
Digital light processing 3D printing with thiol–acrylate vitrimers†
Elisabeth
Rossegger
a,
Rita
Höller
a,
David
Reisinger
a,
Jakob
Strasser
a,
Mathias
Fleisch
a,
Thomas
Griesser
b and
Sandra
Schlögl
 *a
*a
aPolymer Competence Center Leoben GmbH, Roseggerstrasse 12, A-8700 Leoben, Austria. E-mail: sandra.schloegl@pccl.at
bInstitute of Chemistry of Polymeric Materials, Montanuniversitaet Leoben, Otto Glöckel-Strasse 2, A-8700 Leoben, Austria
First published on 14th December 2020
Abstract
Dynamic covalent bonds endow polymer networks with advanced functions such as self-healability, recyclability, malleability and shape memory. Currently, the most attractive dynamic networks are vitrimers, which rely on thermo-activated exchange reactions such as the catalyzed transesterification of hydroxyl ester moieties. However, the introduction of dynamic covalent bonds into 3D printable photopolymers is challenging, as commonly used transesterification catalysts are poorly soluble and compromise on cure rate and pot life of photocurable resins. Herein, a mono-functional methacrylate phosphate is presented as new transesterification catalyst, which overcomes these limitations and unlocks a new toolbox of photocurable vitrimers. Applied in thiol–acrylate vitrimer systems, the fast photopolymerization together with a high storage stability enables the successful additive manufacturing of precise 3D objects with features of 500 μm using bottom-up digital light processing (DLP). Once photo-cured, the dynamic thiol-click networks are able to rapidly undergo thermo-activated rearrangements of their network topology as shown by stress relaxation experiments. The DLP printing of soft active structures with triple-shape memory and thermal mendability is demonstrated. Its versatility makes this unique class of material an ideal candidate for 3D printing of structural and fast acting functional devices in soft robotics, biomedicine and electronics.
With the advent of additive manufacturing techniques (AMTs), digitalization and personalization have found their way into material science. Virtual 3D models are conveniently translated into physical objects by digitally slicing computer-aided designs and building 3D objects layer-by-layer.1 AMTs benefit from freedom in design and radically changed the way polymers are produced.2
Recently, soft active materials have been integrated within AMTs to precisely create polymer-based objects with additional/improved functionality, since they are capable of undergoing large elastic deformation in response to environmental stimuli.3 Currently, there are mainly two types of soft active materials being used: (i) hydrogels that swell when solvent molecules diffuse into the polymer network and (ii) shape memory polymers (SMPs) that are capable of fixing temporary shapes and recovering to the permanent shape upon an external stimulus such as temperature,4 magnetic fields,5 and light.6 Whilst hydrogels typically provide only a low modulus and also exhibit a slow response in a time scale of a few minutes, hours, and even days, 3D printing of SMPs is suitable for the manufacture of structural and fast acting devices.7 Examples of customized functional devices are actuators for soft robotics,8 self-evolving structures9 or electric and electronic devices.10
Another promising route to increase the functionality of additively manufactured objects is the 3D printing with thermosets comprising dynamic bonds, which are associative in nature. Associative covalent adaptable networks maintain their network connectivity at elevated temperature, since thermally induced bond breakage and reformation reactions occur simultaneously.11 Above the topology freezing transition temperature (Tv), the exchange reactions become significantly fast and induce a macroscopic flow of the polymer, which follows an Arrhenius trend analogous to silica-based glasses. Due to this unique behavior, Leibler and co-workers coined this class of networks vitrimers.12 Whilst the chemistry of vitrimers has recently been expanded to numerous materials and exchange reactions, the most intensively studied ones are still epoxy-based networks relying on reversible transesterification reactions.13 In these systems, the addition of an appropriate catalyst is crucial to accelerate thermo-activated bond exchange reactions. Commonly used transesterification catalysts for vitrimeric networks are Brønsted acids, organo–metallic complexes and organic bases.14 Based on the kinetics of the bond exchange reactions, the networks can be reprocessed, welded, reshaped and healed at temperatures well above the Tv.15
Shi and co-workers used thermo-activated transesterification to impart recycling properties in 3D printed parts.16 However, the printed objects suffered from a rather low resolution and poor surface quality due to the applied filament extrusion printing technique. In contrast, Zhang et al. transferred the concept to photo-curable resins printable by digital light processing (DLP).17 Compared to filament extrusion, DLP offers several advantages as it enables the fabrication of 3D objects with high resolution and surface quality and comparably high throughput rates.18 DLP is based on a layer-by-layer photopolymerization process, which is locally carried out in a vat containing a photo-curable resin formulation (Fig. 1a).1 During the printing process, each layer is illuminated all at once using a selectively masked light source comprising binary patterns presented by a digital micro-mirror device. Zhang and co-workers used a photo-curable resin formulation with hydroxyl-functional mono- and diacrylates, a Norrish Type I photoinitator and Zn(OAc)2 for catalyzing the bond exchange reactions, which rendered the network self-healable and reprocessable.17 However, this approach lacks from a versatility in network design as Zn(OAc)2 is insoluble in the majority of common acrylate monomers. Another well-known transesterification catalyst is triazabicyclodecene, which was applied by Bowman et al. for the preparation of covalent adaptable thiol–ene networks.19 However, photocurable resins containing triazabicyclodecene are not applicable to manufacture 3D objects with an adequate speed, as triazabicyclodecene acts as radical scavenger and retards radically induced photopolymerization reactions.
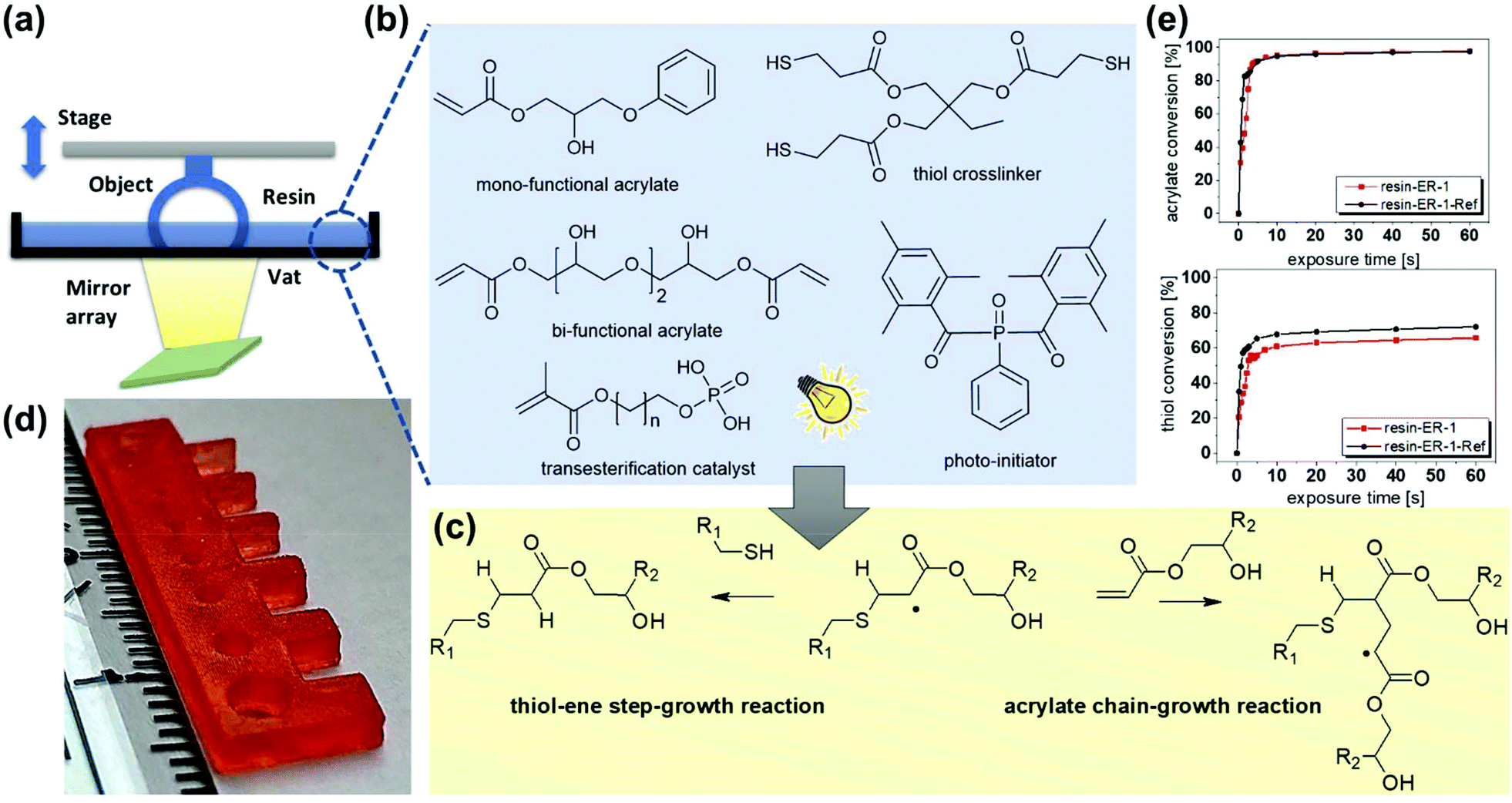 | ||
| Fig. 1 (a) Schematic representation of DLP process. (b) Monomers and transesterification catalyst used for the preparation of covalent adaptable thiol–acrylate photopolymers. (c) Schematic representation of the photo-curing reaction. (d) Photograph of a 3D printed test structure with a length of 2.6 cm. (e) Curing kinetics as obtained from FTIR data. The lines are a guide for the eye. | ||
Herein, we unlock a new toolbox of functional monomers suitable for the DLP printing of vitrimers by introducing an oligomeric methacrylate phosphate as efficient transesterification catalyst. The catalyst is liquid, easily soluble in a wide range of acrylate monomers and covalently incorporated into the network across its methacrylate group. Another key property is it inertness in thiol-click formulations. Whilst acrylate-based photopolymers are well known for their heterogeneous network structure and inferior mechanical properties (e.g. low impact resistance), the addition of functional thiols, acting as chain transfer agents, shifts the gel point towards higher conversions and reduces shrinkage stress.20 At higher thiol concentrations, the photopolymerization of acrylate monomers is dominated by a chain-growth mechanism (Fig. 1c) leading to improved mechanical properties and higher network homogeneity.21 However, prominent transesterification catalysts such as Zn(OAc)2, triazabicyclodecene or triphenylphosphine promote thiol-Michael addition reactions, which compromise on the storage stability of the resins.22 We observed fast gelation under dark conditions, which makes it impossible to print those formulations via DLP (Fig. S1, ESI†).
To demonstrate the salient features of the new catalyst, a photocurable thiol-click formulation (resin-ER-1) was prepared, which consisted of mono- and bi-functional acrylate monomers with –OH groups (Fig. 1b). A high number of –OH groups is beneficial as they facilitate exchange reactions in the photopolymer network.23 25 mol% thiol was added as crosslinker together with 5 wt% catalyst and 2 wt% phenylbis(2,4,6-trimethylbenzoyl)phosphine oxide as photoinitiator.
Rheological measurements revealed that the viscosity of resin-ER-1 amounted to 340 mPa s, directly after mixing, and it did not significantly change during a storage at room temperature for one week (415 mPa s). Along with a high storage stability, the formulation is characterized by a fast cure rate, which is crucial for the layer-by-layer build-up of 3D objects during DLP. The conversion of the acrylate and thiol groups was determined by FTIR spectroscopy, following the time-dependent depletion of the characteristic absorption bands of acrylate and thiol groups at 1635 and 2570 cm−1, respectively. FTIR spectra prior to and after UV exposure are shown in Fig. S2 (ESI†). The cure kinetics is comparable to the non-catalysed reference system (resin-ER-1-Ref) and maximum conversion of thiol and acrylate groups is observed upon 10 s UV exposure (Fig. 1e). The fast photopolymerization enabled the DLP printing of 3D objects with high precision as demonstrated by printing a test structure with various substructures (Fig. 1d). All features with their thickness decreasing from 3 mm to 500 μm were replicated successfully. Subsequently, discs with a diameter of 10 mm were printed for rheological experiments, to determine the stress relaxation of the network as a function of temperature (160–200 °C). Under the applied conditions, thermal degradation was avoided, as thermogravimetric analysis revealed a thermal stability of cured resin-ER-1 well above 200 °C (Fig. S3, ESI†). Moreover, a high mobility of the chain segments was ensured as the network's Tg amounted to 0 °C (DSC curve is shown in Fig. S4a, ESI†). Fig. 2a provides the time-dependent evolution of the relaxation modulus at 180 °C for both non-catalyzed and catalyzed systems. Resin-ER-1-Ref showed a slight stress relaxation, which is explained by the thermal release of volumetric shrinkage stresses arising during network evolution.24 In contrast, a very fast stress relaxation was observed for resin-ER-1, with 63% (1/e) of the initial stress being relaxed within 31.5 min. In contrast, lower crosslinked acrylate networks with Zn(OAc)2 require a 4 times longer relaxation time (120 min) under the same conditions.17 The stress relaxation is clearly temperature-dependent and indicates a rising bond exchange rate at increasing temperature.
 | ||
| Fig. 2 (a) Normalised stress relaxation curves of resin-ER-1 versus temperature in comparison to the non-catalysed resin-ER-Ref. (b) Arrhenius plot of resin-ER-1 derived from measured relaxation times. (c) Stress–strain curves of DLP printed dumbbell specimens of resin-ER-1 prior to and after a thermal annealing at 180 °C for 4 h. (d) Normalised stress relaxation curves of thiol–acrylate vitrimers obtained at 180 °C as a function of the catalyst content. (e) Schematic representation of thermo-activated exchange reactions in covalent adaptable thiol–acrylate photopolymers. | ||
Along with temperature, the stress relaxation kinetics of vitrimers is also governed by the amount of catalyst, with faster relaxation rates being observed at higher catalyst concentration.23 In further experiments, we gradually increased the amount of the oligomeric methacrylate phosphate in the thiol–acrylate resin from 5 to 15 wt%. The results show that the higher catalyst content indeed accelerates the stress relaxation kinetics (Fig. 2d). Whilst at 5 wt%, the thiol–acrylate network requires 31.5 min to reach 63% relaxation of the initial stress, the relaxation time amounts to 11.3 and 7.5 min in the presence of 10 and 15 wt% of the catalyst, respectively.
Whilst mineral phosphates are widely used heterogeneous catalysts for transesterifications in solution,25 the stress relaxation data clearly evidences that organic counterparts are able to efficiently catalyze transesterifications in solid polymer networks. Mono and diester of the phosphoric acid contain free –OH groups and are strong Brønsted acids, which are able to catalyse both esterifications and transesterifications.26 In acid-catalysed transesterifications, the oxygen of the carbonyl ester group is protonated, thereby leading to an increase of the electrophilicity of the adjoining carbon atom.27
Once protonated, the carbonyl group is more susceptible to a nucleophilic attack and forms a tetrahedral intermediate with an available –OH group in the network. A new ester bond is formed by subsequent proton transfer, departure of the leaving group and regeneration of the acidic catalyst (H+) by deprotonation.
Applying the Maxwell Model, the characteristic relaxation times (τ*) were determined as the time required to relax to 1/e of the initial stress at temperatures between 160 and 200 °C.28Fig. 2b demonstrates that the data satisfies the Arrhenius law, confirming the vitrimeric nature of the photopolymer. From the slope (m = −Ea/R) of the straight line fitted to the data, an activation energy (Ea) of 65.6 kJ mol−1 was obtained. The Tv value was derived by extrapolation of the fitted data to a relaxation time of 106 s and amounted to 59 °C.
Above Tv, the exchange reactions become macroscopically relevant and the network changes from an elastic solid to a viscoelastic liquid (Fig. 2e).12 Along with topological rearrangements, additional crosslink sites are formed by hydrogen bonding during prolonged treatment at 180 °C involving a decrease in –OH groups (FTIR spectra are shown in Fig. S5, ESI†) and a distinctive shift of the Tg from 0 to 20 °C (DSC curves are provided in Fig. S4, ESI†).29 The reduced availability of functional groups slows down the stress relaxation kinetics (Fig. S6, ESI†). However, topological rearrangements and additional crosslink sites lead to a substantial improvement of the mechanical properties of printed test specimens. After a thermal treatment at 180 °C for 4 h, both strain and stress increase from 23 to 47% and from 0.26 to 3.1 N, respectively (Fig. 2c).
Whilst thermally annealed acrylic dynamic networks suffered from a brittle behavior with a strain about 8%,17 resin-ER-1 benefits from an adequate stretchability, which is crucial for achieving large shape changes in 3D printed devices. Due to its dynamic bonds, resin-ER-1 features triple shape memory, as it is capable to undergo a controlled and active macroscopic deformation upon heating and programming the network above its two transition temperatures, Tg and Tv. As shown in Fig. 3a, the original/permanent shape of a printed and thermally annealed sample is changed by heating it above its highest thermal transition temperature, which is the Tv at 59 °C, and by applying an external force for deformation. The first temporary shape is fixed by cooling the sample to 40 °C, which is above the network's Tg and enables the programming of a second temporary shape, which is fixed by cooling the network to 0 °C. Subsequent heating of the sample facilitates a sequential recovery of the two shapes. A fast response time (40 s) is observed for the recovery of the second temporary shape, which is shown in video 1 (ESI†). The fast response time combined with the freedom in design makes these networks interesting candidates for the fabrication of customized active materials for soft actuator and soft robotics, as shown by the example of a grabber in Fig. 3b.
 | ||
| Fig. 3 (a) Photographs monitoring the triple-shape memory of DLP printed samples of resin-ER-1 using the Tv to fix the first temporary shape, and the Tg to fix the second temporary shape and sequential recovery of the shapes upon heating. (b) DLP printed gripper showing the potential of this new class of material for the customized production of structural and fast acting devices. (c) Thermally triggered healing of 3D printed test specimens at 180 °C for 4 h. (d) Stress–strain curves and photographs of DLP printed test specimens prior to and after a thermal mending at 180 °C for 4 h. (e) DLP printed test specimen for thermal mending experiments: (i) DLP printed test specimen with a circular-shaped hole in the centre and DLP printed circular shaped counterpart. (ii) Test specimen after fitting the circular-shaped counterpart in the hole and subsequent thermal treatment at 180 °C for 4 h. (iii) DLP printed control sample. | ||
The dynamic nature of the bonds and the macroscopic reflow of the network above Tv, endows the soft active materials with additional functions such as intrinsic healability. Broken parts of 3D printed structures can be conveniently healed by a thermal treatment at 180 °C (Fig. 3c). The healing efficiency was determined by performing uniaxial tensile tests of 3D printed and thermally annealed dumbbell test specimen taking the ratio of the tensile strength prior to and after healing. The corresponding stress–strain curves are depicted in Fig. 3d. Since proper aligning of the thin broken test bars was challenging, we printed samples with a circular-shaped hole in the centre (Fig. 3e). For the repair step, the circular-shaped counterpart was printed and fitted in the hole, followed by a thermal annealing at 180 °C for 4 h. After the repair, the boundaries between the two parts efficiently welded together and the original tensile strength (defect-free bar) could be fully recovered.
Conclusions
Summing up, a new transesterification catalyst was introduced, which radically expands the toolbox of monomers suitable for the DLP printing of vitrimeric photopolymers. Compared to commonly used catalysts, the mono-functional methacrylate phosphate is superior in terms of solubility and stress relaxation. Moreover, it is covalently incorporated into the network and does not compromise on shelf life or cure kinetics of thiol-click photopolymers, which paves the way towards the DLP printing of complex soft active devices with triple-shape memory and additional functions such as self-healability.Experimental section
Materials
The catalyst (Miramer A99) was obtained from Miwon Specialty Chemical (Korea). All other chemicals were purchased from Sigma-Aldrich and used as received.Preparation of resin-ER-1
2-Hydroxy-2-phenoxypropyl acrylate (50 mol%) was mixed with glycerol 1,3-diglycerolate diacrylate (25 mol%) and 5 wt% Miramer A99. 0.05 wt% Sudan II was added and the formulation was ultra-sonicated until the photoabsorber was dissolved. 2 wt% phenylbis(2,4,6-trimethylbenzoyl)phosphine oxide and 25 mol% trimethylolpropane tri(3-mercaptopropionate) were added and dissolved by stirring the formulation at room temperature.Characterization
Light-induced curing of resin-ER-1 was monitored by FTIR spectroscopy utilizing a Vertex 70 spectrometer (Bruker, USA). 16 scans were cumulated in transmission mode from 4000 to 700 cm−1 with a resolution of 4 cm−1 and the absorption peak areas were calculated with OPUS software. 1.5 μL of resin were drop-cast between two CaF2 discs and cured with a light emitting diode lamp (zgood® wireless LED curing lamp) comprising a power density of 3.3 mW cm−2 (λ = 420–450 nm).The viscosity of the resins was determined by using a modular compact rheometer MCR 102 from Anton Paar (Austria) with a CP60-0.5/TI cone (49.97 mm diameter and 1.982° opening angle). Each measurement was carried out with 1 mL resin at room temperature and a shear rate of 300 s−1. Thermal gravimetrical analysis was performed with a Mettler Toledo (USA) TGA/DSC thermogravimetric analyzer. The measurements were carried out under oxygen atmosphere by heating the sample from 23 to 900 °C with a heating rate of 10 °C min−1. Differential scanning calorimetry measurements were carried out with a Mettler-Toledo DSC 821e instrument (USA). A temperature program from −20 to 150 °C with a heating rate of 20 K min−1 was applied under nitrogen atmosphere. The Tg was calculated from the second heating run by using the midpoint in heat capacity. Stress relaxation experiments at temperatures between 160 and 200 °C were carried out on an Anton Paar Physica MCR 501 rheometer (Austria) with parallel plate geometry. The samples were equilibrated to the selected measurement temperature and the specified constant normal force of 20 N for 20 min. Subsequently, 3% step strain was applied and the decreasing stress was recorded over time. For the Arrhenius plot, three measurements were taken for each data point. Tensile tests were performed on a ZwickRoell (Germany) Z1.0 static materials testing machine with a crosshead speed of 250 mm min−1. Dumbbell specimens with the dimensions of 2 × 12.5 × 75 mm were 3D printed.
DLP 3D printing
3D printing was performed on an Anycubic Photon S printer (China) with a LED 405 nm light source. Two bottom layers were exposed for 20 s, whereas the other layers were illuminated for 8 s. The layer height was set to 50 μm with a building speed of 6 mm s−1 and a retracting speed of 1 mm s−1.Self-healing and shape memory experiments
For self-healing experiments, the control sample (30 × 10 × 1.5 mm), the sample with a hole and the corresponding disc (d = 5 mm) were prepared via DLP 3D printing of resin-ER-1. To demonstrate the self-healing ability, the printed disc was fitted inside the hole and all samples were heated to 180 °C for 4 h. Shape memory experiments were carried out with 3D printed grippers (d = 50 mm), which were heated to 180 °C for 4 h. The first shape was fixed by heating the sample to 80 °C for 2 h. For the second shape, the sample was cooled down to 40 °C, re-shaped and cooled down with an ice bath below room temperature. By heating to 80 °C, the first shape could be regained within 40 s. To restore the permanent shape, the sample was heated to 120 °C for 2 h.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research work was performed within the COMET-Module “Chemitecture” (project-no.: 21647048) at the Polymer Competence Center Leoben GmbH (PCCL, Austria) within the framework of the COMET-program of the Federal Ministry for Transport, Innovation and Technology and the Federal Ministry for Digital and Economic Affairs with contributions by Montanuniversitaet Leoben (Institute of Chemistry of Polymeric Materials). The PCCL is funded by the Austrian Government and the State Governments of Styria, Upper and Lower Austria.References
- S. C. Ligon, R. Liska, J. Stampfl, M. Gurr and R. Mülhaupt, Chem. Rev., 2017, 117, 10212–10290 CrossRef CAS.
- M. Nadgorny and A. Ameli, ACS Appl. Mater. Interfaces, 2018, 10, 17489–17507 CrossRef CAS PubMed.
- (a) A. S. Gladman, E. A. Matsumoto, R. G. Nuzzo, L. Mahadevan and J. A. Lewis, Nat. Mater., 2016, 15, 413–418 CrossRef PubMed; (b) Z. X. Khoo, J. E. M. Teoh, Y. Liu, C. K. Chua, S. Yang, J. An, K. F. Leong and W. Y. Yeong, Virtual Phys. Prototyping, 2015, 10, 103–122 CrossRef.
- A. Lendlein and S. Kelch, Angew. Chem., Int. Ed., 2002, 41, 2034 CrossRef CAS.
- A. M. Schmidt, Macromol. Rapid Commun., 2006, 27, 1168–1172 CrossRef CAS.
- J. Ryu, M. D'Amato, X. Cui, K. N. Long, H. Jerry Qi and M. L. Dunn, Appl. Phys. Lett., 2012, 100, 161908 CrossRef.
- M. D. Hager, S. Bode, C. Weber and U. S. Schubert, Prog. Polym. Sci., 2015, 49–50, 3–33 CrossRef CAS.
- Y. Mao, K. Yu, M. S. Isakov, J. Wu, M. L. Dunn and H. Jerry Qi, Sci. Rep., 2015, 5, 13616 CrossRef PubMed.
- D. Raviv, W. Zhao, C. McKnelly, A. Papadopoulou, A. Kadambi, B. Shi, S. Hirsch, D. Dikovsky, M. Zyracki, C. Olguin, R. Raskar and S. Tibbits, Sci. Rep., 2014, 4, 7422 CrossRef CAS PubMed.
- J. Lee, H.-C. Kim, J.-W. Choi and I. H. Lee, Int. J. Precis. Eng. Manuf.-Green Tech., 2017, 4, 373–383 CrossRef.
- C. J. Kloxin and C. N. Bowman, Chem. Soc. Rev., 2013, 42, 7161–7173 RSC.
- D. Montarnal, M. Capelot, F. Tournilhac and L. Leibler, Science, 2011, 334, 965–968 CrossRef CAS PubMed.
- J. M. Winne, L. Leibler and F. E. Du Prez, Polym. Chem., 2019, 10, 6091–6108 RSC.
- W. Alabiso and S. Schlögl, Polymers, 2020, 12, 1660 CrossRef CAS PubMed.
- (a) S. Kaiser, J. N. Jandl, P. Novak and S. Schlögl, Soft Matter, 2020, 16, 8577–8590 RSC; (b) S. Kaiser, S. Wurzer, G. Pilz, W. Kern and S. Schlögl, Soft Matter, 2019, 15, 6062–6072 RSC; (c) M. Giebler, C. Sperling, S. Kaiser, I. Duretek and S. Schlögl, Polymers, 2020, 12, 1148 CrossRef CAS PubMed.
- Q. Shi, K. Yu, X. Kuang, X. Mu, C. K. Dunn, M. L. Dunn, T. Wang and H. Jerry Qi, Mater. Horiz., 2017, 4, 598–607 RSC.
- B. Zhang, K. Kowsari, A. Serjouei, M. L. Dunn and Q. Ge, Nat. Commun., 2018, 9, 1831 CrossRef PubMed.
- B. C. Gross, J. L. Erkal, S. Y. Lockwood, C. Chen and D. M. Spence, Anal. Chem., 2014, 86, 3240–3253 CrossRef CAS PubMed.
- G. B. Lyon, L. M. Cox, J. T. Goodrich, A. D. Baranek, Y. Ding and C. N. Bowman, Macromolecules, 2016, 49, 8905–8913 CrossRef CAS.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- M. Sahin, S. Ayalur-Karunakaran, J. Manhart, M. Wolfahrt, W. Kern and S. Schlögl, Adv. Eng. Mater., 2017, 19, 1600620 CrossRef.
- (a) K. R. Rohit, S. M. Ujwaldev, K. K. Krishnan and G. Anilkumar, Asian J. Org. Chem., 2018, 7, 85–102 CrossRef CAS; (b) D. P. Nair, M. Podgórski, S. Chatani, T. Gong, W. Xi, C. R. Fenoli and C. N. Bowman, Chem. Mater., 2013, 26, 724–744 CrossRef.
- M. Hayashi and R. Yano, Macromolecules, 2019, 53, 182–189 CrossRef.
- J. Chen, S. Jiang, Y. Gao and F. Sun, J. Mater. Sci., 2018, 53, 16169–16181 CrossRef CAS.
- (a) F. Bazi, H. El Badaoui, S. Sokori, S. Tamani, M. Hamza, S. Boulaajaj and S. Sebti, Synth. Commun., 2006, 36, 1585–1592 CrossRef CAS; (b) K. Thinnakorn and J. Tscheikuna, Appl. Catal., A, 2014, 476, 26–33 CrossRef CAS.
- A. Streitwieser, C. H. Heathcock and E. M. Kosower, Introduction to organic chemistry, 4th edn, 1998 Search PubMed.
- (a) P. A. Alaba, Y. M. Sani and W. M. Ashri Wan Daud, RSC Adv., 2016, 6, 78351–78368 RSC; (b) E. Lotero, Y. Liu, D. E. Lopez, K. Suwannakarn, D. A. Bruce and J. G. Goodwin, Ind. Eng. Chem. Res., 2005, 44, 5353–5363 CrossRef CAS.
- M. Capelot, M. M. Unterlass, F. Tournilhac and L. Leibler, ACS Macro Lett., 2012, 1, 789–792 CrossRef CAS.
- S. Wang, N. Teng, J. Dai, J. Liu, L. Cao, W. Zhao and X. Liu, Polymer, 2020, 210, 123004 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: FTIR spectra and normalized stress relaxation curves of the thiol–acrylate vitrimer. See DOI: 10.1039/d0py01520b |
| This journal is © The Royal Society of Chemistry 2021 |