
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Structure dependent photostability of ITIC and ITIC-4F†
Laura
Ciammaruchi
 *a,
Osnat
Zapata-Arteaga
a,
Edgar
Gutiérrez-Fernández
b,
Jaime
Martin
bc and
Mariano
Campoy-Quiles
*a
*a,
Osnat
Zapata-Arteaga
a,
Edgar
Gutiérrez-Fernández
b,
Jaime
Martin
bc and
Mariano
Campoy-Quiles
*a
aInstitute of Materials Science of Barcelona (ICMAB-CSIC), Campus of the UAB, 08193, Bellaterra, Spain. E-mail: lciammaruchi@icmab.es; mcampoy@icmab.es
bPOLYMAT and Polymer Science and Technology Department, Faculty of Chemistry, University of the Basque Country UPV/EHU, Manuel de Lardizabal 3, 20018, Donostia – San Sebastián, Spain
cIkerbasque, Basque Foundation for Science, E-48011, Bilbao, Spain
First published on 7th October 2020
Abstract
Strong synthetic and engineering efforts have taken the efficiency of non-fullerene acceptor (NFA) based organic solar cells above 18% in a few years. Nonetheless, a deep understanding of the fundamental properties of this class of molecules is still missing. Here, we systematically investigated the morphological properties of two high efficient indacenodithienothiophene-based NFAs – namely ITIC and ITIC-4F – in order to correlate the hydrogen/fluorination substitutions with the materials structural and stability properties. We confirm that each NFA structurally evolves with increasing temperature into several polymorphs, identifying through spectroscopy their corresponding narrow temperature ranges. We demonstrate that the materials’ response to accelerated stress tests (ASTs) is both substitution and polymorph dependent. ASTs underlined that the most vulnerable molecular segment corresponds to the thienothiophene C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond along the central backbone, together with the CC linkage between the electron-rich donor and the electron-deficient acceptor moieties, with a degradation process triggered by oxygen and light. ITIC-4F showed lower oxidation capability and a higher bond strength retaining effect compared to ITIC. Lastly, the AST approach employed here allowed for the extrapolation of morphological and stability-related features within a high-throughput framework, and can be considered as a valuable methodological tool for future stability-related studies.
C bond along the central backbone, together with the CC linkage between the electron-rich donor and the electron-deficient acceptor moieties, with a degradation process triggered by oxygen and light. ITIC-4F showed lower oxidation capability and a higher bond strength retaining effect compared to ITIC. Lastly, the AST approach employed here allowed for the extrapolation of morphological and stability-related features within a high-throughput framework, and can be considered as a valuable methodological tool for future stability-related studies.
Introduction
Last decade has witnessed the potential of organic photovoltaics (OPV) as an efficient, lightweight, flexible, and semi-transparent solar technology fabricated through a series of low-cost processes1–3 – like deposition from solution by printing and/or coating techniques – which could replace standard and costly vacuum-based fabrication processes. These advantages come from the unique characteristics of the organic semiconductors used as the photoactive layer (PAL), typically being a conjugated electron-donor polymer or small molecule, in combination with a molecule based either on fullerene acceptors (FAs) or non-fullerene acceptors (NFAs). The latter, showing a broader solar absorption spectrum and the possibility to finely tune their energy levels, have allowed to break the 18% power conversion efficiency (PCE) benchmark for an OPV in ternary configuration.4 Morphology is a key parameter strongly influencing the OPV performance and stability, and due to the huge variety of existing NFAs, rather differing conclusions are being drawn at present, in terms of its impact on the device efficiency. In addition, record efficiencies of NFA-based OPV and an in-depth understanding of the basic properties related to such high-efficiency materials, barely go hand in hand. As an example, Ye et al.5 demonstrated that solvent additives like 1,8-diiodooctane (DIO), together with thermal treatments, would improve the NFA-based blend multilength scale morphology and induce a higher degree of molecular ordering and crystallinity, with a resultant high-efficiency OPV. Nevertheless, as a lesson learned from the fullerene-based OPV world, it is now well known that DIO leads to an accelerated device degradation.6,7 Therefore, an in-depth understanding of the basic morphological properties related to such novel highly efficient acceptors, and how these can ultimately impact the device efficiency and stability, seems essential.Among the most high-efficiency NFAs, indacenodithienothiophene (IDTT)-based ITIC (2,2′-[[6,6,12,12-tetrakis(4-hexylphenyl)-6,12-dihydrodithieno[2,3-d:2′,3′-d′]-s-indaceno[1,2-b:5,6-b′]dithiophene-2,8-diyl]-bis[methylidyne(3-oxo-1H-indene-2,1(3H)-diylidene)]]bis[propanedinitrile]) was first synthesized in 2015,8 and since then many ITIC derivatives were designed, achieving PCEs steadily growing over time.9–11 ITIC acceptor belongs to the family of acceptor–donor–acceptor (A–D–A)-type small molecules, in which a donor core, comprising aromatic fused rings with extended conjugation, is connected to the sides to electron-deficient end groups, which favor the intermolecular π–π overlap and enhance the carrier mobility (Fig. 1b, inset).
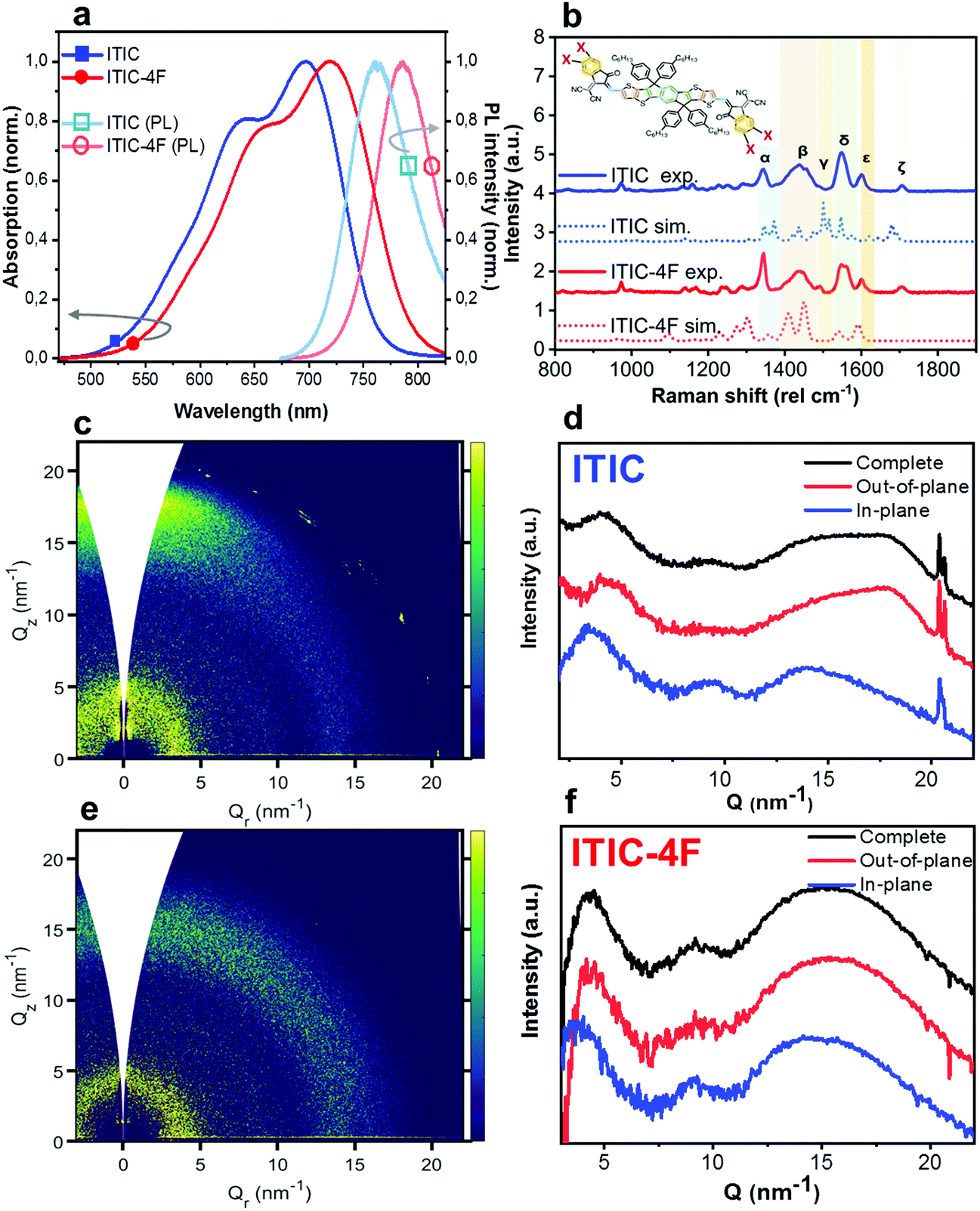 | ||
| Fig. 1 Characterization of as-cast NFAs. Absorption (filled symbols) and PL (unfilled symbols) spectra of ITIC (square) and ITIC-4F (circle) (a). Normalized experimental as-cast (solid lines) and DFT-calculated (dotted lines) ITIC (top 2) and ITIC-4F (bottom 2) spectra (b). Peaks and main vibrational modes assignments are discussed in the main text, and are guided by DFT simulations as well as comparison with the literature. GIWAXS 2D patterns and 1D in-plane and out-of-plane profiles for as-cast ITIC (c and d) and ITIC-4F (e and f). | ||
One of the most remarkable ITIC derivatives is the tetrafluorinated ITIC-4F, in which the end-group 2-(3-oxo-2,3-dihydro-1H-inden-1-ylidene) malononitrile (INCN) is symmetrically fluorine-substituted (Fig. 1b inset, XF). By enhancing the fluorination density, both the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) levels are pulled down simultaneously. While fluorination decreases the open-circuit voltage (VOC), it also induces an enhancement in the short-circuit current (JSC) by contracting the NFA optical bandgap, therefore compensating for the VOC loss.12–14 Fluorine addition also showed to improve the acceptor's inter- and intra-molecular interactions. As well, inclusion of new moieties at the ITIC end-groups can significantly change its temperature-dependent morphology and crystallinity.15 Since thermal treatments are some of the essential processes related to OPV fabrication, a proper knowledge of the relationship between ITIC fluorination and its temperature-dependent structural and optical properties becomes crucial.
While new NFA materials are constantly being designed,16,17 the majority of research groups is mostly focused on the search for the best achievable efficiencies. Top performing but unsuitable (i.e. unstable) materials would, however, not make for a good “real” solar device. In this respect, whereas the scientific problem related to the stability and degradation of standard (fullerene-based) OPV has opened up specific research pathways and is being steadily tackled,18–21 the same debate is starting to arise only now around NFA-based OPV. Interestingly, some disagreement is emerging on whether NFA-based OPVs show worse or better stability compared to their fullerene-based counterparts, and on which is the most relevant factor triggering instability. (Part of such discrepancy could likely come from the quite different testing conditions applied by each research group.) Zhao et al.22 reported joint superior efficiency and thermal stability for OPVs based on PBDB-T poly[(2,6-(4,8-bis(5-(2-ethylhexyl)thiophen-2-yl)-benzo[1,2-b:4,5-b′]dithiophene))-alt-(5,5-(1′,3′-di-2-thienyl-5′,7′-bis(2-ethylhexyl)benzo[1′,2′-c:4′,5′-c′]dithiophene-4,8-dione)] donor and ITIC acceptor, compared to the PBDB-T:PCBM-based counterpart. In contrast, Brabec's group23 showed only similar photoconversion efficiency (PCE) aging trends for PCBM- and for two specific ITIC derivatives- (i.e. fluorine and thiophene substituted ITIC) based cells, owing to the lack of conjugation breaking of the latter upon light soaking. These authors explained the poorer photostability results of the other ITIC derivative-based OPVs tested (ITIC, ITIC-M) in view of the increased number of energetic traps generated upon the NFA reorientation. In parallel, also Nutifafa et al.24 demonstrated the higher photostability of PCBM-based devices over the ITIC-based ones. Nevertheless, they discarded a trap-assisted recombination as the main reason behind the observed photodegradation, instead a mobility loss upon light exposure was identified as the main contributor to the difference in the OPV photodegradation. In any case, the NFA blending with a donor polymer seems to slow-down/alter the degradation pathway of the bare NFA. ITIC-based OPV instability under light soaking was additionally verified by Park and Son,25 but in this case the NFA/zinc oxide interface was identified as the most chemically vulnerable cell's portion, and the need for stable aromatic structures was underlined in order to achieve an acceptable device stability. In the same spirit, Guo et al.26 proved that introducing stabilizers, like nickel chelate S6, would improve the photo-oxidation stability of many NFAs. Whereas the environmental molecules interaction with NFAs can be mitigated with the introduction of anti-oxidants, Kim's group27 investigated how the molecular structure of two different NFAs, i.e. o-IDTBR and IDFBR, could influence their photostability in air. They found a three-phase process for which light and ambient contact triggered an initial conformational change, followed by fragmentation and chromophore disruption, lastly closed by a complete photo-bleaching. Interestingly, the less planar IDFBR showed worse degradation rate with respect to the more crystalline o-IDTBR.
From the literature results presented above it is apparent that there is a lack of general understanding of the stability at the material and device level. A more fundamental understanding of the interaction between the bare NFA morphology and its chemical structure (specific for each NFA family) with its stability under light and/or air, would surely help to disentangle the different instability sources at the device level. Therefore, in here we report a study on thin films of two NFAs, namely ITIC and ITIC-4F, investigated using photoluminescence (PL), absorption and Raman scattering. First we evaluate the annealing temperature effect on the structural and optical properties of the two materials, comparing the influence of fluorination on the type of polymorph as well as on the phase transition temperatures. Moreover, we discuss how the specific end-group substitution affects stability of the NFA polymorphic states, in response to conditions of accelerated stress tests (ASTs). We find that the photostability of ITIC is markedly dependent on the specific polymorphic phase and the fluorine decoration. Fluorination is able to slow down the molecule degradation induced via AST, likely due to a stronger chemical stability inducing a retaining effect. Finally, we point out that this study has been carried out by introducing annealing temperature gradients within the very same samples, thus we assess the NFA morphological characteristics – as well as the AST effects on them – in a high-throughput screening fashion. The advantage of this approach in stability studies is two-fold, as it allows for a significant saving of lab time and material use.19,28–30
Experimental section
Materials
ITIC and ITIC-4F acceptors were purchased from 1-Material, ZnO nanoparticles dispersion was acquired from Avantama, while indium tin oxide (ITO)-covered substrates were purchased from Ossila.Samples preparation
In order to produce an as realistic as possible PV interphase, a uniform ZnO layer was blade coated on the ITO-substrate, before the NFA deposition. ZnO blade coating was performed in air, using an automatic Zehntner ZAA 2300 with an aluminum applicator Zehntner ZUA 2000.29 After the deposition, samples were annealed in ambient conditions at 105 °C for 30 minutes. Both acceptors were dissolved in chlorobenzene (CB) in a concentration of ∼13 mg mL−1. After an overnight stirring at 80 °C and 1000 rpm, the acceptor materials were blade coated inside a nitrogen-filled and dry glove box, using a second blade coater equipment of similar brand and model, kept at 80 °C. According to the selected conditions of speed (70 mm s−1) and blade gap (200 μm), we obtained films roughly 70 nm thick. After deposition, NFA-covered samples were thermally annealed inside the same glove box for 30 minutes within the range (90–250 °C), using a calibrated Kofler bench (Wagner & Munz). This type of hot stage produces a controlled temperature gradient which is then transferred to the sample, enabling the production of films with a lateral thermal annealing gradient.28Characterization
UV-vis absorption spectra were recorded using a Hitachi U-3000 spectrophotometer. Optical microscopy images were obtained using an OPTIKA B-600 MET microscope in crossed polarizer configuration. Raman scattering and PL signals were acquired using a WITec alpha 300 RA+ confocal Raman setup, coupled to an Olympus objective with 10× magnification. 488 nm and 633 nm centered lasers were employed for Raman and PL measurements, respectively. For The 488 nm-based AST on annealed samples, several measurements at different powers were performed on nominally similar samples, in order to plot the Raman features evolution against the dose. The laser powers used were 0.25 mW, 0.50 mW, 0.75 mW, 1 mW, and 1.5 mW. For 633 nm-based PL measurements, we fixed the power to 0.25 mW. All raw data were collected and treated using WITec Project FIVE and the Origin 2019 pieces of software, respectively. In order to make a rough estimation of the power density impinging on the samples during ASTs, we considered to have a laser spot with an approx. 20 μm radius. Remembering the power density definition (W m−2), for a 0.25 mW power we calculate a power density of ≈1.2 × 105 W m−2, while for the maximum 1.5 mW power used, we get ≈1.2 × 106 W m−2. GIWAXS experiments were conducted at the BL11 NCD-SWEET beamline at ALBA Synchrotron Radiation Facility (Spain). The thin films were typically exposed for 1–5 s to an X-ray beam with a wavelength of 0.998 Å at an incidence angle 0.12° to ensure surface sensitivity. 2D scattering patterns were collected by employing a WAXS LX255-HS detector from Rayonix (pixel size = 44 × 44 μm2). DFT calculations were performed using the ORCA ab initio DFT and semi-empirical self- consistent-field molecular orbital theory package.73 Both geometry and Raman calculations were run using the long-range separated ωB97X-D3 DFT functional together with Ahlrichs’ triple-split-valence basis set augmented by polarization functions (TZVP) along an auxiliary def2-TZVP basis set. Fully optimized and equilibrium geometries were validated via force constant calculations (no imaginary frequencies were found).Results and discussion
Characterization of as-cast ITICs
Thin films of NFAs were deposited by blade coating on ZnO/ITO substrates (see experimental section for details). The as-cast films were first characterized by UV-visible optical absorption, PL and Raman spectroscopy techniques, and representative spectra of both ITIC and ITIC-4F are shown in Fig. 1. UV-vis spectra (Fig. 1a) of both ITICs exhibit a stronger 0–0 transition at higher wavelengths and a less intense 0–1 transition at shorter wavelengths. Fluorine substitution in the INCN end group enhances the electron push–pull effect, and is the origin of the slightly red-shifted UV-vis absorption and PL emission onsets compared to the non-fluorinated ITIC (Fig. 1a). On the other hand, experimental Raman spectra (Fig. 1b) of both ITIC types show a general match in their main vibrational modes. In fact, the fluorine substitution in the ITIC end group – which should correspond to a C–F Raman spectral feature falling between 500 cm−1 and 800 cm−1 – could be detected only by using laser powers of ∼200 mW, i.e. at least two order of magnitude higher than the maximum one employed in this work.31,32 We used DFT simulations (Fig. 1b, dashed) to assign the six strongest vibration modes falling between 1300 cm−1 and 1700 cm−1 at different molecular sites. For detailed assignment see Table 1.| Raman shift (cm−1) | Assignment (main vibration) |
|---|---|
| ∼1345 (α) | C–C & C–H (INCN group) |
| ∼1440 (β) | CC (thiophene) |
| CC (alkenes) |
|
| C–H linkage (IDTT and INCN) | |
| ∼1490 (γ) | C–H central ring |
| ∼1550 (δ) | CC (IDTT central benzene) |
| ∼1600 (ε) | CC (phenyl INCN) |
| ∼1705 (ζ) | CN linkage (INCN) |
Two-dimensional Grazing Incidence Wide Angle X-ray Scattering (2D GIWAXS) patterns for both as-cast ITICs (Fig. 1c–f) show poor molecular order, suggesting largely low-crystalline structures. Narrow peaks near Q = 20 nm−1 come from the silicon substrate scattering.
AST on as-cast ITICs
Having realized the initial characterization of both fluorinated and non-fluorinated ITICs in terms of their main structural, optical and vibrational features, we wanted to understand how fluorination would influence the NFA stability. For this, we first performed in situ photodegradation of as-cast ITICs thin-films, using the lasers’ excitations as a way to accelerate the degradation process. The experimental setup we used allowed us to collect real-time Raman and PL signals as a function of the laser irradiation time.27We used a monochromatic 488 nm (Raman) and 633 nm (PL) lasers to perform AST with power densities between two and three orders of magnitude higher than the standard AM1.5G (details in Experimental section). Fig. 2a depicts the integrated PL intensity dependence of as-cast ITICs upon degradation time. In the inset, the spectral evolution of normalized PL spectra selected at specific degradation points, is represented.
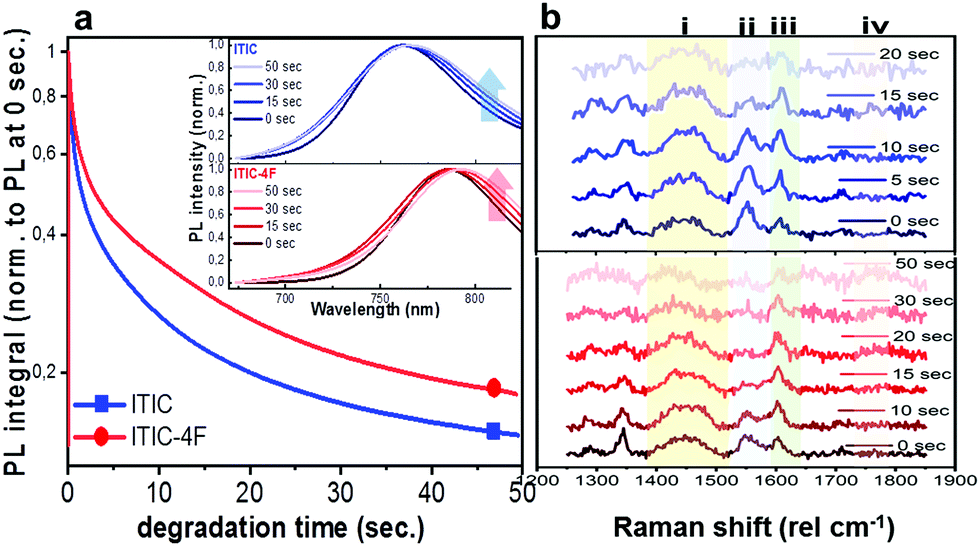 | ||
| Fig. 2 AST on as-cast ITICs. In situ integrated PL intensity decay of as-cast ITIC (square) and ITIC-4F (circle) upon 633 nm laser excitation. Inset: Normalized PL spectra taken at different degradation times, for as-cast ITIC (upper panel) and ITIC-4F (lower panel) (a). In situ Raman spectra of as-cast ITIC (upper panel) and ITIC-4F (lower panel), corresponding to different degradation times upon 488 nm excitation (b). | ||
Both materials show heavy PL quenching over time, with ITIC-4F displaying a slight red-shift over the annealing time (from 785 nm to 793 nm). With increasing illumination time, the main peaks Raman intensity signal decreases (Fig. 2b, taken at 0.5 mW), suggesting a chromophore loss during the photo-oxidation process.
The PL quenching intensity has a direct relationship with the density of quenching sites33 and for samples measured in ambient, air photo-oxidation is known to induce a rapid increase of such density.34 In fact, active radicals can be often formed as a result of photosensitization in the presence of oxygen, metal catalysts (ITO/ZnO) and other residues25,35,36 Also other mechanisms can trigger photochemical bleaching, even in the absence of oxygen.26 In our study, the higher PL intensity signal retained by ITIC-4F at the end of the experiment compared to ITIC, is an indication of a higher stability to photo-oxidation. This can be due to the fluorine-substituted end groups favoring F–S and F–H interactions. This hypothesis seems aligned with fluorine substituent being capable of forming electrostatically driven and stabilizing F–π noncovalent interactions with aromatic molecular species, in donor:acceptor blends.37,38
Alternatively, the slight different energy levels could also be playing a role. In particular, the LUMO of ITIC-4F is −4.07 eV,39 compared to −3.92 eV for ITIC.40 The former is farther from the electron affinity of molecular oxygen (−3.75 eV), thus it is expected to result in a more stable material, as it was very recently shown for other NFA based blends.41In situ Raman-based AST were realized under several powers/exposure time conditions, with the intention of establishing a relationship between degrading trend of relevant peaks and dose intake (defined as power density![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) × exposure time). For each laser power used, a degradation time-dependent Raman spectrum was first produced. Subsequently, in order to perform a more in-depth spectral analysis, time-dependent integrated intensities for peaks i, ii, iii and iv were computed. Fig. 3a and b shows combined data representing the degrading trends for peaks i, ii and iii, against the dose intake. Each panel is a collection of measurement points realized at different powers (0.25 mW – 0.50 mW – 0.75 mW – 1.0 mW – 1.5 mW).
× exposure time). For each laser power used, a degradation time-dependent Raman spectrum was first produced. Subsequently, in order to perform a more in-depth spectral analysis, time-dependent integrated intensities for peaks i, ii, iii and iv were computed. Fig. 3a and b shows combined data representing the degrading trends for peaks i, ii and iii, against the dose intake. Each panel is a collection of measurement points realized at different powers (0.25 mW – 0.50 mW – 0.75 mW – 1.0 mW – 1.5 mW).
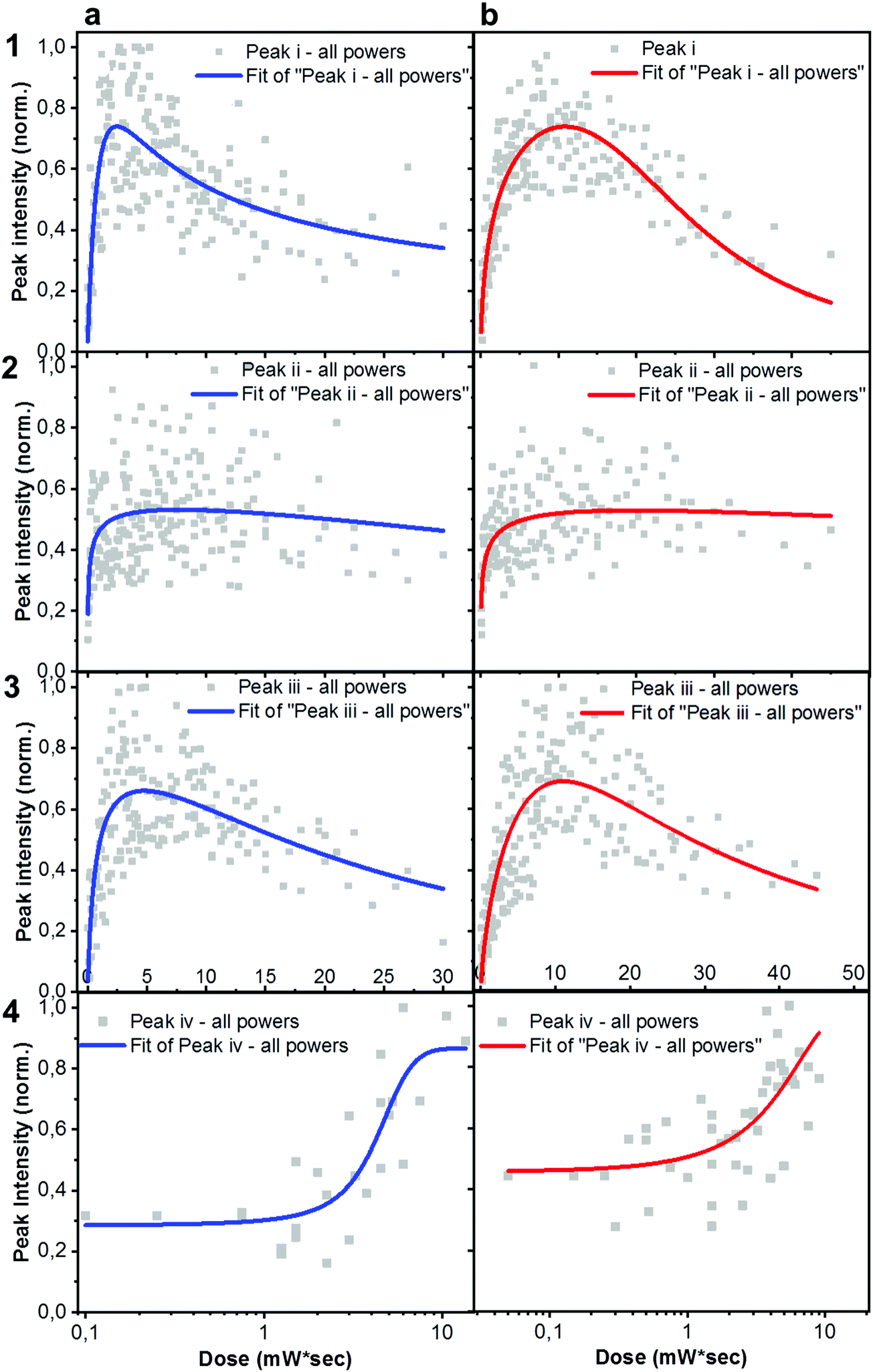 | ||
| Fig. 3 Detailed Raman analysis of peaks evolution upon AST. Normalized integrated intensity of i (〈1450〉 cm−1), ii (〈1550〉 cm−1), iii (〈1600〉 cm−1) and iv (〈1750〉 cm−1) Raman peaks, extracted from Raman time series plots as a function of dose, for as-cast ITIC (column a) and ITIC-4F (column b) upon 488 nm excitation. | ||
Plots represented in Fig. 3a and b and related to peaks i, ii and iii were best fitted with a biphasic Hill equation, while peak iv was best fitted by using a dose–response curve. Despite certain scattering in the data points – which we ascribe to possible local non-linear heat-related effects – general tendencies can still be inferred the increasing dose intake. Raman measurements confirm that ITIC-4F related peaks double the times taken by ITIC to reach half of their maximum intensity under constant 488 nm laser illumination, also in line with PL results showing a more stable ITIC-4F under 633 nm laser degradation.
In principle, ITICs molecules offer various potentially weak chemical points, like the alkene- and the cyclopentadiene- chain-based units, just to name a few. Our experimental results (Fig. 3), together with DFT-based peak identification, allow us spot out which – out of the many possible reaction sites – are actually the most likely degrading candidates. Indeed, the main vibronic Raman modes suffering intensity drop correspond to the double C bond on the thienothiophene along the central backbone, together with the CC linkage between the electron-rich donor and the electron-deficient acceptor moieties, (labeled i); the CC stretching of the benzene central ring (together with the double C bond on the thiophene) (ii), the double C of the end-group fused phenyl ring (iii).
Out of the three peaks monitored, peak i shows the fastest decay curve, followed right after by peak iii, and eventually peak ii. Therefore, ASTs seem to identify the weakest molecular site in correspondence with the CC linkage at the thiophene bonds, while the benzene central ring (CC stretching mode of the central ring) appears as the most stable NFA site. Under our experimental settings, a photochemically induced autoxidation yielding to the formation of carbonyl compounds, seems the most likely pathway for the NFAs degradation and the consequent bonds intensity loss, rather than a chain break due to direct photon impact. In fact, the UV light component of the experimental conditions is practically absent. Second, the acceptors’ absorption in that region is almost absent, and cannot be the trigger of any significant chromophores break.42 In this framework, we suggest the autoxidation cycle as the main NFAs degrading process, via the formation of free radicals.42 This process can be initiated by heat, radiation and oxygen presence, as well as any metal catalyst residues coming, for example, from the synthesis process of the material, or from the conductive substrate itself.42,43 Therefore, during the in situ AST oxygen, light and metal catalyst residues (if any) will all contribute to trigger the formation of free NFA radicals (R˙) through a charge transfer complex between the electron rich donor sites (alkene and cyclopentadiene based units) and O2. Such radical cations (R˙) are, in turn, highly reactive and short-lived species which may rapidly react with triplet ground state oxygen, to create new oxygen-centered reactive species, including peroxy radicals and hydroperoxides (O2 + R˙ → ROO˙ + RH → ROOH + R˙) as well as other fragment species (H2O, H2, H2O2). The hydroperoxides (ROOH), being highly reactive themselves, can also create new free radical species, such as hydroxy and alkoxy radicals (ROOH → ˙OH + RO˙). Since the radicals can keep reacting with alkenes to generate other radical species, the radical cation chain reaction will continue.44 We claim that under the specific experimental conditions of light and oxygen exposure employed in this study, the radical cation chain reaction can easily initiate, as the high HOMO level of alkenes can facilitate the electron transfer from alkenes to oxygen. Other mechanisms involving the presence of photosensitized production of singlet oxygen may also contribute to degradation of organic photovoltaic materials, as recently reported by Turkovic et al.45 On the other side, fluorine substitution in the end-group of the ITIC molecule increases the non-covalent interactions with the consequent introduction of a favored cell packing.46 Moreover, fluorination lowers the HOMO/LUMO levels, increases the electron affinity47 and enhances the molecular electron-pulling effect, with the result of a more efficient delocalization of the electron cloud over the electron-withdrawing site, and a reduced oxidative effect. Similar results on fullerene- and non-fullerene based blends have inversely correlated the higher polymer photobleaching of the photo active layers, with smaller electron affinity.41,48 Moreover, the oxidative cleavage of alkenes to carbonyl compounds has been already reported,49 even in the presence of sole molecular oxygen.44,50
An in-depth analysis of the collected Raman spectra readily reflects our claim, not only by revealing the CC bond break under the in situ degradation conditions (Fig. 3, i), but also with the raise of a new broad vibrational mode in the (1700–1850 cm−1) range (Fig. 3, iv and Fig. S7, ESI† for Raman spectra showing the raise of new vibrational feature), commonly assigned to the carbonyl stretching mode.51,52 The integrated peak area of this new feature increases almost twice as much for ITIC than for ITIC-4F, confirming the higher oxidation capability of the former under the same degrading conditions. Also, ITIC-4F starts reacting/oxidizing after receiving a slightly higher dose intake, compared to non-fluorinated ITIC, and the overall growth process appears much faster for ITIC than for ITIC-4F. Due to the same mechanism, we also explain the almost doubled times for the ITIC-4F Raman modes to reach the same degradation level upon similar dose amounts received, with respect to ITIC (Fig. 3a and b, i–iii).
It has been recently shown53 that, when ITIC is put in a basic environment, OH− anions can react as nucleophiles and attack the linkage between D and A moieties of the NFA, with the direct consequence of a conjugation break and the alteration of the charge transfer properties of the chain. These results provide extra proof of the ITIC molecule weakness mostly localized in the CC linkage between D and A groups. In the same work, Zhu et al. also verified that while the photovoltaic performances of OH− added ITIC devices hugely dropped compared to the OH− free reference – with short circuit current (JSC) and efficiency (PCE) only retaining 5% and 2% of their reference values – OH−-added ITIC-4F based solar cells held almost 50% and 15% of their original JSC and PCE.
Previous experiments have already shown that NFAs are responsible for the largest fraction of the active layer degradation in NFA-based OPV devices.25 Indeed, we confirm that under degradation conditions similar to those reported in this work, the typical donor polymer PBDB-T degrades more than twice as slow as ITIC (see Fig. S10, ESI†). Therefore, in this work we narrow our focus on the NFAs stability.
Exploring ITICs phases to understand stability
As already discussed, fluorination results in changes in the energy levels, and thus in the single molecule chemical stability, as well as in the solid state packing. Which of these two effects dominates? The NFA morphology evolves upon heating, which is a common condition during solar cell fabrication and actual operation.54,55 Therefore, we believe that understanding ITIC thermal evolution upon annealing is a valuable way to infer on its structural stability, as well as to better interpret the differences highlighted by the AST protocols (Fig. 3). It is important to stress here that, throughout this work, all measurements were realized at room temperature (RT), once the materials underwent thermal annealing and subsequent cool-down in inert atmosphere. We consider this experimental condition especially meaningful in order to correlate to typical processing conditions in OPVs. Moreover, it has been recently pointed out that the resulting ITIC film structure of annealed samples compares well with in situ temperature measurements.15Annealing temperature (Ta)-dependent UV-vis absorption and PL emission contour maps of both ITICs (Fig. 4a and b) highlight the presence of four main regions with different solid-state microstructures, according to the scanned temperature range. Each region is clearly material-dependent. Starting from literature results56 relative to the determination of the two main ITIC's polymorphic states – below and above its glass transition temperature (Tg ∼ 180 °C) – we were able to further break-down and distinguish such temperature-related phases of the materials. Indeed, within the T range corresponding to the low-temperature polymorph (i.e. polymorph I* and I**), the UV-vis main absorption peak (A0–0, Fig. 4a) shows an intensity increase up to Ta ∼ 100 °C. After 100 °C, A0–0 experiences a drop until 150 °C. For Ta ∼ 150 °C, the A0–0 intensity sharply increases again and reaches its maximum. While the A0–0 peak position does no show a substantial shift in such T range (Fig. 5a), its FWHM (Fig. S1, ESI†) broadens and decreases between 100 °C and 160 °C, before reaching a relative minimum for Ta ∼ 180 °C. This suggests that within the temperature region of polymorph I, the material passes through a region of looser (I*, Fig. 4a) and one of stronger (I**, Fig. 4a) molecular order, with ∼150 °C as the temperature turning point. Together with optical microscopy data (Fig. S2, ESI†), our results match especially well with Muller's group finding about ITIC capability to start a diffusion-limited crystallization process, once annealed at temperatures ∼145 °C and well below its Tg (Fig. S2b, ESI†). As well, the A0–0/A0–1 ratio trend seen in Fig. 5b (and inset) endorses this explanation, as a lower (higher) level of interchain coupling would correspond to a less (more) ordered crystalline structure, resulting in a higher (lower) A0–0/A0–1 value.57,58
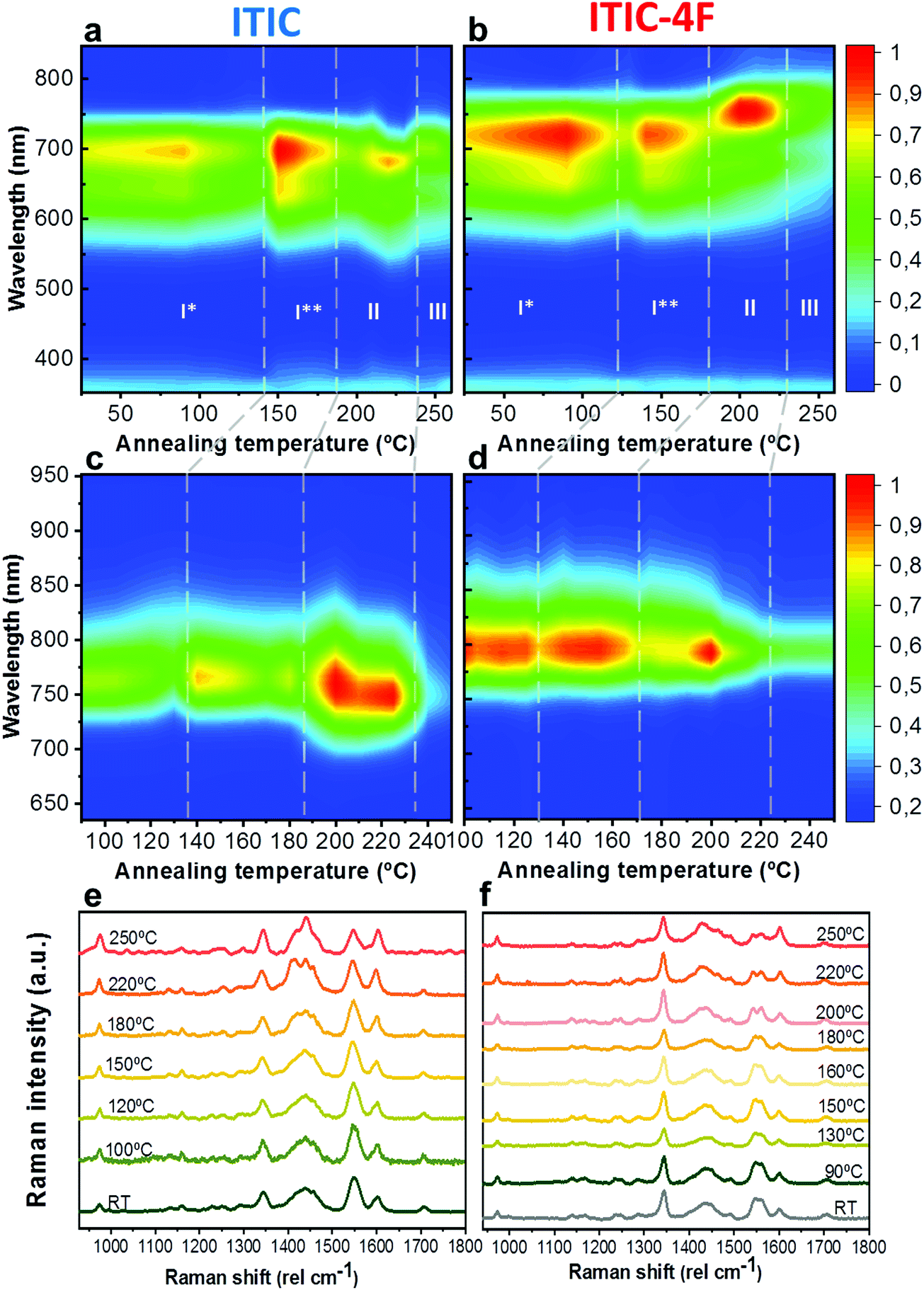 | ||
| Fig. 4 UV-vis, PL and Raman evolution upon materials annealing. Phase-dependent ITIC (left column) and ITIC-4F (right column) UV-vis absorption contour maps (top row, a and b), PL contour maps (middle row, c and d) and normalized Raman spectra (bottom row, e and f) as function of the annealing temperature. Four regions are identified in the contour maps for each NFA, according to their polymorphic phase. All measurements were realized at room temperature, after the materials underwent annealing and subsequent cool-down in inert atmosphere. | ||
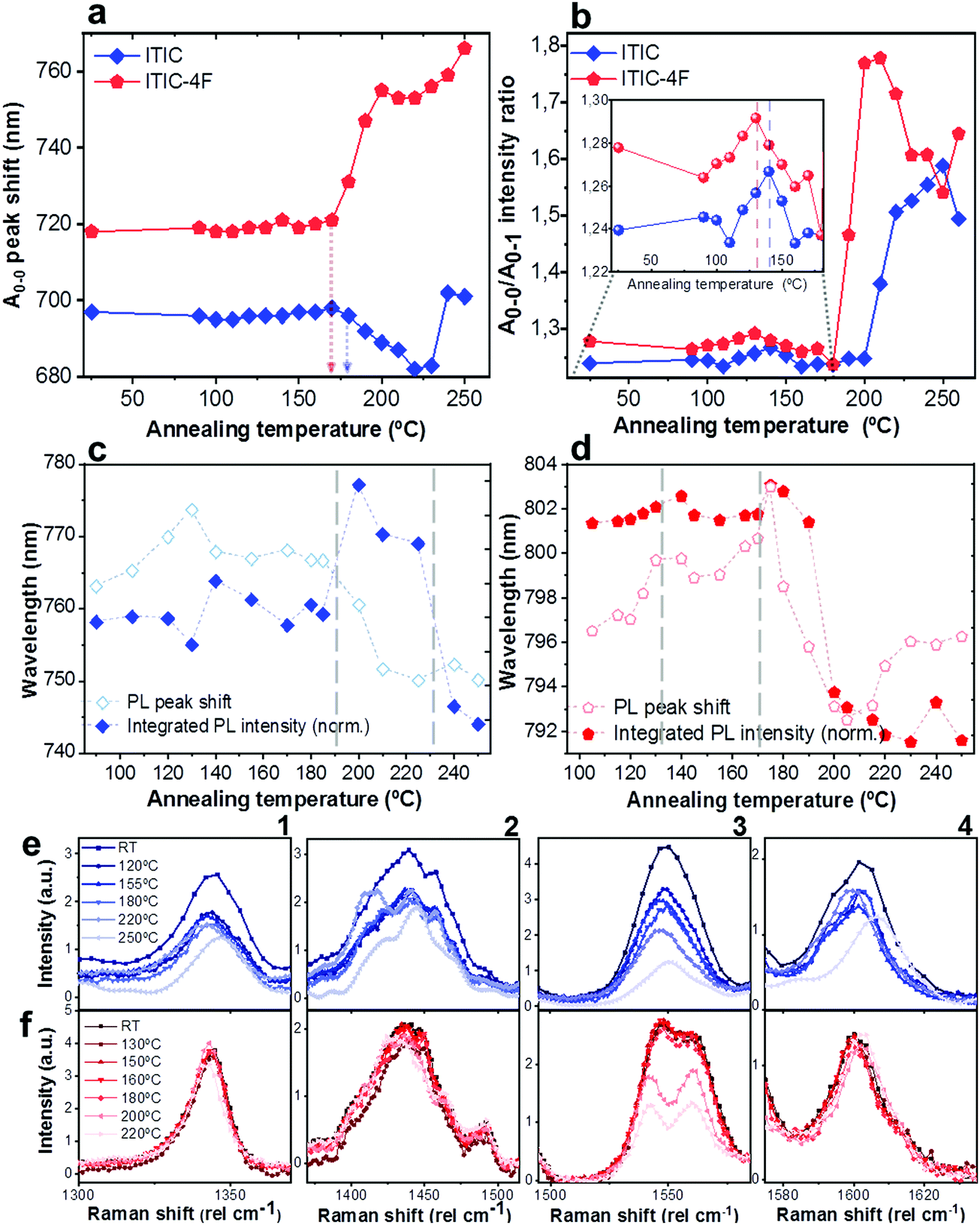 | ||
| Fig. 5 Quantitative analysis extrapolated from UV-vis, PL and Raman results presented in Fig. 4. A0–0 peak shift (a) and A0–0/A0–1 intensity ratio (b) of ITIC (rhombus) and ITIC-4F (pentagon) absorption spectra, as function of the annealing temperature. Integrated normalized PL intensity (filled symbols) and PL peak shift (unfilled symbols) of ITIC (rhombus, c) and ITIC-4F (pentagon, d) NFA, as function of the annealing temperature. Detail of the Ta-dependent evolution of Raman peaks (1: ∼1350 cm−1; 2: ∼1450 cm−1; 3: ∼1550 cm−1; 4: ∼1600 cm−1) for ITIC (e) and ITIC-4F (f). | ||
Annealing both ITICs at T ≥ Tg produces further conformational changes, reflected by the appearance of specific absorption features, as revealed in Fig. 4a, b and 5a, b. In more detail, when ITIC is annealed at temperatures between 180 °C and 230 °C, the A0–0 peak intensity increases (Fig. 4a) most probably due to the formation of a different polymorph (phase II). The further raise of the temperature (to 230–250 °C) results in the quick drop of the A0–0 peak intensity, due to a further transition into a phase III polymorph. On the other side, the A0–0 peak shift (Fig. 5a) undergoes a marked blue-shift (∼20 nm) in the phase II temperature region, followed by a back red-shift for phase III. Therefore, ITIC passes through an intermediate phase (polymorph II15) in which the combination of A0–0 blue shift together with its increased A0–0/A0–1 ratio, indicates that the crystallization process evolves through a temporary shorter conjugation-length. Such conjugation seems to be recovered for T > 230 °C. Indeed, it has been reported that for ideal H(J)-aggregates, increasing disorder (180–230 °C region in our experiments) leads to an increase (decrease) in the vibronic A0–0/A0–1 intensity ratio.59
Nevertheless, in a molecular π-stack a competition always exists between interchain (H-like) and intrachain (J-like) interactions, and some crossover from H- to J-aggregate behavior can easily show up.58 Unlike ITIC, ITIC-4F A0–0 peak shows a strong red-shift when crossing its Tg (∼170 °C), an indication that the small molecule might be acquiring stronger conjugation along the backbone with increased temperature. A closer look at the ITIC-4F A0–0/A0–1 peak ratio reveals that such phase change is accompanied by a transitory increased disorder, briefly showing up between 180 °C and 200 °C. GIWAXS measurements for materials annealed at 200 °C show more in-plane crystallinity features for ITIC-4F, than ITIC (Fig. 6 and 7e, f). The ITIC-4F phase transformation seems to happen in a much smaller temperature window, compared to ITIC. Ta-dependent PL contour plots (Fig. 4c) and PL peak shifts (Fig. 5c) confirm the presence of distinct polymorphic phases, while GIWAXS patterns (Fig. 6 and Fig. 7a, c, e) and scattering profiles (Fig. 6 and 7b, d, f) reveal the different level of crystallinity associated to them. Note that the use of temperature dependent PL measurements has been successfully used to probe the signatures of thermal transitions in conjugated polymers.60
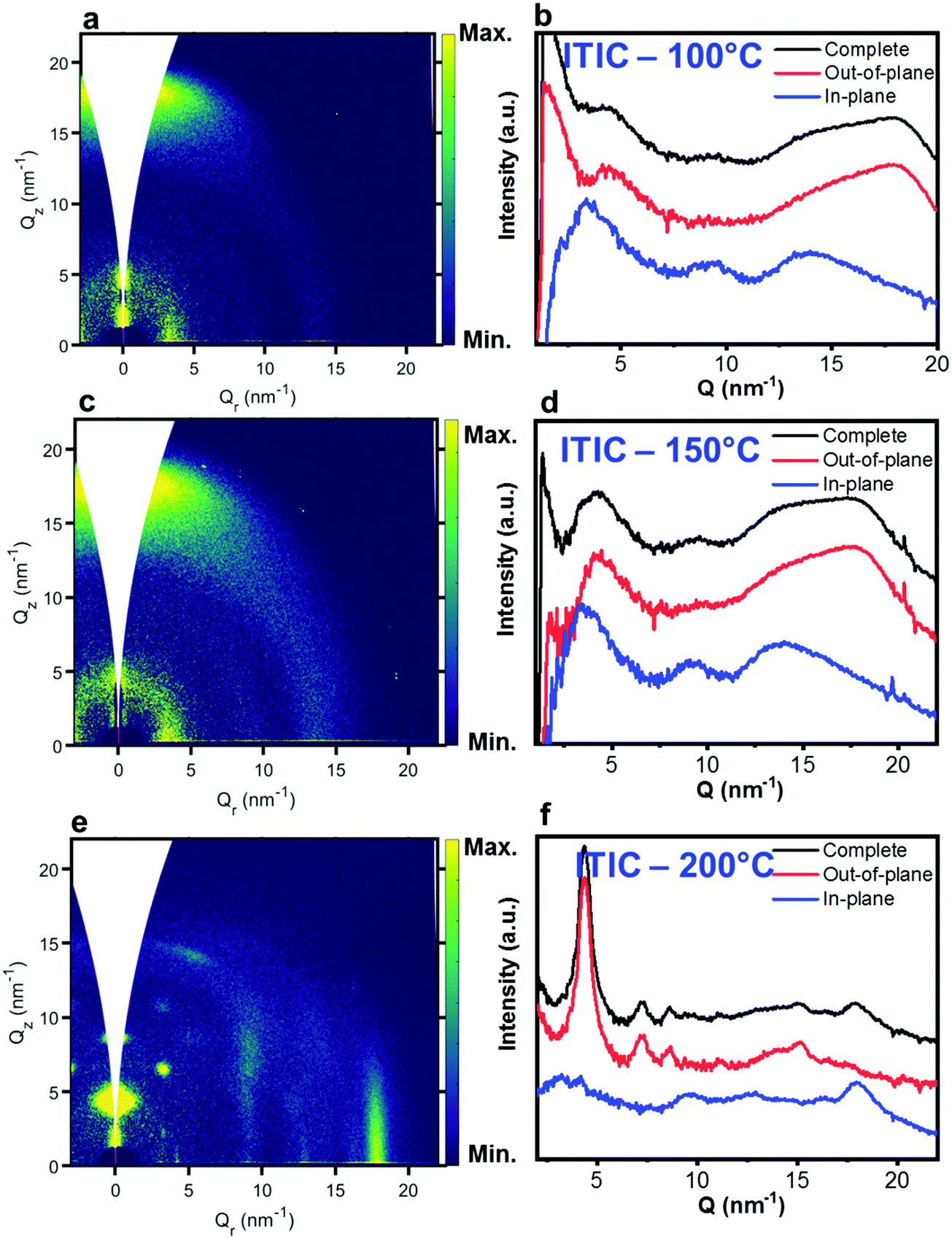 | ||
| Fig. 6 GIWAXS 2D patterns (a, c and e) and 1D in-plane and out-of-plane profiles (b, d and f) for ITIC annealed at different temperatures. | ||
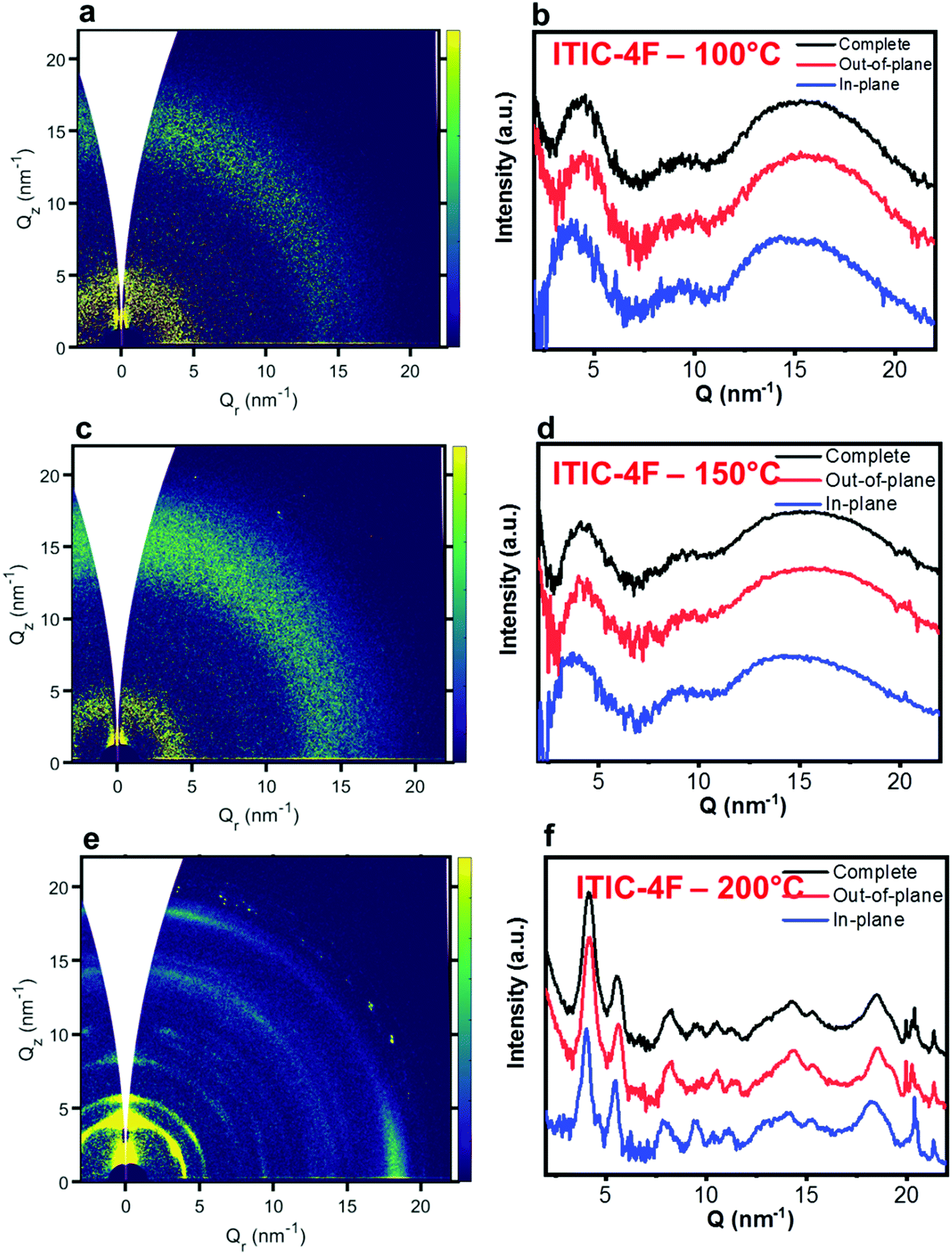 | ||
| Fig. 7 GIWAXS 2D patterns (a, c and e) and 1D in-plane and out-of-plane profiles (b, d and f) for ITIC-4F annealed at different temperatures. | ||
To further understand the effect of fluorine substitution on the ITIC temperature-dependent molecular order, we measured Raman spectra under 488 nm excitation. Fig. 4e and f show the complete normalized Raman spectra of ITIC and ITIC-4F at different Ta, while in Fig. 5e and f the detail of the four main vibrational modes, in the range 1300 cm−1–1700 cm−1, is reported. Since – in very broad terms – the Raman signal is based on the interaction between the material's electronic cloud and the external electric field induced by the measurement laser light,61,62 a general look at Fig. 5e and f already suggests that ITIC-4F may retain its structural features through a wider annealing temperature range, as compared to ITIC. Indeed, the ITIC-4F C–C intraring (in-plane skeleton) stretch modes (∼1340 cm−1, 1) position and intensity seem more unaffected by the specific phase than those of ITIC. On the other hand, the peak at ∼1600 cm−1 (4) assigned to the end-group fused phenyl ring,63 shows a higher variability for ITIC than for ITIC-4F. NFA end-groups have been identified as the ones responsible to create charge transport channels via end-group interactions between different molecules.64–66 Therefore, depending on their planarity and π–π stacking, they could either facilitate – or hinder – such intermolecular interactions and the charge transport across neighboring molecules.67 π–π stacking values of ∼3.5 Å and 3.35 Å have been recently reported for ITIC67 and ITIF-4F,47 respectively. Therefore, these results seem to suggest that fluorination, favoring a higher molecular planarization and closer packing – likely via the formation of the intramolecular noncovalent interactions38,47 – could grant higher molecular stiffness toward annealing temperature, with respect to ITIC. While peaks 2 and 3 (Fig. 5e and f) are commonly associated to collective CC bond stretching modes of the conjugated molecule backbone, based on DFT calculation and literature reports68–70 we could distinguish and assign the ∼1450 cm−1 vibrations (2) mostly to the thiophene-related CC stretching mode, while the ∼1550 cm−1 (3) to the CC central ring stretching mode. The biggest shape change registered for ITIC happens at the CC stretch peak (∼1450 cm−1) for T > 200 °C, when a well pronounced shoulder at ∼1420 cm−1 appears, whose presence has been reportedly associated to a more ordered phase of the molecular species.71 This matches GIWAXS results for ITIC annealed at 200 °C, where the material shows its highest crystalline phase (truncated rods), compared to patterns annealed at 100 °C and 150 °C (Fig. 6e and f).
Also ITIC-4F Raman shape and intensity exhibit the most striking change for >200 °C, mostly in correspondence with ∼1550 cm−1 (peaks become sharper and split, FWHM drops), with the GIWAXS patterns showing as well the highest crystalline phase for Ta = 200 °C (Fig. 7e and f). GIWAXS measurements generally show a higher ITIC-4F conjugation over ITIC, possibly confirming the stabilizing effect on the structure, coming from the multiple intermolecular non-covalent interactions occurring in the fluorinated acceptor.47,72 Nevertheless, until the 200 °C annealing temperature is reached, other structural changes highlighted by GIWAXS for ITIC-4F annealed at T < 200 °C, do not seem to be detectable by the Raman signal (Fig. 7b and d), maybe suggesting a larger Raman scattering cross section for the disordered phase of ITIC-4F, compared to the ordered phase.
Table 2 briefly summarizes the main annealing temperature-dependent phases identified, with their associated crystallinity features. Through this characterization, we have shown that both ITIC and ITIC-4F exhibit three well-marked structural phases, clearly dependent on the annealing temperature applied. Interestingly, within polymorph I – delimited within the RT–Tg range – our detailed temperature mapping allowed us to unveil two sub-phases (I* and I**) for each NFA. Moreover, Raman analysis showed that for each NFA, the processing temperature leads to phase-specific vibrational modes evolution.
| Phase | T range (°C) | Crystallinity features (aggregate type) | |
|---|---|---|---|
| ITIC | I* | RT to ∼140 | Loose crystallinity (HJ-type) |
| I** | ∼140 to ∼180 | Beginning of crystallization process (towards more J-type) | |
| II | ∼180 to ∼230 | Conjugation loss (more H-type) | |
| III | ∼230 to ∼250 | More ordered phase (more J-type) | |
| ITIC-4F | I* | RT to ∼130 | Loose crystallinity (HJ-type) |
| I** | ∼130 to ∼170 | Beginning of crystallization process (towards more J-type) | |
| II | ∼170 to ∼220 | A large crystallinity is acquired (strong J-like character) | |
| III | ∼220 to ∼250 | Crystallinity reduces |
Phase-dependent ITICs stability
Having established a relationship between Ta and the structural features of fluorinated vs. non fluorinated ITIC – i.e. their thermal transition temperatures and how these may affect their molecular bonding – we next study the NFA stability in thermally annealed materials. In fact, thermal treatments are among the most fundamental fabrication steps of a solar cell throughout the entire production process. For this, investigating not just the as cast-, but also the polymorphic phase-dependent ITICs stability becomes especially relevant in order to simultaneously achieve efficient and durable solar devices. Therefore, we screened the different polymorphic ITICs phases under similar accelerated degrading conditions. Fig. 8a and b report the log–log integrated PL decay of the 4 ITICs structural phases as a function of the degrading time; Fig. 8c and d show the semi-log degrading trends of Raman peak i, against the dose intake (peaks ii and iii displayed in Fig. S3, ESI†). First, we notice that PL and Raman measurements coherently verify that stability depends on the specific NFA investigated. In detail, ITIC shows its least stable regimes below Tg, while its most stable polymorph appears for Ta > 200 °C (phases II and III). From the Raman measurements, we also determine that among phases II and III, the longest lasting integrated Raman peak i signal comes from phase III, when – in agreement with results from Martin's group15 – the material reaches its lowest free energy, and presumably its highest thermodynamical stability. The relative stability of ITIC-4F polymorphs instead, does not seem to correlate directly with the order of the material's crystallization phases. In fact, unlike ITIC, the highest annealing temperature (∼230 °C, phase III) for ITIC-4F does not bring the material to the most stable phase, but rather the opposite.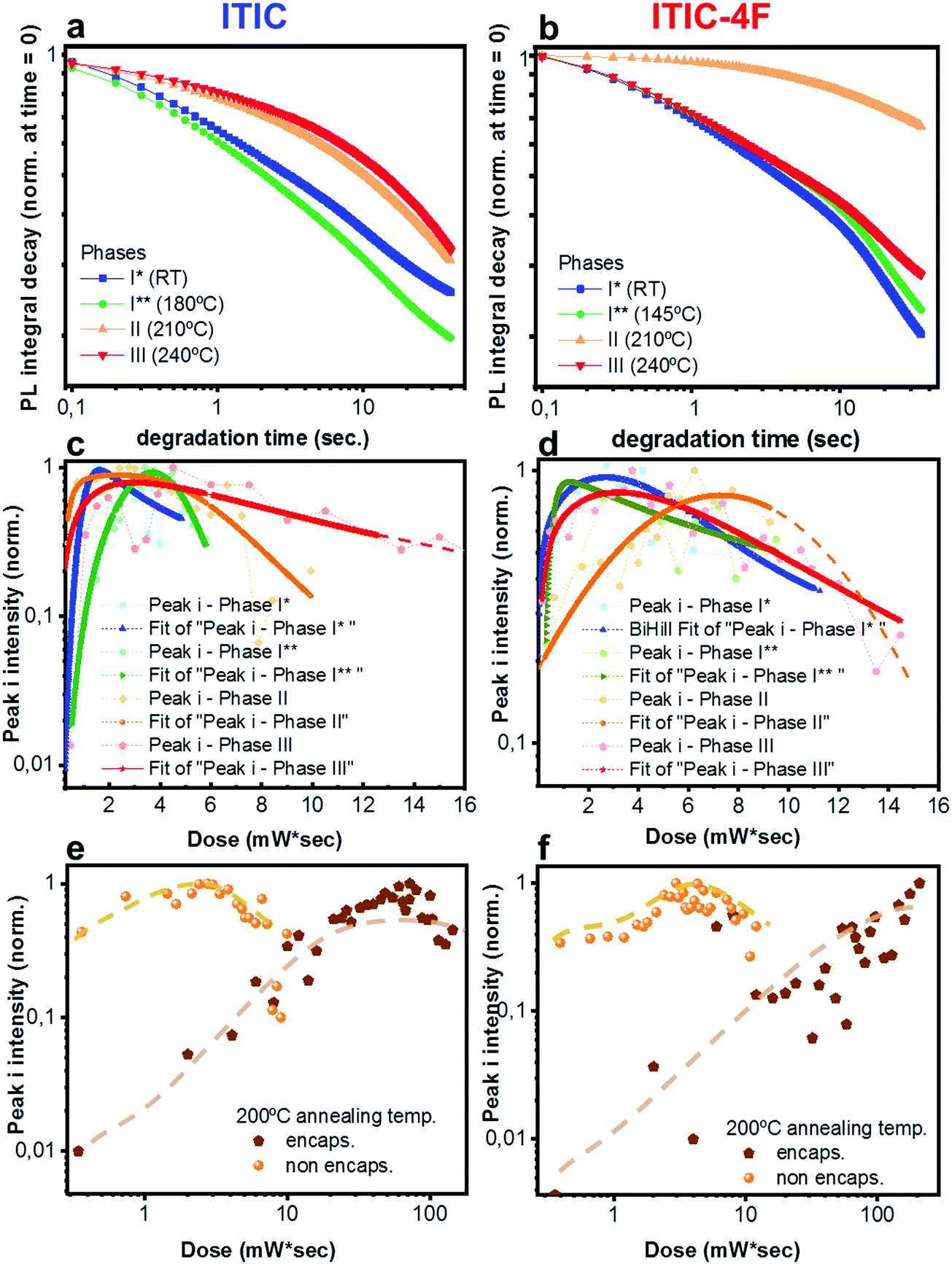 | ||
| Fig. 8 In situ integrated PL intensity decay of ITIC (a) and ITIC-4F (b) in their different polymorphic phases (I* ∼ RT, I** ∼ 180 °C for ITIC and 150° for ITIC-4F, II ∼ 210 °C, III ∼ 240 °C) upon 633 nm laser excitation, as a function of degradation time. Normalized integrated Raman intensity of peak i (〈1450〉 cm−1) as a function of dose for non-encapsulated ITIC (c) and ITIC-4F (d) in their different polymorphic states, upon 488 nm excitation. Normalized integrated Raman intensity of peak i (〈1450〉 cm−1) as a function of dose for encapsulated ITIC (e) and ITIC-4F (f) annealed at 200 °C, upon 488 nm excitation. Results for non-encapsulated ITICs are also reported for comparison. Dashed lines are only a guide for the eye. | ||
It is worth noting that the integrated [630–650 nm] absorption range plotted against Ta (Fig. S4, ESI†) proves that the observed stability variation between phases is not an artifact associated to different phases having different absorption coefficients, which would in turn vary the total absorbed 633 nm laser power. ITIC-4F Raman peak i signal for phases I and III show the quickest intensity drops. Yet, slight differences in the degradation time scales between PL and Raman might be due to the different illumination laser wavelengths used to perform the AST and collect the signals.
In order to further prove the oxidative cleavage of the alkenes-based units as the major trigger for the degradation process of ITIC-based NFAs – as well as to check whether different phases would differently respond to the AST – we repeated similar in situ degradation experiments on annealed and encapsulated samples. Fig. 8e and f show Raman peak i results relative to ITIC (Fig. 8e) and ITIC-4F (Fig. 8f) annealed at 200 °C (Raman peak ii and iii results for samples annealed at 200 °C, as well as Raman peaks i–iii results for samples annealed at 150 °C reported in Fig. S5 and S6, ESI†). Our results make clear that in the absence of oxygen (and water), the degradation process of both NFAs experiences a huge delay. It is also clear that the formation of free radicals, responsible for initiating the autoxidation cycle, is jointly triggered by light. But while ITIC starts to drop in intensity for a dose intake of ca. 80 mW s, ITIC-4F peak i intensity does not show signs of decrease even after a dose intake as high as 110 mW s.
Lastly, PL intensity profiles of Ta-dependent ITICs degraded under 633 nm laser could be accurately fitted using decreasing exponential functions (Fig. S8 and S9, ESI† for fitting equations). The identified trends point at the presence of two main radiative recombination channels in the materials, with the first one (k1, cross +) being preeminent over the second one (k2, tilted cross ×) (Fig. 9). Yet, a closer look at Fig. 9 reveals that, whilst for ITIC-4F (Fig. 9b) k1 and k2 follow the same profile throughout the temperature range – with zones of higher and lower decay rates dependent on the Ta in accordance with the material's response to AST (Fig. 8b) – for ITIC this is not the case. Indeed, while the k1 decay rate shows a drop in the (125–155 °C) annealing region, the second radiative channel k2 shows an increase, even though slight, as if in this region the material underwent a structural transition which could temporarily open up local recombination pathways.
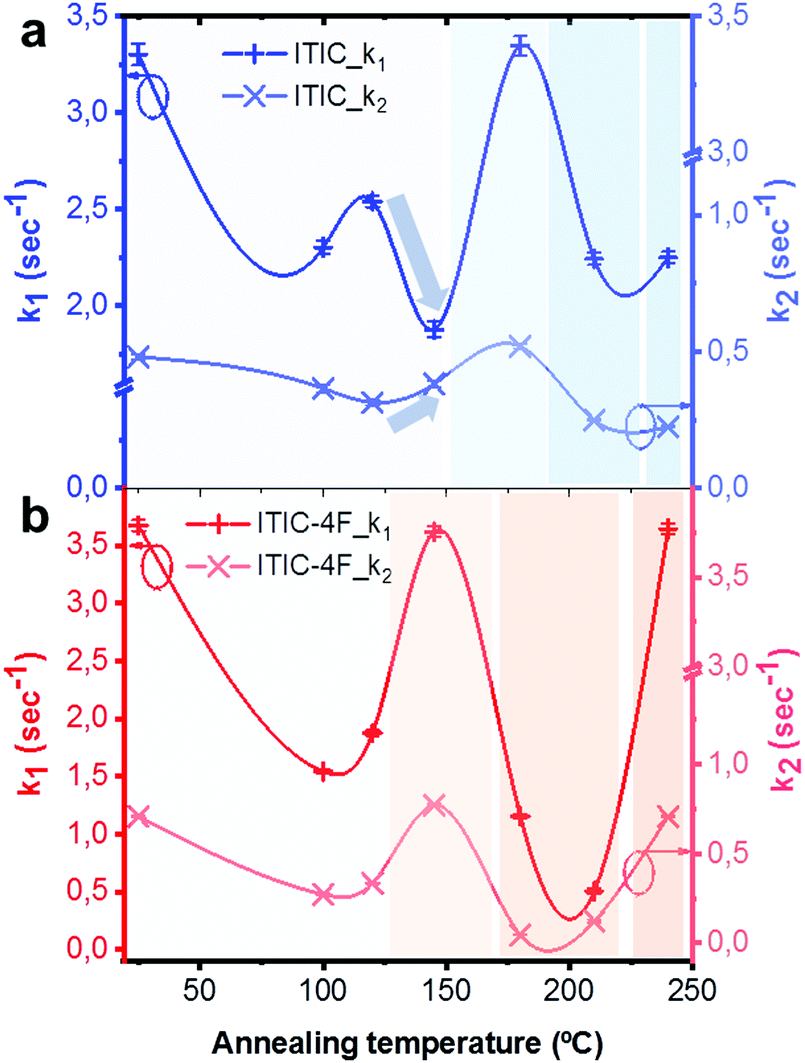 | ||
| Fig. 9 Decay rates k1 and k2 extrapolated from the bi-exponential fitting of ITIC (a) and ITIC-4F (b) PL quenching profiles upon 633 nm laser irradiation, for different annealing temperatures of the materials. | ||
Such structural rearrangements in the Ta range 130–150 °C was also detected by Yu et al.,56 using FSC analysis on annealed ITIC. According to the extrapolated decay rate coefficients of Fig. 9, we see that in the temperature region below Tg each material shows a decay rate minimum, associated to a highly stable phase. The degradation rate upturn shown right before each Tg instead, seems to be likely due to a local amorphization process taking place in the structure.15 For annealing temperatures well above Tg, the two behaviors markedly differ, as ITIC enters a region of high stability while ITIC-4F evolves toward a region of fewer crystallinity, which in turns affects its stability response. Interestingly, as different isomers of ITIC molecular family may result in different molecular arrangement,73 our polymorph dependent stability suggest that there might be also an isomer-dependent stability, which will be the topic of future investigations.
Conclusions
ITIC and ITIC-4F NFAs were blade-coated and subsequently annealed in the temperature range (90–250 °C). By using several spectroscopic techniques we were able to identify distinct phases with different polymorphs and varying degree of crystallinity, depending on the annealing temperature and the fluorine presence. With the help of DFT calculations, we could infer a relationship between the main NFA molecular bonds, and their Ta dependent morphological evolution.ASTs were realized with two different monochromatic lasers in order to assess the materials’ chemical stability. While CC of alkene and thiophene based groups showed to be the weakest chemical points for both ITICs, fluorine-substituted NFA displayed a slower the degradation process, likely due to its more favorable energy levels together with its higher degree of order.
Annealed materials were also degraded under 488 nm and 633 mn lasers. For ITIC, we verified that in the temperature region T < Tg, the highest stability to dose intake takes place for Ta ∼ 150 °C, consistently with literature.56 Still, phases II and III for Ta > Tg show the highest stability. For ITIC-4F instead, in the T < Tg range temperatures as low as ∼120 °C produced the most stable phase against degradation. When passing Tg, the material undergoes a structural evolution marking a phase of high stability (phase II) and a subsequent pretty amorphous one (phase III). While the more stable phases obtained could be associated to higher degree of crystallinity in the NFA materials, this happened at the expense of higher processing temperatures. This may need to be taken into serious consideration, especially when fabricating OPV on flexible substrates. Indeed, usual flexible OPV processing temperatures have to be kept below 150 °C.74 Therefore, our findings could serve as an indication of the optimal processing temperature to be used in such ITIC-based solar cells. We will obviously need to verify our hypotheses in a complete OPV device. Degrading annealed and encapsulated samples proved that oxygen is the major trigger of the autoxidation cycle, and that ITIC-4F performs better (i.e. degrades less) for both Ta tested (150 °C and 200 °C). Having identified the alkene- and the thiophene-related CC bonds as the most sensitive molecular portion, we verify that in the fluorine-substituted ITIC, the vinyl moieties are less exposed to oxygen attack and that – depending on the applied annealing temperatures – such bonds may adjust in orientations more or less favorable to external stimuli.
Considering our findings, specific materials and applications could best suit particular processing temperature conditions and fabrication techniques. In this frame, ITIC-based OPVs might be a good choice for delivering high efficiencies, even though their capability of dose intake could be more limited than that of ITIC-4F based ones. On the other hand, ITIC-4F based devices could grant longer-lasting performance. In addition, designing molecules with larger electron affinities, as well as introducing antioxidants that suppress the autoxidation cycle via the formation of stable radicals, could be both considered as strategies to make the NFA more robust to radicals’ attack. This may result in more intrinsically stable devices. Such accomplishment may loosen, in turn, the present requirements on encapsulation, based on the need to block outer oxygen/moisture percolation.36,48,75 As a last remark, we note that since OSC performance and stability depend jointly on the NFA morphology and fluorination, the photo-active layer composition (e.g. in ternaries blends) and the annealing temperature should be both adjusted, in order to achieve a stable energy output.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Dr Tommaso Salzillo and Dr Valentina Bulova for very fruitful scientific discussions. The authors kindly acknowledge the financial support from Ministerio de Economía y Competitividad of Spain through the “Severo Ochoa” Programme for Centres of Excellence in R&D and projects PGC2018-095411-B-I00 and PGC2018-094620-A-I00, as well as the European Research Council (ERC) under grant agreement no. 648901.References
- T. R. Andersen, H. F. Dam, M. Hösel, M. Helgesen, J. E. Carlé, T. T. Larsen-Olsen, S. A. Gevorgyan, J. W. Andreasen, J. Adams, N. Li, F. Machui, G. D. Spyropoulos, T. Ameri, N. Lemaître, M. Legros, A. Scheel, D. Gaiser, K. Kreul, S. Berny, O. R. Lozman, S. Nordman, M. Välimäki, M. Vilkman, R. R. Søndergaard, M. Jørgensen, C. J. Brabec and F. C. Krebs, Energy Environ. Sci., 2014, 7, 2925–2933 RSC.
- J. E. Carlé, M. Helgesen, O. Hagemann, M. Hösel, I. M. Heckler, E. Bundgaard, S. A. Gevorgyan, R. R. Søndergaard, M. Jørgensen, R. García-Valverde, S. Chaouki-Almagro, J. A. Villarejo and F. C. Krebs, Joule, 2017, 1, 274–289 CrossRef.
- I. Burgués-Ceballos, M. Stella, P. Lacharmoise and E. Martínez-Ferrero, J. Mater. Chem. A, 2014, 2, 17711–17722 RSC.
- Q. Liu, Y. Jiang, K. Jin, J. Qin, J. Xu, W. Li, J. Xiong, J. Liu, Z. Xiao, K. Sun, S. Yang, X. Zhang and L. Ding, Sci. Bull., 2020, 65(4), 272–275 CrossRef CAS.
- L. Ye, W. Zhao, S. Li, S. Mukherjee, J. H. Carpenter, O. Awartani, X. Jiao, J. Hou and H. Ade, Adv. Energy Mater., 2017, 7, 1602000 CrossRef.
- B. J. Tremolet De Villers, K. A. O’Hara, D. P. Ostrowski, P. H. Biddle, S. E. Shaheen, M. L. Chabinyc, D. C. Olson and N. Kopidakis, Chem. Mater., 2016, 28(3), 876–884 CrossRef CAS.
- N. Y. Doumon, G. Wang, X. Qiu, A. J. Minnaard, R. C. Chiechi and L. J. A. Koster, Sci. Rep., 2019, 9(1), 1–14 CrossRef CAS.
- Y. Lin, J. Wang, Z. G. Zhang, H. Bai, Y. Li, D. Zhu and X. Zhan, Adv. Mater., 2015, 27(7), 1170–1174 CrossRef CAS.
- C. Yan, S. Barlow, Z. Wang, H. Yan, A. K. Y. Jen, S. R. Marder and X. Zhan, Nat. Rev. Mater., 2018, 3(3), 1–19 Search PubMed.
- J. Hou, O. Inganas, R. H. Friend and F. Gao, Nat. Mater., 2018, 17(2), 119–128 CrossRef CAS.
- A. Wadsworth, M. Moser, A. Marks, M. S. Little, N. Gasparini, C. J. Brabec, D. Baran and I. McCulloch, Chem. Soc. Rev., 2019, 48(6), 1596–1625 RSC.
- W. Li, L. Ye, S. Li, H. Yao, H. Ade and J. Hou, Adv. Mater., 2018, 30(16), 1707170 CrossRef.
- Z. Zheng, Q. Hu, S. Zhang, D. Zhang, J. Wang, S. Xie, R. Wang, Y. Qin, W. Li, L. Hong, N. Liang, F. Liu, Y. Zhang, Z. Wei, Z. Tang, T. P. Russell, J. Hou and H. Zhou, Adv. Mater., 2018, 30(34), 1801801 CrossRef.
- W. Zhao, S. Li, H. Yao, S. Zhang, Y. Zhang, B. Yang and J. Hou, J. Am. Chem. Soc., 2017, 139, 7148–7151 CrossRef CAS.
- S. Marina, A. Scaccabarozzi, E. Solano, A. Khirbat, A. Iturrospe, A. Balzer, L. Yu, E. Gabirondo, X. Monnier, T. Anthopoulsos, M. Caironi, M. Campoy, C. Müller, D. Cangialosi, N. Stingelin and J. Martin, 2020, unpublished work.
- P. Cheng, G. Li, X. Zhan and Y. Yang, Nat. Photonics, 2018, 12, 131–142 CrossRef CAS.
- G. Zhang, J. Zhao, P. C. Y. Chow, K. Jiang, J. Zhang, Z. Zhu, J. Zhang, F. Huang and H. Yan, Chem. Rev., 2018, 118(7), 3447–3507 CrossRef CAS.
- S. A. Gevorgyan, N. Espinosa, L. Ciammaruchi, B. Roth, F. Livi, S. Tsopanidis, S. Züfle, S. Queirós, A. Gregori, G. A. D. R. Benatto, M. Corazza, M. V. Madsen, M. Hösel, M. J. Beliatis, T. T. Larsen-Olsen, F. Pastorelli, A. Castro, A. Mingorance, V. Lenzi, D. Fluhr, R. Roesch, M. Maria Duarte Ramos, A. Savva, H. Hoppe, L. S. A. Marques, I. Burgués, E. Georgiou, L. Serrano-Luján and F. C. Krebs, Adv. Energy Mater., 2016, 6(22), 1600910 CrossRef.
- Q. Burlingame, B. Song, L. Ciammaruchi, G. Zanotti, J. Hankett, Z. Chen, E. A. Katz and S. R. Forrest, Adv. Energy Mater., 2016, 6(21), 1601094 CrossRef.
- L. Ciammaruchi, F. Brunetti and I. Visoly-Fisher, Sol. Energy, 2016, 137, 490–499 CrossRef CAS.
- M. O. Reese, S. A. Gevorgyan, M. Jørgensen, E. Bundgaard, S. R. Kurtz, D. S. Ginley, D. C. Olson, M. T. Lloyd, P. Morvillo, E. A. Katz, A. Elschner, O. Haillant, T. R. Currier, V. Shrotriya, M. Hermenau, M. Riede, K. R. Kirov, G. Trimmel, T. Rath, O. Inganäs, F. Zhang, M. Andersson, K. Tvingstedt, M. Lira-Cantu, D. Laird, C. McGuiness, S. Gowrisanker, M. Pannone, M. Xiao, J. Hauch, R. Steim, D. M. DeLongchamp, R. Rösch, H. Hoppe, N. Espinosa, A. Urbina, G. Yaman-Uzunoglu, J.-B. Bonekamp, A. J. J. M. van Breemen, C. Girotto, E. Voroshazi and F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2011, 95, 1253–1267 CrossRef CAS.
- W. Zhao, D. Qian, S. Zhang, S. Li, O. Inganäs, F. Gao and J. Hou, Adv. Mater., 2016, 28(23), 4734–4739 CrossRef CAS.
- X. Du, T. Heumueller, W. Gruber, A. Classen, T. Unruh, N. Li and C. J. Brabec, Joule, 2019, 3(1), 215–226 CrossRef CAS.
- N. Y. Doumon, F. V. Houard, J. Dong, H. Yao, G. Portale, J. Hou and L. J. A. Koster, Org. Electron., 2019, 69, 255–262 CrossRef CAS.
- S. Park and H. J. Son, J. Mater. Chem. A, 2019, 7(45), 25830–25837 RSC.
- J. Guo, Y. Wu, R. Sun, W. Wang, J. Guo, Q. Wu, X. Tang, C. Sun, Z. Luo, K. Chang, Z. Zhang, J. Yuan, T. Li, W. Tang, E. Zhou, Z. Xiao, L. Ding, Y. Zou, X. Zhan, C. Yang, Z. Li, C. J. Brabec, Y. Li and J. Min, J. Mater. Chem. A, 2019, 7(43), 25088–25101 RSC.
- J. Luke, E. M. Speller, A. Wadsworth, M. F. Wyatt, S. Dimitrov, H. K. H. Lee, Z. Li, W. C. Tsoi, I. McCulloch, D. Bagnis, J. R. Durrant and J. S. Kim, Adv. Energy Mater., 2019, 9(15), 1803755 CrossRef.
- E. Pascual-San-José, X. Rodríguez-Martínez, R. Adel-Abdelaleim, M. Stella, E. Martínez-Ferrero and M. Campoy-Quiles, J. Mater. Chem. A, 2019, 7(35), 20369–20382 RSC.
- A. Harillo-Baños, X. Rodríguez-Martínez and M. Campoy-Quiles, Adv. Energy Mater., 2020, 10(1), 1902417 CrossRef.
- R. K. Misra, L. Ciammaruchi, S. Aharon, D. Mogilyansky, L. Etgar, I. Visoly-Fisher and E. A. Katz, ChemSusChem, 2016, 9(18), 2572–2577 CrossRef CAS.
- C. M. Sharts, V. S. Gorelik, A. M. Agoltsov, L. I. Zlobina and O. N. Sharts, in Proceeding SPIE, ed. M. Fallahi, R. J. Nordstrom and T. R. Todd, 1999, vol. 3537, pp. 309–318.
- F. Menaa, B. Menaa and O. Sharts, Faraday Discuss., 2011, 149(1), 269–278 RSC.
- M. Knaapila and S. Guha, Rep. Prog. Phys., 2016, 79(6), 066601 CrossRef.
- M. Nikolka, I. Nasrallah, B. Rose, M. K. Ravva, K. Broch, A. Sadhanala, D. Harkin, J. Charmet, M. Hurhangee, A. Brown, S. Illig, P. Too, J. Jongman, I. McCulloch, J. L. Bredas and H. Sirringhaus, Nat. Mater., 2017, 16(3), 356–362 CrossRef CAS.
- H. S. Silva, A. Tournebize, D. Bégué, H. Peisert, T. Chassé, J. L. Gardette, S. Therias, A. Rivaton and R. C. Hiorns, RSC Adv., 2014, 4(97), 54919–54923 RSC.
- M. Salvador, N. Gasparini, J. D. Perea, S. H. Paleti, A. Distler, L. N. Inasaridze, P. A. Troshin, L. Luer, H.-J. Egelhaaf and C. Brabec, Energy Environ. Sci., 2017, 10(9), 2005–2016 RSC.
- P. Li, J. M. Maier, E. C. Vik, C. J. Yehl, B. E. Dial, A. E. Rickher, M. D. Smith, P. J. Pellechia and K. D. Shimizu, Angew. Chem., Int. Ed., 2017, 129(25), 7315–7318 CrossRef.
- S. Feng, M. Li, N. Tang, X. Wang, H. Huang, G. Ran, Y. Liu, Z. Xie, W. Zhang and Z. Bo, ACS Appl. Mater. Interfaces, 2020, 12(4), 4638–4648 CrossRef CAS.
- Q. Fan, Q. Zhu, Z. Xu, W. Su, J. Chen, J. Wu, X. Guo, W. Ma, M. Zhang and Y. Li, Nano Energy, 2018, 48, 413–420 CrossRef CAS.
- Z. Fei, F. D. Eisner, X. Jiao, M. Azzouzi, J. A. Röhr, Y. Han, M. Shahid, A. S. R. Chesman, C. D. Easton, C. R. McNeill, T. D. Anthopoulos, J. Nelson and M. Heeney, Adv. Mater., 2018, 30(8), 1705209 CrossRef.
- E. M. Speller, A. J. Clarke, N. Aristidou, M. F. Wyatt, L. Francàs, G. Fish, H. Cha, H. K. H. Lee, J. Luke, A. Wadsworth, A. D. Evans, I. McCulloch, J. S. Kim, S. A. Haque, J. R. Durrant, S. D. Dimitrov, W. C. Tsoi and Z. Li, ACS Energy Lett., 2019, 4(4), 846–852 CrossRef CAS.
- H. J. Heller and H. R. Blattmann, Pure Appl. Chem., 1973, 36(1–2), 141–162 CAS.
- M. Tolinski, Additives for Polyolefins, 2009 Search PubMed.
- Y. Hayashi, M. Takeda, Y. Miyamoto and M. Shoji, Chem. Lett., 1985, 2002(3), 414–415 CrossRef.
- V. Turkovic, M. Prete, M. Bregnhøj, L. Inasaridze, D. Volyniuk, F. A. Obrezkov, J. V. Grazulevicius, S. Engmann, H. G. Rubahn, P. A. Troshin, P. R. Ogilby and M. Madsen, ACS Appl. Mater. Interfaces, 2019, 11(44), 41570–41579 CrossRef CAS.
- S. Feng, M. Li, N. Tang, X. Wang, H. Huang, G. Ran, Y. Liu, Z. Xie, W. Zhang and Z. Bo, ACS Appl. Mater. Interfaces, 2020, 12(4), 4638–4648 CrossRef CAS.
- T. J. Aldrich, M. Matta, W. Zhu, S. M. Swick, C. L. Stern, G. C. Schatz, A. Facchetti, F. S. Melkonyan and T. J. Marks, J. Am. Chem. Soc., 2019, 141(7), 3274–3287 CrossRef CAS.
- E. T. Hoke, I. T. Sachs-Quintana, M. T. Lloyd, I. Kauvar, W. R. Mateker, A. M. Nardes, C. H. Peters, N. Kopidakis and M. D. McGehee, Adv. Energy Mater., 2012, 2(11), 1351–1357 CrossRef CAS.
- J. Mulzer, Angew. Chem., Int. Ed., 1987, 99(4), 377–377 Search PubMed.
- L. Ciammaruchi, C. Wang, Y. Gao and C. W. Tang, J. Appl. Phys., 2015, 117(24), 245504 CrossRef.
- F. Génin, F. Quilès and A. Burneau, Phys. Chem. Chem. Phys., 2001, 3(6), 932–942 RSC.
- A. Raj, K. Raju, H. T. Varghese, C. M. Granadeiro, H. I. S. Nogueira and C. Yohannan Panicker, J. Braz. Chem. Soc., 2009, 20(3), 549–559 CrossRef CAS.
- X. Zhu, L. Hu, W. Wang, X. Jiang, L. Hu and Y. Zhou, ACS Appl. Energy Mater., 2019, 2(10), 7602–7608 CrossRef CAS.
- X. Yang and J. Loos, Macromolecules, 2007, 40(5), 1353–1362 CrossRef CAS.
- C. Müller, J. Bergqvist, K. Vandewal, K. Tvingstedt, A. S. Anselmo, R. Magnusson, M. I. Alonso, E. Moons, H. Arwin, M. Campoy-Quiles and O. Inganäs, J. Mater. Chem., 2011, 21(29), 10676–10684 RSC.
- L. Yu, D. Qian, S. Marina, F. A. A. Nugroho, A. Sharma, S. Hultmark, A. I. Hofmann, R. Kroon, J. Benduhn, D. M. Smilgies, K. Vandewal, M. R. Andersson, C. Langhammer, J. Martín, F. Gao and C. Müller, ACS Appl. Mater. Interfaces, 2019, 11(24), 21766–21774 CrossRef CAS.
- H. Yamagata and F. C. Spano, J. Chem. Phys., 2012, 136(18), 184901 CrossRef CAS.
- J. M. Granadino-Roldán, N. Vukmirović, M. Fernández-Gómez and L. W. Wang, Phys. Chem. Chem. Phys., 2011, 13(32), 14500–14509 RSC.
- F. C. Spano, Acc. Chem. Res., 2010, 43(3), 429–439 CrossRef CAS.
- M. Sims, K. Zheng, M. C. Quiles, R. Xia, P. N. Stavrinou, D. D. C. Bradley and P. Etchegoin, J. Phys.: Condens. Matter, 2005, 17(41), 6307 CrossRef CAS.
- T. Salzillo, S. D’Agostino, A. Rivalta, A. Giunchi, A. Brillante, R. G. Della Valle, N. Bedoya-Martínez, E. Zojer, F. Grepioni and E. Venuti, J. Phys. Chem. C, 2018, 122(32), 18422–18431 CrossRef CAS.
- C. Cappuccino, P. P. Mazzeo, T. Salzillo, E. Venuti, A. Giunchi, R. G. Della Valle, A. Brillante, C. Bettini, M. Melucci and L. Maini, Phys. Chem. Chem. Phys., 2018, 20(5), 3630–3636 RSC.
- S. Pagliara, M. S. Vitiello, A. Camposeo, A. Polini, R. Cingolani, G. Scamarcio and D. Pisignano, J. Phys. Chem. C, 2011, 115(42), 20399–20405 CrossRef CAS.
- D. He, F. Zhao, L. Jiang and C. Wang, J. Mater. Chem. A, 2018, 6(19), 8839–8854 RSC.
- Y. Lin and X. Zhan, Mater. Horiz., 2014, 1(5), 470–488 RSC.
- V. A. Trukhanov and D. Y. Paraschuk, Polym. Sci., Ser. C, 2014, 56(1), 72–83 CrossRef CAS.
- J. J. Rech, N. Bauer, D. DIrkes, J. Kaplan, Z. Peng, H. Zhang, L. Ye, S. Liu, F. Gao, H. Ade and W. You, Mater. Chem. Front., 2019, 3(8), 1642–1652 RSC.
- A. A. Farah, S. A. Rutledge, A. Schaarschmidt, R. Lai, J. P. Freedman and A. S. Helmy, J. Appl. Phys., 2012, 112(11), 113709 CrossRef.
- S. Garreau, G. Louarn, J. P. Buisson, G. Froyer and S. Lefrant, Macromolecules, 1999, 32(20), 6807–6812 CrossRef CAS.
- S. H. Chang, C. H. Chiang, F. S. Kao, C. L. Tien and C. G. Wu, IEEE Photonics J., 2014, 6(4), 1–7 Search PubMed.
- W. C. Tsoi, D. T. James, E. B. Domingo, J. S. Kim, M. Al-Hashimi, C. E. Murphy, N. Stingelin, M. Heeney and J. S. Kim, ACS Nano, 2012, 6(11), 9646–9656 CrossRef CAS.
- C. A. Morgado, J. P. McNamara, I. H. Hillier, N. A. Burton and M. A. Vincent, J. Chem. Theory Comput., 2007, 3(5), 1656–1664 CrossRef CAS.
- H. Lai, H. Chen, J. Zhou, J. Qu, P. Chao, T. Liu, X. Chang, N. Zheng, Z. Xie and F. He, iScience, 2019, 17, 302–314 CrossRef CAS.
- S. I. Na, Y. H. Seo, Y. C. Nah, S. S. Kim, H. Heo, J. E. Kim, N. Rolston, R. H. Dauskardt, M. Gao, Y. Lee and D. Vak, Adv. Funct. Mater., 2019, 29(6), 1805825 CrossRef.
- K. U. Ingold and D. A. Pratt, Chem. Rev., 2014, 114(18), 9022–9046 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00458h |
| This journal is © The Royal Society of Chemistry 2020 |