
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGeneration of oligonucleotide conjugates via one-pot diselenide-selenoester ligation–deselenization/alkylation†
Christopher
Liczner
 a,
Cameron C.
Hanna
bc,
Richard J.
Payne
bc and
Christopher J.
Wilds
*a
a,
Cameron C.
Hanna
bc,
Richard J.
Payne
bc and
Christopher J.
Wilds
*a
aDepartment of Chemistry and Biochemistry, Concordia University, 7141 Rue Sherbrooke Ouest, Montréal, Québec H4B 1R6, Canada. E-mail: chris.wilds@concordia.ca
bSchool of Chemistry, The University of Sydney, Sydney, NSW 2006, Australia
cAustralian Research Council Centre of Excellence for Innovations in Peptide and Protein Science, The University of Sydney, Sydney, NSW 2006, Australia. E-mail: richard.payne@sydney.edu.au
First published on 19th November 2021
Abstract
A breadth of strategies are needed to efficiently modify oligonucleotides with peptides or lipids to capitalize on their therapeutic and diagnostic potential, including the modulation of in vivo chemical stability and for applications in cell-targeting and cell-permeability. The chemical linkages typically used in peptide oligonucleotide conjugates (POCs) have limitations in terms of stability and/or ease of synthesis. Herein, we report an efficient method for POC synthesis using a diselenide-selenoester ligation (DSL)-deselenization strategy that rapidly generates a stable amide linkage between the two biomolecules. This conjugation strategy is underpinned by a novel selenide phosphoramidite building block that can be incorporated into an oligonucleotide by solid-phase synthesis to generate diselenide dimer molecules. These can be rapidly ligated with peptide selenoesters and, following in situ deselenization, lead to the efficient generation of POCs. The diselenide within the oligonucleotide also serves as a flexible functionalisation handle that can be leveraged for fluorescent labelling, as well as for alkylation to generate micelles.
Introduction
Owing to their intrinsic instability in biological settings and their inability to diffuse through cell membranes, synthetic oligonucleotides must be modified in order to be used for therapeutic applications, such as for gene silencing through antisense1 or RNA interference mechanisms.2 Modification is typically achieved via two main strategies: the chemical modification of the sugar-phosphate backbone3 or through conjugation to other species, such as lipids4,5 and peptides.6 Peptide-oligonucleotide conjugates (POCs) are an important category of modified oligonucleotides as specific peptides can be installed, which engender cell-penetrating7,8 or cell-targeting9,10 properties to the oligonucleotide cargo. Indeed, very recently, a peptide was used to enhance the nuclear delivery of an oligonucleotide drug in the lungs of mice.11 Beyond therapeutics, POCs have also found applications in promoting the self-assembly of artificial proteins12 and as functional biomaterials.13,14While POCs can be prepared by in-line solid-phase synthesis (i.e. by sequential elongation of each species en bloc),15–18 this strategy is hampered by incompatibilities between the peptide and oligonucleotide protection and assembly chemistries, e.g. cleavage and/or depurination of oligonucleotides can occur under the acidic conditions used for peptide deprotection. These limitations have inspired the development of more robust post-synthetic strategies to fuse the peptide and oligonucleotide components. Specifically, by installing complementary reactive groups on the peptide and oligonucleotide fragments, the two pure components can be conjugated, thus circumventing the challenges of in-line synthesis. Some common POCs generated via this approach possess disulfide,19,20 thiosuccinimide,21–23 triazole24,25 and amide linkages.26,27 While each of these conjugates have found use for a number of applications, the majority of these linkages suffer from specific limitations. For example, disulfides are susceptible to reducing conditions in in vitro assays or within cells, thiosuccinimides have been shown to be labile under physiological conditions,28 triazoles are typically installed using copper catalysis, which can damage oligonucleotides29 and amides are generated with poor chemoselectivity, requiring the use of partially or fully protected peptides. Native-chemical ligation (NCL) has emerged as a powerful method to overcome the chemoselectively issues in preparing stable amide-linked POCs.30 Specifically, the reaction occurs between a peptide thioester and an oligonucleotide with a 5′-cysteinyl in the presence of thiophenol and tris(carboxyethyl)phosphine (TCEP) additives (Fig. 1A). The reaction proceeds via transthioesterification between the 5′-thiol and the thioester, followed by a rapid S-to-N acyl shift to afford the desired POC, typically in 1–2 days. In recent years, diselenide-selenoester ligation (DSL), a reaction inspired by the NCL method but with superior kinetics, has been developed for the synthesis of modified proteins.31–34 This seminal methodology enables the rapid and efficient fusion of peptides bearing C-terminal selenoester functionalities with peptides bearing an N-terminal selenocystine residue (the oxidised form of the 21st amino acid selenocysteine/Sec) in the absence of any ligation additives. Without purification, the diphenyl diselenide (DPDS) byproduct can be removed by extraction with hexanes or diethyl ether and deselenization of the Sec residue at the ligation junction to alanine achieved through the addition of excess TCEP and dithiothreitol (DTT) or glutathione (GSH) (Fig. 1B). The DSL reactions proceed rapidly at low to high millimolar concentrations of reacting peptide fragments, with reactions often reaching completion in a few minutes. However, below this concentration poor kinetics are observed owing to the bimolecular nature of the rate limiting step and the low steady state concentration of selenol present under the additive-free conditions. Recently, a modified reaction manifold dubbed the reductive DSL (rDSL) was developed. This variant of the DSL reaction employs TCEP and DPDS in the ligation buffer which enables efficient ligation reactions even down to nanomolar concentrations of reacting peptide fragments.35
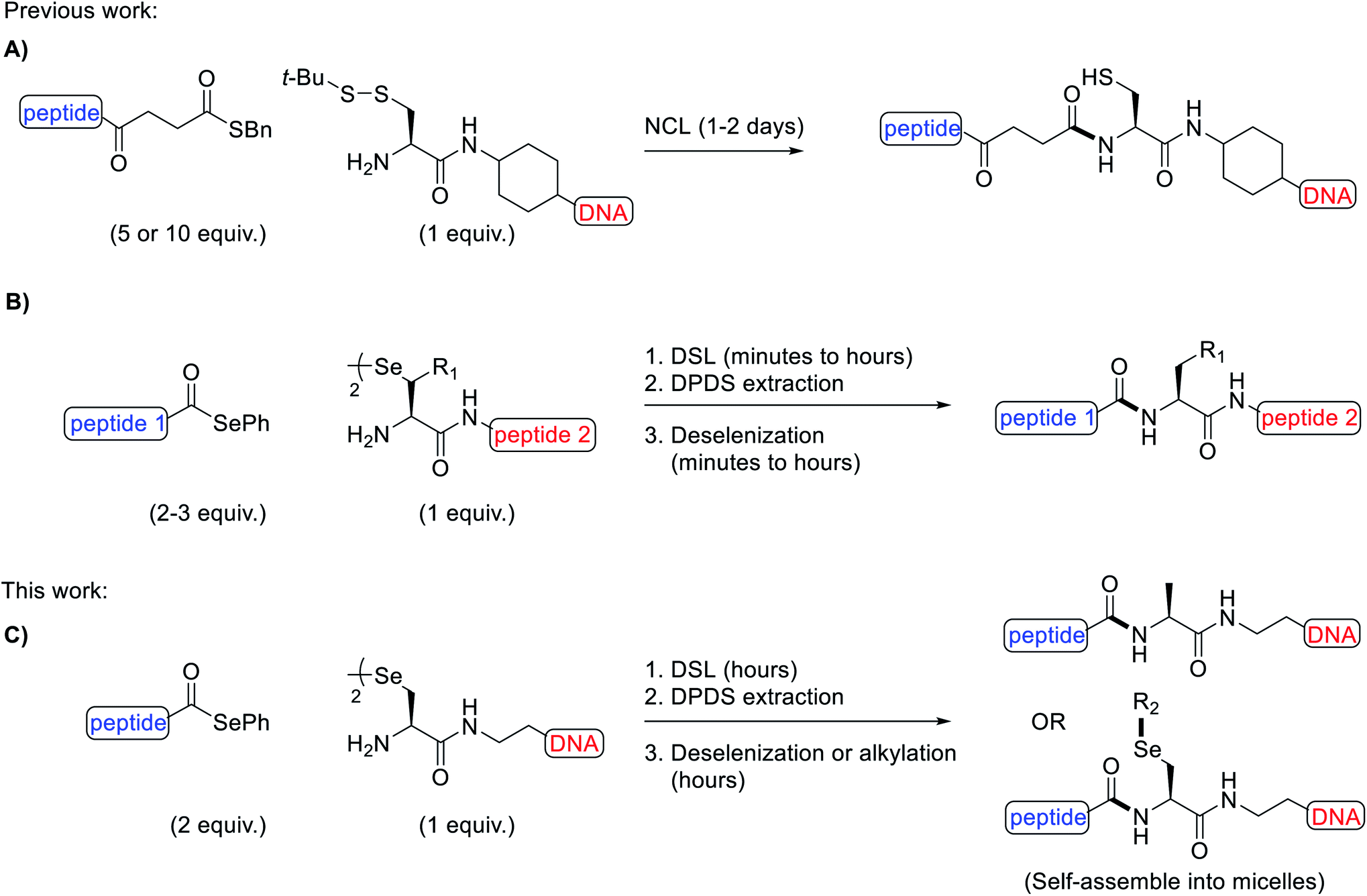 | ||
| Fig. 1 Methods for the chemoselective amidation of biomolecules. (A) Native chemical ligation to generate peptide-oligonucleotide conjugates bearing a free thiol. (B) One-pot diselenide-selenoester ligation–deselenization to generate native or post-synthetically modified proteins. (C) One-pot diselenide-selenoester ligation–deselenization/alkylation to generate peptide-oligonucleotide conjugates or micelles (bonds formed are shown in bold; Bn = benzyl; Ph = phenyl; t-Bu = tert-butyl; DPDS = diphenyl diselenide; R1 = H or amino acid side chain; R2 = decyl chain). | ||
We envisaged that the application of DSL for the rapid and high yielding synthesis of POCs at low concentrations, in a chemoselective and untemplated manner, would be highly desirable for nucleic acid research. Utilizing the diselenide handle for the late stage site-selective alkylation of oligonucleotides was also expected to be synthetically valuable. To the best of our knowledge, there is only one previous example of a diselenide-bridged oligonucleotide in the literature,36 however, this species would be unsuitable for DSL owing to the unfavourable positioning of its reactive functional groups.
Herein, we report the synthesis of a novel selenide phosphoramidite and its incorporation, by automated solid-phase synthesis (SPS), at the 5′-end of both a known i-motif forming sequence37 (5′-dTCCCGTTTCCA-3′) and the mixed-base sequence 5′-dACTCGTTCGTA-3′. Once deprotected and cleaved from the controlled pore glass (CPG) solid-support, spontaneous dimerization generated the desired diselenide-bridged oligonucleotides. Furthermore, we demonstrate the scope of one-pot rDSL-deselenization chemistry for the preparation of POCs, using C-terminal selenoester peptides and 5′-diselenide-bridged oligonucleotides (Fig. 1C). The thermal stability and circular dichroism (CD) profiles were measured for the i-motifs of the unmodified oligonucleotide, the diselenide-bridged oligonucleotide and a POC. The applicability of the 5′-group of the oligonucleotide was highlighted by in situ lipidation of a POC with a decyl chain following additive-free DSL, leading to self-assembly of the conjugate into micelles (Fig. 1C). This late stage alkylation protocol was also applied towards the synthesis of oligonucleotide micelles as well as fluorescently labelled oligonucleotides. Finally, we show that oligonucleotide micelles can be disassembled through H2O2-triggered dealkylation.
Results and discussion
Synthesis of selenide phosphoramidite
Selenide phosphoramidite 6 (Scheme 1) was designed as a key building block for incorporation into oligonucleotides. The reactive diselenide and amine functional groups were positioned three bonds apart in 6, allowing the intramolecular Se-to-N acyl shift in the proposed DSL reactions to proceed through a 5-membered cyclic transition state, known to lead to the most facile ligation rates in NCL.38 Protecting groups were carefully considered for the amine and selenol functional groups to ensure that they would be orthogonal for the solid-phase assembly of the oligonucleotide. A fluorenylmethoxycarbonyl-protected amine was avoided as this group would be removed under the basic oligonucleotide deprotection conditions, which would lead to potential acrylonitrile adduct formation and/or transamidation at the free amine, rendering it inert for subsequent DSL and limiting sequence selection (primary amines can “deprotect” adjacent adenine and guanine nucleobases). As such, base stable trityl (Tr) protection of the amine was selected. The cyanoethyl group was chosen for protection of the selenol moiety, which could be removed during the basic deprotection steps in oligonucleotide isolation, thus eliminating the need for an extra step in the workflow. | ||
Scheme 1 Synthesis of selenide phosphoramidite 6. Reagents and conditions: (a) LiOH (1.1 equiv.), 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (v/v) H2O/MeOH, reflux, 4 h; (b) TBSOCH2CH2NH2 (1.5 equiv.), PyBOP (1.2 equiv.), DMF, RT, 20 h; (c) CBr4 (1.3 equiv.), PPh3 (1.1 equiv.), DCM, 0 °C to RT, 2 h; (d) di(2-cyanoethyl) diselenide (2.1 equiv.), NaBH4 (2.1 equiv.), EtOH, 0 °C to RT, 2 h; (e) TREAT-HF (3.0 equiv.), TEA (2.0 equiv.), THF, RT, 3 h; (f) Cl–P(OCE)N(iPr)2 (3.0 equiv.), DIPEA (4.0 equiv.), THF, RT, 20 min. (Tr = trityl; TBS = tert-butyldimethylsilyl; PyBOP = benzotriazole-1-yl-oxy-tris-pyrrolidino-phosphonium hexafluorophosphate; TREAT-HF = triethylamine trihydrofluoride; TEA = triethylamine; CE = 2-cyanoethyl; DIPEA = N,N-diisopropylethylamine). :1 (v/v) H2O/MeOH, reflux, 4 h; (b) TBSOCH2CH2NH2 (1.5 equiv.), PyBOP (1.2 equiv.), DMF, RT, 20 h; (c) CBr4 (1.3 equiv.), PPh3 (1.1 equiv.), DCM, 0 °C to RT, 2 h; (d) di(2-cyanoethyl) diselenide (2.1 equiv.), NaBH4 (2.1 equiv.), EtOH, 0 °C to RT, 2 h; (e) TREAT-HF (3.0 equiv.), TEA (2.0 equiv.), THF, RT, 3 h; (f) Cl–P(OCE)N(iPr)2 (3.0 equiv.), DIPEA (4.0 equiv.), THF, RT, 20 min. (Tr = trityl; TBS = tert-butyldimethylsilyl; PyBOP = benzotriazole-1-yl-oxy-tris-pyrrolidino-phosphonium hexafluorophosphate; TREAT-HF = triethylamine trihydrofluoride; TEA = triethylamine; CE = 2-cyanoethyl; DIPEA = N,N-diisopropylethylamine). | ||
The synthesis of selenide phosphoramidite 6 started from commercially available N-trityl-L-serine methyl ester 1, which was first saponified to the lithium carboxylate salt intermediate. This was followed by a reaction with tert-butyldimethylsilyl (TBS)-protected ethanolamine (see ESI† for synthesis) using PyBOP as the coupling reagent to afford amide 2 in 67% yield over two steps. Next, conversion of the free hydroxyl in 2 to the corresponding iodide was attempted by treatment with iodine, triphenylphosphine (PPh3) and imidazole, as previously reported39 (see ESI† for full procedure). However, in our hands, this caused formation of the aziridine as the major product. Aziridine formation could be overcome through the use of a base-free Appel reaction using carbon tetrabromide and PPh3, affording alkyl bromide 3 in 71% yield. From here, di(2-cyanoethyl) diselenide (see ESI† for synthesis) was reduced with sodium borohydride to the selenolate, which was treated with alkyl bromide 3 to afford protected selenide 4 in 74% yield. Chemoselective desilylation in the presence of a cyanoethyl group was achieved by treatment with triethylamine trihydrofluoride as the fluoride source,40 affording alcohol 5 in 88% yield. Finally, phosphitylation of the primary alcohol in 5 generated selenide phosphoramidite 6 in 85% yield. Overall, this compound was prepared in 26% yield over 6 steps.
Synthesis and purification of a 5′-diselenide-bridged oligonucleotide
Selenide phosphoramidite 6 was first introduced by automated SPS into the 5′-end of the sequence 5′-dTCCCGTTTCCA-3′, an oligonucleotide known to form a dimeric head-to-head i-motif structure at acidic pH (structure shown in Fig. S1†). This sequence was chosen as a model system to explore ligation via DSL as the 5′-ends of both i-motif monomer oligonucleotides are in close proximity, thus, a 5′-diselenide bridge between the interacting strands was not expected to hinder i-motif formation. Additionally, the influence of the diselenide bridge on the thermal stability and structure of the i-motif could be studied.A trityl-on SPS was performed in order to retain the trityl group on the 5′-amine (to avoid byproduct formation that can occur with a free amine, vide supra). Notably, prior to deprotection, treatment of the CPG-bound oligonucleotide with 10 vol% diethylamine in acetonitrile could not be performed to remove the potent electrophilic acrylonitrile, as this caused premature deselenization of the resulting oligonucleotide. Deprotection was instead achieved by overnight incubation with ammonium hydroxide/ethanol (3:1, v/v) at 37 °C. The supernatant was subsequently lyophilised and the crude 5′-modified oligonucleotide dissolved in 18 MΩ H2O and analyzed by ion-exchange (IEX) HPLC (compared to the unmodified oligonucleotide, Fig. S2†). This demonstrated quantitative coupling of selenide phosphoramidite 6, which could not otherwise be evaluated by the in-line conductivity cell of the ABI synthesiser due to the trityl-on synthesis that was performed (i.e. there was no trityl cation released to accurately measure the coupling efficiency). It should be noted that the oligonucleotide had a later than normal HPLC retention time for a tritylated 12-mer sequence, providing early evidence that the diselenide bridge had formed between the two oligonucleotide strands. Minor thymine acrylonitrile adduct formation was observed by mass spectrometry (MS) analysis of the crude sample (Fig. S3†) and was also observed as the small shoulder adjacent to the target product in the IEX chromatogram (Fig. S2B†). The crude oligonucleotide was then purified by IEX HPLC and loaded onto a C-18 cartridge for detritylation (three successive 2 vol% trifluoroacetic acid (TFA) in H2O washes) and desalting. The purified oligonucleotide was then analyzed by IEX HPLC (Fig. S5†) and MS (Fig. S6†), which confirmed the preparation of the desired deprotected diselenide-bridged oligonucleotide 7. It is important to note that a very small amount of 2′-deoxyadenosine depurination occurred from the acidic detritylation washes (as observed in the mass spectrum: Fig. S6†), however, this small impurity was deemed inconsequential for the proposed DSL optimization studies.
rDSL-deselenization for the preparation of POCs
Peptide selenoesters were next prepared using a side chain anchoring solid-phase strategy as previously reported41 (see ESI† for synthesis and purification). We next attempted to fuse the oligonucleotide diselenide dimer and peptide selenoester via one-pot rDSL-deselenization chemistry to generate POCs. We initially performed reactions on a small scale using rDSL conditions that have been reported to proceed efficiently at low concentrations of reacting fragments. Before initiating rDSL, it was necessary to sonicate the ligation buffer containing TCEP and DPDS for 30 minutes in order to ensure optimal dissolution of phenylselenolate, as this reagent is sparingly soluble in aqueous media. Furthermore, the buffers were all sparged with argon for 10 minutes before use in order to remove any dissolved O2, as aerobic deselenization has been reported to cause β-hydroxylation at amino acids.34,42,43An initial rDSL reaction was attempted by dissolving lyophilised diselenide oligonucleotide 7 (50 nmol, 1 equiv.) and selenoester peptide 8 (200 nmol, 2 equiv. with respect to monomer oligonucleotide) in an aqueous 2-(N-morpholino)ethanesulfonic acid (MES) buffer (pH 5.5) containing 30 mM TCEP and 20 mM DPDS and the resulting mixture was incubated at 37 °C (500 μM oligonucleotide and 2 mM peptide final concentrations, Fig. 2). It should be noted that slightly acidic pH was used in order to minimize undesirable selenoester hydrolysis over the reaction period. Tracking the reaction progress by IEX HPLC indicated minimal starting material remained after just 2 hours (Fig. 2d). Each aliquot was treated with hydrazine before injection in order to confirm the presence of the desired amide and to hydrazinolyse any selenoester intermediates that remained in the reaction mixture. After only 5 minutes, the excess selenoester peptide 8 present in the reaction at this early time point was not completely hydrazinolyzed and was still observed in the HPLC trace (Fig. 2b). Furthermore, the intermediate species were identified by analyzing the 1 h aliquot directly by MS (Fig. S10†; no hydrazine treatment), confirming the presence of symmetrical diselenide 9 and asymmetrical diselenides 11 and 12, generated through diselenide exchange of 9 with starting material 7 and with phenylselenolate liberated during the reaction, respectively (Fig. 2). Intermediate selenoester 10 was also identified, but was no longer present after 3 h by MS analysis (Fig. S11†). The reaction was then extracted with diethyl ether to remove any dissolved diphenyl diselenide or phenylselenolate, which would interfere with the deselenization step owing to their radical scavenging properties. Additional DTT (50 equiv. instead of 5 equiv.) was then added to facilitate deselenization. Pleasingly, following 3 h of incubation, all intermediates coalesced to afford the target POC 13, as observed in the IEX HPLC trace (Fig. 3d; no hydrazine treatment) and confirmed by MS analysis (Fig. S12†). Furthermore, the presence of excess selenoester peptide 8 (Fig. 3b) did not prevent efficient deselenization under these reaction conditions. Thus, rDSL (3 h) and in situ deselenization (3 h) generated POC 13 in 97% yield as judged by analytical HPLC (Fig. S13†), with no β-hydroxylated byproduct observed (Fig. S12†). These results demonstrate that DSL involving oligonucleotides can tolerate secondary structures, as at the temperature and pH used for ligation, the i-motif was formed (see Biophysical study section for confirmation).
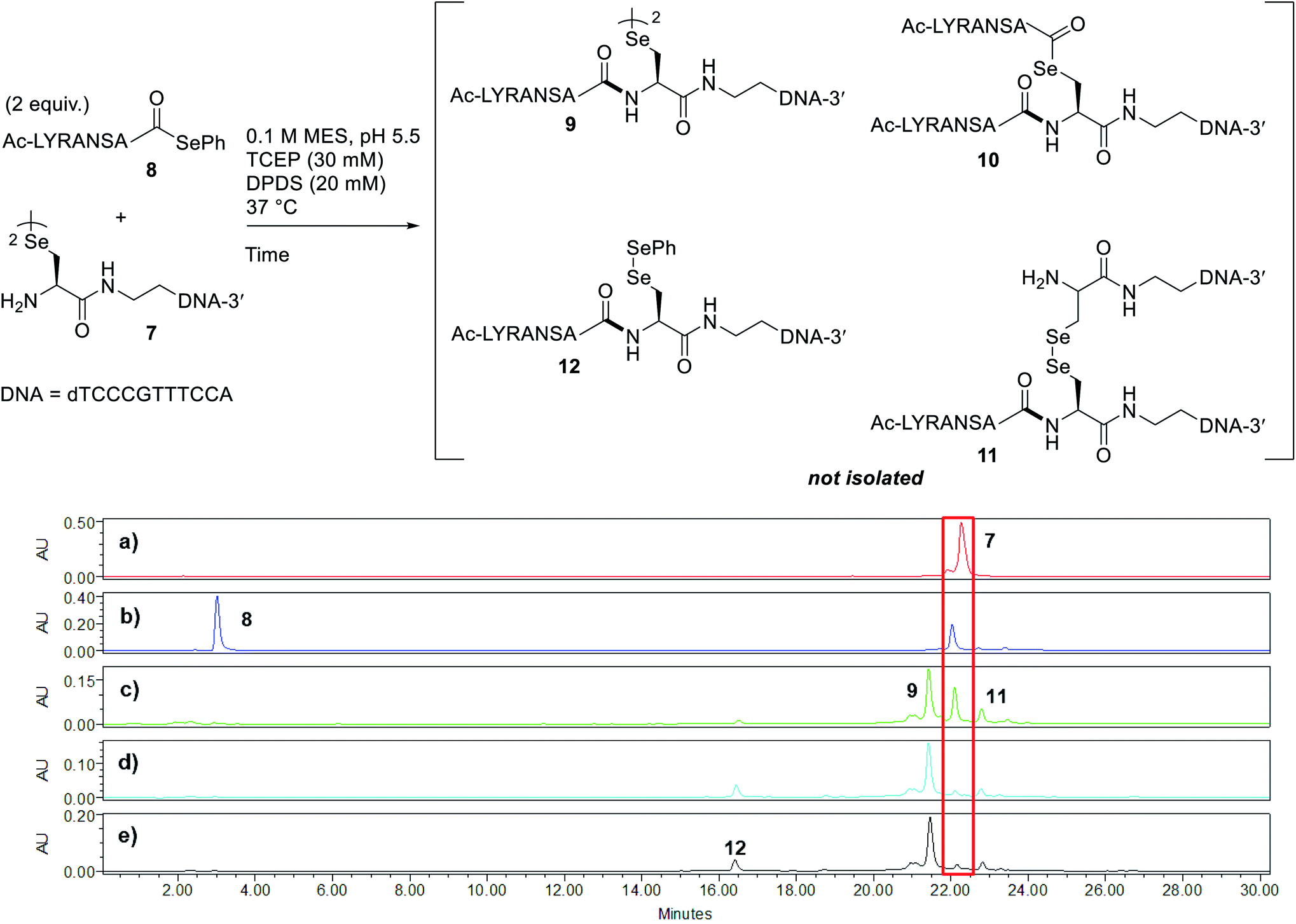 | ||
| Fig. 2 Tracking rDSL between diselenide oligonucleotide 7 and selenoester peptide 8 by IEX HPLC. Traces depict reaction progress after (a) 0 minutes (diselenide 7 prior to mixing) (b) 5 minutes (c) 1 hour (d) 2 hours and (e) 3 hours. The red box is highlighting the disappearance of starting material 7 over time. See ESI† for HPLC conditions and aliquot preparation. Nucleotides of oligonucleotide 7 and amino acid residues of peptide selenoester 8 are provided in one letter code. | ||
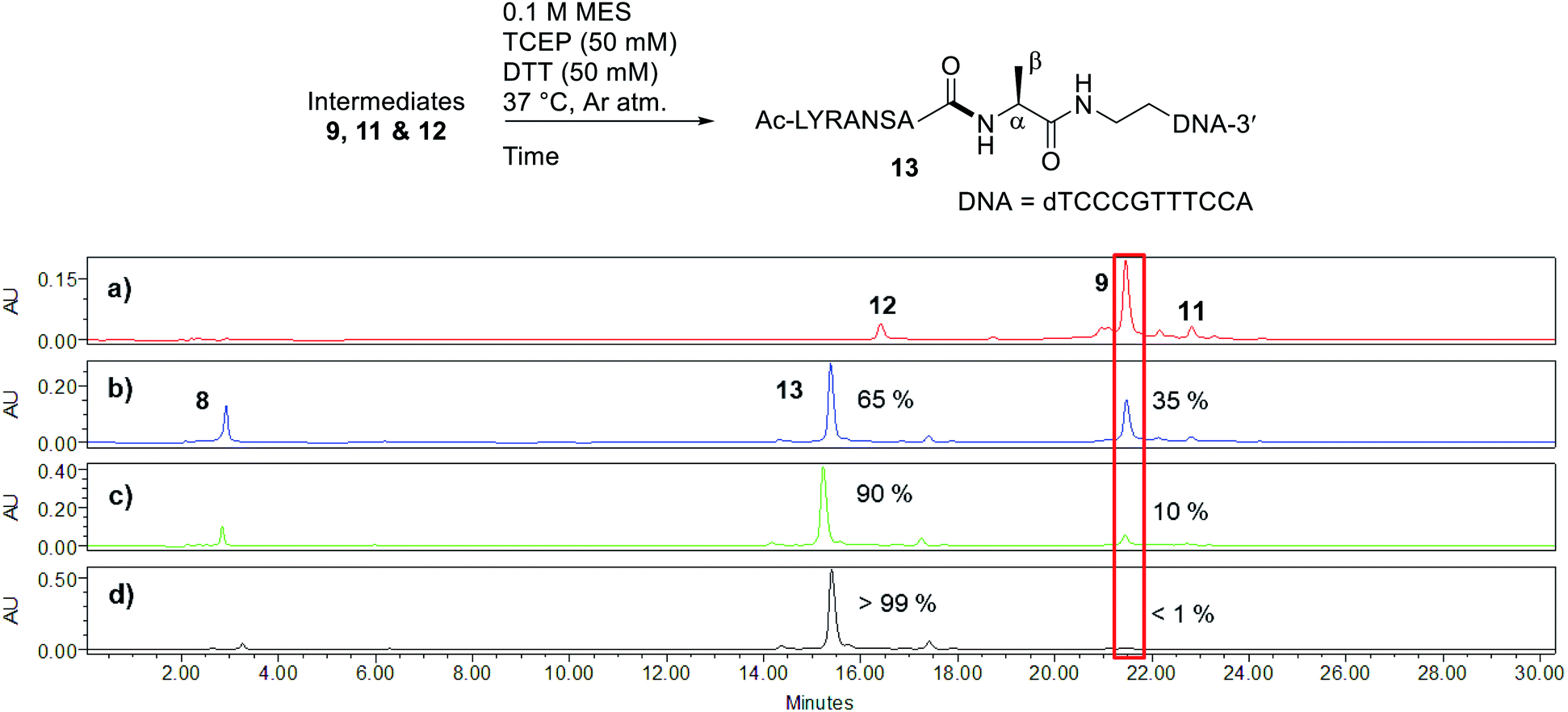 | ||
| Fig. 3 Tracking deselenization of intermediates 9, 11 and 12 after rDSL by IEX HPLC. Traces depict reaction progress after (a) 0 minutes (b) 1 hour (c) 2 hours and (d) 3 hours. The red box is highlighting the disappearance of intermediate 9 over time. The percentages are the relative amounts of POC 13 and intermediate 9 present, determined from the area of the peaks. See ESI† for HPLC conditions and aliquot preparation. | ||
By IEX HPLC, POC 13 and the deselenized product of starting material 7 co-elute (Fig. S14A†), making it necessary to purify the product by reverse-phase (RP) HPLC. This also allowed for a more accurate determination of the HPLC yield of the rDSL-deselenization process, as baseline resolution of the two species is achieved (Fig. S13†). It should be noted that under the RP HPLC conditions used (pH 5.8), a proportion of the POC forms the i-motif in situ and thus, it is necessary to collect the broad peak that elutes after the single-stranded oligonucleotide in order to maximise the yield of POC. With the RP-purified conjugate in hand (Fig. S14B†), further purification by IEX HPLC, followed by C-18 cartridge desalting, yielded an ultra-pure POC (Fig. S14C†), which was used in the subsequent UV thermal denaturation and CD studies.
Biophysical studies of i-motif oligonucleotides
The stability of the pure unmodified oligonucleotide (5′-dTCCCGTTTCCA-3′), diselenide oligonucleotide 7 and POC 13 were each evaluated by UV thermal denaturation at pH 5.5. The reverse sigmoidal profiles observed for each species at 295 nm (Fig. S15†) were indicative of i-motif formation. Furthermore, diselenide oligonucleotide 7 was found to have a melting temperature (Tm) of 52 °C, 22 °C higher than the native structure, demonstrating a significant stabilization by linking the 5′-ends. Conversely, POC 13 had a 3 °C lower Tm value compared to the unmodified i-motif structure. CD profiles were also measured for each species at pH 5.5. All species displayed very similar CD characteristics, exhibiting a positive peak at 287 nm and a negative peak at 242 nm (Fig. S16†). The positive peak at 287 nm is characteristic of i-motif structures, however, typically the negative peak observed is at approximately 260 nm. This deviation is likely due to the GTTT hairpin loops found in this structure, that interact through a T:G:G:T tetrad, as these hairpins are not commonly found in other previously studied i-motifs.Investigating the scope of POC formation via rDSL-deselenization
Next, the focus shifted towards examining the scope of the rDSL-deselenization methodology for POC generation. To this end, selenide phosphoramidite 6 was incorporated at the 5′-end of the sequence 5′-dACTCGTTCGTA-3'. This sequence was chosen in order to demonstrate that i-motif formation is not a prerequisite for efficient DSL between oligonucleotides and peptides, as it is not known to form any secondary structures at acidic pH, nor is it self-complementary. Furthermore, 2′-deoxyadenosine was placed next to the reactive 5′-group in order to confirm that the proximal exocylic amine of adenine does not interfere with the DSL reaction. Finally, this oligonucleotide was designed to clearly demonstrate that the implemented protecting group and purification strategies prevent transamidation between the 5′-amine and adjacent protected adenine (vide supra). The SPS and purification of diselenide oligonucleotide 14 were performed as described above (see Fig. S8† for the IEX HPLC trace of purified product). It is important to note that on this occasion, a more dilute solution was used during deprotection in order to minimize acrylonitrile adduct formation that was observed in the synthesis of oligonucleotide 7. Under these conditions there was no evidence of acrylonitrile adducts as judged by IEX HPLC and MS analysis (see Fig. S7† for comparison). A further modification was also made in the detritylation process. Since this sequence contained more purines than diselenide oligonucleotide 7 (and a small amount of depurination occurred during detritylation of this species), we sought to suppress this side reaction altogether. Gratifyingly, by performing a water wash after each of the 2 vol% TFA/H2O detritylation treatments, as well as the inclusion of 0.5% NH4OH in the eluent (to neutralize any residual TFA), depurination of diselenide oligonucleotide 14 was completely avoided, as observed by MS analysis (Fig. S9†).The one-pot rDSL-deselenization chemistry was next attempted on diselenide oligonucleotide 14 with a range of peptide selenoesters (8, 15–17) each bearing different amino acid residues near the C-terminus. The reactions were performed using the same reaction times, concentrations and equivalencies of peptide selenoester and oligonucleotide components successfully employed above (Table 1, entries 2–5). Pleasingly, clean conversions to the target POCs were obtained as judged by HPLC analysis (HPLC yields >92% over the two steps). In these reactions, the deselenized starting material and target POC products did not co-elute by IEX and thus, could be purified solely by IEX HPLC. Each of the POC products were isolated with purities of 97% or greater. These results indicate that oligonucleotide secondary structure is not required for DSL to proceed efficiently and that a nearby exocyclic amine at the 5′-end is well tolerated.
|
|
||||
|---|---|---|---|---|
| Entry | Peptide | Oligonucleotide (DNA) | Yielda (%) | Purityb (%) |
| a Yield determined by RP HPLC analysis of the crude samples using the areas of the POC product and deselenized/reduced oligonucleotide starting material (see ESI for conditions). b Determined from the areas in the pure IEX HPLC traces (minor impurity: β-hydroxylated species). | ||||
| 1 | Ac-LYRANSA (8) | dTCCCGTTTCCA (7) | 97 | 100 |
| 2 | Ac-LYRANSA (8) | dACTCGTTCGTA (14) | 93 | 99 |
| 3 | Ac-LYRANYF (15) | dACTCGTTCGTA (14) | 92 | 100 |
| 4 | Ac-LYRANQF (16) | dACTCGTTCGTA (14) | 97 | 99 |
| 5 | Ac-LYRANM (17) | dACTCGTTCGTA (14) | 96 | 97 |
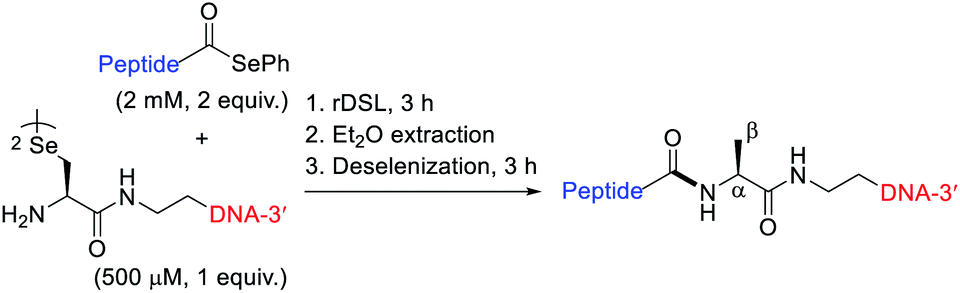
Additive-free DSL-Alkylation for the preparation of POC micelles
Having demonstrated that POCs can be rapidly and efficiently generated using one-pot rDSL-deselenization chemistry, we next sought to apply DSL chemistry to the generation of POC micelles. We envisaged that this could be achieved by omitting the deselenization step and capitalising on the presence of a selenol moiety for late stage lipidation chemistry.35 To this end, we performed additive-free DSL (i.e. with no TCEP and DPDS in the ligation buffers) between diselenide oligonucleotide 7 and selenoester peptide 8. Gratifyingly, this led to clean fusion of the fragments after 4 h (confirmed by IEX HPLC; see Fig. S33A†). Following removal of the DPDS generated in the reaction, in situ alkylation with bromodecane was performed. Specifically, the crude DSL reaction mixture was treated with aqueous 0.1 M MES buffer (pH 6.7) containing 150 mM DTT (150 equiv.), followed by an equal volume of bromodecane (500 equiv.) in DMF, and the resulting mixture was incubated at 37 °C (Scheme 2). Excess DTT was used to reduce, but not deselenize the intermediate diselenides, revealing the highly nucleophilic selenolate for alkylation with bromodecane. Interestingly, at no time interval was the single-stranded alkylated POC observed in the IEX HPLC traces (Fig. S33†). Instead, total disappearance of the intermediate POC peaks occurred over time and were completely absent from the chromatogram in the 23 h IEX HPLC trace. This suggested that self-assembly to larger aggregates was occurring, owing to its amphiphilic character and the lack of observable precipitate in the reaction solution. Importantly, the peptide used for the generation of the POC does not form secondary structures (confirmed by the CD data, Fig. S16†). This supported the idea that micelles were generated following the alkylation instead of other previously reported POC nanostructures where the peptide is integral for proper structure formation, such as hollow44 or multi-lamellar spheres.25 Once lyophilised and redissolved in 0.1 M aqueous MES buffer, pH 6.7, dynamic light scattering (DLS) measurements of a 100 μM solution confirmed the presence of species with an average size of 84 nm (Fig. S34†). In contrast, the unmodified oligonucleotide displayed significantly smaller aggregates, averaging 1.5 nm in size at the same concentration (Fig. S37†). The morphology of the alkylated POC was determined by negative-stain transmission electron microscopy (Fig. S38†), confirming that spherical micelles were indeed prepared. | ||
| Scheme 2 Synthesis of POC micelles by additive-free DSL-alkylation. | ||
Alkylation of the diselenide oligonucleotide dimer
As a control experiment to assess the importance of the peptide for micelle formation, and to further demonstrate the synthetic utility of the 5′-group, diselenide oligonucleotide 7 was alkylated using bromodecane, without prior installation of a peptide (see ESI† for details). The reaction was performed at pH 6.7 and, on this occasion, the single-stranded alkylated oligonucleotide was observed after just 1 h (Fig. S35B†). After 22 h, the HPLC signal for this alkylated species decreased significantly (Fig. S35F†), suggesting that micelles were still forming under the reaction conditions. Aggregate formation was subsequently confirmed by DLS measurements at pH 6.7 (Fig. S36†), indicating they were on average 77 nm, roughly the same size as the POC micelles.In order to more thoroughly characterise this late stage alkylation, an alkyl bromide with a shorter carbon chain (bromopentane) was reacted with diselenide oligonucleotide 7 to minimize self-assembly (see ESI† for details). The alkylation was complete after 1 h, with the Se-alkylated oligonucleotide confirmed to be the major product by IEX HPLC (Fig. S40B†) and MS analysis (Fig. S41†). Identification of the Se-alkylated oligonucleotide by MS was facilitated by the characteristic isotopic distribution for selenium-containing compounds (Fig. S42†). Importantly, there was no evidence of N-alkylation under these conditions, despite the presence of the free 5′-amine.
The alkylation conditions developed above were subsequently applied to the synthesis of a fluorescent dimethoxycoumarin labelled oligonucleotide (see ESI† for details). The reaction was once again complete in 1 h, confirmed by IEX HPLC (Fig. S43†). Following purification, the fluorescence properties of the oligonucleotide were determined, with characteristic absorbance at 350 nm (Fig. S46A†) and emission at 435 nm (Fig. S46B†), further confirming successful alkylation. Taken together, a mild, base-free alkylation method for diselenide oligonucleotides has been developed that is rapid, chemoselective and high yielding. This is in contrast to the previously developed selenol/selenolate alkylation approach, which only resulted in 34% fluorescent labelling.45
Oxidative dealkylation of oligonucleotide micelles by selenoxide elimination
Inspired by a polymer micelle drug delivery system that was shown to efficiently disassemble upon oxidation of its selenide linkage by hydrogen peroxide,46 and another report that showed excess hydrogen peroxide treatment of a phenyl selenide-modified RNA strand promoted selenoxide elimination without undesirable side reactions to the oligonucleotide,47 we explored the application of an oxidative elimination strategy to induce dealkylation and thus disassembly of the oligonucleotide micelles. To this end, a 100 mM H2O2 solution (160 equiv.) was freshly prepared and added to a solution containing 50 nmol of the oligonucleotide micelles (without a conjugated peptide), which was incubated at 37 °C (Scheme 3). An excess of peroxide was used in this reaction due to the presence of remaining DTT from the alkylation step, which would quench the H2O2. Gratifyingly, MS analysis confirmed the presence of the expected dicarbonyl oligonucleotide 18 after just 10 minutes (Fig. S47†). The isotopic distribution of this species (Fig. S48†) was altered compared to that of the Se-alkylated oligonucleotide, corroborating the loss of selenium. The other oligonucleotide species present likely resulted from side reactions with excess hydrogen peroxide. Elimination was expected to be quite facile, as the generated double bond of the enamine intermediate (Scheme 3) is in conjugation with the adjacent carbonyl.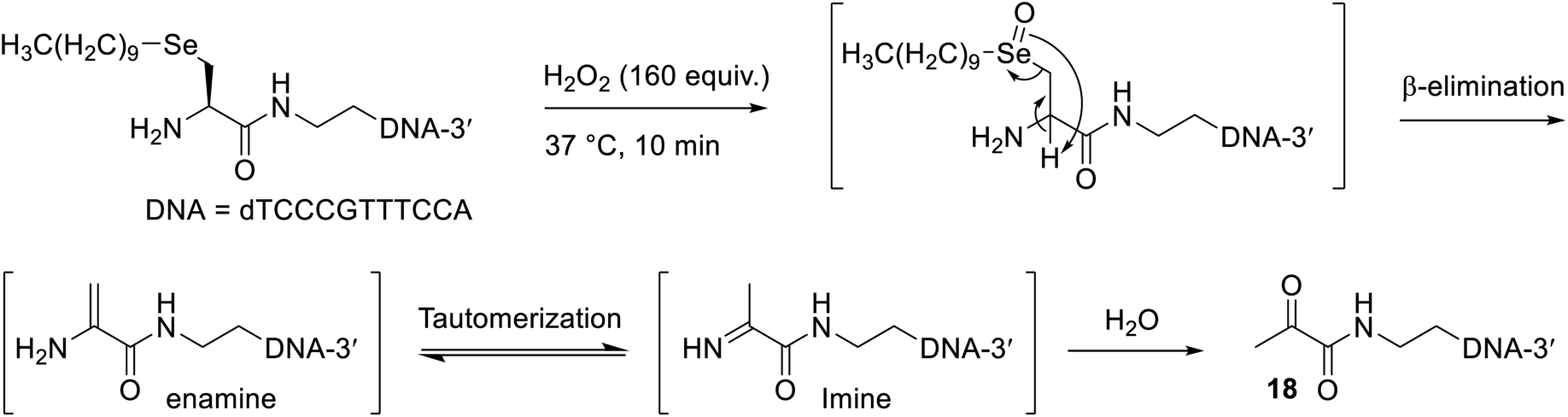 | ||
| Scheme 3 Proposed pathway for the hydrogen peroxide promoted dealkylation of oligonucleotide micelles resulting in dicarbonyl oligonucleotide 18. For clarity, only one monomer of the micelle is shown. The β-elimination mechanism is also shown for clarity. | ||
This finding is particularly valuable, as many pathologies are associated with high levels of reactive oxygen species, such as hydrogen peroxide. Consequently, the present work provides preliminary evidence that micelles, prepared as described with β-selenylated carbonyl groups, could potentially be used as oligonucleotide drug carriers that selectively break apart once adequately oxidised in diseased cells.
Experimental
All experimental procedures, HPLC chromatographs, MS, CD, and NMR spectra, UV thermal denaturation profiles, DLS measurements and TEM images are found in the ESI.†Conclusions
In summary, we have designed and synthesised selenide phosphoramidite 6, which was successfully installed at the 5′-end of two oligonucleotides by SPS. After deprotection, target diselenide oligonucleotides dimers could be generated in excellent yields. Remarkably, the prepared diselenide-bridged i-motif had a 22 °C higher Tm than the native, unmodified structure, where both had identical CD profiles. Two diselenide-bridged oligonucleotides, 7 and 14, were used to demonstrate the scope and efficiency of one-pot rDSL-deselenization for the rapid and efficient synthesis of POCs. The reaction manifold was shown to tolerate a proximal exocyclic amine as well as hybridised (i-motif) or unhybridised (single-stranded) oligonucleotides. The utility of the developed methodology was exemplified by synthesising micelles through either one-pot additive-free DSL-alkylation, generating POC micelles, or simply alkylation, generating oligonucleotide micelles. The alkylation conditions were also used to rapidly synthesise a fluorescently labelled oligonucleotide in high yield. Lastly, evidence for the H2O2-responsiveness of the oligonucleotide micelles was provided, where the C–Se bond was cleaved through an elimination pathway, producing dicarbonyl oligonucleotide 18.Taken together, the present study highlights the power of late stage chemoselective modification chemistry at a highly versatile functional group. This was exemplified through the rapid and efficient generation of a panel of POCs, POC micelles, oligonucleotide micelles and alkylated oligonucleotides from a single reactive 5′-functionality on an oligonucleotide precursor.
Given the speed and efficiency of the DSL methodology for POC synthesis, it stands to reason that this strategy will also find utility in the bioconjugation of other systems, for example, between nanoparticles and peptides/oligonucleotides. This is an especially enticing prospect, as carboxylic acids can be converted to the corresponding selenoester analogue in one synthetic step,48 and the preparation of carboxylic acid functionalised nanoparticles is already well established.49–51 Future work will focus on the production of a library of alkylated POCs, varying the amino acid content and length of the peptide as well as the alkyl chain length, in order to interrogate their encapsulation and H2O2 disassembly potentials. Furthermore, attempts are being made at growing crystals of the diselenide-bridged i-motif to provide high-resolution structural insights on how the 5′-functional group imparts the high thermal stability observed in this work.
Data availability
The datasets supporting this article have been uploaded as part of the ESI.†Author contributions
C. L. proposed the synthetic routes, carried out all synthesis and characterisation of the small molecules, oligonucleotides and oligonucleotide conjugates. C. H. carried out all synthesis and characterisation of the peptides. R. J. P. and C. J. W. supervised the project. All authors contributed to writing and editing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by grants from the Natural Sciences and Engineering Research Council of Canada (NSERC) (RGPIN-2017-06683) to C. J. W. and the Australian Research Council Centre of Excellence for Innovations in Peptide and Protein Science (CE200100012) to R. J. P. C. C. H. is the recipient of a Research Training Program PhD scholarship and John A. Lamberton Scholarship. C. L. is the recipient of a NSERC Alexander Graham Bell Canada Graduate Scholarship – Doctoral (CGS-D), a Fonds de recherche du Québec – Nature et technologies bourse de doctorat and a Miriam Aaron Roland Graduate Fellowship. We thank Dr Anne Noronha for assistance with solid-phase synthesis and Cynthia Messina for helpful discussions.References
- C. Rinaldi and M. J. A. Wood, Antisense oligonucleotides: the next frontier for treatment of neurological disorders, Nat. Rev. Neurol., 2018, 14(1), 9–21 CrossRef CAS.
- A. Fire, S. Xu, M. K. Montgomery, S. A. Kostas, S. E. Driver and C. C. Mello, Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans, Nature, 1998, 391(6669), 806–811 CrossRef CAS PubMed.
- C. Liczner, K. Duke, G. Juneau, M. Egli and C. J. Wilds, Beyond ribose and phosphate: Selected nucleic acid modifications for structure-function investigations and therapeutic applications, Beilstein J. Org. Chem., 2021, 17, 908–931 CrossRef CAS PubMed.
- C. Lorenz, P. Hadwiger, M. John, H. P. Vornlocher and C. Unverzagt, Steroid and lipid conjugates of siRNAs to enhance cellular uptake and gene silencing in liver cells, Bioorg. Med. Chem. Lett., 2004, 14(19), 4975–4977 CrossRef CAS.
- K. Nishina, T. Unno, Y. Uno, T. Kubodera, T. Kanouchi, H. Mizusawa and T. Yokota, Efficient in vivo delivery of siRNA to the liver by conjugation of α-tocopherol, Mol. Ther., 2008, 16(4), 734–740 CrossRef CAS.
- G. McClorey and S. Banerjee, Cell-penetrating peptides to enhance delivery of oligonucleotide-based therapeutics, Biomedicines, 2018, 6(2), 1–15 CrossRef PubMed.
- H. Yin, H. M. Moulton, Y. Seow, C. Boyd, J. Boutilier, P. Iverson and M. J. Wood, Cell-penetrating peptide-conjugated antisense oligonucleotides restore systemic muscle and cardiac dystrophin expression and function, Hum. Mol. Genet., 2008, 17(24), 3909–3918 CrossRef CAS.
- R. Abes, H. M. Moulton, P. Clair, S. T. Yang, S. Abes, K. Melikov, P. Prevot, D. S. Youngblood, P. L. Iversen, L. V. Chernomordik and B. Lebleu, Delivery of steric block morpholino oligomers by (R-X-R)4 peptides: structure-activity studies, Nucleic Acids Res., 2008, 36(20), 6343–6354 CrossRef CAS PubMed.
- X. Ming, M. R. Alam, M. Fisher, Y. Yan, X. Chen and R. L. Juliano, Intracellular delivery of an antisense oligonucleotide via endocytosis of a G protein-coupled receptor, Nucleic Acids Res., 2010, 38(19), 6567–6576 CrossRef CAS PubMed.
- M. R. Alam, X. Ming, M. Fisher, J. G. Lackey, K. G. Rajeev, M. Manoharan and R. L. Juliano, Multivalent cyclic RGD conjugates for targeted delivery of small interfering RNA, Bioconjugate Chem., 2011, 22(8), 1673–1681 CrossRef CAS PubMed.
- Y. Dang, C. van Heusden, V. Nickerson, F. Chung, Y. Wang, N. L. Quinney, M. Gentzsch, S. H. Randell, H. M. Moulton, R. Kole, A. G. Ni, R. L. Juliano and S. M. Kreda, Enhanced delivery of peptide-morpholino oligonucleotides with a small molecule to correct splicing defects in the lung, Nucleic Acids Res., 2021, 49(11), 6100–6113 CrossRef CAS.
- C. Lou, M. C. Martos-Maldonado, C. S. Madsen, R. P. Thomsen, S. R. Midtgaard, N. J. Christensen, J. Kjems, P. W. Thulstrup, J. Wengel and K. J. Jensen, Peptide-oligonucleotide conjugates as nanoscale building blocks for assembly of an artificial three-helix protein mimic, Nat. Commun., 2016, 7, 1–9 Search PubMed.
- C. Li, A. Faulkner-Jones, A. R. Dun, J. Jin, P. Chen, Y. Xing, Z. Yang, Z. Li, W. Shu, D. Liu and R. R. Duncan, Rapid formation of a supramolecular polypeptide-DNA hydrogel for in situ three-dimensional multilayer bioprinting, Angew. Chem., Int. Ed., 2015, 54(13), 3957–3961 CrossRef CAS PubMed.
- N. Stephanopoulos, R. Freeman, H. A. North, S. Sur, S. J. Jeong, F. Tantakitti, J. A. Kessler and S. I. Stupp, Bioactive DNA-peptide nanotubes enhance the differentiation of neural stem cells into neurons, Nano Lett., 2015, 15(1), 603–609 CrossRef CAS.
- J. C. Truffert, O. Lorthioir, U. Asseline, N. T. Thuong and A. Brack, Online solid-phase synthesis of oligonucleotide-peptide hybrids using silica supports, Tetrahedron Lett., 1994, 35(15), 2353–2356 CrossRef CAS.
- S. Soukchareun, G. W. Tregear and J. Haralambidis, Preparation and characterization of antisense oligonucleotide-peptide hybrids containing viral fusion peptides, Bioconjugate Chem., 1995, 6(1), 43–53 CrossRef CAS PubMed.
- F. Bergmann and W. Bannwarth, Solid-phase synthesis of directly linked peptide-oligodeoxynucleotide hybrids using standard synthesis protocols, Tetrahedron Lett., 1995, 36(11), 1839–1842 CrossRef CAS.
- J. Robles, M. Beltran, V. Marchan, Y. Perez, I. Travesset, E. Pedroso and A. Grandas, Towards nucleopeptides containing any trifunctional amino acid, Tetrahedron, 1999, 55(46), 13251–13264 CrossRef CAS.
- J. P. Bongartz, A. M. Aubertin, P. G. Milhaud and B. Lebleu, Improved biological activity of antisense oligonucleotides conjugated to a fusogenic peptide, Nucleic Acids Res., 1994, 22(22), 4681–4688 CrossRef CAS PubMed.
- J. J. Turner, A. A. Arzumanov and M. J. Gait, Synthesis, cellular uptake and HIV-1 Tat-dependent trans-activation inhibition activity of oligonucleotide analogues disulphide-conjugated to cell-penetrating peptides, Nucleic Acids Res., 2005, 33(1), 27–42 CrossRef CAS.
- C. H. Tung, M. J. Rudolph and S. Stein, Preparation of oligonucleotide-peptide conjugates, Bioconjugate Chem., 1991, 2(6), 464–465 CrossRef CAS.
- N. J. Ede, G. W. Tregear and J. Haralambidis, Routine preparation of thiol oligonucleotides: Application to the synthesis of oligonucleotide-peptide hybrids, Bioconjugate Chem., 1994, 5(4), 373–378 CrossRef CAS PubMed.
- J. G. Harrison and S. Balasubramanian, Synthesis and hybridization analysis of a small library of peptide-oligonucleotide conjugates, Nucleic Acids Res., 1998, 26(13), 3136–3145 CrossRef CAS.
- K. Gogoi, M. V. Mane, S. S. Kunte and V. A. Kumar, A versatile method for the preparation of conjugates of peptides with DNA/PNA/analog by employing chemo-selective click reaction in water, Nucleic Acids Res., 2007, 35(21), e139 CrossRef PubMed.
- A. Chotera, H. Sadihov, R. Cohen-Luria, P. A. Monnard and G. Ashkenasy, Functional assemblies emerging in complex mixtures of peptides and nucleic acid-peptide chimeras, Chem.–Eur. J., 2018, 24(40), 10128–10135 CrossRef CAS.
- S. M. Viladkar, Guanine rich oligonucleotide-amino acid/peptide conjugates: Preparation and characterization, Tetrahedron, 2002, 58(3), 495–502 CrossRef CAS.
- G. Cesarone, O. P. Edupuganti, C. P. Chen and E. Wickstrom, Insulin receptor substrate 1 knockdown in human MCF7 ER+ breast cancer cells by nuclease-resistant IRS1 siRNA conjugated to a disulfide-bridged D-peptide analogue of insulin-like growth factor 1, Bioconjugate Chem., 2007, 18(6), 1831–1840 CrossRef CAS.
- S. C. Alley, D. R. Benjamin, S. C. Jeffrey, N. M. Okeley, D. L. Meyer, R. J. Sanderson and P. D. Senter, Contribution of linker stability to the activities of anticancer immunoconjugates, Bioconjugate Chem., 2008, 19(3), 759–765 CrossRef CAS PubMed.
- C. J. Burrows and J. G. Muller, Oxidative nucleobase modifications leading to strand scission, Chem. Rev., 1998, 98(3), 1109–1152 CrossRef CAS.
- D. A. Stetsenko and M. J. Gait, Efficient conjugation of peptides to oligonucleotides by “native ligation”, J. Org. Chem., 2000, 65(16), 4900–4908 CrossRef CAS PubMed.
- N. J. Mitchell, L. R. Malins, X. Liu, R. E. Thompson, B. Chan, L. Radom and R. J. Payne, Rapid additive-free selenocystine-selenoester peptide ligation, J. Am. Chem. Soc., 2015, 137(44), 14011–14014 CrossRef CAS.
- J. Sayers, P. M. T. Karpati, N. J. Mitchell, A. M. Goldys, S. M. Kwong, N. Firth, B. Chan and R. J. Payne, Construction of challenging proline-proline junctions via diselenide-selenoester ligation chemistry, J. Am. Chem. Soc., 2018, 140(41), 13327–13334 CrossRef CAS.
- S. S. Kulkarni, E. E. Watson, B. Premdjee, K. W. Conde-Frieboes and R. J. Payne, Diselenide-selenoester ligation for chemical protein synthesis, Nat. Protoc., 2019, 14(7), 2229–2257 CrossRef CAS.
- X. Wang, L. Corcilius, B. Premdjee and R. J. Payne, Synthesis and utility of β-selenophenylalanine and β-selenoleucine in diselenide-selenoester ligation, J. Org. Chem., 2020, 85(3), 1567–1578 CrossRef CAS.
- T. S. Chisholm, S. S. Kulkarni, K. R. Hossain, F. Cornelius, R. J. Clarke and R. J. Payne, Peptide ligation at high dilution via reductive diselenide-selenoester ligation, J. Am. Chem. Soc., 2020, 142(2), 1090–1100 CrossRef CAS.
- C. Liczner, V. Grenier and C. J. Wilds, Reversible diselenide cross-links are formed between oligonucleotides containing 2'-deoxy-6-selenoinosine, Tetrahedron Lett., 2018, 59(1), 38–41 CrossRef CAS.
- J. Gallego, S. H. Chou and B. R. Reid, Centromeric pyrimidine strands fold into an intercalated motif by forming a double hairpin with a novel T:G:G:T tetrad: Solution structure of the d(TCCCGTTTCCA) dimer, J. Mol. Biol., 1997, 273(4), 840–856 CrossRef CAS PubMed.
- S. S. Panda, C. D. Hall, A. A. Oliferenko and A. R. Katritzky, Traceless chemical ligation from S-, O-, and N-acyl isopeptides, Acc. Chem. Res., 2014, 47(4), 1076–1087 CrossRef CAS.
- V. Swali, M. Matteucci, R. Elliot and M. Bradley, The stereospecific synthesis of 'orthogonally' protected lanthionines, Tetrahedron, 2002, 58(44), 9101–9109 CrossRef CAS.
- H. Saneyoshi, K. Seio and M. Sekine, A general method for the synthesis of 2'-O-cyanoethylated oligoribonucleotides having promising hybridization affinity for DNA and RNA and enhanced nuclease resistance, J. Org. Chem., 2005, 70(25), 10453–10460 CrossRef CAS PubMed.
- C. C. Hanna, S. S. Kulkarni, E. E. Watson, B. Premdjee and R. J. Payne, Solid-phase synthesis of peptide selenoesters via a side-chain anchoring strategy, Chem. Commun., 2017, 53(39), 5424–5427 RSC.
- S. Dery, P. S. Reddy, L. Dery, R. Mousa, R. N. Dardashti and N. Metanis, Insights into the deselenization of selenocysteine into alanine and serine, Chem. Sci., 2015, 6(11), 6207–6212 RSC.
- L. R. Malins, N. J. Mitchell, S. McGowan and R. J. Payne, Oxidative deselenization of selenocysteine: applications for programmed ligation at serine, Angew. Chem., Int. Ed. Engl., 2015, 54(43), 12716–12721 CrossRef CAS PubMed.
- N. Gour, D. Kedracki, I. Safir, K. X. Ngo and C. Vebert-Nardin, Self-assembling DNA-peptide hybrids: Morphological consequences of oligonucleotide grafting to a pathogenic amyloid fibrils forming dipeptide, Chem. Commun., 2012, 48(44), 5440–5442 RSC.
- W. Zhang and Z. Huang, Synthesis of the 5'-Se-thymidine phosphoramidite and convenient labeling of DNA oligonucleotide, Org. Lett., 2011, 13(8), 2000–2003 CrossRef CAS.
- N. Ma, Y. Li, H. F. Ren, H. P. Xu, Z. B. Li and X. Zhang, Selenium-containing block copolymers and their oxidation-responsive aggregates, Polym. Chem., 2010, 1(10), 1609–1614 RSC.
- J. L. Sloane and M. M. Greenberg, Interstrand cross-link and bioconjugate formation in RNA from a modified nucleotide, J. Org. Chem., 2014, 79(20), 9792–9798 CrossRef CAS.
- A. Temperini, F. Piazzolla, L. Minuti, M. Curini and C. Siciliano, General, mild, and metal-free synthesis of phenyl selenoesters from anhydrides and their use in peptide synthesis, J. Org. Chem., 2017, 82(9), 4588–4603 CrossRef CAS.
- C. C. You, M. De, G. Han and V. M. Rotello, Tunable inhibition and denaturation of α-chymotrypsin with amino acid-functionalized gold nanoparticles, J. Am. Chem. Soc., 2005, 127(37), 12873–12881 CrossRef CAS.
- Y. An, M. Chen, Q. Xue and W. Liu, Preparation and self-assembly of carboxylic acid-functionalized silica, J. Colloid Interface Sci., 2007, 311(2), 507–513 CrossRef CAS.
- R. K. Delong, C. M. Reynolds, Y. Malcolm, A. Schaeffer, T. Severs and A. Wanekaya, Functionalized gold nanoparticles for the binding, stabilization, and delivery of therapeutic DNA, RNA, and other biological macromolecules, Nanotechnol., Sci. Appl., 2010, 3, 53–63 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: All experimental procedures, HPLC chromatographs, MS, CD, and NMR spectra, UV thermal denaturation profiles DLS measurements and TEM images. See DOI: 10.1039/d1sc04937b |
| This journal is © The Royal Society of Chemistry 2022 |