
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHighly efficient and stable deep-blue OLEDs based on narrowband emitters featuring an orthogonal spiro-configured indolo[3,2,1-de]acridine structure†
Guoyun
Meng
a,
Dongdong
Zhang
*a,
Jinbei
Wei
b,
Yuewei
Zhang
a,
Tianyu
Huang
a,
Ziyang
Liu
a,
Chen
Yin
a,
Xiangchen
Hong
a,
Xiang
Wang
a,
Xuan
Zeng
a,
Dezhi
Yang
 c,
Dongge
Ma
c,
Guomeng
Li
a and
Lian
Duan
*ad
c,
Dongge
Ma
c,
Guomeng
Li
a and
Lian
Duan
*ad
aKey Lab of Organic Optoelectronics and Molecular Engineering of Ministry of Education, Department of Chemistry, Tsinghua University, Beijing, P. R. China. E-mail: duanl@mail.tsinghua.edu.cn; ddzhang@mail.tsinghua.edu.cn
bBeijing National Laboratory for Molecular Sciences, CAS Research/Education Center for Excellence in Molecular Sciences, Institute of Chemistry Chinese Academy of Sciences, Beijing, 100190, P. R. China
cInstitute of Polymer Optoelectronic Materials and Devices, State Key Laboratory of Luminescent Materials and Devices, South China University of Technology, Guangzhou, 510640, P. R. China
dCenter for Flexible Electronics Technology, Tsinghua University, Beijing, 100084, P. R. China
First published on 20th April 2022
Abstract
High-efficiency and stable deep-blue bottom-emitting organic light-emitting diodes with Commission Internationale de l'Eclairage y coordinates (CIEys) < 0.08 remain exclusive in the literature owing to the high excited-state energy of the emitters. Here, we propose the utilization of narrowband emitters to lower the excited-state energy for stable deep-blue devices by taking advantage of their high color purity. Two proof-of-concept deep-blue emitters with nitrogen-containing spiro-configured polycyclic frameworks are thereafter developed to introduce a multi-resonance effect for narrow emissions and sterically orthogonal configurations for alleviated molecular interactions. Both emitters show bright ultrapure deep-blue emissions with an extremely small full-width-at-half-maxima of only 18–19 nm, which can be maintained even in heavily doped films. Small CIEys of 0.054 and 0.066 are therefore measured from the corresponding electroluminescence devices with peak energies of only 2.77 eV (448 nm) and 2.74 eV (453 nm), accounting for the remarkably long LT80s (lifetime to 80% of the initial luminance) of 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 900 and 43470 hours at 100 cd m−2, respectively. Furthermore, by adopting a thermally activated delayed fluorescence sensitizer, impressive maximum external quantum efficiencies of 25% and 31% are recorded respectively, representing state-of-the-art performances for deep-blue devices.
900 and 43470 hours at 100 cd m−2, respectively. Furthermore, by adopting a thermally activated delayed fluorescence sensitizer, impressive maximum external quantum efficiencies of 25% and 31% are recorded respectively, representing state-of-the-art performances for deep-blue devices.
Introduction
Organic light-emitting diodes (OLEDs) have been successfully commercialized owing to the rapid development of organic semiconductors and device technologies. However, robust blue OLEDs with high efficiency and stability are still an ongoing pursuit as their performances largely lag behind the green and red ones.1 Substantial efforts thereby have been devoted to improving the performances of blue OLEDs. Recently, with conceptual advancements in molecular design, sky-blue OLEDs based on materials with phosphorescence or thermally activated delayed fluorescence (TADF) as emitters or sensitizers for fluorophores have witnessed rapid development in both efficiencies and lifetimes.1b,2 Blue emitters with hybridized local and charge-transfer (HLCT) states have also made great achievements in device efficiencies utilizing the spin-flip transition from a higher triplet state (Tn, n ≥ 2) to a singlet state.3 Despite these efforts, the most successful are still the conventional fluorophores in the view of practical applications as they are solely the ones that are being adopted in commercialized devices. And the blue OLEDs based on them have also made significant improvement in efficiency by virtue of triplet–triplet annihilation (TTA) to recycle triplet excitons.4For commercialized displays, the National Television Standards Committee (NTSC) recommended Commission Internationale de l'Eclairage coordinates of (0.14, 0.08) for a blue point in 1954. Nowadays, demands have emerged for cutting-edge ultra-high-definition (UHD) displays, which brings new requirements for blue OLEDs. In 2012, the International Telecommunication Union Radiocommunication Sector (ITUR) recommended a new color gamut standard for UHD displays, referred to as BT.2020, which required blue CIE coordinates of (0.131, 0.046).5 To satisfy the above demands, numerous deep-blue emitters with a CIEy < 0.08 have been developed, including conventional fluorophores,6 TADF emitters7 and phosphors,2b,d,8 which have afforded significant efficiency improvement with maximum external quantum efficiencies (EQEs) of over 25%. Nevertheless, to the best of our knowledge, the stabilities of bottom-emitting deep-blue OLEDs with a CIEy < 0.08 have not been reported yet, even for devices with conventional fluorophores.9 One of the main reasons should be attributed to the high excited-state energy of deep-blue emitters as an extremely short emission wavelength is required to satisfy the CIEy requirements. Commonly, the onset emission wavelengths of the reported deep-blue emitters are shorter than 400 nm, corresponding to an energy of over 3.10 eV. Such high excited energies would significantly increase the possibility of molecular degradation and thus go against good long-term operation stability. The realization of stable deep-blue devices therefore faces formidable challenges and needs new conceptual advancements in molecular design.10
With this in mind, we presented a strategical concept of utilizing emitters with ultra-narrowband emission to lower the energy of the excited state for good operation stabilities of deep-blue OLEDs by taking advantage of their high color purity. As shown in Scheme 1, bearing the same emission peaks, the narrower the emission band is, the smaller the CIEy value that can be obtained. This character allows an emitter bearing a small full-width-at-half-maxima (FWHM) to achieve the target CIEy with relatively red-shifted emission peaks compared with the one possessing a large FWHM. Both significantly reduced peaking and onset energy can therefore be obtained, which means a much lower excited state energy and thus naturally facilitating good long-term stabilities. Based on this, two proof-of-concept deep-blue emitters with nitrogen-containing spiro-configured polycyclic frameworks were developed. These skeletons not only endow a multi-resonance (MR) effect for extremely narrowband deep-blue emissions, but also possess a sterically orthogonal configuration to alleviate molecular interactions for an eliminated concentration quenching effect. Both emitters afforded an extremely narrowband of only ∼18 nm and photoluminescence quantum yields (PLQYs) of >80% in toluene. More importantly, in doped films or even pristine films, small FWHMs of ∼19 nm were maintained due to the reduced molecular interactions. The corresponding electroluminescent devices realized deep-blue emission colors with CIEy of 0.054 and 0.066 that is close to the BT.2020 blue point with only moderately high emission-peak energies of 2.77 eV (448 nm) and 2.74 eV (453 nm), respectively, owing to the intrinsically small FWHMs. What is truly remarkable is that long LT80s (lifetime to 80% of the initial luminance) of ∼100 and ∼230 hours at 2000 cd m−2 were obtained, corresponding to 18900 and 43470 hours at 100 cd m−2. To the best of our knowledge, these values represented the longest one ever reported for deep-blue bottom-emitting OLEDs, evidencing the concept proposed above. Moreover, ultrahigh EQEmaxs of 25% and 31% were recorded for these two emitters when adopting TADF as a sensitizer, also representing state-of-the-art performances for deep-blue OLEDs. This work not only proposes the advanced concept of breaking the limitation on the stability of deep-blue OLEDs by narrowing the bandwidth of emission but also demonstrates the strategic implementation of two highly efficient and stable N-containing MR emitters with the ability to suppress molecular interactions, providing a new paradigm for deep-blue emitters.
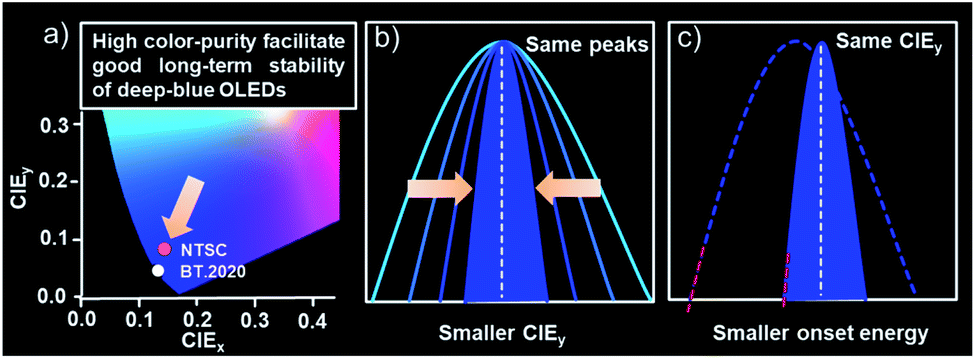 | ||
| Scheme 1 (a) The chromaticity coordinates of the blue point for NTSC and BT.2020. (b and c) The relationship between the CIEy and onset energy with FWHMs for stable deep-blue OLEDs. | ||
Results and discussion
Molecular design and theoretical calculations
This concept of improving the stability of deep-blue devices strongly relies on ultra-narrowband emitters. Multi-resonance (MR) fluorophores with frontier energy levels being located alternatively on single atoms of a rigid π-conjugation scaffold have proven to be effective for high purity, narrowband emissions by minimizing bonding/antibonding characters and suppressing vibrational coupling and relaxation.11 One design strategy for MR emitters is embedding heteroatoms with opposite resonance effects into polycyclic aromatic hydrocarbons (PAHs), including boron/nitrogen (B/N),2f,11a,12 boron/oxygen (B/O),7a,13 nitrogen/carbonyl (N/C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O),14etc. Nevertheless, owing to their charge transfer (CT) characteristics, these MR emitters face formidable challenges in realizing deep-blue emission, let alone ultra-narrowband emission with a FWHM < 20 nm. Alternatively, easily accessible nitrogen (N)-atom embedded PAHs (N-PAHs) such as indolocarbazole (ICz) fusions are more favorable for deep-blue emission due to their quite limited CT characteristics.15 More importantly, the narrowest spectrum having been reported until now was based on N-PAH derivatives.15b Therefore, in this manuscript, the N-PAH skeleton was adopted. Another unignorable fact is that though the intrinsic small FWHMs in solution, the inevitable intermolecular aggregation and packing between host-dopant and dopant themselves arising from the rigid plane structures of MR emitters would always lead to spectral broadening and emission quenching, particularly at a high dopant concentration, which greatly sacrifices the color purity.16 This has been a commonly observed phenomenon for MR emitters as summarized in Scheme S1,† which compares the wavelength and FWHMs of photoluminescence (PL) and electroluminescence (EL) spectra for the representative blue MR emitters. Therefore, to obtain high color-purity deep-blue emission under EL excitation, the molecular interactions in the doped films must be suppressed.17 With this in mind, two emitters based on a spiro-configured indolo[3,2,1-de]acridine fusion structure were developed and the structure of the targeted molecules, 2′,12′-dimethyldispiro[spirofluorene-9,10′-dibenzo[2,3:5,6]indolizino[1,8-ab]indolo[3,2,1-de]acridine-20′,9′′-spirofluorene] (pSFIAc1) and 8′,18′-di-tert-butyl-2′,12′-dimethyldispiro[fluorene-9,10′-dibenzo[2,3:5,6]indolizino[1,8-ab]indolo[3,2,1-de]acridine-20′, 9′′-fluorene] (pSFIAc2) are shown in Fig. 1a. The two emitters were developed, on one hand, to inherit the framework of N-PAH type MR emitters for ultra-narrowband deep-blue emission. On the other hand, the spiro-structure can introduce an orthogonal configuration to alleviate potential intermolecular packing to maintain the intrinsic FWHMs in doped films of the electroluminescent devices. For pSFIAc2, incorporation of bulky tert-butyl groups on the indolo[3,2,1-de]acridine fusion structure can effectively increase the intermolecular distance with nearby molecules and increase the PLQY.
O),14etc. Nevertheless, owing to their charge transfer (CT) characteristics, these MR emitters face formidable challenges in realizing deep-blue emission, let alone ultra-narrowband emission with a FWHM < 20 nm. Alternatively, easily accessible nitrogen (N)-atom embedded PAHs (N-PAHs) such as indolocarbazole (ICz) fusions are more favorable for deep-blue emission due to their quite limited CT characteristics.15 More importantly, the narrowest spectrum having been reported until now was based on N-PAH derivatives.15b Therefore, in this manuscript, the N-PAH skeleton was adopted. Another unignorable fact is that though the intrinsic small FWHMs in solution, the inevitable intermolecular aggregation and packing between host-dopant and dopant themselves arising from the rigid plane structures of MR emitters would always lead to spectral broadening and emission quenching, particularly at a high dopant concentration, which greatly sacrifices the color purity.16 This has been a commonly observed phenomenon for MR emitters as summarized in Scheme S1,† which compares the wavelength and FWHMs of photoluminescence (PL) and electroluminescence (EL) spectra for the representative blue MR emitters. Therefore, to obtain high color-purity deep-blue emission under EL excitation, the molecular interactions in the doped films must be suppressed.17 With this in mind, two emitters based on a spiro-configured indolo[3,2,1-de]acridine fusion structure were developed and the structure of the targeted molecules, 2′,12′-dimethyldispiro[spirofluorene-9,10′-dibenzo[2,3:5,6]indolizino[1,8-ab]indolo[3,2,1-de]acridine-20′,9′′-spirofluorene] (pSFIAc1) and 8′,18′-di-tert-butyl-2′,12′-dimethyldispiro[fluorene-9,10′-dibenzo[2,3:5,6]indolizino[1,8-ab]indolo[3,2,1-de]acridine-20′, 9′′-fluorene] (pSFIAc2) are shown in Fig. 1a. The two emitters were developed, on one hand, to inherit the framework of N-PAH type MR emitters for ultra-narrowband deep-blue emission. On the other hand, the spiro-structure can introduce an orthogonal configuration to alleviate potential intermolecular packing to maintain the intrinsic FWHMs in doped films of the electroluminescent devices. For pSFIAc2, incorporation of bulky tert-butyl groups on the indolo[3,2,1-de]acridine fusion structure can effectively increase the intermolecular distance with nearby molecules and increase the PLQY.
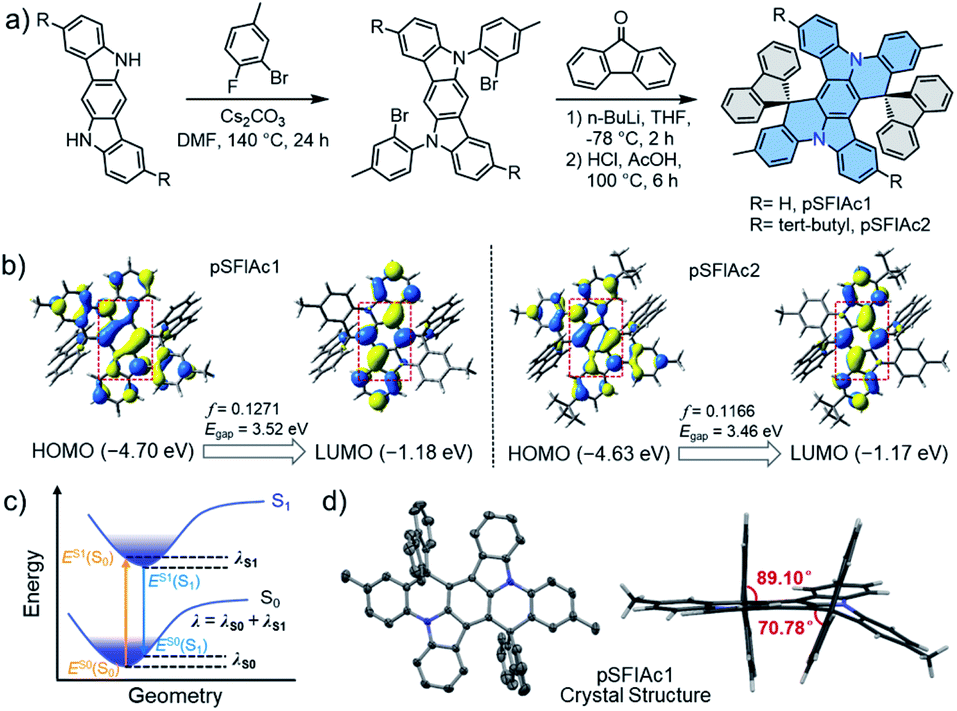 | ||
| Fig. 1 (a) Synthesis procedure and structure of pSFIAc1 and pSFIAc2. (b) The calculated distribution of HOMOs and LUMOs (isovalue = 0.03), oscillator strength (f) and energy gap. (c) Schematic potential energy diagram of the single point energies (E) and reorganization energy (λ). (d) Single-crystal X-ray structure of pSFIAc1. One molecule as an ORTEP drawing with a probability of ellipsoids of 50% and a side view. Hydrogen atoms are omitted for clarity. | ||
To understand the electronic and physical characteristics of the designed emitters, density functional theory (DFT) and time-dependent-DFT (TD-DFT) calculations using the B3LYP/6-31G(d) method were carried out. As shown in Fig. 1b, both emitters exhibit a remarkable atomically separated frontier molecular orbital (FMO) distribution on the skeletons, validating their MR effect. It is noted that the short-range charge transfers were observed between N-substituted methylbenzene and the central backbone. Moreover, the HOMO and LUMO distribution of the two emitters displayed π-orbitals with bonding/antibonding characters at the centre of the phenyl rings, which suggest the strong coupling of electrons on para-positioned N or C atoms, similar to the previous reports.18 Through these hybridized orbitals, extremely small reorganization energies (λ) of 0.206 and 0.187 eV were obtained for pSFIAc1 and pSFIAc2, respectively (Fig. S1†), smaller than that of pICz (0.239 eV).19 It is expected from the molecular orbital distribution and reorganization energy that the vibronic coupling and relaxation of the molecules would be suppressed. As shown in Fig. S1,† emission spectra simulated by Franck–Condon analysis on the S1–S0 transitions were also obtained, which exhibited significantly narrow emission bandwidths and maximum emissions of 17 nm and 454 nm for pSFIAc1, and 14 nm and 458 nm for pSFIAc2, respectively, evidencing ultra-narrowband deep-blue emission for these two compounds. Moreover, both pSFIAc1 and pSFIAc2 exhibited a significantly large oscillator strength (f) of >0.1, benefitting from a fast radiative decay process. The HOMO/LUMO energy values were calculated to be −4.70/−1.18 eV for pSFIAc1 and −4.63/−1.17 eV for pSFIAc2, respectively. The HOMO energy level of pSFIAc2 became shallower compared to pSFIAc1 due to tert-butyl substitution on the indolo[3,2,1-de]acridine fusion structure. Thus, we can expect that pSFIAc2 will show red-shifted emission compared to pSFIAc1. Notably, the calculated singlet (S1) and triplet (T1) energy differences exhibit a non-negligibly large ΔEST value of ∼0.48 eV for both emitters, an indication that the upconversion process from the T1 to the S1 state is intrinsically hindered. The details of the TD-DFT data are provided in the ESI.†
Material synthesis and the single crystal structure
The synthesis protocols for the nitrogen-containing emitters are described in the ESI† and obtained in a two-step reaction from commercially available starting materials in good yields, involving a sequence of nucleophilic substitution and tandem nucleophilic addition/acid-catalyzed annulation reactions. The precise chemical structures of pSFIAc1 and pSFIAc2 were fully characterized using NMR spectroscopy, high-resolution mass spectrometry, and elemental analysis (see the ESI†). In addition, a single crystal of pSFIAc1 suitable for X-ray diffraction analysis was also obtained by vacuum sublimation. As shown in Fig. 1d, the anticipated rigid plane conformation of a hexagonal ring fused backbone was validated in the solid-state crystal structure. Obviously, the two fluorene groups attached to the sp3 C atoms are aligned nearly orthogonally (torsion angles: 70.78–89.10°) to the hexagonal ring fused backbone, which is likely to reduce the impact of possible π–π interactions in the packing patterns. Thus, it can be observed that only a partial π–π stacking involves the head phenyl rings of the backbone with a distance from 3.488 to 3.621 Å. Furthermore, pSFIAc1 displays an extended one-dimensional structure in the crystal lattice via C–H⋯π interactions with distances between 2.765 and 2.892 Å (Fig. S2†). The orthogonal packing structures can not only alleviate the intramolecular nonradiative decay but also restrict the intermolecular interactions, which effectively suppresses molecular aggregation and luminescence concentration quenching, contributing to the high EL efficiency.Thermal, electrochemical and photophysical properties
The thermal stability and electrochemical properties of pSFIAc1 and pSFIAc2 were investigated by thermal analysis and cyclic voltammetry (CV) measurement (Fig. S3–S5†), respectively. It can be observed that both two emitters displayed excellent thermal properties with 5% weight-loss decomposition temperatures of 382 °C (pSFIAc1) and 474 °C (pSFIAc2), respectively. The glass transitions were not observed during differential scanning calorimetry analysis. Based on the electrochemical data, the estimated HOMO and LUMO energy levels were −5.58 and −2.37 eV for pSFIAc1, and −5.49 and −2.32 eV for pSFIAc2, respectively. Notably, pSFIAc2 showed a 0.09 eV shallower HOMO energy level and a 0.05 eV deeper LUMO energy level than pSFIAc1, leading to a reduced HOMO–LUMO energy gap.The ultraviolet-visible (UV-vis) absorption and PL spectra of the two emitters were measured in toluene solution and are shown in Fig. 2a and Table 1. Thus, two emitters showed intense absorption bands below 370 nm, attributed to the π–π* transition of indolo[3,2,1-de]acridine backbones, while the broad absorption bands between 370 and 450 nm should be caused by the n–π* transitions of fluorene fused N-phenylcarbazole.7d,20 In the fluorescence spectra, the sharp emission bands with peak wavelengths (λem) at 443 nm (ΦPL = 84%) and 450 nm (ΦPL = 88%) were recorded for pSFIAc1 and pSFIAc2, respectively. Interestingly, compared to the reported ICz-based MR emitters,7d,15b,19 both emitters showed a narrowed FWHM of 18 nm (0.12 eV) and a small Stokes shift (10 nm), demonstrating a minor vibronic coupling between the ground state and excited state as well as the rigid π-conjugated framework. The weak shoulder peaks that appeared in the region of low energy are assigned to the vibronic split band. In addition, only slight changes in emission spectra are observed upon modulation of the solvent polarity (Fig. S6†), which can be assigned to the more localized FMO distributions. The transient PL decay curves of the two emitters were also measured in degassed toluene solution as shown in Fig. S7,† showing only a single exponential component with fluorescence lifetimes of 7.7 ns for pSFIAc1 and 8.0 ns for pSFIAc2, and high radiative decay rates (krs) of 10.9 × 107 s−1 and 11.0 × 107 s−1 were obtained, respectively. The energy differences (ΔESTs) between the singlet and triplet states of the two emitters were calculated from the peaks of the fluorescence and phosphorescence spectra at 298 K and 77 K, respectively, which are estimated to be 0.29–0.31 eV. These results validate the effectiveness of these two compounds as ultra-narrowband deep-blue emitters, which is in line with expectations of molecular design purposes.
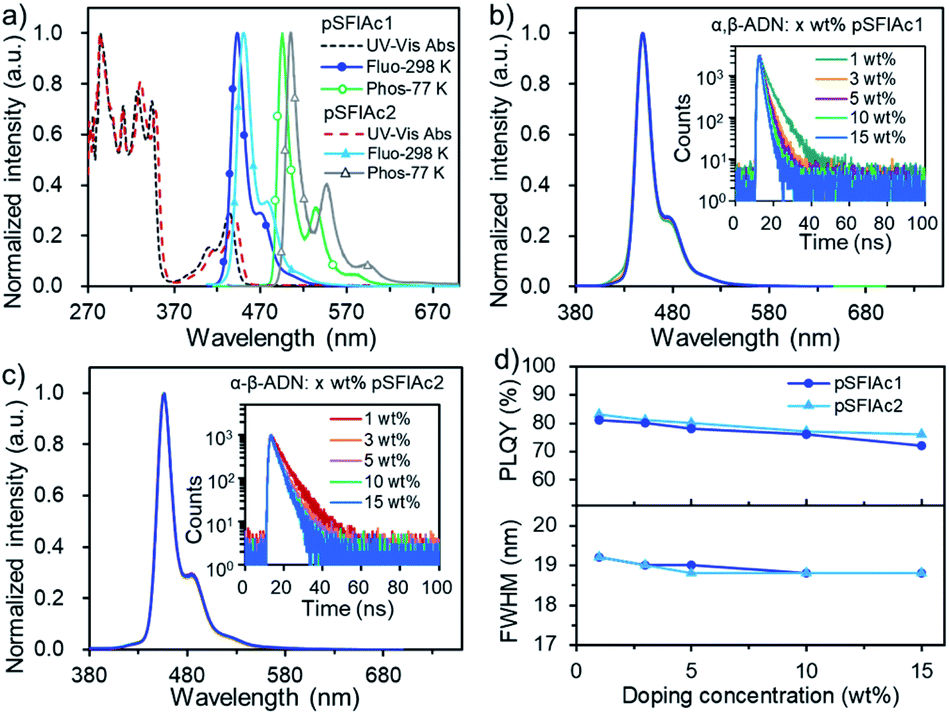 | ||
| Fig. 2 (a) The absorption (left axis), fluorescence and phosphorescence (right axis) spectra of pSFIAc1 and pSFIAc2 in toluene (10−5 M). (b and c) The PL spectra and transient PL (inset) curves with different concentrations in α,β-ADN: x wt% MR emitters (x = 1, 3, 5, 10, 15). (d) The PLQYs and FWHMs of the doped films with different concentrations in α,β-ADN: x wt% MR emitters (x = 1, 3, 5, 10, 15). | ||
| Compound | λ abs [nm], 298 K | λ PL [nm], 298 K | Stokes shifta [nm] | FWHMb [nm eV−1] | Φ PL [%], 298 K | E S1/ET1d [eV] | ΔESTd [eV] | τ PF [ns] | k r [107 s−1] | HOMO/LUMOg [eV] |
|---|---|---|---|---|---|---|---|---|---|---|
| a Measured in toluene (10−5 M) at room temperature. b Full-width at half-maximum of the PL spectrum given in wavelength and energy. c Absolute PL quantum yield evaluated using an integrating sphere. d Lowest excited singlet (ES1) and triplet (ET1) energies estimated from peaks of the fluorescence and phosphorescence spectra recorded at 298 K and 77 K, respectively. ΔEST = ES1 − ET1. e Emission lifetime of fluorescence. f Rate constant of fluorescence radiative decay, Kr = ΦPF/τPF. g The HOMO and LUMO energies were determined according to EHOMO/LUMO = −(Eox/Ered + 4.8) eV. | ||||||||||
| pSFIAc1 | 284, 311, 327, 344, 413, 433 | 443 | 10 | 18/0.12 | 84 | 2.80/2.51 | 0.29 | 7.7 | 10.9 | −5.58/−2.37 |
| pSFIAc2 | 284, 310, 331, 348, 416, 440 | 450 | 10 | 18/0.12 | 88 | 2.76/2.45 | 0.31 | 8.0 | 11.0 | −5.49/−2.32 |
We further evaluated the performances of both emitters in suppressing molecular interactions by dispersing them into a host matrix, 9-(naphthalen-1-yl)-10-(naphthalen-2-yl)anthracene (α,β-ADN), which has been applied widely for P-type delayed fluorescence OLEDs.21 Interestingly, in a wide range of doping concentrations (1, 3, 5, 10, 15 wt%), identical deep-blue emissions peaking at ∼448 and 455 nm were observed for pSFIAc1 and pSFIAc2, respectively, with a high ΦPL ranging from 72% to 83% (Fig. 2). Particularly, both emitters possessed extremely narrowband spectra (FWHM = ∼19 nm or ∼0.12 eV) and showed intrinsically luminescence properties as they were in diluted solution. These behaviors not only suggest efficient energy transfer from the host to dopant but also make clear that the two emitters can greatly suppress molecular interactions in doped films. The reason should be assigned to the steric hindrance effect introduced by the orthogonal spiro-configuration, which prevents the intermolecular aggregation and packing between the host-dopant and dopant themselves to keep the essential emission bandwidth and photo-luminescence efficiency. The transient PL decay curves of all the films also showed single exponential prompt fluorescence decays. Thus, the radiative decay rate constants (krs) of these films were calculated to be approximately 108 s−1 orders of magnitude (Tables S5 and S6†). We also accessed the PL behaviors of pSFIAc1 and pSFIAc2 doped in other common wide energy gap compounds, such as 3,3-di(9H-carbazol-9-yl)biphenyl (mCBP) and bis[2-(diphenylphosphino)phenyl] ether oxide (DPEPO). Similarly, identical narrowband emissions and high ΦPLs were recorded for these films with increased dopant concentrations (Fig. S8 and S9†). Particularly, due to the strong steric effect of diphenylphosphine oxide groups of DPEPO, the ΦPLs of the two emitters in DPEPO can maintain high values of 87–80% when the doping concentration increased from 1 wt% to 15 wt%. These results further evidence the ability of such an orthogonal structure to prevent molecular interactions. It also deserves to be mentioned that even in pristine films, small FWHMs of ∼20 nm can also be maintained for both emitters with only quite weak excimer emission in the long-wavelength region, suggesting that intermolecular packing can be suppressed even in a neat film (Fig. S10†). This result suggests that the strategic introduction of orthogonal fluorene groups into the indolo[3,2,1-de]acridine fusion structure enables intriguing photoelectric properties especially in constructing high color purity MR emitters. How to suppress the molecular interactions for MR emitters has been an exigent task in this field, and the strategy here may provide an effective solution.
Efficiency and stability of deep-blue P-type OLEDs
Benefitting from the excellent photophysical properties of these deep-blue emitters, OLED devices were fabricated using an optimized device structure of indium tin oxide (ITO)/1,4,5,8,9,11-hexaazatriphenylene-hexacarbonitrile (HATCN, 5 nm)/4,4′-N,N′-bis[N-(1-naphthyl)-N-phenylamino]biphenyl (NPB, 30 nm)/9,9′-diphenyl-9H,9′H-3,3′-bicarbazole (BCzPh, 10 nm)/α,β-ADN: x wt% MR emitters (30 nm)/4,6-bis(3-(9H-carbazol-9-yl)phenyl)pyrimidine (CzPhPy, 10 nm)/9,10-bis(6-phenylpyridin-3-yl)anthracene (DPPyA, 20 nm)/LiF (0.5 nm)/Al (150 nm). This device structure was chosen as the OLED industry still relies on P-type delayed fluorescence (also known as TTA4a) to provide blue emission. In the emission layer, the MR emitters were co-deposited with the α,β-ADN host at various doping concentrations (x = 1, 3, 5, 10, 15 wt%, devices A1–A5 for pSFIAc1 and devices B1–B5 for pSFIAc2) to study the ability of this emitter in suppressing molecular packing. The energy level diagram and the molecular structures of the materials used in the devices are shown in Fig. 3 and S11.† The detailed device performances are depicted in the ESI† and summarized in Table 2.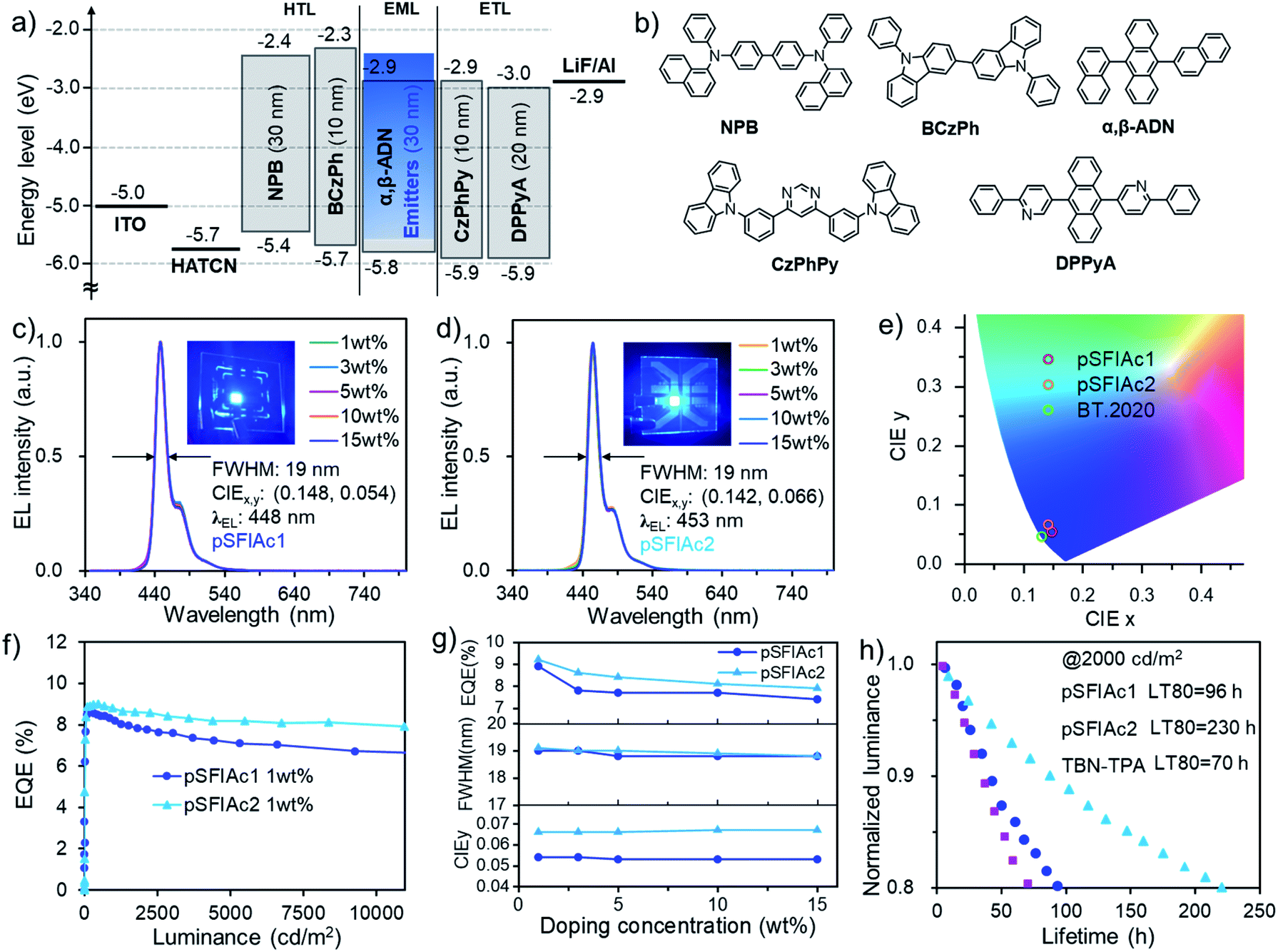 | ||
| Fig. 3 (a) Device configuration with an energy-level diagram and (b) the molecular structures of the materials used in this device. (c and d) The EL spectra of the devices, and (e) EQE versus luminance curves of devices A1 and B1. (f) Luminescence and current density versus voltage. (g) The EQEmax, FWHMs and CIEy-dopant concentration relationships in devices. (h) The device luminance versus lifetime curves measured at 2000 cd m−2. | ||
| Device type | x wt% | λ EL [nm] | FWHM b [nm eV−1] | V on [V] | L max [cd m−2] | PEmaxe [lm W−1] | EQEmaxf [%] | CIE (x, y)a |
|---|---|---|---|---|---|---|---|---|
| a Value recorded at a luminance around 1000 cd cm−2. b Full width at half maximum of electroluminescence. c Turn-on voltage at a luminance of 1 cd m−2. d Maximum luminescence (L). e Maximum power efficiency (PE), value at 1000 and 5000 cd cm−2 for devices A1, B1 and C, and maximum PE, value at 100 and 1000 cd cm−2 for devices A6 and B6. f Maximum EQE, value at 1000 and 5000 cd cm−2 for devices A1, B1 and C, and maximum EQE, value at 100 and 1000 cd cm−2 for devices A6 and B6. | ||||||||
| Device A1 | 1.0 | 448 | 19.0 (0.12 eV) | 2.6 | 14040 |
4.6/2.9/2.0 | 8.9/8.2/7.3 | (0.147, 0.054) |
| Device B1 | 1.0 | 453 | 19.0 (0.12 eV) | 2.7 | 16250 |
5.2/4.0/3.0 | 9.1/8.7/8.2 | (0.142, 0.066) |
| Device C | 1.0 | 453 | 23.0 (0.13 eV) | 2.9 | 10660 |
3.7/2.6/1.8 | 8.4/8.0/7.2 | (0.142, 0.054) |
| Device A6 | 1.0 | 446 | 21.0 (0.13 eV) | 3.4 | 2002 | 14.9/1.2/0.6 | 24.9/4.1/3.2 | (0.148, 0.058) |
| Device B6 | 1.0 | 451 | 21.0 (0.13 eV) | 3.5 | 2253 | 23.0/3.0/1.4 | 31.4/5.6/4.3 | (0.146, 0.078) |
Fig. 3c and d show the EL spectra of all these devices. It is interesting to note that in the dopant range of 1–15 wt%, all devices showed the same EL spectra with emission peaks of 448 and 453 nm for pSFIAc1 and pSFIAc2, and both small FWHMs of ∼19 nm (0.12 eV) as shown in Fig. 3g. The independence of emission spectra on the concentration can be assigned to the alleviated molecular interactions owing to the orthogonal spiro-structure of these emitters as discussed above. Even non-doped devices using neat films as the emitting layer (Fig. S12 and S13†) also exhibit bright deep blue EL at 448 and 453 nm with both small FWHMs ∼20 nm for pSFIAc1 and pSFIAc2, respectively, in agreement with the PL results. It should be emphasized is that though the corresponding moderately high emission-peak energies of 2.77 eV and 2.74 eV and CIE coordinates of (0.148, 0.054) and (0.142, 0.066) were obtained for pSFIAc1 and pSFIAc2, respectively, being close to the BT.2020 standard (0.131, 0.046) without using microcavities or color filters as shown in Fig. 3e. Particularly, the onset wavelengths of pSFIAc1 and pSFIAc2 were only 428 nm and 432 nm, corresponding to the energies of 2.90 eV and 2.87 eV, respectively. These onset energies were significantly lower than those of previously reported emitters with similar colors but much wider emission bands.
The device EQE–luminance characteristics are shown in Fig. 3f. Notably, both EQEmaxs of ∼9.0% were obtained for the devices A1 and B1 with a maximum brightness of 14040 and 16250 cd m−2, respectively. It is also worth noting that the efficiency roll-off of the devices was small with the EQEmaxs remaining at 8.0% and 7.1% for device A1 and 8.7% and 8.2% for device B1 at a brightness of 1000 and 5000 cd m−2, respectively. These EQEs greatly exceed the theoretical upper limit of conventional fluorescent OLEDs and should partly benefit from the TTA mechanism based delayed fluorescence owing to the adoption of the anthracene-based host material. To prove this, the voltage-dependent EL transient curves of devices A1 and B1 were measured as shown in Fig. S18.† An obvious EL delayed component in the order of microseconds can be observed after switching off the electrical pulse, confirming the contribution of the TTA process in the devices. The delayed emission ratios of approximately 26% for the pSFIAc1- and pSFIAc2-based devices correspond to the total radiative exciton ratios of about 35% (=[0.26/(1 − delayed emission ratio)]).4b Meanwhile, with the voltages being increased from 4 V to 6 V, shorter delayed lifetimes were observed, which indicated the accelerated TTA process from the high triplet density at high current densities, further evidencing this emission mechanism. We also provided the dependence of EQEs on the dopant concentration in Fig. 3g and found only slightly varied EQEmax values (9.1–7.4%) in a wide range of dopant concentrations (1–15 wt%) for both series of devices, benefiting from the greatly relieved concentration caused emission quenching issues due to the orthogonal structure of both MR emitters as evidenced by the slightly varied ΦPLs of these doped films.
We further evaluated the EL operational stability of pSFIAc1 and pSFIAc2 in the 1 wt% doped devices at a constant current density with an initial luminance of 2000 cd m−2. As shown in Fig. 3h, remarkably long LT80s (lifetime to 80% of the initial luminance) of 96 and 230 hours were realized for device A1 and device B1, respectively. With a degradation acceleration factor (n) of 1.75, the extrapolated LT80s of these devices at an initial luminance of 100 cd m−2 can be calculated to be 18900 and 43470 hours for devices A1 and B1, respectively. The relatively longer lifetime of pSFIAc2 in the device compared to pSFIAc1 should arise from its red-shifted emission. To the best of our knowledge, these lifetimes represent the record-long values for deep-blue OLEDs with a CIEy < 0.1. The good stabilities should arise from the high color-purity of these emitters. As aforementioned, benefiting from their ultra-narrowband emissions, both emitters feature reduced onset- and peak-energies, naturally facilitating alleviation of the degradation possibility of molecules in excited states and finally accounting for the remarkably long lifetimes. To further evaluate the stability of our compounds, we synthesised a B/N-type MR TADF emitter with 2,12-di-tert-butyl-5,9-bis(4-(tert-butyl)phenyl)-7-(3,6-di-tert-butyl-9H-carbazol-9-yl)-5,9-dihydro-5,9-diaza-13b-boranaphtho[3,2,1-de]anthracene (TBN-TPA)12c as the reference emitter for comparison (Fig. S20†). The contrast device C with TBN-TPA as the emitter afforded deep-blue EL spectra with an emission peak of 453 nm, an FWHM of 23 nm (0.13 eV) and CIE coordinates of (0.142, 0.054) simultaneously (Fig. S21† and Table 2). However, device C only exhibited an LT80 of 70 hours at an initial luminance of 2000 cd m−2, much shorter than the device with pSFIAc1 as the emitter despite their same CIEy values (Fig. 3h). One plausible reason can be ascribed to the relatively longer lifetimes of the delayed component, resulting in more significant exciton quenching at a high current density (Fig. S18†), which has been taken as the main reason for device degradation.22 It is also speculated that the orthogonal spiro-configured structure of pSFIAc1 should also benefit device stability by alleviating molecular interactions. Though numerous studies on the stability issues of B/N-type MR emitters have been carried out, this is the first time that the stability of N-embedded MR emitters was evaluated as far as we know. And the results here reveal good long-term operation stability of such emitters, which will arouse interest from both academic and industrial fields.
TSF-OLED device performances
Besides the TTA mechanism, an alternative strategy to assist the recycling of triplet excitons in fluorescent devices is the so-called TADF-sensitized fluorescence (TSF), whereby a TADF material is adopted as a sensitizer for fluorophores.23 To unlock the full potential of the device efficiencies of pSFIAc1 and pSFIAc2, we thereby constructed TSF based devices. Given that an efficient Förster energy transfer (FET) between the sensitizer and fluorophore is the prerequisite to promote the TSF process, a deep-blue TADF compound, m4TCzPhBN18a was chosen here as a sensitizer for both the emitters. Previous studies have revealed the blue TADF emission of m4TCzPhBN, making it a potential TADF sensitizer for deep-blue emitters.Fig. 4a shows the significant overlap between the emission spectra of m4TCzPhBN and the absorption spectra of both the emitters. Large Förster energy transfer radii of 2.9 and 3.0 nm were thereby recorded for m4TCzPhBN → pSFIAc1 and m4TCzPhBN → pSFIAc2, respectively. Furthermore, both PL spectra of mCPB: 30 wt% m4TCzPhBN: 1.0 wt% emitter-doped films exhibited narrow deep-blue emission with identical FWHMs of 21 nm and high ΦPLs of >80%, which nearly coincide with that of the mCPB: 1.0 wt% emitter-doped films, suggesting complete energy transfer from the sensitizer to dopants (Fig. S22†). We also measured the PL decay curves of these doped films, unveiling clearly delayed components from dopant emission in the mCPB: 30 wt% m4TCzPhBN: 1.0 wt% emitter-doped films (Fig. S23†). In contrast, the PL transient decay curve of mCBP: 1.0 wt% emitter-doped films only exhibited a single exponential component. These results suggest that a part of the dopant emission originates by energy transfer from the up-converted triplets of the sensitizer in the tri-component films via the TSF process.22
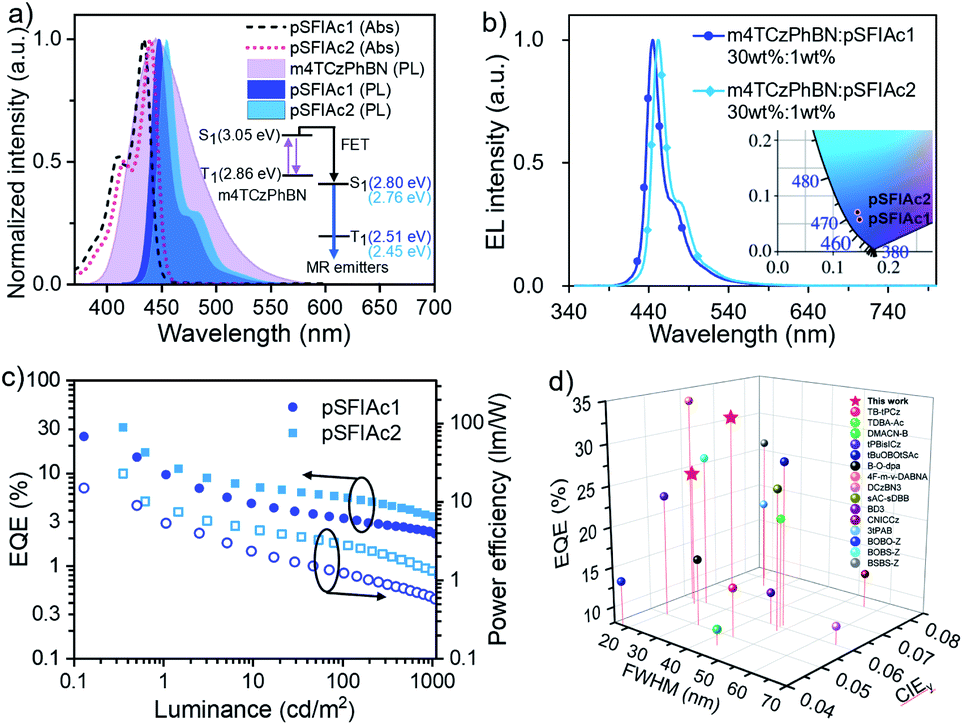 | ||
| Fig. 4 (a) The absorption spectra of MR emitters and the PL spectra of the doped films of mCBP: 30 wt% m4TCzPhBN; mCBP: 30 wt% m4TCzPhBN: 1 wt% MR emitters. (b) The EL spectra of devices were recorded at 1000 cd m−2. (c) The EQE and PE versus luminance curves of the devices. (d) Summary of the EQE versus FWHMs and CIEy color coordinates plot of deep-blue materials reported in the literature. The red star represents the results of the present work. | ||
Then, the TSF devices were constructed with structures of ITO/TAPC (30 nm)/TCTA (5 nm)/mCP (5 nm)/EMLs (30 nm)/PPF (5 nm)/Bphen (30 nm)/LiF (0.5 nm)/Al (150 nm), where EMLs stand for mCPB: 30 wt% m4TCzPhBN: x wt% pSFIAc1 (x = 1, 2, 3 wt%, devices A6–A8) and mCPB: 30 wt% m4TCzPhBN: x wt% pSFIAc2 (x = 1, 2, 3 wt%, devices B6–B8), respectively. Detailed energy diagrams of each functional layer, as well as the molecular structures, are shown in Fig. S24.† The EL spectra of the devices with increased dopant concentrations are shown in Fig. 4b, S25 and S26,† which exhibited identical deep-blue peaks at 446 and 451 nm along with small FWHMs of ∼21 nm (∼0.13 eV) for devices A6–A8 and B6–B8, respectively. CIE coordinates of (0.148, 0.058) and (0.146, 0.078) were also recorded for pSFIAc1 and pSFIAc2 based devices. Besides, these EL spectra remain unchanged with increased luminances, suggesting complete energy transfer.
Strikingly, the TSF-devices showed excellent efficiencies with EQEmaxs of 24.9% and 31.4% and power efficiencies (PE) of 14.9 and 23.0 lm W−1 for devices A6 and B6 (Fig. 4c), respectively. Owing to the rigid π-conjugated skeleton, moderate emitting dipole orientations (Θ‖) of 71% and 72% are fitted for pSFIAc1 and pSFIAc2 doped films (Fig. S27†), respectively, which mean that both emitters exhibit almost an isotropic dipole orientation and an improved outcoupling. For pSFIAc1 and pSFIAc2 based devices, the EQEmaxs slightly decreased with increasing dopant ratios, which could be attributed to the carrier trapping by the dopants and enhanced Dexter energy transfer at high dopant concentrations. The lifetimes of devices A6 and B6 were also estimated and the corresponding lifetimes (LT50) at an initial luminance of 1000 cd m−2 were 13 and 20 h (Fig. S28†), respectively. Both devices displayed a shorter operation lifetime than the TTA devices, which is in agreement with previous reports.24 As shown in Fig. 4d, we summarized the device efficiencies (lifetimes) versus FWHMs and CIEy color coordinates based on the reported deep-blue (blue) OLEDs, which are listed in the ESI (Tables S13 and S14†). These state-of-the-art performances further validate the superiority of pSFIAc1 and pSFIAc2 as deep-blue narrowband emitters. However, efficiency roll-off was observed for both TSF devices, and revealing a similar overall performance to the previously reported work.7d,19,25 We speculate that the plausible direct carrier trapping on the emitter molecules at a high current density should be responsible. Ideally, charge recombination should be on the sensitizer to guarantee the sensitizing process. The device energy levels are shown in Fig. S24,† revealing relatively shallower LUMO levels of the emitter than the sensitizer. Therefore, at a low voltage, electrons tend to be directly injected into the LUMO levels of the sensitizer and then recombine with holes. At a high voltage, electrons can overcome the energy barrier to be injected into the LUMO levels of the emitter and then combine with holes on the emitter directly. Thus, at low brightness, the sensitizing process is efficient to afford a high EQE. But at high brightness, the sensitizing process is disabled and thus results in a low EQE, accounting for the significant efficiency roll-off. The direct charge trapping on the emitter can be avoided by optimizing the energy levels of the sensitizer and emitter. But at this moment, efficient deep-blue TADF sensitizers are still rare. We believe that by further optimizing the device structures and with the development of more efficient deep-blue TADF sensitizers, the performances of TSF devices based on N-embedded MR emitters can be further improved.
Conclusions
In conclusion, we presented an advanced strategy of using emitters with ultra-narrowband spectra to reduce the excited-state energy to break the stability limitation of deep-blue OLEDs. Based on this concept, two deep-blue N-containing emitters featuring a spiro-configuration were developed, affording bright ultrapure deep-blue emissions with an extremely small full-width-at-half-maxima of only 18–19 nm, which are maintained even in heavily doped films due to the alleviated molecular interactions. The corresponding electroluminescent devices realized deep-blue emission colors with CIEys of 0.054 and 0.066, close to the BT.2020 blue point, though with moderate peak energies of 2.77 eV (448 nm) and 2.74 eV (453 nm). Remarkably long LT80s of 18900 and 43470 hours at 100 cd m−2 were thereby obtained respectively, representing the longest lifetimes of deep-blue OLEDs in the literature. We also adopted a TADF sensitizer to recycle excitons for both the emitters, realizing a high EQEmax of 25% and 31% respectively, which also represent state-of-the-art performances for deep-blue devices. This work not only opens a new avenue towards stable deep-blue OLEDs but also provides a new paradigm of N-embedded MR emitters with the ability to suppress molecular interactions, which will surely arouse a lot of attention and motivate the production of more efficient deep-blue emitters and devices.
Data availability
All data supporting this study are available from article and ESI.†Author contributions
L. D. conceived and supervised the project. D. Z. and L. D. designed the experiments. G. M. synthesized and characterized the deep-blue emitters, OLED fabrication and measurement, and wrote the manuscript. G. L. synthesized the TBN-TPA emitter and performed the electrochemical measurements of the deep-blue emitters. X. H. and X. Z. helped in theoretical calculations. X. W. and J. W. measured the single crystal structure. Y. Z., Z. L., C. Y. and T. H. provided suggestions on device fabrication and experiments. D. Y. and D. M. measured the dipole orientations of the emitters. L. D. and D. Z. supervised and reviewed and revised the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Fund of China (Grant No. 22135004, 51903137 and 61890942), the National Key Basic Research and Development Program of China (Grant No. 2020YFA0715000 and 2017YFA0204501), the Guangdong Major Project of Basic and Applied Basic Research (Grant No. 2019B030302009), the Foshan Xianhu Laboratory of the Advanced Energy Science and Technology Guangdong Laboratory XHT2020-005, the China Postdoctoral Science Foundation (Grant No. 2021M701865) and the Guangdong Basic and Applied Basic Research Foundation (2021B1515120041).Notes and references
- (a) S. O. Jeon, K. H. Lee, J. S. Kim, S.-G. Ihn, Y. S. Chung, J. W. Kim, H. Lee, S. Kim, H. Choi and J. Y. Lee, Nat. Photonics, 2021, 15, 208 CrossRef CAS; (b) C.-Y. Chan, M. Tanaka, Y.-T. Lee, Y.-W. Wong, H. Nakanotani, T. Hatakeyama and C. Adachi, Nat. Photonics, 2021, 15, 203 CrossRef CAS; (c) D. Zhang and L. Duan, Nat. Photonics, 2021, 15, 173 CrossRef CAS; (d) T.-L. Wu, M.-J. Huang, C.-C. Lin, P.-Y. Huang, T.-Y. Chou, R.-W. Chen-Cheng, H.-W. Lin, R.-S. Liu and C.-H. Cheng, Nat. Photonics, 2018, 12, 235 CrossRef CAS; (e) Y. Chen, D. Zhang, Y. Zhang, X. Zeng, T. Huang, Z. Liu, G. Li and L. Duan, Adv. Mater., 2021, 33, e2103293 CrossRef PubMed; (f) Y. L. Zhang, Q. Ran, Q. Wang, Y. Liu, C. Hanisch, S. Reineke, J. Fan and L. S. Liao, Adv. Mater., 2019, 31, 1902368 CrossRef CAS PubMed; (g) K. R. Naveen, K. Prabhu Cp, R. Braveenth and J. H. Kwon, Chem.–Eur. J., 2022, 28, e202103532 Search PubMed.
- (a) X. Li, J. Zhang, Z. Zhao, L. Wang, H. Yang, Q. Chang, N. Jiang, Z. Liu, Z. Bian, W. Liu, Z. Lu and C. Huang, Adv. Mater., 2018, 30, e1705005 CrossRef PubMed; (b) A. K. Pal, S. Krotkus, M. Fontani, C. F. R. Mackenzie, D. B. Cordes, A. M. Z. Slawin, I. D. W. Samuel and E. Zysman-Colman, Adv. Mater., 2018, 30, e1804231 CrossRef PubMed; (c) J. Lee, H. F. Chen, T. Batagoda, C. Coburn, P. I. Djurovich, M. E. Thompson and S. R. Forrest, Nat. Mater., 2016, 15, 92 CrossRef CAS PubMed; (d) K. W. Lo, G. S. M. Tong, G. Cheng, K. H. Low and C. M. Che, Angew. Chem., Int. Ed., 2022, 61, e20211551 CrossRef PubMed; (e) S. Nam, J. W. Kim, H. J. Bae, Y. M. Maruyama, D. Jeong, J. Kim, J. S. Kim, W. J. Son, H. Jeong, J. Lee, S. G. Ihn and H. Choi, Adv. Sci., 2021, 8, e2100586 CrossRef PubMed; (f) Y. Kondo, K. Yoshiura, S. Kitera, H. Nishi, S. Oda, H. Gotoh, Y. Sasada, M. Yanai and T. Hatakeyama, Nat. Photonics, 2019, 13, 678 CrossRef CAS; (g) X. Cai and S.-J. Su, Adv. Funct. Mater., 2018, 28, 1802558 CrossRef.
- (a) Y. Xu, P. Xu, D. Hu and Y. Ma, Chem. Soc. Rev., 2021, 50, 1030 RSC; (b) X. Lv, M. Sun, L. Xu, R. Wang, H. Zhou, Y. Pan, S. Zhang, Q. Sun, S. Xue and W. Yang, Chem. Sci., 2020, 11, 5058 RSC.
- (a) C. Gao, W. W. H. Wong, Z. Qin, S. C. Lo, E. B. Namdas, H. Dong and W. Hu, Adv. Mater., 2021, 33, e2100704 CrossRef PubMed; (b) H. Lim, S. J. Woo, Y. H. Ha, Y. H. Kim and J. J. Kim, Adv. Mater., 2021, e2100161 Search PubMed.
- M. Sugawara, S.-Y. Choi and D. Wood, IEEE Signal Process. Mag., 2014, 31, 170 Search PubMed.
- (a) R. Guo, W. Liu, S. Ying, Y. Xu, Y. Wen, Y. Wang, D. Hu, X. Qiao, B. Yang, D. Ma and L. Wang, Sci. Bull., 2021, 66, 2090 CrossRef CAS; (b) L. Peng, J.-W. Yao, M. Wang, L.-Y. Wang, X.-L. Huang, X.-F. Wei, D.-G. Ma, Y. Cao and X.-H. Zhu, Sci. Bull., 2019, 64, 774 CrossRef CAS; (c) Y. Takita, K. Takeda, N. Hashimoto, S. Nomura, T. Suzuki, H. Nakashima, S. Uesaka, S. Seo and S. Yamazaki, J. Soc. Inf. Disp., 2018, 26, 55 CrossRef CAS; (d) J.-Y. Hu, Y.-J. Pu, F. Satoh, S. Kawata, H. Katagiri, H. Sasabe and J. Kido, Adv. Funct. Mater., 2014, 24, 2064 CrossRef CAS.
- (a) D. H. Ahn, S. W. Kim, H. Lee, I. J. Ko, D. Karthik, J. Y. Lee and J. H. Kwon, Nat. Photonics, 2019, 13, 540 CrossRef CAS; (b) A. Khan, X. Tang, C. Zhong, Q. Wang, S. Y. Yang, F. C. Kong, S. Yuan, A. S. D. Sandanayaka, C. Adachi, Z. Q. Jiang and L. S. Liao, Adv. Funct. Mater., 2021, 31, 2009488 CrossRef CAS; (c) H. J. Kim, H. Kang, J. E. Jeong, S. H. Park, C. W. Koh, C. W. Kim, H. Y. Woo, M. J. Cho, S. Park and D. H. Choi, Adv. Funct. Mater., 2021, 31, 2102588 CrossRef CAS; (d) V. V. Patil, H. L. Lee, I. Kim, K. H. Lee, W. J. Chung, J. Kim, S. Park, H. Choi, W. J. Son, S. O. Jeon and J. Y. Lee, Adv. Sci., 2021, 8, e2101137 CrossRef PubMed; (e) I. S. Park, M. Yang, H. Shibata, N. Amanokura and T. Yasuda, Adv. Mater., 2021, e2107951 Search PubMed.
- (a) T. Fleetham, G. Li, L. Wen and J. Li, Adv. Mater., 2014, 26, 7116 CrossRef CAS PubMed; (b) Y. Wu, C. Yang, J. Liu, M. Zhang, W. Liu, W. Li, C. Wu, G. Cheng, Q. Yang, G. Wei and C. M. Che, Chem. Sci., 2021, 12, 10165 RSC.
- (a) S. K. Jeon, H. L. Lee, K. S. Yook and J. Y. Lee, Adv. Mater., 2019, 31, e1803524 CrossRef PubMed; (b) M. Sarma, W. L. Tsai, W. K. Lee, Y. Chi, C. C. Wu, S. H. Liu, P. T. Chou and K. T. Wong, Chem, 2017, 3, 461 CrossRef CAS.
- A. Monkman, ACS Appl. Mater. Interfaces, 2021 DOI:10.1021/acsami.1c09189.
- (a) T. Hatakeyama, K. Shiren, K. Nakajima, S. Nomura, S. Nakatsuka, K. Kinoshita, J. Ni, Y. Ono and T. Ikuta, Adv. Mater., 2016, 28, 2777 CrossRef CAS PubMed; (b) S. Madayanad Suresh, D. Hall, D. Beljonne, Y. Olivier and E. Zysman-Colman, Adv. Funct. Mater., 2020, 30, 1908677 CrossRef CAS; (c) J.-M. Teng, Y.-F. Wang and C.-F. Chen, J. Mater. Chem. C, 2020, 8, 11340 RSC; (d) J. M. Ha, S. H. Hur, A. Pathak, J.-E. Jeong and H. Y. Woo, NPG Asia Mater., 2021, 13, 53 CrossRef CAS.
- (a) K. Matsui, S. Oda, K. Yoshiura, K. Nakajima, N. Yasuda and T. Hatakeyama, J. Am. Chem. Soc., 2018, 140, 1195 CrossRef CAS PubMed; (b) Y. Zhang, D. Zhang, J. Wei, Z. Liu, Y. Lu and L. Duan, Angew. Chem., Int. Ed., 2019, 58, 16912 CrossRef CAS PubMed; (c) S. Oda, W. Kumano, T. Hama, R. Kawasumi, K. Yoshiura and T. Hatakeyama, Angew. Chem., Int. Ed., 2021, 60, 2882 CrossRef CAS PubMed; (d) Y. Liu, X. Xiao, Y. Ran, Z. Bin and J. You, Chem. Sci., 2021, 12, 9408 RSC.
- (a) N. Ikeda, S. Oda, R. Matsumoto, M. Yoshioka, D. Fukushima, K. Yoshiura, N. Yasuda and T. Hatakeyama, Adv. Mater., 2020, 32, e2004072 CrossRef PubMed; (b) H. Hirai, K. Nakajima, S. Nakatsuka, K. Shiren, J. Ni, S. Nomura, T. Ikuta and T. Hatakeyama, Angew. Chem., Int. Ed., 2015, 54, 13581 CrossRef CAS PubMed.
- (a) H. Min, I. S. Park and T. Yasuda, Angew. Chem., Int. Ed., 2021, 60, 7643 CrossRef CAS PubMed; (b) Y. Yuan, X. Tang, X. Y. Du, Y. Hu, Y. J. Yu, Z. Q. Jiang, L. S. Liao and S. T. Lee, Adv. Opt. Mater., 2019, 7, 1801536 CrossRef; (c) X. Li, Y. Z. Shi, K. Wang, M. Zhang, C. J. Zheng, D. M. Sun, G. L. Dai, X. C. Fan, D. Q. Wang, W. Liu, Y. Q. Li, J. Yu, X. M. Ou, C. Adachi and X. H. Zhang, ACS Appl. Mater. Interfaces, 2019, 11, 13472 CrossRef CAS PubMed; (d) X. Qiu, G. Tian, C. Lin, Y. Pan, X. Ye, B. Wang, D. Ma, D. Hu, Y. Luo and Y. Ma, Adv. Opt. Mater., 2020, 9, 2001845 CrossRef.
- (a) T. Taniguchi, Y. Itai, Y. Nishii, N. Tohnai and M. Miura, Chem. Lett., 2019, 48, 1160 CrossRef CAS; (b) H. L. Lee, W. J. Chung and J. Y. Lee, Small, 2020, 16, 1907569 CrossRef CAS PubMed.
- (a) D. Hall, S. M. Suresh, P. L. dos Santos, E. Duda, S. Bagnich, A. Pershin, P. Rajamalli, D. B. Cordes, A. M. Z. Slawin, D. Beljonne, A. Köhler, I. D. W. Samuel, Y. Olivier and E. Zysman-Colman, Adv. Opt. Mater., 2019, 8, 1901627 CrossRef; (b) K. Stavrou, A. Danos, T. Hama, T. Hatakeyama and A. Monkman, ACS Appl. Mater. Interfaces, 2021, 13, 8643 CrossRef CAS PubMed; (c) Y. Xu, Z. Cheng, Z. Li, B. Liang, J. Wang, J. Wei, Z. Zhang and Y. Wang, Adv. Opt. Mater., 2020, 1902142 CrossRef CAS.
- (a) Y. Zhang, J. Wei, D. Zhang, C. Yin, G. Li, Z. Liu, X. Jia, J. Qiao and L. Duan, Angew. Chem., Int. Ed., 2022, 61, e202113206 CAS; (b) P. Jiang, J. Miao, X. Cao, H. Xia, K. Pan, T. Hua, X. Lv, Z. Huang, Y. Zou and C. Yang, Adv. Mater., 2021, e2106954 Search PubMed.
- (a) D. Zhang, X. Song, A. J. Gillett, B. H. Drummond, S. T. E. Jones, G. Li, H. He, M. Cai, D. Credgington and L. Duan, Adv. Mater., 2020, 32, 1908355 CrossRef CAS PubMed; (b) T. Hosokai, H. Matsuzaki, H. Nakanotani, K. Tokumaru, T. Tsutsui, A. Furube, K. Nasu, H. Nomura, M. Yahiro and C. Adachi, Sci. Adv., 2017, 3, e1603282 CrossRef PubMed.
- J. Wei, C. Zhang, D. Zhang, Y. Zhang, Z. Liu, Z. Li, G. Yu and L. Duan, Angew. Chem., Int. Ed., 2021, 60, 12269 CrossRef CAS PubMed.
- S. Thiery, D. Tondelier, B. Geffroy, O. Jeannin, J. Rault-Berthelot and C. Poriel, Chem.–Eur. J., 2016, 22, 10136 CrossRef CAS PubMed.
- (a) Z. Q. Gao, B. X. Mi, C. H. Chen, K. W. Cheah, Y. K. Cheng and W.-S. Wen, Appl. Phys. Lett., 2007, 90, 123506 CrossRef; (b) D. Zhang, D. Zhang and L. Duan, ACS Appl. Mater. Interfaces, 2016, 8, 23197 CrossRef CAS PubMed.
- J. U. Kim, I. S. Park, C. Y. Chan, M. Tanaka, Y. Tsuchiya, H. Nakanotani and C. Adachi, Nat. Commun., 2020, 11, 1765 CrossRef CAS PubMed.
- (a) D. Zhang, L. Duan, C. Li, Y. Li, H. Li, D. Zhang and Y. Qiu, Adv. Mater., 2014, 26, 5050 CrossRef CAS PubMed; (b) H. Nakanotani, T. Higuchi, T. Furukawa, K. Masui, K. Morimoto, M. Numata, H. Tanaka, Y. Sagara, T. Yasuda and C. Adachi, Nat. Commun., 2014, 5, 4016 CrossRef CAS PubMed.
- (a) S. G. Ihn, D. Jeong, E. S. Kwon, S. Kim, Y. S. Chung, M. Sim, J. Chwae, Y. Koishikawa, S. O. Jeon, J. S. Kim, J. Kim, S. Nam, I. Kim, S. Park, D. S. Kim, H. Choi and S. Kim, Adv. Sci., 2021, e2102141 Search PubMed; (b) K. R. Naveen, H. Lee, R. Braveenth, D. Karthik, K. J. Yang, S. J. Hwang and J. H. Kwon, Adv. Funct. Mater., 2021, 2110356 Search PubMed.
- D. Hall, K. Stavrou, E. Duda, A. Danos, S. Bagnich, S. Warriner, A. M. Z. Slawin, D. Beljonne, A. Kohler, A. Monkman, Y. Olivier and E. Zysman-Colman, Mater. Horiz., 2022, 9, 1068 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2107659. For ESI and crystallographic data in CIF or other electronic format see https://doi.org/10.1039/d2sc01543a |
| This journal is © The Royal Society of Chemistry 2022 |