Molecular selective photocatalytic decomposition of alkylanilines by crystalline TiO2 particles and their nanocomposites with mesoporous silica
Masataka
Yasui
,
Kiyofumi
Katagiri
,
Shoji
Yamanaka
and
Kei
Inumaru
*
Department of Applied Chemistry, Institute of Engineering, Hiroshima University, 1-4-1, Higashi-Hiroshima, Hiroshima 739-8527, Japan. E-mail: inumaru@hiroshima-u.ac.jp
First published on 17th September 2012
Abstract
Molecular selective photocatalytic decomposition of alkylanilines in water was investigated by using crystalline TiO2 particles (P-25) and their nanocomposites with mesoporous silica. In the nanocomposites, pre-formed TiO2 particles were embedded into surfactant-templated mesoporous silica with tuned pore sizes (1.4–5.3 nm) as described in our previous reports (Chem. Commun. 2005, 2131–2133; J. Mater. Chem. 2011, 21, 12117–12125). Contrary to the generally accepted knowledge of TiO2 photocatalysts, pristine TiO2 particles showed molecule selective photocatalysis: decomposition of 4-n-heptylaniline (NHA) was much faster than that of 4-n-butylaniline (NBA) or p-toluidine (4-methylaniline, PT) in a mixed aqueous solution. After NHA had almost disappeared, decomposition of NBA accelerated. When the TiO2 particles were surrounded by mesoporous silica, the molecular selectivity was strongly modified: the nanocomposites showed high molecular selectivity for NBA decomposition and the reaction of PT was considerably suppressed. The origin of molecular selectivity can basically be ascribed to preferential molecular adsorption, and the difference in selectivity between alkylanilines and alkylphenols is discussed in terms of the interaction of the molecules with the nanocomposite surfaces.
Introduction
Molecular selectivity is becoming increasingly important in environmental protection, because it provides an effective way to remove dilute pollutants. One strategy is molecular selective adsorption using nanoscale-designed organic–inorganic materials.1–4 Another important strategy is to design molecule selective photocatalysts. TiO2 is a well known, versatile photocatalyst.5–19 Environmental purification is a promising and extensively studied fields for the application of TiO2 photocatalysts. However, TiO2 photocatalysts are known not to be molecule selective. A promising way towards molecule selective photocatalysis is to combine molecule selective adsorption and photocatalysis.20–22 Although many researchers have studied the enhancement of photocatalytic activity by dispersing TiO2 onto various adsorbents,23–31 few studies have reported molecule selective photocatalysis.Surfactant-templated mesoporous silica32–41 has been a subject of great interest because of its potential applications as host structures, adsorbents, and catalysts for reactions involving large molecules.42–47 To develop new functions such as catalysis, many studies have been carried out by combining transition metal oxides with mesoporous structures.48–58 The most frequently used method is to disperse transition metal oxide particles onto the pore-walls of mesoporous silica. To improve the activity of the photocatalysts, many researchers have attempted to combine TiO2 with mesoporous silica by simple methods such as impregnation, sol–gel methods, and surface grafting. However, it seems that materials of this type generally have poorer photocatalytic performance than well-crystallized pure TiO2 particles such as commercially available “P-25”. This is probably because of the low crystallinity of the highly-dispersed TiO2 particles on the mesoporous supports, and the limited TiO2 loading level (ca. 10–25 wt%). To overcome these limitations, some papers have reported crystalline mesoporous TiO2 materials and their applications in photocatalysis.59,60
We reported an approach to combine preformed crystalline TiO2 and surfactant-templated mesoporous silica61–63 to achieve molecule selective photocatalysis for the decomposition of organic contaminants in water. We employed preformed well-crystallized TiO2 particles (P-25) directly as the source material for nanocomposite synthesis. These nanocomposites have a unique structure in which TiO2 crystalline particles are surrounded by mesoporous silica.62,63 After our reports, a trial of selective organic transformation was also made using similar TiO2–mesoporous silica nanocomposites, which indicates the potential applications of this composite material.64 In our previous study, we investigated the decomposition of alkylphenols.62,63 The molecular selectivity of the photocatalytic decomposition was ascribed to the molecular selectivity of preferential adsorption into the nanospaces of the mesoporous silica. Since phenolic hydroxyl groups are known to show acidity, examining the behaviour of molecules such as anilines, containing basic groups, is the next stage.
Here we report molecule selective photocatalytic decomposition of alkylanilines by TiO2 and TiO2–mesoporous silica nanocomposites. The differences between the behaviour of alkylanilines and that of alkylphenols were revealed and discussed in terms of molecule–photocatalyst surface interactions.
Experimental methods
Sample preparation
Detailed sample preparation procedures were described in our previous reports.62,63n-Octadecyl-grafted TiO2 (C18-TiO2) particles were prepared by the following procedure: 2 g TiO2 powder (Degussa P-25) was dehydrated in vacuo at 473 K for 1 h in a glass high vacuum system, and then refluxed with 5 g n-octadecyltriethoxysilane in freshly-distilled toluene for 48 h. After refluxing, the reaction mixture was filtered and the residue was washed with distilled toluene and anhydrous methanol. Using a CHN analyser (PerkinElmer 2400II), the number of grafted alkyl groups on the TiO2 particles was determined to be 2.5 × 10−4 mol g−1, corresponding to a surface density of 2.9 molecules nm−2.Mesoporous silica (MPS(2.7)) and the composite catalyst (NC(2.7)) were prepared by the following procedure: 4.4 g hexadecyltrimethylammonium bromide was dissolved in ca. 230 g hot water (333 K). Then 17 g of 35% ammonia solution was added. A weighed amount of TiO2 (Degussa P-25, 7.3 g) or C18–TiO2 was ultrasonically dispersed in the solution. After adjusting the pH to 11.8 by adding ammonia solution, 17 g tetraethoxysilane (TEOS) was quickly added to the mixture, under vigorous stirring. After aging for 1 h, the precipitate was filtered, washed with water, and dried at 353 K for one day. The materials were calcined at 813 K for 6 h to obtain the mesoporous composite materials. The nanocomposites prepared from unmodified TiO2 and C18–TiO2 are denoted as NC(2.7) and C18-NC(2.7), respectively. “2.7” represents the pore diameter (nm), as determined by nitrogen adsorption at 77 K. All nanocomposites contain 60 wt% TiO2. MPS(2.7) was prepared according to the same procedure, without the addition of TiO2.
To investigate the effect of pore size on the molecular selectivity of the photocatalysts, we prepared a series of samples using surfactants with different alkyl-chain lengths. The preparation procedures were the same as those used to prepare NC(2.7), except that C8H17N(CH3)3Br (3.2 g) or C10H21N(CH3)3Br (4.4 g) was used as the template. To expand the pores, 3.68 g tridecane was added to a solution of C16H33N(CH3)3Br (3.7 g dissolved in 34 g water),65 and then the nanocomposite was prepared in a similar way to that used for NC(2.7). In this case, 6.36 g TiO2 and 14.7 g triethoxysilane were used.
Characterization of nanocomposites
Nitrogen adsorption isotherms were measured at 77 K using a Belsorp-mini (BEL Japan Inc., Osaka, Japan). TEM and SEM images were taken with a JEM-2010 transmission electron microscope and a JSM-6320FS scanning electron microscope, respectively (JEOL, Tokyo, Japan). X-ray powder diffraction patterns were measured with X-ray diffractometers M18XHF (MacScience, Japan; low angle regions) and D8 Advance (Bruker AXS, Germany; high angle regions) using Cu-Kα radiation.Photocatalytic test
Photocatalytic tests were carried out as follows: typically 30 mg of the composite photocatalyst was added to 300 g of a mixed aqueous solution of 4-n-heptylaniline (NHA), 4-n-butylaniline (NBA), and p-toluidine (4-methylaniline, PT). Their initial concentrations were 2.4–2.6 × 10−5 mol dm−3. After adsorption of the phenols onto the catalysts had reached equilibrium, the mixture was irradiated with a 500 W Xe lamp, under stirring. The solution was analysed using an HPLC equipped with a UV detector (Shimadzu LC-10).Results and discussion
Structure of the nanocomposites
The nanocomposites used in this study are the same as those reported in our previous study. The X-ray powder diffraction patterns of selected samples63 are shown in Fig. 1. Mesoporous silica (MPS(2.7)), without TiO2 incorporation, showed intense diffraction peaks in the low angle region, attributed to hexagonally ordered mesopores. The nanocomposites gave much weaker diffraction peaks than mesoporous silica, and also showed peaks from crystalline TiO2 (mixture of anatase and rutile types) at higher angles. The TiO2 diffraction peaks for the nanocomposites were identical to the TiO2 peaks in the original TiO2 (P-25), indicating that there was no phase change in the TiO2 crystals. The nitrogen adsorption isotherms of the nanocomposites showed a step from nitrogen capillary condensation, indicating the presence of mesopores templated by the surfactants. Table 1 summarizes the properties derived from the nitrogen adsorption isotherms.63 According to TEM and SEM images, which were presented in our previous paper,63 a large fraction of the TiO2 particles was embedded into the mesoporous silica. In the case of C18-NC(2.7), almost all the TiO2 particles were embedded into mesoporous silica. | ||
| Fig. 1 X-ray diffraction patterns of mesoporous silica and nanocomposites: (a), TiO2 (P-25); (b) mesoporous silica (MPS(2.7)); (c) NC(1.4); (d) NC(1.9); (e) NC(2.7); (f) NC(5.3); (g) C18-NC(2.7). See ref. 63. | ||
| Sample | Templateb | S BET c(m2 g−1) | V p d(cm3 g−1) | Pore sizee (nm) | |
|---|---|---|---|---|---|
| d K | d C | ||||
| a See ref. 63. b Cn represents CnH2n+1(CH3)3NBr. c BET surface area. d Total pore volume. e Diameters. dK was determined using Kelvin's equation and multilayer adsorption thickness. dC = 4Vp/SBET. | |||||
| MPS(2.7) | C16 | 1016 | 0.87 | 2.7 | 3.4 |
| NC(5.3) | C16 + tridecane | 482 | 0.60 | 5.3 | 5.0 |
| NC(2.7) | C16 | 405 | 0.36 | 2.7 | 3.5 |
| NC(1.9) | C10 | 452 | 0.24 | (1.9) | 2.1 |
| NC(1.4) | C8 | 310 | 0.15 | (1.4) | 1.9 |
| C18-NC(2.7) | C16 | 444 | 0.37 | 2.7 | 3.3 |
Molecular selective photocatalysis of pristine TiO2 and the nanocomposite photocatalysts
First we checked the behaviour of the mixed molecules under irradiation without photocatalysts (blank test). Fig. 2a shows the time courses for the blank test. After the photo-irradiation started, the concentrations of the three compounds decreased, indicating that decomposition of these molecules occurred without photocatalysts, but the decomposition rates were slow. When MPS(2.7) was added to the solution, a large decrease in NHA concentration was observed because of preferential adsorption of the NHA molecules to MPS(2.7) prior to the start of irradiation (Fig. 2b). A decrease in the concentration of NBA was also observed, but it was much smaller than that observed for NHA. The adsorption of PT on MPS was small, similar to that of NBA. | ||
| Fig. 2 Time courses of concentrations of alkylanilines in aqueous solutions under irradiation. (a) Blank test, (b) mesoporous silica (MPS(2.7)) only, without TiO2 addition. | ||
Fig. 3 shows the time courses during the photocatalytic reactions for pristine TiO2 and for some nanocomposite photocatalysts. It should be noted here, as shown in Fig. 3a, that pristine TiO2 showed considerable molecular selectivity for decomposition of NHA; when TiO2 was added to a mixed solution of alkylanilines, the concentration of NHA decreased significantly because of adsorption onto TiO2, while the adsorption of NBA was quite small, and the adsorption of PT was much smaller again. After the irradiation started, NHA decomposed much faster than NBA or PT. The first-order plot for this reaction (Fig. 3b) clearly shows the difference in the decomposition rates between these molecules. In Fig. 3a and 3b, it is clearly observed that decomposition of NBA accelerated just after almost all NHA disappeared. A slight acceleration of PT decomposition was also observed once NBA decomposition had largely proceeded. These trends indicate that pristine TiO2 preferentially decomposed the alkylanilines in the order NHA ≫ NBA > PT. The molecule selective photocatalysis of pristine TiO2 will be discussed in a later section.
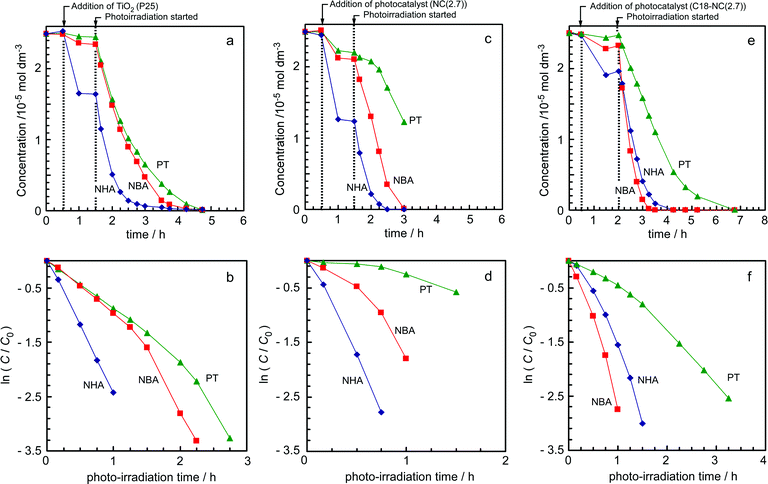 | ||
| Fig. 3 Time courses of concentrations of alkylanilines in aqueous solutions under irradiation. (a), (b) Pristine TiO2 (P25); (c), (d) NC(2.7); (e), (f) C18-NC. (b), (d), and (f) are the corresponding first-order plots. | ||
In the case of NC(2.7) (Fig. 3c and 3d), there was a much clearer difference in the molecular selectivity of photocatalytic decomposition between NBA and PT. Their initial reaction rate constants were very different, as can be seen from the initial stages of the reaction time courses. This point is clearly indicated in the first order plots of this reaction (Fig. 3d), in contrast to the case of pristine TiO2 (Fig. 3b). The reaction rates were in the order NHA > NBA ≫ PT, based on the initial reaction stage.
Fig. 4 compares the rate constants derived from the first order plots. The rate constant k for NHA was clearly larger in the presence of the nanocomposite photocatalyst NC(2.7) than in the presence of pristine TiO2 (P25). This means that the photocatalytic reaction was accelerated by incorporating the TiO2 into a composite with mesoporous silica. This acceleration may be ascribed to concentration of the NHA molecules into the nanospaces of the mesoporous silica surrounding the TiO2 particles; the effective concentration around the TiO2 surfaces could be increased by this preferential adsorption of NHA. On the other hand, the decomposition rate of PT was significantly lower in the presence of NC(2.7), compared with that in the presence of pristine TiO2, while the rate of decomposition of NBA was similar with both catalysts.
 | ||
| Fig. 4 First-order rate constants for photocatalytic decomposition of alkylanilines. The constants were determined from the initial 30 min of the first-order plots. | ||
Fig. 4 also contains results obtained with pore-size controlled nanocomposites. As the pore size increased to 5.3 nm (NC(5.3)) the decomposition selectivity decreased slightly. Although the results of NC(1.9) were similar to those of NC(2.7); for NC(1.4) the reaction rates of all compounds were significantly smaller than those obtained with the other nanocomposites. This may be ascribed to slow diffusion rates of the molecules in the narrow pores of NC(1.4) and/or access to the TiO2 surfaces being partly hindered by silica overlayers.
Fig. 3e and f represent the time courses of the reactions and the first order plots for C18-NC(2.7), which was prepared using surface organo-modified TiO2. In this case, the molecular selectivity for NBA was significantly higher than for the other nanocomposites: the kinetic constant as well as the decomposition rate of NBA exceeded those of NHA under these conditions.
The factors governing molecular selectivity
It has been widely considered that TiO2 photocatalysts show no molecular selectivity. In our previous study,62,63 we demonstrated that a TiO2 particle–mesoporous silica nanocomposite showed molecule selective decomposition of nonylphenol in a mixed aqueous solution of alkylphenols. In that case, nonylphenol was preferentially adsorbed into the nanospaces of the mesoporous silica and then was decomposed selectively. In the present decomposition experiment with mixed alkylanilines, however, the pristine TiO2 (P-25) showed molecular selectivity: NHA decomposed much faster than NBA or PT in the mixed aqueous solution. Since it was obvious that NHA was preferentially adsorbed onto TiO2 (see Fig. 3a), the molecule selective decomposition of NHA can reasonably be ascribed to selective adsorption on the TiO2 surface. As shown in Fig. 3a and b, decomposition of NBA accelerated after NHA was almost completely decomposed, and decomposition of PT accelerated after the concentration of NBA had decreased. This order may be the same as the order of preferential adsorption of molecules from aqueous solution onto TiO2 surfaces. It is notable that the order (NHA > NBA > PT) coincides with the order of hydrophobicity. There are some reports that preferential molecular adsorption onto TiO2 leads to molecule selective photocatalysis.20,66 Recently, Liu et al. reported that anatase particles with exposed {001} facets showed molecule selective photocatalysis towards the decomposition of dyes.66In contrast, for the nanocomposites such as NC(2.7) and C18-NC, in which TiO2 particles were surrounded by mesoporous silica, selectivity of NBA against PT was greatly increased compared to the selectivity found for pristine TiO2. For NC(2.7), the reaction rate of PT was much smaller than that of NBA, while for pristine TiO2, the reaction rates of NBA and PT are comparable. For C18-NC(2.7), the enhancement of selectivity for the decomposition of NBA was significant: the order of the molecular selectivity was reversed, although the amount of NHA adsorbed was much larger than that of NBA (Fig. 3e). That is, it appeared that the NBA molecules concentrated into the nanospaces of the nanocomposites such as C18-NC(2.7) were more efficiently decomposed than the NHA molecules. Such a phenomenon was not observed for alkylphenols; the order of decomposition rates of alkylphenols can be simply explained by the order of the amounts adsorbed onto the composite photocatalysts.61–63 It should be noted here again that, in the case of pristine TiO2, the order of reaction rates of alkylphenols could also be simply explained by the amounts adsorbed. At present the reason for these differences between alkylanilines and alkylphenols are not clearly proven. It is likely, however, that molecular interactions between silica surfaces and the adsorbed molecules play an important role. As was shown in our previous study,4 amino groups of alkylanilines strongly interact with SiOH groups on the walls of mesoporous silica via hydrogen bonding and/or weak acid–base interactions. In the case of the TiO2–mesoporous silica nanocomposites, a large amount of NHA was adsorbed onto the silica walls; Fig. 2b shows this very high affinity of NHA molecules for mesoporous silica surfaces. A strong interaction between NHA molecules and Si–OH might retard the mobility of NHA molecules in the mesoporous silica channels. Of course, increasing the effective concentration of NHA molecules around TiO2 must contribute to the reaction rate of NHA molecules. The strong adsorption of a molecule onto the silica walls has trade-off effects on the reaction rates: the concentration of the molecules around the TiO2 increases, but the mobility of the molecules is decreased by the strong adsorption. In contrast, NBA molecules showed moderate adsorption onto the silica walls. This is likely to both increase the concentration of NBA molecules around TiO2 and maintain high mobility of the molecules in the mesoporous channels.
Here, the diffusion of active species such as hydroxyl radicals from the TiO2 surfaces should be considered. The lifetime and diffusion distance of hydroxyl radicals depend greatly on the conditions. It was reported that, in ambient air, hydroxyl radicals diffuse as far as 70–125 μm (diffusion coefficient D = 0.22 × 10−4 m2 s−1, and their lifetime was estimated to be 170 μs).67 In water, the diffusion is much slower (D = 2.8 × 10−9 m2 s−1)68,69 and the diffusion distance is significantly shorter as a result of the presence of reactant molecules or radical scavengers; for example, the diffusion distance can be as short as 6 nm in cells.70 In the case of our composite photocatalysts, organic reactants were concentrated into the nanospaces of the mesoporous silica around the TiO2 particles, and it is likely that the reaction of hydroxyl radicals and the reactants occurs inside the pores. Under such conditions, even if the hydroxyl radicals diffuse faster than the reactant molecules, the molecular selectivity is expected to be governed by the adsorption selectivity of the reactant molecules.
The significant difference between NC(2.7) and C18-NC(2.7) (Fig. 4) is interesting, and the reasons for the difference are not yet clear but are probably concerned with the structure of the composites. A possibility is that although C18-NC(2.7) contains a very high concentration of TiO2 particles embedded in the mesoporous silica, the surface organo-modifications on the TiO2 may produce some gaps or voids between the TiO2 surfaces and the walls of the silica mesopores, resulting in slow diffusion of the molecules strongly adsorbed onto the silica surfaces to the TiO2 surfaces. This point will be an interesting subject for future studies.
Conclusions
Molecule selective photocatalytic decomposition of alkylanilines in water was investigated using crystalline TiO2 particles (P-25) and their nanocomposites with mesoporous silica. Pristine TiO2 particles showed molecule selective photocatalysis: decomposition of 4-n-heptylaniline (NHA) was much faster than that of 4-n-butylaniline (NBA) or p-toluidine (4-methylaniline, PT) in a mixed aqueous solution. When the TiO2 particles were surrounded by mesoporous silica, the molecular selectivity was strongly modified. The nanocomposites with mesoporous silica showed much higher molecular selectivity for NBA decomposition. The reason for the molecular selectivity is likely to be the effect of concentration and mobility of the molecules in the mesoporous channels; it is likely that a crucial factor for the molecular selectivity is chemical interactions (such as hydrogen bonding and weak acid–base interaction) between the organic molecules and silica surfaces.Acknowledgements
This study was partly supported by a Grant-in-Aid on Priority Areas (Area No. 417, Grant No. 17029043) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), by Grants-in-Aid for Scientific Research (B) (Nos. 20350095 and 17360389), and for Creative Scientific Research (No. 17GS0206), and the Funding Program for World-Leading Innovative R&D on Science and Technology (FIRST) Program from Japan Society for the Promotion of Science (JSPS).References
- K. Inumaru, Y. Inoue, S. Kakii, T. Nakano and S. Yamanaka, Phys. Chem. Chem. Phys., 2004, 6, 3133–3139 RSC.
- K. Inumaru, J. Kiyoto and S. Yamanaka, Chem. Commun., 2000, 903–904 RSC.
- K. Inumaru, S. Kakii, H. Yoshida and S. Yamanaka, Chem. Lett., 2010, 39, 1215–1217 CrossRef CAS.
- K. Inumaru, Y. Inoue, S. Kakii, T. Nakano and S. Yamanaka, Chem. Lett., 2003, 32, 1110–1111 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37 CrossRef CAS.
- D. F. Ollis, E. Pelizzetti and N. Serpone, Environ. Sci. Technol., 1991, 25, 1522 CrossRef CAS.
- M. R. Hoffman, S. T. Martin, W. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69 CrossRef.
- M. A. Fox and M. T. Dulay, Chem. Rev., 1993, 93, 341 CrossRef CAS.
- A. Hagfeldt and M. Gratzel, Chem. Rev., 1995, 95, 49 CrossRef CAS.
- M. Gratzel, Inorg. Chem., 2005, 44, 6841–6851 CrossRef.
- L. I. Halaoui, N. M. Abrams and T. E. Mallouk, J. Phys. Chem. B, 2005, 6334–6342 CrossRef CAS.
- M. Suzuki, C. C. Waraksa, T. E. Mallouk, H. Nakayama and K. Hanabusa, J. Phys. Chem. B, 2002, 106, 4227–4231 CrossRef CAS.
- A. L. Linsebigler, G. Lu and J. T. Yates Jr., Chem. Rev., 1995, 95, 735 CrossRef CAS.
- B. Kraeutler and A. J. Bard, J. Am. Chem. Soc., 1979, 100, 4317 CrossRef.
- H. G. Yu, H. Irie and K. A. Hashimoto, J. Am. Chem. Soc., 2010, 132, 6898–6899 CrossRef CAS.
- A. A. Ismail and D. W. Bahnemann, J. Phys. Chem. C, 2011, 115, 5784–5791 CAS.
- H. Irie, T. Shbanuma, K. Kamiya, S. Miura, T. Yokoyama and K. Hashimoto, Appl. Catal., B, 2010, 96, 142–147 CrossRef CAS.
- R. Wang, K. Hashimoto, A. Fujishima, M. Chikuni, E. Kojima, A. Kitamura, M. Shimohigoshi and T. Watanabe, Nature, 1997, 388, 431–432 CrossRef CAS.
- H. H. Mohamed, C. B. Mendive, R. Dillert and D. W. Bahnemann, J. Phys. Chem. A, 2011, 115, 2139–2147 CrossRef CAS.
- K. Inumaru, M. Murashima, T. Kasahara and S. Yamanaka, Appl. Catal., B, 2004, 52, 275–280 CrossRef CAS.
- T. Kasahara, K. Inumaru and S. Yamanaka, Microporous Mesoporous Mater., 2004, 76, 123–130 CrossRef CAS.
- Y. Paz, C. R. Chim., 2006, 9, 774–787 CrossRef CAS.
- C. Anderson and A. J. Bard, J. Phys. Chem., 1995, 99, 9882–9885 CrossRef CAS.
- C. Anderson and A. J. Bard, J. Phys. Chem. B, 1997, 101, 2611–2616 CrossRef CAS.
- T. Torimoto, S. Ito, S. Kuwabata and H. Yoneyama, Environ. Sci. Technol., 1996, 30, 1275–1281 CrossRef CAS.
- S. Usseglio, P. Calza, A. Damin, C. Minero, S. Bordiga, C. Lamberti, E. Pelizzetti and A. Zecchina, Chem. Mater., 2006, 18, 3412–3424 CrossRef CAS.
- W. G. Fan, L. Gao and J. Sun, J. Am. Ceram. Soc., 2006, 89, 731–733 CrossRef CAS.
- K. Mogyoroshi, A. Farkas, I. Dekany and A. Dombi, Environ. Sci. Technol., 2006, 36, 3618–3624 CrossRef.
- J. Chen, L. Eberlein and C. H. Langford, J. Photochem. Photobiol., A, 2002, 148, 183–189 CrossRef CAS.
- S. Fukahoi, H. Ichiura, T. Kitaoka and H. Tanaka, Environ. Sci. Technol., 2003, 37, 1048–1051 CrossRef.
- W. A. Adams, M. G. Bakker, T. Macias and I. A. Jefcoat, J. Hazardous Mater, 2004, B112, 25 Search PubMed.
- T. Kresge, M. E. Leonowicz, W. J. Roth, J. T. Vartuli and J. S. Beck, Nature, 1992, 359, 710 CrossRef.
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge, K. D. Schmitt, C. T.-W. Chu, D. H. Olson, E. W. Sheppard, S. B. McCullen, J. B. Higgins and J. L. Schlenker, J. Am. Chem. Soc., 1992, 114, 10834 CrossRef CAS.
- T. Yanagisawa, T. Shimizu, K. Kuroda and C. Kato, Bull. Chem. Soc. Jpn., 1990, 63, 988 CrossRef CAS.
- S. Inagaki, A. Koiwai, N. Suzuki, Y. Fukushima and K. Kuroda, Bull. Chem. Soc. Jpn., 1996, 69, 1449 CrossRef CAS.
- P. T. Tanev and T. J. Pinnavaia, Science, 1995, 267, 865–867 CAS.
- S. A. Bagshaw, E. Prouzet and Pinnavaia, Science, 1995, 269, 1242–1244 Search PubMed.
- D. Y. Zhao, J. L. Feng, Q. S. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548–552 CrossRef CAS.
- D. Y. Zhao, Q. S. Huo, J. L. Feng, B. F. Chmelka and G. D. Stucky, J. Am. Chem. Soc., 1998, 120, 6024–6036 CrossRef CAS.
- K. M. McGrath, D. M. Dabbs, N. Yao, I. A. Aksay and S. M. Gruner, Science, 1998, 279, 1289–1289 CAS.
- K. M. McGrath, D. M. Dabbs, N. Yao, K. J. Edler, I. A. Aksay and S. M. Gruner, Langmuir, 2000, 16, 398–406 CrossRef CAS.
- A. Stein, Adv. Mater., 2003, 15, 763 CrossRef CAS.
- A. Sayari and S. Hamoudi, Chem. Mater., 2001, 13, 3151 CrossRef CAS.
- R. Anwander, Chem. Mater., 2001, 13, 4419 CrossRef CAS.
- A. P. Wight and M. E. Davis, Chem. Rev., 2002, 102, 3589 CrossRef CAS.
- V. Dufaud and M. E. Davis, J. Am. Chem. Soc., 2003, 125, 9403–9413 CrossRef CAS.
- A. Sayari, S. Hamoudi and Y. Yang, Chem. Mater., 2005, 17, 212–216 CrossRef CAS.
- D. R. Rolison, Science, 2003, 299, 1698 CrossRef CAS.
- M. Ziolek, I. Kilos, B. Nowak, I. Sobczak, P. Decyk, M. Trejda and J. C. Volta, J. Phys. Chem. Solids, 2004, 65, 571 CrossRef CAS.
- K. Takehira, Y. Ohishi, T. Shishido, T. Kawabata, K. Takaki, Q. H. Zhang and Y. Wang, J. Catal., 2004, 224, 404 CrossRef CAS.
- W. Zhang and T. J. Pinnavaia, Catal. Lett., 1996, 38, 261 CrossRef CAS.
- E. P. Reddy, L. Davydov and P. Smirniotis, Appl. Catal., B, 2003, 42, 1 CrossRef CAS.
- A. Corma, M. J. Domine, A. Gaona, J. L. Jorda, M. T. Navarro, F. Rey, J. Perez-Pariente, J. Tsuji, B. McCulloch and L. T. Nemeth, Chem. Commun., 1998, 2211 RSC.
- B. J. Aronson, C. F. Blanford and A. Stein, Chem. Mater., 1997, 9, 2842 CrossRef CAS.
- K. A. Koyano and T. Tatsumi, Chem. Commun., 1996, 145 RSC.
- M. D. Alba, Z. H. Luan and J. Klinowski, J. Phys. Chem., 1996, 100, 2178 CrossRef CAS.
- R. Raja, J. M. Thomas, M. D. Jones, B. F. G. Johnson and D. E. W. Vaughan, J. Am. Chem. Soc., 2003, 125, 14982 CrossRef CAS.
- V. Ayala, A. Corma, M. Iglesias, J. A. Rincon and F. Sanchez, J. Catal., 2004, 224, 170 CrossRef CAS.
- Y. Shiraishi, N. Saito and T. Hirai, J. Am. Chem. Soc., 2005, 127, 12820–12822 CrossRef CAS.
- H. Shibata, T. Ogura, T. Mukai, T. Ohkubo, H. Sakai and M. Abe, J. Am. Chem. Soc., 2005, 127, 16396–16397 CrossRef CAS.
- (a) T. Kasahara, K. Inumaru, S. Yamanaka, The 42nd Symposium on Basic Science of Ceramics, Jan. 2004, 1F04, Nagaoka, Japan Search PubMed; (b) K. Inumaru, S. Yamanaka, T. Kasahara and M. Yasui, JP2005-314208 Search PubMed.
- K. Inumaru, T. Kasahara, M. Yasui and S. Yamanaka, Chem. Commun., 2005, 2131–2133 Search PubMed.
- K. Inumaru, M. Yasui, T. Kasahara, Y. Kubota, K. Yamaguchi, A. Yasuda and S. Yamanaka, J. Mater. Chem., 2011, 21, 12117–12125 RSC.
- Y. Shiraishi, Y. Sugano, D. Inoue and T. Hirai, J. Catal., 2009, 264, 175–182 CrossRef CAS.
- S. K. Jana, R. Nishida, K. Shindo, T. Kugita and S. Namba, Microporous Mesoporous Mater., 2004, 68, 133 CrossRef CAS.
- (a) S. Liu, J. Yu and M. Jaroniec, J. Am. Chem. Soc., 2010, 132, 11914–11916 CrossRef CAS; (b) Q. Xiang, J. Yu and M. Jaroniec, Chem. Commun., 2011, 47, 4532–4534 RSC.
- K. Naito, T. Tachikawa, M. Fujitsuka and T. Majima, J. Phys. Chem. C, 2008, 112, 1048–1059 CAS.
- H. A. Schwarz, J. Phys. Chem., 1969, 73, 1928–1937 CrossRef CAS.
- E. Codorniu-Hernández and P. G. Kusalik, J. Am. Chem. Soc., 2011, 134, 532–538 CrossRef.
- R. Roots and S. Okada, Radiat. Res., 1975, 64, 306–320 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |