
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn electron-delocalized sp2-N hybridized organic electrode enables sustainable and high-efficiency electrochemical ammonium removal†
Haoyuan
Qiu
a,
Minjie
Shi
 *a,
Peipei
Zhang
a,
Yueheng
Tao
a,
Xinyue
Zhang
a,
Jun
Yang
a,
Jingxin
Zhao
*bc and
Huan
Pang
*d
*a,
Peipei
Zhang
a,
Yueheng
Tao
a,
Xinyue
Zhang
a,
Jun
Yang
a,
Jingxin
Zhao
*bc and
Huan
Pang
*d
aSchool of Materials Science and Engineering, Jiangsu University of Science and Technology, Zhenjiang, Jiangsu 212003, P. R. China. E-mail: shiminjie@just.edu.cn
bNanotechnology Center, School of Fashion and Textiles, The Hong Kong Polytechnic University, Hung Hom, Kowloon, Hong Kong 999077, P. R. China. E-mail: JingxinZhao@tju.edu.cn
cDepartment of Mathematics and Information Technology, The Education University of Hong Kong, Tai Po, New Territories, Hong Kong SAR, 999077, P. R. China
dSchool of Chemistry and Chemical Engineering, Yangzhou University, Yangzhou, Jiangsu 225009, P. R. China. E-mail: panghuan@yzu.edu.cn
First published on 15th April 2025
Abstract
Water scarcity emerges as a critical global challenge, with the purity of aquatic ecosystems intimately linked to ammonium concentrations. The removal of ammonium ions (NH4+) is vital for mitigating ammonium contamination and promoting the sustainability of nitrogenous resources. Capacitive deionization (CDI) utilizing organic electrodes offers a promising electrochemical solution through a unique “ion coordination” mechanism; however, its efficacy is hindered by the presence of electrochemically inert units within the molecular framework for ion capture. Here, we introduce a rod-shaped DHPZ organic compound designed as a CDI electrode, distinguished by four imine rings and lone pair electrons in sp2 orbitals. This configuration establishes a hybridized sp2-N framework that exhibits significant electron delocalization and an exceptionally low HOMO–LUMO gap of 1.18 eV, enhancing its affinity for fast, stable, and efficient NH4+ capture. The DHPZ-based CDI device achieves an impressive NH4+ removal capacity of 136.6 mg g−1 at 1.2 V, a swift removal rate of 4.55 mg g−1 min−1, and outstanding regeneration (95.76% retention after 200 cycles), positioning it among the leading technologies in current CDI devices for NH4+ adsorption. Furthermore, we have developed interconnected CDI devices for targeting NH4+ removal from real wastewater, highlighting a sustainable and innovative approach to water remediation.
Introduction
The scarcity of freshwater, comprising a mere 2.53% of the Earth's total water supply, with only 0.34% readily accessible for human use, poses a substantial global challenge.1–4 Ammonium contamination from sources such as agricultural runoff and industrial wastewater represents a significant threat to freshwater ecosystems. The unintentional discharge of ammonium-laden wastewater leads to acidification and eutrophication, adversely affecting aquatic life and jeopardizing the safety of drinking water.5–9 Typical domestic wastewater contains approximately 40 to 80 mg L−1 of total nitrogen, with ammonium ions (NH4+) constituting around 82%. In light of NH4+ serving simultaneously as a beneficial resource and an environmental contaminant, the removal of NH4+ from wastewater becomes essential for curtailing nitrogen pollution and advancing practices of sustainability.10–13 A variety of techniques, including struvite crystallization, ion exchange, and reverse osmosis, are utilized for NH4+ removal from wastewater; yet, each method comes with its own set of limitations, including ecological impacts and steep costs. Struvite crystallization requires the addition of chemicals to regulate pH and is effective mainly at high ammonium levels. Ion exchange and adsorption processes involve lengthy treatment times and generate significant amounts of secondary waste. Additionally, reverse osmosis depends on energy-intensive pumps and does not specifically target NH4+.14,15 Therefore, the development of cost-effective, energy-conserving, and environmentally friendly technologies for the targeted extraction of NH4+ poses a significant challenge, especially in cases of wastewater with low ammonium content.Capacitive deionization (CDI), recognized as an electrochemical technique, is emerging as a promising solution for desalination, water purification, and the elimination of pollutants. This approach offers a multitude of advantages, such as reduced operational expenses, energy conservation, ecological compatibility, and the prevention of secondary contaminants.16–19 There is growing interest in applying CDI systems for extracting NH4+ from wastewater with low ammonium levels, wherein the use of pseudocapacitive electrodes has shown significant potential.19,20 These electrodes generate considerable electrochemical capacitance via redox processes, enhancing ion adsorption and selectivity in CDI applications. Although progress has been made with a range of pseudocapacitive electrodes, especially those composed of metal compounds such as MnO2, TiO2, MXenes, and Prussian blue analogues, intrinsic shortcomings remain.9,14,20,21 The dependence of these metal compounds on limited mineral resources, notably metal ores and geologic deposits, greatly limits their accessibility. Moreover, the pseudocapacitive reactions of these metal compounds are characterized by the formation and alteration of intermediate species, with some undergoing nonreversible changes that impede reaction rates and reduce long-term efficiency.22
Organic materials are progressively being identified as viable contenders for pseudocapacitive electrodes in CDI applications.23–29 A notable advantage lies in their minimal environmental footprint, as they can be synthesized from renewable resources, thereby alleviating the risks associated with the disposal of heavy metals typically present in metal compounds.25,30–33 More impressively, the inherent pseudocapacitive characteristics of organic materials enable them to capture ions through direct interactions with their functional groups, utilizing a distinctive “ion coordination” mechanism that transpires without any phase transition during the electrochemical process.22,34 Moreover, their versatile structures provide adaptable frameworks that are unimpeded by the lattice expansion and structural strain associated with metal compounds. These attributes render organic matters suited as promising pseudocapacitive electrodes in CDI technologies. Notwithstanding their promising attributes, the application of organic electrodes in electrochemical deionization is still in its formative stages. The realization of their full potential is encumbered by the significant presence of electrochemically inert units within the organic molecular framework, resulting in an insufficient density of accessible active sites that undermines the ion removal efficacy of the CDI devices.
In this study, we have pioneered an innovative organic material, denoted as DHPZ, meticulously engineered as a CDI electrode with exceptional NH4+ capture capability. The DHPZ molecule features an extraordinary orbital architecture, incorporating four imine rings that contain highly accessible C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N redox-active sites and lone pair electrons positioned within an sp2 orbital. This innovative design engenders a distinctive non-equivalent hybridized sp2-N framework, characterized by extensive electron delocalization and an extremely narrow HOMO–LUMO gap of merely 1.18 eV. Such a configuration endows the DHPZ electrode with a high affinity for rapid, stable, and efficient NH4+ capture, as validated by in situ characterization and theoretical calculations. When integrated into a CDI device, the DHPZ electrode demonstrates remarkable performance, achieving a high NH4+ removal capacity of 136.6 mg g−1, a rapid average rate of 4.55 mg g−1 min−1 and impressive regeneration performance, retaining 95.76% of its initial efficiency even after 200 cycles, outperforming current CDI devices in NH4+ removal. COMSOL simulations further corroborate the high NH4+ penetration and distribution throughout the DHPZ electrode. Bridging the gap between laboratory innovation and practical application, we have successfully integrated the DHPZ-based CDI device into an electrochemical system, demonstrating its efficacy in removing NH4+ from real wastewater and paving the way for sustainable environmental remediation solutions.
N redox-active sites and lone pair electrons positioned within an sp2 orbital. This innovative design engenders a distinctive non-equivalent hybridized sp2-N framework, characterized by extensive electron delocalization and an extremely narrow HOMO–LUMO gap of merely 1.18 eV. Such a configuration endows the DHPZ electrode with a high affinity for rapid, stable, and efficient NH4+ capture, as validated by in situ characterization and theoretical calculations. When integrated into a CDI device, the DHPZ electrode demonstrates remarkable performance, achieving a high NH4+ removal capacity of 136.6 mg g−1, a rapid average rate of 4.55 mg g−1 min−1 and impressive regeneration performance, retaining 95.76% of its initial efficiency even after 200 cycles, outperforming current CDI devices in NH4+ removal. COMSOL simulations further corroborate the high NH4+ penetration and distribution throughout the DHPZ electrode. Bridging the gap between laboratory innovation and practical application, we have successfully integrated the DHPZ-based CDI device into an electrochemical system, demonstrating its efficacy in removing NH4+ from real wastewater and paving the way for sustainable environmental remediation solutions.
Results and discussion
Molecular configuration and electronic structure
The DHPZ molecule was synthesized using an efficient method that involved a solvent-free condensation reaction between 2,5-dihydroxy-1,4-benzoquinone (DHBQ) and phenazine-2,3-diyldiamine (TTFQ) precursors, as shown in Fig. 1a. The successful synthesis of the DHPZ molecule can be confirmed through Nuclear Magnetic Resonance (NMR) analyses. The solid-state 13C NMR spectrum (Fig. 1b) unveils distinct signals corresponding to specific carbon environments: two peaks at 127.9 ppm and 140.2 ppm are assigned to the carbon atoms in CN bonds of the imine rings, while a peak at 144.3 ppm is attributed to the carbon atoms in C–N bonds of the imine rings. Additionally, a peak at 129.3 ppm belongs to the carbon atoms in CC bonds of the benzene rings. In the liquid-state 1H NMR spectrum, two resonances at 7.98 ppm and 7.72 ppm are designated for the hydrogen atoms in the benzene rings and a resonance at 7.64 ppm is associated with the hydrogen atoms in the imine rings (Fig. S1†). The orbital configuration of the resultant DHPZ molecule exhibits unique characteristics, including four imine rings and lone pair electrons situated in sp2 orbitals, leading to a non-equivalent hybridized sp2-N framework. The π electrons of the neutral state DHPZ molecule are 38, aligning with the 4n + 2 Hückel's rule (where n = 1, 2, 3, etc.), which signifies a high degree of aromaticity.35 The π-electron localization function (ELF-π) illustrated in Fig. 1c reveals that both the DHBQ and TTFQ precursors exhibit fragmented ELF-π isosurfaces, indicative of their low levels of conjugation. Remarkably, upon condensing DHBQ and TTFQ to create the DHPZ molecule, there is a considerable enhancement in conjugation levels, with the isosurface rings extending throughout the entire molecular framework, encompassing both benzene and imine rings within the DHPZ molecule. As observed from the scanning electron microscope (SEM) image, the DHPZ molecule displays rod-shaped morphological characteristics, each measuring a few micrometers in length (Fig. S2†). This distinctive one-dimensional rod-like configuration creates longer and uninterrupted channels for ion diffusion, thereby enhancing the accessibility of active sites within the DHPZ molecule. More characterization studies about the structural characteristics of the DHPZ molecule are provided in Fig. S3–S6.†
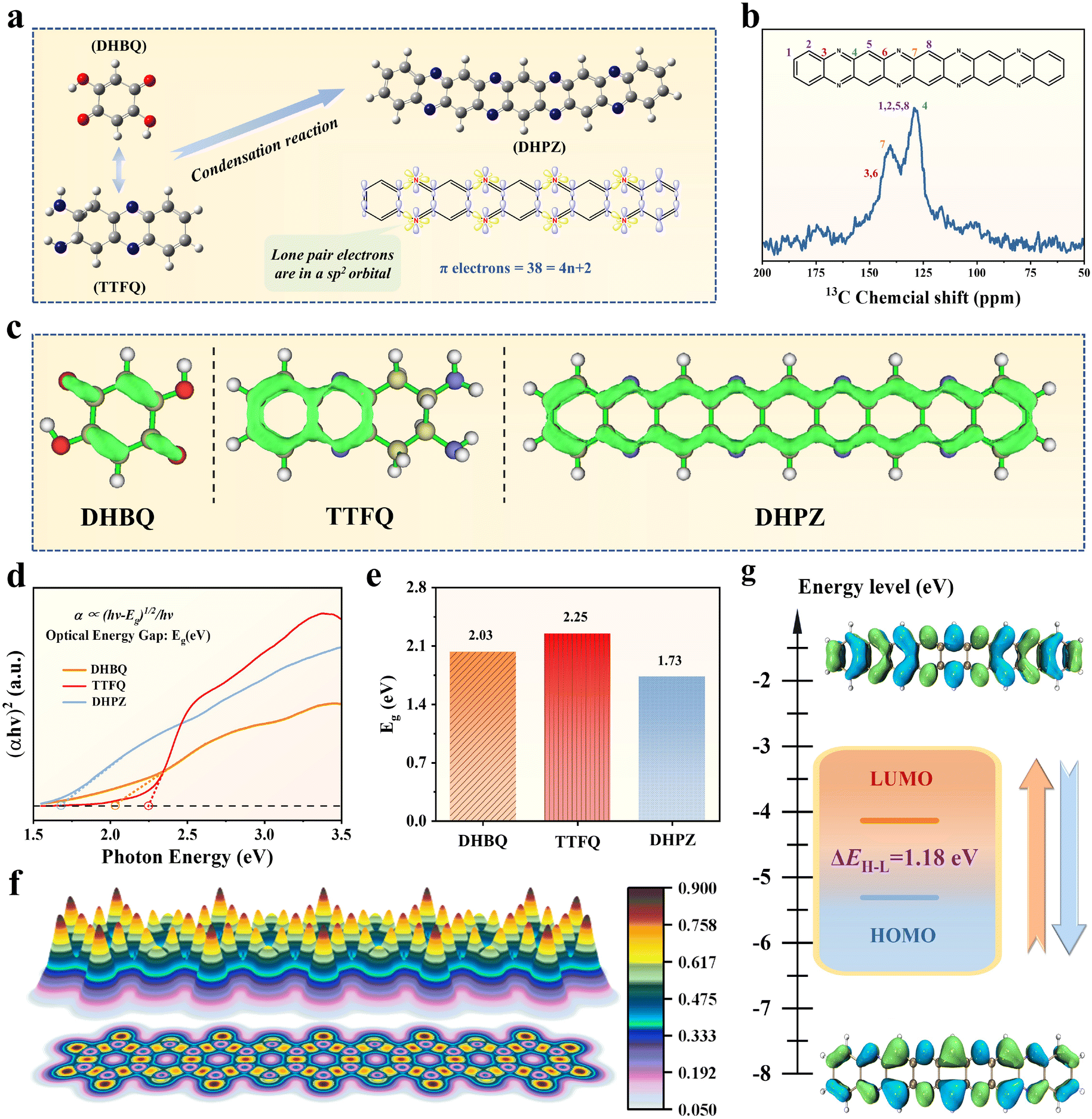 | ||
| Fig. 1 Structural characterization of the DHPZ organic molecule. (a) Molecular structure diagram and (b) solid-state 13C NMR spectrum of DHPZ synthesized from DHBQ and TTFQ precursors. (c) ELF-π plots, (d) UV-vis absorption spectra and (e) the calculated Eg values of DHBQ, TTFQ and DHPZ molecules. (f) LOL-π color-filled map and (g) HOMO–LUMO distributions of the DHPZ molecule. | ||
Investigating the optical properties of the DHPZ molecule provides valuable information about its electronic behaviors. A key factor in this regard is the optical band gap (Eg), which represents the energy difference between electronic levels within the organic molecule (Fig. 1d). The Eg value is a crucial parameter influencing the electrochemical activity of the organic materials. A smaller Eg generally corresponds to improved conductivity and faster electrochemical reaction rates.36 As illustrated in Fig. 1e, the DHPZ molecule exhibits a remarkably small Eg of 1.73 eV, significantly lower than the Eg values of its precursors, DHBQ (2.03 eV) and TTFQ (2.25 eV). This small Eg value signifies that the DHPZ molecule requires less energy for electron excitation, significantly enhancing its redox kinetics and promoting efficient charge transfer properties by substantially lowering the kinetic barrier within the DHPZ molecule. To probe the kinetic behavior of the DHPZ molecule, specifically its capacity for electron delocalization, the localized orbital locator-π (LOL-π) method was employed. This method utilizes a LOL value of 0.5 as a demarcation: values below indicating localized electrons and those above signifying delocalization.37 As evidenced in Fig. 1f, the LOL-π profile for the DHPZ molecule presents values consistently ranging from 0.621 to 0.862 along its molecular backbone. This observation confirms substantial π-electron delocalization, underscoring the highly conjugated arrangement of the DHPZ structure. When examining the partial density of states (Fig. S7†), one can observe a dense population of electrons near the Fermi energy level, indicating a considerable degree of electron delocalization and a natural propensity for charge movement in the DHPZ molecule. Additionally, the orbital energy profile illustrates a low-energy unoccupied molecular orbital (LUMO) alongside a high-energy occupied molecular orbital (HOMO), as shown in Fig. 1g. The computational results demonstrate that the DHPZ molecule exhibits an exceptionally small HOMO–LUMO gap of merely 1.18 eV. Such a narrow energy gap greatly lowers the activation energy required for electron transfer, promoting efficient oxidation and reduction processes. This electronic configuration enhances the redox activity while simultaneously boosting charge transport properties of the DHPZ molecule through facilitated interorbital electron mobility37,38 It is important to note that this energy gap is significantly smaller than those found in other electroactive organic materials (Fig. S8†).
Electrochemical measurements and the absorption mechanism
Further investigations into the electrochemical characteristics of the DHPZ molecule were conducted in NH4Cl aqueous solution. A three-electrode setup was employed for the study, which included the use of the synthesized DHPZ as the working electrode, a graphite rod serving as the counter electrode, and a saturated Ag/AgCl electrode functioning as the reference electrode. Cyclic voltammetry (CV) was performed on the DHPZ electrode over a range of scan rates from 5 to 50 mV s−1. As illustrated in Fig. 2a, the resulting voltammograms display clear pseudocapacitive features, indicative of faradaic processes associated with the NH4+ uptake. The discerned escalation in peak currents with increasing scan rates, combined with the uniform morphology of the CV curves, alongside the slight variations in peak potentials (Fig. S9†) with minimal electrode polarization, further corroborates the remarkable electrochemical responsiveness and efficient redox kinetics of the DHPZ electrode. The b values depicted in Fig. 2b are quantitatively determined to be 0.79 and 0.88 during the oxidation phase and 0.76 and 0.93 during the reduction phase, confirming that the DHPZ electrode is predominantly governed by surface-controlled capacitive contributions (Fig. S10†), as opposed to the more sluggish diffusion-controlled processes. As illustrated in Fig. 2c, the galvanostatic charge/discharge (GCD) profiles for the DHPZ electrode, obtained at various current densities ranging from 1 to 8 A g−1, display a high degree of symmetry, underscoring the reversibility of the electrochemical processes involved. Calculations based on these curves reveal a remarkable specific capacitance of 361.05 F g−1 at a current density of 1 A g−1. Notably, even when subjected to a tenfold increase in current density, the DHPZ electrode maintains a considerable specific capacitance of 178.8 F g−1 (Fig. 2f), underscoring its exceptional rate capability for NH4+ capture. Furthermore, it is evident that the increase in “IR drop” remains negligible across various current densities, further affirming the outstanding electron transfer efficiency of the DHPZ electrode for NH4+ capture. | ||
| Fig. 2 Electrochemical properties of the DHPZ electrode tested in NH4Cl aqueous electrolyte. (a) CV curves and (b) the calculated b values at different scan rates. (c) GCD plots, (f) the corresponding specific capacitances and “IR drops” at various current densities. (d) Isosurface representation of ICSSZZ, (e) color-filled contour map of ICSSZZ in the slice plane perpendicular to the symmetry plane, and (g) the corresponding ELF image of the DHPZ molecule. (h) Ex situ Bode plots. (i) Long-term cycling stability of the DHPZ electrode during repeated NH4+ uptake/release. (j) The calculated HOMA values of the reduced DHPZ state upon NH4+ adsorption. | ||
To evaluate the electronic structure and aromatic properties of the DHPZ electrode, we employed the iso-chemical shielding surface (ICSS) methodology. Fig. 2e illustrates the Z-component of the ICSS (ICSSZZ) corresponding to the symmetrical supramolecular plane. Distinct shielding regions (depicted in red) project outward perpendicular to the plane, encircled by a closed and deshielded isosurface (represented in blue). This characteristic ICSSZZ distribution pattern offers compelling evidence in support of the π-aromaticity of the DHPZ molecule.39,40 A more profound comprehension of the ICSSZZ can be achieved by scrutinizing the slice plane that is perpendicular to the symmetry plane (Fig. 2d). This visualization uncovers a significant magnetic shielding effect, characterized by a relatively smooth gradient within the ring's interior. In quantitative terms, the ICSSZZ values at the center of the ring (ICSSZZ (0)) and 1 Å above this point (ICSSZZ (1)) are 13.88 ppm and 8.91 ppm, respectively (Fig. 2d). These results accentuate the intricate in-plane aromatic characteristics of the DHPZ molecule. The delocalization of π-electrons throughout the conjugated superstructures facilitates the injection of electrons into the DHPZ molecule with a reduced energy barrier, thereby enhancing structural stability. The electron localization function (ELF) of the DHPZ molecule is illustrated in Fig. 2g. The pronounced brightness surrounding the carbon and nitrogen atoms within the benzene and imine rings indicates a significant accumulation of electrons, further confirming the complete delocalization of π-electrons in the DHPZ molecule. Consequently, the freely delocalized π-electrons and the extended conjugation considerably lower the energy barrier for the NH4+ diffusion in the DHPZ electrode.
Fig. 2h showcases the Bode plots of the DHPZ electrode at various stages throughout the charging and discharging process. The dashed line, representing a phase angle of 45°, delineates the eigenfrequency ranging from 0.34 to 1.70 Hz. This range corresponds to a rapid frequency response, characterized by time constants spanning from 0.59 to 2.94 s. In the Nyquist plot (Fig. S11†), the characteristic near-vertical lines in the low-frequency domain, associated with a low charge transfer resistance of 2.17 Ω in the high-frequency domain, suggest efficient NH4+ diffusion and charge transfer across the DHPZ electrode. Calculations indicate a high diffusion coefficient for the DHPZ electrode, measuring 1.77 × 10−12 cm2 s−1 (Fig. S12†), thereby highlighting the remarkable electrochemical kinetics of the electrode and its capacity for swift NH4+ capture.
As shown in Fig. 2i, the DHPZ electrode exhibits exceptional stability, retaining an impressive 95.93% of its capacity after over 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 charge–discharge cycles and achieving nearly 100% coulombic efficiency at 5 A g−1. When compared to a range of other NH4+-capturing electrodes, including transition metal oxides, Prussian blue analogs, and layered double hydroxides (Table S1†), the DHPZ electrode demonstrates superior cycling stability, both in terms of capacitance retention and overall cycle life. Fig. 2j illustrates the indices of the harmonic oscillator model of aromaticity (HOMA) for the DHPZ molecule in its reduced state. As the HOMA index approaches 1, it indicates a considerable improvement in both the aromatic character and structural stability of the organic molecule. Significantly, the HOMA values near 1 upon the complete adsorption of NH4+ highlight the exceptional stability of the DHPZ molecule for NH4+ capture.40 The results, presented in Fig. S13,† further demonstrate exceptional electrochemical stability, with the DHPZ electrode retaining 96.27% of its initial specific capacitance after 10000 cycles at a high current density of 10 A g−1.
000 charge–discharge cycles and achieving nearly 100% coulombic efficiency at 5 A g−1. When compared to a range of other NH4+-capturing electrodes, including transition metal oxides, Prussian blue analogs, and layered double hydroxides (Table S1†), the DHPZ electrode demonstrates superior cycling stability, both in terms of capacitance retention and overall cycle life. Fig. 2j illustrates the indices of the harmonic oscillator model of aromaticity (HOMA) for the DHPZ molecule in its reduced state. As the HOMA index approaches 1, it indicates a considerable improvement in both the aromatic character and structural stability of the organic molecule. Significantly, the HOMA values near 1 upon the complete adsorption of NH4+ highlight the exceptional stability of the DHPZ molecule for NH4+ capture.40 The results, presented in Fig. S13,† further demonstrate exceptional electrochemical stability, with the DHPZ electrode retaining 96.27% of its initial specific capacitance after 10000 cycles at a high current density of 10 A g−1.
In situ Raman spectra (Fig. S14†) of the DHPZ electrode were collected during the electrochemical testing, as presented in Fig. 3a. During the discharge process, the peak intensity of the CN bonds (1589 cm−1) significantly decreases with the absorption of NH4+, while the peak intensity of the C–N bonds (1374 cm−1) gradually increases accordingly. When the potential is reversed, the signal for the C–N bonds gradually weakens, coinciding with the reappearance of the CN bonds as a result of the oxidation reaction during NH4+ removal. As illustrated in Fig. S15,† the ratio of peak intensities for the CN and C–N bonds (ICN/IC–N) was graphed against potential. This analysis uncovers a distinct trend: the ICN/IC–N ratio progressively diminishes during the discharging phase (NH4+ uptake) and increases during the charging phase (NH4+ removal). This behavior indicates that a reversible redox reaction involving the transformation of CN into C–N bonds takes place within the DHPZ electrode during the NH4+ uptake/release process. Ex situ XPS analysis at its initial, discharged, and charged states (Fig. 3b) reveals shifts in the N 1s spectra that suggest reversible bond transformations. During the discharging phase, the CN bond (398.88 eV) predominantly shifts to 400.98 eV, indicating the formation of C–N bonds. This alteration returns to its original state upon charging, confirming the reversibility of the electrochemical process. Fig. 3c illustrates the molecular electrostatic potential (MESP) representation, emphasizing the eight nitrogen atoms situated within the four imine rings. These nitrogen atoms are surrounded by areas of elevated electronegativity, indicated by significant negative restrained electrostatic potential (RESP) values (approximately −0.60 eV at each imine location, Fig. 3d). This finding suggests that the CN bonds in the imine rings are capable of interacting with NH4+ ions during the redox process. Analyzing the charge density differentials further confirms notable charge depletion and accumulation between NH4+ and imine sites within the DHPZ molecule (Fig. 3e), reinforcing the assertion that the DHPZ electrode can efficiently capture NH4+ ions. Based on these observations, a schematic illustration of the electrochemical transformations within the DHPZ electrode is provided in Fig. 3f, where NH4+ ions are adsorbed and desorbed through redox reaction involving the eight CN bonds of the DHPZ electrode, with the CN ↔ C–N transformations upon NH4+ uptake/release.
 | ||
| Fig. 3 Electrosorption mechanism of the DHPZ electrode for NH4+ capture. (a) In situ Raman spectra during the electrochemical process. (b) Ex situ N 1s XPS spectra at pristine, fully charged and discharged states. (c) MESP surface and (d) the calculated RESP values of DHPZ molecule. (e) Charge differential distribution of the DHPZ molecule upon NH4+ adsorption. (f) Schematic illustration of the proposed NH4+ adsorption–desorption process within the DHPZ electrode. | ||
CDI performances and applications for NH4+ removal
To maximize the electrochemical capacity generated by redox reactions, we have adopted an asymmetric design strategy, which combines a rod-shaped DHPZ organic electrode with exceptional NH4+ adsorption capability and commercial activated carbon (AC) with high surface area for Cl− uptake. The synergistic integration of these electrode materials enables the construction of a hybrid CDI device with significantly improved overall electrosorption capacity. As shown in Fig. 4a, a hybrid CDI device has been fabricated employing DHPZ as the anode and AC as the cathode. An anion exchange membrane is positioned adjacent to the AC cathode to prevent cation adsorption during voltage reversal. The assembled CDI device was investigated at various applied voltages within 10 mM NH4Cl aqueous solution. Fig. 4b shows the associated transient current curves, which demonstrate the real-time variations in current throughout the adsorption and desorption phases. The conductivity of the solution was continuously monitored using an online conductivity meter, and the NH4+ concentration can be determined based on a calibration curve derived from the conductivity data (Fig. S16†). When different voltages are applied to the CDI device, a noticeable initial current is generated, which quickly drops and then stabilizes. At the same time, the solution conductivity also shows a substantial decline (Fig. S17†). It is evident that the enhanced electrostatic forces improve the NH4+ absorption within the DHPZ electrode,41,42 leading to a significant increase in the specific capacity for NH4+ removal as the voltage increases (Fig. 4c). When subjected to the tested voltage of 1.2 V, the CDI device achieves exceptional NH4+ removal performance, achieving a considerably large capacity of 136.6 mg g−1 at 1.2 V with a high charge efficiency of 81.94%. However, an examination of charge efficiency uncovers a significant declining trend as applied voltages increase (Fig. S18†), suggesting an elevated probability of side reactions at voltages exceeding 1.2 V. When operated at 1.2 V, the CDI device delivers a rapid average NH4+ removal rate of 4.55 mg g−1 min−1 (Fig. 4d), highlighting its superior adsorption efficiency for NH4+ removal. To assess the regeneration stability of the assembled CDI device, continuous adsorption–desorption experiments were conducted in constant voltage mode for 200 cycles at 1.2 V. As illustrated in Fig. 4e, the NH4+ removal capacity of the CDI device achieves an impressive retention rate of 95.76% after 200 cycles, outperforming other CDI devices incorporating pseudocapacitive materials (Fig. S19†). Chemical oxygen demand (COD) analysis further indicates negligible dissolution of organic matter from the DHPZ electrode during CDI operation (Fig. S20†). Significantly, the specific energy consumption (SEC) per cycle remains consistently low, ranging from 54.25 to 57.94 kJ mol−1, confirming the minimal occurrence of side reactions during the cycling process. | ||
| Fig. 4 CDI performances for NH4+ removal. (a) A schematic representation of the hybrid CDI device incorporating a rod-like DHPZ anode paired with an AC cathode. (b) Current response profiles; (c and d) NH4+ removal capacities as a function of removal times and removal rates at varying applied voltages. (e) Regeneration stability and the corresponding SEC values across 200 cycles at 1.2 V. (f) Finite element simulations of the NH4+ distributions within rod, block, and granule electrode models and (g) the simulated NH4+ concentrations and diffusion rates. (h) Schematic illustration of rapid NH4+ diffusion through the rod-like DHPZ structure. (i) A comparative analysis of the achieved NH4+ removal capacity against existing CDI devices utilizing various electrode materials. | ||
Finite element simulations were performed using COMSOL Multiphysics to examine the NH4+ diffusion behavior within the DHPZ electrode in the CDI device. For comparative analysis, three distinct electrode configurations—rod, block, and granule—were simulated in a 10 mM NH4Cl solution at an applied voltage of 1.2 V. The simulation results reveal notable differences in NH4+ concentration profiles across the three electrode models over identical time intervals. Specifically, the rod-shaped electrode model exhibits more pronounced NH4+ concentration variations compared to the block and granule models, with substantially higher NH4+ concentrations observed in its deeper regions (Fig. 4f). Further analysis of the simulation data demonstrates that the rod-shaped electrode model possesses a remarkably higher ion diffusion coefficient of 1.77 mol m−2 s−1, which is approximately three- and four-fold greater than that of block and granule models, respectively. Additionally, the rod-shaped electrode model exhibits the highest NH4+ concentration (9.8 mol m−3) among the three configurations (Fig. 4g), confirming its superior ability to facilitate the penetration and uniform distribution of NH4+ ions. These findings underscore the advantages of the rod-shaped design in optimizing NH4+ diffusion and adsorption efficiency in CDI devices, as depicted in Fig. 4h. As a result, the NH4+ removal capability of the CDI device shows a remarkable enhancement compared to reported CDI devices for NH4+ removal that utilize a range of electrode materials (Fig. 4i and Table S2†).7,8,13–15,20,21,43,44 The experimental findings strongly indicate that the DHPZ organic material demonstrates significant potential as a faradaic electrode for CDI applications. Its advantages over conventional inorganic materials are multifaceted, encompassing not only superior environmental compatibility, including enhanced sustainability, reduced ecological impact, and lower carbon emissions, but also outstanding electrosorption performance. These properties stem from its unique structural characteristics, such as abundant redox-active sites, extensive π-electron conjugation, and a highly stable framework that facilitates exceptional and long-term NH4+ adsorption.
The selective extraction of NH4+ is of importance for the efficient capture of ammonium resources (Fig. 5a), particularly in addressing environmental concerns and enhancing nitrogenous recovery from wastewater systems. To systematically evaluate the selectivity of the CDI device for NH4+ amidst competing cations, a series of experiments were conducted using mixed solutions. The findings from these experiments are presented in Fig. S21 and S22,† which illustrate the competitive behavior of NH4+ against other cations. As illustrated in Fig. 5b, the selectivity coefficients of the CDI device for NH4+ are 4.9 and 8.65 when competing with Na+ and Ca2+ in binary solutions, respectively. Furthermore, when these three cations are present at equal concentrations, the selectivity of the CDI device for NH4+ is greater than that for Na+ and Ca2+, with selectivity coefficients of NH4+/Na+ and NH4+/Ca2+ being 1.9 and 11.4, respectively. To gain a deeper insight into the differences in selectivity, the adsorption energies of various ions on the DHPZ electrode were computed. The results indicate that, in comparison to Na+ and Ca2+, the adsorption energy of NH4+ is the most negative (Fig. 5c), suggesting that the DHPZ electrode in the CDI device provides remarkable selectivity for the NH4+ capture.
 | ||
| Fig. 5 CDI applications for NH4+ removal from wastewater. (a) Schematic illustration of the selective NH4+ removal process in the assembled CDI device. (b) Selectivity coefficients of NH4+ over competing cations in binary and multicomponent solutions. (c) Calculated adsorption energies of the DHPZ electrode coordinated with different cations (Na+, Ca2+ and NH4+). (d) An integrated system utilizing three interconnected CDI devices and (e) the corresponding reductions in NH4+ concentrations in actual ammonium wastewaters before and after system processing. (f) The NH4+ concentrations in the treated effluent following successive system processing cycles. (g) A schematic representation of the assembled CDI device with considerable potential for practical applications in domestic wastewater treatment. | ||
To illustrate practical implementation, we have crafted interconnected CDI devices that enable multi-stage removal processes. As depicted in Fig. 5d, ammonium wastewater flows into a three-stage CDI treatment system, ultimately yielding purified water. The system performance was evaluated using authentic ammonium-containing wastewater collected from a municipal plant in Jiangsu Province, China, with an NH4+ concentration of 52.69 mg L−1 and conductivity of 1482 μS cm−1 (see the Experimental section for details). Fig. 5e shows that the integrated system effectively achieves consistent NH4+ removal throughout each stage of the electrosorption process. Even with the existence of competing ions in the actual wastewater, the NH4+ concentration can be lowered to 8.44 mg L−1 after three rounds of CDI treatment (only ∼90 min), exceeding the stringent requirements of China's Class I wastewater discharge standard (GB8978-1996). Moreover, the integrated system has been employed in the continuous treatment of actual wastewater. It is evident that, following a three-stage purification process lasting 90 min, the NH4+ concentrations in the treated effluent can be diminished to less than 12 mg L−1 (Fig. 5f), thereby successfully complying with the wastewater discharge standards. This outcome substantiates the excellent electrochemical ammonium capture capabilities of the CDI-based systems for practical applications. It visually illustrates the variation in conductivity from wastewater to the final output of the three-stage purification process, reflecting a significant reduction from 1482 to 691 μS cm−1, with nearly negligible NH4+ content (Fig. S23†). Besides, the CDI device demonstrates sustainable energy recovery capability,45 as shown in Fig. S24 and S25.† Due to its efficiency, energy-saving properties, recyclability, and high selectivity for NH4+ removal, the developed organic-based CDI device holds considerable potential for real-world applications in water purification and contaminant removal, especially in the treatment of domestic wastewater (Fig. 5g).
Conclusion
In summary, a novel DHPZ organic material has been crafted for a CDI electrode with superior NH4+ adsorption capacity, which showcases a specialized orbital structure comprising four imine rings and lone pair electrons in an sp2 orbital, forging a unique non-equivalent hybridized sp2-N framework. The presence of available CN redox-active sites within the imine rings is complemented by the highly aromatic DHPZ framework, characterized by extensive electron delocalization and featuring a minimal HOMO–LUMO gap of merely 1.18 eV. These distinctive characteristics enhance its ability for rapid, consistent, and effective NH4+ adsorption. Consequently, the DHPZ-based CDI device achieves an impressive NH4+ removal capacity of 136.6 mg g−1 at 1.2 V, alongside a swift average adsorption rate of 4.55 mg g−1 and exceptional regenerative performance, maintaining 95.76% efficiency after 200 cycles. This remarkable and consistent removal capability surpasses those of existing CDI devices for NH4+ adsorption. For applied scenarios, we have devised a series of interconnected CDI units specifically tailored for the NH4+ elimination from real wastewater, embodying a sustainable and cutting-edge solution for wastewater treatment and water purification.
Data availability
The authors confirm that the data supporting the findings of this study are available within the article [and/or its ESI†]. More detailed data are available on request from the corresponding author, upon reasonable request.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. Minjie Shi, Jingxin Zhao and Huan Pang conceived the idea. Haoyuan Qiu, Peipei Zhang and Yueheng Tao prepared materials and theoretical calculations. Haoyuan Qiu, Jun Yang, and Xinyue Zhang conducted the performance test and material characterization. Haoyuan Qiu, Peipei Zhang, Yueheng Tao and Xinyue Zhang conducted electrochemical performance test of integrated device. Haoyuan Qiu and Peipei Zhang analyzed the experimental data and wrote the manuscript instructed by Minjie Shi, Jun Yang, Jingxin Zhao and Huan Pang with input from all the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the China Postdoctoral Science Foundation (2022M711686) and the Science and Technology Planning Social Development Project of Zhenjiang City (SJC20240100056).Notes and references
- L. Xiang, X. Xu, Y. Liu, H. Zhang, R. Xu, C. Li, F. Xu, Y. Yamauchi and Y. Mai, Nat. Mater., 2024, 2, 1195–1206 CAS
.
- B. R. Scanlon, S. Fakhreddine, A. Rateb, I. de Graaf, J. Famiglietti, T. Gleeson, R. Q. Grafton, E. Jobbagy, S. Kebede, S. R. Kolusu, L. F. Konikow, D. Long, M. Mekonnen, H. M. Schmied, A. Mukherjee, A. MacDonald, R. C. Reedy, M. Shamsudduha, C. T. Simmons, A. Sun, R. G. Taylor, K. G. Villholth, C. J. Vörösmarty and C. Zheng, Nat. Rev. Earth Environ., 2023, 4, 87–101 CrossRef
- T. Ying, Y. Xiong, H. Peng, R. Yang, L. Mei, Z. Zhang, W. Zheng, R. Yan, Y. Zhang, H. Hu, C. Ma, Y. Chen, X. Xu, J. Yang, D. Voiry, C. Y. Tang, J. Fan and Z. Zeng, Adv. Mater., 2024, 36, 2403385 CrossRef CAS PubMed
- F. Yu, Y. Yang, P. Liu and J. Ma, Chem. Sci., 2025, 16, 4635–4645 RSC
- C. Zhang, J. Ma, D. He and T. D. Waite, Environ. Sci. Technol. Lett., 2018, 5, 43–49 CrossRef CAS
- C. Li, B. Liu, D. Fang, P. Zhang, F. Li, X. Qiu, X. Mo and K. Li, J. Environ. Chem. Eng., 2023, 11, 110857 CrossRef CAS
- J. Jin, R. Wang, K. Yu, Y. Tao, P. Zhang, L. Ke, J. Yang and M. Shi, Sep. Purif. Technol., 2025, 353, 128290 CrossRef CAS
- Y. Wimalasiri, M. Mossad and L. Zou, Desalination, 2015, 357, 178–188 CrossRef CAS
- Q. Liu, F. Ye, K. Guan, Y. Yang, H. Dong, Y. Wu, Z. Tang and L. Hu, Adv. Energy Mater., 2023, 13, 2202908 CrossRef CAS
- H. Sakar, I. Celik, C. Balcik Canbolat, B. Keskinler and A. Karagunduz, Sep. Sci. Technol., 2017, 52, 2591–2599 CrossRef CAS
- B. Xu, K. Jiang, Y. Gan, K. Zhang, J. Zhang, J. Luo, H. Xu, Z. Chen, W. Yang, H. Li and X. Lu, J. Water Process Eng., 2024, 58, 104912 CrossRef
- T. Kim, C. A. Gorski and B. E. Logan, Environ. Sci. Technol. Lett., 2018, 5, 578–583 CrossRef CAS
- S. Wang, L. Zhao, Y. Lei, Z. Li and G. Wang, Sep. Purif. Technol., 2024, 329, 125204 CrossRef CAS
- S. Wang, S. Du, Y. Lei, L. Zhao and G. Wang, Chem. Eng. J., 2024, 500, 157248 CrossRef CAS
- S. Wang, Z. Pan, Z. Li, H. Zhuang, L. Zhao, Z. Li, Y. Lei and G. Wang, Chem. Eng. J., 2023, 466, 143163 CrossRef CAS
- J. Li, R. Wang, L. Han, T. Wang, Z. M. El-Bahy, Y. Mai, C. Wang, Y. Yamauchi and X. Xu, Chem. Sci., 2024, 15, 11814–11824 RSC
- Z. Liu, D. Wang, H. Yang, L. Feng, X. Xu, W. Si, Y. Hou, G. Wen, R. Zhang and J. Qiu, Angew. Chem., Int. Ed., 2024, 63, e202409204 CAS
- S. Choi, B. Chang, S. Kim, J. Lee, J. Yoon and J. W. Choi, Adv. Funct. Mater., 2018, 28, 1802665 CrossRef
- C. Zhang, J. Ma, J. Song, C. He and T. D. Waite, Environ. Sci. Technol., 2018, 52, 14275–14285 CrossRef CAS PubMed
- S. Tsai, D. V. Cuong and C. Hou, Water Res., 2022, 221, 118786 CrossRef CAS PubMed
- Q. Wang, Q. Wu, M. Zhao, S. Lu and D. Liang, Chem. Eng. J., 2024, 482, 148923 CrossRef CAS
- Y. Tao, Y. Cui, H. Wang, Z. Li, Z. Qian, P. Zhang, H. Zhou and M. Shi, Adv. Funct. Mater., 2024, 2414805 Search PubMed
- D. Liu, X. Ning, Y. Hong, Y. Li, Q. Bian and J. Zhang, Electrochim. Acta, 2019, 296, 327–334 CrossRef CAS
- W. Wei, W. Zou, D. Yang, R. Zheng, R. Wang and H. Chen, J. Mater. Chem. A, 2020, 8, 11811–11817 RSC
- Q. Li, X. Xu, J. Guo, J. P. Hill, H. Xu, L. Xiang, C. Li, Y. Yamauchi and Y. Mai, Angew. Chem., Int. Ed., 2021, 60, 26528–26534 CrossRef CAS PubMed
- S. Zhang, X. Xu, X. Liu, Q. Yang, N. Shang, X. Zhao, X. Zang, C. Wang, Z. Wang, J. G. Shapter and Y. Yamauchi, Mater. Horiz., 2022, 9, 1708–1716 RSC
- A. Fombona-Pascual, N. Patil, E. García-Quismondo, N. Goujon, D. Mecerreyes, R. Marcilla, J. Palma and J. J. Lado, Chem. Eng. J., 2023, 461, 142001 CrossRef CAS
- L. Xu, Y. Liu, Z. Ding, X. Xu, X. Liu, Z. Gong, J. Li, T. Lu and L. Pan, Small, 2024, 20, 2307843 CrossRef CAS PubMed
- Y. Li, R. Li, M. Wu, L. Shi, X. Wang, P. Wang and W. Wang, ACS ES&T Eng., 2024, 4, 956–965 Search PubMed
- D. Jiang, R. Xu, L. Bai, J. P. Hill, J. Henzie, L. Zhu, W. Xia, R. Bu, Y. Zhao, Y. Kang, T. Hamada, R. Ma, N. Torad, J. Wang, T. Asahi, X. Xu and Y. Yamauchi, Adv. Funct. Mater., 2024, 34, 2407479 CrossRef CAS
- Z. Zhuang, L. Sun, Y. Tao, J. Shao, J. Yang, P. Yu, H. Chen, J. Zhou, J. Xiao, K. Yin, M. Shi and P. Xiao, Energy Environ. Mater., 2024, e12852 CrossRef
- S. Dutta, R. F. de Luis, J. Goscianska, A. Demessence, R. Ettlinger and S. Wuttke, Adv. Funct. Mater., 2024, 34, 2304790 CrossRef CAS
- X. Xu, M. Eguchi, Y. Asakura, L. Pan and Y. Yamauchi, Energy Environ. Sci., 2023, 16, 1182–1815 Search PubMed
- Y. Tao, J. Jin, Y. Cui, H. Wang, Z. Qian and M. Shi, ACS Sustain. Chem. Eng., 2024, 12, 16434–16443 CrossRef CAS
- J. Ning, X. Zhang, D. Xie, Q. He, J. Hu, J. Tang, R. Li, H. Meng and K. X. Yao, Angew. Chem., Int. Ed., 2024, 63, e202319796 CrossRef CAS PubMed
- J. Lee, H. Lim, J. Park, M. Kim, J. Jung, J. Kim and I. Kim, Adv. Energy Mater., 2023, 13, 2300442 CrossRef CAS
- A. Kubaib and P. M. Imran, J. Mater. Sci., 2023, 58, 4005–4019 CrossRef CAS
- T. Sun, W. Zhang, Z. Zha, M. Cheng, D. Li and Z. Tao, Energy Storage Mater., 2023, 59, 102778 CrossRef
- Z. Song, L. Miao, L. Ruhlmann, Y. Lv, L. Li, L. Gan and M. Liu, Angew. Chem., Int. Ed., 2023, 62, e202219136 CrossRef CAS PubMed
- R. Wang, J. He, C. Yan, R. Jing, Y. Zhao, J. Yang, M. Shi and X. Yan, Adv. Mater., 2024, 36, 2402681 CrossRef CAS PubMed
- X. Zhang, Y. Li, Z. Yang, P. Yang, J. Wang, M. Shi, F. Yu and J. Ma, Sep. Purif. Technol., 2022, 297, 121510 CrossRef CAS
- M. Yu, D. Li, G. Sui, D. Guo, D. Chu, Y. Li, D. Chai and J. Li, Adv. Funct. Mater., 2024, 2416963 Search PubMed
- X. Zhao, J. Yang, J. Peng, Y. Liu, H. Zhang, C. Yan and M. Shi, Desalination, 2024, 592, 118120 CrossRef CAS
- T. K. A. Nguyen, N. T. N. Anh, M. D. Nguyen, V. T. Nguyen and R. Doong, Sep. Purif. Technol., 2023, 327, 124934 CrossRef CAS
- F. Chen, Y. Huang, L. Guo, L. Sun, Y. Wang and H. Y. Yang, Energy Environ. Sci., 2017, 10, 2081–2089 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5sc02192h |
| This journal is © The Royal Society of Chemistry 2025 |