
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiscovery of a monomeric green fluorescent protein sensor for chloride by structure-guided bioinformatics†
Weicheng
Peng
 ab,
Caden C.
Maydew
a,
Hiu
Kam
a,
Jacob K.
Lynd
a,
Jasmine N.
Tutol
a,
Shelby M.
Phelps
a,
Sameera
Abeyrathna
a,
Gabriele
Meloni
a and
Sheel C.
Dodani
*a
ab,
Caden C.
Maydew
a,
Hiu
Kam
a,
Jacob K.
Lynd
a,
Jasmine N.
Tutol
a,
Shelby M.
Phelps
a,
Sameera
Abeyrathna
a,
Gabriele
Meloni
a and
Sheel C.
Dodani
*a
aDepartment of Chemistry and Biochemistry, The University of Texas at Dallas, Richardson 75080, Texas, USA. E-mail: sheel.dodani@utdallas.edu
bDepartment of Biological Sciences, The University of Texas at Dallas, Richardson 75080, Texas, USA
First published on 6th October 2022
Abstract
Chloride is an essential anion for all forms of life. Beyond electrolyte balance, an increasing body of evidence points to new roles for chloride in normal physiology and disease. Over the last two decades, this understanding has been advanced by chloride-sensitive fluorescent proteins for imaging applications in living cells. To our surprise, these sensors have primarily been engineered from the green fluorescent protein (GFP) found in the jellyfish Aequorea victoria. However, the GFP family has a rich sequence space that could already encode for new sensors with desired properties, thereby minimizing protein engineering efforts and accelerating biological applications. To efficiently sample this space, we present and validate a stepwise bioinformatics strategy focused first on the chloride binding pocket and second on a monomeric oligomerization state. Using this, we identified GFPxm163 from GFPxm found in the jellyfish Aequorea macrodactyla. In vitro characterization shows that the binding of chloride as well as bromide, iodide, and nitrate rapidly tunes the ground state chromophore equilibrium from the phenolate to the phenol state generating a pH-dependent, turn-off fluorescence response. Furthermore, live-cell fluorescence microscopy reveals that GFPxm163 provides a reversible, yet indirect readout of chloride transport via iodide exchange. With this demonstration, we anticipate that the pairing of bioinformatics with protein engineering methods will provide an efficient methodology to discover and design new chloride-sensitive fluorescent proteins for cellular applications.
Introduction
The ability to clone, express, and visualize GFP from the jellyfish Aequorea victoria (avGFP) in living cells catalyzed the green fluorescent protein revolution.1 Since then, new GFP family members from a range of marine organisms found in diverse ecological niches have been identified, characterized, and engineered as reporters of cellular activities.2–16 Members of this family are typically oligomeric and have a β-barrel structure with a buried tripeptide chromophore primarily based on tyrosine.9,17 Even though the chromophore is enclosed, crystal structures show that it is connected to bulk water that allows for the coordinated passage and recognition of small ions, including protons, halides, and oxyanions, in a nearby pocket.18–20 Wachter and Remington first demonstrated this in dimeric avGFP variants such as avYFP-H148Q where the binding of anions increases the chromophore pKa, generating a pH-dependent, turn-off fluorescence response (Fig. S1†).18,21 Since chloride is the most abundant biologically relevant anion, this intrinsic mechanism was exploited by Jayaraman and Verkman to apply avYFP-H148Q (Kd = 100 mM at pH 7.5) as the first genetically encoded sensor to image the uptake and exchange of intracellular chloride with exogenously supplemented iodide and nitrate in mammalian cells.18,19,21–23Building off this starting point, rational design and mutagenesis strategies have provided access to new sensors with improved properties such as dynamic range, binding affinity, pH dependence, oligomerization state, and/or excitation/emission profiles.19,22,24–33 While these advances have enabled the discovery of new roles for chloride in normal physiology and disease, sensor design has been largely derived from the avGFP scaffold.24,34–38 Notably, the red-fluorescent protein EqFP578 from the sea anemone Entacmaea quadricolor has been used as a scaffold to generate mBeRFP-based sensors.39,40 Given the biodiversity of fluorescent proteins, the GFP family has a rich yet undersampled sequence space. To explore this, we are using bioinformatics to identify new scaffolds that can ultimately be applied for imaging cellular chloride pools over a wide concentration range ∼3–110 mM.23,41–44 These efforts are part of our larger program aimed at decoding the sequence level determinants of biological anion recognition.45–47
In our first report in 2019, we used a global similarity analysis with reference to avYFP-H148Q to identify a homologous sequence in the jellyfish Phialidium sp. that encodes for a dimeric yellow fluorescent protein called phiYFP.13,42,48 Surprisingly, this naturally occurring protein and the engineered avYFP-H148Q have identical residues similarly arranged in the anion binding pocket: Q69, R94 (96 in avYFP-H148Q), Q183, and Y203 (Fig. 1A and B). The latter residue still forms an anti-parallel π–π interaction with the tyrosine-based chromophore, whereby anion binding generates an optical change. Like avYFP-H148Q, the binding of chloride to phiYFP increases the chromophore pKa, shifting the equilibrium from the fluorescent phenolate state to the non-fluorescent phenol state. Interestingly, excitation of the neutral chromophore generates a turn-on fluorescence response corresponding to the anionic chromophore with a weak binding affinity (Kd = 290 mM ± 44 mM at pH 5). This excitation ratiometric response is indicative of an excited state proton transfer and is unique to phiYFP.49 However, the operational pH and binding affinity for chloride must be engineered to directly monitor the uptake and cellular distributions of chloride. Moreover, it will also need to be monomerized to prevent aggregation and enable fusions to signal peptide/proteins for subcellular targeting or to secondary fluorescent proteins for ratiometric/multianalyte imaging.50 The monomerization process could in turn affect the anion sensing properties. To minimize these protein engineering efforts, we have utilized our prior knowledge of the chloride-binding pocket described above to develop a stepwise bioinformatic strategy. This has allowed us to rapidly mine the GFP sequence space for monomeric chloride-sensitive fluorescent proteins as new starting points. Herein, we have validated this strategy and report the identification, in vitro characterization, and cellular application of GFPxm163 as a monomeric, turn-off fluorescent sensor for chloride.
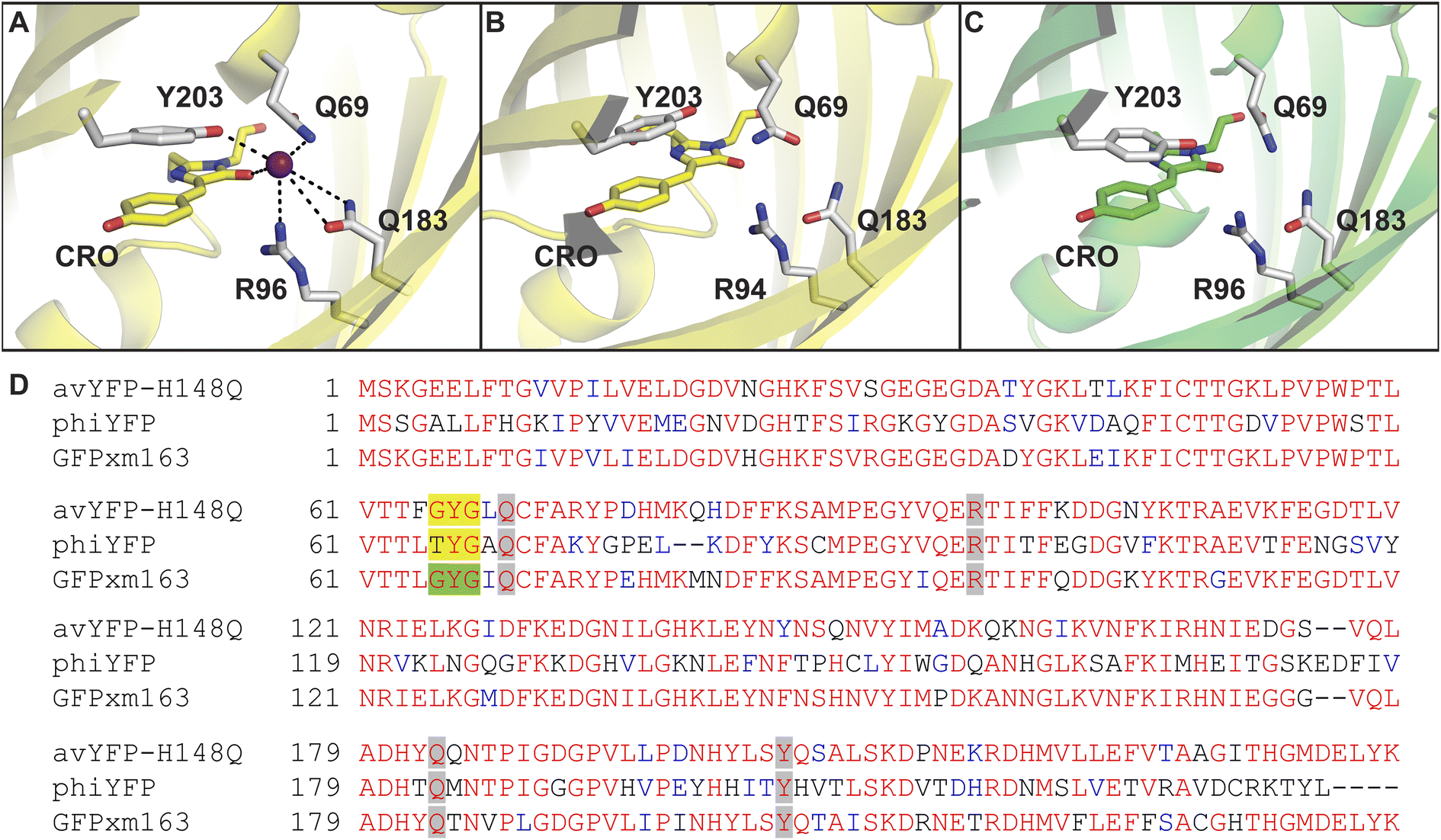 | ||
| Fig. 1 The anion binding pockets for (A) avYFP-H148Q bound to iodide (PDB ID: 1F09), (B) phiYFP (homology model derived from phiYFPv, PDB ID: 4HE4), and (C) GFPxm163 (homology model derived from avYFP-H148Q, PDB ID: 1F09) are conserved. The chromophores are shown as sticks in yellow or green and abbreviated as CRO. The residues in the anion binding pockets are shown as sticks in gray and labeled with the corresponding single letter amino acid code and residue number. Oxygen atoms are shown in red, and nitrogen atoms are shown in blue. The iodide ion is shown as a purple sphere. (D) Multiple sequence alignment of avYFP-H148Q, phiYFP, and GFPxm163. Residues are defined as follows: conserved and identical (red), conserved and similar (blue), and non-conserved (black). In each sequence, the residues that make up chromophore are highlighted in yellow or green, and the binding pocket residues are highlighted in gray. The alignment gaps are shown as dashes (–). | ||
Results and discussion
To identify a monomeric, chloride-sensitive fluorescent protein, we first analyzed all the sequences in the GFP family (Pfam: PF01353). Sequences with identical residues at the positions that define the chloride binding pocket in both avYFP-H148Q and phiYFP were selected (Fig. 1A and B).19,42,48 Since the analyzed sequence space encodes for both naturally occurring and engineered fluorescent proteins with different oligomerization states, the third filter was restricted to known monomers. This approach led us to identify GFPxm163 (UniProt ID: Q8WTC8), which is derived from GFPxm (UniProt ID: Q8WP95) that is naturally found in the jellyfish Aequorea macrodactyla (Fig. 1C and D).51,52 Based on a global sequence alignment, GFPxm163 is 83% and 51% identical to avYFP-H148Q and phiYFP, respectively (Fig. S2†). Moreover, based on a structural model of GFPxm163, the residues in the putative chloride binding pocket are arranged in a similar fashion near the tyrosine-based chromophore (Fig. 1C). With this comparison, we were encouraged to characterize if GFPxm163 could also be sensitive to chloride.To date, GFPxm163 has only been purified and characterized in 25 mM Bis-Tris buffer at pH 6.5 with 100 mM NaCl.51 To test our hypothesis, we first prepared and characterized the chloride-free monomer (Fig. S3–S5†). Like other fluorescent proteins, GFPxm163 has a pH-dependent absorption profile corresponding to the equilibrium of the tyrosine-based chromophore between the phenolate and phenol states. At pH 9, GFPxm163 has an absorption maximum at 512 nm with a shoulder at 480 nm. These spectral features are indicative of the phenolate state (Fig. S6 and S7†). Upon lowering the pH to 4, the absorbance intensity at 512 nm decreases with the appearance of an isosbestic point at 440 nm and a new absorption maximum at 392 nm, indicating the formation of the phenol state (pKa = 5.39 ± 0.06). To our delight, similar spectral changes are also observed in the presence of 512 mM chloride with the largest changes across a physiological range at pH 6 > 6.5 > 7 (Fig. S6 and S7†). Interestingly, the binding of chloride increases the chromophore pKa to 7.13 ± 0.12, favoring the phenol state to a greater extent. Based on these results, chloride titrations at pH 6, 6.5, and 7 show that the chromophore equilibrium can be tuned in a dose-dependent manner (Fig. 2A, S8 and S9†). Moreover, this response does not arise from ionic strength effects as indicated by a negative control with gluconate (Fig. S10 and S11†).
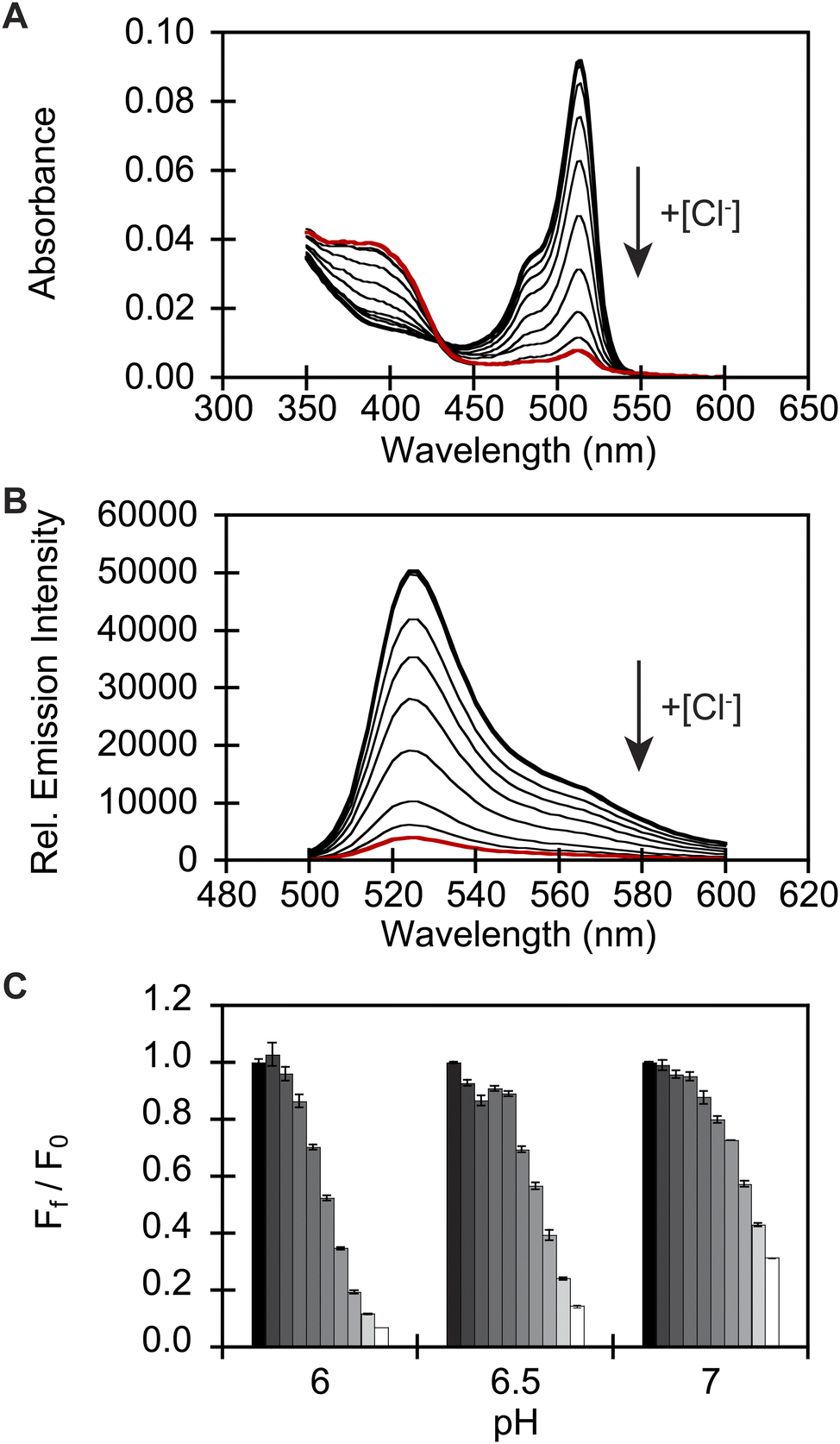 | ||
| Fig. 2 In vitro spectroscopic characterization reveals that GFPxm163 is a pH-dependent, turn-off fluorescent sensor for chloride. (A) Absorbance and (B) emission spectra of GFPxm163 titrated with 0 mM (bold black) to 512 mM Cl− (red) at pH 6. (C) Fluorescence response of GFPxm163 at 524 nm to [Cl−] from 0 mM (black) to 512 mM (white) at pH 6, 6.5, and 7. In panels A and B, the arrow direction corresponds to increasing pH or [Cl−]. Data is shown for one of two protein preparations each measured in triplicate and reported as an average with the standard error of the mean (S.E.M.). All experiments were carried out at room temperature (24–26 °C) with 2.3 μM of protein in 50 mM sodium acetate buffer for pH 4–5.5 or 50 mM sodium phosphate buffer for pH 5.5–9. Absorbance was collected from 350–600 nm. Excitation was provided at 480 nm, and emission was collected from 500–600 nm. | ||
Next, we characterized the emission response of GFPxm163 to chloride with steady-state and time-resolved fluorescence spectroscopy at pH 6, 6.5, and 7. Excitation of the phenolate state (λex = 480 nm) in the absence of chloride at pH 6, results in an emission maximum at 524 nm (Fig. 2B, S8 and S9†). The addition of chloride does not shift the emission maximum but quenches the intensity by 93% with 512 mM chloride. Using a single-site binding model, the apparent dissociation constant (Kd) for chloride binding to GFPxm163 was determined to be 34.4 ± 0.2 mM (Table 1, Fig. S18 and S19†). We note that excitation of the phenol state does not generate a fluorescent signal above background and was not further evaluated in this study. Similar, albeit attenuated, fluorescence quenching and weaker chloride binding affinities are observed at pH 6.5 (86%, Kd = 79.6 ± 0.4 mM) and pH 7 (69%, Kd = 230.1 ± 19.7) (Table 1, Fig. 2C, S8, S9, S18 and S19†). Notably, the molar brightness is more significantly affected by the addition of chloride rather than differences in pH (Table 1, Fig. S20–S26†). We can further connect these data to the kinetics of chloride binding determined with stopped-flow measurements under pseudo-first order conditions (Fig. S27–S29†). Fitting of the kinetic traces to a biexponential model reveals two components as reported previously for avYFP-H148Q (Fig. S33†).21,53 The major and faster on-rate can be ascribed to the binding of chloride whereas the minor and slower on-rate could possibly correspond to a rearrangement in the chromophore environment or weak secondary interactions with the β-barrel but was not investigated in greater detail.54 We find that the on-rate of chloride binding decreases from pH 6 to 7 as follows: 2.5 ± 0.3 M−1 s−1 > 0.8 ± 0.1 M−1 s−1 > 0.3 ± 0.03 M−1 s−1 (Table 1, Fig. S33†).
| pH 6 | pH 6.5 | pH 7 | ||
|---|---|---|---|---|
| Molar extinction coefficient (M−1 cm−1) | Apo | 57![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 067 ± 5930 067 ± 5930 |
65939 ± 2706 |
67845 ± 2818 |
| Cl− | 3574 ± 253 | 10784 ± 627 |
29453 ± 2130 |
|
| Quantum yield | Apo | 0.97 ± 0.03 | 0.92 ± 0.03 | 0.89 ± 0.03 |
| Cl− | 0.08 ± 0.001 | 0.08 ± 0.003 | 0.08 ± 0.001 | |
| Molar brightness | Apo | 54.45 ± 5.71 | 60.99 ± 4.02 | 61.04 ± 4.13 |
| Cl− | 0.29 ± 0.02 | 0.89 ± 0.05 | 2.31 ± 0.15 | |
| K d (mM) | Cl− | 34.4 ± 0.2 | 79.6 ± 0.4 | 230.1 ± 19.7 |
| Br− | 7.9 ± 0.3 | 18.6 ± 0.5 | 46.2 ± 5.3 | |
| I− | 0.7 ± 0.03 | 1.7 ± 0.1 | 6.4 ± 0.1 | |
| NO3− | 3.8 ± 0.1 | 8.9 ± 0.3 | 24.1 ± 0.8 | |
| k on (M−1 s−1) | Cl− | 2.5 ± 0.3 | 0.8 ± 0.1 | 0.3 ± 0.03 |
| Br− | 4.1 ± 0.6 | 1.3 ± 0.2 | 0.8 ± 0.01 | |
| I− | 9.7 ± 0.7 | 3.0 ± 0.2 | 1.6 ± 0.06 | |
| NO3− | 24.0 ± 2.0 | 6.5 ± 0.5 | 2.6 ± 0.3 | |
| k off (s−1) | Cl− | 85 ± 10 | 63 ± 4 | 70 ± 9 |
| Br− | 32 ± 5 | 25 ± 3 | 37 ± 4 | |
| I− | 7.1 ± 0.6 | 5.2 ± 0.4 | 10.2 ± 0.4 | |
| NO3− | 93 ± 7 | 58 ± 4 | 63 ± 7 | |
Given the known plasticity of anion-sensitive fluorescent proteins, the selectivity of GFPxm163 was evaluated with a panel of halides and oxyanions under the same pH conditions tested above. Based on the absorption profiles, bromide, iodide, and nitrate can tune chromophore equilibrium from the phenolate to the phenol state; thus, providing evidence that, like chloride, these anions bind near the chromophore (Fig. S12–S17†).19–22,42,43 Indeed, titrations from pH 6 to 7 show all three anions quench the fluorescence intensity by more than 95% with clear differences in the binding affinities: iodide > nitrate > bromide > chloride (Table 1, Fig. S12–S19†). One noteworthy observation is that this ranking tracks with increasing ion dehydration enthalpies; however, this is not the case for the on-rates of binding: nitrate > iodide > bromide > chloride (Table 1, Fig. S30–S36†).55 Both trends are maintained with increasing pH for each anion, as the degree of fluorescence quenching, binding affinity, and on-rate of binding decrease. Interestingly, the calculated off-rates of binding track in the opposite direction of the ionic radii: iodide > bromide > nitrate ≈ chloride (Table 1).55
We exploited affinity of GFPxm163 for chloride versus iodide to validate it as a sensor in live cells. To do so, we selected the well-established Fischer rat thyroid (FRT) cell line overexpressing the cAMP-activated cystic fibrosis transmembrane conductance regulator (CFTR).22,56 This cell model enables a real-time anion exchange assay that relies on the endogenously expressed sodium-iodide symporter (NIS) and the activation of CFTR to effectively transport both chloride and iodide but not gluconate to the same extent.57,58 Cells transiently overexpressing GFPxm163 and incubated in buffer with chloride show endogenous fluorescence signal in the cytoplasm and nucleus (Fig. 3A–D, S37, ESI Movie 1–4†). The replacement of extracellular chloride with iodide, results in an influx of iodide through the NIS quenching the intracellular fluorescence (∼40%), which is in line with the fact that GFPxm163 is more sensitive to iodide than chloride at pH 7 (Fig. 3E). This response is further enhanced (∼5%) through the activation of CFTR with forskolin (FSK) and readily reversed to baseline levels by replenishing extracellular chloride, suggesting that chloride could displace iodide bound to the sensor. To further probe this, a chloride–gluconate exchange assay was carried out in a similar fashion, however, the intracellular fluorescence does not significantly change (Fig. 3F, ESI Movie 5–10†). Since the binding affinity for chloride to GFPxm163 is relatively weak at pH 7 (Kd = 230.1 ± 19.7 mM), the baseline fluorescence could correspond to the apo form or a partially bound form that does not alter the fluorescence signal. We do note that avYFP-H148Q produces only a 12% increase in the intracellular fluorescence under similar conditions. As such a higher affinity sensor could be used to monitor an efflux of endogenous chloride.21 Moreover, the intracellular pH does not change under the assay conditions tested as indicated by the pH-sensitive dye BCECF (Fig. S38, S39, ESI Movie 11–22†).59–61
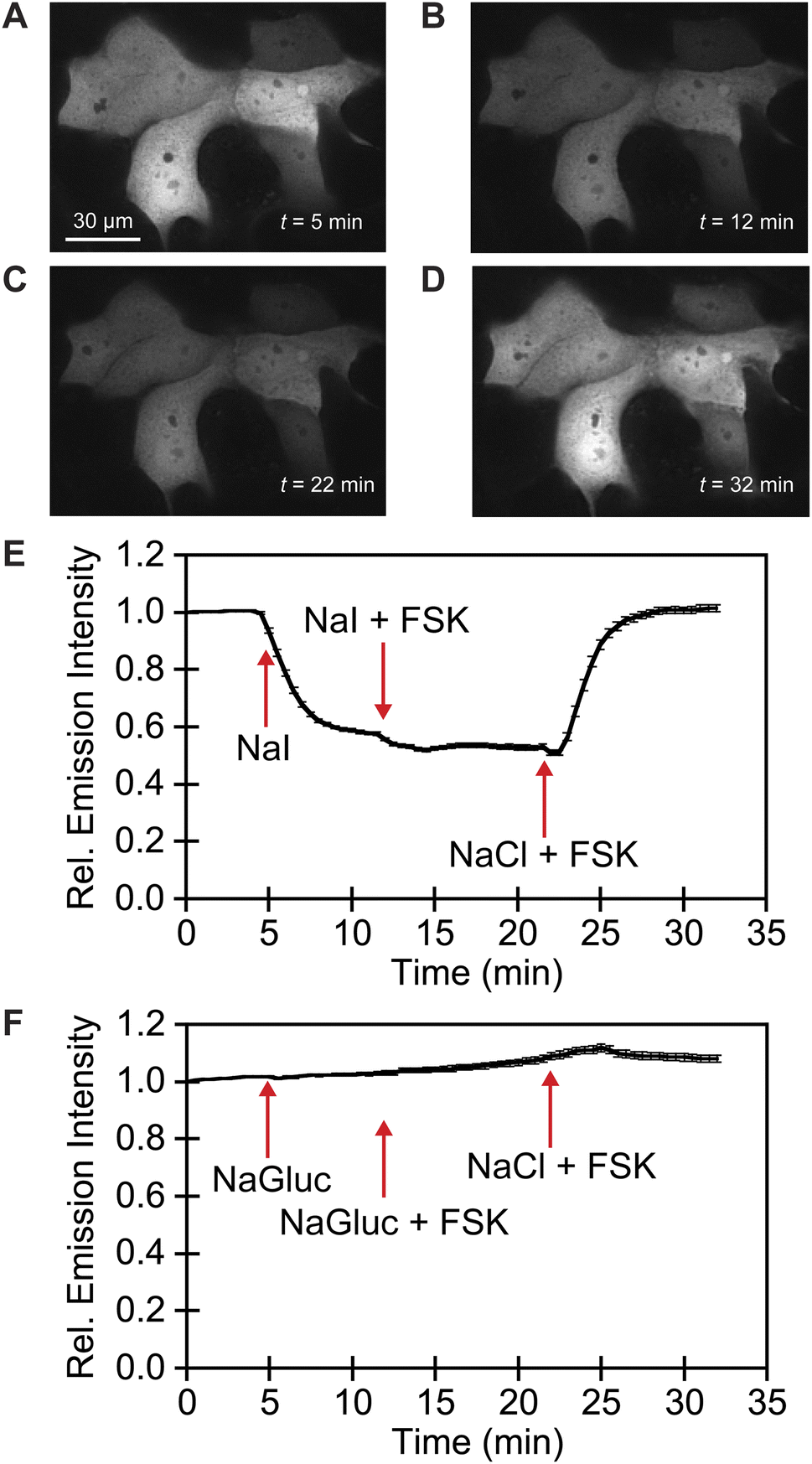 | ||
| Fig. 3 Fluorescence microscopy reveals that GFPxm163 is sensitive to halides in live FRT cells overexpressing CFTR. Representative fluorescence images of cells at 37 °C in a modified live cell imaging solution at pH 7.4 containing (A) 140 mM Cl−, (B) 40 mM Cl−/100 mM I−, (C) 40 mM Cl−/100 mM I− with 20 μM FSK, and (D) 140 mM Cl− with 20 μM FSK. (E) Plot of the normalized median fluorescence intensity over time for the Cl−/I− exchange. The average of all regions of interest (n = 69) from four biological replicates with the S.E.M. is reported. (F) Plot of the normalized median fluorescence intensity over time for the Cl−/Gluc exchange. The average of all regions of interest (n = 168) from six biological replicates with the S.E.M. is reported. The corresponding time-lapse fluorescence microscopy data are shown in ESI Movie 1–10.† Abbreviation: Fischer rat thyroid, FRT; Cystic fibrosis transmembrane conductance regulator, CFTR; Gluconate, Gluc; Forskolin, FSK. | ||
Finally, we evaluated the oligomeric state of GFPxm163 by anchoring the protein to the cytosolic face of the endoplasmic reticulum (ER) through an N-terminal fusion to a transmembrane segment from a rabbit cytochrome P450 (Fig. S40†).62,63 Homo-oligomerization of the fusion protein drives restructuring of the ER from a reticular architecture into an organized smooth ER (OSER). Analysis of 10000 cells reveals that 89.7 ± 1.1% of cells retain normal ER morphology, indicating that GFPxm163 has a low tendency to oligomerize (Fig. 4).50 Taken together, this application showcases that GFPxm163 is a monomeric sensor that provides a reversible yet indirect readout of intracellular chloride.
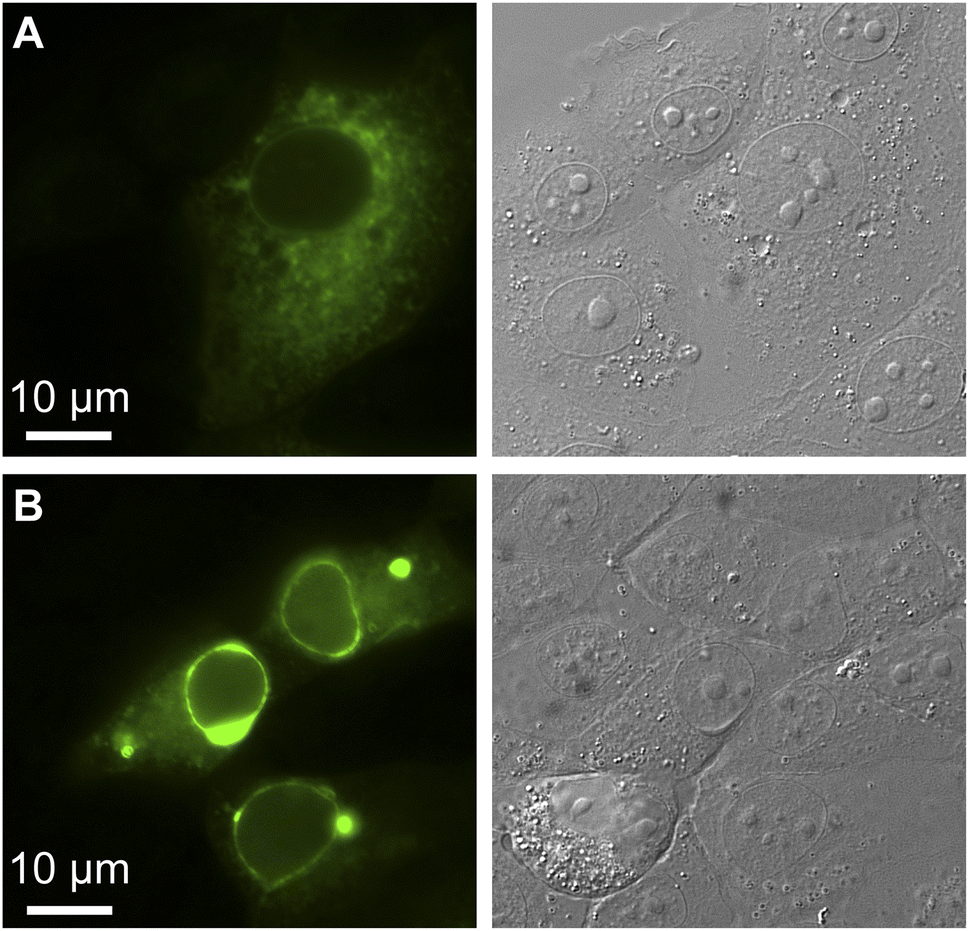 | ||
| Fig. 4 The organized smooth endoplasmic reticulum (OSER) assay reveals that GFPxm163 is a monomer in FRT-CFTR cells (n = 10000, Score = 89.7). Representative images are shown for live FRT-CFTR cells overexpressing CytERM-GFPxm163 with (A) normal and (B) whorled ER structures at 37 °C in a modified live cell imaging solution at pH 7.4. | ||
Conclusion
In summary, we have presented and validated a stepwise bioinformatics-guided strategy to discover GFPxm163 as a new monomeric fluorescent protein sensor for chloride. Spectroscopic characterization shows that the binding of not only chloride but also bromide, iodide, and nitrate rapidly tunes the ground state chromophore equilibrium from the phenolate to the phenol state generating a pH-dependent, turn-off fluorescence response. Furthermore, fluorescence microscopy reveals that GFPxm163 provides a reversible, yet indirect readout of intracellular chloride via iodide exchange in the FRT-CFTR model. In the context of anion sensitive GFPs, GFPxm163 is the first derived from the GFP found in the jellyfish Aequorea macrodactyla. Even though GFPxm163 has a weaker chloride binding affinity at physiological pH than avYFP-H148Q and other widely used sensors, it has a higher molar brightness and is a monomer.22 Because of these properties, GFPxm163 is an ideal starting point for engineering new sensors. With respect to the parent GFPxm, GFPxm163 has only three mutations and thus an underexplored mutational landscape.51,52 To this end, we are using directed evolution to enrich and sample this sequence space for improved and new sensor properties to feedback into our bioinformatics pipeline. Ultimately, this will allow us not only to image cellular chloride but also to identify mutational hotspots to inform the design and discovery of this novel function within the GFP family.Experimental methods
General
Reagents and chemicals were purchased from Sigma-Aldrich, Thermo Fisher Scientific, or VWR International unless otherwise stated.Multiple sequence alignment and percent identity matrix
A global sequence alignment was generated using the sequences from the green fluorescent protein family (GFP; Pfam: PF01353) in Pfam. This was analyzed using Python to identify the only known monomer GFPxm163 (Uniprot ID: Q8WTC8), an engineered variant of GFPxm (Uniprot ID: Q8WP95) that is naturally found in Aequorea macrodactyla.64 The multiple sequence alignment in Fig. 1 and percent identity matrix in Fig. S2† were generated using the Clustal Omega software V1.2.4.65Crystal structures and homology models
The homology models of phiYFP and GFPxm163 were generated using MODELLER V10.1 with the crystal structures of phiYFPv (PDB ID: 4HE4) and avYFP-H148Q (PDB ID: 1F09), respectively.66 The images in Fig. 1 were generated using PyMol V2.5.0.Construction of plasmids
All plasmids used in this study were commercially synthesized by GenScript. For recombinant protein expression, the nucleotide sequence encoding wild-type GFPxm163 was codon-optimized for Escherichia coli and cloned between the NdeI and BamHI restriction sites with an N-terminal polyhistidine tag and C-terminal stop codon in the pET-28a(+)-TEV vector. For live cell fluorescence imaging experiments, the DNA sequence encoding wild-type GFPxm163 was codon-optimized for expression in human cell lines and cloned between the BamHI and EcoRI restriction sites with a C-terminal stop codon in the pcDNA3.1(+) vector. The GFPxm163-pcDNA3.1(+) plasmid will be deposited in Addgene. For the Organized Smooth Endoplasmic Reticulum (OSER) assay, the first 29 amino acids of the endoplasmic reticulum signal anchor membrane protein CytERM with a 17 amino acid linker were added to the N-terminal end of GFPxm163 to anchor the fused protein onto the cytoplasmic side of the ER.62,63 This gene was cloned between the BamH1 and EcoR1 restriction sites with a C-terminal stop codon in the pcDNA3.1(+) vector. The nucleotide and amino acid sequences for each plasmid are shown in Fig. S3, S37, and S40†.Expression of GFPxm163
E. cloni EXPRESS BL21 (DE3) competent cells (Lucigen) were transformed with the GFPxm163-pET-28a(+)-TEV plasmid by electroporation (MicroPulser Electroporator, Bio-Rad Laboratories), plated onto a LB (lysogeny broth; 10 g L−1 tryptone, 5 g L−1 yeast extract, and 10 g L−1 NaCl, Research Products International) agar plate with 50 μg mL−1 kanamycin, and incubated at 37 °C for 18 h. For protein expression, a single colony was picked into 50 mL of LB with 50 μg mL−1 kanamycin and incubated overnight at 37 °C with shaking at 250 rpm. The next day, 24 mL of the overnight culture was inoculated into 600 mL of LB with 50 μg mL−1 kanamycin for protein expression. The culture was incubated at 37 °C with shaking at 250 rpm until the OD600 reached ∼0.7. Following this, protein expression was induced by adding 600 μL of 1 M isopropyl β-D-thiogalactopyranoside (IPTG, Gold Biotechnology) to a final concentration of 1 mM. After incubation at 37 °C with shaking at 250 rpm for 24 h, cells were harvested by centrifugation at 3,000g for 25 min at 4 °C, resuspended in 30 mL of lysis buffer (20 mM Tris at pH 7.5 containing 200 mM NaCl, 2 mM MgCl2, 20 μg mL−1 DNase, and 1 capsule of protease inhibitor per 500 mL of buffer (Pierce)), and stored at −20 °C until further use.Purification of GFPxm163
The frozen cell pellet was thawed in a water bath at room temperature. Once thawed, the cell suspension was sonicated at 30% power amplitude with a 15 s and 45 s on-off pulse cycle for a total of 20 min in an ice bath, followed by ultracentrifugation at 18,000g for 35 min at 4 °C (Optima XPN, Beckman Coulter). The clarified supernatant was loaded through a sample pump of an NGC Quest 10 Chromatography System (Bio-Rad Laboratories) onto a pre-equilibrated 5 mL nickel nitrilotriacetic acid affinity column (HisTrap HP, GE Healthcare) with a running buffer (20 mM Tris buffer at pH 7.5 containing 200 mM NaCl and 30 mM imidazole). The column was washed with the running buffer over 10 column volumes, and then the protein was eluted with a 0–100% linear gradient of an elution buffer (20 mM Tris at pH 7.5 containing 200 mM NaCl and 500 mM imidazole) over 10 column volumes. The fractions with an absorbance at 512 nm were combined and loaded as described above onto a pre-equilibrated desalting column (HiPrep 26/10 Desalting, GE Healthcare) with a desalt buffer (200 mM sodium phosphate buffer at pH 7.5). The resulting fractions with an absorbance at 512 nm were combined and loaded as described above onto a pre-equilibrated size-exclusion column (HiLoad 26/600 Superdex 200 prep grade) using the desalt buffer. A representative size-exclusion chromatography chromatogram is shown in Fig. S4† with a gel filtration protein standard (Bio-Rad Laboratories). The fractions from the single monomer peak were combined and diluted ten-fold and buffer exchanged three times into 10 mM sodium phosphate buffer at pH 6.5 with an Amicon Ultra-15 Centrifugal Filter Unit Ultracel-10 (Millipore) by centrifugation at 3,000g at 4 °C. The resulting protein stock solution was stored at −20 °C until further use. The purity of the buffer-exchanged fractions was determined using sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) as previously described.42 Briefly, an aliquot of the purified protein was diluted to 1 mg mL−1 with distilled water, and 20 μL of the sample was mixed with 5 μL of a 5X Laemmli sample buffer containing 10% β-mercaptoethanol (Bio-Rad Laboratories). The sample was boiled for 5 min at 95 °C and loaded into a 12% polyacrylamide gel. The gel was run in 1X Tris-Glycine-SDS Buffer (TGS 10X solution, Research Products International) at 150 V for 45 min, and then visualized with Coomassie Blue stain. The Coomassie-stained SDS-PAGE gel with all four protein preparations used in this study is shown in Fig. S5.†Protein concentration determination
Stock solutions of GFPxm163 were serially diluted with 50 mM sodium phosphate buffer at pH 7, and a 200 μL aliquot of each solution was transferred to a 96-well UV-Star microtiter plate (Greiner) for analysis with a microplate reader (Spark, Tecan) at room temperature (24–26 °C). For each well, the absorbance intensity was collected from 250–600 nm (2 nm step size, 3.5 nm bandwidth) and corrected by subtracting the absorbance intensity at 600 nm at each point. The dilution factors (d) were plotted versus the corrected absorbance intensities at 280 nm and fitted with a linear regression (R2 > 0.99) to determine the slope for each sample (Gs). To maintain the linearity of the fitting, corrected absorbance values less than 0.1 were plotted. Based on this analysis, the protein concentrations were calculated using the following equations based on the Beer–Lambert law (A = εlc) where A is the absorbance, ε is the extinction coefficient, l is the optical path length and c is the concentration. The extinction coefficient of 23380 M−1 cm−1 was determined using the ProtParam tool in ExPASy using the sequence shown in Fig. S3†.67 The optical pathlength of 0.59 cm was determined by collecting the absorbance intensities at 975 nm (A975) and 900 nm (A900) for a 200 μL sample of water in a well of a 96-well microtiter plate using the microplate reader and an 800 μL sample of water in a 1 cm quartz cuvette using a UV-Vis-NIR spectrophotometer (Cary 7000, Agilent) in the equation below. For each biological replicate, an average of three measurements was used to determine the concentration.| A=Gs × d |
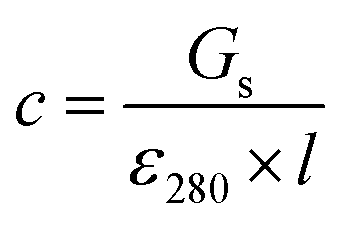
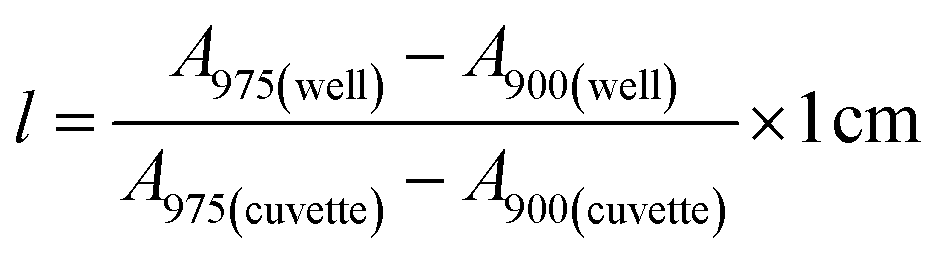
pKa determination
The stock solutions of GFPxm163 were diluted in 10 mM sodium phosphate buffer at pH 6.5 to a final protein concentration of 9.6 μM or 18.4 μM. These new stock solutions were further diluted 8-fold with 50 mM sodium acetate buffer at pH 4, 4.5, 5, and 5.5 and 50 mM sodium phosphate buffer at pH 5.5, 6, 6.5, 7, 7.5, 8, 8.5, and 9 in the absence and presence of sodium chloride to a final protein concentration of 1.2 μM or 2.3 μM protein and 0 or 512 mM sodium chloride. A 200 μL aliquot of each solution was transferred to a 96-well microtiter plate for analysis using a microplate reader at room temperature (24–26 °C). For each well, the absorbance intensity was collected from 350–650 nm (2 nm step size, 3.5 nm bandwidth). The absorbance spectra were corrected by subtracting the absorbance intensity at 600 nm for each point. Fluorescence excitation was provided at 480 nm with (5 nm bandwidth), and the emission intensity was collected from 500–600 nm (2 nm step size, 5 nm bandwidth, 30 flashes). To calculate the pKa values, each pH was plotted versus the corrected absorbance intensity at 512 nm (A512) and fitted to the equation below where Amax is A512 at pH 9, Amin is A512 at pH 4, P− is the deprotonated chromophore, and HP is the protonated chromophore.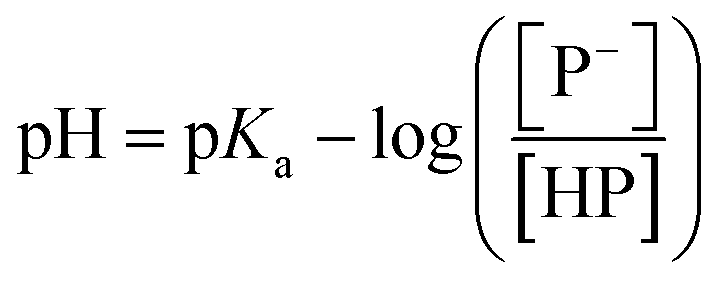
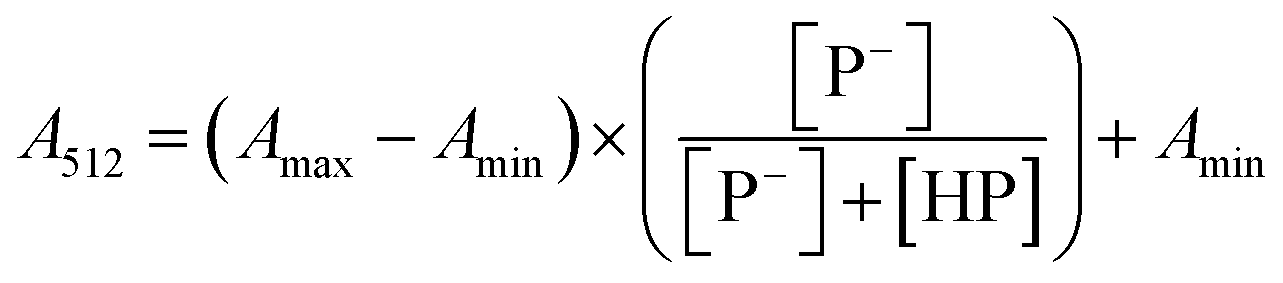
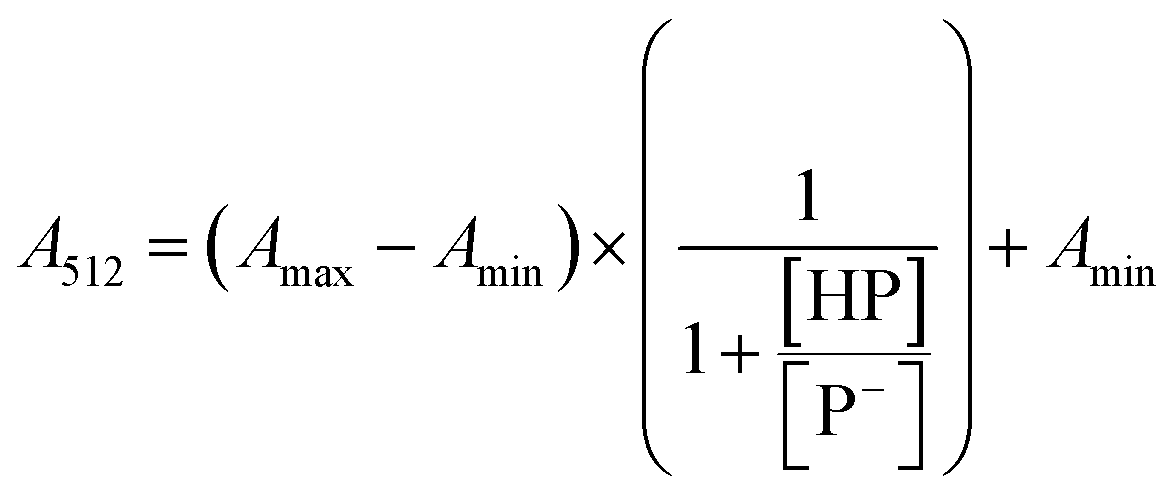
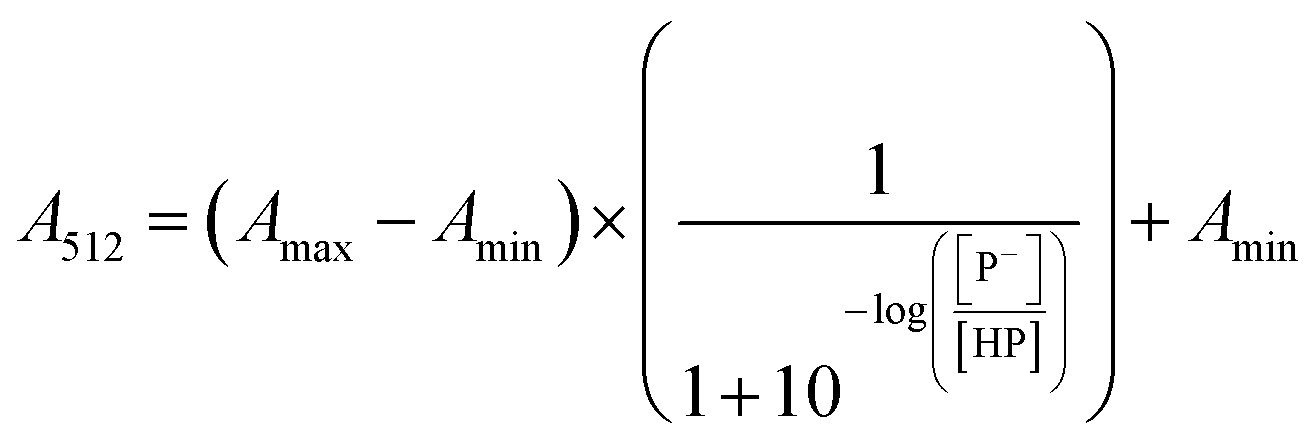
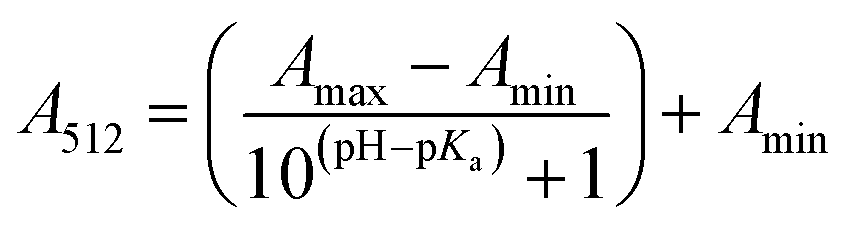
The pKa values were also calculated by plotting the normalized emission intensity at 524 nm (Fn) versus the pH and fitted to the equation below where F is the raw emission intensity at 524 nm, Fmax is F at pH 9, and Fmin is F at pH 4.
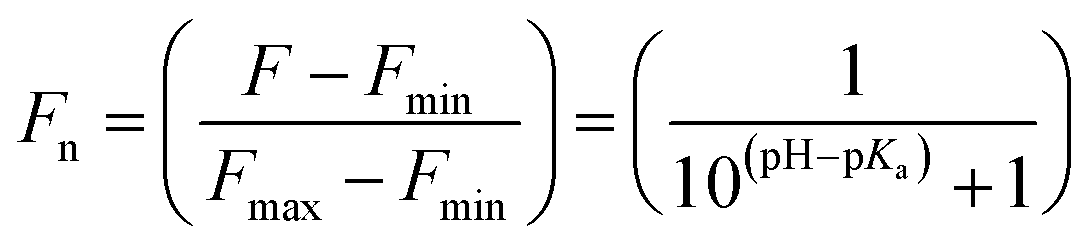
Data is shown for two protein preparations each measured in triplicate and reported as an average with the S.E.M. (Fig. S6 and S7†). The average of the two protein preparations with S.E.M. is reported in Table 1. All errors were propagated by a grouped data standard deviation.
Apparent dissociation constant (Kd) determination
The stock solutions of GFPxm163 were diluted with 50 mM sodium phosphate buffer at pH 6, 6.5, and 7 to a final protein concentration of 8 μM or 13.6 μM. These new stock solutions of GFPxm163 were further diluted 8-fold with 50 mM phosphate at the same pH in the presence of sodium chloride, bromide, iodide, nitrate, and gluconate to make a final concentration of 1 μM or 2.3 μM with 0, 2, 4, 8, 16, 32, 64, 128, 256, 512 mM of the sodium salt for each anion. A 200 μL aliquot of each solution was transferred to a 96-well microtiter plate for analysis using a microplate reader at room temperature (24–26 °C). For each well, the absorbance intensity was collected from 350–650 nm (2 nm step size, 3.5 nm bandwidth) and corrected by subtracting the absorbance intensity at 600 nm for each point. Fluorescence excitation was provided at 480 nm with (5 nm bandwidth), and emission intensity was collected from 500–600 nm (2 nm step size, 5 nm bandwidth, 30 flashes). For each anion at each concentration, the fluorescence intensity (F) at 524 nm was first normalized (Fn), where Fmax is F with 0 mM anion and Fmin is F with 512 mM of iodide at the corresponding pH. Then the normalized fluorescence intensity was used to determine the apparent dissociation constant (Kd) by fitting with SciPy to a single binding site model using the equation below.68 Data is shown for two protein preparations each measured in triplicate and reported as an average with the S.E.M. (Fig. S8–S19†). The average of the two protein preparations with S.E.M. is reported in Table 1. All errors were propagated by a grouped data standard deviation.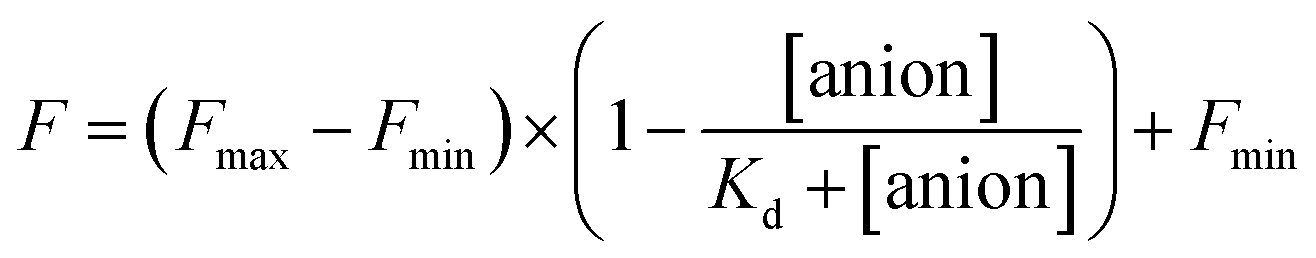
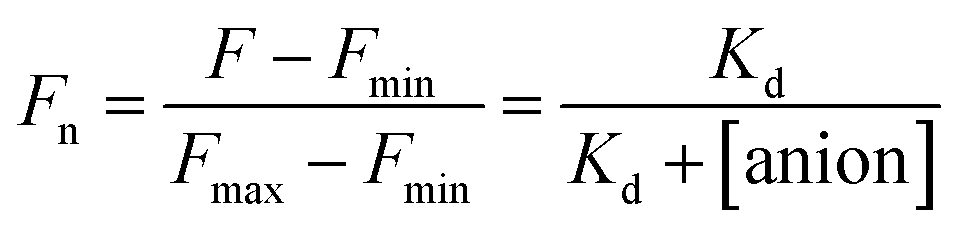
Quantum yield and extinction coefficient determination
Coumarin 153 was used as the reference standard (Φc = 0.25 in 50% ethanol).69 A 1 mM stock solution of Coumarin 153 was prepared and serially diluted in 50% ethanol eight times, and a 200 μL aliquot of each solution was transferred to a 96-well microtiter plate for analysis using a microplate reader at room temperature (24–26 °C). For each well, the absorbance intensity was collected from 400–600 nm (2 nm step size, 3.5 nm bandwidth) and corrected by subtracting the absorbance intensity at 600 nm for each point. Fluorescence excitation was provided at 425 nm with (5 nm bandwidth), and the emission intensity was collected from 440–800 nm (2 nm step size, 5 nm bandwidth, 30 flashes). Then each emission spectrum was integrated from 440–800 nm in Microsoft Excel. The integrated emission responses were plotted versus the corrected absorbance intensities at 425 nm and fitted with a linear regression (R2 > 0.99) to determine the slope for Coumarin 153 (Sc) (Fig. S20†). To maintain the linearity of the fitting, corrected absorbance intensities less than 0.1 were plotted.The stock solution of GFPxm163 was diluted in 50 mM sodium phosphate buffer at pH 6, 6.5, and 7 containing 0 or 512 mM NaCl and then serially diluted with the same buffer eight times, and a 200 μL aliquot of each solution was transferred to a 96-well UV-Star microtiter plate for analysis using a microplate reader at room temperature (24–26 °C). For each well, the absorbance intensity was collected from 250–600 nm (2 nm step size, 2.5 nm bandwidth) and corrected by subtracting the absorbance intensity at 600 nm for each point. Fluorescence excitation was provided at 480 nm with (5 nm bandwidth), and emission intensity was collected from 500–600 nm (2 nm step size, 5 nm bandwidth, 30 flashes). Then each emission spectrum was integrated from 500–600 nm. The integrated emission responses were plotted versus the corrected absorbance intensities at 480 nm and fitted with a linear regression (R2 > 0.99) to determine the slope for each sample (Ss) (Fig. S21–S23†). To maintain the linearity of the fitting, corrected absorbance intensities less than 0.1 were plotted.
Given the refractive indices of the 50% ethanol (ηc = 1.36) and the water (ηs = 1.33), the fluorescence quantum yields of GFPxm163 (Φs) were calculated using the equation below.70,71 The average of three protein preparations each measured in triplicate with S.E.M. is reported in Table 1. All errors were propagated by a grouped data standard deviation.
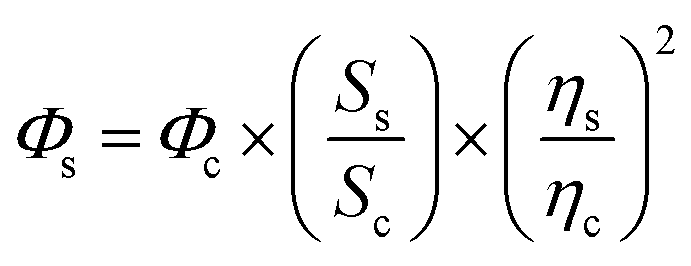
The extinction coefficients of apo and chloride bound GFPxm163 were calculated for the phenolate form at 512 nm (ε512) based on the extinction coefficient of GFPxm163 at 280 nm (ε280). For these calculations, the corrected absorbance intensities at 512 nm and 280 nm for each sample above were plotted versus each other and fitted with a linear regression (R2 > 0.99) to determine the slope for each sample (ΔA512/ΔA280) (Fig. S24–S26†). Based on this analysis, the extinction coefficients were calculated based on the Beer–Lambert law. Since the optical pathlength (l) and protein concentration (c) in each sample were the same, the equation can be simplified as shown below. The average of three protein preparations each measured in triplicate with S.E.M. is reported (Table 1). All errors were propagated by a grouped data standard deviation.
| ΔA = ε × l × Δc |
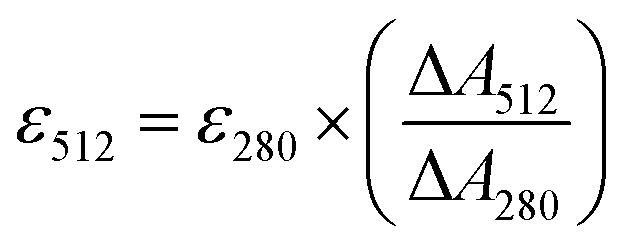
Kinetic rate constant determination
The stock solutions of GFPxm163 were diluted in 50 mM sodium phosphate buffer at pH 6, 6.5, and 7 to a final concentration of ∼2 μM and kept on ice until further use. Stock solutions of sodium chloride, bromide, iodide, nitrate, and gluconate were prepared at five different concentrations in 50 mM sodium phosphate buffer at pH 6, 6.5, and 7. The concentrations of the stock solutions for each anion were minimally at the Kd and spread over a three-fold range from the lowest to highest concentrations. Sodium chloride was tested at the following series of final concentrations: 32, 48, 64, 80, and 96 mM or 64, 96, 128, 160, and 192 mM at pH 6; 80, 120, 160, 200, and 240 mM at pH 6.5; 250, 375, 500, 625, and 750 mM at pH 7. Sodium bromide was tested at the following series of final concentrations: 8, 12, 16, 20, and 24 mM or 16, 24, 32, 40, and 48 mM at pH 6; 20, 30, 40, 50, and 60 mM at pH 6.5; 75, 150, 225, 300, 375, 450, 525, 600, 675, and 750 mM at pH 7. Sodium iodide was tested at the following series of final concentrations: 1, 2, 4, 6, and 8 mM or 2, 4, 8, 12, and 16 mM at pH 6; 20, 30, 40, 50, and 60 mM at pH 6.5; 8, 16, 32, 48, 64, 80, 96, 112, 128, 144, and 160 mM at pH 7. Sodium nitrate was tested at the following series of final concentrations: 4, 6, 8, 10, and 12 mM or 8, 12, 16, 20, and 24 mM at pH 6; 20, 30, 40, 50, and 60 mM for pH 6.5; 24, 36, 48, 60, and 72 mM at pH 7.These experiments were carried out using a stopped-flow spectrometer (SX20, Applied Photophysics) equipped with a 7 liter refrigerated circulator (Polyscience) at 22 °C. A 1.5 mL aliquot of the protein stock solution was loaded into one syringe and 1.5 mL of phosphate buffer, at the same pH, was loaded into the second syringe. The syringe temperature was allowed to equilibrate for 5 min. After this, at least seven co-injections of both syringes were performed to purge and equilibrate the fluidics and sample cell in the system. Following this, the detector baseline fluorescence emission signal of the apo protein was adjusted to 8 V using the auto PM function of the instrument. For each sample, three equal volume co-injections were measured for the kinetics of the apo protein. When necessary, the syringe containing the protein was reloaded from the same protein stock solution. The syringe containing phosphate buffer was exchanged starting with the anion stock solution with the lowest concentration, and the equilibration, injections, and measurements were repeated to test the kinetics. This process was repeated for all concentrations of a given anion and pH, with thorough cleaning between condition tested. Data was collected for two different protein preparations. Excitation was provided at 500 nm (9.3 nm bandwidth), and the emission signal was collected with a 515 nm cutoff filter placed between the sample cell and the fluorescence photomultiplier. The fluorescence was measured until the kinetic process was completed.
All data analysis was carried out and plotted using Python.72 For each fluorescence trace, the first 10 ms was removed to account for the dead time of the instrument (∼2 ms). Then, the fluorescence intensity of each kinetic trace was normalized from 1 to 0 by applying the following function, where S is the emission fluorescence signal.
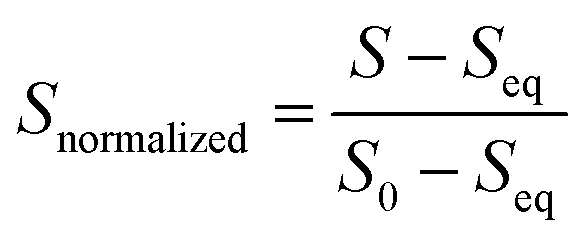
The average and standard deviation for the three co-injections were calculated. This was repeated for each technical replicate, and the overall average was determined with a grouped data standard deviation.
These data were fitted to the following double exponential model, where S is emission fluorescence signal and F is the fractional change of fluorescence signal for the first kinetic process, with a weighted least-squares minimization, where initial parameters were calculated based on the solution to a simple linear regression arising from an integral transformation of the unweighted data for each anion at each concentration and pH:73–75
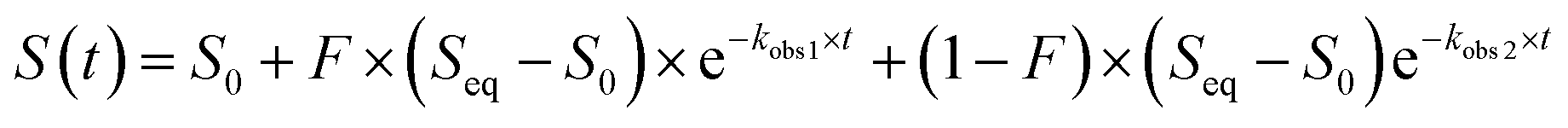
Since all measurements were carried out under pseudo-first order conditions, the kobs1 rate constants were linearly related to concentration. As such, the on (k1) rate of anion binding was determined using the following relationship:53
| kobs1 = k1 × [anion] + k−1 |
The on-rate of binding was determined for each protein preparation. The off-rate was calculated based on the on-rate from stopped-flow measurements and the Kd determined from the microplate reader measurements as follows:
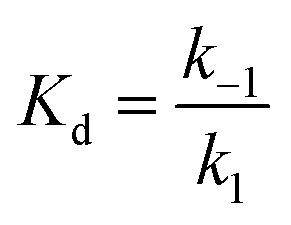
For Fig. S27–S36,† data is shown for two protein preparations across three technical replicates each with three co-injections. Data from each protein preparation is reported as an average with the standard deviation. For Table 1, k1 and k−1 constants are reported as an average with S.E.M. between the two protein preparations. All errors were propagated by a grouped data standard deviation.
Preparation of cell cultures
Fischer rat thyroid cells overexpressing the cystic fibrosis transmembrane conductance regulator (FRT-CFTR) were kindly gifted by Dr Jeong S. Hong at Emory University and cultured according to procedures from the Cystic Fibrosis Foundation (https://www.cff.org/cell-model-resources). Briefly, cells were maintained in Coon's modified Ham's F-12 medium supplemented with 10% fetal bovine serum (Corning), 100 μg mL−1 hygromycin B (Gibco), and 100 μg mL−1 Zeocin (Gibco) in a T-25 culture flask at 37 °C, 5% CO2. For passaging and plating, cells were trypsinized with 3 mL 0.05% trypsin–EDTA (Gibco) for 15 min at 37 °C, 5% CO2. The trypsin was neutralized with 6 mL media, and then the cells were collected by centrifugation at 200g for 5 min at room temperature. Then the media was aspirated, and the cell pellets were resuspended in fresh media. An aliquot of the collected cells was stained with Trypan blue for counting using a hemocytometer prior to plating. All experiments were carried out with cells at a passage number less than twelve.Ion exchange assays for GFPxm163 with fluorescence microscopy
For reverse transfection, 1 μg of the industrial grade GFPxm163-pcDNA3.1(+) plasmid (GenScript), 1.5 μL of Lipofectamine 3000, and 2 μL of P3000 reagent were complexed in 250 μL of Opti-MEM media for 15 min at room temperature and added into a 35 mm glass-bottom dishes (10 mm micro-well, #1.5 coverslip, Cellvis). Cells were plated at a seeding density of ∼2.5 × 105 cells per dish with 1.75 mL of media and were incubated for 3 days at 37 °C, 5% CO2.Before imaging, the cells were washed twice with 2 mL of the pre-warmed modified live cell imaging solution (20 mM HEPES buffer at pH 7.4 containing 140 mM NaCl, 2.5 mM KCl, 1.8 mM CaCl2,1 mM MgCl2, and 10 mM glucose) and incubated for 30 min at 37 °C on the stage top incubator (Tokai Hit) prior to imaging at 37 °C. All modified live cell imaging solutions were adjusted to ∼300 mOsm with distilled water using a vapor pressure osmometer (VAPRO, ELITechGroup). All fluorescence and differential interference contrast (DIC) images were collected using an inverted fluorescence microscope (IX83, Olympus) equipped with a light engine (Spectra X, Lumencor) at 37 °C. For image acquisition, a 40X oil immersion objective with a numerical aperture of 1.4 and a working distance of 0.17 mm and a custom filter set for EGFP (Chroma) were used. This filter set consists of a 470/40 nm excitation filter for excitation at 480 nm, a 495 nm dichroic beamsplitter, and a 525/50 nm emission filter to detect the emission at 524 nm. Excitation was provided with 25% LED power at level 1 and an exposure time of 60–100 ms. The resolution of the camera was set to 64 × 64 pixels with 4 × 4 binning to enhance signal-to-noise ratio.
The following methods were adapted from previous studies.21 At the start of the chloride–iodide exchange experiment, the imaging solutions were pre-warmed and exchanged using an automated perfusion system. First, 8 mL of modified live cell imaging solution supplemented with iodide (20 mM HEPES buffer at pH 7.4 containing 40 mM NaCl, 100 mM NaI, 2.5 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, and 10 mM glucose) was perfused into the dish at 4 mL min−1, followed by an incubation step for 5 min. Next, 8 mL of the modified live cell imaging solution supplemented with iodide and 20 μM forskolin (FSK, EMD Millipore; 20 mM HEPES buffer at pH 7.4 containing 40 mM NaCl, 100 mM NaI, 2.5 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, 10 mM glucose, and 20 μM FSK from a 20 mM stock in DMSO) was perfused into the dish at 4 mL min−1, followed by an incubation step for 7 min. Finally, 8 mL of modified live cell imaging solution supplemented with FSK (20 mM HEPES buffer at pH 7.4 containing 140 mM NaCl, 2.5 mM KCl, 1.8 mM CaCl2, 1 mM MgCl2, 10 mM glucose, and 20 μM FSK) was perfused into the dish at 4 mL min−1, followed by an incubation step for 7 min All fluorescence and DIC images were collected every 30 s with a ZDC at 37 °C. Four biological replicates from varying cell passage numbers were imaged. The chloride–gluconate exchange experiments were carried out using the same protocol, but the modified live cell imaging solution was supplemented with 140 mM sodium gluconate in place of sodium iodide (20 mM HEPES buffer at pH 7.4 containing 140 mM NaGluc, 2.5 mM KCl, 1.8 mM CaCl2, 1.0 mM MgCl2, and 10 mM glucose). Six biological replicates from varying cell passage numbers were imaged. All images were analyzed as described below (Fig. 3 and ESI Movie 1–10†).
Dye-based pH calibration assay with fluorescence microscopy
Cells were plated at a seeding density of ∼5 × 105 cells in 35 mm glass-bottom dishes (10 mm micro-well, #1.5 coverslip, Cellvis) with 2 mL of media and grown for at least 1 day at 37 °C, 5% CO2. The next day, cells were washed twice with 2 mL of the pre-warmed modified live cell imaging solution and stained with 2 mL of the same buffer supplemented with 5 μM BCECF-AM (1 mM stock in DMSO) for 1 h at 37 °C on the stage top incubator. After staining, the cells were washed twice with 2 mL of the pre-warmed modified live cell imaging solution. The cells were then incubated in a clamping buffer at pH 8 (10 mM HEPES buffer containing 137 mM KCl, 0.8 mM CaCl2, 5 μM nigericin from a 20 mM stock in DMSO, and 5 μM valinomycin from a 10 mM stock in DMSO) for 30 min at 37 °C on the stage top incubator, prior to imaging at 37 °C. All clamping buffers were adjusted to ∼288 mOsm with distilled water using the vapor pressure osmometer before adding the ionophores. For image acquisition, a 20× air objective with a numerical aperture of 0.7 and a working distance of 1.6 mm and a custom filter set for BCECF (Chroma) were used. This filter set consists of two excitation filters, 434/21 nm and 495/10 nm, for excitation at 440 nm and 490 nm, a 510 nm dichroic beamsplitter, and a 540/40 nm emission filter to detect the emission at 535 nm. Excitation was provided with 25% power at level 1 and an exposure time of 200 ms. The resolution of the camera was set to 1024 × 1024 pixels.The following methods were adapted from previous studies.57 In solution, BCECF is excitation ratiometric (440 nm and 490 nm) and has a single emission maximum (535 nm).56 At the start of the experiment, the offset for Z-Drift Compensation (ZDC) was set based on the fluorescence signal for the 490 nm excitation. Following this, the buffer was exchanged on stage with 2 mL of each clamping buffer in the following order: pH 7.5, 7, 6.5, 6, and 8. The cells were incubated for 5 min at 37 °C between each buffer, followed by collecting fluorescence and DIC images with ZDC at 37 °C. Six biological replicates from varying cell passage numbers were imaged. All images were analyzed as described below (Fig. S38, ESI Movie 11–16†).
Dye-based pH assay for Cl−/I− exchange with fluorescence microscopy. Cells were plated and stained with BCECF-AM as described above. After staining, the cells were washed twice with 2 mL of the pre-warmed modified live cell imaging solution and incubated for 30 min at 37 °C on the stage top incubator, prior to imaging at 37 °C. For image acquisition, the 20× air objective and the BCECF filter set described above were used. Excitation was provided with 25% LED power at level 1 and an exposure time of 200 ms. The resolution of the camera was set to 1024 × 1024 pixels. At the start of the experiment, the imaging solutions were pre-warmed and exchanged as described above in the ion exchange assay for GFPxm163. All fluorescence and DIC images were collected every 1 min with a ZDC at 37 °C. Six biological replicates from varying cell passage numbers were imaged. All images were analyzed as described below (Fig. S39 and ESI Movie 17–22†).
OSER assay for CytERM-GFPxm163 with fluorescence microscopy
Cells were plated at a seeding density of ∼5 × 105 cells in 35 mm glass-bottom dishes (20 mm micro-well, #1.5 coverslip, MatTek) with 2 mL of media and grown for at least 1 day at 37 °C, 5% CO2 prior to transfection. The next day, 1 μg of the industrial grade CytERM-GFPxm163-pcDNA3.1(+) plasmid (Genscript), 1.5 μL of Lipofectamine 3000, and 2 μL of the P3000 reagent were complexed in 250 μL of Opti-MEM media for 15 min at room temperature. Following this, cells were treated with the complex and incubated for 24 h at 37 °C, 5% CO2. Prior to fluorescence microscopy, cells were washed twice with 2 mL of a pre-warmed modified live cell imaging solution and incubated for 10 min at 37 °C on a stage top incubator. For image acquisition, the 40× oil immersion objective and the EGFP filter set were used. Excitation was provided with 100% LED power at level 1 and an exposure time of ∼70 ms. The resolution of the camera was set to 1024 × 1024 pixels. Cells that were flat, mononuclear, and nonmitotic were selected for counting (until 104 fluorescent cells) as previously described using the built-in Cellsens Software (Olympus). Six biological replicates from varying passage numbers were processed (Fig. 4).Image process and analysis
Fiji is Just ImageJ software (Fiji v2.0) was used to process and analyze all fluorescence imaging experiments. First, images collected over time were visualized with the DragDrop function in the Olympus Viewer plugin and concatenated into one stack. All channels were separated by the Split Channels function, and the fluorescence channels were processed by the Sharpen function. The sharpened image stacks were aligned using the StackReg function in the Registration plugin. For the fluorescence channel, a mask was created based on the maximum intensity Z-projection. The threshold on each mask was manually adjusted to filter out background and oversaturated signals. The resulting mask was analyzed by the Analyze Particles function to mark the cells as regions of interest (ROIs) for analysis. ROIs greater than 5 μm with circularity between 0 and 0.5, excluding the edges of the field were selected. The DIC images were used to confirm the selection of the ROIs. Specifically, cells that were overlapping or in close contact were considered as one ROI and not selected by the software were manually added as an ROI. The selected ROIs were applied to the stacks with the ROI manager and analyzed through Multi Measure to calculate the median fluorescence intensity of each ROI over time. For the BCECF experiments, the images were processed as described above except all aligned stacks were processed through Subtract Background function before creating the mask. The mask corresponding to the 490 nm excitation channel was used to mark the ROIs on both fluorescence channels.For GFPxm163 analysis, the median fluorescence intensity of each ROI over time was normalized to the median of its initial fluorescence intensity for that biological replicate. The normalized ROI fluorescence intensities from all biological replicates were averaged for reporting the fluorescence change over time in Fig. 3. For the BCECF analysis, the median fluorescence intensity of each ROI from the 490 nm excitation channel over time was divided by its median fluorescence intensity from the 440 nm excitation channel over time to calculate the emission ratio (Ex490/Ex440). To create a calibration curve, the pH was plotted versus the averaged emission ratio for a linear fit (R2 = 0.99) (Fig. S38†). This curve corresponds to the linear part of the sigmoidal pKa curve. The average intracellular pH change over time from the dye-based pH assay for Cl−/I− exchange is shown in Fig. S39† based on the calibration curve above.
Data availability
The data for this manuscript is provided in the main text and ESI† and can be directly requested from the corresponding author.Author contributions
W. P., C. C. M., H. K., J. K. L., and S. C. D. designed and carried out experiments and data analysis. J. N. T. and S. M. P. contributed technical expertise for live-cell imaging experiments and data analysis. S. A. and G. M. contributed technical expertise for stopped-flow experiments and data analysis. W. P. and S. C. D. wrote the manuscript with input from all co-authors.Conflicts of interest
A provisional patent application has been filed.Acknowledgements
We thank members of the Dodani Lab and Dr Faruck Morcos for helpful discussions. We also thank Dr Jeong Hong from Emory University and the Cystic Fibrosis Foundation for providing stocks of the FRT-CFTR cells. G. M. acknowledges support from the Welch Foundation (AT-1935-20170325 and AT-2073-20210327) and the National Institute of General Medical Sciences of the National Institutes of Health (R35GM128704). S. C. D. acknowledges support from UT Dallas, the Welch Foundation (AT-1918-20170325, AT-2060-20210327), and the National Institute of General Medical Sciences of the National Institutes of Health (R35GM128923). This work is the sole responsibility of the authors and does not represent the views of the funding agencies.References
- R. Y. Tsien, The Green Fluorescent Protein, Annu. Rev. Biochem., 1998, 67, 509–544 CrossRef CAS PubMed.
- M. V. Matz, A. F. Fradkov, Y. A. Labas, A. P. Savitsky, A. G. Zaraisky, M. L. Markelov and S. A. Lukyanov, Fluorescent Proteins from Nonbioluminescent Anthozoa Species, Nat. Biotechnol., 1999, 17, 969–973 CrossRef CAS PubMed.
- A. Salih, A. Larkum, G. Cox, M. Kühl and O. Hoegh-Guldberg, Fluorescent Pigments in Corals Are Photoprotective, Nature, 2000, 408, 850–853 CrossRef CAS PubMed.
- T. J. F. P. Lambert, A Community-Editable Fluorescent Protein Database, Nat. Methods, 2019, 16, 277–278 CrossRef CAS PubMed.
- K. Lu, C. Q. Vu, T. Matsuda and T. Nagai, Fluorescent Protein-Based Indicators for Functional Super-Resolution Imaging of Biomolecular Activities in Living Cells, Int. J. Mol. Sci., 2019, 20, 5784 CrossRef CAS PubMed.
- A. Pal and L. Tian, Imaging Voltage and Brain Chemistry with Genetically Encoded Sensors and Modulators, Curr. Opin. Chem. Biol., 2020, 57, 166–176 CrossRef CAS PubMed.
- E. P. S. Pratt, L. J. Damon, K. J. Anson and A. E. Palmer, Tools and Techniques for Illuminating the Cell Biology of Zinc, Biochim. Biophys. Acta, Mol. Cell Res., 2021, 1868, 118865 CrossRef CAS PubMed.
- Y. Nasu, Y. Shen, L. Kramer and R. E. Campbell, Structure- and Mechanism-Guided Design of Single Fluorescent Protein-Based Biosensors, Nat. Chem. Biol., 2021, 17, 509–518 CrossRef CAS PubMed.
- M. Zimmer, Green Fluorescent Protein (GFP): Applications, Structure, and Related Photophysical Behavior, Chem. Rev., 2002, 102, 759–781 CrossRef CAS PubMed.
- Y. A. Labas, N. G. Gurskaya, Y. G. Yanushevich, A. F. Fradkov, K. A. Lukyanov, S. A. Lukyanov and M. V. Matz, Diversity and Evolution of the Green Fluorescent Protein Family, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 4256–4261 CrossRef CAS PubMed.
- J. Wiedenmann, A. Schenk, C. Röcker, A. Girod, K. D. Spindler and G. U. Nienhaus, A Far-Red Fluorescent Protein with Fast Maturation and Reduced Oligomerization Tendency from Entacmaea Quadricolor (Anthozoa, Actinaria), Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 11646–11651 CrossRef CAS PubMed.
- S. Karasawa, T. Araki, M. Yamamoto-Hino and A. Miyawaki, A Green-Emitting Fluorescent Protein from Galaxeidae Coral and Its Monomeric Version for Use in Fluorescent Labeling, J. Biol. Chem., 2003, 278, 34167–34171 CrossRef CAS PubMed.
- D. A. Shagin, E. V. Barsova, Y. G. Yanushevich, A. F. Fradkov, K. A. Lukyanov, Y. A. Labas, T. N. Semenova, J. A. Ugalde, A. Meyers, J. M. Nunez, E. A. Widder, S. A. Lukyanov and M. V. Matz, GFP-like Proteins as Ubiquitous Metazoan Superfamily: Evolution of Functional Features and Structural Complexity, Mol. Biol. Evol., 2004, 21, 841–850 CrossRef CAS PubMed.
- S. Karasawa, T. Araki, T. Nagai, H. Mizuno and A. Miyawaki, Cyan-Emitting and Orange-Emitting Fluorescent Proteins as a Donor/Acceptor Pair for Fluorescence Resonance Energy Transfer, Biochem. J., 2004, 381, 307–312 CrossRef CAS PubMed.
- N. O. Alieva, K. A. Konzen, S. F. Field, E. A. Meleshkevitch, M. E. Hunt, V. Beltran-Ramirez, D. J. Miller, J. Wiedenmann, A. Salih and M. V. Matz, Diversity and Evolution of Coral Fluorescent Proteins, PLoS One, 2008, 3, e2680 CrossRef PubMed.
- E. C. Greenwald, S. Mehta and J. Zhang, Genetically Encoded Fluorescent Biosensors Illuminate the Spatiotemporal Regulation of Signaling Networks, Chem. Rev., 2018, 118, 11707–11794 CrossRef CAS PubMed.
- A. A. Pakhomov and V. I. Martynov, GFP Family: Structural Insights into Spectral Tuning, Chem. Biol., 2008, 15, 755–764 CrossRef CAS PubMed.
- R. M. Wachter and S. J. Remington, Sensitivity of the Yellow Variant of Green Fluorescent Protein to Halides and Nitrate, Curr. Biol., 1999, 9, 628–629 CrossRef.
- R. M. Wachter, D. Yarbrough, K. Kallio and S. J. Remington, Crystallographic and Energetic Analysis of Binding of Selected Anions to the Yellow Variants of Green Fluorescent Protein, J. Mol. Biol., 2000, 301, 157–171 CrossRef CAS PubMed.
- D. Arosio, G. Garau, F. Ricci, L. Marchetti, R. Bizzarri, R. Nifosì and F. Beltram, Spectroscopic and Structural Study of Proton and Halide Ion Cooperative Binding to GFP, Biophys. J., 2007, 93, 232–244 CrossRef CAS PubMed.
- S. Jayaraman, P. Haggie, R. M. Wachter, S. J. Remington and A. S. Verkman, Mechanism and Cellular Applications of a Green Fluorescent Protein-Based Halide Sensor, J. Biol. Chem., 2000, 275, 6047–6050 CrossRef CAS PubMed.
- L. J. V. Galietta, P. M. Haggie and A. S. Verkman, Green Fluorescent Protein-Based Halide Indicators with Improved Chloride and Iodide Affinities, FEBS Lett., 2001, 499, 220–224 CrossRef CAS PubMed.
- P. Bregestovski, Genetically Encoded Optical Sensors for Monitoring of Intracellular Chloride and Chloride-Selective Channels Activity, Front. Mol. Neurosci., 2009, 2, 15 Search PubMed.
- M. Zajac, K. Chakraborty, S. Saha, V. Mahadevan, D. T. Infield, A. Accardi, Z. Qiu and Y. Krishnan, What Biologists Want from Their Chloride Reporters - A Conversation between Chemists and Biologists, J. Cell Sci., 2020, 133, jcs240390 CrossRef CAS PubMed.
- K. Baek, K. Ji, W. Peng, S. M. Liyanaarachchi and S. C. Dodani, The Design and Evolution of Fluorescent Protein-Based Sensors for Monoatomic Ions in Biology, Protein Eng., Des. Sel., 2021, 34, gzab023 CrossRef PubMed.
- T. Kuner and G. J. Augustine, A Genetically Encoded Ratiometric Indicator for Chloride: Capturing Chloride Transients in Cultured Hippocampal Neurons, Neuron, 2000, 27, 447–459 CrossRef CAS PubMed.
- R. Bizzarri, C. Arcangeli, D. Arosio, F. Ricci, P. Faraci, F. Cardarelli and F. Beltram, Development of a Novel GFP-Based Ratiometric Excitation and Emission pH Indicator for Intracellular Studies, Biophys. J., 2006, 90, 3300–3314 CrossRef CAS PubMed.
- O. Markova, M. Mukhtarov, E. Real, Y. Jacob and P. Bregestovski, Genetically Encoded Chloride Indicator with Improved Sensitivity, J. Neurosci. Methods, 2008, 170, 67–76 CrossRef CAS PubMed.
- D. Arosio, F. Ricci, L. Marchetti, R. Gualdani, L. Albertazzi and F. Beltram, Simultaneous Intracellular Chloride and pH Measurements Using a GFP-Based Sensor, Nat. Methods, 2010, 7, 516–518 CrossRef CAS PubMed.
- J. V. Raimondo, B. Joyce, L. Kay, T. Schlagheck, S. E. Newey, S. Srinivas and C. J. Akerman, A Genetically-Encoded Chloride and pH Sensor for Dissociating Ion Dynamics in the Nervous System, Front. Cell. Neurosci., 2013, 7, 202 CAS.
- J. S. Grimley, L. Li, W. Wang, L. Wen, L. S. Beese, H. W. Hellinga and G. J. Augustine, Visualization of Synaptic Inhibition with an Optogenetic Sensor Developed by Cell-Free Protein Engineering Automation, J. Neurosci., 2013, 33, 16297–16309 CrossRef CAS PubMed.
- S. Zhong, D. Navaratnam and J. Santos-Sacchi, A Genetically-Encoded YFP Sensor with Enhanced Chloride Sensitivity, Photostability and Reduced pH Interference Demonstrates Augmented Transmembrane Chloride Movement by Gerbil Prestin (SLC26a5), PLoS One, 2014, 9, e99095 CrossRef PubMed.
- J. M. Paredes, A. I. Idilli, L. Mariotti, G. Losi, L. R. Arslanbaeva, S. S. Sato, P. Artoni, J. Szczurkowska, L. Cancedda, G. M. Ratto, G. Carmignoto and D. Arosio, Synchronous Bioimaging of Intracellular pH and Chloride Based on LSS Fluorescent Protein, ACS Chem. Biol., 2016, 11, 1652–1660 CrossRef CAS PubMed.
- Z. Qiu, A. E. Dubin, J. Mathur, B. Tu, K. Reddy, L. J. Miraglia, J. Reinhardt, A. P. Orth and A. Patapoutian, SWELL1, a Plasma Membrane Protein, Is an Essential Component of Volume-Regulated Anion Channel, Cell, 2014, 157, 447–458 CrossRef CAS PubMed.
- J. Yang, J. Chen, M. Del Carmen Vitery, J. Osei-Owusu, J. Chu, H. Yu, S. Sun and Z. Qiu, PAC, an Evolutionarily Conserved Membrane Protein, Is a Proton-Activated Chloride Channel, Science, 2019, 364, 395–399 CrossRef CAS PubMed.
- F. Ullrich, S. Blin, K. Lazarow, T. Daubitz, J. P. von Kries and T. J. Jentsch, Identification of TMEM206 Proteins as Pore of PAORAC/ASOR Acid-Sensitive Chloride Channels, Elife, 2019, 8, e49187 CrossRef CAS PubMed.
- C. Lodovichi, G. M. Ratto, A. J. Trevelyan and D. Arosio, Genetically Encoded Sensors for Chloride Concentration, J. Neurosci. Methods, 2022, 368, 109455 CrossRef CAS PubMed.
- J. N. Schellinger, Q. Sun, J. M. Pleinis, S.-W. An, J. Hu, G. Mercenne, I. Titos, C.-L. Huang, A. Rothenfluh and A. R. Rodan, Chloride Oscillation in Pacemaker Neurons Regulates Circadian Rhythms through a Chloride-Sensing WNK Kinase Signaling Cascade, Curr. Biol., 2022, 32, 1429–1438 CrossRef CAS PubMed.
- J. Yang, L. Wang, F. Yang, H. Luo, L. Xu, J. Lu, S. Zeng and Z. Zhang, MBeRFP, an Improved Large Stokes Shift Red Fluorescent Protein, PLoS One, 2013, 8, e64849 CrossRef CAS PubMed.
- R. Salto, M. D. Giron, V. Puente-Muñoz, J. D. Vilchez, L. Espinar-Barranco, J. Valverde-Pozo, D. Arosio and J. M. Paredes, New Red-Emitting Chloride-Sensitive Fluorescent Protein with Biological Uses, ACS Sensors, 2021, 6, 2563–2573 CrossRef CAS PubMed.
- K. Chakraborty, K. Leung and Y. Krishnan, High Lumenal Chloride in the Lysosome Is Critical for Lysosome Function, Elife, 2017, 6, e28862 CrossRef PubMed.
- J. N. Tutol, W. Peng and S. Dodani, Discovery and Characterization of a Naturally Occurring, Turn-on Yellow Fluorescent Protein Sensor for Chloride, Biochemistry, 2019, 58, 31–35 CrossRef CAS PubMed.
- J. N. Tutol, H. C. Kam and S. C. Dodani, Identification of mNeonGreen as a pH-Dependent, Turn-On Fluorescent Protein Sensor for Chloride, ChemBioChem, 2019, 20, 1759–1765 CAS.
- A. S. Verkman and L. J. V. Galietta, Chloride Transport Modulators as Drug Candidates, Am. J. Physiol., 2021, 321, C932–C946 CrossRef CAS PubMed.
- J. N. Tutol, J. Lee, H. Chi, F. N. Faizuddin, S. S. Abeyrathna, Q. Zhou, F. Morcos, G. Meloni and S. C. Dodani, A Single Point Mutation Converts a Proton-Pumping Rhodopsin into a Red-Shifted, Turn-on Fluorescent Sensor for Chloride, Chem. Sci., 2021, 12, 5655–5663 RSC.
- H. Chi, Q. Zhou, J. N. Tutol, S. M. Phelps, J. Lee, P. Kapadia, F. Morcos and S. C. Dodani, Coupling a Live Cell Directed Evolution Assay with Coevolutionary Landscapes to Engineer an Improved Fluorescent Rhodopsin Chloride Sensor, ACS Synth. Biol., 2022, 11, 1627–1638 CrossRef CAS PubMed.
- K. Ji, K. Baek, W. Peng, K. A. Alberto, H. Torabifard, S. O. Nielsen and S. C. Dodani, Biophysical and in Silico Characterization of NrtA: A Protein-Based Host for Aqueous Nitrate and Nitrite Recognition, Chem. Commun., 2022, 58, 965–968 RSC.
- N. V. Pletneva, V. Z. Pletnev, E. Souslova, D. M. Chudakov, S. Lukyanov, V. I. Martynov, S. Arhipova, I. Artemyev, A. Wlodawer, Z. Dauter and S. Pletnev, Yellow Fluorescent Protein phiYFPv (Phialidium): Structure and Structure-Based Mutagenesis, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2013, 69, 1005–1012 CrossRef CAS PubMed.
- C. Chen, J. N. Tutol, L. Tang, L. Zhu, W. S. Y. Ong, S. C. Dodani and C. Fang, Excitation Ratiometric Chloride Sensing in a Standalone Yellow Fluorescent Protein Is Powered by the Interplay between Proton Transfer and Conformational Reorganization, Chem. Sci., 2021, 12, 11382–11393 RSC.
- N. C. Shaner, G. G. Lambert, A. Chammas, Y. Ni, P. J. Cranfill, M. A. Baird, B. R. Sell, J. R. Allen, R. N. Day, M. Israelsson, M. W. Davidson and J. Wang, A Bright Monomeric Green Fluorescent Protein Derived from Branchiostoma Lanceolatum, Nat. Methods, 2013, 10, 407–409 CrossRef CAS PubMed.
- N.-S. Xia, W.-X. Luo, J. Zhang, X.-Y. Xie, H.-J. Yang, S.-W. Li, M. Chen and M.-H. Ng, Bioluminescence of Aequorea Macrodactyla, a Common Jellyfish Species in the East China Sea, Mar. Biotechnol., 2002, 4, 155–162 CrossRef CAS PubMed.
- W. X. Luo, T. Cheng, B. Q. Guan, S. W. Li, J. Miao, J. Zhang and N. S. Xia, Variants of Green Fluorescent Protein GFPxm, Mar. Biotechnol., 2006, 8, 560–566 CrossRef CAS PubMed.
- X. Zheng, C. Bi, Z. Li, M. Podariu and D. S. Hage, Analytical Methods for Kinetic Studies of Biological Interactions: A Review, J. Pharm. Biomed. Anal., 2015, 113, 163–180 CrossRef CAS PubMed.
- H. E. Seward, J. Basran, R. Denton, M. Pfuhl, F. W. Muskett and C. R. Bagshaw, Halide and Proton Binding Kinetics of Yellow Fluorescent Protein Variants, Biochemistry, 2013, 52, 2482–2491 CrossRef CAS PubMed.
- Y. Marcus, Ions in Solution and Their Solvation, John Wiley & Sons, Inc, Hoboken, NJ, 2015 Search PubMed.
- D. N. Sheppard, M. R. Carson, L. S. Ostedgaard, G. M. Denning and M. J. Welsh, Expression of Cystic Fibrosis Transmembrane Conductance Regulator in a Model Epithelium, Am. J. Physiol., Cell Physiol., 1994, 266, L405–L413 CAS.
- P. Linsdell, J. A. Tabcharani and J. W. Hanrahan, Multi-Ion Mechanism for Ion Permeation and Block in the Cystic Fibrosis Transmembrane Conductance Regulator Chloride Channel, J. Gen. Physiol., 1997, 110, 365–377 CrossRef CAS PubMed.
- D. C. Dawson, S. S. Smith and M. K. Mansoura, CFTR: Mechanism of Anion Conduction, Physiol. Rev., 1999, 79, 47–75 CrossRef PubMed.
- T. I. Rink, R. Y. Tsien and T. Pozzan, Cytoplasmic pH and Free Mg2+ in Lymphocytes, J. Cell Biol., 1982, 95, 189–196 CrossRef CAS PubMed.
- X. Wang and I. Kurtz, H+/Base Transport in Principal Cells Characterized by Confocal Fluorescence Imaging, Am. J. Physiol., 1990, 259, C365–C373 CrossRef CAS PubMed.
- K. J. Rhoden, S. Cianchetta, V. Stivani, C. Portulano, L. J. V. Galietta and G. Romeo, Cell-Based Imaging of Sodium Iodide Symporter Activity with the Yellow Fluorescent Protein Variant YFP-H148Q/I152L, Am. J. Physiol. Cell Physiol., 2007, 292, 814–823 CrossRef PubMed.
- E. Szczesna-Skorupa, C.-D. Chen, S. Rogers and B. Kemper, Mobility of Cytochrome P450 in the Endoplasmic Reticulum Membrane, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 14793–14798 CrossRef CAS PubMed.
- E. L. Snapp, R. S. Hegde, M. Francolini, F. Lombardo, S. Colombo, E. Pedrazzini, N. Borgese and J. Lippincott-Schwartz, Formation of Stacked ER Cisternae by Low Affinity Protein Interactions, J. Cell Biol., 2003, 163, 257–269 CrossRef CAS PubMed.
- G. Van Rossum and F. L. Drake, Python 3 Reference Manual, CreateSpace, Scotts Valley, CA, 2009 Search PubMed.
- H. McWilliam, W. Li, M. Uludag, S. Squizzato, Y. M. Park, N. Buso, A. P. Cowley and R. Lopez, Analysis Tool Web Services from the EMBL-EBI, Nucleic Acids Res., 2013, 41, 597–600 CrossRef PubMed.
- B. Webb and A. Sali, Comparative Protein Structure Modeling Using MODELLER, Curr. Protoc. Bioinf., 2016, 54, 5.6.1–5.6.37 Search PubMed.
- M. R. Wilkins, E. Gasteiger, A. Bairoch, J.-C. Sanchez, K. L. Williams, R. D. Appel and D. F. Hochstrasser, Protein Identification and Analysis Tools in the ExPASy Server, in 2-D Proteome Analysis Protocols, Humana Press, New Jersey, 1999, pp. 531–552 Search PubMed.
- P. Virtanen, R. Gommers, T. E. Oliphant, M. Haberland, T. Reddy, D. Cournapeau, E. Burovski, P. Peterson, W. Weckesser, J. Bright, S. J. van der Walt, M. Brett, J. Wilson, K. J. Millman, N. Mayorov, A. R. J. Nelson, E. Jones, R. Kern, E. Larson, C. J. Carey, İ. Polat, Y. Feng, E. W. Moore, J. VanderPlas, D. Laxalde, J. Perktold, R. Cimrman, I. Henriksen, E. A. Quintero, C. R. Harris, A. M. Archibald, A. H. Ribeiro, F. Pedregosa, P. van Mulbregt, A. Vijaykumar, A. P. Bardelli, A. Rothberg, A. Hilboll, A. Kloeckner, A. Scopatz, A. Lee, A. Rokem, C. N. Woods, C. Fulton, C. Masson, C. Häggström, C. Fitzgerald, D. A. Nicholson, D. R. Hagen, D. V. Pasechnik, E. Olivetti, E. Martin, E. Wieser, F. Silva, F. Lenders, F. Wilhelm, G. Young, G. A. Price, G. L. Ingold, G. E. Allen, G. R. Lee, H. Audren, I. Probst, J. P. Dietrich, J. Silterra, J. T. Webber, J. Slavič, J. Nothman, J. Buchner, J. Kulick, J. L. Schönberger, J. V. de Miranda Cardoso, J. Reimer, J. Harrington, J. L. C. Rodríguez, J. Nunez-Iglesias, J. Kuczynski, K. Tritz, M. Thoma, M. Newville, M. Kümmerer, M. Bolingbroke, M. Tartre, M. Pak, N. J. Smith, N. Nowaczyk, N. Shebanov, O. Pavlyk, P. A. Brodtkorb, P. Lee, R. T. McGibbon, R. Feldbauer, S. Lewis, S. Tygier, S. Sievert, S. Vigna, S. Peterson, S. More, T. Pudlik, T. Oshima, T. J. Pingel, T. P. Robitaille, T. Spura, T. R. Jones, T. Cera, T. Leslie, T. Zito, T. Krauss, U. Upadhyay, Y. O. Halchenko and Y. Vázquez-Baeza, SciPy 1.0: Fundamental Algorithms for Scientific Computing in Python, Nat. Methods, 2020, 17, 261–272 CrossRef CAS PubMed.
- G. Jones II, W. R. Jackson and A. M. Halpern, Medium Effects on Fluorescence Quantum Yields and Lifetimes for Coumarin Laser Dyes, Chem. Phys. Lett., 1983, 72, 391 CrossRef.
- G. M. Hale and M. R. Querry, Optical Constants of Water in the 200-nm to 200-μm Wavelength Region, Appl. Opt., 1973, 12, 555 CrossRef CAS PubMed.
- G. Nelu and R. Sonia, Ewing's Analytical Instrumentation Handbook, CRC Press, Boca Raton, FL, 4th edn, 2019 Search PubMed.
- J. D. Hunter, Matplotlib: A 2D Graphics Environment, Comput. Sci. Eng., 2007, 9, 90–95 Search PubMed.
- W. McKinney, Data Structures for Statistical Computing in Python, in Proceedings of the 9th Python in Science Conference, ed. S. van der Walt and J. Millman, 2010, pp. 56–61 Search PubMed.
- C. R. Harris, K. J. Millman, S. J. van der Walt, R. Gommers, P. Virtanen, D. Cournapeau, E. Wieser, J. Taylor, S. Berg, N. J. Smith, R. Kern, M. Picus, S. Hoyer, M. H. van Kerkwijk, M. Brett, A. Haldane, J. del Río, M. Wiebe, P. Peterson, P. Gérard-Marchant, K. Sheppard, T. Reddy, W. Weckesser, H. Abbasi, C. Gohlke and T. E. Oliphant, Array Programming with {NumPy}, Nature, 2020, 585, 357–362 CrossRef CAS PubMed.
- P. Lecca, M. Lecca, C. A. Maestri and M. Scarpa, Biexponential Fitting for Noisy Data with Error Propagation, Math. Methods Appl. Sci., 2021, 44, 10154–10171 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sc03903f |
| This journal is © The Royal Society of Chemistry 2022 |