
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective desaturation of amides: a direct approach to enamides†
Xinwei
Li‡
a,
Zengrui
Cheng‡
a,
Jianzhong
Liu
a,
Ziyao
Zhang
a,
Song
Song
 *a and
Ning
Jiao
*ab
*a and
Ning
Jiao
*ab
aState Key Laboratory of Natural and Biomimetic Drugs, Chemical Biology Center, School of Pharmaceutical Sciences, Peking University, Xue Yuan Rd. 38, Beijing 100191, China. E-mail: jiaoning@pku.edu.cn; ssong@bjmu.edu.cn
bShanghai Key Laboratory of Green Chemistry and Chemical Processes, East China Normal University, Shanghai 200062, China
First published on 6th July 2022
Abstract
C(sp3)–H bond desaturation has been an attractive strategy in organic synthesis. Enamides are important structural fragments in pharmaceuticals and versatile synthons in organic synthesis. However, the dehydrogenation of amides usually occurs on the acyl side benefitting from enolate chemistry like the desaturation of ketones and esters. Herein, we demonstrate an Fe-assisted regioselective oxidative desaturation of amides, which provides an efficient approach to enamides and β-halogenated enamides.
Introduction
The enamide is a vital functional group in pharmaceuticals featuring excellent biological and physiological properties (Fig. 1).1 Moreover, enamide compounds are also important and versatile synthons in organic synthesis.2 Thus, the synthesis of enamides has attracted great interest from chemists for several decades.3 Recently, selective C(sp3)–H bond desaturation has been widely used in organic synthesis, including oxidative desaturation,4 transition-metal catalyzed desaturation,5 photochemical desaturation6 and electrochemical desaturation.7 However, compared to the dehydrogenation reaction of carbonyl compounds,7a,b,e,g,8 such as ketones,9 the regioselective dehydrogenation of N-alkanoylamides on the N-alkyl side with higher C–H bond dissociation energy (BDE)10 is still a tremendous challenge (Scheme 1a). Generally, the desaturation of esters11 and amides12 on the acyl side is much preferred benefitting from enolate chemistry. So far, the regioselective N-α,β-desaturation of amides remains unknown (Scheme 1a).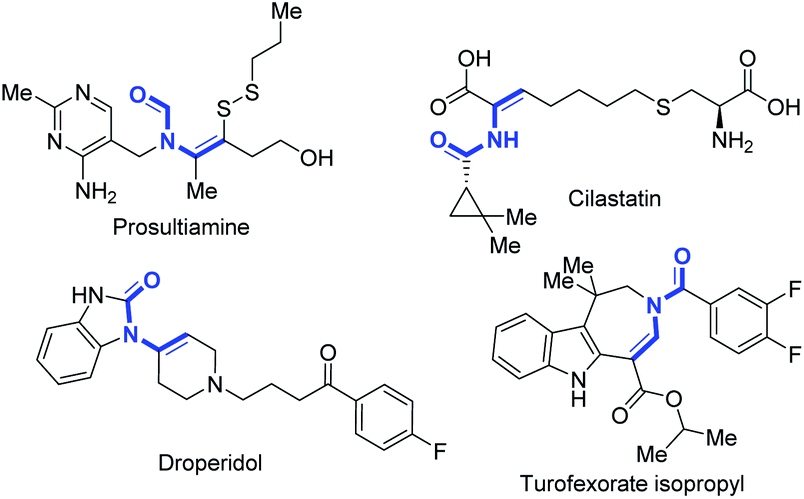 | ||
| Fig. 1 Representative drugs containing enamide fragments. | ||
 | ||
| Scheme 1 The significance and challenges for the synthesis of enamides from amides. | ||
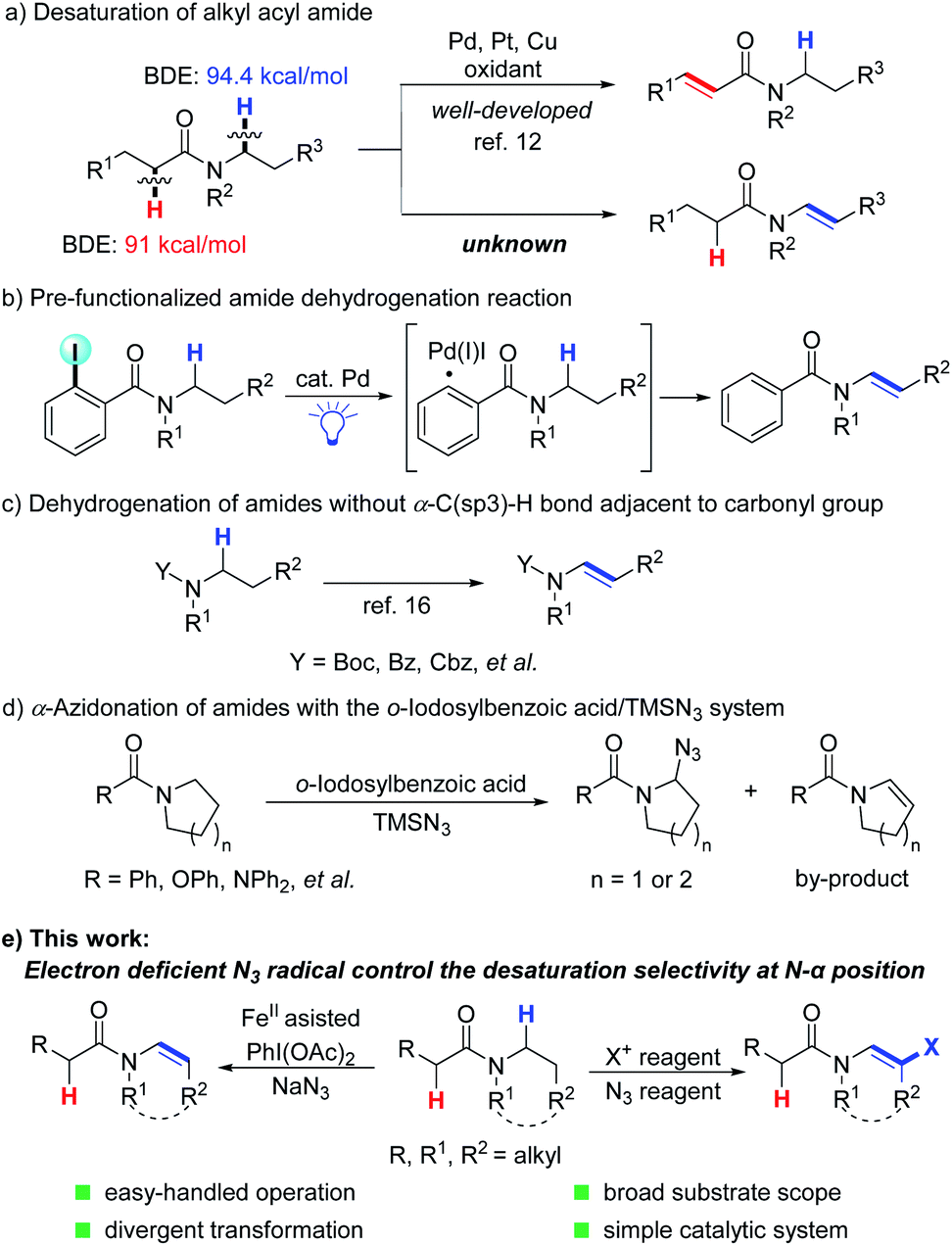
Considering that the desaturation of amides is one of the most efficient and ideal approaches to enamides, many efforts have been made to realize the desaturation of amides. Early in the 1980s, Shono first accomplished this goal via a two-step reaction. Amides were initially oxidized to N-α-methoxyl amides13 and subsequently went through a methanol elimination process to yield the enamide product.14 Recently, trailblazing work regarding the direct desaturation of amides has been significantly achieved by the Gevorgyan group, which includes the generation of a hybrid aryl Pd-radical species under visible light irradiation and a HAT (hydrogen atom transfer) process to realize the desaturation reactions (Scheme 1b).15 Meanwhile, the desaturation of N-Boc, Cbz and Bz protected amides through manganese-catalyzed α-C(sp3)–H hydroxylation and electrophilic activation of amides was demonstrated by the groups of Groves and Maulide, respectively (Scheme 1c).16 Magnus's group reported the first example of α-azidation of amides and their analogues using TMSN3 and 2-iodosobenzoic acid, with enamides as by-products (Scheme 1d).17 Despite the significance, a limitation is that substrates containing an acidic C(sp3)–H bond adjacent to the carbonyl group are not amenable for selective desaturation. Thus, general and efficient methods to furnish enamides from amides are highly desired and unpresented. Herein, we report a general method to realize regioselective oxidative N-α,β-desaturation of amides to generate enamides. This transformation shows very broad substrate tolerance, and aroyl-, alkanoyl-, cyclic-, and acyclic-amides are all compatible. Besides, β-halogenated enamides can also be generated under slightly adjusted conditions, which can be further modified by a coupling reaction (Scheme 1e).
Results and discussion
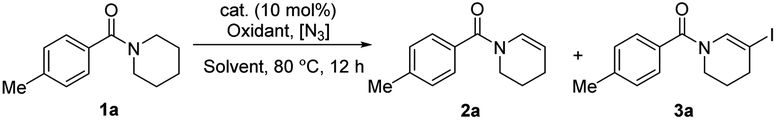
The azide radical, which was an electron-deficient N-centered radical, was proved to be an efficient hydrogen-abstracting species in C(sp3)–H bond functionalization18 and plays versatile roles in different transformations.17–19 Hence, we attempted to use the azide radical to abstract more electron-rich N-α-C–H bonds to realize selective oxidation reaction of amides.20 At the very beginning, we tried to utilize NIS and azide to synthesize the highly reactive reagent IN3in situ,19e initiating amide oxidative desaturation via a radical process. As initial attempts to realize the desaturation of amides, N-(4-methylbenzoyl)piperidine 1a was selected as the model substrate, using NIS as the oxidant and NaN3 as the azide source. To our delight, after screening solvents, we found that enamide 2a was generated in ethyl acetate (EA) with moderate yield (Table 1, entry 2). NaN3 was essential for this transformation (Table 1, entry 1). The iodine ion is one of the by-products in this reaction, but this conversion was inhibited when an excessive amount of NaI was used (Table 1, entry 3). To avoid the inhibiting effect of the excessive amount of by-product NaI, the oxidant NIS was replaced by PhI(OAc)2 (PIDA), and the yield was reduced to 23% (Table 1, entry 4). During the screening of the amount of NaI and various catalysts (for more condition screening, see the ESI†), the yield was further raised to 64% when 30 mol% NaI and 10 mol% FeCl2 was added (Table 1, entry 5). It is very interesting that the desaturated iodination product 3a was obtained in 40% yield when more soluble TMSN3 was added instead of NaN3 (Table 1, entry 6). 2.5 equiv. of TMSN3 and NIS with CCl4 as the solvent could increase the isolated yield of 3a to 65% (entry 7). Besides, no doubly dehydrogenated products were produced with a large excess of the N3 reagent and oxidant. The rotamerism was observed from many enamide and N-β-halogenated enamide products, which is confirmed by variable-temperature 1H NMR experiments (see ESI, Fig. S1†).21|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Cat. | [N3] | Oxidant | Solvent | 2a [%] | 3a [%] |
| a Reaction conditions: 1a (0.4 mmol) and additives in EA (4.0 mL) under Ar, 80 °C, and 12 h. Determined by 1H NMR analysis using 1,1,2,2-tetrachloroethane as the internal standard. Isolated yield in parentheses. b NaI (2.0 equiv.) as the additive. c NaI (30 mol%) as the additive. d 1a (0.2 mmol) and open air reaction. | ||||||
| 1 | — | — | NIS (3.0 equiv.) | EA | 0 | 0 |
| 2 | — | NaN3 (3.0 equiv.) | NIS (3.0 equiv.) | EA | 54 | Trace |
| 3b | — | NaN3 (3.0 equiv.) | NIS (3.0 equiv.) | EA | 15 | 0 |
| 4 | — | NaN3 (3.0 equiv.) | PIDA (1.8 equiv.) | EA | 23 | 0 |
| 5 | FeCl2 | NaN 3 (3.6 equiv.) | PIDA (1.8 equiv.) | EA | 71 (64) | 0 |
| 6d | — | TMSN3 (2.0 equiv.) | NIS (2.0 equiv.) | DCE | 0 | 40 |
| 7 | — | TMSN 3 (2.5 equiv.) | NIS (2.5 equiv.) | CCl 4 | 0 | 68 (65) |
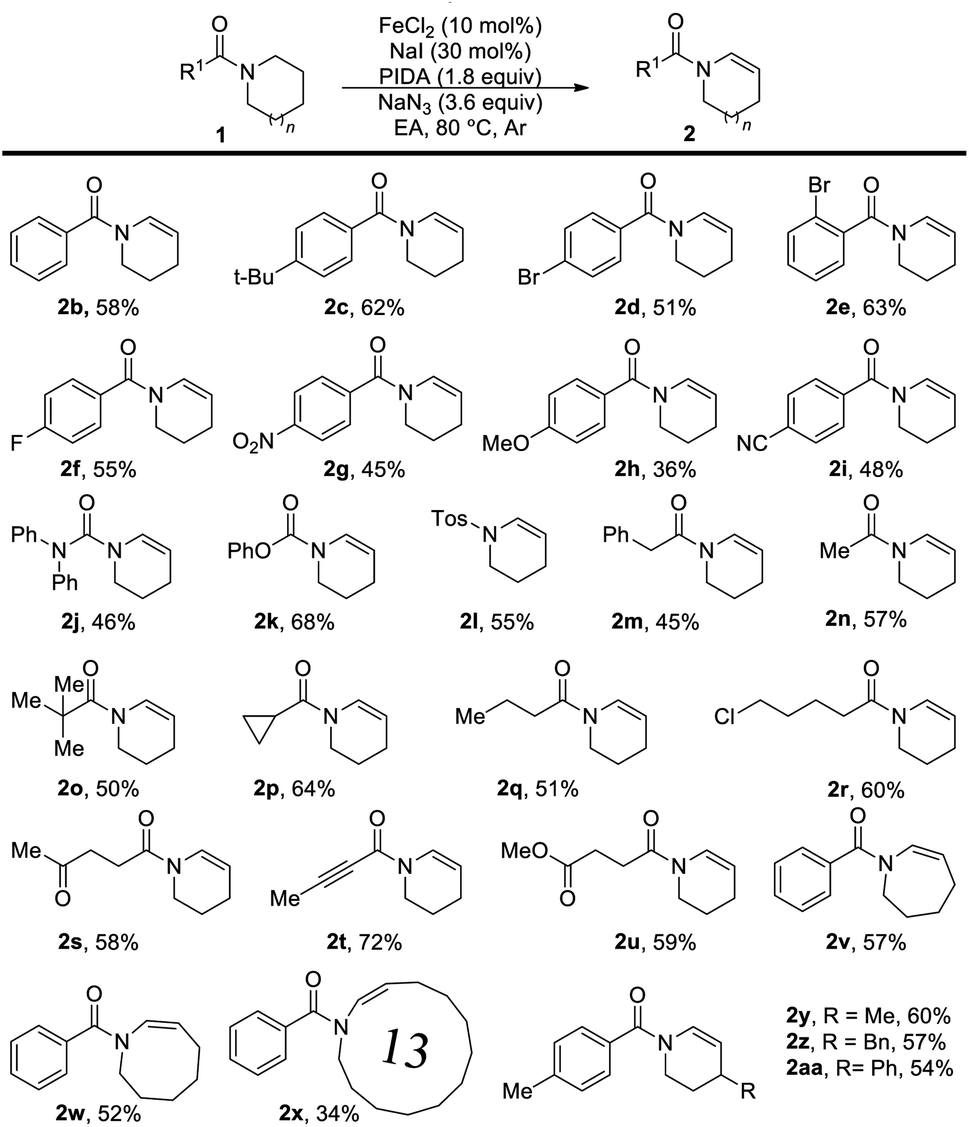
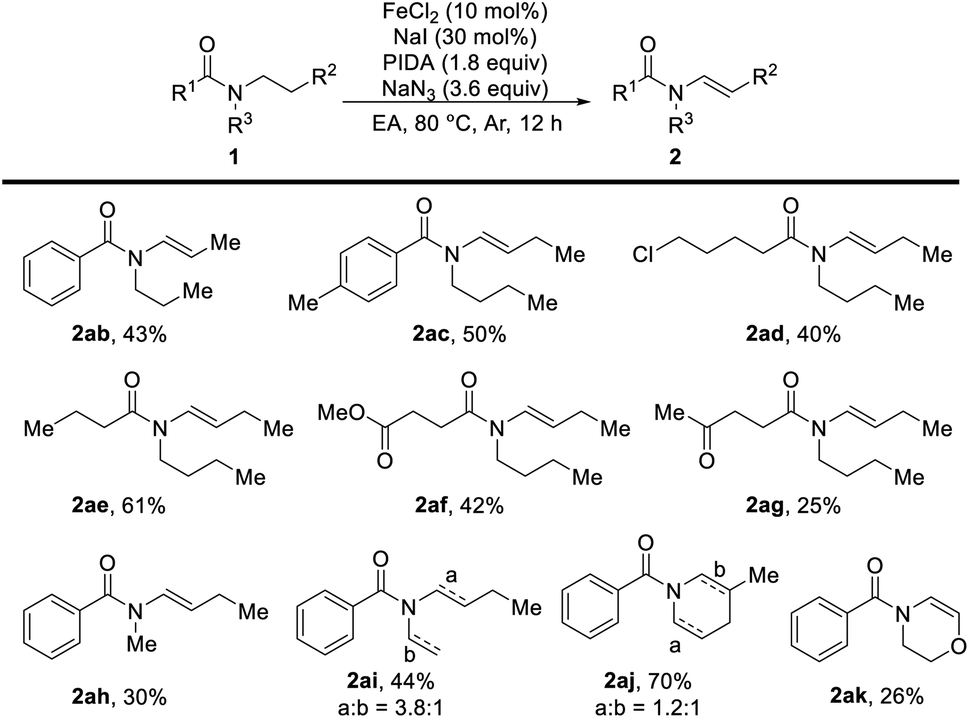
With the optimal conditions in hand (Table 1, entry 5), we commenced investigating the scope of this reaction. At first, the influence of the N-acyl substituent was tested. As shown, this method is compatible for broad functional groups (Table 2). Simple N-aroyl amides underwent selective desaturation to furnish corresponding enamides in good yields. Urea (2j), N-carboxylate compound (2k), and tosyl amine (2l) were also applicable and provided the product in acceptable to good yields. It is noteworthy that N-alkanoyl amides worked well in this reaction with good yield. Benzyl amide (2m), acetamide (2n) and tert-butylamide (2o) could afford the desired product in good yields. Cyclopropyl amide was also dehydrogenated successfully in good yield (2p). Chlorinated amide underwent this transformation in good yield with the chlorine functional group untouched (2r) without the detection of the SN2 product. Especially, the ketone group (2s) and alkyne group (2t) can also be reserved under these oxidative conditions with good yields. Notably, high selective desaturation was observed in the presence of other carbonyl functional groups, for instance, ketone (2s) and ester (2u). Moreover, the desaturation of amides bearing seven- and eight-membered rings produced the corresponding enamides (2v and 2w) in moderate yields, and this method was able to apply in the synthesis of a thirteen-membered cyclic enamide (2x). The substituents on the piperidine showed little influence on the desaturation reaction, and substituted enamides (2y–2aa) were obtained in good yields.
| a Reaction conditions (Method A): reactions were conducted on a 0.4 mmol scale using 10 mol% FeCl2, 30 mol% NaI, 1.8 equiv. PIDA, 3.6 equiv. NaN3, in 4.0 mL EA, 80 °C, under an Ar atmosphere, and 12 h. Isolated yields. |
|---|
|
We next tested the desaturation reaction of acyclic amides (Table 3). To our delight, when there were two identical substituents on amides, both N-alkanoyl and N-benzoyl acyclic amides were converted to the desired products in acceptable yields with high selectivity (2ab–2ae). Even in the presence of ester and ketone groups, no conjugated alkenyl product was observed in this oxidative desaturation reaction (2af and 2ag). For unsymmetrical tert-amides, lower regioselectivity was obtained when the steric effect was similar to alkyl groups (2ai and 2aj).
| a Reaction conditions (Method A): reactions were conducted on a 0.4 mmol scale using 10 mol% FeCl2, 30 mol% NaI, 1.8 equiv. PIDA, 3.6 equiv. NaN3, in 4.0 mL EA, 80 °C, under Ar atmosphere, and 12 h. Isolated yields. |
|---|
|
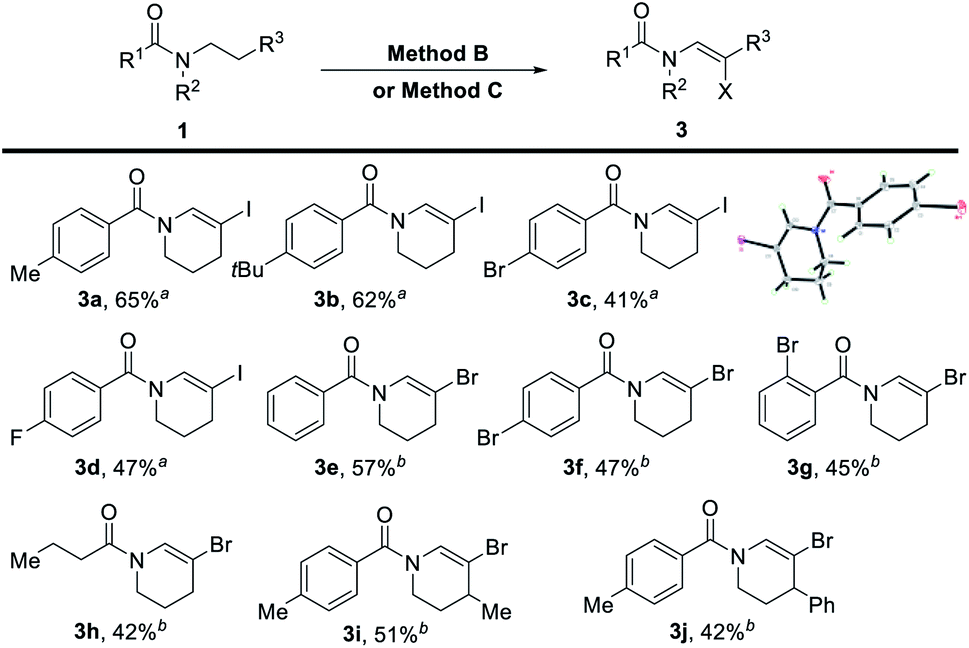
The β-halogenated enamides can be further modified to synthesize more sophisticated compounds via a well-developed transition metal-catalyzed cross-coupling reaction. The direct synthesis of β-halogenated enamides from amides has not been reported yet. We then tested the dehydrogenative halogenation of amides with NIS as the oxidant, TMSN3 as the additive and CCl4 as the solvent (Table 4). The scope of this transformation was set out for further investigation (Table 4). The structure of the β-iodine enamide was confirmed by X-ray single crystal diffraction (3c, CCDC 2097260). Additionally, the dehydrogenative N-β-bromination of amides was achieved with 1,3-dibromo-5,5-dimethylhydantoin (DBDMH) as the brominating reagent and TogniN3 (ref. 18a) as the oxidant. The yields of the desired products were acceptable (Table 4). Among them, not only aroyl amide could furnish this transformation, but also alkanoyl amide could produce the desired product (3h). The N-β-halogenated enamide can be used to synthesize more complicated enamides by utilizing the classic coupling reaction.
| a Reaction conditions (Method B): reactions were conducted on a 0.2 mmol scale using 2.5 equiv. NIS, 2.5 equiv. TMSN3 in 2.0 mL CCl4, 80 °C, under an Ar atmosphere, and 7 h. Isolated yields. b Reaction conditions (Method C): reactions were conducted on a 0.3 mmol scale using 0.55 equiv. 1,3-dibromo-5,5-dimethylhydantoin (DBDMH), 1.1 equiv. TogniN3 in 2.0 mL DCE, 80 °C, under an Ar atmosphere, and 12 h. Isolated yields. |
|---|
|
In order to get a better insight into the mechanism, some control experiments were designed and carried out (Scheme 2). At first, the reaction was inhibited after adding 2,2,6,6-tetramethyl-piperidine-N-oxyl (TEMPO) or 2,6-di-tert-butyl-4-methyl-phenol (BHT) into the reaction system (Method A), and the starting materials remained (Scheme 2a), indicating that the radical process might be part of the dehydrogenation transformation. The radical clock experiment was also carried out, but no ring opening dehydrogenation product was detected under the standard conditions (Scheme 2b). This result may not deny the free radical process because the radical with the intact cyclopropane is likely more stable than the corresponding homoallylic primary radical. Moreover, five-membered ring amide (1al) and secondary amide (1am) did not produce the dehydrogenation product but N-α-azide substituted amides were produced (4al and 4am), which are very stable compounds.17 It reveals that the carbocation intermediates may be key intermediates in this process (Scheme 2c). In addition, intermediate 5 was isolated under the standard conditions (Method B, Scheme 2d). Furthermore, we monitored the reaction process by in situ1H-NMR (see ESI, Fig. S2†). With the extension of reaction time, the intermediate 5 gradually increased and reached the peak in 4 hours, and then it was gradually converted to 3a. This demonstrated that the N-α-azide-β-halogenated amide is the key intermediate in the dehydrogenative halogenation reaction.
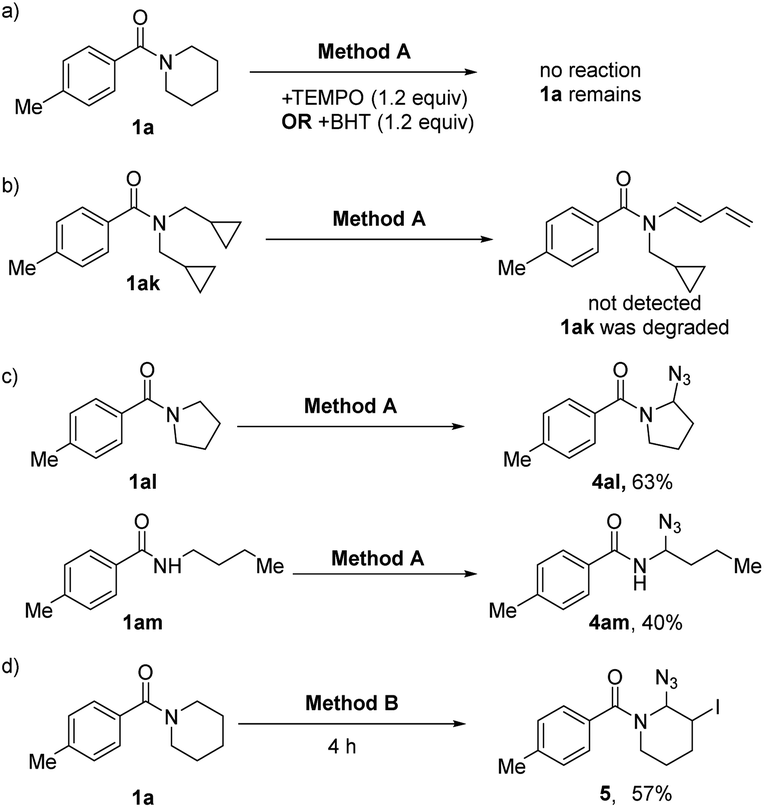 | ||
| Scheme 2 Mechanistic studies. | ||
On the basis of the above experiments, a plausible mechanism was proposed (Scheme 3). The azide radical is generated in situ through two pathways under different conditions. Initial ligand exchange between PIDA and NaN3 would afford intermediate A, which undergoes thermal homolytic cleavage owing to a weak I–N bond to generate an azide radical.19g,n Another pathway to generate the azide radical is the decomposition of IN3, which is produced from NIS and TMSN3.19e Then the radical intermediate B is produced via hydrogen abstraction by the azide radical.18a,19v Subsequently, intermediate B is converted to cationic intermediate C through a single electron oxidation process promoted by FeCl2.22 The enamide can be also generated via two pathways from intermediate B. In path a, the nucleophilic addition of intermediate C by the azide anion occurs to generate intermediate D, which produces product 2via the elimination of HN3 (path a). Alternatively, enamide 2 can be generated directly from intermediate Cvia deprotonation (path b). Regarding the dehydrogenative N-β-halogenation of amide process, intermediate B combines with the iodine radical to obtain intermediate E,19o which will further transform into 2 by elimination. Then, enamide 2 undergoes addition quickly with IN3 affording the key intermediate F. Finally, the desired product N-β-halogenated enamide 3 is formed with the concomitant elimination of the HN3 unit. The different electrophilicity of NIS and the different active azide intermediates in different solvents might account for the selective formation of the terminal product.
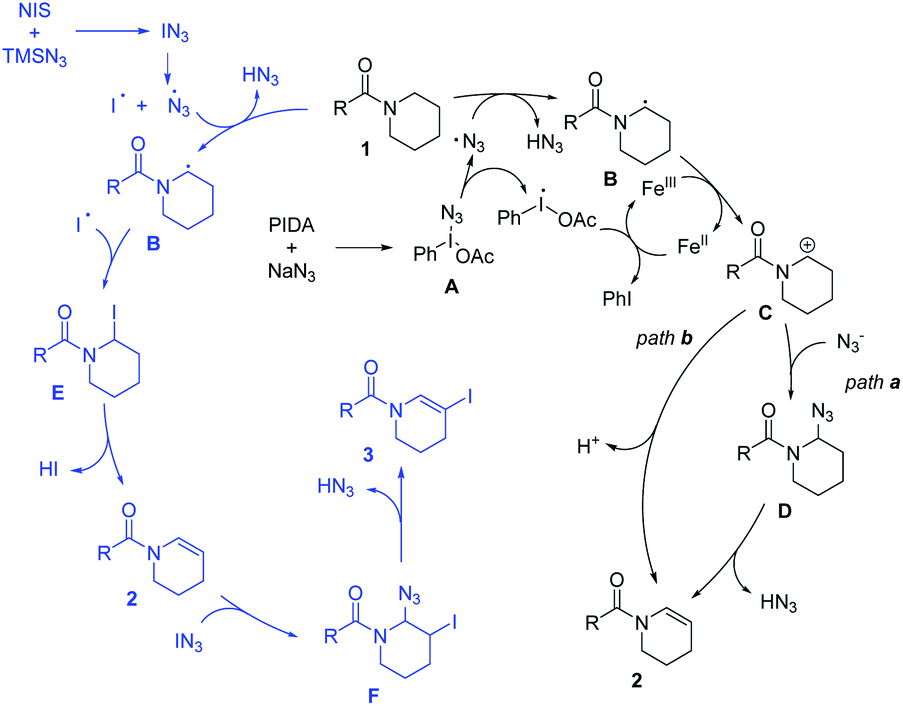 | ||
| Scheme 3 Mechanistic investigations. | ||
Conclusions
In summary, we described a novel, general and regioselective oxidative N-α,β-desaturation and dehydrogenative N-β-halogenation of amides. This chemistry features excellent functional group compatibility, and thus provides efficient and general access to enamides and β-halogenated enamides, which are biologically important scaffolds and versatile organic building blocks in pharmaceutical sciences and synthetic organic chemistry.Author contributions
X. L., Z. C., S. S. and N. J. conceived and designed the experiments. X. L. and Z. C. carried out most of the experiments. X. L., Z. C., J. L., Z. Z., S. S. and N. J. analyzed the data. X. L., Z. C., S. S. and N. J. wrote the paper. N. J. directed the research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the National Key R&D Program of China (No. 2021YFA1501700), the NSFC (No. 22131002, 22071005, and 22161142019), the Beijing Nova Program (No. Z201100006820099), and the Tencent Foundation for financial support. We thank Cheng Zhang in this group for reproducing the reactions of 2q and 3e.Notes and references
- M. Perez-Sayans, J. M. Somoza-Martin, F. Barros-Angueira, J. M. Rey and A. Garcia-Garcia, Cancer Treat. Rev., 2009, 35, 707–713 CrossRef CAS PubMed.
- (a) D. R. Carbery, Org. Biomol. Chem., 2008, 6, 3455–3460 RSC; (b) K. Gopalaiah and H. B. Kagan, Chem. Rev., 2011, 111, 4599–4657 CrossRef CAS PubMed; (c) G. Masson and G. Bernadat, Synlett, 2014, 25, 2842–2867 CrossRef; (d) G. Masson, T. Courant and G. Dagousset, Synthesis, 2015, 47, 1799–1856 CrossRef; (e) M. X. Wang, Chem. Commun., 2015, 51, 6039–6049 RSC.
- (a) F. Pohlki and S. Doye, Chem. Soc. Rev., 2003, 32, 104–114 RSC; (b) J. R. Dehli, J. Legros and C. Bolm, Chem. Commun., 2005, 973–986 RSC.
- (a) R. P. Linstead, E. A. Braude, P. W. D. Mitchell, K. R. H. Wooldridge and L. M. Jackman, Nature, 1952, 169, 100–103 CrossRef CAS; (b) E. A. Mamedov and V. Cortés Corberán, Appl. Catal., A, 1995, 127, 1–40 CrossRef CAS; (c) H. H. Kung and M. C. Kung, Appl. Catal., A, 1997, 157, 105–116 CrossRef CAS; (d) L. M. Madeira and M. F. Portela, Catal. Rev., 2002, 44, 247–286 CrossRef CAS; (e) X. Zhang, A. Fried, S. Knapp and A. S. Goldman, Chem. Commun., 2003, 2060–2061 RSC; (f) S. Wang and Z. H. Zhu, Energy Fuels, 2004, 18, 1126–1139 CrossRef CAS; (g) M. B. Ansari and S.-E. Park, Energy Environ. Sci., 2012, 5, 9419 RSC; (h) M. T. Schümperli, C. Hammond and I. Hermans, ACS Catal., 2012, 2, 1108–1117 CrossRef; (i) A.-F. Voica, A. Mendoza, W. R. Gutekunst, J. O. Fraga and P. S. Baran, Nat. Chem., 2012, 4, 629–635 CrossRef CAS PubMed; (j) S. Wertz and A. Studer, Green Chem., 2013, 15, 3116 RSC; (k) B. Chen, L. Wang and S. Gao, ACS Catal., 2015, 5, 5851–5876 CrossRef CAS; (l) R. A. Sheldon, Catal. Today, 2015, 247, 4–13 CrossRef CAS; (m) A. E. Wendlandt and S. S. Stahl, Angew. Chem., Int. Ed., 2015, 54, 14638–14658 CrossRef CAS PubMed; (n) M. J. Zhou, S. F. Zhu and Q. L. Zhou, Chem. Commun., 2017, 53, 8770–8773 RSC.
- (a) I. Göttker-Schnetmann, P. White and M. Brookhart, J. Am. Chem. Soc., 2004, 126, 1804–1811 CrossRef PubMed; (b) A. D. Bolig and M. Brookhart, J. Am. Chem. Soc., 2007, 129, 14544–14545 CrossRef CAS PubMed; (c) R. Giri, N. Maugel, B. M. Foxman and J.-Q. Yu, Organometallics, 2008, 27, 1667–1670 CrossRef CAS; (d) G. E. Dobereiner and R. H. Crabtree, Chem. Rev., 2010, 110, 681–703 CrossRef CAS PubMed; (e) T. C. Johnson, D. J. Morris and M. Wills, Chem. Soc. Rev., 2010, 39, 81–88 RSC; (f) J. Choi, A. H. MacArthur, M. Brookhart and A. S. Goldman, Chem. Rev., 2011, 111, 1761–1779 CrossRef CAS PubMed; (g) C. Gunanathan and D. Milstein, Science, 2013, 341, 1229712 CrossRef PubMed; (h) C. B. Bheeter, R. Jin, J. K. Bera, P. H. Dixneuf and H. Doucet, Adv. Synth. Catal., 2014, 356, 119–124 CrossRef CAS; (i) W. Yao, Y. Zhang, X. Jia and Z. Huang, Angew. Chem., Int. Ed., 2014, 53, 1390–1394 CrossRef CAS PubMed; (j) S. Werkmeister, J. Neumann, K. Junge and M. Beller, Chemistry, 2015, 21, 12226–12250 CrossRef CAS PubMed; (k) A. Kumar, T. M. Bhatti and A. S. Goldman, Chem. Rev., 2017, 117, 12357–12384 CrossRef CAS PubMed; (l) L. Huang, A. Bismuto, S. A. Rath, N. Trapp and B. Morandi, Angew. Chem., Int. Ed., 2021, 60, 7290–7296 CrossRef CAS PubMed.
- (a) K. Nomura and Y. Saito, J. Chem. Soc., Chem. Commun., 1988, 161–162 RSC; (b) S. Toshiyasu, S. Touru, T. Yuko and T. Masato, Chem. Lett., 1988, 17, 263–264 CrossRef; (c) J. A. Maguire, W. T. Boese and A. S. Goldman, J. Am. Chem. Soc., 1989, 111, 7088–7093 CrossRef CAS; (d) A. J. Esswein and D. G. Nocera, Chem. Rev., 2007, 107, 4022–4047 CrossRef CAS PubMed; (e) X. Sun, J. Chen and T. Ritter, Nat. Chem., 2018, 10, 1229–1233 CrossRef CAS PubMed; (f) W. M. Cheng, R. Shang and Y. Fu, Nat. Commun., 2018, 9, 5215 CrossRef PubMed; (g) J. G. West, D. Huang and E. J. Sorensen, Nat. Commun., 2015, 6, 10093 CrossRef PubMed.
- (a) T. Shono, Y. Matsumura and Y. Nakagawa, J. Am. Chem. Soc., 1974, 96, 3532–3536 CrossRef CAS; (b) T. Shono, M. Okawa and I. Nishiguchi, J. Am. Chem. Soc., 1975, 97, 6144–6147 CrossRef CAS; (c) M. Okimoto, Y. Takahashi, K. Numata and G. Sasaki, Heterocycles, 2005, 65, 371–375 CrossRef CAS; (d) Y. Wu, H. Yi and A. Lei, ACS Catal., 2018, 8, 1192–1196 CrossRef CAS; (e) R. J. Perkins, R. Feng, Q. Lu and K. D. Moeller, Chin. J. Chem., 2019, 37, 672–678 CrossRef CAS; (f) M. Bugnola, K. Shen, E. Haviv and R. Neumann, ACS Catal., 2020, 10, 4227–4237 CrossRef CAS; (g) S. Gnaim, Y. Takahira, H. R. Wilke, Z. Yao, J. Li, D. Delbrayelle, P. G. Echeverria, J. C. Vantourout and P. S. Baran, Nat. Chem., 2021, 13, 367–372 CrossRef CAS PubMed.
- (a) H. J. Reich and S. Wollowitz, Org. React., 2004, 1–296 Search PubMed; (b) J. Muzart, Eur. J. Org. Chem., 2010, 2010, 3779–3790 CrossRef; (c) S. S. Stahl and T. Diao, in Comprehensive Organic Synthesis II, ed. P. Knochel, Elsevier, Amsterdam, 2nd edn, 2014, pp. 178–212 Search PubMed; (d) T. Newhouse, A. Turlik and Y. Chen, Synlett, 2015, 27, 331–336 CrossRef.
- (a) R. J. Theissen, J. Org. Chem., 1971, 36, 752–757 CrossRef CAS; (b) R. C. Larock, T. R. Hightower, G. A. Kraus, P. Hahn and D. Zheng, Tetrahedron Lett., 1995, 36, 2423–2426 CrossRef CAS; (c) T. Diao and S. S. Stahl, J. Am. Chem. Soc., 2011, 133, 14566–14569 CrossRef CAS PubMed; (d) T. Diao, T. J. Wadzinski and S. S. Stahl, Chem. Sci., 2012, 3, 887–891 RSC; (e) T. Diao, D. Pun and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 8205–8212 CrossRef CAS PubMed; (f) X. Jie, Y. Shang, X. Zhang and W. Su, J. Am. Chem. Soc., 2016, 138, 5623–5633 CrossRef CAS PubMed; (g) Y. Sakamoto, T. Amaya, T. Suzuki and T. Hirao, Chem.–Eur. J., 2016, 22, 18686–18689 CrossRef CAS PubMed; (h) Y. Shang, X. Jie, K. Jonnada, S. N. Zafar and W. Su, Nat. Commun., 2017, 8, 2273 CrossRef PubMed; (i) D. Huang, Y. Zhao and T. R. Newhouse, Org. Lett., 2018, 20, 684–687 CrossRef CAS PubMed; (j) H. Li, Q. Jiang, X. Jie, Y. Shang, Y. Zhang, L. J. Goossen and W. Su, ACS Catal., 2018, 8, 4777–4782 CrossRef CAS.
- Y.-R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, Boca Raton, 2007 Search PubMed.
- (a) Y. Chen, J. P. Romaire and T. R. Newhouse, J. Am. Chem. Soc., 2015, 137, 5875–5878 CrossRef CAS PubMed; (b) S. M. Szewczyk, Y. Zhao, H. A. Sakai, P. Dube and T. R. Newhouse, Tetrahedron, 2018, 74, 3293–3300 CrossRef CAS.
- (a) Y. Chen, A. Turlik and T. R. Newhouse, J. Am. Chem. Soc., 2016, 138, 1166–1169 CrossRef CAS PubMed; (b) M. Chen and G. Dong, J. Am. Chem. Soc., 2017, 139, 7757–7760 CrossRef CAS PubMed; (c) M. Chen, A. J. Rago and G. Dong, Angew. Chem., Int. Ed., 2018, 57, 16205–16209 CrossRef CAS PubMed; (d) Z. Wang, Z. He, L. Zhang and Y. Huang, J. Am. Chem. Soc., 2018, 140, 735–740 CrossRef CAS PubMed; (e) M. Chen and G. Dong, J. Am. Chem. Soc., 2019, 141, 14889–14897 CrossRef CAS PubMed; (f) C. J. Teskey, P. Adler, C. R. Goncalves and N. Maulide, Angew. Chem., Int. Ed., 2019, 58, 447–451 CrossRef CAS PubMed.
- T. Shono, H. Hamaguchi and Y. Matsumura, J. Am. Chem. Soc., 1975, 97, 4264–4268 CrossRef CAS.
- T. Shono, Y. Matsumura, K. Tsubata, Y. Sugihara, S. Yamane, T. Kanazawa and T. Aoki, J. Am. Chem. Soc., 1982, 104, 6697–6703 CrossRef CAS.
- (a) P. Chuentragool, M. Parasram, Y. Shi and V. Gevorgyan, J. Am. Chem. Soc., 2018, 140, 2465–2468 CrossRef CAS PubMed; (b) M. Parasram, P. Chuentragool, D. Sarkar and V. Gevorgyan, J. Am. Chem. Soc., 2016, 138, 6340–6343 CrossRef CAS PubMed; (c) P. Chuentragool, D. Kurandina and V. Gevorgyan, Angew. Chem., Int. Ed., 2019, 58, 11586–11598 CrossRef CAS PubMed; (d) M. Ratushnyy, N. Kvasovs, S. Sarkar and V. Gevorgyan, Angew. Chem., Int. Ed., 2020, 59, 10316–10320 CrossRef CAS PubMed.
- (a) G. Li, P. A. Kates, A. K. Dilger, P. T. Cheng, W. R. Ewing and J. T. Groves, ACS Catal., 2019, 9, 9513–9517 CrossRef CAS; (b) P. Spiess, M. Berger, D. Kaiser and N. Maulide, J. Am. Chem. Soc., 2021, 143, 10524–10529 CrossRef CAS PubMed.
- P. Magnus, C. Hulme and W. Weber, J. Am. Chem. Soc., 1994, 116, 4501–4502 CrossRef CAS.
- (a) Y. Wang, X. Hu, C. A. Morales-Rivera, G. X. Li, X. Huang, G. He, P. Liu and G. Chen, J. Am. Chem. Soc., 2018, 140, 9678–9684 CrossRef CAS PubMed; (b) T. Liu, M. C. Myers and J. Q. Yu, Angew. Chem., Int. Ed., 2017, 56, 306–309 CrossRef CAS PubMed; (c) P. T. Rabet, G. Fumagalli, S. Boyd and M. F. Greaney, Org. Lett., 2016, 18, 1646–1649 CrossRef CAS PubMed; (d) T. Liu, T. S. Mei and J. Q. Yu, J. Am. Chem. Soc., 2015, 137, 5871–5874 CrossRef CAS PubMed.
- (a) K. Gollnick, D. Haisch and G. Schade, J. Am. Chem. Soc., 1972, 94, 1747–1748 CrossRef CAS; (b) D. A. Armstrong, M. Farahani and P. S. Surdhar, Can. J. Chem., 1990, 68, 1974–1978 CrossRef CAS; (c) M. S. Workentin, B. D. Wagner, J. Lusztyk and D. D. M. Wayner, J. Am. Chem. Soc., 1995, 117, 119–126 CrossRef CAS; (d) V. V. Zhdankin, A. P. Krasutsky, C. J. Kuehl, A. J. Simonsen, J. K. Woodward, B. Mismash and J. T. Bolz, J. Am. Chem. Soc., 1996, 118, 5192–5197 CrossRef CAS; (e) A. Hassner, L. Marinescu and M. Bols, Encyclopedia of Reagents for Organic Synthesis, 2005 Search PubMed; (f) A. G. Griesbeck, M. Reckenthaler and J. Uhlig, Photochem. Photobiol. Sci., 2010, 9, 775–778 CrossRef CAS PubMed; (g) K. Matcha and A. P. Antonchick, Angew. Chem., Int. Ed., 2013, 52, 2082–2086 CrossRef CAS PubMed; (h) G. Fumagalli, P. T. Rabet, S. Boyd and M. F. Greaney, Angew. Chem., Int. Ed., 2015, 54, 11481–11484 CrossRef CAS PubMed; (i) M. Z. Lu, C. Q. Wang and T. P. Loh, Org. Lett., 2015, 17, 6110–6113 CrossRef CAS PubMed; (j) A. Sharma and J. F. Hartwig, Nature, 2015, 517, 600–604 CrossRef CAS PubMed; (k) B. Olofsson, in Hypervalent Iodine Chemistry, ed. T. Wirth, Springer International Publishing, Cham, 2016, pp. 135–166 Search PubMed; (l) Y. Wang, G. X. Li, G. Yang, G. He and G. Chen, Chem. Sci., 2016, 7, 2679–2683 RSC; (m) Y. A. Yuan, D. F. Lu, Y. R. Chen and H. Xu, Angew. Chem., Int. Ed., 2016, 55, 534–538 CrossRef CAS PubMed; (n) P. Magnus and J. Lacour, J. Am. Chem. Soc., 1992, 114, 767–769 CrossRef CAS; (o) J. Barluenga, E. Campos-Gomez, D. Rodriguez, F. Gonzalez-Bobes and J. M. Gonzalez, Angew. Chem., Int. Ed., 2005, 44, 5851–5854 CrossRef CAS PubMed; (p) N. Fu, G. S. Sauer, A. Saha, A. Loo and S. Lin, Science, 2017, 357, 575–579 CrossRef CAS PubMed; (q) C. E. Hendrick, K. J. Bitting, S. Cho and Q. Wang, J. Am. Chem. Soc., 2017, 139, 11622–11628 CrossRef CAS PubMed; (r) Y. Wang, N. Wang, J. Zhao, M. Sun, H. You, F. Fang and Z.-Q. Liu, ACS Catal., 2020, 10, 6603–6612 CrossRef CAS; (s) T. G. Driver, Org. Biomol. Chem., 2010, 8, 3831–3846 RSC; (t) K. Shin, H. Kim and S. Chang, Acc. Chem. Res., 2015, 48, 1040–1052 CrossRef CAS PubMed; (u) P. Sivaguru, Y. Ning and X. Bi, Chem. Rev., 2021, 121, 4253–4307 CrossRef CAS PubMed; (v) W. Zhou, L. Zhang and N. Jiao, Angew. Chem., Int. Ed., 2009, 48, 7094–7097 CrossRef CAS PubMed; (w) T. Wang and N. Jiao, J. Am. Chem. Soc., 2013, 135, 11692–11695 CrossRef CAS PubMed; (x) T. Wang and N. Jiao, Acc. Chem. Res., 2014, 47, 1137–1145 CrossRef CAS PubMed; (y) X. Sun, X. Li, S. Song, Y. Zhu, Y. F. Liang and N. Jiao, J. Am. Chem. Soc., 2015, 137, 6059–6066 CrossRef CAS PubMed; (z) J. Liu, X. Qiu, X. Huang, X. Luo, C. Zhang, J. Wei, J. Pan, Y. Liang, Y. Zhu, Q. Qin, S. Song and N. Jiao, Nat. Chem., 2019, 11, 71–77 CrossRef CAS PubMed.
- F. Parsaee, M. C. Senarathna, P. B. Kannangara, S. N. Alexander, P. D. E. Arche and E. R. Welin, Nat. Rev. Chem., 2021, 5, 486–499 CrossRef CAS.
- R. E. Quinones, C. M. Glinkerman, K. Zhu and D. L. Boger, Org. Lett., 2017, 19, 3568–3571 CrossRef CAS PubMed.
- K. Nakajima, Y. Miyake and Y. Nishibayashi, Acc. Chem. Res., 2016, 49, 1946–1956 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2097260. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc02210a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2022 |