
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Triazenide based metal precursors for vapour deposition
Nathan J.
O'Brien
 * and
Henrik
Pedersen
*
* and
Henrik
Pedersen
*
Department of Physics, Chemistry and Biology, Linköping University, SE-581 83 Linköping, Sweden. E-mail: nathan.o.brien@liu.se; henrik.pedersen@liu.se
First published on 13th January 2025
Abstract
Molecules featuring a metal centre in a positive valence surrounded by 1,3-dialkyltrianzenide ligands, Mx+[R–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–N–R′]x, were shown to have both high thermal stability and volatility, making them interesting as precursors in chemical vapour deposition (CVD) and atomic layer deposition (ALD). So far, metals from groups 11–14 and lanthanoids form stable triazenides and the In and Ga triazenides have proven to be excellent precursors for InN, In2O3, GaN and InGaN. We believe the exploration of the triazenides as CVD and ALD precursors has only begun and hope to inspire further research with this perspective.
N–N–R′]x, were shown to have both high thermal stability and volatility, making them interesting as precursors in chemical vapour deposition (CVD) and atomic layer deposition (ALD). So far, metals from groups 11–14 and lanthanoids form stable triazenides and the In and Ga triazenides have proven to be excellent precursors for InN, In2O3, GaN and InGaN. We believe the exploration of the triazenides as CVD and ALD precursors has only begun and hope to inspire further research with this perspective.
 Nathan J. O'Brien | Nathan O'Brien completed his B.Sc. with First-Class Honours in Chemistry (2009) and PhD in Organic Chemistry (2014) from La Trobe University, Melbourne, Australia. Following three years of postdoctoral research with Prof. Naokazu Kano at the University of Tokyo, Japan (2014–2017), he undertook a second postdoctoral position in the Department of Physics, Chemistry and Biology at Linköping University, Sweden (2017–2019), working with Prof. Henrik Pedersen. He was appointed Assistant Professor in 2019 and has held the position of Associate Professor within the department since 2024. His research focuses on the structure, reactivity and surface chemistry of metal–organic compounds for applications in catalysis and materials chemistry. |
 Henrik Pedersen | Henrik Pedersen received his M.Sc. in Chemistry in 2004 and his Ph.D. in Materials Science in 2008, both from Linköping University in Sweden. After a stint as industrial researcher at Sandvik Tooling Research and Development center in Stockholm, Sweden, he returned to academia and is today Professor of Inorganic Chemistry at Linköping University. He was visiting Professor at the Department of Chemistry at Carelton University, Ottawa, Canada 2016. His research is focused on understanding and developing new and better chemical vapor deposition methods with controlled surface chemistry. |
Introduction
Semiconductor chips are found in all parts of our lives. They are the hearts of our computers and smartphones and are required in everything from cars to refrigerators. The ability to deposit thin layers of various types of materials, with great precision, is at the centre of chip production.1 The complexity of the deposition varies at different steps of the production process. Materials need to be deposited onto large planar surfaces, such as full 300 mm wafers, or into narrow holes with sub-micrometre width and height with aspect ratios of >50. Different materials also set challenges during their deposition. For example, highly crystalline compound semiconductors, such as Al1−xGaxN, require precise control of the elemental composition, doping concentration, and crystallinity, whilst amorphous dielectric oxides are usually less complicated to deposit.The deposition process is often based on chemical reactions and the more challenging the process, the more precise control over the chemistry is required. Chemical vapor deposition (CVD) is the workhorse for depositing thin films of materials and has been used since the early days of chipmaking.2 CVD is based on chemical reactions between volatile precursor molecules, containing the atoms needed to form the film material, in the gas-phase and the surface where the film is depositing.3 While CVD is mainly known and used for depositing films over large areas of flat surfaces, high precision methods have been developed for filling more complex architecture, such as holes or trenches.4 The technology that excels at precision is the time-resolved form of CVD known as atomic layer deposition (ALD).5 ALD involves sequential, self-limiting adsorption of alternating precursor pulses onto a surface. Unlike traditional CVD, ALD introduces the metal and non-metal precursors separately into the reactor. This separation ensures that the precursors react primarily with the surface, rather than with each other in the gas-phase, and builds a film of material with atomic layer precision. ALD, patented in 1974,6 has been instrumental in the semiconductor industry since the early 2000s when ALD proved to be the enabler for the high-k transistors, where HfO2 deposited by ALD replaced thermally grown SiO2 as the gate oxide in silicon-based transistors.7 ALD is now used to deposit a wide range of materials, for example oxides, nitrides, sulphides, metals, and even organic and polymer films.
A significant aspect of ALD research involves developing new precursor molecules, primarily metal precursors, that possess favourable physical and chemical properties for film deposition.8 These advancements aim to enable material deposition at lower temperatures and produce films with reduced impurity levels.9 An ALD precursor must be sufficiently volatile to be transported to the reaction chamber and growth surface. It should not decompose in the gas phase in a way that leads to continuous CVD growth.9 At the surface, the precursor should react in a self-limiting manner to form a stable monolayer and unreactive by-products that can be easily removed by purging.10 After purging, the monolayer should react with the second precursor (e.g. H2O, H2S, NH3) in another self-limiting reaction. Additionally, using precursors with the same oxidation state as the target material eliminates the need for redox chemistry during the process, reducing the risk of forming undesired mixed-phase materials. This consideration is not applicable to the deposition of most metallic films, such as Cu films for interconnects, where reduction to the ground state is required. Although the surface chemistry requirements of precursors are well understood, creating a metal precursor with all these desired properties has been challenging to date.
Herein, we give our perspective on a class of precursors with metals from groups 11–14 and lanthanoids surrounded by the 1,3-dialkyltriazenide ligands and their use in CVD and ALD. These new triazenide precursors are easy to synthesise, purify and derivatise the ligand steric bulk. The group 13 triazenides have demonstrated ALD of both semiconductor and conductor materials, whilst the other examples are yet to be deployed in a deposition process. We believe that we have only seen a fraction of the potential of the triazenide precursors, and we hope that this perspective can stimulate more research on their deposition chemistry.
M–N bonded ligands
The most widely used ALD precursors are monodentate ligated metal chlorides, alkyls, cyclopentadienyls, alkoxides and amides due to their high reactivity and volatility.10 While sufficient volatility can be achieved for most precursors, there is always a trade-off between reactivity and thermal stability. Strong M–Cl, M–C or M–O bonds provide precursors with high thermal stability but make it challenging to completely remove their ligands from the deposited precursor at low temperatures, leading to high impurity levels in the deposited films. Polarised M–N bonds are more reactive, making it easier to remove their surface ligands at low temperatures and thus useful for a broader range of materials. However, the low thermal stability of the deposited surface species leads to impurities in the film and thus sets upper limits for the deposition temperatures. This has led to the use of M–N bonded bidentate ligands, which form two bonds to the metal centre, thereby improving the thermal stability of the precursor and its surface species.The monoanionic amidinate and guanidinate bidentate ligands (Fig. 1a and b) have been used to create a large family of volatile M–N bonded ALD precursors.10,11 These compounds exhibit higher thermal stability than amides and generally produce high-quality films. However, they remain susceptible to thermolysis via β-hydride elimination or carbodiimide disinsertion (CDI).9,12 These thermolysis processes have hindered the deposition of first-row transition metal metallic films by ALD, as their decomposition temperatures are much lower than those required for successful deposition.13 Additionally, high-valent metal amidinates and guanidinates suffer from low volatility and slow surface reactivity, or both, due to the crowding of the metal centre by the bulky bidentate ligands.14–19 We observed this when depositing InN by ALD, as multiple consecutive pulses of these precursors were required to saturate the surface.20 This study also revealed that smaller and less electron-donating substituents on the central substituted carbon led to higher quality InN films. This led us to the monoanionic triazenide ligand, which is isoelectronic to amidinates and guanidinates but differs by a nitrogen atom instead of a substituted carbon (Fig. 1c). Replacing the substituted carbon with a nitrogen would also effectively block the CDI disinsertion pathway.
 | ||
| Fig. 1 The (a) amidinate, (b) guanidinate and (c) triazenide bidentate ligands, and (d) an alkyl azide for comparison. | ||
Triazenides
In comparison to amidinates and guanidinates,21,22 the coordination chemistry of triazenides is far less explored. Most of the reported metal triazenides feature the 1,3-diphenyltriazenide ligand with no volatility data.23–29 Until our recent work, only a few examples of 1,3-dialkyltriazendies had been produced whilst none had been used in an ALD process.30–32 The reason for lack of research on 1,3-dialkyltriazenides is unknown, but most likely stems from the assumption that these compounds are explosive due to their resemblance to organic azides (Fig. 1d). Making such presumptions is misguided as these two catenated nitrogen compounds differ in their structure and bonding, and most likely decomposition pathways. From the first series of 1,3-dimethyltriazenides reported, only one titanium-magnesium chloride triazenide side product showed “explosive tendencies”.30 Although the decomposition pathway of metal triazenides has not been studied, a comprehensive review states that “Triazenide ligands when bound to transition metals are very stable entities and the N3 system does not fragment except under the most vigorous conditions”.33 Our experiences reflect the latter statement as we have not observed any concerning behaviour from these compounds when synthesising, scratching or heating to high temperatures for long periods of time (e.g., sublimation and decomposition studies, and ALD precursor source). Nevertheless, it is ensured that the C/N ratio is kept at >1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1,34 standard operational procedures are in place and all safety equipment such as blast shields, face shields, thermal gloves is being used when heating these compounds.
:1,34 standard operational procedures are in place and all safety equipment such as blast shields, face shields, thermal gloves is being used when heating these compounds.
The 1,3-dialkyltriazenide ligand is easy to synthesise and derivatise through strategic choice of the alkylazide and alkyllithium reagents (eqn (1)). The ligand can be isolated as a lithium salt or reacted directly with a metal chloride to give the desired metal 1,3-dialkyltriazenide (eqn (2)). These compounds are soluble in n-hexane, toluene, diethyl ether and THF, and are easily purified by either low temperature recrystallisation or sublimation. The simple synthesis and purification of these metal triazenides suggest that they are suited for industrial production and applications in both ALD and CVD.
| R–NNN + R′–Li → Li[R–NN–N–R′]; | (1) |
| MCln + nLi[R–NN–N–R′] → Mn+[R–NN–N–R′]n + nLiCl. | (2) |
Triazenides used in thin films deposition
The first triazenide precursor we reported and demonstrated for ALD was tris(1,3-diisopropyltriazenide)indium(III), In(triaz)3 (Fig. 2a), used together with an NH3 plasma, to deposit indium nitride (InN).35 The crystal structure of In(triaz)3 showed three sets of triazenide ligands chelating the central In atom in a distorted octahedral coordination geometry. This new In precursor was found to be volatile (sub. 80 °C at 0.5. mbar) and thermally stable (decomp. 212–214 °C). Thermogravimetric analysis (TGA) showed a single step volatilisation curve with an onset temperature of 145 °C and negligible residual mass (Fig. 2b). | ||
| Fig. 2 (a) X-ray crystal structure of In(triaz)3 with thermal ellipsoids at the 50% probability level. All hydrogen atoms were removed for clarity. (b) TGA of In(triaz)3 showing near ideal single step volatilisation with negligible residue mass. Images are reprinted from O'Brien et al.35 under a CC-BY license. | ||
The ALD process exhibited unusual behaviour with two temperature intervals where the InN film growth per ALD cycle (GPC) was constant with temperature (Fig. 3a). The first temperature interval at 220–250 °C showed a GPC of 0.4 Å per cycle and second at 300–350 °C with a GPC of 1.2 Å per cycle. The deposition chemistry was shown to be self-limiting for both temperature intervals (Fig. 3b and c) and linear film growth with number of ALD cycles (Fig. 3d). It was speculated that an intact In(triaz)3 molecule was the growth species for first temperature interval, rendering the low GPC. At the higher temperature interval, In(triaz)3 decomposes in the gas phase forming a smaller tricoordinated amide complex, which rendered a higher GPC due to lower steric hindrance and therefore higher surface coverage of the deposited species. This is unusual behaviour for an ALD precursor, as gas-phase decomposition of the precursor is usually an unwanted property. The InN films deposited at 300 °C grew epitaxially on 4H-SiC (0001) and were found to have an In/N ratio of 1.0 and no detectable carbon impurities by XPS.
 | ||
| Fig. 3 ALD characteristics of the In(triaz)3 with NH3 plasma. (a) Dependence on growth per cycle (GPC) with process temperature, (b) saturation curves at 220 °C, (c) saturation curves at 300 °C, (d) film thickness as a function of number of ALD cycles. Reprinted from O'Brien et al.35 under a CC-BY license. | ||
In(triaz)3 was also used for ALD of In2O3 in a thermal ALD process with H2O as the oxygen precursor.36 This process demonstrated stable growth with a wide temperature interval between 270 and 385 °C and a GPC of ∼1 Å per cycle. The In2O3 films deposited at the higher end of this temperature interval were polycrystalline on Si (100) and showed no C or N impurities by XPS. The temperature interval is higher than that of previously reported tris(N,N′-diisopropylformamidinato)indium(III), In(famd)3, which showed a temperature growth interval of 150–275 °C.14 This was at least partly ascribed to differences in ALD reactors, as attempts to reproduce the reported results with In(famd)3 rendered a temperature window of 245–315 °C.
Following the success with In(triaz)3, the gallium analogue tris(1,3-diisopropyltriazenide)gallium(III), Ga(triaz)3, was synthesised and demonstrated to deposit gallium nitride (GaN) with NH3 plasma.37 Ga(triaz)3 showed a similar solid-state structure, volatility (90 °C at 0.5 mbar) and thermal stability (decomp. 228–230 °C) to In(triaz)3. It also showed a near identical TGA graph with a slightly higher onset volatilisation temperature (155 °C) and negligible residual mass. These results were quite remarkable when compared to the Ga amidinates and guanidinates, which are unsuitable for ALD due to their lack of volatility.16,18 This makes Ga(triaz)3 the first example of a volatile hexacoordinated M–N bonded precursor. The ALD chemistry of Ga(triaz)3 produced several temperature windows with stable, self-limiting deposition of GaN. The ALD process chemistry was self-limiting up to at least 400 °C. A similar gas-phase decomposition mechanism as In(triaz)3, rendering a tricoordinated Ga amide, was suggested to explain this behaviour. Films grown at 350 °C afforded GaN that grew epitaxially on 4H-SiC (0001) with an atomically smooth interface (Fig. 4), a Ga/N ratio of 1.05 and no detectable C by ToF-ERDA. The bandgap of these films was measured to 3.41 eV, very close to the value of 3.40 eV for single-crystalline GaN.38
 | ||
| Fig. 4 Overview of the STEM-HAADF image from the GaN film with a thickness of approximately 60 nm on 4H-SiC. HR-STEM-HAADF image of the film–substrate interface and (c) top part of the film. The FFT patterns are shown as insets in (a–c) with orange as GaN and cyan as the 4H-SiC substrate. Reprinted from Rouf et al.37 under a CC-BY license. | ||
Given the similar sublimation temperatures of In(triaz)3 and Ga(triaz)3, mixing them in the precursor source was tested for ALD of the ternary nitride In1−xGaxN.39 Co-evaporation of the precursors into the ALD reactor resulted in In1−xGaxN films, with the composition (value of x) being tuneable based on deposition temperature, sublimation temperature, and mixing ratio. In1−xGaxN films with x-values ∼0.5 are known to be metastable, precipitating out InN or In metal. This presents a challenge for crystal growth, which must occur at temperatures below 500 °C.40 Consequently, ALD is particularly intriguing for these meta-stable materials, as it operates at temperatures where the chemisorbed monolayers remain stable. The In1−xGaxN films grew epitaxially on 4H-SiC (0001) and investigations by TEM show no signs of precipitation of InN (Fig. 5). A gradient on the composition was, however, noted, rendering approximately In0.18Ga0.82N towards the substrate, and In0.82Ga0.18N towards the film surface (Fig. 5e and f). This was speculated to be due to stress minimization with a smaller lattice mismatch to 4H-SiC with higher Ga-content. The bandgap of a In0.55Ga0.45N film deposited on Si (100) was measured to be 1.94 eV.
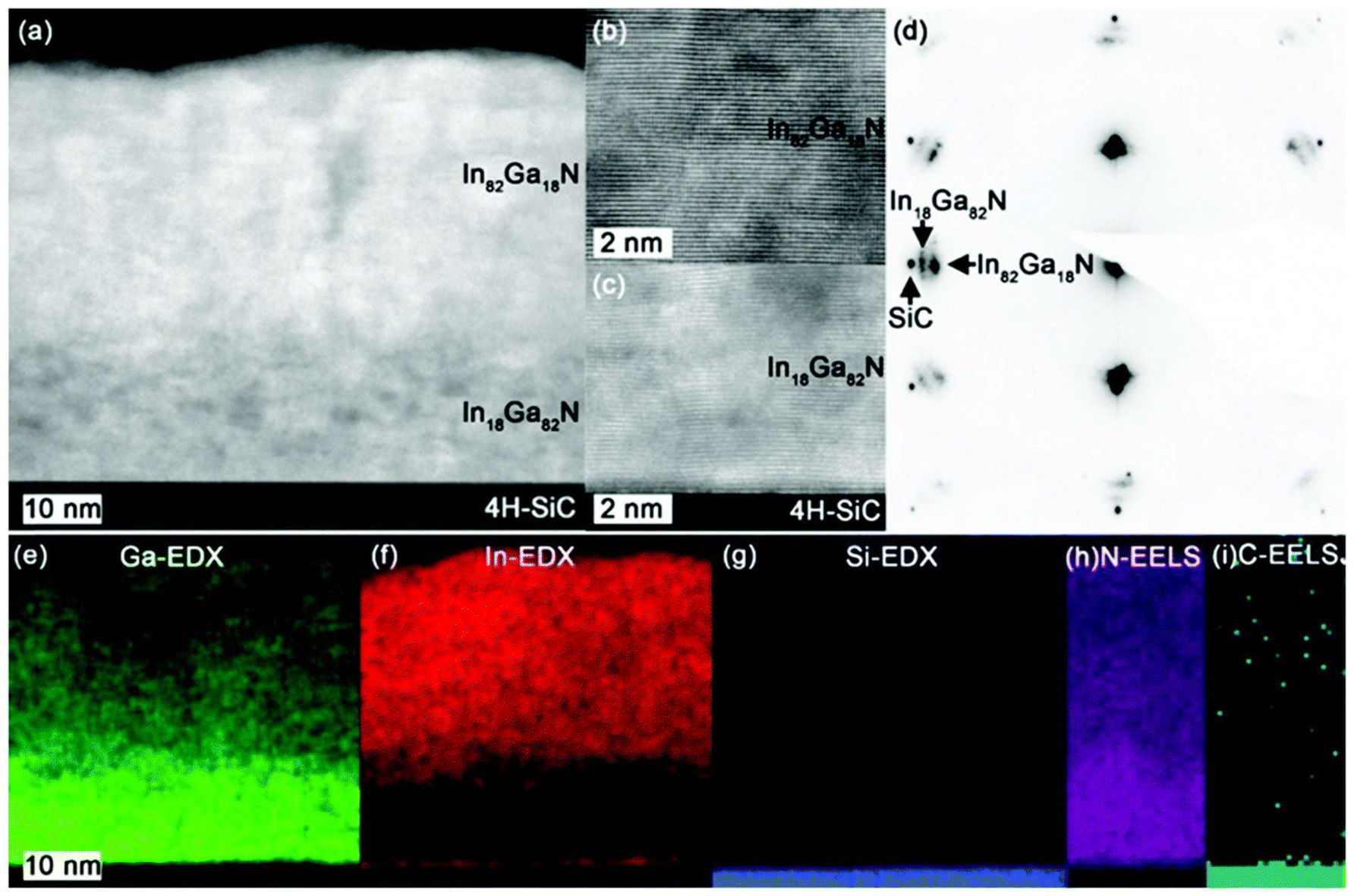 | ||
| Fig. 5 (a) cross-sectional STEM-HAADF image of the ∼60 nm In1−xGaxN film on the 4H-SiC substrate with a zoomed in image of the (b) In82Ga18N and (c) In18Ga82N layers. (d) SAED pattern from the film and substrate. EDX maps of (e) Ga, (f) In and (g) Si. EELS maps on (h) N and (i) C. Reprinted from Rouf et al.39 under a CC-BY license. | ||
Other reported volatile triazenides
The success of In(triaz)3 and Ga(triaz)3 led us to synthesise structural analogues by employing combinations of isopropyl, sec- or tert-butyl alkyl groups, both symmetrically and unsymmetrically, on the triazenide ligand.41 These compounds were all volatile (80–120 °C, 0.5 mbar), showed very good thermal stability (200–300 °C) and a majority with one step volatilisation cure (Fig. 6a and b). This provides a range of compounds in which the thermal stability and steric bulk of the precursor can be tailored to strategic choice. The same structural analogues were also synthesised for Al, rendering a total of six tris(1,3-dialkyltriazenide)aluminum(III) compounds.42 Especially when tert-butyls, or a mixture of sec- and tert-butyls were used, these Al triazenides showed sublimation temperatures of 90–125 °C with no residual masses by TGA (Fig. 6c) and thermal stability >200 °C. Unfortunately, attempts to deposit aluminium nitride (AlN) using these triazenides and NH3, both with and without plasma activation, showed limited success, poor repeatability, and primarily aluminium oxynitride films.43 | ||
| Fig. 6 The TGA graphs from structural analogues (a) In triazenides, (b) Ga triazenides and (c) Al triazenides where 1 = iPrN3iPr, 2 = iPrN3sBu, 3 = iPrN3tBu, 4 = sBuN3sBu, 5 = sBuN3tBu and 6 = tBuN3tBu. Reprinted from Samii et al.41,42 under a CC-BY license. | ||
We have since extended this initial chemistry to group 11, 12 and 14 metals. Using the 1,3-di-tert-buyltriazenide ligand, divalent Ge, Sn and Pb compounds were synthesised.44 Similarly to the group 13 triazenides, these compounds showed mononuclear structures with the triazenide ligand in a chelating binding mode to the metal centre (Fig. 7a). The compounds sublimed quantitatively between 60–75 °C (0.5 mbar) with impressive thermal stability (170–280 °C). TGA of the compounds showed single step volatilisation curves with onset temperatures between 137–152 °C and negligible residual mass (Fig. 7b). The divalent Ge and Pb triazenides were the first examples of tetracoordinated and exclusively M–N bonded compounds with high volatility and thermal stability. The Sn derivative showed similar volatility and thermal stability to its amidinate analogue.45
 | ||
| Fig. 7 (a) X-ray crystal structure of the Ge trianzenide precursor with thermal ellipsoids at the 50% probability level. All hydrogen atoms were removed for clarity. (b) The TGA graphs of Ge, Sn and Pb triazenides. Reprinted from Samii et al.44 under a CC-BY license. | ||
Volatile and thermally stable group 11 triazenides were also synthesised.46 Reaction of 1,3-di-tert-butyltriazenide ligand with MCl (M = Cu, Au and Ag) gave dinuclear Cu and Au and tetranuclear Ag triazenides (Fig. 8a and b). Heating the dinuclear Au triazenide led to dimerization and furnished the tetranuclear Au triazenide. For all these compounds, the triazenide ligand preferred a bridging binding mode instead of chelating. These compounds showed volatility (∼120 °C at 0.5 mbar) and thermal stability (180–230 °C) that rivalled the previously reported precursors bearing M–N bonded bidentate ligands. Thermolysis of these compounds by prolonged heating to 150 °C led to metallic films of Cu, Ag and Au by XRD analysis (Fig. 8c). The triazenide ligand provided the electrons for the reduction of the metal atoms and therefore these compounds can be seen as “single-source” precursors. This was a substantial result as usually a reducing agent, for example H2 gas, is required to reduce the ionic metal atom to the ground state. So far we have tested the Cu triazenide in CVD with some promise, but further work is required to fully evaluate its potential as a precursor.47
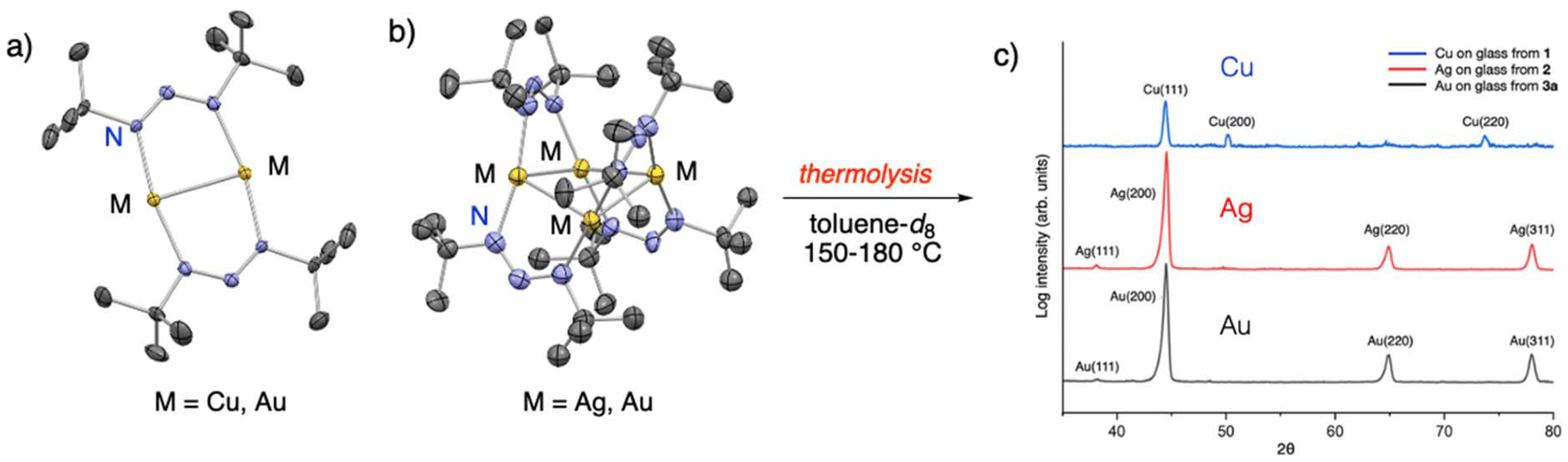 | ||
| Fig. 8 The crystal structure of (a) dinuclear Cu and Au, and (b) tetranuclear Ag and Au triazenides. (c) The XRD graph of the metallic films deposited by thermolysis of the Cu, Ag and Au triazenides. Reprinted from Samii et al.46 under a CC-BY license. | ||
We recently synthesised a dinuclear Zn triazenide, Zn2(triaz)4, as a potential vapour deposition precursor.48 Its crystal structure showed two Zn atoms each with a terminal chelating ligand and two ligands bridging the metal centres (Fig. 9a). It was found to be volatile (80 °C at 0.5 mbar), thermally stable (decomp. 145 °C) and gave a single step volatilisation curve (onset at 125 °C) by TGA with ∼5% residual mass. To mimic an ALD surface during ZnS film growth, Zn2(traiz)4 was reacted in ligand exchange reactions with two and four equivalents of triphenylsilanethiol to give dimeric and monomeric zinc thiolates, respectively (Fig. 9b and c). The high volatility and reactivity of this Zn triazenide gives it great promise to be used for ALD of ZnS.
 | ||
| Fig. 9 (a) The crystal structure of dinuclear Zn triazenide and its ligand exchange reactions with (b) two and (c) four equivalents of triphenylsilylthiol to replicate a bulky ALD surface. Reprinted from Samii et al.48 under a CC-BY license. | ||
We are happy to note that the triazenide ligand has been recently explored by another research group. Caroff and Girolami reported the synthesise of volatile lanthanide 1,3-di-tert-butyltriaznides for Nd, Eu and Er rare-earth metals.49 X-ray crystallography of the Nd and Eu compounds showed three sets of triazenide ligands chelating to the central metal atom in addition to a coordinated THF molecule (Fig. 10a). The Er derivative, however, crystallised without a solvent molecule (Fig. 10b). These compounds were thermally stable (∼280 °C) and sublimed at reasonable temperatures (90–120 °C at ∼0.01 Torr). TGA of these compounds produced smooth volatilization curves with ∼10–15% residual mass, although the Nd and Eu triazenides showed small events at ∼100–125 °C for the loss of THF (Fig. 10c). These new lanthanide triazenides have great potential as precursors for deposition of lanthanide nitride thin films.
 | ||
| Fig. 10 The crystal structure of (a) the Nd triazenide with a coordinated THF molecule and (b) the Er triazenide. (c) The TGA graphs of the lanthanide triazenides. Reprinted from Caroff and Girolami, Copyright 2022 American Chemical Society.49 | ||
Summary and outlook
In summary, volatile and thermally stable 1,3-dialkyltriazenides suitable as CVD and ALD precursors for Cu, Ag, Au, Zn, Al, Ga, In, Ge, Sn, Pb, Nd, Eu, and Er have been synthesised. So far, the In and Ga triazenides have been used to deposit high-quality and purity InN, GaN, InGaN and In2O3 films by ALD. The Al triazenides have not successfully deposited AlN but should be tested for other Al-containing materials. Cu, Ag and Au show promising behaviour as precursors for depositing the elemental films, but full deposition studies are yet to be investigated. The Zn triazenide shows promise for ZnS deposition, but again, a proper deposition study is not presented. The group 14 and lanthanide precursors are not yet tested for deposition, to our knowledge. We believe the volatility and thermal stability of these triazenides, along with their interesting thermal properties, makes them highly promising to future CVD and ALD processes. We hope that the works summarized here can spark interest in further exploring the triazenide ligand, both with the presented metals and those yet to be reported.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this frontier article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We wish to express our deep appreciation for the work by our students Rouzbeh Samii, Polla Rouf, Karl Rönnby, Anton Fransson, Pamburayi Mpofu, Robin Björnfot, and Peggy Bagherzadeh Tabrizi for their work on developing and understanding the trianzenide precursors. This work was financially supported by the Swedish Foundation for Strategic Research through the project “Time-resolved low temperature CVD for III-nitrides” (No. SSF-RMA 15-0018).References
- R. Clark, K. Tapily, K.-H. Yu, T. Hakamata, S. Consiglio, D. O'Meara, C. Wajda, J. Smith and G. Leusink, APL Mater., 2018, 6, 058203 CrossRef.
- R. D. Dupuis, J. Vac. Sci. Technol., B: Nanotechnol. Microelectron.:Mater., Process., Meas., Phenom., 2023, 41, 060803 CAS.
- H. Pedersen and S. D. Elliott, Theor. Chem. Acc., 2014, 133, 1476 Search PubMed.
- J. R. Abelson and G. S. Girolami, J. Vac. Sci. Technol., A, 2020, 38, 030802 CrossRef CAS.
- S. M. George, Chem. Rev., 2010, 110, 111–131 CrossRef CAS PubMed.
- R. L. Puurunen, Chem. Vap. Deposition, 2014, 20, 332–344 CrossRef CAS.
- M. T. Bohr, R. S. Chau, T. Ghani and K. Mistry, IEEE Spectrum, 2007, 44, 29–35 Search PubMed.
- T. Hatanpää, M. Ritala and M. Leskelä, Coord. Chem. Rev., 2013, 257, 3297–3322 CrossRef.
- S. E. Koponen, P. G. Gordon and S. T. Barry, Polyhedron, 2016, 108, 59–66 CrossRef CAS.
- R. G. Gordon, in Atomic Layer Deposition for Semiconductors, ed. C. S. Hwang and C. Y. Yoo, Springer US, New York, 2014, pp. 15–46 Search PubMed.
- A. Kurek, P. G. Gordon, S. Karle, A. Devi and S. T. Barry, Aust. J. Chem., 2014, 67, 989–996 CrossRef CAS.
- S. T. Barry, Coord. Chem. Rev., 2013, 257, 3192–3201 CrossRef CAS.
- T. J. Knisley, L. C. Kalutarage and C. H. Winter, Coord. Chem. Rev., 2013, 257, 3222–3231 CrossRef CAS.
- S. B. Kim, A. Jayaraman, D. Chua, L. M. Davis, S.-L. Zheng, X. Zhao, S. Lee and R. G. Gordon, Chem. – Eur. J., 2018, 24, 9525–9529 CrossRef CAS PubMed.
- M. Gebhard, M. Hellwig, H. Parala, K. Xu, M. Winter and A. Devi, Dalton Trans., 2014, 43, 937–940 RSC.
- A. L. Brazeau, G. DiLabio, K. A. Kreisel, W. Monillas, G. P. A. Yap and S. T. Barry, Dalton Trans., 2007, 3297–3304 RSC.
- A. L. Brazeau, Z. Wang, C. N. Rowley and S. T. Barry, Inorg. Chem., 2006, 45, 2276–2281 CrossRef CAS PubMed.
- A. P. Kenney, G. P. A. Yap, D. S. Richeson and S. T. Barry, Inorg. Chem., 2005, 44, 2926–2933 CrossRef CAS PubMed.
- P. de Rouffignac, A. P. Yousef, K. H. Kim and R. G. Gordon, Electrochem. Solid-State Lett., 2006, 9, 45–48 CrossRef.
- P. Rouf, N. J. O'Brien, K. Rönnby, R. Samii, I. G. Ivanov, L. Ojamäe and H. Pedersen, J. Phys. Chem. C, 2019, 123, 25691–25700 CrossRef CAS.
- F. T. Edelmann, Adv. Organomet. Chem., 2008, 57, 183–352 CrossRef CAS.
- F. T. Edelmann, Adv. Organomet. Chem., 2013, 61, 55–374 CrossRef CAS.
- M. Corbett and B. F. Hoskins, Chem. Commun., 1968, 1602–1604 RSC.
- M. Corbett and B. F. Hoskins, Aust. J. Chem., 1974, 27, 665–670 CrossRef CAS.
- J. T. Leman and A. R. Barron, Polyhedron, 1989, 8, 1909–1912 CrossRef CAS.
- N. Nimitsiriwat, V. C. Gibson, E. L. Marshall, P. Takolpuckdee, A. K. Tomov, A. J. P. White, D. J. Williams, M. R. Elsegood and S. H. Dale, Inorg. Chem., 2007, 46, 9988–9997 CrossRef CAS PubMed.
- A. L. Johnson, A. M. Willcocks and S. P. Richards, Inorg. Chem., 2009, 48, 8613–8622 CrossRef CAS PubMed.
- K. R. Flanagan, J. D. Parish, M. A. Fox and A. L. Johnson, Inorg. Chem., 2019, 58, 16660–16666 CrossRef CAS PubMed.
- I. A. Guzei, L. M. Liable-Sands, A. L. Rheingold and C. H. Winter, Polyhedron, 1997, 16, 4017–4022 CrossRef CAS.
- F. E. Brinckman, H. S. Haiss and R. A. Robb, Inorg. Chem., 1965, 4, 936–942 CrossRef CAS.
- K. Soussi, S. Mishra, E. Jeanneau, J.-M. Millet and S. Daniele, Dalton Trans., 2017, 46, 13055–13064 RSC.
- K. Soussi, S. Mishra, E. Jeanneau, A. Mantoux and D. Stéphane, Polyhedron, 2018, 152, 84–89 CrossRef CAS.
- D. S. Moore and S. D. Robinson, Adv. Inorg. Chem., 1986, 30, 1–68 CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- N. J. O'Brien, P. Rouf, R. Samii, K. Rönnby, S. C. Buttera, C.-W. Hsu, I. G. Ivanov, V. Kessler, L. Ojamäe and H. Pedersen, Chem. Mater., 2020, 32, 4481–4489 CrossRef.
- P. Mpofu, P. Rouf, N. J. O'Brien, U. Forsberg and H. Pedersen, Dalton Trans., 2022, 51, 4712–4719 RSC.
- P. Rouf, R. Samii, K. Rönnby, B. Bakhit, S. C. Buttera, I. Martinovic, L. Ojamäe, C.-W. Hsu, V. Kessler, J. Palisaitis, V. Kessler, H. Pedersen and N. J. O'Brien, Chem. Mater., 2021, 33, 3266–3275 CrossRef CAS.
- A. A. Burk, M. J. O'Loughlin, R. R. Siergiej, A. K. Agarwal, S. Sriram, R. C. Clarke, M. F. MacMillan, V. Balakrishna and C. D. Brandt, Solid-State Electron., 1999, 43, 1459–1464 CrossRef CAS.
- P. Rouf, J. Palisaitis, B. Bakhit, N. J. O'Brien and H. Pedersen, J. Mater. Chem. C, 2021, 9, 13077–13080 RSC.
- S. V. Ivanov, T. V. Shubina, T. A. Komissarova and V. N. Jmerik, J. Cryst. Growth, 2014, 403, 83–89 CrossRef CAS.
- R. Samii, S. C. Buttera, V. Kessler and N. J. O'Brien, Eur. J. Inorg. Chem., 2022, e202200161 CrossRef CAS.
- R. Samii, D. Zanders, S. C. Buttera, V. Kessler, L. Ojamäe, H. Pedersen and N. J. O'Brien, Inorg. Chem., 2021, 60, 4578–4587 CrossRef CAS PubMed.
- R. Samii, PhD Thesis, Linköping University, 2022.
- R. Samii, D. Zanders, A. Fransson, G. Bačić, S. T. Barry, L. Ojamäe, V. Kessler, H. Pedersen and N. J. O'Brien, Inorg. Chem., 2021, 60, 12759–12765 CrossRef CAS PubMed.
- S. B. Kim, P. Sinsermsuksakul, A. S. Hock, R. D. Pike and R. G. Gordon, Chem. Mater., 2014, 26, 3065–3073 CrossRef CAS.
- R. Samii, A. Fransson, P. Mpofu, P. Niiranen, L. Ojamäe, V. Kessler and N. J. O'Brien, Inorg. Chem., 2022, 61, 20804–20813 CrossRef CAS PubMed.
- P. Bagherzadeh, BSc Thesis, Linköping University, 2023.
- R. Samii, D. Zanders, E. Barkas, A. Fransson, M. Lahtinen, V. Kessler, H. M. Tuononen, J. O. Moilanen and N. J. O'Brien, Dalton Trans., 2024, 53, 5911–5916 RSC.
- C. M. Caroff and G. S. Girolami, Inorg. Chem., 2022, 61, 16740–16749 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |