
A para-azaquinodimethane integrated quinoidal conjugated microporous polymer†
Aswani Raj
K
and
Rajeswara Rao
M
 *
*
Department of Chemistry, IIT Dharwad, Karnataka-580011, India. E-mail: rajesh@iitdh.ac.in
First published on 27th October 2023
Abstract
Quinoidal compounds own unique properties that make them a promising platform for optoelectronic applications. This has resulted in the development of a plethora of small and one-dimensional (1D) polymers. Surprisingly, there are currently no two-dimensional (2D) analogues available. In this paper, we report the synthesis of a 2D quinoidal-conjugated microporous polymer (Q1) derived from p-azaquinodimethane via Knoevenagel condensation of N,N-diacetyl-piperazine-2,5-dione and tris(4-formylphenyl)amine followed by O-alkylation. The presence of quinoidal p-azaquinodimethane in Q1 improves the π-delocalization within the framework, resulting in a deep red colour, low energy absorption (red edge ∼650 nm) and a bandgap of 1.9 eV. The polymer also enables halochromism to tune the optical bandgap to 1 eV. The p-doped (using iodine vapours) polymer (Q1+˙) exhibits high electrical conductivity up to 0.08 s m−1. Most critically, Q1+˙ maintains its stability for six days (and beyond) and exhibits stable electrical conductivity, overcoming the low intrinsic stability of such doped materials. This is due to the formation of stable delocalized mixed-valence species assisted by pyrazine formation. This report will spur the development of more quinoidal CMPs and pave the way for overcoming the inherent limitation of poor intra-sheet electron delocalization in 2D polymers.
Introduction
π-conjugated organic compounds have drawn increased attention due to their unique properties and outstanding applications.1–5 The π-conjugated quinoidal systems, in particular, are a subclass with carbon–carbon double bonds (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) in each aryl-ring.6,7 The π-bond arrangement in quinoidal systems is fundamentally different from that of conventional aromatic-ring based compounds. It improves π-delocalization in the conjugated backbone significantly, bestowing interesting properties such as narrow band gaps, low lowest unoccupied molecular orbital (LUMO) energy and long-wavelength light absorption characteristics, among others.6,8–10 The special properties of these compounds has prompted the active development of several quinoidal-based small molecules11–13 and one-dimensional polymers9,14,15 for optoelectronic applications such as field-effect transistors, organic photovoltaics, photodetectors and photofilters etc.16–19 Furthermore, quinoidal π-conjugated systems with large π-systems or D–A units exhibit biradical character (open shell), which may have applications in spintronics20 and photonics, among others.15,16,21,22
C) in each aryl-ring.6,7 The π-bond arrangement in quinoidal systems is fundamentally different from that of conventional aromatic-ring based compounds. It improves π-delocalization in the conjugated backbone significantly, bestowing interesting properties such as narrow band gaps, low lowest unoccupied molecular orbital (LUMO) energy and long-wavelength light absorption characteristics, among others.6,8–10 The special properties of these compounds has prompted the active development of several quinoidal-based small molecules11–13 and one-dimensional polymers9,14,15 for optoelectronic applications such as field-effect transistors, organic photovoltaics, photodetectors and photofilters etc.16–19 Furthermore, quinoidal π-conjugated systems with large π-systems or D–A units exhibit biradical character (open shell), which may have applications in spintronics20 and photonics, among others.15,16,21,22
Conjugated microporous polymers (CMPs)2,23–25 are a class of porous materials that combine the π-conjugated framework with a porous structure. As a result, these compounds enjoy both the functional properties of π-conjugated compounds and the functional properties of porous systems. The π-electron delocalization within the 2D framework contributes to the unique optoelectronic properties of CMPs and is felicitous for a wide range of applications, including solar cells, chemical sensors, photocatalysis and light-emitting diodes.26–29 The properties of the CMPs can be readily tuned by varying the building units or the linkers as desirable to a specific application. The amenability for structural modification combined with special properties of the CMPs resulted in a surge in the development of various CMPs via incorporation of a variety of π-conjugated building blocks (pyrene, tetraphenyl ethene, porphyrin, triaryl amine, etc.) and linkers (boronate ester/boroxine, imine, ketoenamine, cyano vinylene, vinylene, ethynylene, phenylene, etc.).2,30–32 However, the π-delocalization in the CMPs is less efficient than in 1D polymers. The polarizable linkers and cross-conjugated trifold symmetry building blocks are primarily responsible for this.2 As a result, the 2D inplane π-delocalization in CMPs is limited, resulting in wider band gaps, high-energy light absorption and inferior semiconducting properties (low electrical- and photo-conductivity, electron/hole mobility). Strategies such as (a) integrating donor (D)–acceptor (A) units,33,34 and (b) incorporation of cationic or radical building blocks35 have been employed to improve the electron delocalization. While these strategies are generally effective in achieving low bandgap CMPs (absorption red edge ≥ 650 nm), selecting the appropriate D–A units and stabilizing the radical building units can be difficult. Also, strong D and A building blocks are in short supply. To truly capitalize on the 2D-π-electron delocalization in CMPs and to realize the envisioned superior applications, a new class of conjugated backbones must be developed that can overcome the intrinsic limitations of these materials. Introducing quinoidal building units is one solution that can improve the π-delocalization within the framework. However, 2D-quinoidal polymers have been developed so far. Although Yildrim et al. recently developed a quinoid-oligothiophene incorporated covalent organic polymer (COP), the polymer was used for iodine uptake,36 with no discussion about the optoelectronic properties/applications.
Herein we report the development of a p-azaquinodimethane integrated 2D-quinoidal conjugated microporous polymer (Q1) via Knoevenagel condensation of N-diacetyl-piperazine-2,5-dione and tris(4-formylphenyl)amine followed by O-alkylation. p-azaquinodimethane is a highly stable quinoidal building block with efficient π-delocalization but has been scarcely explored.37 The incorporation of the p-azaquinodimethane moiety in Q1 significantly improved the π-delocalization resulting in low energy absorption (red edge ∼650 nm) and a bandgap of 1.9 eV. When doped, the polymer exhibits high electrical conductivity reaching up to 0.08 s m−1. The presence of p-azaquinodimethane improves the stability of the doped polymer (radical species) and results in comparable conductivity for at least six days.
Results and discussion
The targeted quinoidal polymer (Q1) was synthesized through a two-step synthetic strategy (Scheme 1). In the first step, N,N-diacetyl-piperazine-2,5-dione (AP) and tris(4-formylphenyl)amine (TTA) are subjected to Knoevenagel condensation in ethanol in the presence of K2CO3 at 100 °C to yield poly(3,6-bis(4-(diphenylamino)benzylidene)piperazine-2,5-dione) (nQ1). The intermediate CMP was subsequently alkylated under basic conditions using bromopropane in DMF at RT to produce poly(4,4′-(3,6-dipropoxypyrazine-2,5-diylidene)bis(methaneylylidene))bis(N,N-diphenylaniline)) Q1 in 90% yield. Using a similar synthetic procedure, a soluble model compound (MQ1) was also developed‡ (Scheme 1) to benefit from the solubility and evaluate the extended π-delocalization in the polymer.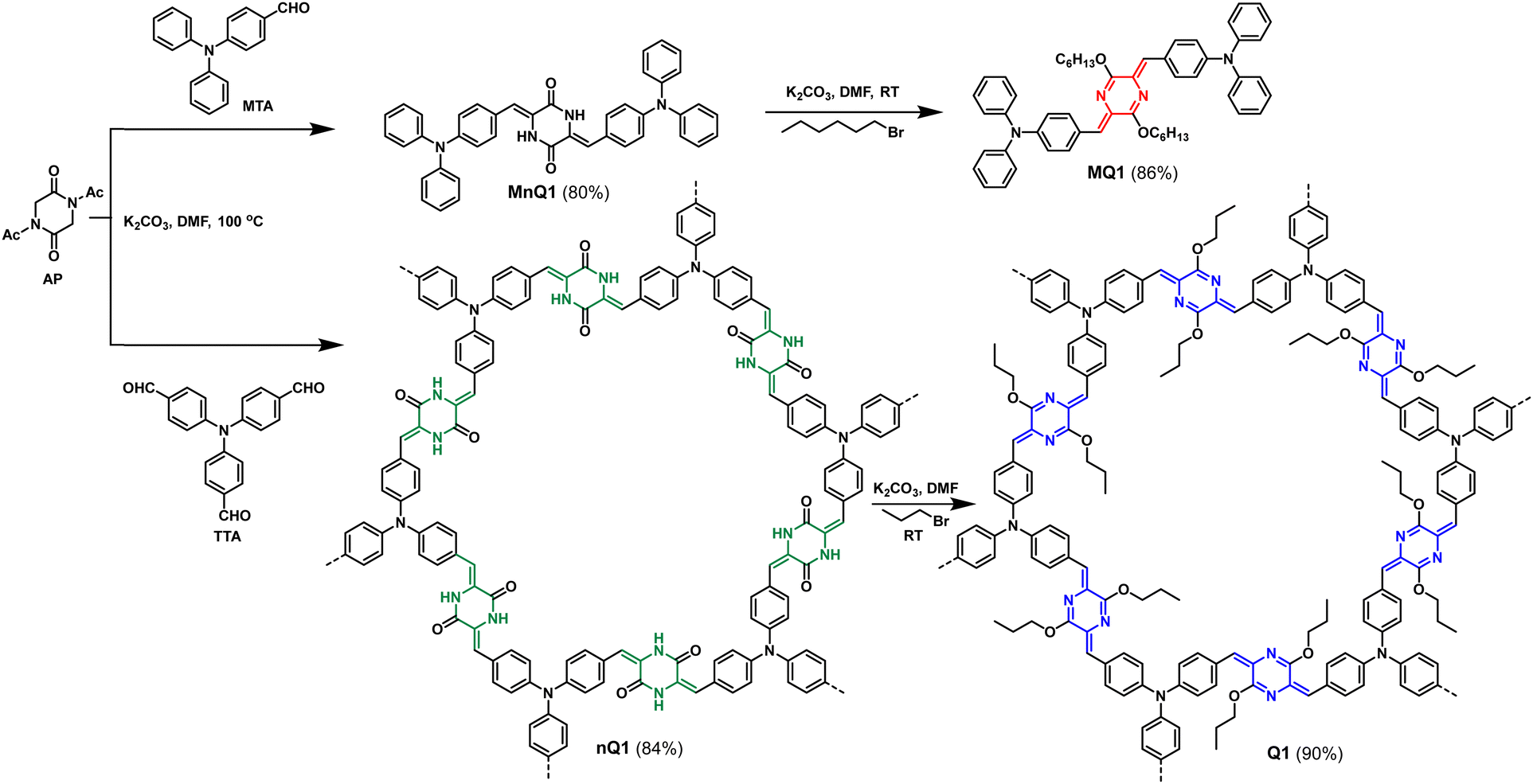 | ||
| Scheme 1 Synthesis of the quinoidal polymer (Q1) and the model compound (MQ1). | ||
The formation of the polymers was confirmed by CP/MAS 13C-NMR, Fourier transform Infrared (FT-IR) and UV-Visible absorption spectroscopy. In 13C NMR, nQ1 showed a characteristic signal at 160 ppm corresponding to carbonyl (–NH–CO) and the other key signals belonged to alkene (CC), and triphenylamine around 135 ppm and 110–130 ppm, respectively. On the other hand, for the quinoidal polymer (Q1), the signal for the carbonyl carbon completely vanished, and a new peak belonging to imine carbon (–CN) appeared at 152 ppm. This denotes the complete transformation of nQ1 to Q1. Moreover, one of the vinylene carbons showed a significant downfield shift (Fig. 1a) from 135 ppm to 147 ppm and the presence of the alkyl signals at 68 ppm (–OCH2–), 22 ppm (–CH2–) and 11 ppm (–CH3) supports the formation of the quinoidal polymer. The 13C NMR resonances of Q1 have also matched closely with that of the solution state 13C NMR of the model compound (MQ1). The FT-IR spectrum of nQ1 showed characteristic –CO and –C–NH stretching frequencies at ∼1674 cm−1 and ∼1396 cm−1, respectively, attributed to cyclic amide or the piperazine dione (lactam). In addition, the CC stretching of the aromatic rings appears at 1630 cm−1. The model compound (MnQ1) also exhibits similar frequencies confirming the formation of the polymer. In addition, the carbonyl stretching of nQ1 is significantly different from that of the starting material AP (1697 cm−1), confirming the consumption/absence of AP (Fig. S2a, ESI†). For Q1, the –CO and –C–NH stretching frequencies disappeared and new signals for –C–O- and –CN bands at ∼1275 cm−1 and ∼1654 cm−1, respectively, were observed, assuring the O-alkylation of the polymer. Furthermore, the stretching frequency of CC was also shifted from 1630 cm−1 to 1581 cm−1 (Fig. 1b). The bands observed in the FT-IR have been further confirmed by Raman spectroscopy (Fig. S2b, ESI†).
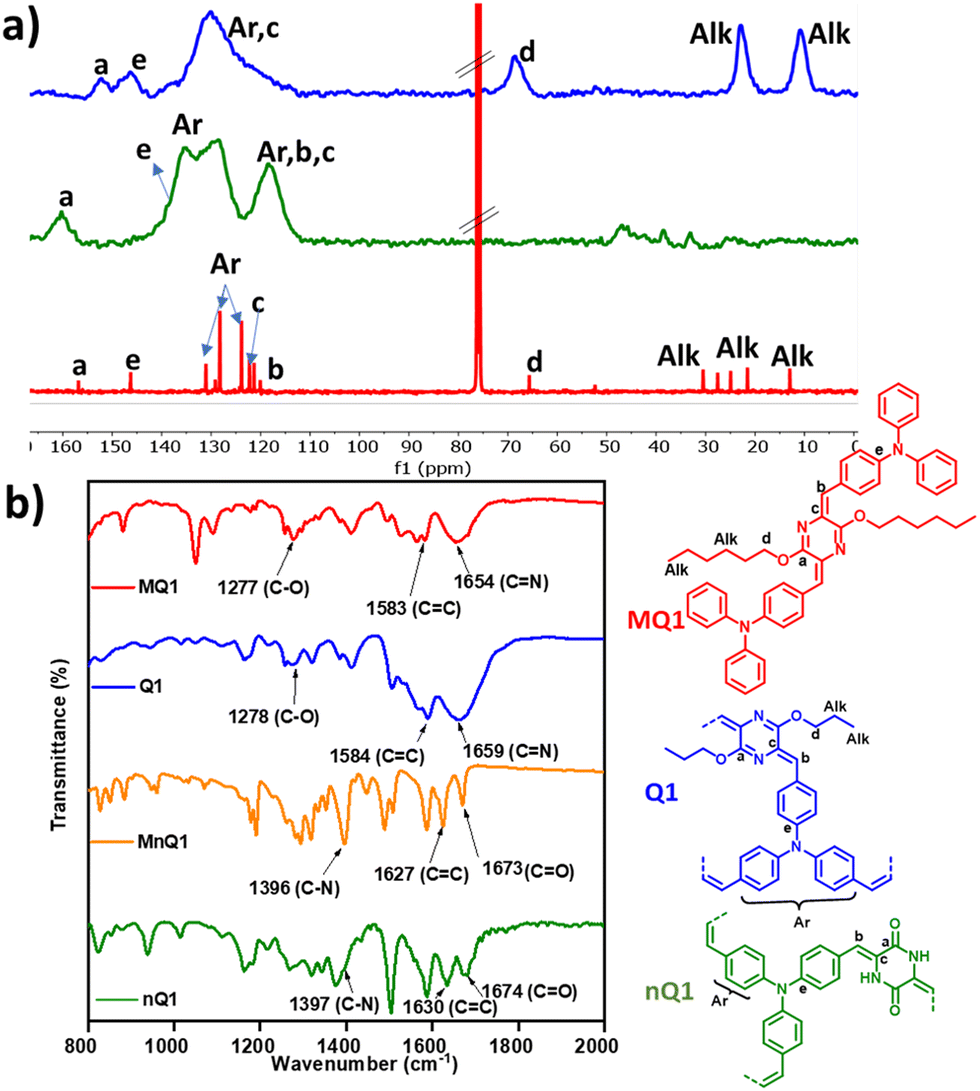 | ||
| Fig. 1 (a) CP/MAS 13C NMR and (b) FT-IR spectra of nQ1, Q1 and MQ1. | ||
The morphology of both polymers was studied by field emission scanning electron microscopy (FE-SEM). nQ1 exhibits globular/spherical morphology (200 nm to one μm dimensions), while Q1 exhibits small flake-like morphology (several μm dimensions) (Fig. S3a and S3b, ESI†). The 2D-morphology of Q1 has been studied using high resolution transmission electron microscopy (HR-TEM) and atomic force microscopy (AFM). The TEM images of Q1 clearly indicated the presence of large 2D polymeric sheets (and stacks) with sizes varying from 200–500 nm. Similarly, AFM also showed similar sheets piling up together to yield large 2D stacks having an average height of 100–120 nm. These two studies have clearly demonstrated that Q1 indeed possess a 2D network (Fig. S3c, ESI†). The stability of the two polymers was studied using thermogravimetric analysis. It is observed that both the polymers are thermally stable up to ∼250 °C, and the first decomposition temperatures of nQ1 and Q1 are 251 and 292 °C, respectively (Fig. S5, ESI†). Powder X-ray diffraction (PXRD) of nQ1 and Q1 revealed a broad spectrum implying that the materials are amorphous in nature (Fig. S6, ESI†). The permanent porosities of both polymers were determined by the Brunauer–Emmett–Teller (BET) method using N2 adsorption at 77 K. The polymers (nQ1 and Q1) exhibit the BET surface areas of 56.58 m2 g−1 and 72.79 m2 g−1 and a total pore volume of 0. 283 cm3 g−1 and 0.4251 cm3 g−1, respectively (Fig. S7, ESI†). Both the polymers possess a reversible pseudo type IV isotherm as nQ1 and Q1 show hysteresis and gas uptake at low pressure, however, with low uptake. This indicates that the network possesses micro pores with small sized macro pores due to the amorphous nature of the polymer.
DFT studies
Density functional theory (DFT) calculations on a single 2D-polymeric sheet of the polymers and their small molecular counterparts (MnQ1 and MQ1) were performed to better understand the structural and electronic properties of nQ1 and Q1. The polymers were optimised using the B3LYP functional under periodic boundary conditions (PBC). The plots of nQ1 and Q1 frontier orbitals (HOMO and LUMO) revealed nearly identical surfaces: the HOMOs are evenly distributed throughout the molecule, with relatively high electron density on the TPA moieties (Fig. 2 and Fig. S8, ESI†). Simultaneously, the LUMO is mostly delocalized on the p-azaquinodimethane units, indicating the presence of D–A interactions in the polymers. However, one significant difference in the surfaces of the polymers is the presence of a node at the carbonyl group in the HOMO of nQ1, which severely limits the electron communication between the TPA units. However, due to the quinoidalization, the node disappears in Q1 and allows a facile electron delocalization. It also had a significant impact on the HOMO and LUMO energies of the polymers: nQ1 exhibits −4.5 eV and −2.9 eV (band gap of 1.52 eV) while for Q1, the energies have been found to be −3.8 eV and −2.8 eV (band gap of 0.97 eV), respectively. The computed electronic surfaces and the HOMO–LUMO energies of the model compounds agree with those of the polymers, however with high HOMO and low LUMO energies, giving rise to enlarged bandgaps (Fig. 2).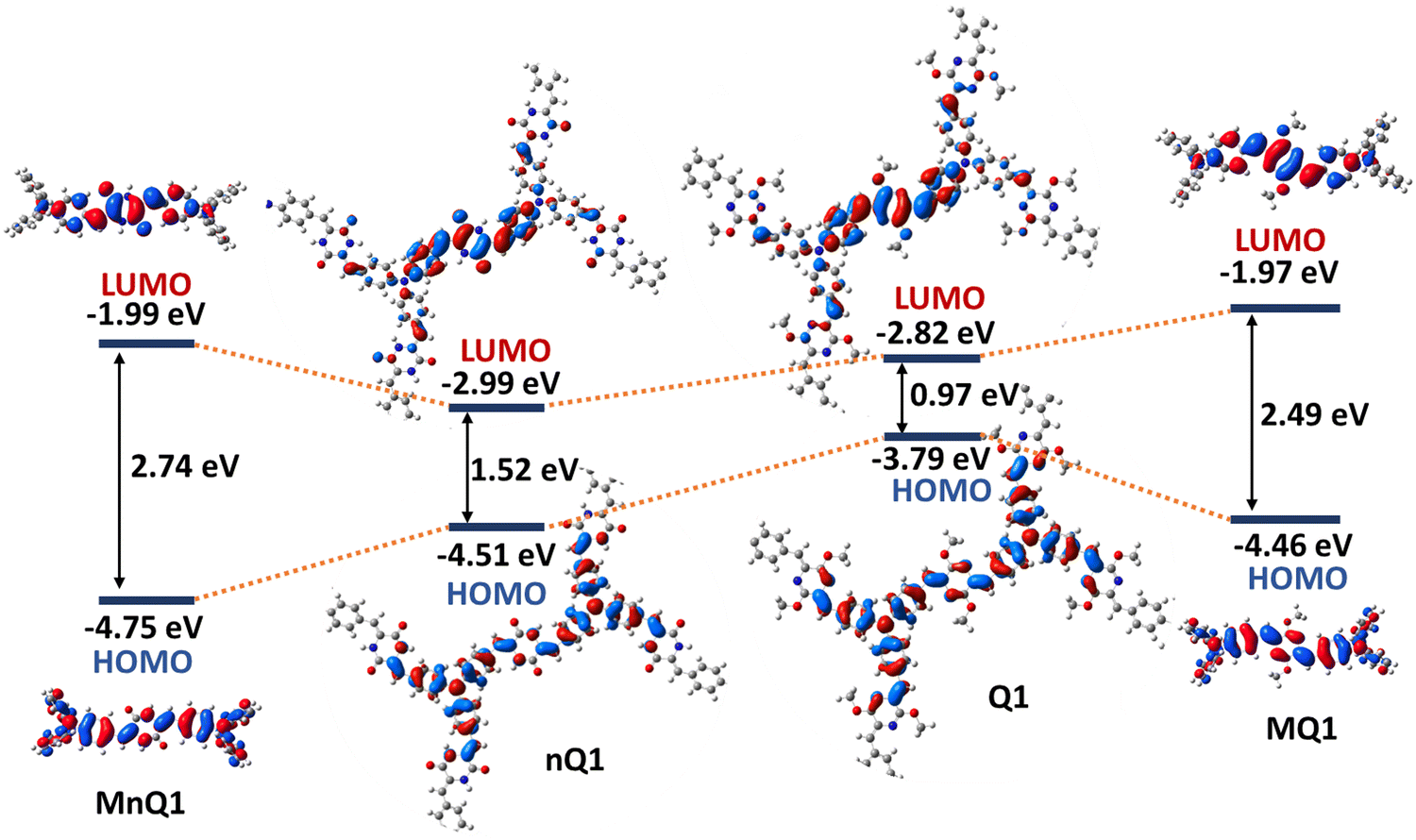 | ||
| Fig. 2 DFT-optimized frontier molecular orbitals (FMOs) and energy level diagram of the model compounds and polymers [B3LYP/6-31G(d), gaseous state]. | ||
To assess the effect of the quinoidal unit on the electronic properties of the polymers, DFT calculations were performed on a set of small molecules containing various π-electron promoting linkers, such as diimine benzene (M1), divinyl benzene (M2), and quinodimethane (M3) and the results were compared with MQ1 (Fig. 3). Moving from M1 to M3 and to MQ1, the energy of the HOMO decreases while the energy of the LUMO increases. As a result, the bandgaps have shrunk in the order of M1 (3.24 eV) > M2 (2.94 eV) > M3 (2.52 eV) > MQ1 (2.49 eV). This implies that the p-azaquinodimethane units promote π-delocalization far better compared to other commonly used π-conjugated units.
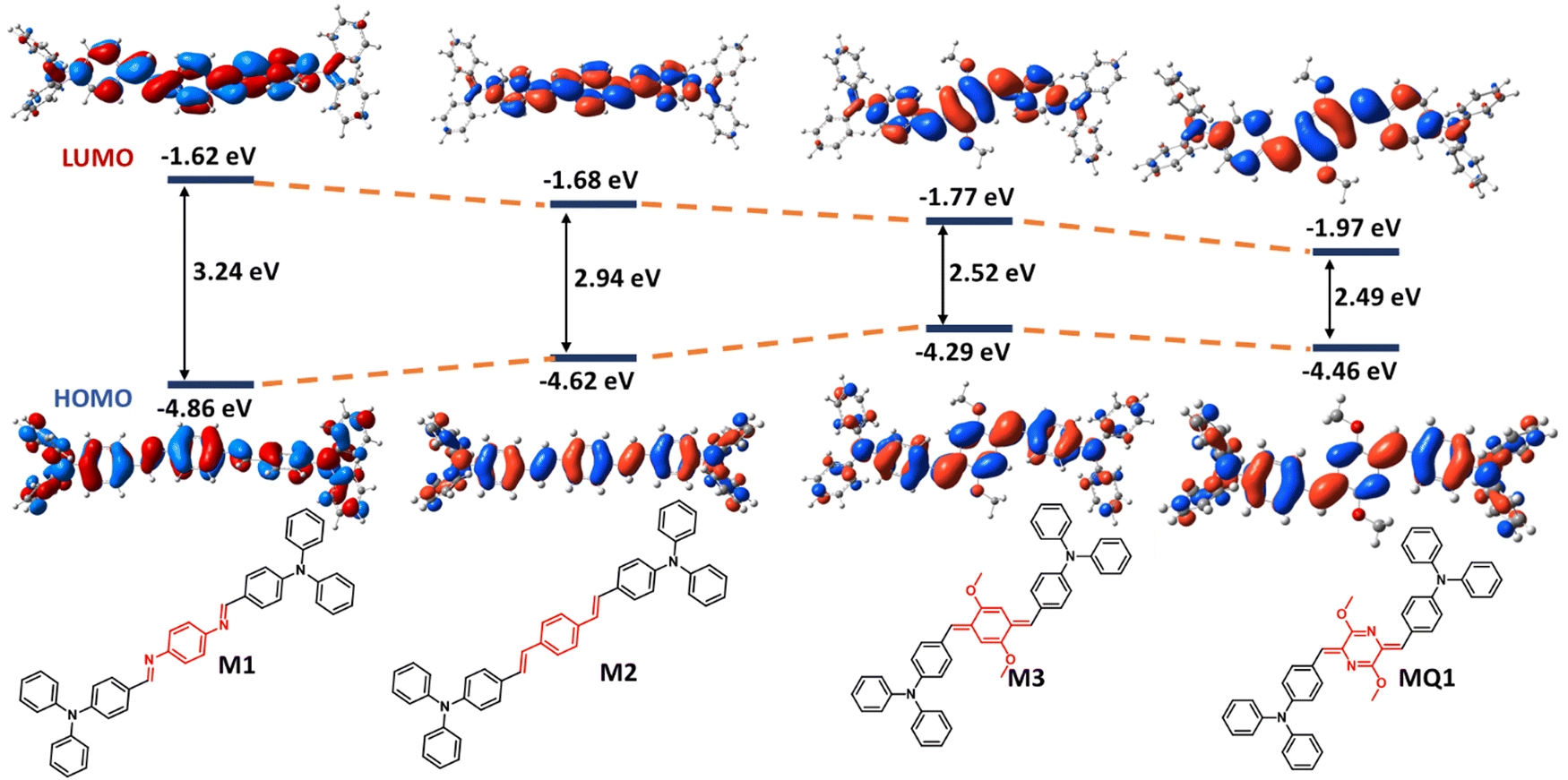 | ||
| Fig. 3 DFT-optimized frontier molecular orbitals (FMOs) and energy level diagram of model compounds M1, M2, M3 and MQ1[B3LYP/6-31G(d), gaseous state]. | ||
The optical properties of the polymers were investigated using UV-Vis-NIR diffuse reflectance absorption spectroscopy (Fig. 4a). The powder of non-alkylated polymer (nQ1) showed a strong absorption red edge at 600 nm. Tautomerism promoted quinoidalization of the central diketopiperazine unit is responsible for nQ1's strong visible absorption. On the other hand, the absorption red edge is redshifted to 650 nm in Q1, which is consistent with the better π-electron delocalization due to quinoidalization. The optical band gaps of nQ1 and Q1 are estimated to be 2.07 eV and 1.91 eV, respectively. The band gap is reduced by ∼0.2 eV due to quinoidal arrangement. A similar trend of redshift has also been observed in the absorption spectra of the model compounds of the polymers MnQ1 (486 nm) and MQ1 (545 nm). However, it is important to note that the polymers vs. their model compounds exhibit a redshift of ∼110 nm due to extended π-delocalization in the polymers. The deep colour of raspberry and red of the quinoidal MQ1 and Q1vs light colours (yellow and brown) of the non-quinoidal congeners also supports the extended π-delocalization (Fig. 4b). It is also worth mentioning that the red edge of Q1 is also ∼120 nm bathochromically shifted compared to the imine-linked structurally similar 2D-polymer (Fig. S9, ESI†).38
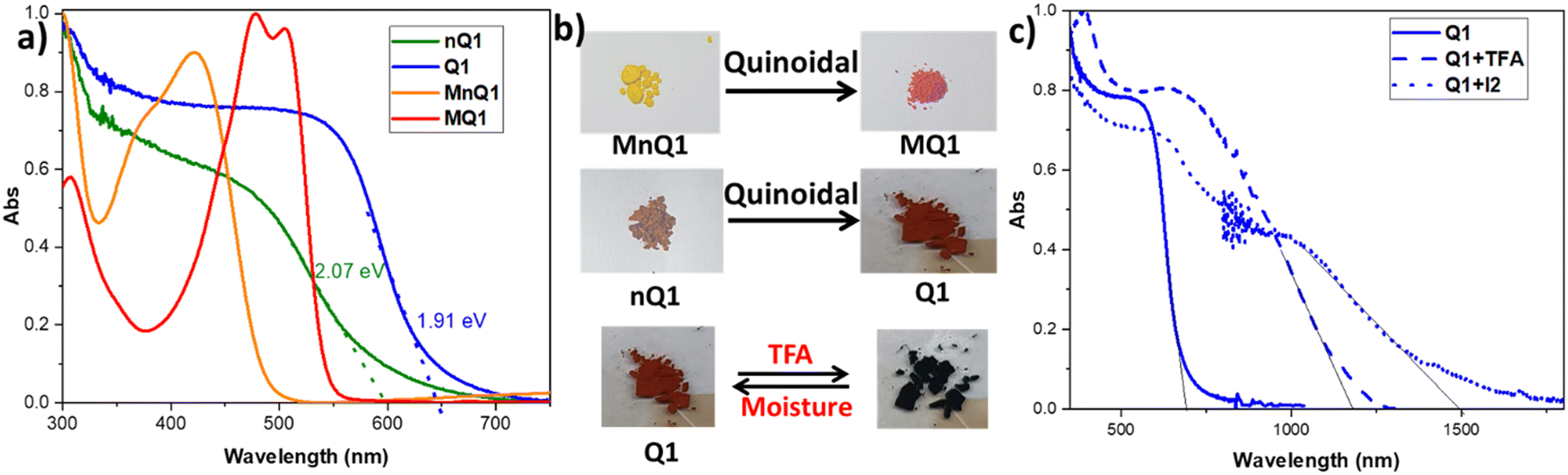 | ||
| Fig. 4 (a) The UV-Vis absorption spectra and (b) photograph of the polymers (nQ1, Q1) and the model compounds (MnQ1, MQ1); (c) Comparison of the UV-Vis absorption spectra of Q1 with Q1 in TFA and iodine. | ||
The polymer (Q1) exhibits halochromic properties in trifluoroacetic acid (TFA). Halochromism allows for reversible tuning of the optical and electronic properties of the molecules without causing structural alterations.39–42 The basic lactam nitrogen in the p-azaquinodimethane can interact with the acid and modulate it's properties. Interestingly, when the polymer is exposed to TFA, the colour of the polymer turned black (Fig. 4b), and the red edge of the absorption shifted from 650 nm to ∼1200 nm (Fig. 4c). The corresponding band gap was calculated to be ∼1 eV. Such a redshift of ∼600 nm upon protonation is unusual for 2D polymers. This could be attributed to the enhanced electron-accepting nature of protonated piperazine moieties. Interestingly, the protonated species will return to its original non-protonated form in ambient conditions in a time frame of 16–24 hours. This could be due to the high energy form of the protonated species, which may transfer the proton to the moisture in the air. nQ1 also displays halochromism with a redshift of ∼250 nm in the absorption. This could be attributed to protonation on the carbonyl site (Fig. S10, ESI†).
The electrochemical properties of the polymers (nQ1 and Q1) along with the model compound (MQ1) have been investigated (Fig. 5). The non-quinoidal polymer (nQ1) exhibits one reversible oxidation peak at 0.26 V vs. Fc/Fc+ corresponding to the TPA unit and a quasi-reversible reduction peak at −1.55 V vs. Fc+/Fc belonging to the diketopiperazine unit. Upon quinoidalization in Q1, the oxidation peak becomes relatively easier and a peak appears at 0.15 V vs. Fc/Fc+ while the reduction becomes slightly harder with a reduction peak at −1.56 V vs. Fc/Fc+. This observation is in agreement with the electron deficient withdrawing character of the piperazine-2,5-dione in nQ1. The corresponding HOMO/LUMO energies of nQ1 and Q1 are calculated to be −5.06/−3.25 eV and −4.95/−3.24 eV, respectively. The model compound (MQ1) has also exhibited similar redox properties; however, with slightly lower HOMO (0.25 V/−5.05 eV) and higher LUMO (−1.70 V/−3.10 eV) energies compared to Q1 due to restricted π-delocalization (Fig. 5).
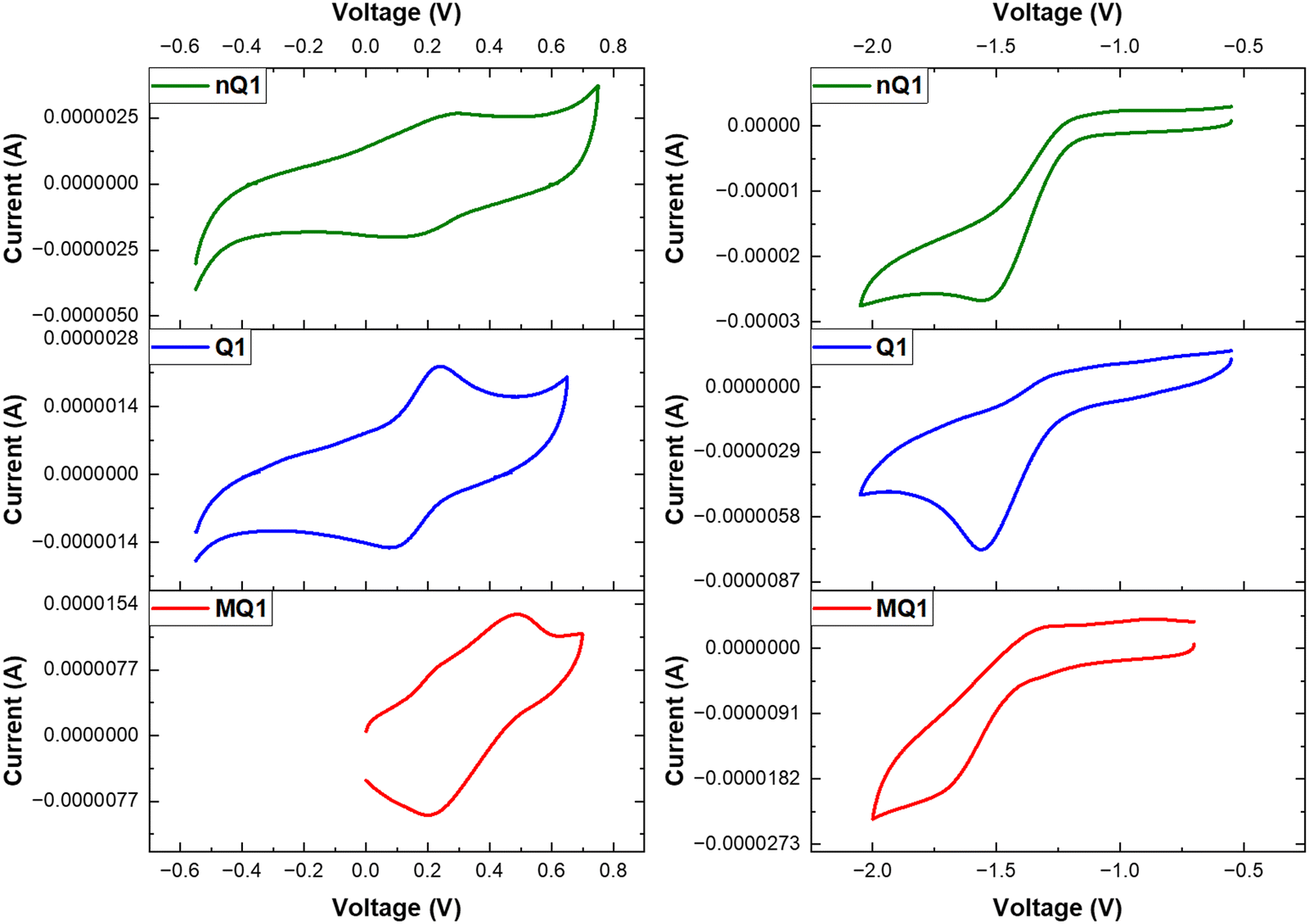 | ||
| Fig. 5 Cyclic voltammograms of nQ1, Q1 and MQ1. | ||
Electrical conductivity
Inspired by the presence of a quinoidal p-azaquinodimethane unit, we investigated the electrical conductivity of Q1. As a first step, the electron-rich triphenylamine units associated with the permanent porosity of Q1 enabled us to develop charge carriers via doping the material with an iodine dopant. The process of doping of organic materials has been a well-established strategy43–46 for introducing charge carriers and increasing the conducting ability of materials. Iodine is the most commonly used oxidant due to its reduction potential of 0.784 V for I3−.47 By exposing the quinoidal and also its precursor polymer, Q1 and nQ1, to iodine vapours, they were successfully doped (oxidizing triphenylamine). The formation of the polymers' charge carriers (cation radicals) has been confirmed by the strong NIR region absorption characteristics (∼1500 nm and bandgap of ∼0.8 eV) (Fig. 4c and Fig. S10, ESI†). The formation of delocalized mixed-valence species within the framework is responsible for such low-energy light absorption. Using the 2-probe method, electrical conductivity measurements were performed on compressed cylindrical pellets of pristine and doped nQ1 and Q1 with dimensions of 0.15 × 0.4 cm. The current–voltage (I–V) curves of the pristine CMPs (nQ1 and Q1) displayed nearly an insulating behaviour (∼10-7 Sm−1). However, the conductivity (σRT) of doped nQ1 and Q1 increased by several orders of magnitude to 2.3 × 10−3 and 8 × 10−2 Sm−1, respectively (Fig. 6a). When compared to nQ1, the facile electron/charge delocalization pathways supported by the quinoidal polymer (Q1) demonstrated 35 times higher conductivity. The conductivity of the Q1 samples doped for different time intervals (4, 8, 12, 16 and 24 hrs) was also tested to determine the optimal doping time (Fig. 6b). The study found that the conductivity was highest when the polymer pellets were exposed to iodine vapours for 12 hours and maintains a similar conductivity with slight drop after that. This indicates that the concentration of the charge carriers is at its highest after 12 hours of exposure.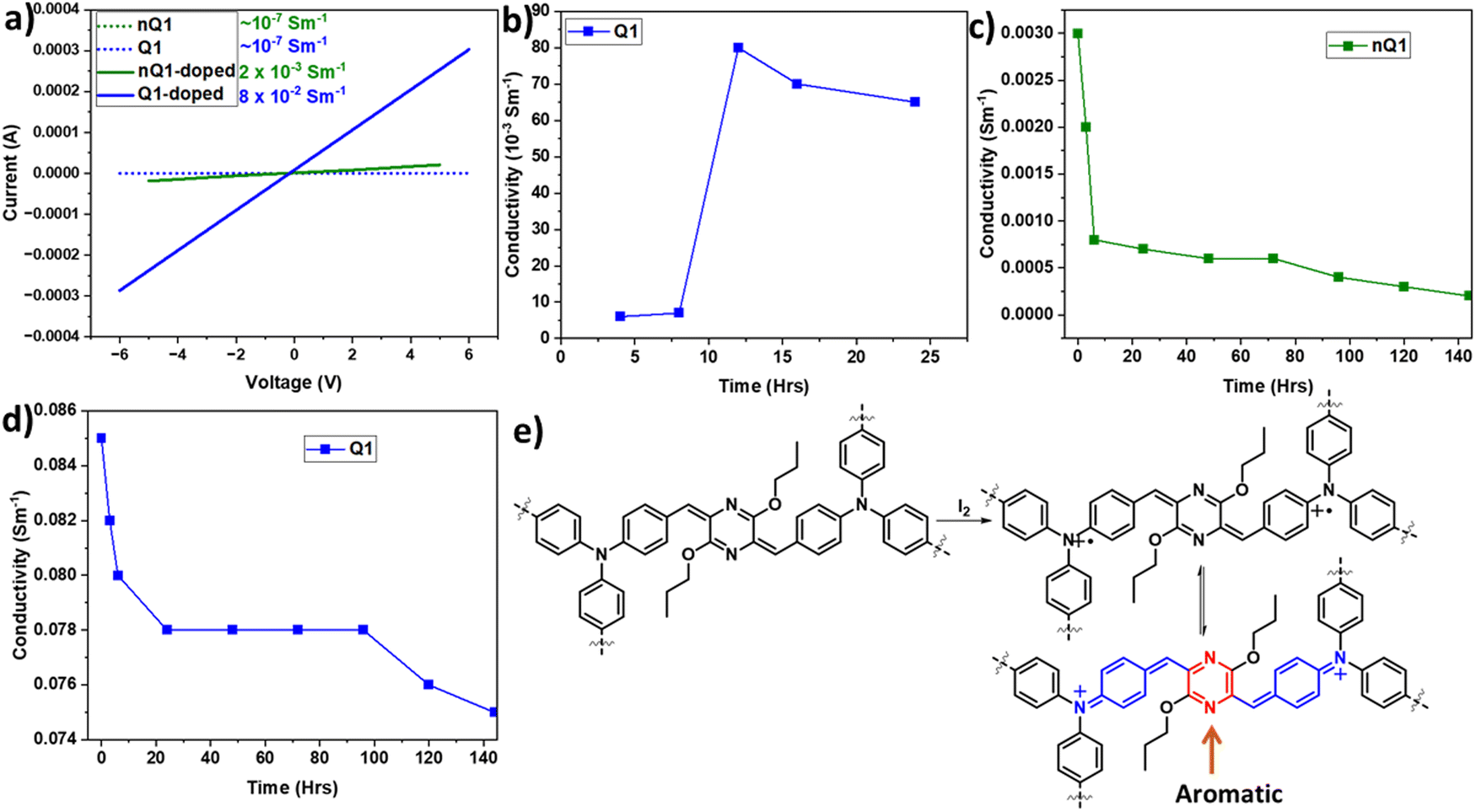 | ||
| Fig. 6 (a) I–V graph of pristine and doped nQ1 and Q1; (b) time dependent doping of Q1; the time dependent stability of nQ1 (c) and Q1 (d); (e) the resonance structures of doped Q1. | ||
Although the doping will greatly enhance the conductivity of the polymer, the resultant radicals often suffer from poor stability. The high reactivity of the charge carriers undergoes unwanted reactions and will decompose. Thus, it is highly important not only to improve the conducting properties of the materials but they should also have high stability. In this line, to assess the stability of doped nQ1 and Q1, we measured the conductivity of the doped polymers at different time intervals (up to 140 hours) (Fig. 6c and d). The conductivity of doped nQ1 dropped consistently (2 × 10−4) as the time progressed, however with a drastic jump from 3 × 10−3 S m−1 to 5 × 10−4 S m−1 within 10 hours. On the other hand, the doped nQ1 showed a slight decline of conductivity of the polymer from ∼0.085 S m−1 to 0.08 S m−1 within 10 hours but later it stabilized and showed a stable conductivity result throughout the measurement (up to 140 hours). The stable conductivity can in turn be assigned to high stability of the doped polymer. This is due to the formation of a neutral aromatic pyrazine ring via combination of the radicals (Fig. 6e). However, the initial drop in the conductivity can be attributed to the high vapour pressure of iodine, which hinders the long-term doping.48
Conclusions
In summary, a synthetic approach was developed to construct a quinoidal conjugated microporous polymer. The CMP was synthesized in a two-step synthetic process. In the first step, N,N-diacetyl-piperazine-2,5-dione and tris(4-formylphenyl)amine were condensed to yield nQ1 which was subsequently O-alkylated under basic conditions to obtain the quinoidal p-azaquinodimethane polymer (Q1). The quinoidal p-azaquinodimethane unit promotes the electron delocalization between the integrated building blocks. Both the polymers have been thoroughly characterized using solid state 13C NMR, FT-IR, UV-Vis-NIR and DFT studies. The quinoidal polymers exhibit strong low energy absorption at 650 nm (red edge) and a narrow band gap (1.9 eV), which experienced a redshift to 1200 nm (1 eV) upon protonation. The p-doped polymer (Q1+˙) exhibited high electrical conductivity of 0.08 S m−1 and high stability up to 120–140 hours.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Rajeswara Rao thanks the Science and Engineering Research Board (SERB), India, for partially supporting this research through its early career research award scheme (ECR/2018/002285). In addition, the authors acknowledge the help of the staff members associated with the sophisticated central instrumentation facility, IIT Dharwad, for carrying out the material characterizations.References
- A. Mateo-Alonso, Chem. Mater., 2023, 35, 1467–1469 CrossRef CAS.
- H. V. Babu, M. G. M. Bai and M. Rajeswara Rao, ACS Appl. Mater. Interfaces, 2019, 11, 11029–11060 CrossRef CAS PubMed.
- W. Wu, Y. Liu and D. Zhu, Chem. Soc. Rev., 2010, 39, 1489–1502 RSC.
- A. Moliton and R. C. Hiorns, Polym. Int., 2004, 53, 1397–1412 CrossRef CAS.
- S. B. Mdluli, M. E. Ramoroka, S. T. Yussuf, K. D. Modibane, V. S. John-Denk and E. I. Iwuoha, Polymers, 2022, 14(4), 716 CrossRef CAS.
- J. Huang and G. Yu, Mater. Chem. Front., 2021, 5, 76–96 RSC.
- X. Shi and C. Chi, Top. Curr. Chem., 2017, 375, 1–39 CrossRef CAS.
- J. Casado, Top. Curr. Chem., 2017, 375, 1–40 CrossRef CAS PubMed.
- K. Kawabata, M. Saito, I. Osaka and K. Takimiya, J. Am. Chem. Soc., 2016, 138, 7725–7732 CrossRef CAS PubMed.
- T. Mikie and I. Osaka, J. Mater. Chem. C, 2020, 8, 14262–14288 RSC.
- F. Valentini, F. Sabuzi, M. Forchetta, V. Conte and P. Galloni, RSC Adv., 2023, 13, 9065–9077 RSC.
- C. Wang, T. Du, Y. Deng, J. Yao, R. Li, X. Zhao, Y. Jiang, H. Wei, Y. Dang, R. Li and Y. Geng, Chem. Sci., 2021, 12, 9366–9371 RSC.
- K. Kitamura, R. Kudo, H. Sugiyama, H. Uekusa and T. Hamura, Chem. Commun., 2020, 56, 14988–14991 RSC.
- X. Liu, B. He, C. L. Anderson, J. Kang, T. Chen, J. Chen, S. Feng, L. Zhang, M. A. Kolaczkowski, S. J. Teat, M. A. Brady, C. Zhu, L. W. Wang, J. Chen and Y. Liu, J. Am. Chem. Soc., 2017, 139, 8355–8363 CrossRef CAS.
- X. Li, J. Guo, L. Yang, M. Chao, L. Zheng, Z. Ma, Y. Hu, Y. Zhao, H. Chen and Y. Liu, Front. Chem., 2019, 7, 1–12 CrossRef.
- B. Dyaga, S. Mayarambakam, O. A. Ibraikulov, N. Zimmermann, S. Fall, O. Boyron, T. Heiser, N. Leclerc, N. Berton and B. Schmaltz, Mater. Adv., 2022, 3, 6853–6861 RSC.
- L. Pan, T. Zhan, J. Oh, Y. Zhang, H. Tang, M. Yang, M. Li, C. Yang, X. Liu, P. Cai, C. Duan, F. Huang and Y. Cao, Chem. – Eur. J., 2021, 27, 13527 CrossRef CAS PubMed.
- J. Huang, S. Lu, P. A. Chen, K. Wang, Y. Hu, Y. Liang, M. Wang and E. Reichmanis, Macromolecules, 2019, 52, 4749–4756 CrossRef CAS.
- K. Hwang, M. H. Lee, J. Kim, Y. J. Kim, Y. Kim, H. Hwang, I. B. Kim and D. Y. Kim, Macromolecules, 2020, 53, 1977–1987 CrossRef CAS.
- T. Kubo, Chem. Lett., 2015, 44, 111–122 CrossRef.
- X. Ji and L. Fang, Polym. Chem., 2021, 12, 1347–1361 RSC.
- Y. Kim, Y. J. Kim, Y. A. Kim, E. Jung, Y. Mok, K. Kim, H. Hwang, J. J. Park, M. G. Kim, S. Mathur and D. Y. Kim, ACS Appl. Mater. Interfaces, 2021, 13, 2887–2898 CrossRef CAS.
- Y. Xu, S. Jin, H. Xu, A. Nagai and D. Jiang, Chem. Soc. Rev., 2013, 42, 8012–8031 RSC.
- J. S. M. Lee and A. I. Cooper, Chem. Rev., 2020, 120, 2171–2214 CrossRef CAS.
- Y. Liao, Z. Cheng, W. Zuo, A. Thomas and C. F. J. Faul, ACS Appl. Mater. Interfaces, 2017, 9, 38390–38400 CrossRef CAS.
- M. G. M. Bai, H. V. Babu, V. Lakshmi and M. R. Rao, Mater. Chem. Front., 2021, 5, 2506–2551 RSC.
- S. Luo, Z. Zeng, G. Zeng, Z. Liu, R. Xiao, P. Xu, H. Wang, D. Huang, Y. Liu, B. Shao, Q. Liang, D. Wang, Q. He, L. Qin and Y. Fu, J. Mater. Chem. A, 2020, 8, 6434–6470 RSC.
- Y. B. Zhou and Z. P. Zhan, Chem. – Asian J., 2018, 13, 9–19 CrossRef CAS.
- K. Amin, N. Ashraf, L. Mao, C. F. J. Faul and Z. Wei, Nano Energy, 2021, 85, 105958 CrossRef CAS.
- A. Acharjya, P. Pachfule, J. Roeser, F.-J. Schmitt and A. Thomas, Angew. Chem., Int. Ed., 2019, 58, 14865 CrossRef CAS.
- X. Guo, M. Baumgarten and K. Müllen, Prog. Polym. Sci., 2013, 38, 1832–1908 CrossRef CAS.
- M. G. Mohamed, T. H. Mansoure, M. M. Samy, Y. Takashi, A. A. K. Mohammed, T. Ahamad, S. M. Alshehri, J. Kim, B. M. Matsagar, K. C. W. Wu and S. W. Kuo, Molecules, 2022, 27(6), 2025 CrossRef CAS PubMed.
- Z. Wang, X. Xing, I. F. Perepichka, Y. Sun, B. Han, Y. Wu, Y. Zhu, J. Ning, M. Wang, J. He, H. Liu, Y. He, C. K. F. Shen and H. Meng, Chem. Mater., 2022, 34, 4896–4909 CrossRef CAS.
- S. Sau and S. K. Samanta, Chem. Commun., 2022, 59, 635–638 RSC.
- M. M. G. Bai, K. Bramhaiah, S. Bhattacharyya and R. M. Rao, Chem. – Eur. J., 2022, 28, e202202023 CrossRef CAS.
- O. Yildirim, A. Tsaturyan, A. Damin, S. Nejrotti, V. Crocellà, A. Gallo, M. R. Chierotti, M. Bonomo and C. Barolo, ACS Appl. Mater. Interfaces, 2023, 15(12), 15819–15831 CrossRef CAS.
- X. Liu, C. L. Anderson and Y. Liu, Acc. Chem. Res., 2023, 56(12), 1669–1682 CrossRef CAS.
- L. D. Tran, K. F. Presley, J. K. Streit, J. Carpena-Núñez, L. K. Beagle, T. A. Grusenmeyer, M. J. Dalton, R. A. Vaia, L. F. Drummy, N. R. Glavin and L. A. Baldwin, Chem. Mater., 2022, 34, 529–536 CrossRef CAS.
- K. Laxman, Y. Che, K. A. Raj, D. F. Perepichka and M. R. Rao, J. Mater. Chem. C, 2023, 11, 2680–2687 RSC.
- H. Phan, T. J. Kelly, A. Zhugayevych, G. C. Bazan, T. Q. Nguyen, E. A. Jarvis and S. Tretiak, J. Phys. Chem. Lett., 2019, 10, 4632–4638 CrossRef CAS PubMed.
- T. Roy, I. Debnath and K. Mahata, Org. Chem. Front., 2022, 9, 3255–3261 RSC.
- P. Gayathri, P. Nag, N. Anand, S. R. Vennapusa, M. Pannipara, A. G. Al-Sehemi, D. Moon and S. P. Anthony, New J. Chem., 2021, 45, 22450–22460 RSC.
- M. L. Tietze, J. Benduhn, P. Pahner, B. Nell, M. Schwarze, H. Kleemann, M. Krammer, K. Zojer, K. Vandewal and K. Leo, Nat. Commun., 2018, 9, 1–8 CrossRef.
- I. Salzmann, G. Heimel, M. Oehzelt, S. Winkler and N. Koch, Acc. Chem. Res., 2016, 49, 370–378 CrossRef CAS PubMed.
- A. L. Dixon, H. Vezin, T. Q. Nguyen and G. N. M. Reddy, Mater. Horiz., 2022, 9, 981–990 RSC.
- P. Raval, M. Dhennin, H. Vezin, T. Pawlak, P. Roussel, T. Q. Nguyen and G. N. Manjunatha Reddy, Electrochim. Acta, 2022, 424, 140602 CrossRef CAS.
- P. H. Qi and J. B. Hiskey, Hydrometallurgy, 1993, 32, 161–179 CrossRef CAS.
- S. L. Cai, Y. B. Zhang, A. B. Pun, B. He, J. Yang, F. M. Toma, I. D. Sharp, O. M. Yaghi, J. Fan, S. R. Zheng, W. G. Zhang and Y. Liu, Chem. Sci., 2014, 5, 4693–4700 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tc02233a |
| ‡ The synthesis of MQ1 is reported in a manuscript which is under review for some other journal. |
| This journal is © The Royal Society of Chemistry 2024 |