
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePrimary trifluoroborate-iminiums enable facile access to chiral α-aminoboronic acids via Ru-catalyzed asymmetric hydrogenation and simple hydrolysis of the trifluoroborate moiety†
Andrej
Šterman
 a,
Izidor
Sosič
a and
Zdenko
Časar
*ab
a,
Izidor
Sosič
a and
Zdenko
Časar
*ab
aFaculty of Pharmacy, University of Ljubljana, Aškerčeva cesta 7, SI-1000 Ljubljana, Slovenia. E-mail: zdenko.casar@ffa.uni-lj.si
bLek Pharmaceuticals d.d., Sandoz Development Center Slovenia, Verovškova ulica 57, SI-1526 Ljubljana, Slovenia. E-mail: zdenko.casar@sandoz.com
First published on 26th January 2022
Abstract
This work describes the first preparation and application of primary trifluoroborate-iminiums (pTIMs) as a new, easily accessible and valuable class of organoboron derivatives. An array of structurally diverse pTIMs was prepared from potassium acyltrifluoroborates in excellent yields. Highly efficient and enantioselective [(R,R)-TethTsDpen-RuCl] complex-catalyzed hydrogenation of pTIMs provided direct access to chiral primary trifluoroborate-ammoniums (pTAMs). Moreover, facile synthesis of a series of structurally diverse chiral α-aminoboronic acids from chiral pTAMs was accomplished through novel, operationally simple and efficient conversion using hexamethyldisiloxane/aqueous HCl. Using no chromatography at any point, this work allowed easy access to chiral α-aminoboronic acids, as exemplified by the synthesis of optically pure anti-cancer drugs bortezomib and ixazomib.
Introduction
Organoboron compounds1 play a pivotal role in synthetic organic chemistry, where they have enabled a myriad of useful transformations2–4 and have also been used in the field of materials science.5 The most advantageous application of organoboron compounds was achieved in medicinal chemistry6 where several boron-containing drugs reached the market in the last two decades. Among those the key role is attributed to proteasome-inhibiting7 anti-cancer drugs bortezomib8 and ixazomib.9 Furthermore, a structural analog delanzomib is in the development phase.10 These drugs possess a unique chemical structure in the sense that they all contain an α-aminoboronic acid moiety (Scheme 1). Beside their application in medicinal chemistry, α-aminoboronic acid derivatives found numerous applications in organic synthesis.11 However, preparation of chiral α-aminoboronates is more challenging compared to their natural counterparts and methods for their preparation are limited. A seminal work on the synthesis of chiral α-aminoboronates was conducted by Matteson, who developed a powerful homologation reaction which provided chiral (α-chloroalkyl)boronic esters that could be transformed into α-aminoboronic acids (Scheme 1).12 In 2008, Ellman reported a novel approach to obtain chiral α-aminoboronic esters using highly diastereoselective Cu-catalyzed addition of bis(pinacolato)diboron to N-tert-butanesulfinyl aldimines (Scheme 1).13 Next, we reported an alternative asymmetric hydrogenation approach to Matteson's methodology to prepare chiral (α-chloroalkyl)boronic- and α-aminoboronic esters (Scheme 1).14 Subsequently, novel methodologies for the synthesis of α-aminoboronates were reported by groups of Yudin,15 Sawamura,16 Morken,17 and others.11,18 In recent years, this field has continued to flourish with many exciting methods being developed.19–33 Among recent methodologies, discoveries of Yudin32 and Bode33 caught our attention. Namely, in these reports trifluoroborate-iminiums (TIMs) and MIDA-protected iminoboronates were prepared from acylboranes,34 and converted to racemic N-substituted α-aminoboronic acid derivatives with borohydride reagents (Scheme 1). This stimulated us to explore Ir-catalyzed hydrogenation of benzyl-protected TIMs (Bn-TIMs) to benzyl-protected trifluoroborate-ammoniums (Bn-TAMs). Although a series of racemic Bn-TAMs was prepared and their conversion to α-aminoboronic acids was demonstrated (Scheme 1), an asymmetric approach was not successful as a consequence of E/Z-isomerism that was observed in Bn-TIMs.35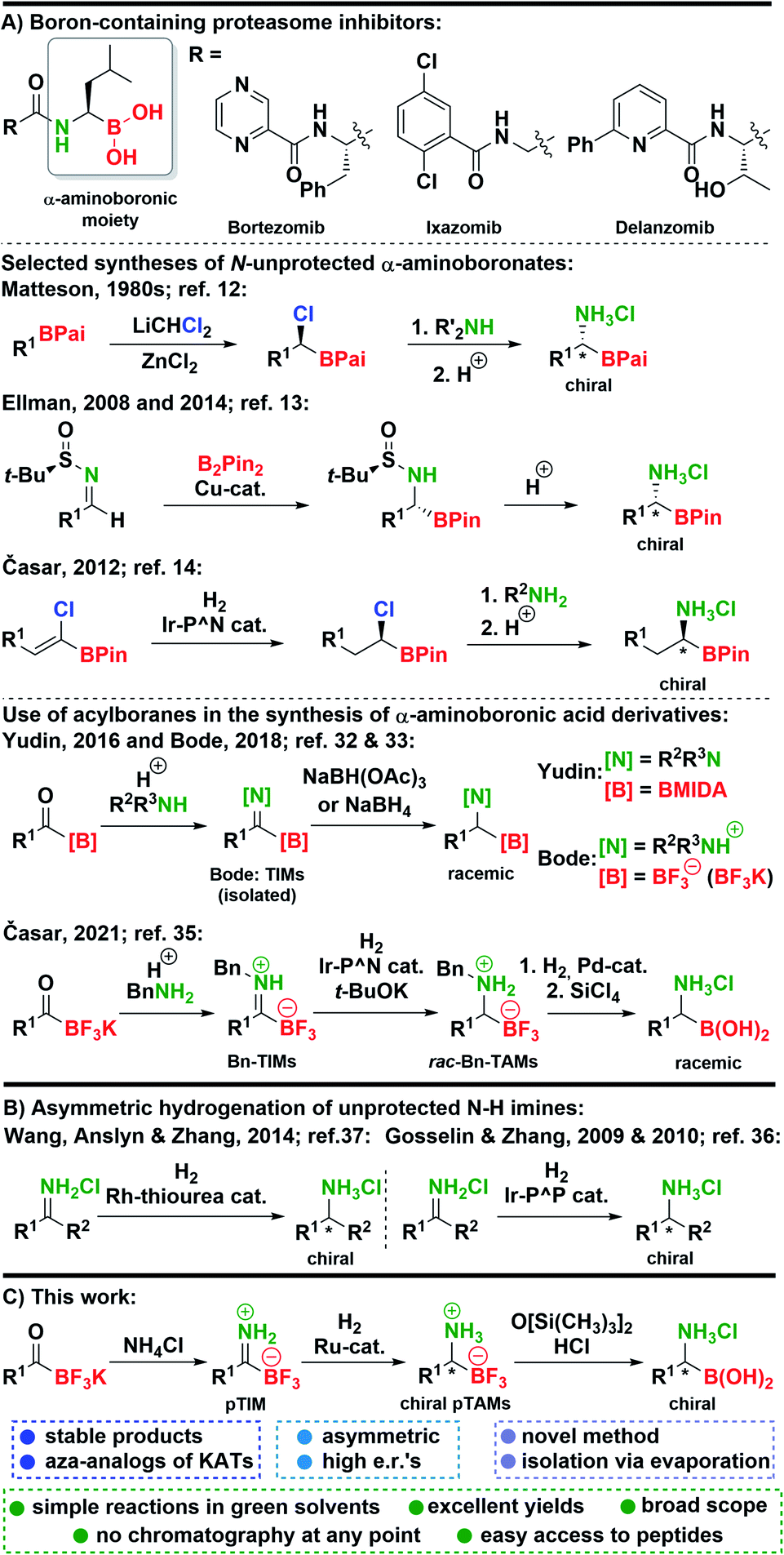 | ||
| Scheme 1 (A) Boron-containing proteasome inhibitors and key syntheses of α-aminoboronates. (B) Asymmetric hydrogenation of unprotected N–H imines. (C) This work. | ||
Despite significant advances in the area, a need for new synthetic routes that would allow the preparation of chiral N-unprotected α-aminoboronic acids exists. Many of the existing methods are either racemic, allow limited scope of substrates, or do not enable the synthesis of N-unprotected derivatives.11,18–33,35
Based on our recent research,35 we envisioned that unsubstituted primary TIMs (pTIMs) could provide access to chiral α-aminoboronic acids using asymmetric hydrogenation. Moreover, the use of pTIMs would eliminate the N-deprotection step to obtain the free amino group, thereby shortening the overall synthetic sequence. However, the unprotected N–H imine functionality is considered challenging and thus underexplored in the area of asymmetric hydrogenation (Scheme 1),36–38 which suggests that asymmetric hydrogenation of pTIMs might be a demanding task. Herein, we present the results of our investigation that started with the preparation of a structurally diverse array of a novel subtype of TIM, i.e. pTIMs, which underwent Ru-catalyzed asymmetric hydrogenation to chiral pTAMs, followed by mild and efficient conversion of the trifluoroborate moiety to boronic acid functionality (Scheme 1).
Results and discussion
At the outset of our studies, we investigated the preparation of pTIMs. Although secondary and tertiary TIMs were prepared before by Bode33 and Bn-TIMs in our group,35 the formation of pTIMs was not an obvious task. We began our research using potassium acyltrifluoroborate (KAT) 1a as a model substrate for the formation of the corresponding pTIM 2a upon reaction with NH4Cl as a simple nitrogen precursor (Scheme S2†).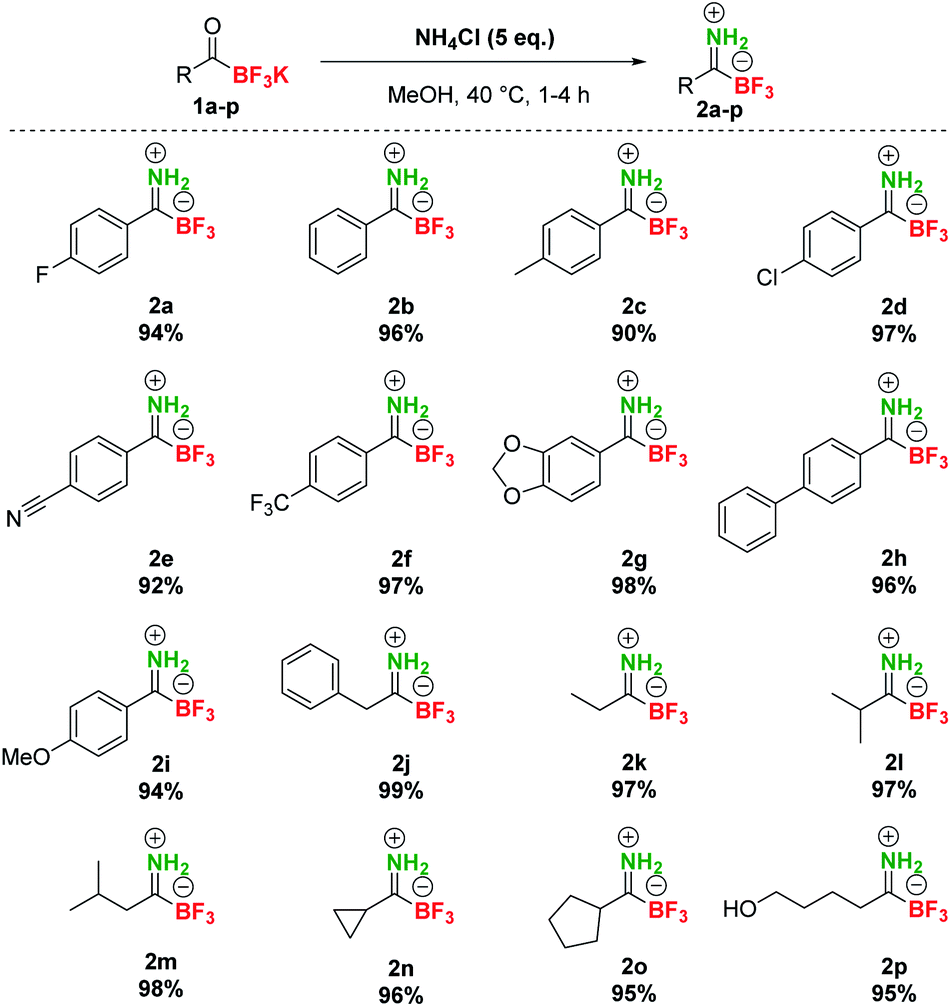 | ||
| Scheme 2 Synthesis of pTIMs 2 from KATs 1. | ||
We initially screened various reaction conditions to establish the viability of the envisioned approach, using NH4Cl as an environmentally benign and non-toxic nitrogen source. Interestingly, no reaction took place between 1a and NH4Cl (2 eq.) in MeCN and DMF even at 60 °C in the presence of molecular sieves (3 Å) (Table S1, entries 1–3†), although these solvents enabled the preparation of secondary and tertiary TIMs.33,35 More encouraging results with 38% yield of 2a in 20 h (0.2 M concentration) were observed in MeOH at room temperature (Table S1, entry 4†). Interestingly, the reaction conducted at 0.05 M concentration afforded 62% conversion due to increased solubility of NH4Cl (Table S1, entry 5†). Increasing the temperature to 40 °C gave full conversion both at 0.2 M and 0.05 M concentration, albeit at the expense of 5–10% of unknown side product formation (Table S1, entries 6 and 7†) and a similar outcome was observed with 1 eq. NH4Cl (Table S1, entry 8†). By increasing the amount of NH4Cl to 5 eq., a clean reaction was obtained with 100% conversion in 4 h (Table S1, entry 9†). Finally, even without molecular sieves, a clean reaction was observed between 1a (0.08 M) and NH4Cl (5 eq.) in MeOH at 40 °C and afforded pTIM 2a after 4 h in 94% yield (Table S1, entry 10†). With optimal reaction conditions in hand, we converted a series of aromatic and aliphatic KATs 1a–2p to pTIMs 2a–2p. Overall, these reactions proceeded smoothly and provided aromatic and aliphatic pTIMs 2 in excellent 90–99% yields and high purity (Scheme 2).
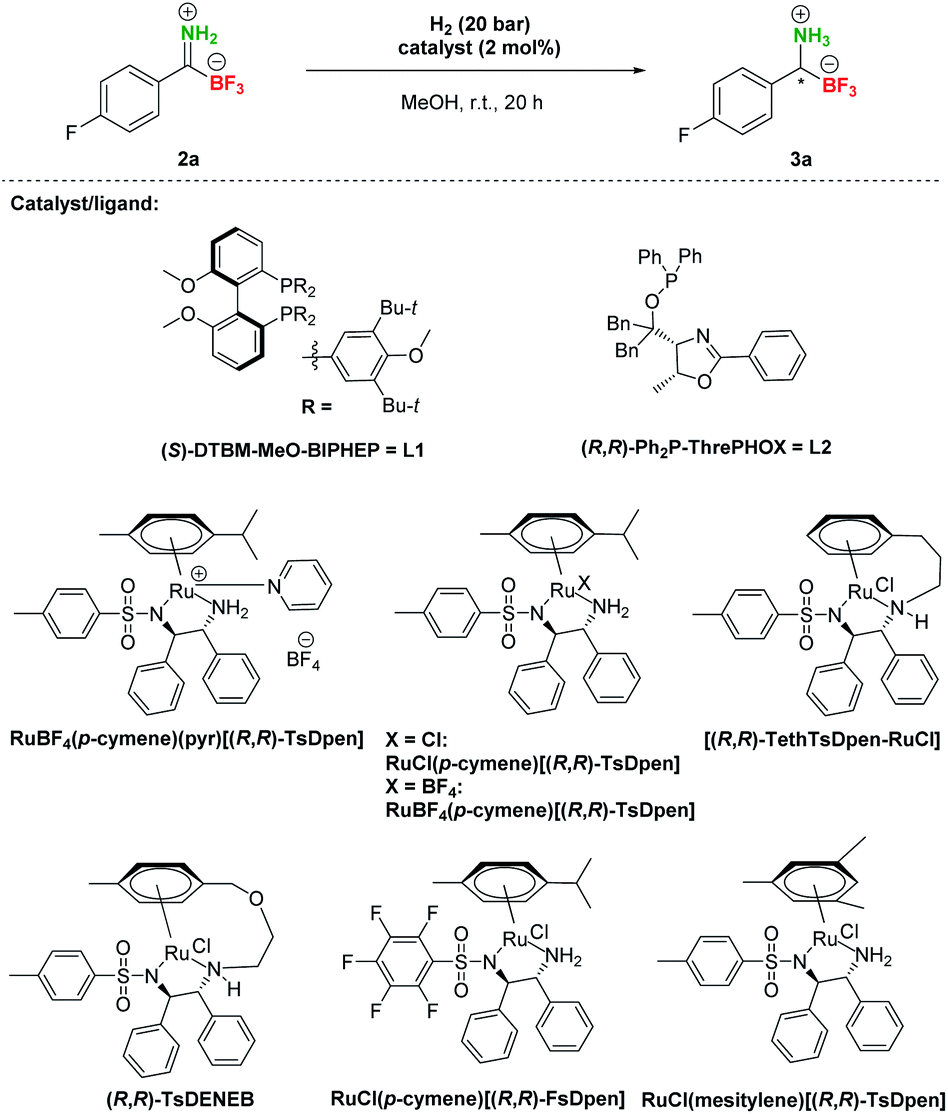
After the preparation of a series of pTIMs 2, we focused our attention on their asymmetric hydrogenation to chiral pTAMs 3 (Table 1). Initially, we decided to explore a diverse set of catalysts based on Ir, Ru and Rh metal precursors in combination with various P^P and P^N-ligands (Fig. S3†), as well as commercially available Noyori–Ikariya-type catalysts (Fig. S4†). The initial hydrogenation screening was done on substrate 2a at 20 bar H2 using Ir, Ru and Rh metal precursors (2 mol%) in combination with (S)-DTBM-MeO-BIPHEP (L1) and (R,R)-Ph2P-ThrePHOX (L2) ligands which provided good conversion in hydrogenation of Bn-TIMs to Bn-TAMs in the presence of t-BuOK base,35 albeit at moderate e.r.s. In contrast to Bn-TIMs, hydrolysis of 2a to 1a or decomposition of 2a was observed with selected catalytic systems when t-BuOK was used as an additive (Table 1, entries 1–4; Table S2, entries 1–6†) or no conversion of pTIM 2a to pTAM 3a was noticed in the absence of t-BuOK (Table 1, entries 5 and 6; Table S2, entries 7–10†). Encouraging first hit was obtained with the catalyst based on [Ru(p-cymene)Cl2]2 and (S)-DTBM-MeO-BIPHEP (L1) which enabled 23% conversion to 3a (racemic) in the absence of t-BuOK (Table 1, entry 7; Table S2, entry 11†). Furthermore, reaction using RuBF4(p-cymene)(pyr)[(R,R)-TsDpen] provided 89% conversion to 3a and a promising e.r. of 79![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :21 when t-BuOK was not used (Table 1, entry 8; Table S2, entry 12†), which is in sharp contrast with a similar reaction using t-BuOK (Table 1, entry 4, Table S2; entry 6†) where only hydrolysis of 2a to 1a was observed. These results indicated that the base additive is detrimental to hydrogenation of pTIMs 2 and that only Ru-based catalysts are feasible for this reaction (Scheme 3). Currently, the price of Ru metal is more than seven times lower than that of Ir metal and thirtyfold lower compared to Rh metal.39 This implies that hydrogenation of pTIMs is not only a more atom economic approach, but also requires a less expensive metal catalyst than in the case of Bn-TIMs. For these reasons, we focused further on Ru-based catalysis.
:21 when t-BuOK was not used (Table 1, entry 8; Table S2, entry 12†), which is in sharp contrast with a similar reaction using t-BuOK (Table 1, entry 4, Table S2; entry 6†) where only hydrolysis of 2a to 1a was observed. These results indicated that the base additive is detrimental to hydrogenation of pTIMs 2 and that only Ru-based catalysts are feasible for this reaction (Scheme 3). Currently, the price of Ru metal is more than seven times lower than that of Ir metal and thirtyfold lower compared to Rh metal.39 This implies that hydrogenation of pTIMs is not only a more atom economic approach, but also requires a less expensive metal catalyst than in the case of Bn-TIMs. For these reasons, we focused further on Ru-based catalysis.
|
|
|||
|---|---|---|---|
| Entryb | Catalyst | 3a [%] | e.r. |
| a Cod = cyclooctadiene, BArF = tetrakis(3,5-bis(trifluoromethyl)phenyl)borate. b Reactions were conducted by using 2a (0.1 mmol) and catalyst (2 mol%) in MeOH (0.05 M) with 20 bar H2 at room temperature for 20 h. The yields were determined by 1H-NMR and the e.r. (S/R) by chiral HPLC. c t-BuOK (1 eq.) as additive. d ∼30% conversion to 1a observed. e No reaction observed. f With 50 bar H2. g Reaction time 44 h. | |||
| 1c | [Ir(cod)]Cl/L1 | 0d | n.a. |
| 2c | [Ir(cod)(L2)]BArF | 0d | n.a. |
| 3c | RuCl[(p-cymene)]Cl/L1 | 0d | n.a. |
| 4c | RuBF4(p-cymene)(pyr)[(R,R)-TsDpen] | 0d | n.a. |
| 5 | [Ir(cod)]Cl/L1 | 0e | n.a. |
| 6 | [Ir(cod)(L2)]BArF | 0e | n.a. |
| 7 | RuCl[(p-cymene)]Cl/L1 | 23 | 50:50 |
| 8 | RuBF4(p-cymene)(pyr)[(R,R)-TsDpen] | 89 | 79:21 |
| 9 | RuCl(p-cymene)[(R,R)-TsDpen] | 81 | 89:11 |
| 10 | RuBF4(p-cymene)[(R,R)-TsDpen] | 100 | 89:11 |
| 11 | [(R,R)-TethTsDpen-RuCl] | 100 | 97:3 |
| 12 | (R,R)-TsDENEB | 88 | 92:8 |
| 13 | RuCl(p-cymene)[(R,R)-FsDpen] | 36 | 91:9 |
| 14 | RuCl(mesitylene)[(R,R)-TsDpen] | 40 | 88:12 |
| 15f | RuBF4(p-cymene)(pyr)[(R,R)-TsDpen] | 100 | 77:23 |
| 16g | RuBF4(p-cymene)(pyr)[(R,R)-TsDpen] | 100 | 80:20 |
| 17g | RuCl(p-cymene)[(R,R)-TsDpen] | 100 | 88:12 |
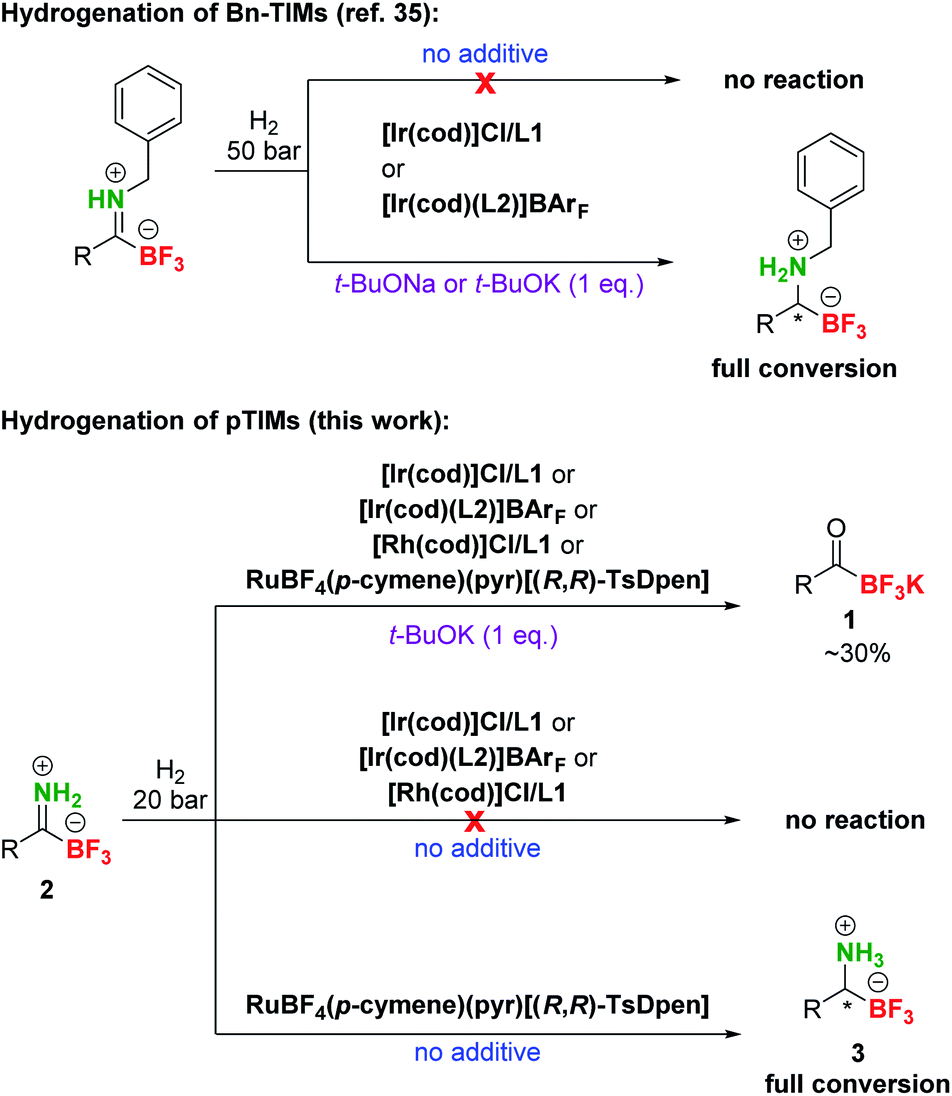 | ||
| Scheme 3 Comparison of different reactivities of Bn-TIMs and pTIMs. Under the conditions that were successful in hydrogenation of the former, only partial hydrolysis to KAT was observed with model pTIM and an altogether different catalytic system was required. Catalyst and ligand structures are presented in Table 1. | ||
Therefore, additional extensive catalyst screening was conducted using Ru-metal precursors in combination with various P^P and P^N-ligands and an extended list of Noyori–Ikariya-type catalysts40 (Table S3†). The study of ruthenium catalysts revealed that Ru–(p-cymene)–phosphine ligand complexes showed very low activity and no enantioselectivity in the hydrogenation of substrate 2a (Table S3, entries 2–14†). The RuOAc2–phosphine complexes showed limited enantioselectivity and also low conversions (Table S3, entries 15 and 16†), while the RuCl–diphosphine–diamine complexes performed slightly better in terms of enantioselectivity, but with similarly meagre conversions (Table S3, entries 17–24†). The dimethylamine adducts of RuCl–phosphine complexes were slightly more active but gave low enantioselectivity (Table S3, entries 25–27†). In contrast to these experiments, the best results for conversion of 2a to 3a were achieved with Noyori–Ikariya-type catalysts (Table 1). RuCl(p-cymene)[(R,R)-TsDpen] performed with slightly lower conversion compared to RuBF4(p-cymene)(pyr)[(R,R)-TsDpen], but gave higher e.r. (Table 1, entry 8 vs. entry 9). Furthermore, a full conversion was achieved with RuBF4(p-cymene)[(R,R)-TsDpen] along with a good e.r. (89:11, Table 1, entry 10). Gratifyingly, Wills's tethered catalyst [(R,R)-TethTsDpen-RuCl]41 provided full conversion and an excellent 97:3 e.r. (Table 1, entry 11). Catalysts (R,R)-TsDENEB®, RuCl(p-cymene)[(R,R)-FsDpen], and RuCl(mesitylene)[(R,R)-TsDpen] also afforded good enantioselectivities, albeit at incomplete conversions (Table 1, entries 12–14). Reactions with incomplete conversion could be completed by using higher hydrogen pressure (50 bar, Table 1, entry 15 vs. entry 8) with marginal loss of enantioselectivity, or by prolonging the reaction time to 44 h with retained e.r. (Table 1, entries 16 and 17 vs. entries 8 and 9).
Based on these screening results, [(R,R)-TethTsDpen-RuCl] was chosen as the optimal catalyst for the asymmetric reduction of TIMs 2 to TAMs 3. Next, the effect of the solvent was carefully examined (Table S4†), and MeOH was confirmed as the optimal choice. In addition, a mixture of MeOH/H2O (1–10% v/v) performed similarly at full conversion and nearly the same enantioselectivity, while the MeOH/H2O = 1:1 mixture or pure H2O promoted notable hydrolysis of 2a to 1a (Table S4, entries 1–9†). Furthermore, we established that concentration had no effect on conversion in the range of 0.025–0.1 M. Of note, the highest enantioselectivities were achieved at 0.05–0.10 M concentration (Table S4, entries 10–12†). Variations in [(R,R)-TethTsDpen-RuCl] loading were also examined (0.1–2.0 mol%) and while the reaction proceeded to completion also at 1 mol% catalyst (Table S4, entries 13–15†), 2 mol% catalyst loading was chosen in hydrogenation of other compounds to ensure full conversions across the whole substrate scope. Next, we proceeded with preparative hydrogenation of pTIMs 2a–p. Hydrogenation of aromatic derivatives 2a–h proceeded smoothly with full conversion at room temperature and 20 bar H2 with 2 mol% of [(R,R)-TethTsDpen-RuCl] in MeOH containing 2% water (v/v) giving the corresponding pTAMs 3a–i in excellent yields (88–95%) and high enantioselectivities with e.r.s ranging from 98:2 for 3h to 90:10 for 3i (Scheme 4). Interestingly, when aliphatic pTIM 2j was subjected to hydrogenation under the same conditions, low conversion (45%) was obtained (Table S5, entry 1†). Further optimization of the reaction conditions by increasing the catalyst loading to 4 mol% and prolongation of the reaction time to 72 hours provided 3j with a 94:6 e.r. at full conversion (Table S5, entry 4†). By applying these conditions, aliphatic pTIMs 2j–p were successfully hydrogenated and provided pTAMs 3j–p in 89–96% yields and excellent e.r.s that were in the range of 99:1 to 93:7, which is remarkable for such small compounds.36,37 Overall, aliphatic substrates gave slightly higher enantioselectivities compared to aromatic counterparts42 and provided the opposite enantiomer43 (Scheme 4).
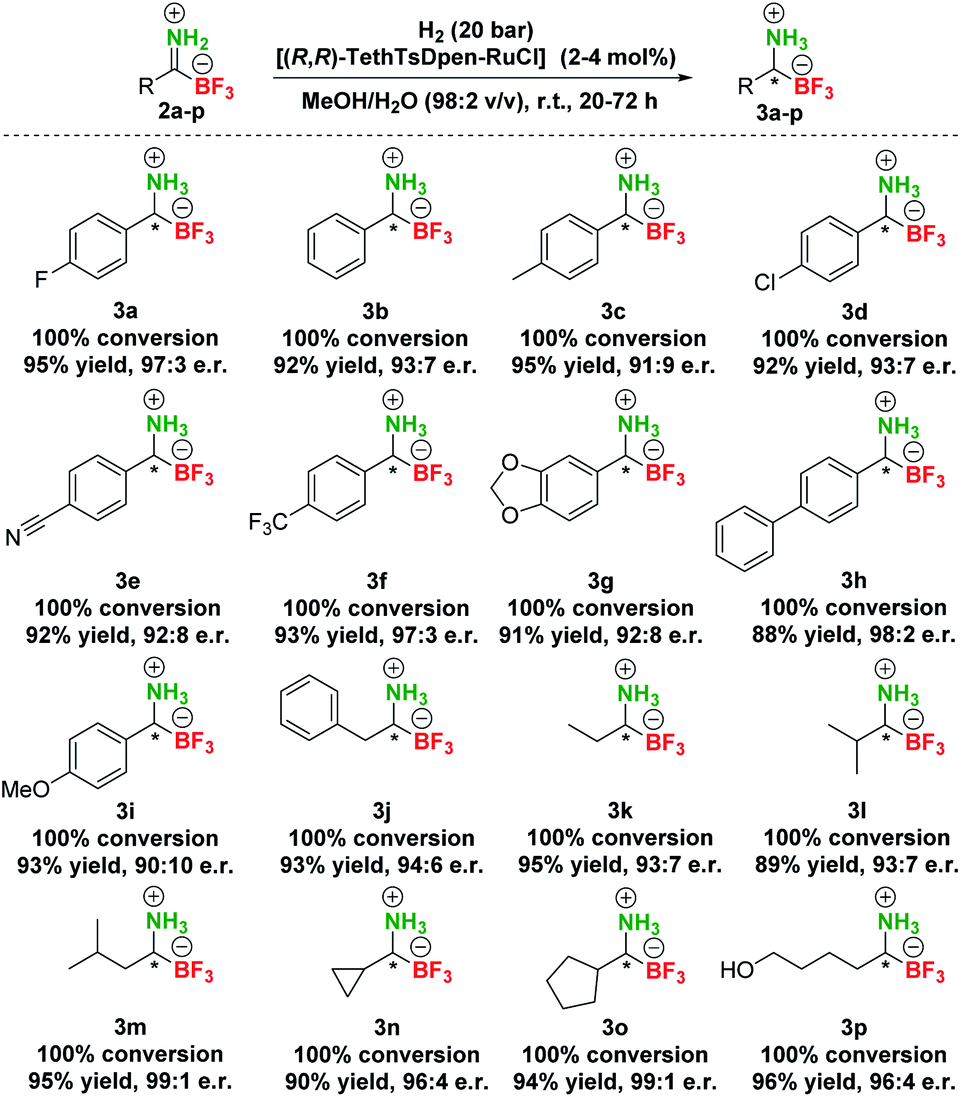 | ||
| Scheme 4 Scope of asymmetric hydrogenation of pTIMs 2. | ||
In the final stage of our investigation, we studied the conversion of the trifluoroborate moiety in pTAMs 3 to the boronic acid functionality. Although hydrolysis of organic trifluoroborates to boronic acids in the presence of Cs2CO3 was extensively studied in connection with Suzuki–Miyaura coupling,44 general methods for the preparative hydrolysis of organic trifluoroborates to boronic acids are scarce and involve TMSCl/H2O,45 TMSCl/K2CO3/MeCN,46 silica gel/H2O,47 FeCl3/THF/H2O,48 alumina/H2O,49 and SiCl4/MeOH + HCl(aq.).33 This is associated with the fact that structurally diverse organic trifluoroborates exhibit notably different hydrolysis rates.50,51 As we have shown recently, only Bode's SiCl4 method33 enabled the conversion of pTAMs to the aminoboronic acids while other methods failed.35 However, huge excess (10 eq.) of difficult-to-handle SiCl4 was needed to transform pTAMs to aminoboronic acids.
Therefore, we decided to investigate other options for conversion of pTAMs 3 to α-aminoboronic acids 4 (Table S6†). Reactions using Cs2CO3 or silica gel/Cs2CO3 in MeOH or MeOH/H2O (95:5 v/v) gave no conversion of 3 to 4 (Table S6, entries 1–4†). The same was observed with TMSCl/Cs2CO3 in MeOH (Table S6, entry 5†). Surprisingly, TMSCl/Cs2CO3 in MeOH/H2O (95:5 v/v) provided 100% conversion of 3a, but the product 4a was contaminated with a side product (Table S6, entry 6†). Finally, replacement of the silicon source with hexamethyldisiloxane (HMDSO, O[Si(CH3)3]2) provided unexpected results (Table S6, entries 7–9†) and to our delight reaction of 3a with HMDSO (3 eq.) in the presence of aqueous HCl (4 eq.) in MeOH provided complete conversion to 4a (Table S6, entry 8†). It should be noted that HMDSO is non-toxic and has been studied as an MRI probe, where no toxic effects were observed when it was injected in mammals.52 Subsequent preparative transformation of pTAMs 3 to the corresponding α-aminoboronic acids 4 proceeded smoothly in excellent 94–100% yields (Scheme 5) without the loss of stereochemistry42 except for 3e where decomposition of the corresponding boronic acid to the benzyl amine was observed (Scheme S7 and Fig. S9†). Importantly, these highly polar and potentially labile compounds were isolated in pure form only by evaporation of volatiles.53
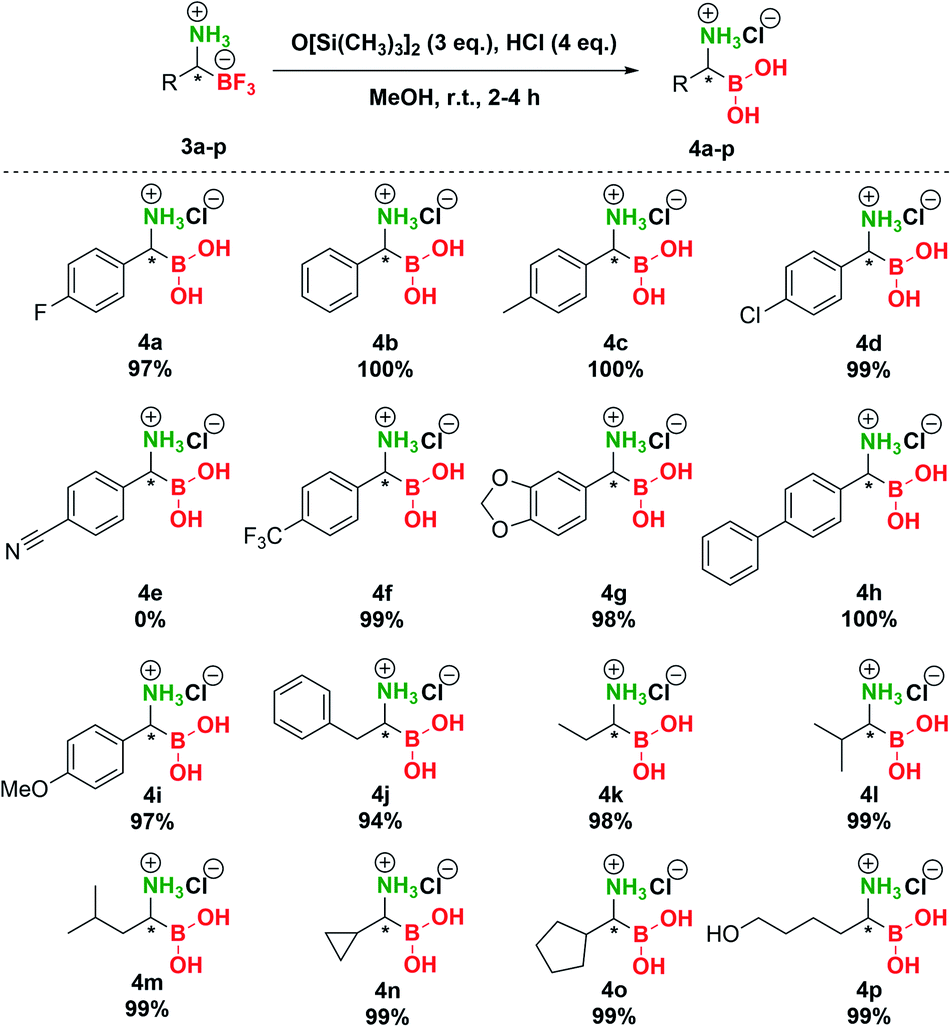 | ||
| Scheme 5 Conversion of trifluoroborates 3 to boronic acids 4. | ||
To demonstrate the significance of the presented methodology, we converted α-aminoboronic acid (R)-4m to bortezomib and ixazomib using a modified solid phase coupling approach (Scheme 6).54 Pure bortezomib and ixazomib were obtained in 44% and 50% yield respectively, which represents overall one of the shortest, simplest and most efficient approaches to these drugs (see Schemes S10–S15†).
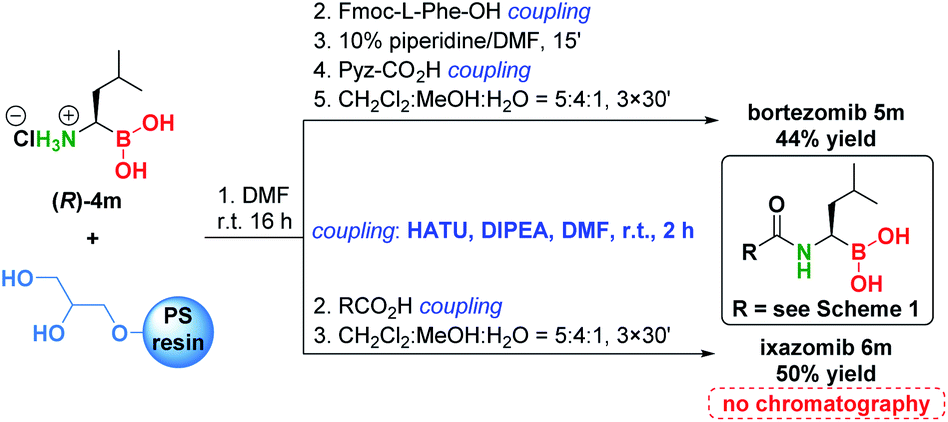 | ||
| Scheme 6 Conversion of (R)-4m to bortezomib and ixazomib. | ||
Conclusions
In summary, we established a novel three-step synthetic strategy to a diverse array of chiral α-aminoboronic acids. The presented synthetic methodology is based on the conversion of KATs to their direct aza-analogs pTIMs 2, which are disclosed for the first time and could prove to be valuable organoboron reagents in their own right. Afterwards, highly stereoselective asymmetric hydrogenation of pTIMs 2 to chiral pTAMs 3 using the [(R,R)-TethTsDpen-RuCl] catalyst was successfully accomplished, followed by a novel and mild conversion of trifluoroborate to the boronic acid functionality in the presence of HMDSO and aqueous HCl. The represented transformations feature various aliphatic and aromatic substrates, proceed efficiently in high to excellent yields, are conducted in MeOH and water as sustainable solvents, and enable protecting-group free access to chiral α-aminoboronic acids. Our methodology towards these valuable building blocks will enable their broad deployment in access to libraries of novel proteasome inhibitors and beyond,11 as demonstrated by the synthesis of stereochemically pure drugs bortezomib and ixazomib.Data availability
Data for this paper, including reactions screening and optimization experiments, detailed experimental procedures, overview of key synthetic procedures to bortezomib and characterization data, are provided in the ESI.†Author contributions
A. Š. and Z. Č. conceived the study. A. Š. performed all experiments and collected the data. I. S. and Z. Č. guided the research. Z. Č. wrote the manuscript. A. Š. and I. S. performed editing of the manuscript. All authors discussed the results and contributed to the finalization of the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Slovenian Research Agency (research program P1-0208, P1-0230 and PhD grant to A. Š.). The authors acknowledge Lek Pharmaceuticals d.d. a Sandoz Company for support in chiral HPLC analysis, Mrs L. Kolenc and Dr N. Žigart for HPLC analyses and Mrs M. Frelih for HRMS analyses.Notes and references
- For selected monographs on organoboron compounds and their chemistry, see: (a) Boronic Acids: Preparation and Applications in Organic Synthesis, Medicine and Materials, ed. D. Hall, Wiley-VCH, Weinheim, 2011, vol. 1 and 2 Search PubMed; (b) Synthesis and Application of Organoboron Compounds, ed. E. Fernández and A. Whiting, Springer International Publishing, Cham, 2015 Search PubMed; (c) Boron-Based Compounds, ed. E. Hey-Hawkins and C. V. Teixidor, John Wiley & Sons, Ltd, Chichester, 2018 Search PubMed.
- For selected and recent reviews on preparation of organoboron compounds and their reactivity, see: (a) N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS; (b) I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890–931 CrossRef CAS PubMed; (c) J. Cid, H. Gulyâs, J. J. Carbó and E. Fernândez, Chem. Soc. Rev., 2012, 41, 3558–3570 RSC; (d) A. J. J. Lennox and G. C. Lloyd-Jones, Chem. Soc. Rev., 2014, 43, 412–443 RSC; (e) A. Ros, R. Fernández and J. M. Lassaletta, Chem. Soc. Rev., 2014, 43, 3229–3243 RSC; (f) J. W. B. Fyfe and A. J. B. Watson, Chem, 2017, 3, 31–55 CrossRef CAS; (g) D. Hemming, R. Fritzemeier, S. A. Westcott, W. L. Santos and P. G. Steel, Chem. Soc. Rev., 2018, 47, 7477–7494 RSC; (h) S. Namirembe and J. P. Morken, Chem. Soc. Rev., 2019, 48, 3464–3474 RSC; (i) S. Roscales and A. G. Csáky, Chem. Soc. Rev., 2020, 49, 5159–5177 RSC; (j) Y. M. Tian, X. N. Guo, H. Braunschweig, U. Radius and T. B. Marder, Chem. Rev., 2021, 121, 3561–3597 CrossRef CAS PubMed; (k) J. Hu, M. Ferger, Z. Shi and T. B. Marder, Chem. Soc. Rev., 2021, 50, 13129–13188 RSC; (l) S. K. Bose, L. Mao, L. Kuehn, U. Radius, J. Nekvinda, W. Santos, S. A. Westcott, P. G. Steel and T. B. Marder, Chem. Rev., 2021, 121, 13238–13341 CrossRef CAS PubMed.
- For selected reviews on specific classes of organoboron compounds and reagents, see: (a) G. A. Molander and N. Ellis, Acc. Chem. Res., 2007, 40, 275–286 CrossRef CAS PubMed; (b) S. Darses and J.-P. Genet, Chem. Rev., 2008, 108, 288–325 CrossRef CAS PubMed; (c) E. C. Neeve, S. J. Geier, I. A. I. Mkhalid, S. A. Westcott and T. B. Marder, Chem. Rev., 2016, 116, 9091–9161 CrossRef CAS PubMed; (d) N. Miralles, R. J. Maza and E. Fernández, Adv. Synth. Catal., 2018, 360, 1306–1327 CrossRef CAS; (e) N. J. de Hiller, N. A. do Amaral e Silva, T. A. Tavares, R. X. Faria, M. N. Eberlin and D. de Luna Martins, Eur. J. Org. Chem., 2020, 2020, 4841–4877 CrossRef; (f) J. He, F. Rauch, M. Finze and T. B. Marder, Chem. Sci., 2021, 12, 128–147 RSC; (g) X. Li and D. G. Hall, Adv. Synth. Catal., 2021, 363, 2209–2223 CrossRef CAS; (h) Y. P. Budiman, S. A. Westcott, U. Radius and T. B. Marder, Adv. Synth. Catal., 2021, 363, 2224–2255 CrossRef CAS; (i) R. J. Maza, J. J. Carbó and E. Fernández, Adv. Synth. Catal., 2021, 363, 2274–2289 CrossRef CAS; (j) S. Kamio and H. Yoshida, Adv. Synth. Catal., 2021, 363, 2310–2324 CrossRef CAS.
- For selected reviews on boron compounds in activation and catalysis, see: (a) D. G. Hall, Chem. Soc. Rev., 2019, 48, 3475–3496 RSC; (b) Y. Su and R. Kinjo, Chem. Soc. Rev., 2019, 48, 3613–3659 RSC; (c) J. L. Carden, A. Dasgupta and R. L. Melen, Chem. Soc. Rev., 2020, 49, 1706–1725 RSC; (d) Y. Ma, S. J. Lou and Z. Hou, Chem. Soc. Rev., 2021, 50, 1945–1967 RSC; (e) J. J. Feng, W. Mao, L. Zhang and M. Oestreich, Chem. Soc. Rev., 2021, 50, 2010–2073 RSC.
- For selected reviews on organoboron compounds in material science, see: (a) G. Alcaraz, M. Grellier and S. Sabo-Etienne, Acc. Chem. Res., 2009, 42, 1640–1649 CrossRef CAS PubMed; (b) E. Galbraith and T. D. James, Chem. Soc. Rev., 2010, 39, 3831–3842 RSC; (c) D. Li, H. Zhang and Y. Wang, Chem. Soc. Rev., 2013, 42, 8416–8433 RSC; (d) R. Kumar, A. Karkamkar, M. Bowden and T. Autrey, Chem. Soc. Rev., 2019, 48, 5350–5380 RSC; (e) S. K. Mellerup and S. Wang, Chem. Soc. Rev., 2019, 48, 3537–3549 RSC; (f) Y. Qin, X. Liu, P. P. Jia, L. Xu and H. B. Yang, Chem. Soc. Rev., 2020, 49, 5678–5703 RSC; (g) Z. Shi, X. Han, W. Hu, H. Bai, B. Peng, L. Ji, Q. Fan, L. Li and W. Huang, Chem. Soc. Rev., 2020, 49, 7533–7567 RSC; (h) A. N. Bismillah and I. Aprahamian, Chem. Soc. Rev., 2021, 50, 5631–5649 RSC; (i) S. Berger and T. B. Marder, Mater. Horiz., 2022, 9, 112–120 RSC; (j) L. Ji, S. Griesbeck and T. B. Marder, Chem. Sci., 2017, 8, 846–863 RSC; (k) Y.-Q. Yan, Y.-B. Li, J.-W. Wang and C.-H. Zhao, Chem.–Asian J., 2013, 8, 3164–3176 CrossRef CAS PubMed; (l) S.-Y. Li, Z.-B. Sun and C.-H. Zhao, Inorg. Chem., 2017, 56, 8705–8717 CrossRef CAS PubMed; (m) J. Miao, Y. Wang, J. Liu and L. Wang, Chem. Soc. Rev., 2022, 51, 153–187 RSC; (n) M. Hirai, N. Tanaka, M. Sakai and S. Yamaguchi, Chem. Rev., 2019, 119, 8291–8331 CrossRef CAS PubMed; (o) E. von Grotthuss, A. John, T. Kaese and M. Wagner, Asian J. Org. Chem., 2018, 7, 37–53 CrossRef CAS; (p) H. Budy, J. Gilmer, T. Trageser and M. Wagner, Eur. J. Inorg. Chem., 2020, 2020, 4148–4162 CrossRef CAS; (q) Z. Huang, S. Wang, R. D. Dewhurst, N. V. Ignat'ev, M. Finze and H. Braunschweig, Angew. Chem., Int. Ed., 2020, 59, 8800–8816 CrossRef CAS PubMed; (r) M. Stępień, E. Gońka, M. Żyła and N. Sprutta, Chem. Rev., 2017, 117, 3479–3716 CrossRef PubMed.
- For selected reviews on organoboron compounds in medicinal chemistry, see: (a) S. J. Baker, C. Z. Ding, T. Akama, Y. K. Zhang, V. Hernandez and Y. Xia, Future Med. Chem., 2009, 1, 1275–1288 CrossRef CAS PubMed; (b) V. M. Dembitsky, A. A. A. Al Quntar and M. Srebnik, Chem. Rev., 2011, 111, 209–237 CrossRef CAS PubMed; (c) S. J. Baker, J. W. Tomsho and S. J. Benkovic, Chem. Soc. Rev., 2011, 40, 4279–4285 RSC; (d) R. Smoum, A. Rubinstein, V. M. Dembitsky and M. Srebnik, Chem. Rev., 2012, 112, 4156–4220 CrossRef CAS PubMed; (e) A. Adamczyk-Woźniak, K. M. Borys and A. Sporzyński, Chem. Rev., 2015, 115, 5224–5247 CrossRef PubMed; (f) D. B. Diaz and A. K. Yudin, Nat. Chem., 2017, 9, 731–742 CrossRef CAS PubMed; (g) G. F. S. Fernandes, W. A. Denny and J. L. Dos Santos, Eur. J. Med. Chem., 2019, 179, 791–804 CrossRef CAS PubMed; (h) J. Plescia and N. Moitessier, Eur. J. Med. Chem., 2020, 195, 112270 CrossRef CAS PubMed; (i) E. Lence and C. González-Bello, Adv. Therap., 2021, 4, 2000246 CrossRef CAS; (j) K. G. Sangu, A. U. Shinde, S. Chopra and H. B. Rode, Future Med. Chem., 2021, 13, 229–232 CrossRef PubMed.
- For proteasome inhibitors, see: (a) B. A. Teicher and J. E. Tomaszewski, Biochem. Pharmacol., 2015, 96, 1–9 CrossRef CAS PubMed; (b) E. E: Manasanach and R. Z. Orlowski, Nat. Rev. Clin. Oncol., 2017, 14, 417–433 CrossRef PubMed; (c) L. D. Fricker, Annu. Rev. Pharmacol. Toxicol., 2020, 60, 457–476 CrossRef CAS PubMed.
- (a) B. A. Teicher, G. Ara, R. Herbst, V. J. Palombella and J. Adams, Clin. Cancer Res., 1999, 5, 2638–2645 CAS; (b) R. Z. Orlowski, T. E. Stinchcombe, B. S. Mitchell, T. C. Shea, A. S. Baldwin, S. Stahl, J. Adams, D.-L. Esseltine, P. J. Elliott and C. S. Pien, et al. , J. Clin. Oncol., 2002, 20, 4420–4427 CrossRef CAS PubMed; (c) P. G. Richardson, P. Sonneveld, M. W. Schuster, D. Irwin, E. A. Stadtmauer, T. Facon, J. L. Harousseau, D. Ben-Yehuda, S. Lonial and H. Goldschmidt, et al. , N. Engl. J. Med., 2005, 352, 2487–2498 CrossRef CAS PubMed.
- (a) E. Kupperman, E. C. Lee, Y. Cao, B. Bannerman, M. Fitzgerald, A. Berger, J. Yu, Y. Yang, P. Hales and F. Bruzzese, et al. , Cancer Res., 2010, 70, 1970–1980 CrossRef CAS PubMed; (b) P. G. Richardson, S. Zweegman, E. K. O'Donnell, J. P. Laubach, N. Raje, P. Voorhees, R. H. Ferrari, T. Skacel, S. K. Kumar and S. Lonial, Expert Opin. Pharmacother., 2018, 19, 1949–1968 CrossRef CAS PubMed.
- (a) R. Piva, B. Ruggeri, M. Williams, G. Costa, I. Tamagno, D. Ferrero, V. Giai, M. Coscia, S. Peola and M. Massaia, et al. , Blood, 2008, 111, 2765–2775 CrossRef CAS PubMed; (b) E. Gallerani, M. Zucchetti, D. Brunelli, E. Marangon, C. Noberasco, D. Hess, A. Delmonte, G. Martinelli, S. Böhm and C. Driessen, et al. , Eur. J. Cancer, 2013, 49, 290–296 CrossRef CAS PubMed.
- W. Ming, H. S. Soor, X. Liu, A. Trofimova, A. K. Yudin and T. B. Marder, Chem. Soc. Rev., 2021, 50, 12151–12188 RSC.
- (a) D. S. Matteson, Chem. Rev., 1989, 89, 1535–1551 CrossRef CAS; (b) D. S. Matteson, Tetrahedron, 1989, 45, 1859–1885 CrossRef CAS; (c) D. S. Matteson, Tetrahedron, 1998, 54, 10555–10607 CrossRef CAS; (d) D. S. Matteson, J. Org. Chem., 2013, 78, 10009–10023 CrossRef CAS PubMed.
- (a) M. A. Beenen, C. An and J. A. Ellman, J. Am. Chem. Soc., 2008, 130, 6910–6911 CrossRef CAS PubMed; (b) A. W. Buesking, V. Bacauanu, I. Cai and J. A. Ellman, J. Org. Chem., 2014, 79, 3671–3677 CrossRef CAS PubMed.
- (a) I. Gazić Smilović, E. Casas-Arcé, S. J. Roseblade, U. Nettekoven, A. Zanotti-Gerosa, M. Kovačevič and Z. Časar, Angew. Chem., Int. Ed., 2012, 51, 1014–1018 CrossRef PubMed; (b) I. Gazić Smilović and Z. Časar, Chim. Oggi, 2013, 31, 20–25 Search PubMed.
- (a) Z. He, A. Zajdlik, J. D. St Denis, N. Assem and A. K. Yudin, J. Am. Chem. Soc., 2012, 134, 9926–9929 CrossRef CAS PubMed; (b) A. Zajdlik, Z. Wang, J. L. Hickey, A. Aman, A. D. Schimmer and A. K. Yudin, Angew. Chem., Int. Ed., 2013, 52, 8411–8415 CrossRef CAS PubMed.
- S. Kawamorita, T. Miyazaki, T. Iwai, H. Ohmiya and M. Sawamura, J. Am. Chem. Soc., 2012, 134, 12924–12927 CrossRef CAS PubMed.
- K. Hong and J. P. Morken, J. Am. Chem. Soc., 2013, 135, 9252–9254 CrossRef CAS PubMed.
- For detailed recent reviews on the synthesis of α-aminoboronic acids, see: (a) P. Andrés, G. Ballano, M. I. Calaza and C. Cativiela, Chem. Soc. Rev., 2016, 45, 2291–2307 RSC; (b) A. Šterman, I. Sosič, S. Gobec and Z. Časar, Org. Chem. Front., 2019, 6, 2991–2998 RSC; (c) F. Thomas and T. L. Pham, Nachr. Chem., 2020, 68, 75–79 CrossRef CAS; (d) D. M. Volochnyuk, A. O. Gorlova and O. O. Grygorenko, Chem.–Eur. J., 2021, 27, 15277–15326 CrossRef CAS PubMed.
- C. B. Schwamb, K. P. Fitzpatrick, A. C. Brueckner, H. C. Richardson, P. H.-Y. Cheong and K. A. Scheidt, J. Am. Chem. Soc., 2018, 140, 10644–10648 CrossRef CAS PubMed.
- X. Y. Bai, W. Zhao, X. Sun and B. J. Li, J. Am. Chem. Soc., 2019, 141, 19870–19878 CrossRef CAS PubMed.
- D. W. Gao, Y. Gao, H. Shao, T. Z. Qiao, X. Wang, B. B. Sanchez, J. S. Chen, P. Liu and K. M. Engle, Nat. Catal., 2020, 3, 23–29 CrossRef CAS PubMed.
- W. Yao, J. Yang and F. Hao, ChemSusChem, 2020, 13, 121–125 CrossRef CAS PubMed.
- W. Ming, X. Liu, A. Friedrich, J. Krebs and T. B. Marder, Org. Lett., 2020, 22, 365–370 CrossRef CAS PubMed.
- R. L. Reyes, M. Sato, T. Iwai and M. Sawamura, J. Am. Chem. Soc., 2020, 142, 589–597 CrossRef CAS PubMed.
- W. Ming, X. Liu, A. Friedrich, J. Krebs, Y. P. Budiman, M. Huang and T. B. Marder, Green Chem., 2020, 22, 2184–2190 RSC.
- (a) D. Fan, J. Zhang, Y. Hu, Z. Zhang, I. D. Gridnev and W. Zhang, ACS Catal., 2020, 10, 3232–3240 CrossRef CAS; (b) Y. Lou, J. Wang, G. Gong, F. Guan, J. Lu, J. Wen and X. Zhang, Chem. Sci., 2020, 11, 851–855 RSC.
- X. Li and D. G. Hall, J. Am. Chem. Soc., 2020, 142, 9063–9069 CrossRef CAS PubMed.
- L. Guo, A. Noble and V. K. Aggarwal, Angew. Chem., Int. Ed., 2021, 60, 212–216 CrossRef CAS PubMed.
- H. A. Sharma, J. Z. Essman and E. N. Jacobsen, Science, 2021, 374, 752–757 CrossRef CAS PubMed.
- L. Yang, D. H. Tan, W. X. Fan, X. G. Liu, J. Q. Wu, Z. S. Huang, Q. Li and H. Wang, Angew. Chem., Int. Ed., 2021, 60, 3454–3458 CrossRef CAS PubMed.
- K. Kubota, D. Miura, T. Takeuchi, S. Osaki and H. Ito, ACS Catal., 2021, 11, 6733–6740 CrossRef CAS.
- D. B. Diaz, C. C. G. Scully, S. K. Liew, S. Adachi, P. Trinchera, J. D. St. Denis and A. K. Yudin, Angew. Chem., Int. Ed., 2016, 55, 12659–12663 CrossRef CAS PubMed.
- T. Shiro, A. Schuhmacher, M. K. Jackl and J. W. Bode, Chem. Sci., 2018, 9, 5191–5196 RSC.
- For recent reviews on KATs, see: (a) Z. He, A. Zajdlik and A. K. Yudin, Acc. Chem. Res., 2014, 47, 1029–1040 CrossRef CAS PubMed; (b) F. K. Scharnagl, S. K. Bose and T. B. Marder, Org. Biomol. Chem., 2017, 15, 1738–1752 RSC; (c) A. Šterman, I. Sosič, S. Gobec and Z. Časar, ACS Omega, 2020, 5, 17868–17875 CrossRef PubMed; (d) D. Wu, J. Taguchi, M. Tanriver and J. W. Bode, Angew. Chem., Int. Ed., 2020, 59, 16847–16858 CrossRef CAS PubMed; (e) A. . Holownia, C. N. Apte and A. Yudin, Chem. Sci., 2021, 12, 5346–5360 RSC.
- A. Šterman, J. Košmrlj, N. Žigart, S. Gobec, I. Sosič and Z. Časar, Adv. Synth. Catal., 2021, 363, 2396–2402 CrossRef.
- (a) G. Hou, F. Gosselin, W. Li, J. C. McWilliams, Y. Sun, M. Weisel, P. D. O'Shea, C.-y. Chen, I. W. Davies and X. Zhang, J. Am. Chem. Soc., 2009, 131, 9882–9883 CrossRef CAS PubMed; (b) G. Hou, R. Tao, Y. Sun, X. Zhang and F. Gosselin, J. Am. Chem. Soc., 2010, 132, 2124–2125 CrossRef CAS PubMed.
- Q. Zhao, J. Wen, R. Tan, K. Huang, P. Metola, R. Wang, E. V. Anslyn and X. Zhang, Angew. Chem., Int. Ed., 2014, 53, 8467–8470 CrossRef CAS PubMed.
- For recent reviews on hydrogenation of imines, see: (a) N. Fleury-Brégeot, V. de la Fuente, S. Castillón and C. Claver, ChemCatChem, 2010, 2, 1346–1371 CrossRef; (b) J.-H. Xie, S.-F. Zhu and Q.-L. Zhou, Chem. Rev., 2011, 111, 1713–1760 CrossRef CAS PubMed; (c) Z. Yu, W. Jin and Q. Jiang, Angew. Chem., Int. Ed., 2012, 51, 6060–6072 CrossRef CAS PubMed; (d) C. S. G. Seo and R. H. Morris, Organometallics, 2019, 38, 47–65 CrossRef CAS; (e) J. Barrios-Rivera, Y. Xu, M. Wills and V. K. Vyas, Org. Chem. Front., 2020, 7, 3312–3342 RSC; (f) K. Morisaki, H. Morimoto and T. Ohshima, ACS Catal., 2020, 10, 6924–6951 CrossRef CAS; (g) I. Peñafiel, J. Mangas-Sánchez and C. Claver, in Asymmetric Hydrogenation and Transfer Hydrogenation, ed. V. Ratovelomanana-Vidal and P. Phansavath, Wiley, 1st edn, 2021, pp. 281–305 Search PubMed.
- Prices of metals are reported as of January 18th, 2022 in US dollars per troy ounce. Rh: 16 500 $, Ir: 4000 $, Ru: 550 $. Source: Johnson Matthey, Price Charts, http://www.platinum.matthey.com/prices/price-charts, accessed January 2022 Search PubMed.
- (a) S. Hashiguchi, A. Fujii, J. Takehara, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1995, 117, 7562–7563 CrossRef CAS; (b) A. Fujii, S. Hashiguchi, N. Uematsu, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1996, 118, 2521–2522 CrossRef CAS.
- (a) J. Hannedouche, G. J. Clarkson and M. Wills, J. Am. Chem. Soc., 2004, 126, 986–987 CrossRef CAS PubMed; (b) A. M. Hayes, D. J. Morris, G. J. Clarkson and M. Wills, J. Am. Chem. Soc., 2005, 127, 7318–7319 CrossRef CAS PubMed; (c) H. G. Nedden, A. Zanotti-Gerosa and M. Wills, Chem. Rec., 2016, 16, 2623–2643 CrossRef PubMed; (d) M. Wills, Top. Curr. Chem., 2016, 374, 14 CrossRef PubMed.
- Stereochemical purity of aliphatic TAMs 3k–p was determined by their conversion to aliphatic α-aminoboronic acids 4k–p followed by the formation of Mosher amides and determination of e.r. with 19F-NMR: D. A. Allen, A. E. Tomaso, O. P. Priest, D. F. Hindson and J. L. Hurlburt, J. Chem. Educ., 2008, 85, 698–700 CrossRef CAS . For the α-aminoboronic acids 4 containing aromatic substituents the enantiomeric purity was also verified with Mosher amides of derivatives 4a and 4j which had comparable e.r. to the corresponding pTAM 3a and 3j (see the ESI†)..
- With [(R,R)-TethTsDpen-RuCl], aliphatic pTIMs 2j–p provide (S)-pTAMs 3j–p as unambiguously confirmed by the synthesis of (S,S)-bortezomib from 4m and determination of optical rotation for 3j–p. Based on the opposite direction of optical rotation, aromatic pTIMs 2a–i provide (R)-pTAMs 3a–i. The correct diastereoisomer of bortezomib was synthesized using [(S,S)-TethTsDpen-RuCl] in conversion of 2m to (R)-3m..
- A. J. J. Lennox, Organotrifluoroborate Preparation, Coupling and Hydrolysis, Springer, Heidelberg, 2013 Search PubMed.
- (a) A. K. L. Yuen and C. A. Hutton, Tetrahedron Lett., 2005, 46, 7899–7903 CrossRef CAS; (b) S. R. Inglis, E. C. Y. Woon, A. L. Thompson and C. J. Schofield, J. Org. Chem., 2010, 75, 468–471 CrossRef CAS PubMed.
- Q. I. Churches, J. F. Hooper and C. A. Hutton, J. Org. Chem., 2015, 80, 5428–5435 CrossRef CAS PubMed.
- G. A. Molander, L. N. Cavalcanti, B. Canturk, P.-S. Pan and L. E. Kennedy, J. Org. Chem., 2009, 74, 7364–7369 CrossRef CAS PubMed.
- D. W. Blevins, M.-L. Yao, L. Yong and G. W. Kabalka, Tetrahedron Lett., 2011, 52, 6534–6536 CrossRef CAS.
- G. W. Kabalka and V. Coltuclu, Tetrahedron Lett., 2009, 50, 6271–6272 CrossRef CAS.
- A. J. J. Lennox and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2012, 134, 7431–7441 CrossRef CAS PubMed.
- I. Omari, L. P. E. Yunker, J. Penafiel, D. Gitaari, A. San Roman and J. S. McIndoe, Chem.–Eur. J., 2021, 27, 3812–3816 CrossRef CAS PubMed.
- (a) V. D. Kodibagkar, W. Cui, M. E. Merritt and R. P. Mason, Magn. Reson. Med., 2006, 55, 743–748 CrossRef CAS PubMed; (b) P. K. Gulaka, U. Rastogi, M. A. McKay, X. Wang, R. P. Mason and V. D. Kodibagkar, NMR Biomed, 2011, 24, 1226–1234 CrossRef CAS PubMed.
- S. P. A. Hinkes and C. D. P. Klein, Org. Lett., 2019, 21, 3048–3052 CrossRef CAS PubMed.
- For the synthesis of bortezomib and ixazomib we used a modified solid phase procedure of Klein et al. and Stivala et al., where Fmoc-protection/deprotection of α-aminoboronic acids was omitted from the synthesis and thus the overall two step shorter solid phase synthetic sequence was obtained and provided even cleaner products without chromatography (see the ESI†): (a) M. A. M. Behnam, T. R. Sundermann and C. D. Klein, Org. Lett., 2016, 18, 2016–2019 CrossRef CAS PubMed; (b) B. E. Daniels and C. E. Stivala, RSC Adv., 2018, 8, 3343–3347 RSC; (c) S. P. A. Hinkes, S. Kämmerer and C. D. P. Klein, Chem. Sci., 2020, 11, 9898–9903 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Preparation of pTIMs, enantioenriched pTAMs, and α-aminoboronic acids, details regarding reaction screening and optimization of reactions, determination of optical purities, and 1H, 13C, 11B, and 19F NMR spectra. See DOI: 10.1039/d1sc07065g |
| This journal is © The Royal Society of Chemistry 2022 |