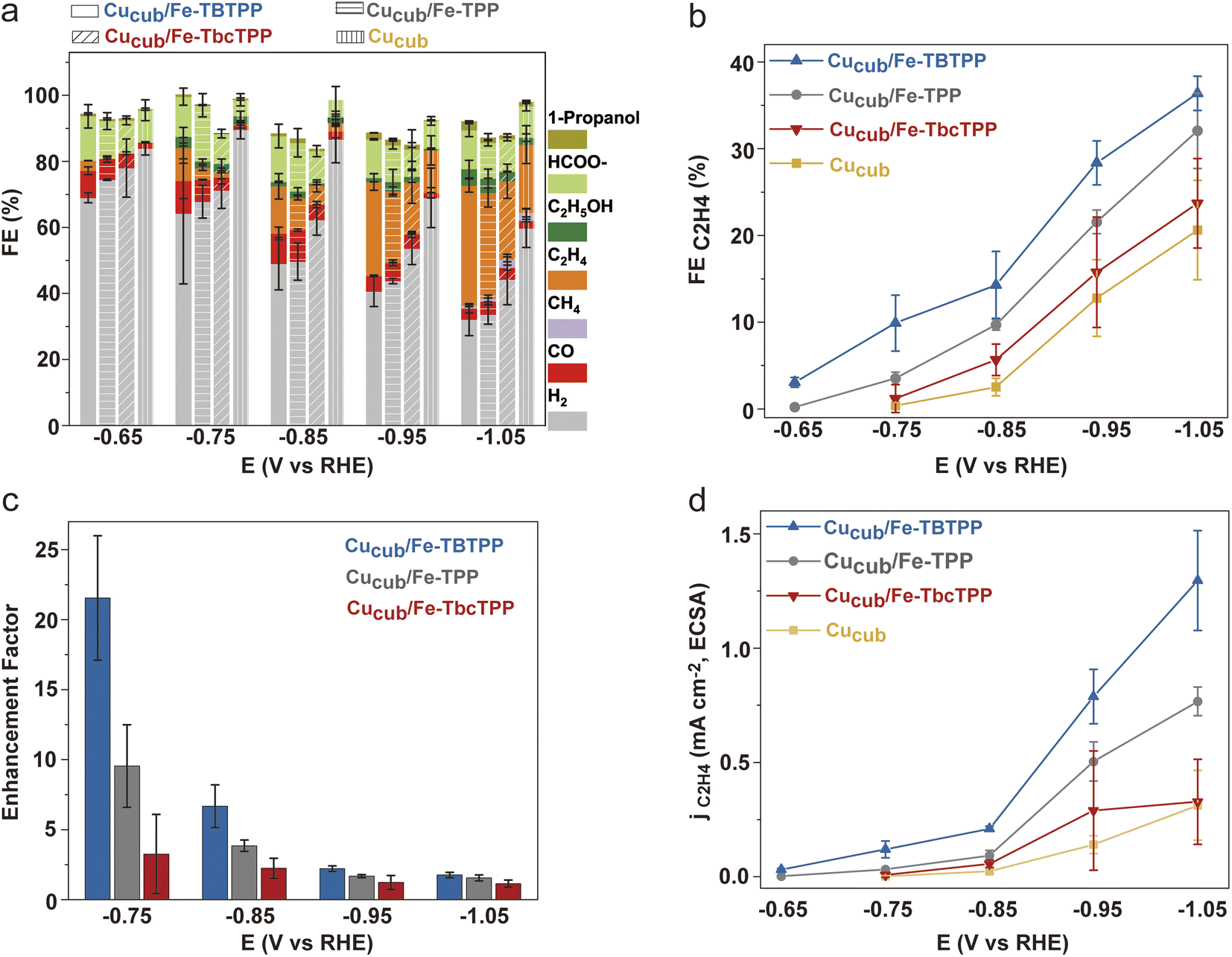 | ||
| Fig. 4 (a) Total FEs and (b) FEC2H4 for Cucub/Fe-TBTPP, Cucub/Fe-TPP, Cucub/Fe-TbcTPP and Cucub as a function of potential; (c) C2H4 product enhancement factor of the Cucub/Fe-Por compared to the pristine Cucub (enhancement factor = (FEC2H4 of Cucub/Fe-Por)/(FEC2H4 of Cucub) as a function of potential; (d) jC2H4/ECSA for Cucub/Fe-TBTPP, Cucub/Fe-TPP, Cucub/Fe-TbcTPP and Cucub as a function of potential. The electrodes for these experiments were prepared by loading 3.12 × 102 nmol cm−2 of Cucub and 8 nmol cm−2 of the Fe-Por onto glassy carbon electrodes. The reported values are an average of three independent experiments with error bars indicating the standard deviations. | ||
To quantify the impact of the Fe-Por on C–C coupling, we define an enhancement factor for C2H4 production as the ratio of FEC2H4 of Cucub/Fe-Por to the FEC2H4 of Cucub at a given potential. The corresponding enhancement factors for Cu/Fe-TBTPP, Cu/Fe-TPP and Cu/Fe-TbcPP are 21.5, 9.5 and 3.2 at −0.75 V vs. RHE, respectively (Fig. 4c). Analysis of the electrochemically active surface area (ECSA) of Cucub/Fe-Por reveals a decreased solid/liquid contact area compared to the Cucub, which is likely a consequence of the blocking of Cu active sites by the molecules (Fig. S10†). However, the ECSA-normalized current density (jC2H4/ECSA) indicates an increase in the intrinsic activity of the unpassivated sites in the presence of the Fe-Por, with a trend that is consistent with the FE and the enhancement factor (Fig. 4d).
To exclude the contribution of C1 product suppression on the observed trends and to provide further evidence for the tandem mechanism, we analyzed the C1 products in greater detail (Fig. 5). The FEC1 and the jC1,ECSA of the Cucub/Fe-Por catalysts are generally higher than those for the pristine Cucub across the entire potential range (Fig. 5a, b and S11†). Furthermore, the trend of FEC1 and jC1/ECSA among the Cucub/Fe-Por follows that of the FEC2H4, which are higher values measured for higher molecular TOFCO. The improved capacity of Cucub to catalyze C–C coupling at more negative potentials results in an increase of FEC2+/FEC1 for all catalysts at higher overpotentials (Fig. 5c). The decrease of FECO (Fig. 5d) and the saturation of jCO (Fig. 5e) with increasing overpotential is consistent with the increased consumption of CO as more ethylene is generated via the tandem catalytic mechanism. In contrast, FECO and jCO remain approximately constant for pure Cucub across all potentials, reflecting the unmodified native activity of the nanocubes in the absence of tandem CO generation.
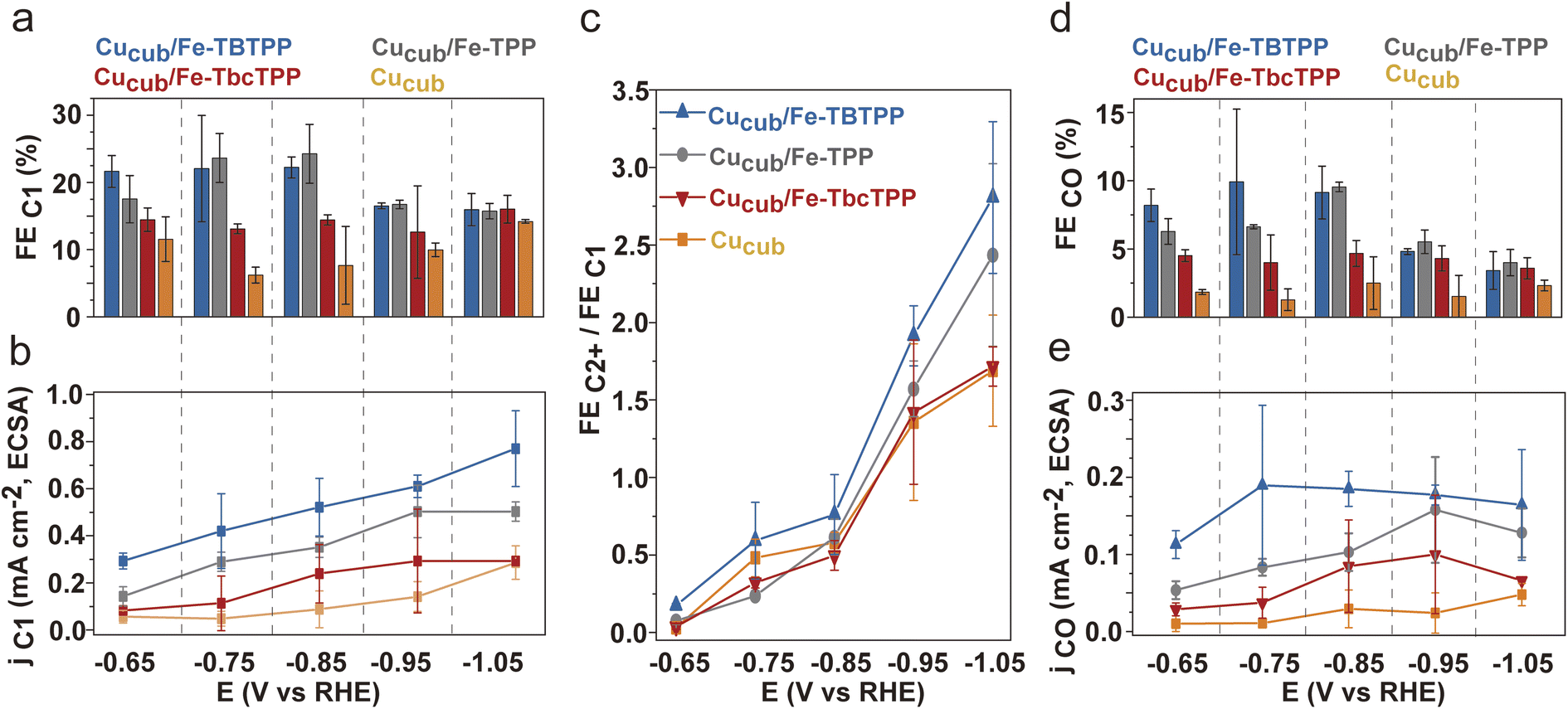 | ||
| Fig. 5 (a) FEC1 and (b) partial current density for the C1 products (jC1) normalized by ECSA, (c) FE ratio of C2+ to C1 products, (d) FEs and (e) partial current density for CO (jCO) normalized by ECSA of Cucub/Fe-TBTPP, Cucub/Fe-TPP, Cucub/Fe-TbcTPP and Cucub at different potentials. The reported values are an average of three independent experiments with error bars indicating the standard deviations. | ||
To investigate possible changes of electronic and chemical properties of Cucub due to interactions with the molecular constituents, which could influence the overall catalytic properties of the Cucub/Fe-Por, we performed X-ray photoelectron spectroscopy (XPS) (Fig. S12†). The analysis of the Cu 2p peaks and Cu LMM spectra indicated that Cu is present in its metallic form in all systems. Furthermore, the absence of any peak shifts in spectra from Cucub/Fe-Por compared to Cucub indicated that there are no changes in surface composition and/or charge transfer effects. This lack of a specific chemical or electronic change of the Cu surface, together with the systematic correlations between C–C coupling efficiency of Cucub/Fe-Por with the TOFCO trends among Fe-Por, indicates that overall selectivity and activity is governed by the tandem catalytic process.
Having established that a tandem mechanism is dominant in defining activity and selectivity of Cucub/Fe-Por, optimization of ethylene production requires understanding of the relationship between the loading of the CO-generating component and the CO conversion rate of the Cu catalyst. With this aim, we tested the impact of Fe-Por loading on enhancing C–C coupling in Cucub/Fe-Por tandem catalysts (Fig. 6). We observe that the FEC2H4 increases with the Fe-Por loading until reaching its maximum value at 8 nmol cm−2 (Fig. 6a). This trend suggests that addition of more Fe-Por results in the local accumulation of CO in the vicinity of Cucub, which promotes C–C coupling. However, a further increase of the Fe-Por coverage leads to decreased C2H4 selectivity. Concomitantly, the FECO increases (Fig. S13†), which indicates that the additionally generated CO escapes the system.
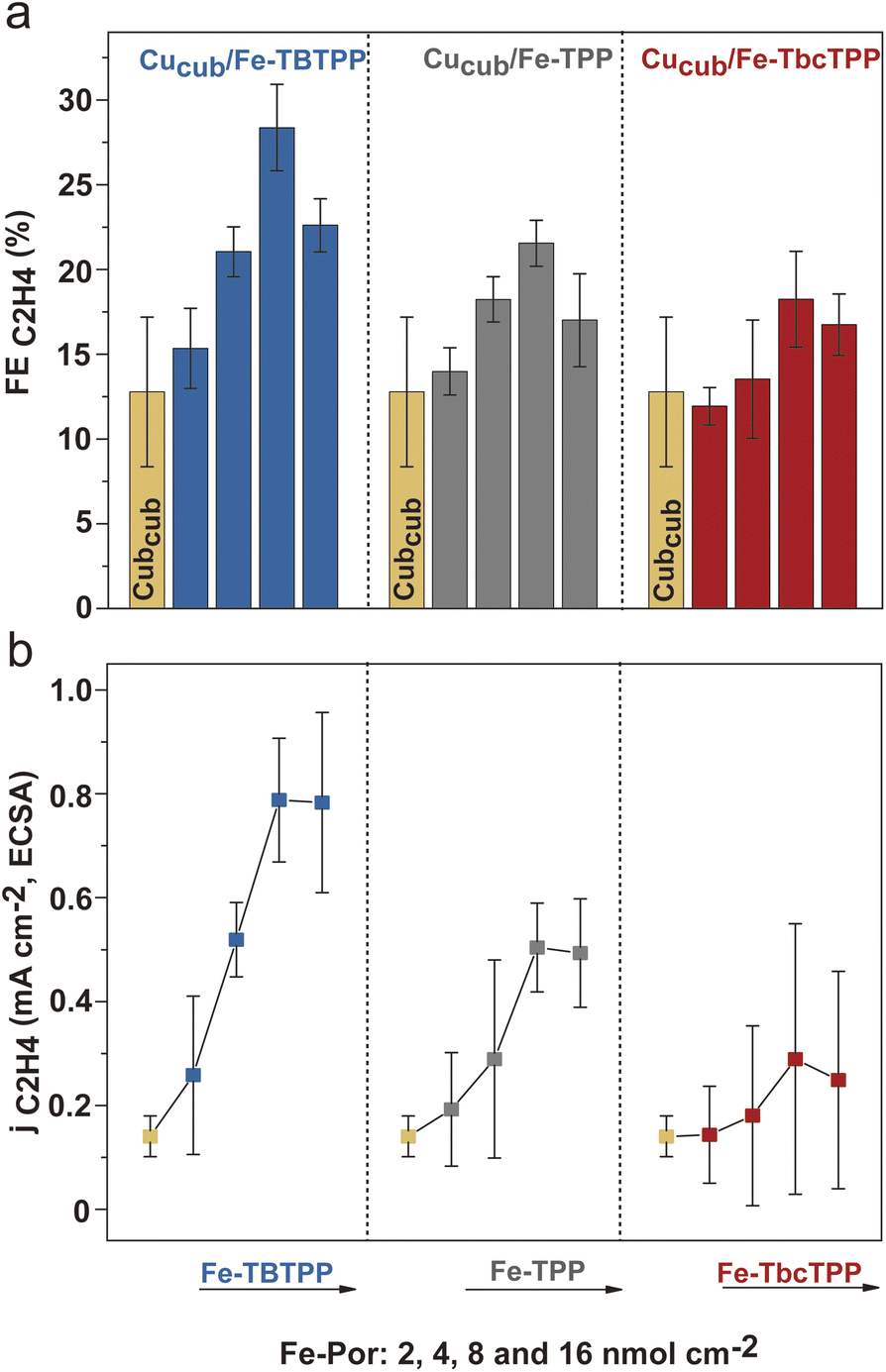 | ||
| Fig. 6 (a) FEC2H4 and (b) jC2H4/ECSA of Cucub/Fe-TBTPP, Cucub/Fe-TPP, Cucub/Fe-TbcTPP and Cucub at different loadings of the Fe-Por (2, 4, 8 and 16 nmol cm−2) at −0.95 V vs. RHE in CO2 saturated 0.1 M KHCO3 H-cell. The reported values are an average of three independent experiments with error bars indicating the standard deviations. | ||
As discussed above, the Fe-Por molecules also block Cu active sites. Consistent with this conclusion, we observe a continuous decrease of the ECSA with increased loading (Fig. S14†). The enhancement in intrinsic activity of the unpassivated sites on the Cucub compensates the decreasing concentration of sites, but only up to a certain loading. Above that point, the C2H4 selectivity is suppressed, which is also reflected in the jC2H4,ECSA (Fig. 6b). In particular, the jC2H4,ECSA in Cucub/Fe-Por increases up to 8 nmol cm−2 and thereafter saturates.
The comparison of different Fe-Por at the same loadings reveals more efficient C2H4 production for the Cucub/Fe-TBTPP. Furthermore, we note that lower loadings of the Fe-TBTPP generate higher FEC2H4 and jC2H4/ECSA compared to higher loadings of the Fe-TbcTPP (e.g. 4 nmol cm−2 of Fe-TBTPPvs. 8 nmol cm−2 of Fe-TbcTPP). As the Fe-TBTPP possesses the highest TOFCO, these data prove that the CO production rate plays a crucial role in achieving optimal C–C coupling in the tandem catalysts.
To test whether the bottleneck for further improving C2H4 generation with the highest Fe–Por loading is the CO consumption, we tested increased Cu-cube loadings for the best performing Cucub/Fe-TBTPP (Fig. 7 and S15†). The Fe-TBTPP loading was kept at 16 nmol cm−2 for comparison with Fig. 6. An increase in Cucub loading from 3.12 × 102 to 6.24 × 102 nmol cm−2 does improve the C2H4 production, with both the FEC2H4 and jC2H4,ECSA increasing (Fig. 7a). A FEC2H4 of 33% is thus measured at −0.95 V vs. RHE. The concomitant decrease FECO and jCO,ECSA (Fig. 7b) indicates that the CO produced by Fe-TBTPP is indeed further consumed by the increased Cucub loading.
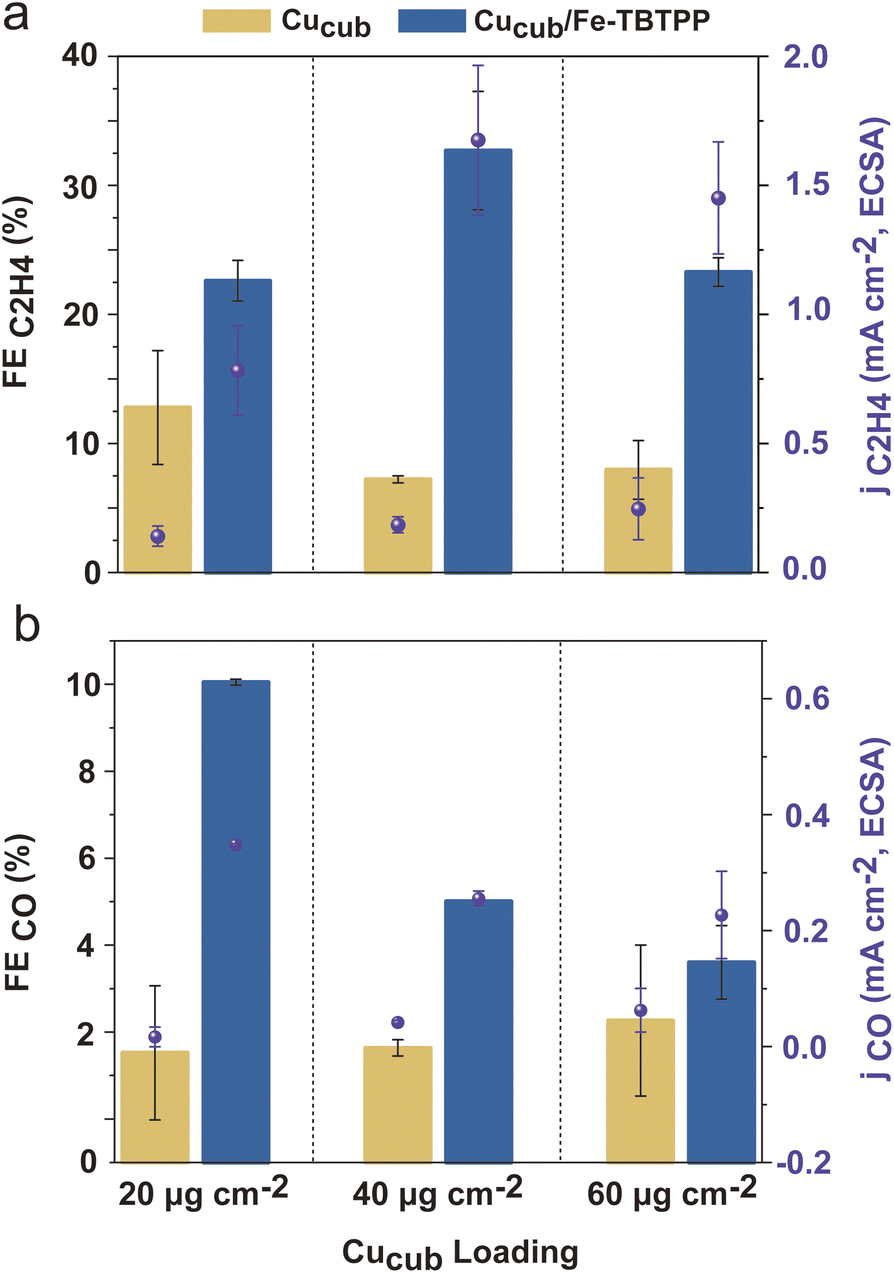 | ||
| Fig. 7 (a) FE and partial current density for the C2H4 product normalized by ECSA (jC2H4,ECSA) and (b) FE and partial current density for the CO product normalized by ECSA (jCO,ECSA) for Cucub/Fe-TBTPP at different loading of Cucub (3.12 × 102, 6.24 × 102 and 9.36 × 102 nmol cm−2) with the same Fe-TBTPP loading (16 nmol cm−2) at −0.95 V vs. RHE in the CO2 saturated 0.1 M KHCO3 H-cell. The reported values are an average of three independent experiments with error bars indicating the standard deviations. | ||
For higher Cucub loading (9.36 × 102 nmol cm−2), the FEC2H4 and jC2H4,ECSA decrease and the FECO and jCO,ECSA don't change further. In a similar manner to the Fe-Por loading experiments, the ratio between the CO-generating component and the CO conversion rate of the Cu catalyst must be balanced for optimal performance.
Finally, we characterized the Fe-Por by Fourier-transform infrared spectroscopy (FT-IR) and ultraviolet-visible spectroscopy (UV-vis) (Fig. S16 and S17†), and the Cucub morphology by TEM (Fig. S18†), which proved the intact structure of Fe-Por and cubic shape of Cucub after 1.5 hours of electrolysis. Furthermore, the best performing tandem catalyst in terms of C2H4 production, which is Cucub/Fe-TBTPP, displayed an excellent stability and selectivity over 10 hours of electrolysis (Fig. S19†). Altogether, these data indicate that the addition of the Fe-Por has a stabilizing effect on the morphology of the Cu cubes. While a dedicated study is needed, we speculatively attribute the origin of this observation to the formation of the CO intermediate on a different surface than Cu.
Conclusions
In summary, we built tandem catalysts including Fe-Por with tunable CO2 to CO selectivity (Fe-TBTPP > Fe-TPP > Fe-TbcTPP) and Cucub. We discovered that the C–C coupling enhancement in these tandem catalysts correlates directly with the Fe-Por selectivity and conversion rates for CO, with the Cucub/Fe-TBTPP being the best performing one.We found a 100 mV anodic potential shift for C2H4 detection, a 22 times enhancement of C2H4 production at −0.75 V vs. RHE and a doubled FEC2H4 of 36% at −1.05 V vs. RHE in the Cucub/Fe-TBTPP compared to the Cucub. This performance is comparable to the state-of-the-art in this class of catalysts.25–27 Going beyond it, these results demonstrate that the rate and potential at which CO is generated plays a defining role for the selectivity and overpotential of multiple-carbon products in tandem schemes (Fig. 8).
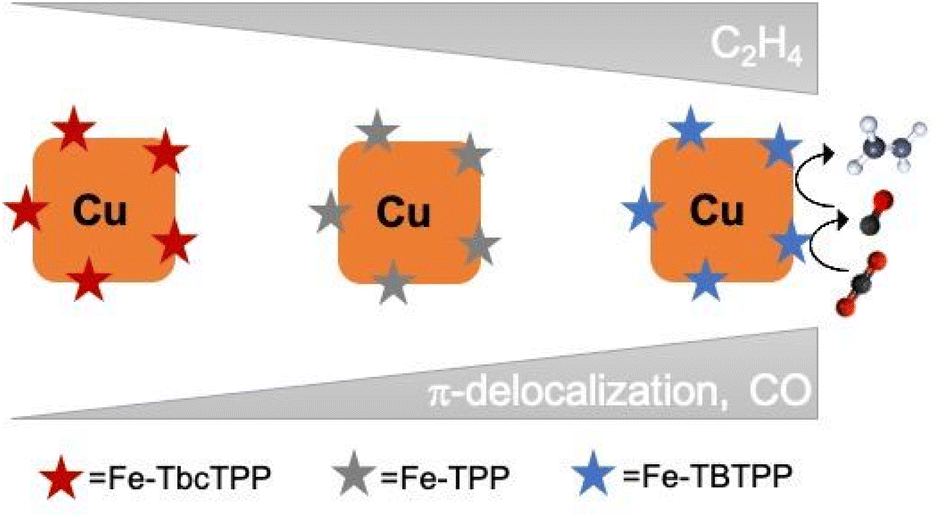 | ||
| Fig. 8 Schematic representation of the proposed mechanism for C2H4 promotion in Cucub/Fe-Por tandem catalysts. The C–C coupling enhancement and C2H4 promotion correlate with the Fe-Por selectivity and conversion rate for CO. | ||
Overall, this study encourages further investigations on the design of the homogenous catalyst as a promising strategy to further optimize molecular-based tandem systems.
Data availability
Data for this paper, including UV-vis and electrochemical characterization, are available at Zenodo at https://doi.org/10.5281/zenodo.7229402.Author contributions
M. W. carried out the synthesis of the tandem catalysts and all electrocatalytic testing for CO2RR. V. N. synthesized and characterized the molecular catalysts. A. L. performed X-ray photoelectron spectroscopy and contributed to daily discussions. A. L. supervised the work on the molecular catalysts. I. S. contributed to discussions. R. B. supervised the work on the tandem catalysts and CO2RR and coordinated the project. All the authors contributed to the writing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the support from EU-funded LICROX FET project (Grant agreement 951843).References
- X. Lim, Nature, 2015, 526, 628–630 CrossRef CAS.
- H.-R. M. Jhong, S. Ma and P. J. A. Kenis, Curr. Opin. Chem. Eng., 2013, 2, 191–199 CrossRef.
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. M. Koper, Nat. Energy, 2019, 4, 732–745 CrossRef CAS.
- T. K. Todorova, M. W. Schreiber and M. Fontecave, ACS Catal., 2020, 10, 1754–1768 CrossRef CAS.
- S. Nitopi, E. Bertheussen, S. B. Scott, X. Liu, A. K. Engstfeld, S. Horch, B. Seger, I. E. L. Stephens, K. Chan, C. Hahn, J. K. Nørskov, T. F. Jaramillo and I. Chorkendorff, Chem. Rev., 2019, 119, 7610–7672 CrossRef CAS.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Nørskov, Energy Environ. Sci., 2010, 3, 1311–1315 RSC.
- J. Li, Z. Wang, C. McCallum, Y. Xu, F. Li, Y. Wang, C. M. Gabardo, C. T. Dinh, T. T. Zhuang, L. Wang, J. Y. Howe, Y. Ren, E. H. Sargent and D. Sinton, Nat. Catal., 2019, 2(12), 1124–1131 CrossRef CAS.
- A. J. Garza, A. T. Bell and M. Head-Gordon, ACS Catal., 2018, 8(2), 1490–1499 CrossRef CAS.
- X. Wang, J. F. de Araújo, W. Ju, A. Bagger, H. Schmies, S. Kühl, J. Rossmeisl and P. Strasser, Nat. Nanotechnol., 2019, 14, 1063–1070 CrossRef CAS PubMed.
- A. Ozden, Y. Wang, F. Li, M. Luo, J. Sisler, A. Thevenon, A. Rosas-Hernández, T. Burdyny, Y. Lum, H. Yadegari, T. Agapie, J. C. Peters, E. H. Sargent and D. Sinton, Joule, 2021, 5(3), 706–719 CrossRef CAS.
- Y. Zhu, X. Cui, H. Liu, Z. Guo, Y. Dang, Z. Fan, Z. Zhang and W. Hu, Tandem catalysis in electrochemical CO2 reduction reaction, Nano Res., 2021, 14, 4471–4486 CrossRef CAS.
- M. K. Birhanu, M. C. Tsai, A. W. Kahsay, C. T. Chen, T. S. Zeleke, K. B. Ibrahim, C. J. Huang, W. N. Su and B. J. Hwang, Adv. Mater. Interfaces, 2018, 5(24), 1800919 CrossRef.
- Y. Lum and J. W. Ager, Energy Environ. Sci., 2018, 11(10), 2935–2944 RSC.
- C. G. Morales-Guio, E. R. Cave, S. A. Nitopi, J. T. Feaster, L. Wang, K. P. Kuhl, A. Jackson, N. C. Johnson, D. N. Abram, T. Hatsukade, C. Hahn and T. F. Jaramillo, Nat. Catal., 2018, 1, 764–771 CrossRef CAS.
- C. Chen, Y. Li, S. Yu, S. Louisia, J. Jin, M. Li, M. B. Ross and P. Yang, Joule, 2020, 4, 1688–1699 CrossRef CAS.
- P. Iyengar, M. J. Kolb, J. R. Pankhurst, F. Calle-Vallejo and R. Buonsanti, ACS Catal., 2021, 11(8), 4456–4463 CrossRef CAS.
- J. Huang, M. Mensi, E. Oveisi, V. Mantella and R. Buonsanti, J. Am. Chem. Soc., 2019, 141, 2490–2499 CrossRef CAS PubMed.
- D. Ren, B. Ang and B. Yeo, ACS Catal., 2016, 6, 8239–8247 CrossRef CAS.
- G. Guisbiers, S. Mejia-Rosales, S. Khanal, F. Ruiz-Zepeda, R. L. Whetten and M. José-Yacaman, Nano Lett., 2014, 14, 6718–6726 CrossRef CAS PubMed.
- S. B. Varandili, D. Stoian, J. Vavra, K. Rossi, J. R. Pankhurst, Y. Guntern, N. Lopez and R. Buonsanti, Chem. Sci., 2021, 12, 14484 RSC.
- Y. C. Li, Z. Wang, T. Yuan, D.-H. Nam, M. Luo, J. Wicks, B. Chen, J. Li, F. Li, F. P. G. de Arquer, Y. Wang, C.-T. Dinh, O. Voznyy, D. Sinton and E. H. Sargent, J. Am. Chem. Soc., 2019, 141, 8584–8591 CrossRef CAS PubMed.
- L. Zaza, K. Rossi and R. Buonsanti, ACS Energy Lett., 2022, 7, 1284 CrossRef CAS.
- A. K. Buckley, M. Lee, T. Cheng, R. V. Kazantsev, D. M. Larson, W. A. Goddard III, F. D. Toste and F. M. Toma, J. Am. Chem. Soc., 2019, 141(18), 7355–7364 CrossRef CAS PubMed.
- J. R. Pankhurst, Y. T. Guntern, M. Mensi and R. Buonsanti, Chem. Sci., 2019, 10, 10356 RSC.
- F. Li, Y. C. Li, Z. Wang, J. Li, D. H. Nam, Y. Lum and E. H. Sargent, Nat. Catal., 2020, 3(1), 75–82 CrossRef CAS.
- X. Kong, J. Zhao, J. Ke, C. Wang, S. Li, R. Si, B. Liu, J. Zeng and Z. Geng, Nano Lett., 2022, 22, 3801–3808 CrossRef CAS PubMed.
- L. Lin, T. Liu, J. Xiao, H. Li, P. Wei, D. Gao, B. Nan, R. Si, G. Wang and X. Bao, Angew. Chem., Int. Ed., 2020, 59(50), 22408–22413 CrossRef CAS PubMed.
- E. E. Benson, C. P. Kubiak, A. J. Sathrum and J. M. Smieja, Chem. Soc. Rev., 2009, 38(1), 89–99 RSC.
- K. E. Dalle, J. Warnan, J. J. Leung, B. Reuillard, I. S. Karmel and E. Reisner, Chem. Rev., 2019, 119(4), 2752–2875 CrossRef CAS PubMed.
- E. Boutin, L. Merakeb, B. Ma, B. Boudy, M. Wang, J. Bonin, E. Anxolabéhère-Mallart and M. Robert, Chem. Soc. Rev., 2020, 49(16), 5772–5809 RSC.
- P. Gotico, W. Leibl, Z. Halime and A. Aukauloo, ChemElectroChem, 2021, 8(18), 3472–3481 CrossRef CAS.
- L. Sun, V. Reddu, A. C. Fisher and X. Wang, Energy Environ. Sci., 2020, 13(2), 374–403 RSC.
- A. Maurin and M. Robert, J. Am. Chem. Soc., 2016, 138(8), 2492–2495 CrossRef CAS PubMed.
- M. Abdinejad, K. Tang, C. Dao, S. Saedy and T. Burdyny, J. Mater. Chem. A, 2022, 10(14), 7626–7636 RSC.
- O. S. Finikova, A. V. Cheprakov, P. J. Carroll, S. Dalosto and S. A. Vinogradov, Inorg. Chem., 2002, 41, 6944–6946 CrossRef CAS.
- K. Chen, M. Cao, G. Ni, S. Chen, H. Liao, L. Zhu, H. Li, J. Fu, J. Hu, E. Cortés and M. Liu, Appl. Catal., B, 2022, 306, 121093 CrossRef CAS.
- S. Yang, Y. Yu, M. Dou, Z. Zhang and F. Wang, J. Am. Chem. Soc., 2020, 142, 17524–17530 CrossRef CAS.
- D. Gao, I. Zegkinoglou, N. J. Divins, F. Scholten, I. Sinev, P. Grosse and B. Roldan Cuenya, ACS Nano, 2017, 11(5), 4825–4831 CrossRef CAS PubMed.
- Y. Wang, H. Shen, K. J. T. Livi, D. Raciti, H. Zong, J. Gregg, M. Onadeko, Y. Wan, A. Watson and C. Wang, Nano Lett., 2019, 19(12), 8461–8468 CrossRef CAS.
- G. L. De Gregorio, T. Burdyny, A. Loiudice, P. Iyengar, W. A. Smith and R. Buonsanti, ACS Catal., 2020, 10, 4854–4862 CrossRef CAS.
- A. Loiudice, P. Lobaccaro, E. A. Kamali, T. Thao, B. H. Huang, J. W. Ager and R. Buonsanti, Angew. Chem., Int. Ed., 2016, 55, 5789–5792 CrossRef CAS.
- A. Fukatsu, M. Kondo, Y. Okabe and S. J. Masaoka, J. Photochem. Photobiol., A, 2015, 313, 143–148 CrossRef CAS.
- J. S. Lindsey, I. C. Schreiman, H. C. Hsu, P. C. Kearney and A. M. Marguerettaz, J. Org. Chem., 1987, 52(5), 827–836 CrossRef CAS.
- P. A. Davethu and S. P. De Visser, J. Phys. Chem. A, 2019, 123(30), 6527–6535 CrossRef CAS PubMed.
- C. Römelt, S. Ye, E. Bill, T. Weyhermüller, M. Van Gastel and F. Neese, Inorg. Chem., 2018, 57(4), 2141–2148 CrossRef.
- P. Gotico, W. Leibl, Z. Halime and Aukauloo, ChemElectroChem, 2021, 8(18), 3472–3481 CrossRef CAS.
- R. Zhang and J. J. Warren, ChemSusChem, 2021, 14(1), 293–302 CrossRef CAS PubMed.
- D. Dedić, A. Dorniak, U. Rinner and W. Schöfberger, Front. Chem., 2021, 9, 1–18 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc04794b |
| ‡ These authors have equally contributed. |
| This journal is © The Royal Society of Chemistry 2022 |