Comparative cytotoxicity of cadmium forms (CdCl2, CdO, CdS micro- and nanoparticles) in renal cells
Béatrice
L'Azou
*a,
Isabelle
Passagne
a,
Sandra
Mounicou
b,
Mona
Tréguer-Delapierre
c,
Igor
Puljalté
a,
Joanna
Szpunar
b,
Ryszard
Lobinski
b and
Céline
Ohayon-Courtès
d
aPharmacochimie FRE CNRS 3396, Université de Bordeaux, Bordeaux Segalen, 146 rue Léo-Saignat, 33076 Bordeaux, France. E-mail: beatrice.lazou@u-bordeaux2.fr; Fax: +33 5 57 57 12 46; Tel: +33 5 57 57 12 45
bLaboratoire de Chimie Analytique Bio Inorganique et Environnement, CNRS UMR 5254, Hélioparc, 2 av. Pr. Angot 64053 Pau cedex, France
cICMCB, CNRS UPR9048, Université de Bordeaux, 87 avenue du Docteur Schweitzer, 33608 Pessac Cedex, France
dLHE, laboratoire hydrologie-Environnement, Université de Bordeaux, Bordeaux Segalen, 146 rue Léo-Saignat, 33076 Bordeaux, France
First published on 20th August 2013
Abstract
Cadmium (Cd) is a potent selective toxicant that preferentially accumulates in the kidneys where it is known to induce cellular injury. The cellular mechanisms explaining toxicity have been well documented by in vivo and in vitro studies. However, experiments have mainly been performed on cadmium chloride (CdCl2), while several other forms, such as cadmium oxide (CdO) and cadmium sulfide (CdS), are of interest regarding occupational exposure. CdS is mainly used in nanotechnology, primarily to construct particles known as quantum dots (QDs). The present study focuses on cadmium forms that differ compared to their solubility and their particle size (CdS micro- and uncoated nanoparticles (NPs)), and investigates their effects on mesangial and proximal tubular kidney cells. Cellular toxicity was evaluated by using Neutral Red, MTT and WST-1 assays. Intracellular Cd content was measured using inductively coupled optical emission spectrometry (ICP-OES) and cellular stress was investigated via the effects of Cd complexing bioligands using size exclusion chromatography ICP-MS (SEC-ICP-MS). Data indicated the variability of cytotoxicity responses after CdCl2, CdO, and CdS exposure that were strongly correlated to the cellular Cd content. Release of Cd2+ linked to solubility and particle degradation were considered to be the cause of these toxicities. The bioinduction of the Cd–MT also evidenced release of Cd2+. Our in vitro data identified heterogeneity of Cd toxicity that was dependent on the physico-chemical properties of the form studied and, when considering particle size, constitute an additional step toward the determination of nanoparticle effects in renal cells.
Introduction
Cadmium (Cd) is a well-known environmental pollutant that affects health adversely through occupational and environmental exposure. Cadmium toxicity is associated with several clinical complications depending on the dose, the route, and mainly duration of exposure. Acute toxicity primarily induces local pulmonary and gastric necrosis. Once absorbed, Cd is distributed by the blood, and tissue deposition is rapid. But Cd has a biological half-life of at least 20 years and the most documented toxicity results from chronic exposure.1,2 Therefore, Cd is considered to be an important cumulative pollutant, especially for the kidneys and all renal structures, i.e. glomeruli and tubules may be affected.3,4In vivo and in vitro toxicity studies have mainly focused on chloride cadmium salt (CdCl2), yet several other forms of cadmium, such as cadmium oxide (CdO) and cadmium sulfide (CdS), are also of concern in relation to occupational exposure. CdO is used as an electroplating chemical and a semiconductor in many applications such as photodiodes, phototransistors, photovoltaic cells, electrodes, and anti-reflective coatings. CdS also has many industrial uses, such as an inorganic photoconductor, pigment, and coloring for soaps. CdS is included in X-ray fluorescent screens in rectifiers, transistors, photovoltaic cells, and in solar cells.5 In nanotechnology, Cd is also primarily used in the construction of particles known as quantum dots (QDs), which are semiconductor crystal structures.6,7 The development of QDs shows great promise for the diagnosis of cancer and targeted drug delivery. However, after intravenous administration of QDs, Cd content has been observed in the liver and the kidneys even after 29 days, suggesting that these particles can pass through the kidneys.6,8 Nanoparticles (NPs) distribution in renal corpuscles is considered to be size-dependent.9,10 QDs smaller than approximately 10 nm, can be excreted by the kidneys, whereas the largest particles (50 nm) penetrate through the endothelial pores, enter the mesangium and accumulate inside mesangial cells. To reduce the risk of Cd toxicity, QDs have been developed with appropriate shells (e.g. CdTe/CdS/ZnS and CdSe/ZnS) and coatings. Nevertheless, the degradation of QDs is not yet fully understood and Cd ions (Cd2+) can be released after surface oxidation.11,12
In this study, we examined the relationship between cytotoxicity, cellular response and intracellular Cd content using different forms of Cd, i.e. soluble (CdCl2), particles at the micro scale (1 μm CdO and 1.8 μm CdS) and nano scale (10 nm CdS). The use of micro- and nanosize CdS gave the opportunity for the role of scale in cytotoxicity to be evaluated.
One of the proposed mechanisms of Cd toxicity is through an increase in the levels of damaging reactive oxygen species (ROS)13–15 and by releasing Cd2+ in cells.16,17 Cd2+ is not a Fenton metal and does not appear to generate ROS by itself but can replace iron and copper in various cytoplasmic and membrane proteins, thus increasing the amount of unbound free or chelated copper and iron ions.18 Cd can also induce lysosomal damage responsible for increased ROS generation.19 In response to Cd exposure, the liver and kidneys synthesize low molecular weight proteins, metallothioneins (MTs), which bind Cd with high affinity to form intracellular cadmium–metallothionein (Cd–MT) complexes that are biologically inert. In association with oxidative stress, over-expression of MTs can be considered to be adaptive cellular mechanisms and their synthesis may be one of several protective cellular mechanisms against the toxic effects of Cd.20,21 Elevation of both glutathione and MTs have been reported in cultured cells exposed to cadmium salts.22,23 In a recent study, Chen et al.24 reported a list of genes differentially expressed in response to Cd-based QD cellular exposure. Compared to controls, 31 up-regulated transcripts and 3 down-regulated transcripts were identified. Among up-regulated genes, many were stress-responsive and involved in the protective process of protein binding and metal ion binding, including that code for MTs.
The present study used various Cd forms and focused on their physicochemical properties, mainly solubility and particle size that can influence toxicity and cellular effects. The assays were conducted using two different renal cell lines, a glomerular mesangial cell line (IP15) and a proximal epithelial tubular cell line (LLC-PK1). Mesangial cells are perivascular pericytes located within the central portion of the glomerular tuft between the capillary loops and are involved in the control of glomerular hemodynamics,25,26 and LLC-PK1 cells retain the functional characteristics of isolated proximal tubular cells. Cytotoxic effects of Cd forms were studied using WST, MTT and Neutral Red (NR) assay. Intracellular Cd content was measured by inductively coupled optical emission spectrometry (ICP-OES). Cellular effects on the induction of metallothioneins (MTs) in response to Cd were screened and investigated by means of size exclusion chromatography coupled with inductively coupled plasma mass spectrometry (SEC-ICP-MS).
Results
Cadmium forms and particle characterization
Differences in metal forms have been shown to play an important role in toxicology. Therefore, we first determined the physicochemical properties of CdCl2, CdO, CdS micro- and nanoparticles in culture medium. Transmission electron microscopy was used to determine morphological information such as the diameter and size distribution of Cd stock solutions. Fig. 1 shows representative transmission electron microscopy images of CdO (Fig. 1a), CdS micro (Fig. 1b) and CdS nano (Fig. 1c). Particle size was calculated using a camera and ImageJ software: the size of CdO was 193.54 ± 10.01 nm, that of CdS micro 1045.27 ± 43.62 nm and of CdS nano 7.7 ± 1.4 nm. These data obtained in RPMI 1640 were different from those provided by the supplier for the powder, in particular for CdO (193.5 nm versus 1 μm from Sigma-Aldrich), thus confirming the solubility of CdO. In contrast, the reduction in size was less substantial for CdS micro (1045 nm versus 1800 nm, from Alfa Aesar). No image of CdCl2 was obtained due its complete solubility. The solubility in culture medium of the different forms of Cd tested was confirmed by ICP-OES. Concentration of [Cd] element was evaluated directly after acid digestion (total concentration noted as [Cd] acid) or after a first step of centrifugation ([Cd] supernatant) or filtration ([Cd] filtrate) on a nylon membrane that retains insoluble particles. At 0.130 μg ml−1 of Cd, CdCl2 and CdO were soluble, while only 10% of both CdS micro and nano were recovered in the supernatant or in the filtrate confirming CdO solubility in the culture medium (Fig. 2). Similar results were obtained with 0.0686 or 0.548 μg ml−1 of Cd (data not shown). Additional parameters (zeta potential and turbidimetry study) were previously described in Pujalté et al.27 and confirmed their dispersion state.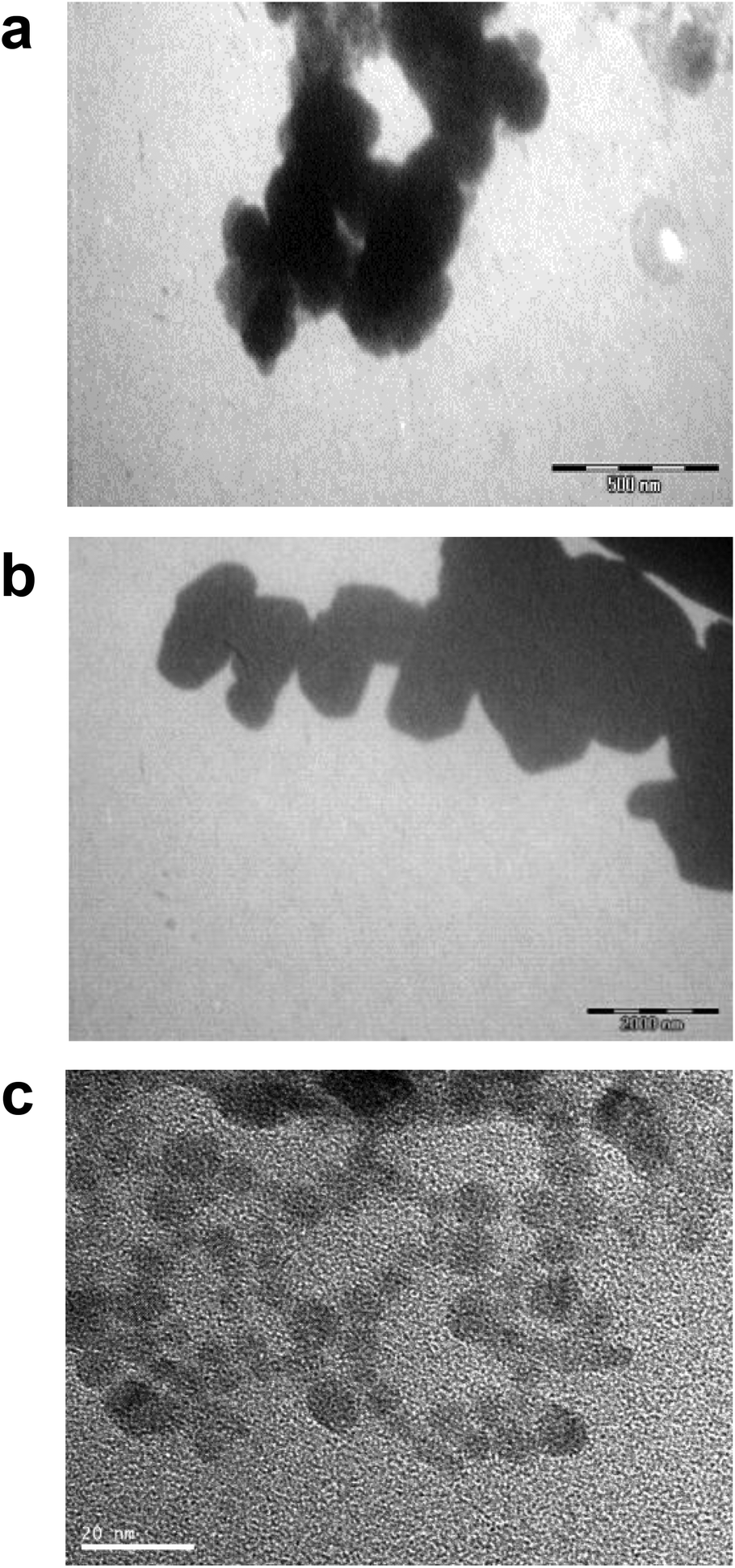 | ||
| Fig. 1 Microscopy characterization of Cd forms. Transmission electron microscopy (TEM) images of stock solutions of CdO (2 mg ml−1) (a), CdS micro- (100 μg ml−1) (b) and CdS nanoparticles (33.7 μg ml−1) (c). TEM scale bars: (a) 500 nm, (b) 2000 nm, (c) 20 nm. | ||
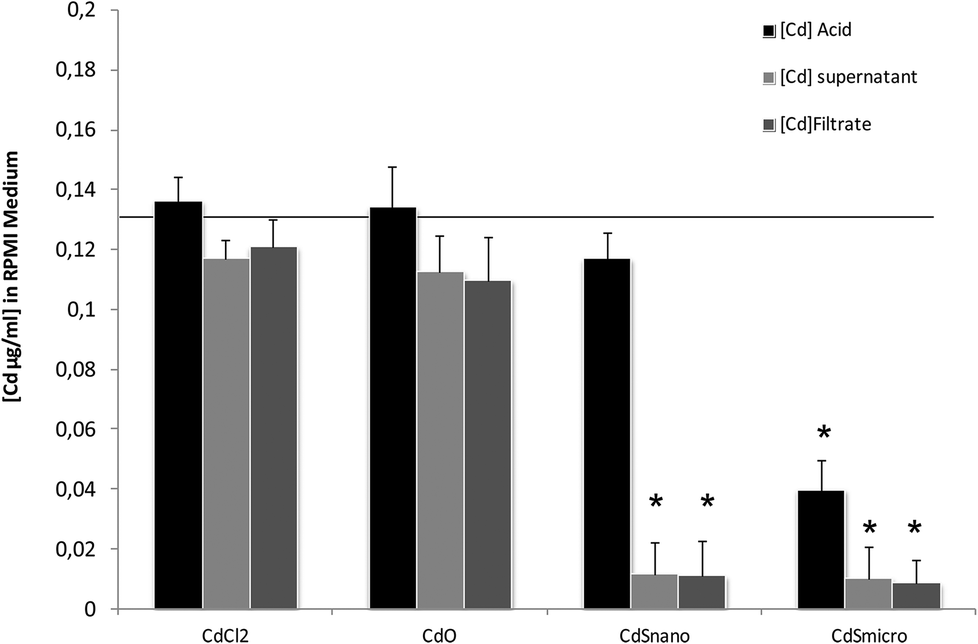 | ||
| Fig. 2 ICP-OES solubility characterization of Cd forms. Total [Cd] concentrations were measured using ICP-OES as described in the Materials and methods section. At 0.130 mg ml−1 the theoretical [Cd] concentration was indicated by the line. In culture medium, the Cd concentration [Cd], was measured directly after acidification ([Cd] acid), after subsequent centrifugation ([Cd] supernatant), or filtration on nylon membrane unit ([Cd] filtrate). | ||
Cadmium toxicity
Effects of CdCl2, CdO, CdS micro, and CdS nano on the mortality of IP15 (Fig. 3) and LLC-PK1 cells (Fig. 4) were assessed using different assays: WST-1, NR and MTT. Sub-confluent cells in 96-well plates were exposed for 24 h to varying concentrations of Cd forms diluted in RPMI 1460 serum-free medium.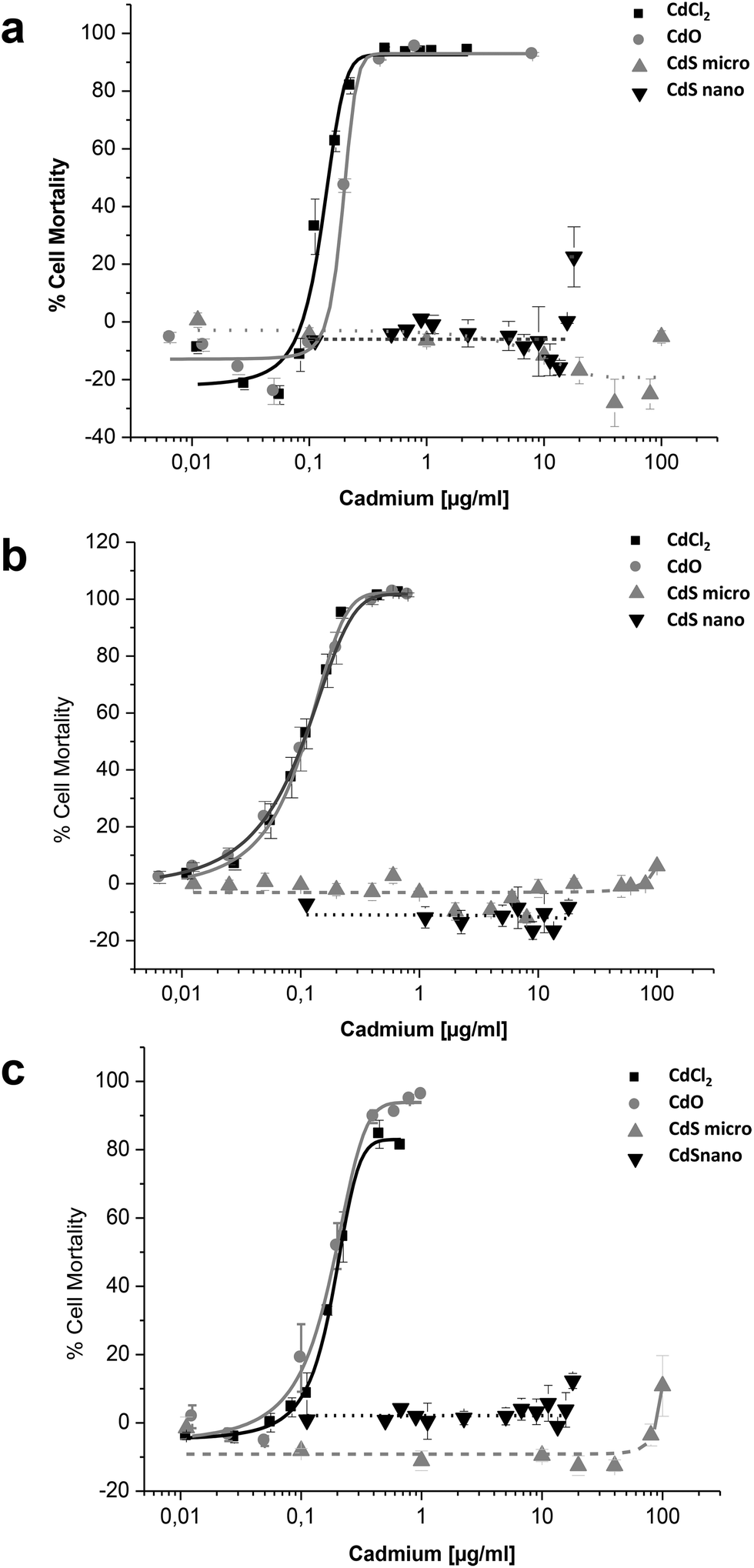 | ||
| Fig. 3 Cytotoxicity of Cd forms on IP15 cells. Effects of CdCl2, CdO, CdS micro- and CdS nanoparticles on the mortality of IP15 using the WST-1 (a), Neutral Red (b) and MTT (c) assays. Cells in RPMI-1640 serum-free medium were exposed to different concentrations of Cd for 24 h, as described in the Materials and methods section. Results are expressed as the percent of cell mortality compared to the control. The data are presented as the mean ± SE of at least three independent experiments. | ||
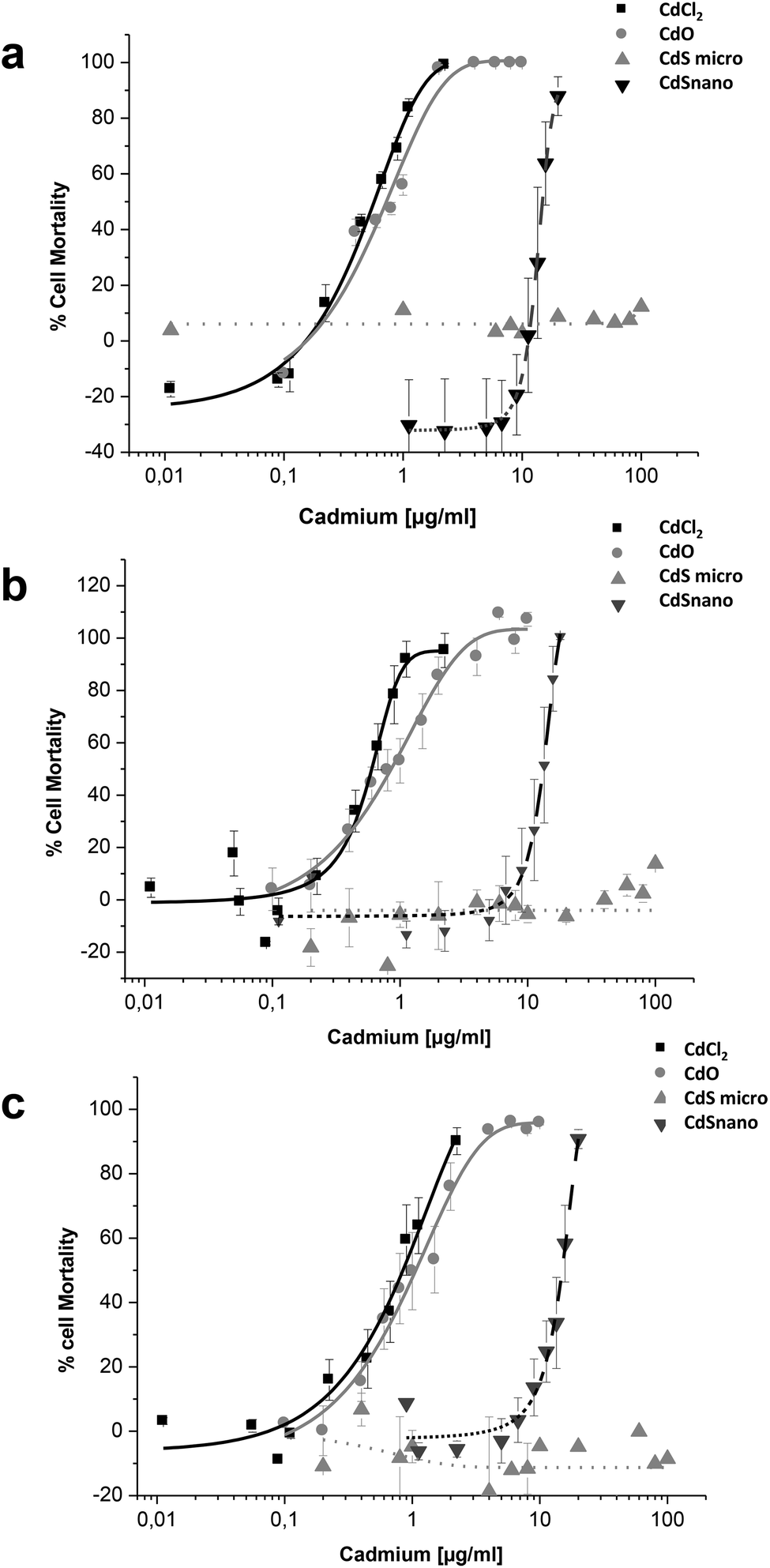 | ||
| Fig. 4 Cytotoxicity of Cd forms on LLC-PK1 cells. Effects of CdCl2, CdO, CdS micro- and CdS nanoparticles on the mortality of LLC-PK1 cells using the WST-1 (a), Neutral Red (b) and MTT (c) assays. Cells were exposed in serum-free medium to different concentrations of cadmium for 24 h, as described in the Materials and methods section. Results are expressed as the percent of cell mortality compared to the control. The data are presented as the mean ± SE of at least three independent experiments. | ||
CdCl2 and CdO exhibited dose-dependent toxicity on IP15 cells (Fig. 3). The CdCl2 toxicity curve obtained with WST-1 (Fig. 3a) was close to that of CdO with respective IC50 of 0.13 ± 0.01 μg ml−1 and 0.19 ± 0.01 μg ml−1. Similar IC50 values were also observed with both experimental NR (Fig. 3b) and MTT (Fig. 3c) assays. No significant toxicity was observed with CdS micro. At the maximal concentration used (18 μg ml−1), CdS nano had only a slight effect (22.54% ± 5 of cell mortality using the WST-1 assay).
Sensitivity of LLC-PK1 cells was different, with a shift in the IC50 of CdCl2 and CdO, and CdS nano-induced cell mortality above 11.24 μg ml−1 (Fig. 4). As an example, using the NR assay IC50 was calculated to be 0.52 ± 0.03 μg ml−1 for CdCl2, 0.703 ± 0.50 μg ml−1 for CdO, and 13.46 ± 1.29 μg ml−1 for CdS nano. No cell mortality was observed with CdS, even above 100 μg ml−1. No significant difference between the tests used was found on LLC-PK1 cells. The CdS nano IC50 index was determined at 13.46 ± 0.39 μg ml−1 for WST-1 (Fig. 4a), 15.76 ± 4.47 μg ml−1 for MTT (Fig. 4b) and 13.70 ± 2.5 μg ml−1 for NR (Fig. 4c), respectively.
Cadmium internalization
CdS nano was observed within cells after 24 h exposure using transmission electron microscopy. Selected images (Fig. 5) demonstrated that CdS nano (11.24 μg ml−1) was taken from LLC-PK1 cells without affecting the morphology (Fig. 5b) as compared to the control conditions (Fig. 5a). Cd internalization into IP15 cells was previously described in Pujalté et al.27 and both studies confirmed that CdS nano were found within cytoplasmic vesicles. The presence of Cd in cells was confirmed by electron dispersive X-ray spectrometry (EDS) (Fig. 5c).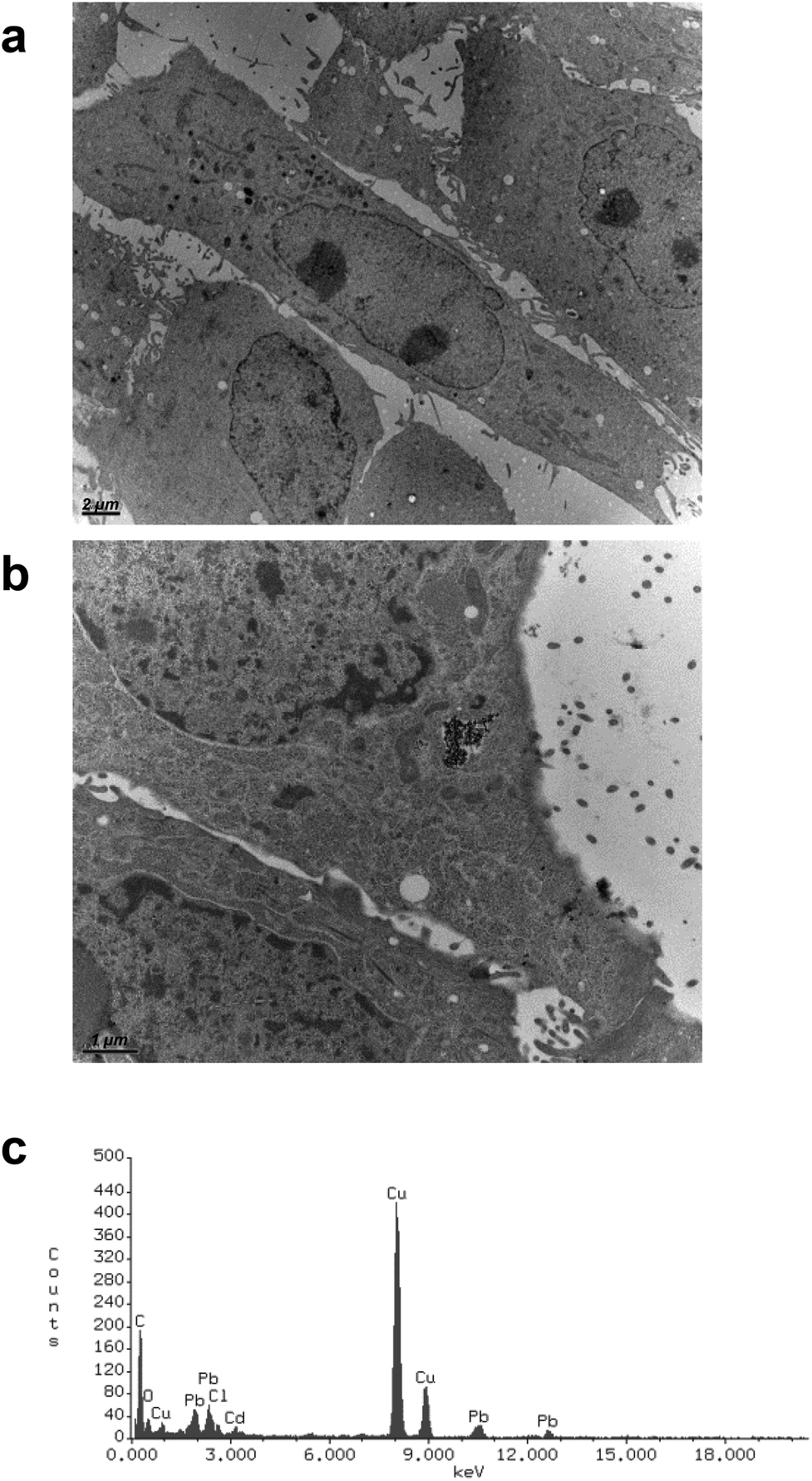 | ||
| Fig. 5 Transmission electronic microscopy observations of Cd nano uptake. Uptake and subcellular localization of CdS nano (arrows) were observed using transmission electron microscopy (TEM): control (a) and exposed LLC-PK1 cells (b) for 24 h to CdS nano (11.24 μg ml−1) (TEM scale bars: (a) 2 μm, (b) 1 μm). The presence of Cd element was confirmed by qualitative analysis using electron dispersive X-ray spectrometry (EDS) performed in cells (c). | ||
Cadmium uptake
Intracellular Cd was quantified after 24 h exposure to the different concentrations of Cd forms. Results are expressed as a ratio of Cd uptake (μg of Cd per mg of protein) by IP15 cells (Fig. 6a) and LLC-PK1 cells (Fig. 6b). ICP-OES analysis found that intracellular Cd concentrations after CdCl2 and CdO exposure were similar. Uptake by IP15 cells was lower when cells were exposed to CdS nano, but the Cd ratio values increased linearly and reached 0.366 ± 0.021 μg mg−1 protein, with 0.244 μg ml−1. Exposure to CdS micro induced a very low increase in the Cd ratio and thus probably explained why CdS under a microparticular form does not induce toxicity. In LLC-PK1 cells, the ratio after CdS nano exposure was as great as that obtained with CdCl2 and CdO.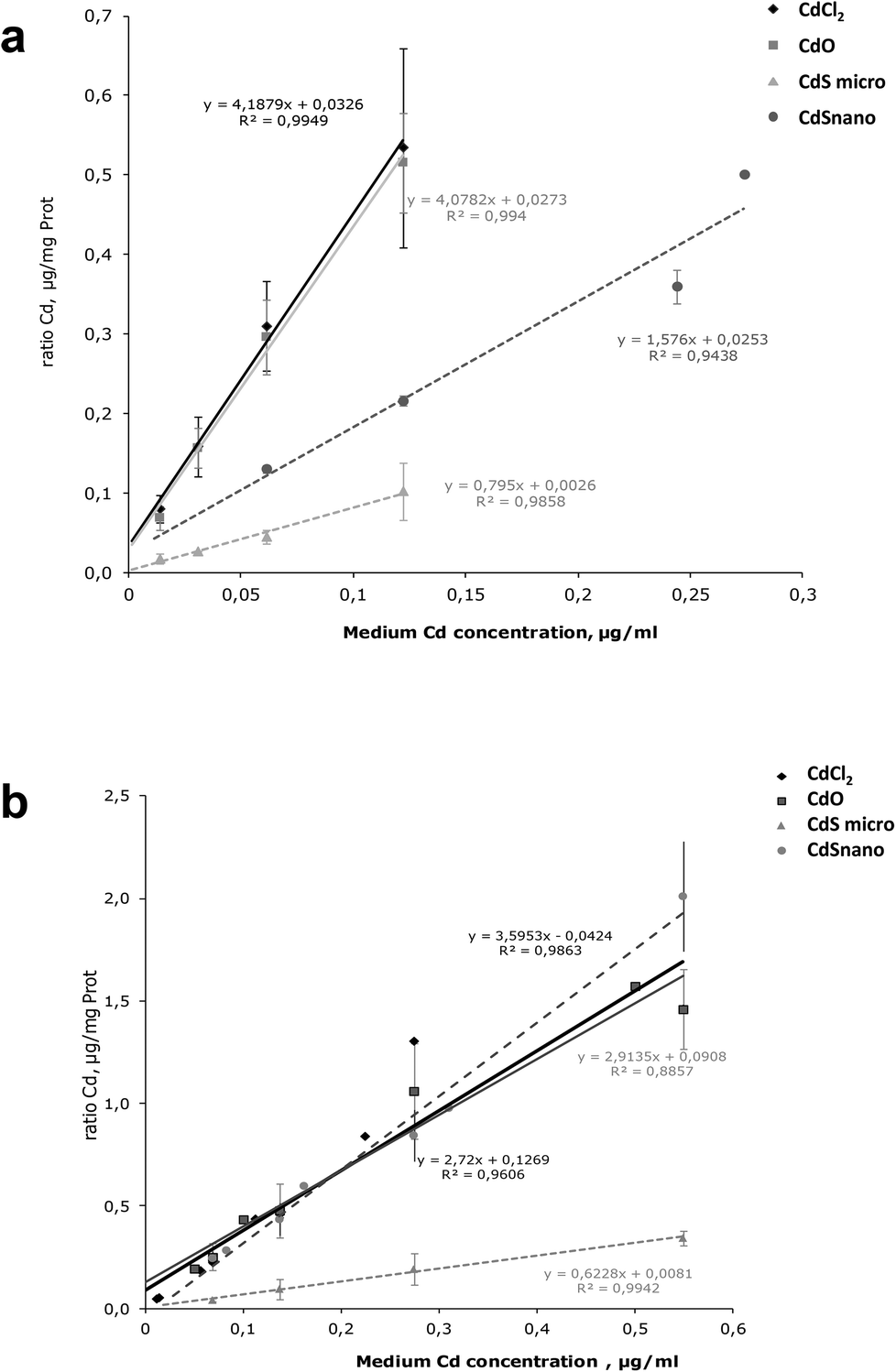 | ||
| Fig. 6 Cd uptake in renal cells. Comparison of Cd uptake in IP15 (a) and LLC-PK1 cells (b) after 24 h exposure to CdCl2, CdO, CdS micro- and CdS nanoparticles. Intracellular Cd was determined in cells by ICP-OES, after treatment as described in the Materials and methods section and expressed as a ratio of Cd μg mg−1 of cell protein. The data are presented as the mean ± SD of at least three independent experiments. | ||
Metallothioneins
The MT fractions of the cytosol of IP15 (Fig. 7a) and LLC-PK1 (Fig. 7b) cells exposed to CdCl2 and CdS nano were first investigated by SEC-ICP-MS for the screening of Cd complexing bioligands. SEC-ICP-MS chromatograms of both cell types exposed to CdCl2 presented the same Cd chromatographic pattern, with a major Cd peak at around 14 min matching the elution time of the MT standard (6000–7000 Da). This dominant Cd peak accounted for approximately 99% of the eluted Cd and was accompanied by three other minor Cd peaks, one in the void of the SEC column (at ca. 10.8 min) corresponding to a Cd-containing biomolecule of molecular weight (MW) ≥ 7000 Da, a second at around 16.5 min still within the MW range of MTs, and a third at around 32 min corresponding to low MW Cd-biomolecules interacting with the stationary phase of the column. As the chromatogram of the control sample was free of the dominant Cd peak, this constituted clear evidence of the formation of Cd-biomolecule complexes, which were most likely MT isoforms as expected. When cells were exposed to CdS nano, similar Cd chromatographic patterns were found but with a lower response of the induced Cd-bioligand (at 14 min). For example, the Cd-bioligand response induced by exposure of IP15 cells to CdS nano was approximately 80 times lower (in terms of peak height) than when cells were exposed to CdCl2 at identical Cd concentrations. A similar decreased bioinduction was observed for LLC-PK1 cells.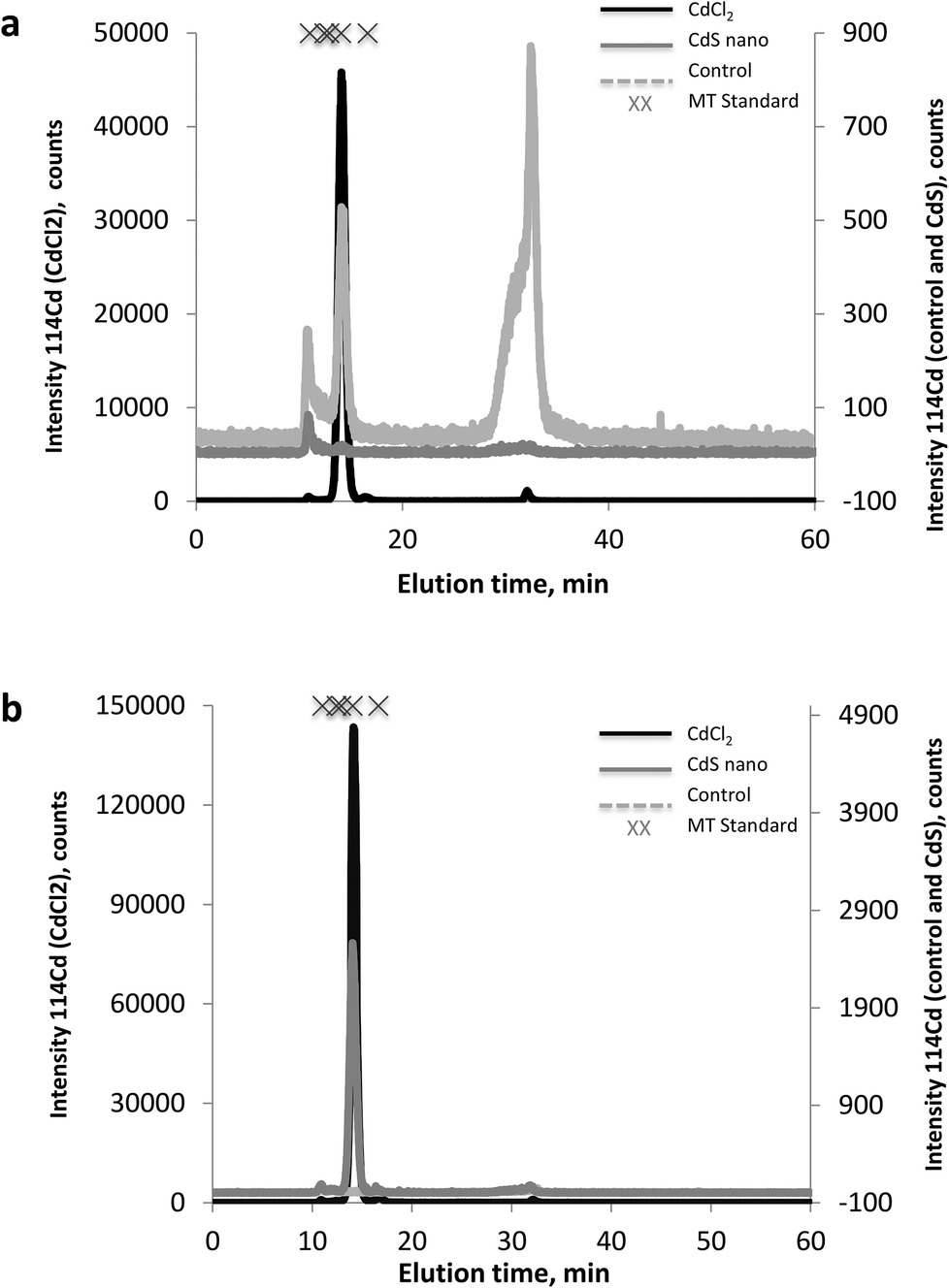 | ||
| Fig. 7 Metallothioneins by SEC-ICP-MS. 114Cd SEC-ICP-MS chromatograms from the cytosol of IP15 (a) and LLC-PK1 (b) cell lines: control (grey dashed line), exposed to CdCl2 (solid line), and to CdS nano (dashed line). | ||
Discussion
The kidney has been identified as the critical organ for Cd-related adverse effects occurring after prolonged exposure or high levels of concentration that result in morphological and functional changes.28–31 Cd deposits have been mainly found in the proximal tubules within the renal cortex. LLC-PK1 cells were previously validated for the assessment of renal tubular toxicity.32,33 Glomerular structures may also be affected and we previously reported that CdCl2 induced direct toxicity on isolated glomeruli and primary mesangial cell cultures.34,35 In this study, we used both glomerular (mesangial IP15 cells) and tubular culture models to document Cd-induced cytotoxicity. LLC-PK1 appeared to be more sensitive to CdS nano than glomerular cells. The impact of cell type could be due to difference in cell function. Proximal tubular cells express divalent transporter 1 in their membrane and such transporters are not described in glomeruli.36,37 LLC-PK1 cells have active and passive cell transports and the transporters were involved in intensive accumulation. Almost all studies on Cd toxicity have used Cd chloride (CdCl2); the present study goes further by also evaluating the toxicity of CdO, CdS micro-, and CdS nanoparticles. These differ by their physicochemical properties, mainly solubility and size.Our data indicated that CdCl2 and CdO had similar cytotoxic effects (IC50 indexes) but were more cytotoxic than CdS nano. Toxicity occurs when Cd2+ ions are released into cells. CdO was considered as an insoluble particle. However, ICP-OES analysis supported CdO solubility at the concentration used and transmission electron microscopy (TEM) did not find CdO particles inside cells at the concentration used, which further confirmed dissolution. Thus, at the concentration used, soluble CdO was sufficient to induce toxicity. CdS nano were found to be partially soluble and the amount of Cd2+ released might not be sufficient to induce a cytotoxic effect similar to that found by CdCl2 or CdO. CdS nano exhibited lower toxicity. Furthermore, they were more toxic than micro-sized CdS. We hypothesized that the cytotoxic effects of Cd are strongly correlated with the intracellular Cd content and can be promoted by a greater and easier solubilization of Cd nano. CdS nano appeared to be approximately hundred times less cytotoxic than CdCl2 or CdO, whereas intracellular Cd content in LLC-PK1 was similar for all forms. This result may be explained by the partial solubility of CdS nano. In the acellular assay, only 10% of Cd was found in the medium as evaluated by ICP-OES after removal of the particle by centrifugation or filtration. Even if lysosomes can promote NP dissolution by acidic pH, the amount of Cd2+ released might not be sufficient to induce cytotoxic effects as observed with CdCl2 and CdO. These findings were also consistent with the study reported by Derfus et al.12 where it was shown that the release of Cd2+ from CdSe NPs was responsible for cytotoxic effects. Since NPs are small, they can diffuse through the cell membrane using the endocytotic pathway and be localized in cytoplasmic vesicles, whereas insoluble micro-sized CdS are too large to penetrate into cells. Other studies using Fe2O3, CuO, ZnO, and platinium NPs have also indicated a higher toxicity as compared to micro-particles, indicating that size is an important parameter for toxicity.38–40 At the nanoscale, CdS releases more Cd2+ metal ions in solution. Moreover, in a cellular environment, the dissolution of NPs can be accentuated by acidification of the extracellular medium. Inside vesicles, digestion systems such as lysosomes decrease the pH of the cellular compartment and induce secretion of degradation enzymes. The present study demonstrated that CdS nano were internalized into cytoplasmic vesicles as observed using TEM microscopy. This is concordant with a previous report that found QDs to be internalized via lipid rafts processes or caveolae-mediated endocytosis.41 Das et al.,42 in a study of hepatoma (HepG2) and fibroblast (NIH3T3) cells, concluded that QD cytotoxicity was due not only to Cd ions but also to the presence of the particles themselves. A similar hypothesis was reported by Su et al.43 when comparing CdTe, CdTe/CdS, and CdCl2 in kidney HEK293 cells. It was also observed by Yeh et al.44 that Cd in kidneys from QD 705 had longer body retention than that of CdCl2, and observed that QD had even greater renal toxicity than CdCl2. Chen et al.24 also observed that with equal intracellular concentrations of Cd2+ ions in cells exposed to CdCl2 and QDs, CdCl2 had a much lower effect on metabolic activity and proliferation. They explained this difference by a “high local concentration” observed around the nuclei or certain organelles inside the cells and suggested that this local release of Cd ions was responsible for the cytotoxicity of Cd-based QDs. Additionally, the Trojan horse mechanism could participate in the toxicity of CdS NPs. Surface oxidation can induce release of Cd2+, thereby increasing the risk of renal toxicity.
The cellular mechanisms explaining Cd toxicity have been documented by in vivo and in vitro studies through the production of ROS and oxidative stress.45,46 As surface per unit mass is larger with NPs, increase of ROS production is higher; CdS NPs are reported to trigger an increase of ROS within cells whereas no oxidative stress was observed with micro-sized CdS.16 A direct correlation between the formation of ROS and cytotoxic effects has already been established for glomerular (IP15) and HK2 proximal tubular renal cells.27 These ROS products can induce oxidative stress, cellular damage (lipid peroxidation, lysosomal membrane damage, apoptosis) and molecular adaptive responses (antioxidant response, response to cellular stress, apoptosis) over short periods. Due to the high affinity of Cd2+ for sulfhydryl groups, complexes of Cd2+ with metal binding proteins, such as MTs, may be formed. The interaction of MTs with metal ions occurs through thiol groups. By binding Cd, MTs induce protection of sensitive target molecules that could be affected by Cd ions. Cd has been reported to be a strong inducer, as shown by an enhancement of the MT1/MT2 content in several tissues, especially the liver and the kidneys.47–49 Cd toxicity may occur when an imbalance exists between these protective proteins and intracellular Cd concentration.14 Cd is known to induce formation of radical oxygen in cells. Moreover, the ability of NPs to induce toxicity has been attributed to their increased surface reactivity. Associated with oxidative stress, expression of MTs was increased. In our study, MT fractions were assessed by SEC-ICP-MS for the screening of Cd complexing biologands in renal cells. Exposure to CdCl2 and CdS nano induced Cd–MT formation, especially in LLC-PK1 cells. We previously identified Cd-containing biomolecules by μHPLC coupled with ICP-MS, and confirmed the presence of known forms and identified two MTs (MT1) isoforms after CdS nano exposure in LLC-PK1 cells.50 Up-expression of MT1 has been described in vivo by Ohba et al.51 and Thijssen et al.52 It has been reported that expression of 73 genes was increased, especially that of MT1a, in rat epithelial cells (NRK-52E) after CdCl2 exposure.53 We observed that even though a lower cytotoxicity was induced by CdS nano, the bioinduction of the Cd-bioligand evidences release of Cd2+. Our results are consistent with those described in the literature. Cd can stimulate MTs in renal tubular cells and protect the cells but when total Cd concentration in cells increases above a certain level, protection becomes insufficient and toxic effects occur.
Experimental
Materials preparation
CdCl2 and CdO were purchased from Sigma-Aldrich (St-Quentin-Fallavier, France). CdS microparticle (CdS micro) was purchased from Alfa Aesar (Schiltigheim, France). Stock solution of CdCl2 was prepared as an aqueous stock solution (10 mM, CdCl2) and kept at 4 °C. Stock suspensions of CdO and CdS micro were prepared (2 mg ml−1 of Cd) in RPMI 1640 serum-free medium, without any additional medium supplement. Stock suspensions were frozen immediately after sonication (20 s, 9 times; Vibracell 75186, 130 W, 56–60 Hz). CdS nanoparticles (CdS nano) were manufactured by the Bordeaux Institute of Chemistry of Condensed Matter Physics (ICMCB, Bordeaux, France). The CdS nanoparticle (CdS nano) were prepared via a colloidal approach in an aqueous solution as previously described by Pujalté et al.27 This procedure yielded a yellow solution consisting of both [Cd] = 300 μM and [(NaPO3)n] = 300 μM.All products used for cell culture (RPMI 1640 and MEM media, PBS (phosphate buffer saline)) were purchased from Lonza (Verviers, Belgium), except for fetal bovine serum (FBS), which was from Eurobio (Les Ullis, France). WST-1 [2-(4-iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)-2H-tetrazolium] was purchased from Roche Diagnostics (Meylan, France). MTT [3-(4,5-dimethyl thiazol-2-yl)-2,5-diphenyltetrazolium bromide], Neutral Red, and bovine serum albumin (BSA), DTT, PMSF, rabbit liver metallothionein standards MT (013K7010) and MT-1 (014K7053) were purchased from Sigma-Aldrich (St-Quentin-Fallavier, France). One mg of each MT was dissolved in 1 ml of ultrapure water and store at −20 °C.
Prior to each cell culture experiment, Cd stock solutions were freshly diluted, after an additional sonication step (20 s, 3 times), in RPMI 1640, without red phenol (RP), serum-free medium (pH 7.3) before exposure. To directly compare Cd element expressed as μg ml−1 or μM, or μg cm−2, we chose to express all concentrations as μg ml−1 of Cd element used.
Physicochemical Cd form characterizations
Characterization of stock solutions of CdO and CdS micro and nano was performed using a Transmission Electronic Microscope (TEM, JEOL 2000FX) to determine particle size, shape and aggregation state (SIS software). Particles in RPMI 1640 serum-free medium were examined after subsequent deposition onto collodion-coated carbon grids. ICP-OES was used to evaluate dissolution of CdO, CdS micro, and CdS nano in RPMI 1640 serum-free medium. In preliminary studies, we investigated the ability of ICP-OES devices for detecting Cd accurately in culture media, with statistical characteristics of the calibrating functions and reliability of the method (sensitivity, limits of detection, limits of quantification, repeatability and reproducibility, accuracy54). Concentration of [Cd] element was evaluated directly, after a first step of centrifugation (100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 20 min) or after filtration on a nylon membrane unit in order to determine the soluble part of the different Cd forms used. All media, obtained directly or after an additional step containing the soluble part, were digested by 5% (v/v) of HNO3 and diluted 5-fold in deionized H2O before analysis by ICP-OES.
000 rpm for 20 min) or after filtration on a nylon membrane unit in order to determine the soluble part of the different Cd forms used. All media, obtained directly or after an additional step containing the soluble part, were digested by 5% (v/v) of HNO3 and diluted 5-fold in deionized H2O before analysis by ICP-OES.
Cell cultures
Glomerular mesangial human cells (IP15) obtained from Dr I. Dubus (University of Rouen, France) were cultured in RPMI 1640 medium containing penicillin (100 U ml−1), streptomycin (100 μg ml−1) and amphotericin B (0.25 μg ml−1), 2 mM L-glutamine, sodium pyruvate, non-essential amino acids and 10 mM Hepes supplemented with 10% (v/v) inactivated FBS.55 This cell line exhibits morphological (smooth muscle α-actin, collagen I) and functional properties (fibronectin synthesis, contractile response to angiotensin II) of mesangial cells, which are smooth muscle-derived cells.25,55,56 LLC-PK1 cells derived from the Hampshire pig were purchased from the European Collection of Cell Cultures (ECACC). These cells constitute an established cell line derived from a normal pig kidney displaying several characteristics of the proximal tubule. Cells were grown in EMEM (Eagle's minimum essential medium) supplemented with 10 mM Hepes, 2 mM L-glutamine, streptomycin (100 U ml−1) and penicillin (100 μg ml−1), with 5% (v/v) FBS. Both cell cultures grew in 75 cm2 culture flasks (Greiner BioOne, Courtaboeuf, France) and were maintained in 5% CO2–95% air (v/v) atmosphere. The media were changed every 2 days and cells were trypsinized when necessary (0.05% [v/v] trypsin–0.53 mM EDTA).Cadmium cytotoxicity
In order to choose appropriate Cd concentrations and to compare cell viability, we used different methods as Neutral Red (NR; 3-amino-7-dimethylamino-2-methylphenazine-hydrochloride), MTT and WST-1 assays. Sub-confluent cells in 96-well plates were exposed for 24 h to varying concentrations of CdCl2, CdO, and CdS particles diluted in RPMI-1640 serum-free medium, ranging from 0.0112 to 2.24 μg ml−1 for CdCl2, 0.0065 or 0.1 to 10 μg ml−1 for CdO, 0.01 to 100 μg ml−1 CdS micro, and 0.112 to 17.98 μg ml−1 CdS nano. In the NR assay, after exposure, cells were incubated with culture medium containing 4% NR, fixed with formaldehyde–CaCl2 solution, and finally extracted by acetic acid–ethanol solution. For the MTT assay, after NP exposure and additional 3 h incubation with culture medium containing MTT (0.5 mg ml−1), formazan salts were solubilized with DMSO. For WST-1 assay, the reagent was added directly to culture medium without additional solubilization. Absorbance was measured at 570 nm for the MTT, 450 nm for the WST-1, and 540 nm for the NR assay compared to a 630 nm reference using a multiscan photometer (Titertek, II, Labsystem). Data from at least 3 independent triplicates were expressed as percentage of dead cells compared to control.Cellular cadmium uptake
Metallothioneins by SEC-ICP-MS
After 24 h Cd exposure, MTs were first investigated for CdCl2 and CdS nano on IP15 (0.25 and 0.5 μg ml−1), and LLC-PK1 (0.112 and 0.224 μg ml−1), respectively. Sub-confluent cells in 100 mm Petri dishes were washed with PBS and scraped off in Tris/HCl (25 mM, pH 7.2) containing 1 mM dithiothreitol (DTT) and 0.1 mM phenylmethyl-sulfonylfluoride (PMSF). Cells were lyzed by a successive freeze–thaw procedure (described above in the Cd uptake section). Cytosols were separated by ultracentrifugation (4 °C, 120000g, 20 min) using a HimaCs 120GX (Hitachi, Tokyo, Japan). The recovered supernatant was heated at 95 °C for 5 min and ultra-centrifuged (4 °C, 120000g, 20 min) for fractionation by size exclusion chromatography. A Superdex peptide HR 10/30 (GE Healthcare, Uppsala, Sweden) column (10 × 300 mm × 5 μm) was used with an optimal molecular weight separation range between 100 and 7000 Da and an exclusion limit of 20000 Da. Before sample injection, the column was cleaned for 2 h with the mobile phase (50 mM Tris/HCl, pH 7.4) containing 5 mM β-mercaptoethanol and then conditioned overnight at 0.2 mL min−1 with the mobile phase. Metallothionein standards (6000–7000 Da) were diluted 350-fold and injected into the column to serve as elution time reference. 100 μl of sample were injected and isocratically fractionated at 0.7 ml min−1 for metallothionein screening. SEC-ICP-MS coupling was achieved by using an Agilent 1100 HPLC system (Agilent, Wilmington, DE, USA) connected to an Agilent 7500 ICP-MS (Agilent, Tokyo, Japan) for element-specific detection on 106Cd, 110Cd, 111Cd, 112Cd, 113Cd, 114Cd et 63Cu, 65Cu, 64Zn, and 66Zn.
Statistics
For cytotoxicity experiments, results were calculated using the formula (100 − (absorbance treated sample × 100/absorbance control sample)) and expressed as percent of cell mortality % ± SE of at least three independent experiments. Non-linear Boltzman regression analysis was performed using the Origin® software (Origin Lab. Corp, Northampton, MA, USA) and the IC50 (defined as concentration which induces 50% cell mortality increase) was calculated. Statistical calculations used for validating ICP-OES were performed using reference AFNOR XP T90-210. Values reported for Cd uptake were means ± SD of data points from triplicate assays and independent experiments. For all experiments, results with p < 0.05 were considered to be statistically significant.Conclusions
With nanomaterials used for biomedical applications, it is important to ensure that they do not have any adverse effects. However, when using Cd-based QDs, the nephrotoxic potential of Cd raises many questions. The present findings provide comparative data on the cytotoxicity of Cd forms. We suggest that toxicity and cellular responses were influenced by Cd size and solubility, which can modify Cd2+ released inside cells. The cytotoxic effects of Cd were correlated to the cellular Cd content, but specific properties of NPs and adaptative cell responses influenced their cytotoxicity. Many questions remain unanswered concerning the behaviour and toxicity of Cd-based QDs and further research concerning the release of Cd from cores and their biological reactivity needs to be carried out. Thus, these in vitro models can act as pre-screening tools and allow mechanistic approaches.Competing interest
The authors report no conflict of interest.Acknowledgements
This work was funded by ANSES (Agence Nationale de Sécurité Sanitaire de l'Alimentation, de l'Environnement et du Travail; EST 2010/2/87) and received support from the CRAq (Conseil Régional d'Aquitaine) for the spectrometry equipment. We gratefully acknowledge C. DePortal and J. Jorly for their help in ICP-OES measurements. Technical help for TEM studies was provided by E. Sellier (CREMEM). We thank the Bordeaux Imaging Center (BIC) for the TEM cell observations.References
- R. A. Goyer and M. G. Cherian, in, Metal toxicology, Academic press, San Diego, CA, 1995, pp. 389–412 Search PubMed.
- C. G. Elinder, C. Edling, E. Lindberg, B. Kagedal and O. Vesterberg, Br. J. Int. Med., 1985, 42, 754–760 CAS.
- D. M. Templeton and Y. Liu, Chem.-Biol. Interact., 2012, 188, 267–275 CrossRef PubMed.
- W. C. Prozialeck and J. R. Edwards, J. Pharmacol. Exp. Ther., 2012, 343, 2–12 CrossRef CAS PubMed.
- IARC, Cadmium and its compounds, in IARC Monograph, 1993, p. 104 Search PubMed.
- B. A. Rzigalinski and J. S. Strobl, Toxicol. Appl. Pharmacol., 2009, 238, 280–288 CrossRef CAS PubMed.
- P. Juzenas, W. Chen, Y. P. Sun, M. A. Coelho, R. Genralov, N. Genralova and I. L. Christensen, Adv. Drug Delivery Rev., 2008, 60, 1600–1614 CrossRef CAS PubMed.
- A. Sadaf, B. Zesham, Z. Wang, R. Zhang, S. Xu, C. Wang and Y. Cui, J. Nanosci. Nanotechnol., 2012, 12, 8287–8292 CrossRef CAS PubMed.
- M. L. Schipper, G. Lyer, A. L. Koh, Z. Cheng, Y. Ebenstein, A. Aharoni, S. Keren, L. A. Bentolila, J. Li, J. Rao, X. Chen, U. Banin, A. M. Wu, R. Sinclair, S. Weiss and S. S. Gambhir, Small, 2009, 5, 126–134 CrossRef CAS PubMed.
- X. Huang, L. Li, T. Liu, N. Hao, H. Liu, D. Chen and F. Tang, ACS Nano, 2011, 5, 5390–5399 CrossRef CAS PubMed.
- C. H. Lin, L. W. Chang, H. Chang, C. S. Yang, W. H. Lai, W. H. Chang and P. Lin, Nanotechnology, 2009, 20, 215101 CrossRef PubMed.
- A. M. Derfus, W. C. W. Chan and S. N. Bhatia, Nano Lett., 2004, 4, 11–18 CrossRef CAS.
- G. Gobe and D. Crane, Toxicol. Lett., 2010, 198, 49–55 CrossRef CAS PubMed.
- F. Thévenod, Nephron Physiol., 2003, 93, 87–93 CrossRef PubMed.
- J. Liu, W. Qu and M. B. Kadiiska, Toxicol. Appl. Pharmacol., 2009, 238, 209–214 CrossRef CAS PubMed.
- K. G. Li, J. T. Chen, S. S. Bai, X. Wen, S. Y. Song, Q. Yu, Y. Li and Y. Q. Wang, Toxicol. in Vitro, 2009, 23, 1007–1013 Search PubMed.
- B. I. Ipe, M. Lehnig and C. M. Niemeyer, Small, 2005, 1, 706–709 CrossRef CAS PubMed.
- B. L'Azou and F. Marano, in Nanoparticle toxicity mechanisms: Oxidative stress and inflammation, ed. P. Houdy, M. Lahmani and F. Marano, Springer Verlag, Berlin, 2011, 87–105 Search PubMed.
- G. Fotakis and J. A. Timbrell, Toxicol. Lett., 2006, 160, 171–177 CrossRef CAS PubMed.
- C. D. Klaassen, J. Liu and B. A. Diwan, Toxicol. Appl. Pharmacol., 2009, 238, 215–220 CrossRef CAS PubMed.
- G. Nordberg, T. Jin, X. Wu, J. Lu, J. Chen, Y. Liang, L. Lei, F. Hong, I. A. Berghadi and M. Norderg, J. Trace Elem. Med. Biol., 2012, 26, 197–200 CAS.
- T. A. Chin and D. M. Templeton, Toxicology, 1993, 77, 145–156 CrossRef CAS.
- Y. M. Xu, Y. Zhou, D. J. Chem, D. Y. Huang, J. F. Chiu and A. T. Y. Lau, Toxicol. Res., 2013, 2, 280–287 RSC.
- N. Chen, Y. He, Y. Su, L. Xiaoming, Q. Huang, H. Wang, X. Zhang, R. Tai and C. Fan, Biomaterials, 2012, 33, 1238–1244 CrossRef CAS PubMed.
- I. Dubus, B. L'Azou, M. Gordien, Y. Delmas, J. P. Labouyrie, J. Bonnet and C. Combe, Hypertension, 2003, 42, 956–961 CrossRef CAS PubMed.
- B. L'Azou, I. Dubus, C. Ohayon-Courtès and J. Cambar, Cell Biol. Toxicol., 2007, 23, 267–278 CrossRef CAS PubMed.
- I. Pujalté, I. Passagne, B. Brouillaud, M. Tréguer, E. Dureand, C. Ohayon-Courtès and B. L'Azou, Part. Fibre Toxicol., 2011, 8, 10 CrossRef PubMed.
- K. J. Ellis, S. H. Cohn and T. J. Smith, J. Toxicol. Environ. Health, 1985, 15, 173–187 CrossRef CAS PubMed.
- G. F. Norberg, Toxicol. Appl. Pharmacol., 2009, 238, 192–200 CrossRef PubMed.
- W. C. Prozialeck, J. R. Edwards, P. C. Lamar, J. Liu and V. S. Vaidya, Toxicol. Appl. Pharmacol., 2009, 238, 306–314 CrossRef CAS PubMed.
- A. Bernard, BioMetals, 2004, 17, 519–523 CrossRef CAS.
- B. L'Azou, M. Hengé-Napoli, L. Minaro, H. Mirto, M. P. Barrouillet and J. Cambar, Cell Biol. Toxicol., 2002, 18, 329–340 CrossRef CAS.
- D. S. Muller, P. Houpert, J. Cambar and M. H. Hengé-Napoli, Toxicol. Sci., 2008, 101, 254–262 CrossRef CAS PubMed.
- M. P. Barrouillet, M. Potier and J. Cambar, Exp. Nephrol., 1999, 7, 251–258 CrossRef CAS PubMed.
- B. L'Azou, I. Dubus, C. Ohayon-Courtès, J. P. Labouyrie, L. Perez, C. Pouvreau, L. Juvet and J. Cambar, Toxicology, 2002, 179, 233–245 CrossRef CAS.
- M. Abouhamed, J. Gburek, W. Liu, B. Torchalski, A. Wilhelm, N. A. Wolff, E. I. Christensen, F. Thévenod and C. P. Smith, Am. J. Physiol.: Renal Physiol., 2006, 290, F1525–F1533 CrossRef CAS PubMed.
- C. J. Ferguson, M. Wareing, D. T. Ward, R. Green, C. P. Smith and D. Riccardi, Am. J. Physiol.: Renal Physiol., 2001, 280, F803–F814 CAS.
- H. L. Karlsson, J. Gustafsson, P. Cronholm and L. Möller, Toxicol. Lett., 2009, 188, 112–118 CrossRef CAS PubMed.
- P. J. Moos, K. Chung, D. Woessner, M. Honeggar, N. S. Cutler and J. M. Veranth, Chem. Res. Toxicol., 2010, 23, 733–739 CrossRef CAS PubMed.
- J. Palka, H. Gehrke, M. Esselen, M. Türk, M. Crone, S. Bräse, T. Muller, H. Blank, W. Send, V. Zibat, P. Brenner, R. Schneider, D. Gerthsen and D. Marko, Chem. Res. Toxicol., 2009, 22, 649–659 CrossRef PubMed.
- L. W. Zhang and N. A. Monteiro-Riviere, Toxicol. Sci., 2009, 110, 138–155 CrossRef CAS PubMed.
- G. K. Das, P. Y. Chan, A. Teo, J. S. C. Loo, J. M. Anderson and T. T. Y. Tan, J. Biomed. Mater. Res., Part A, 2009, 93, 337–346 Search PubMed.
- Y. Su, S. Hu, C. Fan, Y. He, Q. Li, W. Li, L. Wang, P. Shen and Q. Huang, Biomaterials, 2010, 31, 4829–4834 CrossRef CAS PubMed.
- T. H. Yeh, J. P. Wu, L. W. Chang, M. H. Tsai, W. H. Chang, H. T. Stai, C. S. Yang and P. Lin, Nanotoxicology, 2010, 5, 91–97 CrossRef PubMed.
- A. M. Schrand, M. F. Rahman, S. M. Hussain, J. J. Schlager, D. A. Smith and A. F. Syed, Nanomed. Nanobiotechnol., 2010, 2, 544–568 CrossRef CAS PubMed.
- L. Wang, H. Wang, J. Li, D. Chen and Z. Liu, Arch. Environ. Contam. Toxicol., 2011, 61, 500–511 CrossRef CAS PubMed.
- M. P. Waalkes and C. D. Klaassen, Fundam. Appl. Toxicol., 1985, 5, 473–477 CrossRef CAS.
- M. Norberg and G. F. Nordberg, Metal Ions Life Sci., 2009, 5, 1–29 Search PubMed.
- I. Sabolic, D. Breljak, M. Skarica and C. M. Herak-Kramberger, BioMetals, 2010, 23, 897–926 CrossRef CAS PubMed.
- S. Mounicou, L. Ouerdane, B. L'Azou, I. Passagne, C. Ohayon-Courtès, J. Szpunar and R. Lobinski, Anal. Chem., 2010, 82, 6947–6957 CrossRef CAS PubMed.
- K. Ohba, Y. Okawa, Y. Matsumoto, Y. Nakamura and H. Ohta, J. Toxicol. Sci., 2007, 32, 103–105 CrossRef CAS.
- S. Thijssen, A. Cuypers, J. Maringwa, K. Smeets, N. Horemans, I. Lambrichts and E. VanKerkhove, Toxicology, 2007, 236, 29–41 CrossRef CAS PubMed.
- M. Tokumoto, T. Ohtsu, A. Honda, Y. Fujiwara, N. Huisamitsu and M. Satoh, J. Toxicol. Sci., 2011, 36, 127–129 CrossRef CAS.
- C. Ohayon-Courtès, I. Passagne, C. De Portal, C. Pouvreau, J. Cambar and B. L'Azou, J. Toxicol. Environ. Health, Part A, 2007, 70, 750–759 CrossRef PubMed.
- I. Dubus, B. Vendrely, I. Christophe, J.-P. Labouyrie, Y. Delmas, J. Bonnet and C. Combe, Kidney Int., 2002, 62, 857–867 CrossRef CAS PubMed.
- I. Dubus, S. Sena, J. P. Labouyrie, J. Bonnet and C. Combe, Life Sci., 2005, 77, 3366–3374 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |