
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemical degradation of oxygenated polymers: the case of polyethers and polysiloxanes
Shamna
Salahudeen
 ,
Tabea A.
Thiel
and
Esteban
Mejía
*
,
Tabea A.
Thiel
and
Esteban
Mejía
*
Leibniz Institute for Catalysis (LIKAT), Albert-Einstein-Str. 29a, 18059 Rostock, Germany. E-mail: Esteban.mejia@catalysis.de
First published on 27th May 2024
Abstract
Plastics have become ubiquitous and indispensable in our daily lives since their invention at the beginning of the 20th century. As a result, millions of tonnes of plastic waste are produced every year and efforts to reduce their environmental impact are increasing. Most of the efforts have logically been devoted to tackling the plastic wastes with the highest volume, namely polyolefins and polyesters, as is reflected in the scientific and patent literature. In this review we have focused on a less studied class of polymer wastes, polyethers and polysiloxanes, both consisting of monomeric units linked by oxygen atoms. The most representative examples of chemical degradation of these materials at laboratory and industrial scales are presented and their reaction mechanisms are discussed.
 Shamna Salahudeen | Shamna Salahudeen was born in Kerala, India. She received her BSc degree (2017) from the Department of Chemistry, N. S. S College, Pandalam and her MSc. degree (2019) from the School of Chemical Sciences, Mahatma Gandhi University, Kerala. In 2022, she joined the Leibniz Institute for Catalysis (LIKAT), Germany, where she is currently doing her PhD in the group of Esteban Mejía. Her current research interests revolve around development of catalyst and methodologies for depolymerization and value-added polymers synthesis. |
 Tabea A. Thiel | Tabea Angela Thiel studied chemistry in Berlin, where she obtained her bachelor's degree at the Freie Universität and her master's degree at the Technische Universität Berlin. In 2022, she moved to Rostock to pursue her PhD in catalyzed polymerizations at the Leibniz Institute for Catalysis (LIKAT) under the supervision of Esteban Mejía Udo Kragl. Next to her PhD, her research interests also include topics of reaction modeling and engineering, heterogeneous (photo-)catalysis, and, recently, astrochemistry. |
 Esteban Mejía | Esteban Mejía studied chemistry at National University of Colombia in Bogota, where he also obtained his master's degree. In 2008 he moved to Switzerland to pursue his PhD in homogeneous catalysis at the ETH Zurich under the supervision of Antonio Togni. In 2012 he joined the group of Matthias Beller at the Leibniz Institute for Catalysis (LIKAT) in Rostock (Germany) as a postdoc. In 2014 he started his independent career at LIKAT where he completed his Habilitation in 2020 (German equivalent to tenured professorship). He is currently leader of the group of Polymer Chemistry and Catalysis at LIKAT. |
Sustainability spotlightThe continuous accumulation of plastic waste is one of the greatest environmental consequences of human activity on Earth and needs to be addressed urgently. Many researchers, both in industry and academia, have made commendable efforts to chemically recycle plastic waste, focusing mainly on polyolefins and polyesters, as these represent the largest volume. However, another important class of synthetic polymers, namely polyethers and silicones, has escaped the spotlight. Therefore, a literature review is needed to present the latest academic and industrial advances in their chemical degradation. The research efforts summarised in this review are aimed at achieving the UN SDGs: Responsible consumption and production, Sustainable cities and communities, Life below water, Life on land and Climate action. |
1 Introduction
From the development of Bakelite, the first fully synthetic thermosetting plastic, by the Belgian chemist Leo H. Baekeland in 1907,1 the plastics industry grew dramatically over the following four decades. Important milestones were achieved by industrial and academic researchers, such as Wallace H. Carothers, who contributed to the development of polyamides and polyesters, and Karl Ziegler and Giulio Natta, whose discoveries revolutionized the polyolefins field. Furthermore, during World War II, significant efforts were made to replace natural polymers such as rubber, which led to the development of numerous applications.2Since 1950, the global production of plastics has experienced exponential growth, resulting in an enormous amount of plastic waste. It is projected that by 2025, the amount of plastic waste generated will exceed 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 million metric tons.3 Worryingly, around 80% of polymer waste ends up in the environment, either in landfills or contaminating land and marine ecosystems, while a further 10% is incinerated, polluting the atmosphere.4 The ubiquity of plastic waste, in particular of microplastic particles (<5 nm), their resistance to degradation and their high mobility have made them “technofossils” that define the beginning of the Anthropocene, the ongoing unit in the geological time scale that marks the consequences of human activity on Earth.5,6 These activities obviously have had a terrible impact on the environment and need to be addressed urgently.
000 million metric tons.3 Worryingly, around 80% of polymer waste ends up in the environment, either in landfills or contaminating land and marine ecosystems, while a further 10% is incinerated, polluting the atmosphere.4 The ubiquity of plastic waste, in particular of microplastic particles (<5 nm), their resistance to degradation and their high mobility have made them “technofossils” that define the beginning of the Anthropocene, the ongoing unit in the geological time scale that marks the consequences of human activity on Earth.5,6 These activities obviously have had a terrible impact on the environment and need to be addressed urgently.
Less than 10% of all discarded plastic is recycled, mostly mechanically, to manufacture lower quality plastic products. Only a minute fraction of the waste (around 1%) is chemically degraded to produce lower molecular weight compounds or monomers that can be cycled back into the parent polymers or upcycled into products with higher functionality or market value.7
As of 2022, of the 400 million Mt per year of plastics produced globally,8 more than 75% consists of those with a C–C backbone, namely polyolefins, including HDPE, LDPE, LLDPE, PP, PS and PVC. The second most important type of polymer is polyethers with 10.2% of the total.3 A recent algorithmic literature analysis on catalytic plastics treatment by Pérez-Ramírez and colleagues has shown that, as expected, the most targeted plastics are LDPE, HDPE, PP and PET, in line with their high production volumes.7
Other important groups of polymers, namely those consisting of monomers linked by non-carbonylic oxygen atoms (X–O–X), which include polyethers (X = alkyl or aryl) and silicones (X = SiR2), have fallen out of the spotlight.
Polyethers, especially polyols, are the main component of most polyurethane foams and adhesives, which are the third most common type of plastic, with total annual production of around 27 Mt per year in 2019, and an annual growth rate of 4%.7,9 Virtually all chemical recycling methods for thermoset polyurethanes result in the recovery of the parent polyol after solvolysis (glycolysis, acidolysis, hydrolysis or aminolysis) of the urethane hard domains, while further depolymerisation of the polyether is never pursued.9–12
Silicones are organosiloxane polymers that are ubiquitous in our daily lives, widely used in household appliances and many technical sectors.13 The estimated global production of silicones in 2020 was over 8 million tonnes,10 which although only about a third of the global production of PET,7 is still an amount that should not be overlooked. It is important to note that the driving force behind the recycling of silicones is not the environmental impact they may cause at the end of their life, but rather the nature of their raw materials and how they are produced. The precursors of all commercial silicones today are methylchlorosilanes, mainly (CH3)2SiCl2, produced by what is known as “direct synthesis” (also known as the Müller–Rochow process), discovered independently in the 1940s by R. Müller at the Chemische Fabrik von Heyden in Radebeul/Dresden,14 and E. G. Rochow at General Electric Co.15,16 The original process, as described by Rochow, consisted of passing gaseous mixture of hydrogen chloride and chloromethane over a Si/Cu alloy in a oven at 370 °C, and has not changed much since.17 The metallurgical silicon required for this process is obtained by carbothermic reduction of quartz at 2000 °C,18 which has a significant carbon footprint and energy consumption of around 10 kW h kg−1.19 With an estimated volume of 6 million Mt in 2021, China is the largest producer of silicon. Russia, in the second place, is an order of magnitude lower at 580000 Mt.19 Therefore, any geopolitical turmoil that could affect the global distribution chain of this valuable raw material will certainly be followed by a crisis in the silicones sector.
With this in mind, we have compiled the present review of the most representative advances in the chemical degradation of these two important types of oxygenated polymers. We have paid attention not only to academic (laboratory scale) methods, as reported in scientific journals, but also to processes with (potential) industrial relevance, as attested in the patent literature.
It should be noted that this review focuses exclusively on the chemical degradation processes of traditional polymers, excluding those materials in which reactive moieties have been introduced at the monomer stage in order to obtain depolymerisable materials.
2 Depolymerization of polyethers
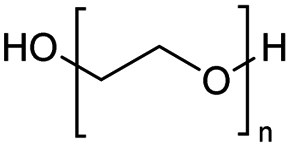
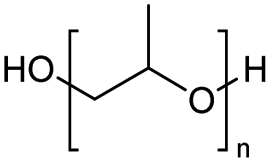
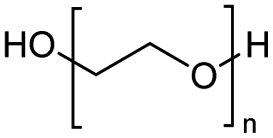
Polyethers, as considered here, are homopolymers or copolymers in which the repeating units are linked by ether linkages, C–O–C. There the carbon atom is either aromatic (sp2) or aliphatic (sp3) and the oxygen atom is of the alkoxy or aryloxy type. Depolymerization methods of polyethers require the activation and cleavage of C–O bonds, which remains a challenge due to the high dissociation energies of single C–O bonds in organic compounds.20,21 Therefore, most reports of ether cleavage involve the use of catalysts.The most prominent polyethers discussed in this review are polyethylene glycol (PEG), polypropylene glycol (PPG), poly(tetrahydrofuran) (PTHF), polyphenylene ether (PPE), polyether ether ketone (PEEK), and epoxy resins. Typically, plastics recycling and depolymerization efforts are driven by environmental concerns. However, prominent polyethers such as PEG and PPG have rarely been investigated as wastewater contaminants due to their low toxicity.22 PEG is widely used in the textile, polishing, toiletry, cosmetic, pharmaceutical, and food industries from some of which it can be easily introduced into wastewater.22,23 However, the pollution by other polyethers such as epoxy resins, which contain additives hazardous to health and environment, are deeply concerning, and have largely motivated the search for greener end-of-life management options.24,25
Topics related to the biodegradation of polyethers, the breakdown of polyethers containing ester groups, and the depolymerization of natural polyethers such as lignin are reviewed elsewhere and are not covered in this review.
Depolymerization into valuable chemicals is more desirable than complete degradation (to the original monomer) because it is economically attractive and less laborious. Important examples were provided by Yang et al. who first reported catalysed depolymerization of polyethers to valuable chemicals via esterification in 1993.26 The Enthaler group has extensively investigated polyether depolymerization using homogeneous iron and zinc catalysts added as inexpensive salts. Their application to the synthesis of chloroesters, diesters, and the reverse polymerization of PTHF was studied.27–32 Following these pioneering reports, the Jitsukawa group investigated not only the application of cation-exchanged montmorillonites to the synthesis of chloroesters and diesters, but also the C–C cleavage to tetralin derivatives.33–36 Recent publications have investigated the depolymerization of arene-containing polyether compounds.37–39 Since catalysis is the key technique for polyether depolymerization, this review will address the substrate scope as well as the mechanism of catalysed depolymerizations.
2.1. Depolymerization via advanced oxidation processes
In a comparative study of physicochemical methods for PEG removal from wastewater by Vocciante et al.,22 Fenton oxidation and ozonization were compared as depolymerization processes in terms of effectiveness, economic cost, and environmental side effects.22 Degradation of PEG-1450 and PEG-8000 was monitored by molecular weight and total carbon content (TOC). TOC is the indicator of the total degradation of contaminants in water to water and carbon dioxide. Although ozone is a powerful oxidant, it cannot be stored and must be generated before use. The molecular weight of PEG is mainly reduced by the ozonization process and rarely completely removed. In Fenton oxidation, the contaminant is reduced by the strong oxidant hydroxyl radicals catalytically formed by Fe(II) species and the consumption of hydrogen peroxide via a well-known reaction mechanism:| FeII + H2O2 → FeIII + OH− + HO˙ |
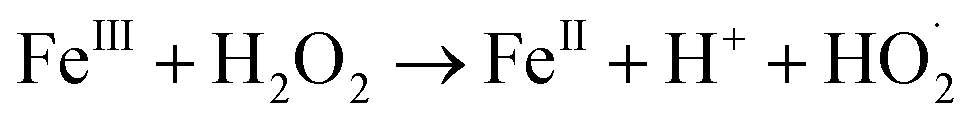
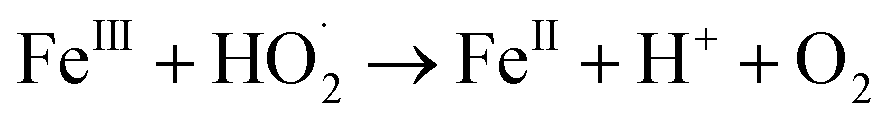
| HO˙ + FeII → OH− + FeIII |
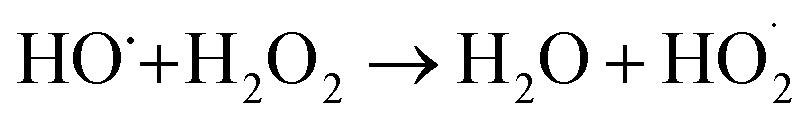
Ferric hydroxide (Fe(OH)3) is formed if the solution is not sufficiently acidic. In Fenton oxidation, 86–96% of 1 g L−1 PEG was degraded within one hour, but high Fe(II) concentrations of 3.5 g L−1 were required. At this relative amount, a significant amount of iron hydroxide sludge was formed, requiring additional purification steps. Both oxidation processes produce many by-products that are more hazardous than PEG, causing further disposal problems. In addition, the by-products pose other hazards that would be caused by accidental loss of containment.22,40
The photoinitiated photodegradation of PEGs (photolysis) with FeCl3 and UV light, among others, was found as a side observation during the investigations of the respective coordination complexes. PEG photolysis is caused by the formation of chlorine radicals that abstract a hydrogen from the methylene groups in the polymer chain (PH), resulting in a polymeric alkyl radical (P˙). These polymeric radical can further react with oxygen to generate polymeric alkylperoxy open-shell species:41
| Fe3+(Cl−)n + hν ⇌ Fe3+(Cl−)n−1·Cl˙ → Fe2+(Cl−)n−1 + Cl˙ |
| PH + Cl˙ → P˙ + HCl |
| P˙ + O2 → POO˙ |
Alkylperoxy radicals can further react with a H donor to form hydroperoxyl species (POOH) which can form photolabile complexes with Fe(II) and Fe(III) ions, releasing alkylperoxy and alkyloxy radicals, leading to the eventual destruction of the polyether chains. As mentioned before, the aim of advanced oxidation processes is to eliminate polyether pollutants rather than convert them into valuable chemicals, which makes it economically unattractive as a depolymerisation method.
2.2. Conversion to chlorinated esters
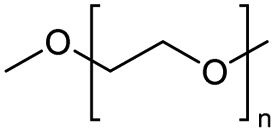
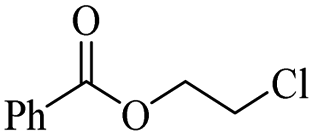
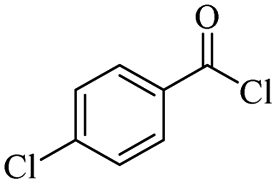
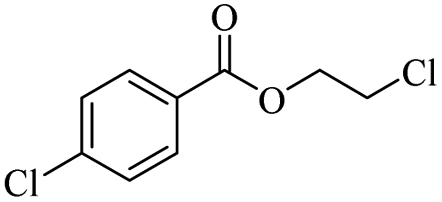
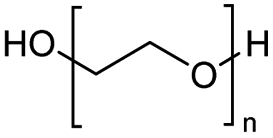
The depolymerisation of polyethers to chloroesters has been the subject of much research. The chloroesters can be converted to vinyl esters, which are valuable monomers for subsequent polymerisations. Enthaler's group first investigated the synthesis of chloroesters from polyethers using zinc and iron salts as precatalysts in homogeneous processes.29–32The use of cheap and abundant transition metals, as well as not having to use solvents or inert gases, are major advantages of these reactions. Depolymerisation with acid chlorides, such as benzoyl chloride derivatives and fatty acid chlorides, resulting in chloroesters, proved to be the most efficient depolymerisation method in their work, giving high yields of chloroesters of up to 95%.30,31 It has been found that a variety of precatalysts provide active zinc and iron catalysts for the depolymerisation of tetraethylene glycol with benzoyl chloride at 130 °C (Table 1) as a model reaction. Catalyst activity is measured by the yields at constant reaction conditions. Zinc(II) chloride (ZnCl2) and iron(II) chloride (FeCl2·4H2O) were found to provide the most active catalysts (entries 1 and 9). Methyl benzoate is obtained as a by-product. Good yields are also obtained with zinc(II) tetrafluoroborate (Zn(BF4)2) and iron(III) chloride (FeCl3). Lower to moderate yields were obtained with the other halide and perchlorate salts. The metallo-organic salts zinc(II) acetate (Zn(OAc)2) and zinc(II) acetylacetonate (Zn(acac)2) provide a working catalyst, although low yields are obtained. With the zinc catalysts the reaction only takes place at a temperature of 130 °C, whereas with the iron catalysts the reaction still takes place at 60 °C with a lower yield, although good yields can be obtained at 100 °C.29,31
|
|
||
|---|---|---|
| Entry | Pre-catalyst | Yield (%) |
| 1 | ZnCl2 | 84 |
| 2 | ZnBr2 | 61 |
| 3 | Zn(ClO4)2·6H2O | 63 |
| 4 | Zn(OAc)2 | 19 |
| 5 | Zn(acac)2·H2O | 17 |
| 6 | Zn(BF4)2 | 82 |
| 7 | ZnO | 71 |
| 8 | Zn(OTf)2 | 75 |
| 9 | FeCl2·4H2O | 87 |
| 10 | FeBr2 | 69 |
| 11 | Fe(ClO4)2·H2O | 71 |
| 12 | Fe(ClO4)3·4H2O | 73 |
| 13 | FeCl3 | 81 |
The Jitsukawa group has reported using montmorillonite as a heterogeneous catalyst to synthesise chloroesters by depolymerising polyethers.33 Montmorillonite (Na0.66(OH)4Si8(Al3.34Mg0.66Fe0.19)O20) is a negatively charged layered clay material with two characteristics that make it attractive for heterogeneous catalysts. First, the interlayer Na cations are readily exchanged for protons and various metal cations with high catalytic activity for various organic transformations. Secondly, in polar media, a confined reaction space is created by the widening interlayer gallery, which allows the intercalation of molecules.42–44 Proton-exchanged montmorillonite (H-mont) is prepared by treating the parent montmorillonite with HCl. H-mont, the parent montmorillonite and other commercially available solid acid catalysts such as Amberlyst 36, Nafion NR 50, H-USY and H-mordenite were tested in the depolymerisation of PEG 350 with benzyl chloride as nucleophile (Table 2, entries 7–12). In terms of yield, H-mordenite is superior to the other solid acid catalysts. The advantage of the heterogeneous H-mordenite over the homogeneous Zn and Fe catalysts is the recyclability of the H-mordenite without loss of activity.33 An interlayer spacing of 2.6–2.8 Å has been determined for H-mont by XRD measurements. Treatment with PEG increases the interlayer distance to 12 Å.33,34,45 It is worth noting that in these experiments the same amount per subunit of PEG was used with Mn = 8000 and Mn = 2000000, resulting in the same interlayer distance.33,34 The authors concluded that the PEG chains extend into the interlayer by intercalation, based on IR spectroscopy measurements.34,46,47
| Entry | Substrate | Mn | Product | (pre)catalyst | Yield (%) |
|---|---|---|---|---|---|
| 1 |
|
n = 1 |
|
ZnCl2 | 72 |
| 2 | FeCl2 | 79 | |||
| 3 | n = 2 | ZnCl2 | 69 | ||
| 4 | FeCl2 | 81 | |||
| 5 | n = 4 | ZnCl2 | 81 | ||
| 6 | FeCl2 | 78 | |||
| 7 | 350 | H-mont | 75 | ||
| 8 | Amberlyst 36 | 72 | |||
| 9 | Nafion NR 50 | 39 | |||
| 10 | H-USY | 12 | |||
| 11 | H-mordenite | 4 | |||
| 12 | H-mont | 0 | |||
| 13 |
|
— | ZnCl2 | 79 | |
| 14 | FeCl2 | 82 | |||
| 15 |
|
n = 3 | ZnCl2 | 83 | |
| 16 | FeCl2 | 73 | |||
| 17 | 8000 | H-mont | 80 | ||
| 18 | 100000 |
ZnCl2 | 70 | ||
| 19 | FeCl2 | 89 | |||
| 20 | 1000000 |
ZnCl2 | 78 | ||
| 21 | FeCl2 | 95 | |||
| 22 | 2000000 |
H-mont | 81 | ||
| 23 |
|
1000 |
|
ZnCl2 | 92 |
| 24 | FeCl2 | 92 | |||
| 25 | 2000 | H-mont | 63 | ||
| 26 |
|
2500 |
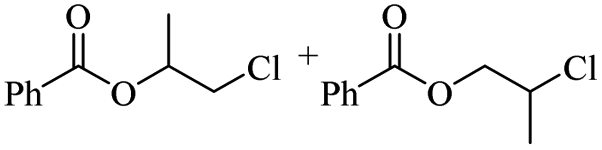
|
ZnCl2 | 81 |
| 27 | FeCl2 | 68 | |||
| 28 | 2700 | H-mont | 71 | ||
| 29 |
|
n = 10 (Triton X-100) |
|
ZnCl2 | 86 |
| 30 | FeCl2 | 91 | |||
| 31 |
|
w + x + y + z = 20 | H-mont | 77 | |
| 32 |
|
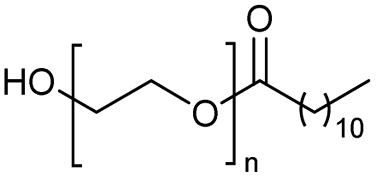
n = 10 | H-mont | 84 |
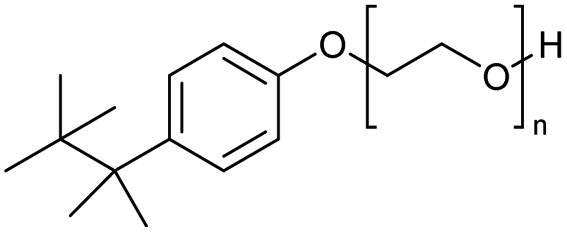
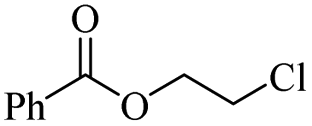
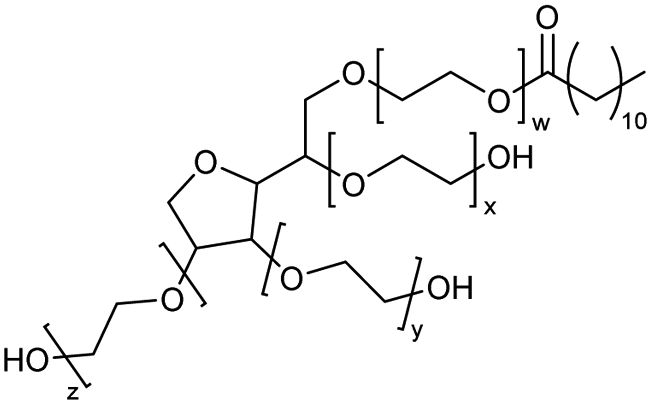
Using benzoyl chloride as a nucleophile and reaction temperatures of 130 °C for the Zn-catalysed, 100 °C for the Fe-catalysed and 100 °C for the H-mont catalysed depolymerisation reaction, several neat polyethers were investigated (Table 2). Regardless of the end group of the polyether, the same chloroester is obtained. The length of the chloroalkyl chain depends on the length of the alkyl chain between the ether linkages. Yields were mostly between 70% and 90% for all catalyst systems. The Enthaler group carried out experiments with Zn and Fe catalysts for the synthesis of chloroesters from oligomeric PEG (entries 1–8 and 14–16). For the synthesis of chloroesters from high molecular weight PEGs (8000–2000000, entries 17–22), Zn catalysts proved to be more effective than Fe catalysts. H-mont gave yields in a similar range. A trend seems to exist where the use of an Fe catalyst in the degradation process of polymeric PEG (entries 19 and 21) gives higher yields of 89–95% compared to oligomeric PEG (73–81%, entries 2, 4, 6 and 10). No similar trend is evident with the use of a Zn catalyst. The Zn catalyst gave higher PPG depolymerisation yields than the Fe or H-mont catalysts (entries 26–28). The homogeneous catalyst systems have demonstrated high efficacy for PTHF depolymerisation (92%), although H-montmorillonite performs relatively poorly for PPG and PEG depolymerisation (63%). As there is no observed correlation for various end groups, such as hydroxyl and methyl groups, the Triton X-100 end group (entries 29 and 30), Polysorbate 20 (entry 31) and polyethylene glycol monolaurate (entry 32), it can be inferred that the esterification process may be tolerant of a range of end groups in polyethers. The ese results demonstrate a lack of general superiority for either catalyst in depolymerization with benzoyl chloride as a nucleophile. Instead, their effectiveness relies on the specific polyether substrate and acid chloride used. Further variations of the latter can be found in Table 3.
| Entry | Polyether | Reagent | Mn | Product | (Pre-) catalyst | Yield (%) |
|---|---|---|---|---|---|---|
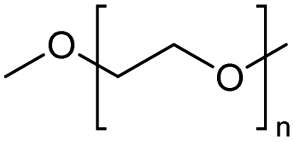
| 1 |
|
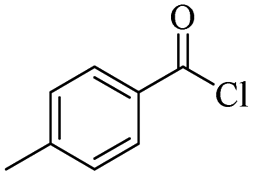
|
350 |
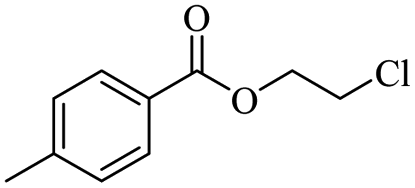
|
H-mont | 78 |
| 2 |
|
100000 |
ZnCl2 | 87 | ||
| 3 | FeCl2 | 91 | ||||
| 4 | ZnCl2 | 91 | ||||
| 5 | FeCl2 | 89 | ||||
| 6 |
|
350 |
|
H-mont | 80 | |
| 7 |
|
2000 |
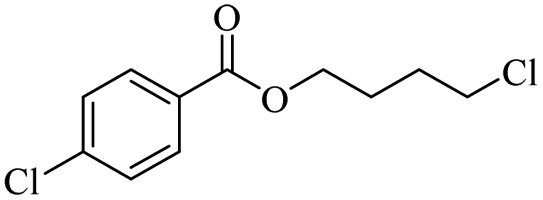
|
H-mont | 81 | |
| 8 | 1000 | ZnCl2 | 85 | |||
| 9 | FeCl2 | 92 | ||||
| 10 |
|
|
350 |
|
H-mont | 76 |
| 11 |
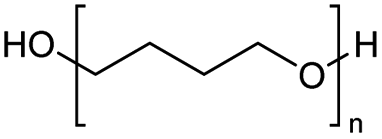
|
2000 |
|
H-mont | 63 | |
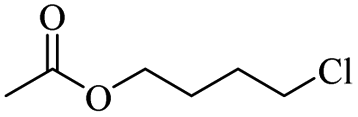
| 12 |
|
|
300 |
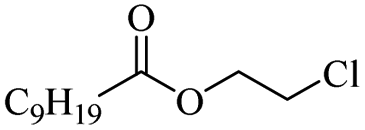
|
ZnCl2 | 92 |
| 13 | FeCl2 | 78 | ||||
| 13 |
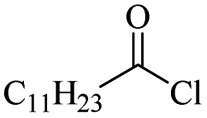
|
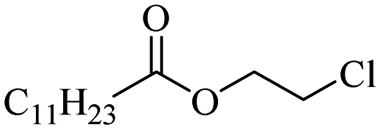
|
ZnCl2 | 92 | ||
| 14 | FeCl2 | 77 | ||||
| 15 |
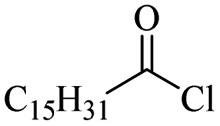
|
|
ZnCl2 | 91 | ||
| 16 | FeCl2 | 72 | ||||
| 17 |
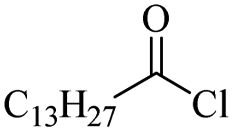
|
|
ZnCl2 | 94 | ||
| 18 | FeCl2 | 82 | ||||
| 19 | 1000 | ZnCl2 | 91 | |||
| 20 | FeCl2 | 69 | ||||
| 21 | 1000000 |
ZnCl2 | 90 | |||
| 22 | FeCl2 | 56 | ||||
| 23 |
|
n = 10 (Triton X-100 | ZnCl2 | 92 | ||
| 24 | FeCl2 | 96 | ||||
| 25 |
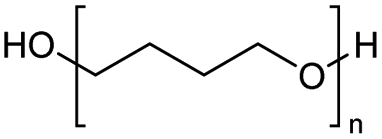
|
1000 |
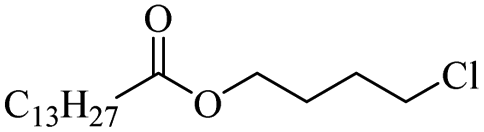
|
ZnCl2 | 90 | |
| 26 | FeCl2 | 57 | ||||
| 27 |
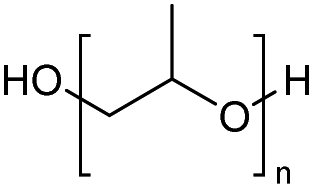
|
2500 |
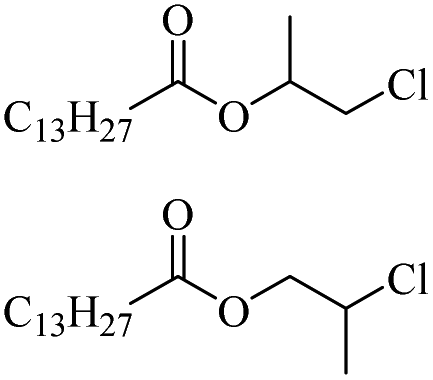
|
ZnCl2 | 83 | |
| 28 | FeCl2 | 77 |
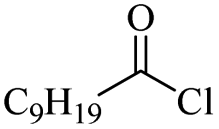
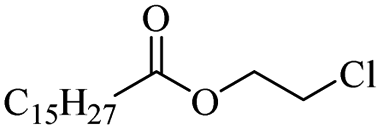
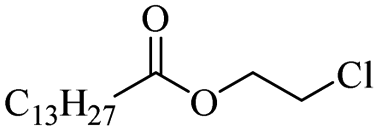
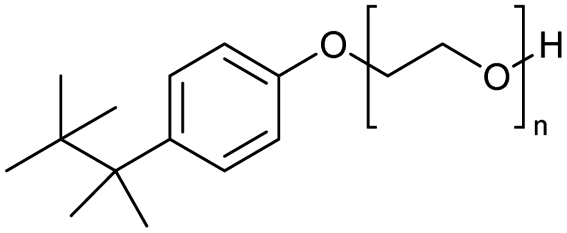
The use of 4-chloro- and 4-methylbenzoyl chloride for the depolymerisation of PEG or PTHF showed almost identical yields for all catalysts compared to the yields resulting from the reaction with benzoyl chloride. The easy accessibility of fatty acid chlorides and their potential to link unsaturated fatty acids made the use of fatty acid chlorides attractive. The Zn and Fe catalysts were examined using both saturated and unsaturated acid chlorides, including capric acid chloride, lauroyl chloride, myristoyl acid, palmitoyl chloride, 10-undecanoyl chloride, oleic acid chloride, and linoleic acid chloride (entries 12–22). Depolymerization catalysis with the Zn-system resulted in higher yields (91–94%) than the Fe system (75–82%) when using saturated fatty acids. The Zn-catalysed system is ineffective for unsaturated fatty acid chlorides, as well as the Fe-catalysed system except for oleic acid chloride (39%). An additional experiment was conducted using polyethers for depolymerization with myristoyl chloride as a nucleophile in homogeneous systems. As for the results of depolymerization with benzoyl chloride, there is a discernible trend concerning catalyst activity for the scope employing myristoyl chloride. Generally, markedly superior yields were achieved by implementing the Zn catalyst (83–92%) in comparison to the Fe catalyst (56–77%), with the exception of the depolymerization of Triton X-100 (96%).
Esterification mechanisms (see Fig. 1 and 2)29,31 have been proposed for both homogeneous catalytic systems. The first step in both cases involves coordinating the oxygen functionality to the metal centre (A), resulting in the activation of the carbon atom bound to oxygen. The bond between the C and O atoms in the ether is cleaved by the nucleophilic attack of a chloride ion on the activated (electrophilic) carbon atom (see Fig. 1C and 2B). In the case of iron, a metal alkoxide and alkyl chloride are formed (see Fig. 2C), whereas an ester compound is formed by reaction between the iron alkoxide and an acid chloride via intermediate D. During this reaction, abstraction of the reactant chlorides regenerates complex A. In both mechanisms, the alkyl chloride and ester coordinate back to the complex to feed the catalytic system until they react to provide monomeric esters.
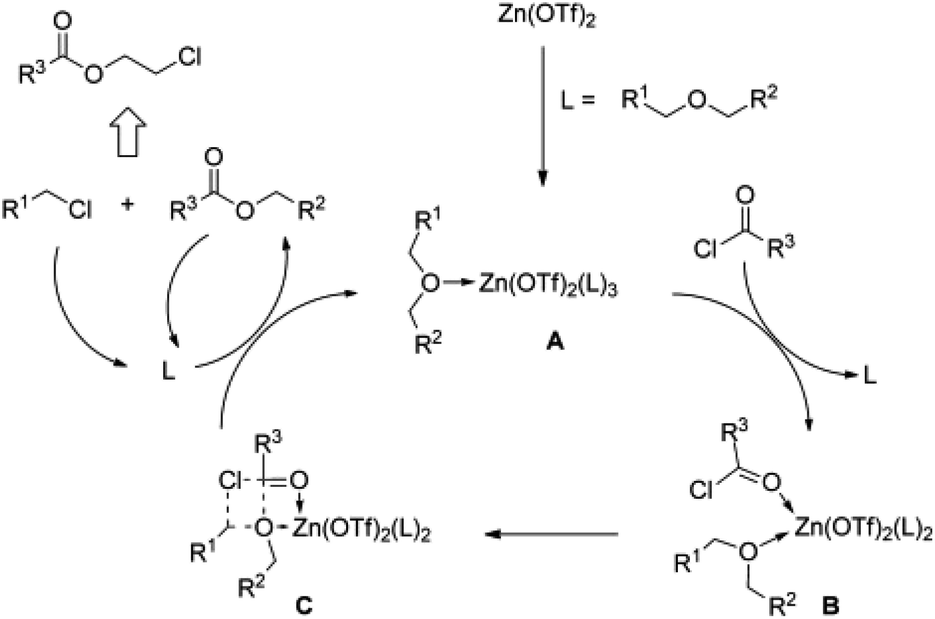 | ||
| Fig. 1 Proposed catalytic cycle for the zinc-catalysed depolymerisation of polyethers to chloroesters (reprinted with permission from Wiley, ©2012).29 | ||
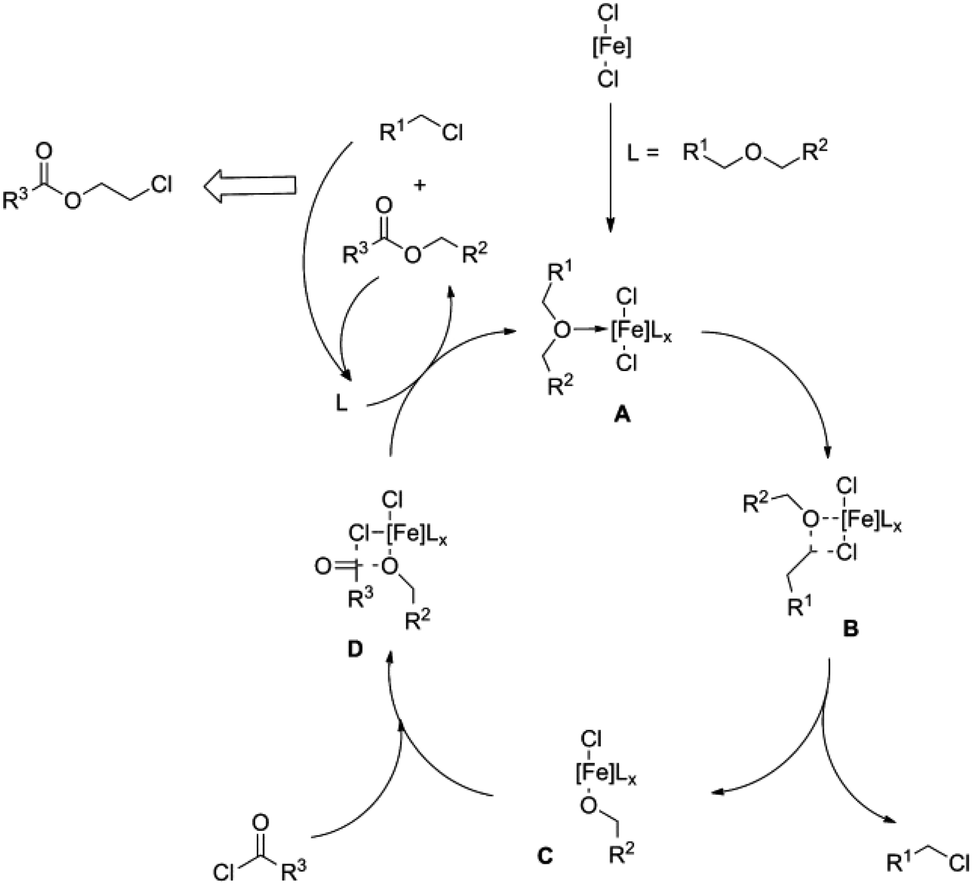 | ||
| Fig. 2 Proposed catalytic cycle for the iron-catalysed depolymerisation of polyethers to chloroesters (reprinted with permission from Wiley, ©2012).13 | ||
The mechanism of the reaction with H-montmorillonite differs from that of its homogeneous counterparts, although, it also relies on the nucleophilic attack of chloride ions to the electrophilic carbon adjacent to the oxygen atom (Fig. 3).33
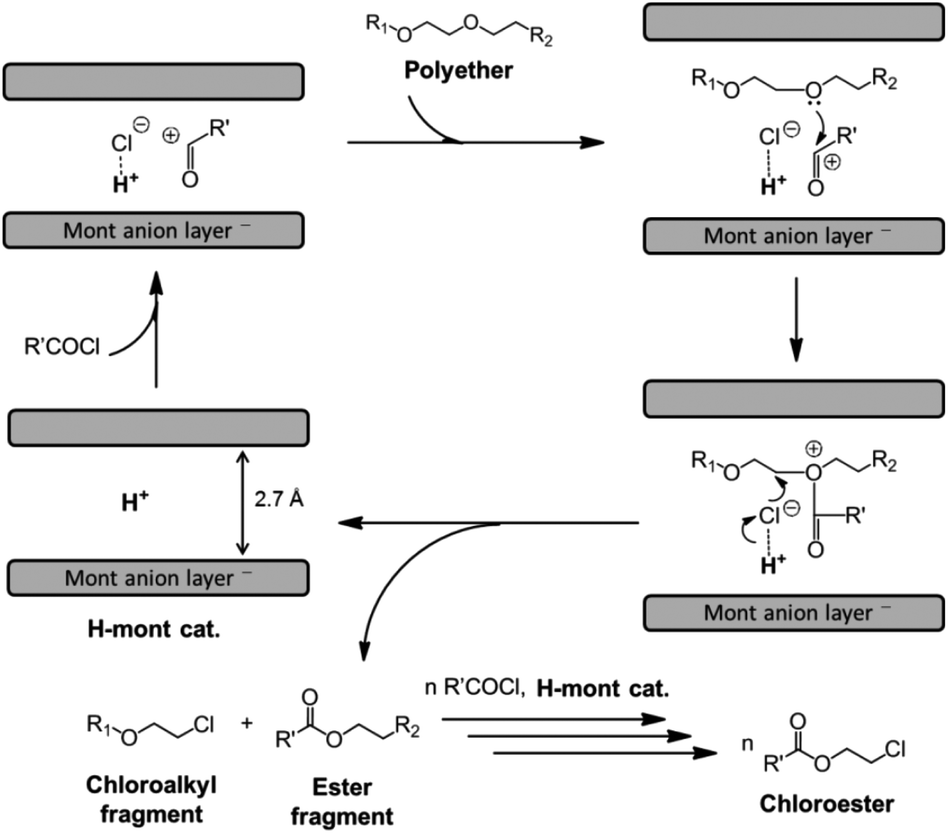 | ||
| Fig. 3 Proposed catalytic cycle for the H-mont-catalysed depolymerisation of polyethers to chloroesters (reprinted with permission from Wiley, ©2016).33 | ||
The active species for catalysis is the interlayered proton in H-montmorillonite. This inference stems from XRD measurements and the suppressed reaction when 2,6-lutidine, which interacts with Brønsted but not Lewis acid sites, is added, thus rendering the parent montmorillonite inactive. The proposed reaction mechanism commences with the activation of acyl chloride into an acyl cation on Brønsted acid sites, as illustrated in Fig. 3. Acyl cations are formed, which are then attacked by the free electron pair of oxygen in the C–O bond of the polyether. This results in the generation of corresponding oxonium cations. The C–O bond of the polyether is cleaved through the attack of the chloride anion, regenerating the montmorillonite interlayer space and forming chloroalkyl and ester fragments, which will eventually produce the corresponding chloroesters via transesterification.
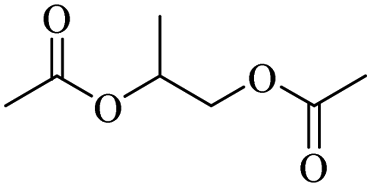
2.3. Conversion to glycol derivatives
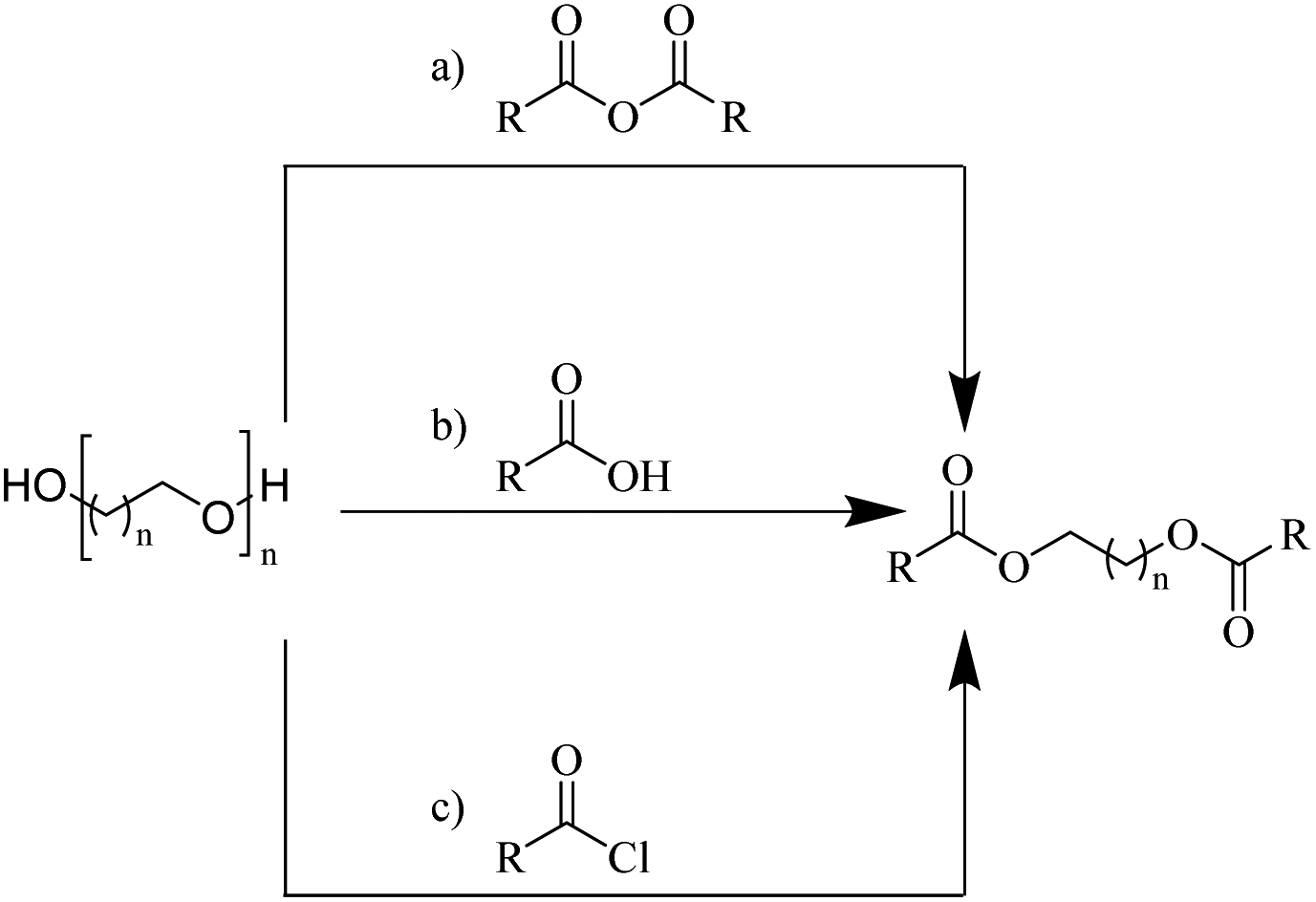 | ||
| Fig. 4 General routes for synthesizing glycol diesters from polyethers (a) with carboxylic anhydrides; (b) with carboxylic acids; (c) with acyl chlorides. | ||
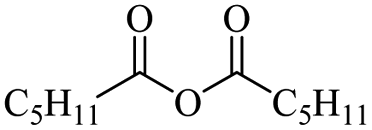
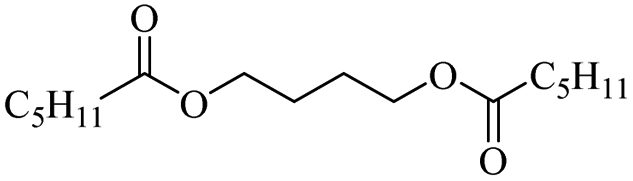
Enthaler and co-workers also reported successful depolymerisations using zinc(II) triflate, Zn(OTf)2, as a precatalyst with acetic anhydride, hexanoic acid and hexanoic acid anhydride to give glycol diesters (Table 4). However, depolymerisation of polyethers to glycol esters with Zn catalyst is less efficient than depolymerisation to chloroesters. Although various Zn salts were examined for the depolymerisation of the tetraglyme oligomer, high yields were only achieved with Zn(OTf)2 (88%) and the next highest yield was with ZnCl2 at 12%. Moderate yields (18–45%) were obtained in the synthesis of glycol ester using PEG and PPG, acetic anhydride, and hexanoic acid. Good yields (59–71%) were obtained in the depolymerisation of PTHF using acetic anhydride (Ac2O), hexanoic anhydride and hexanoic acid as depolymerisation reagents.29
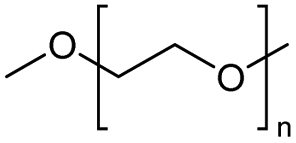
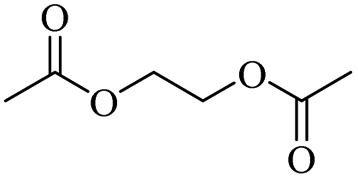
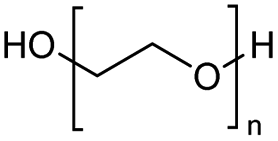
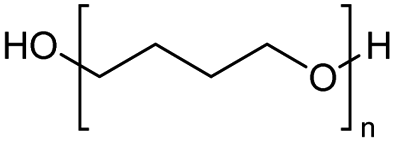
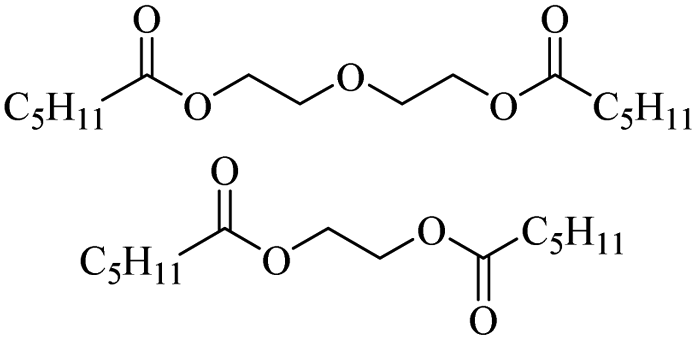
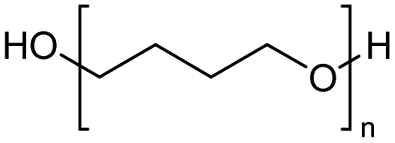
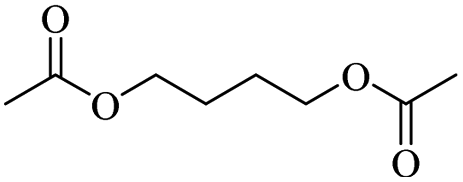
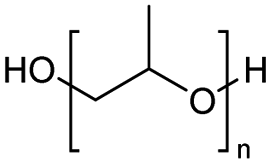
The H-mont catalyst described by Jitsukawa has been extensively studied due to its significantly higher activity in the production of glycol diesters compared to Zn and Fe catalysts. Table 5 shows the results of the tests carried out on commercially available acid catalysts, namely Amberlyst 36, Amberlyst 15, Nafion NR50 and other ion-exchanged montmorillonite catalysts, for the reaction with the oligomer tetraethylene glycol and benzoic anhydride (Ph2O) or acetic acid (AcOH).34,35
|
|
|||
|---|---|---|---|
| Entry | Catalyst | Conditions | Yield (%) |
| 1 | H-mont | Ph2O, 100 °C, 1 h | 57 |
| 2 | H-mont | Ph2O, 100 °C, 5 h | 99 |
| 3 | Amberlyst 36 | Ph2O, 100 °C, 1 h | 19 |
| 4 | Nafion NR50 | Ph2O, 100 °C, 1 h | 15 |
| 5 | Amberlyst 15 | Ph2O, 100 °C, 1 h | 2 |
| 6 | Zn(OTf)2 | Ph2O, 100 °C, 1 h | 2 |
| 7 | H2SO4 | Ph2O, 100 °C, 1 h | 75 |
| 8 | H-mont | AcOH, 160 °C, 2 h | 55 |
| 9 | H-mont | AcOH, 160 °C, 24 h | 89 |
| 10 | Amberlyst 36 | AcOH, 160 °C, 2 h | 45 |
| 11 | Nafion NR50 | AcOH, 160 °C, 2 h | 33 |
| 12 | Al3+-mont | AcOH, 160 °C, 2 h | 22 |
| 13 | Ti4+-mont | AcOH, 160 °C, 2 h | 10 |
It was found that H-mont gave moderate results after 1–2 hours and good yields after 5 hours or 24 hours (entries 1, 2, 8 and 9). Significantly lower yields are obtained using alternative catalysts for the reaction with benzoic acid (entries 3–6). The Zn catalyst used in the experiment by Enthaler et al. also gave low yields (entry 6). In contrast, sulfuric acid produced better results in a control reaction (entry 7). Similarly, H-mont retained its efficacy after multiple cycles in the AcOH reaction, consistent with its use in chloroester synthesis. The application of commercial catalysts Amberlyst 36 and Nafion NR proved more efficacious when reacting with AcOH (entries 10 and 11) compared to Ph2O (entries 3 and 4). However, initial yields were marginally lower than those achieved with H-mont. Exchange of the montmorillonite with Al3+, Ti4+, and Cu2+ ions failed to yield comparable results to H-mont (entries 12–14).
The investigation of identical polyethers (with the exception of Tween 20 instead of Polysorbate 20) for the production of glycol diesters, as well as for the chloroesters, proved to be very effective with exceptionally high yields with Ph2O and a mixture of Ac2O and AcOH, regardless of the end group of the polyether.34,35 However, the process using Ac2O/AcOH mixture required harsh reaction conditions and long time periods (160 °C, 48 h) compared to the mild conditions with Ph2O (100 °C, 12 h).34 Entry 1 showed that the H-mont could be reused three times for the depolymerisation of the PEG without significant loss of activity, demonstrating the remarkable durability of the H-mont. As ethylene glycol diacetate is a widely used slow-evaporating solvent in paints, the researchers demonstrated that a minor increase in the depolymerisation of PEG (Mn = 300, 2 g, 41.5 mmol) can be achieved by using an Ac2O/AcOH mixture and H-mont (3.4 g) at a temperature of 180 °C for 72 hours, resulting in a yield of 81%.
In the proposed mechanism for the depolymerisation of polyether with carboxylic acid derivatives to obtain glycol diesters (Fig. 5), the Brønsted acid site was proven to be the catalytically active site through the addition of 2,6-lutidine, which inhibits the reaction. The catalytic cycle initiates with the intercalation of the polyether between the layers of the H-mont (step i). The carboxylic anhydride is inserted into the interlayer and activated at the Brønsted acid sites to produce an acyl cation (step ii), which uses the same activation mechanism as chloroesters. The electrophilic attack of the acyl cation generates a carbocation-terminated fragment and an ester-terminated fragment (step iii). Afterwards, the carboxylic acid reacts with the carbocation, producing another ester-terminated fragment (step iv). Based on the results of the asymmetric production of glycol diesters, the authors propose that the addition of carboxylic acids increases the overall reaction rate. The produced ester fragments recombine eventually through acid-catalysed transesterification to yield the corresponding monomeric glycol esters. After depolymerisation, the interlayer distance decreased from 12 Å to 3.1 Å, which was measured using XRD.34
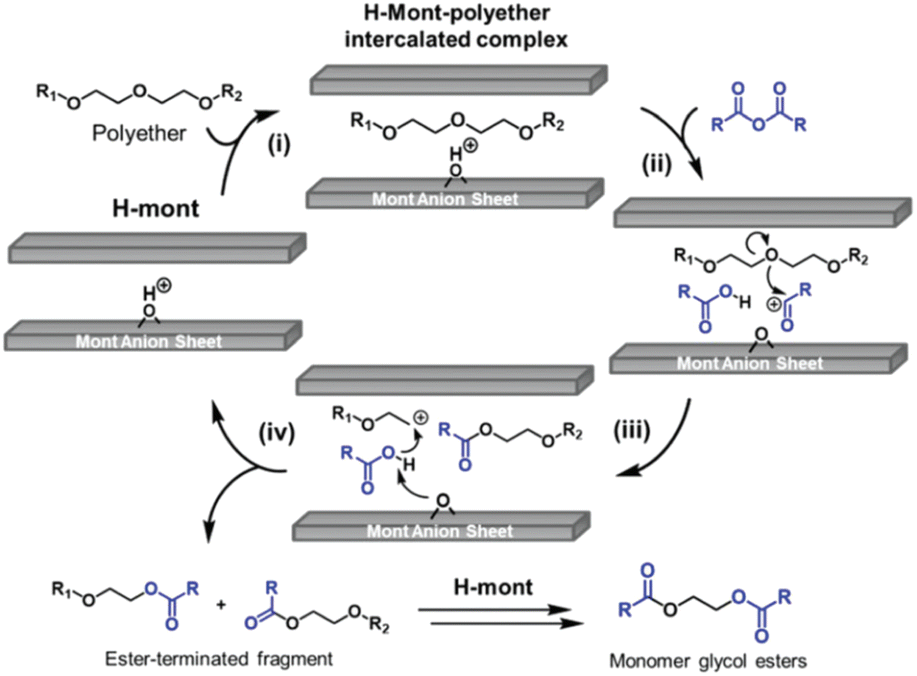 | ||
| Fig. 5 Proposed catalytic cycle for the H-mont-catalysed depolymerisation of polyethers to glycol diesters (reprinted with permission from The Royal society of Chemistry, ©2017).34 | ||
The potential environmental application of polyether depolymerization for wastewater treatment using proton-exchanged montmorillonite catalysts was tested using surfactants as model contaminants.34 The process involves two main steps. Firstly, complete adsorption of polyethers Triton X-100, Tween 20 or polyethylene glycol monolaurate (2.4 mmol per subunit) onto H-mont (400 mg) followed by separation from water via filtration and vacuum drying. The second stage involves the depolymerization of the polyether with an Ac2O/AcOH mixture at 160 °C, giving yields up to 67%. This process could be more economically attractive than the above-described treatments of polyether-contaminated water (Section 2.1), as it would use the polyether contaminants as a resource to recover valuable chemicals.
The hydrosilylation of PEG was a single experiment without deeper investigation in the work of Brookhart et al. which focused on the C–O bond cleavage of alkyl ethers with an iridium pincer catalyst (Ir-pin+, Fig. 6) and triethylsilane to silylethers via hydrosilylation.48 The mechanism of this reaction (Fig. 6A) begins with the formation of an agostic complex between the hydrogen in the triethylsilane and the Ir-pin+, activating the Si–H bond. The Si–H cleavage is followed by the addition of Et2O, resulting in the formation of an oxonium cation. Simultaneously, the hydrogen is transferred to the iridium complex as a hydride. One of the hydrides is transferred to the carbon atom at the β-position of a siloxy group of Et3SiO, resulting in the formation of Et3OSiEt3 and ethane.48 Later on, Cantat et al. applied Brookhart's catalyst for the depolymerization of various oxygenated plastics, including PEG (Fig. 6B).49
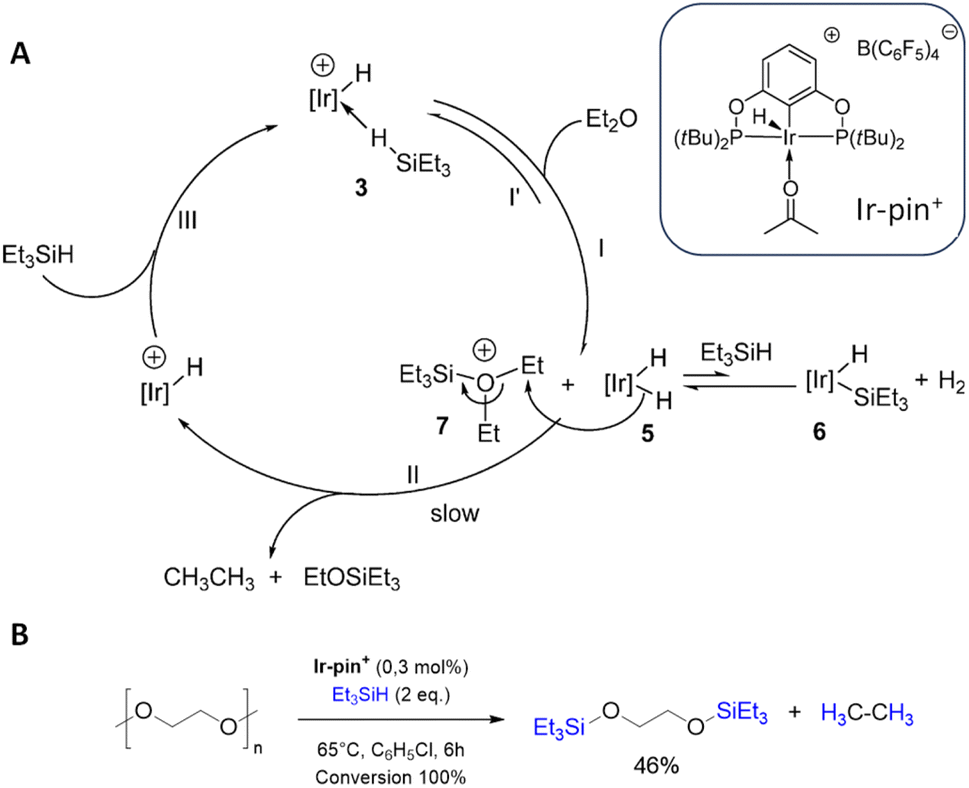 | ||
| Fig. 6 (A) Proposed catalytic cycle for the of the hydrosilylation with of diethylether with the iridium pincer complex Ir-pin+. (adapted with permission from The America Chemical Society, ©2008).48 (B) Reaction conditions for the iridium-catalyzed depolymerization of PEG.49 | ||
The metal-free and well-established hydrosilylation catalyst lithium tetrakis(pentafluorophenyl)borate B(C6F5)4 allows effective PEG depolymerisation at room temperature. With triethylsilane, a mixture of glycol silyl ether and ethane is obtained, while polymethylhydrosiloxane PMHS and tetramethyldisiloxane TMDS provide a complete conversion of PEG and silane to ethene. For hydrosilylation with PMHS and TMDS no intermediates could be identified.50
2.4. Ring-closing depolymerisations
000) and polyether ketone into macrocycles (Fig. 7). The depolymerization process is catalysed by caesium fluoride CsF (20 ppm) at 150–155 °C at low polyether concentrations of 30 ppm in dimethylacetamide (DMA) in 48 h. In the product mixture obtained (95% yield), the molecular weights verified by GPC indicate the formation of macrocycles from the trimer to the nonamer of ether sulfone oligomers and macrocyclic ether ketone oligomers up to the cycloundecamer. However, only the smaller macrocyclic oligomers – such as the trimer, tetramer, pentamer, and hexamer – could be extracted from the depolymerisation product mixture. Upon examining a single crystal of the ether sulfone trimer using XRD, it was discovered that there is a slight ring strain, with all three C–S–C bond angles compressed to 101.6 ± 0.1° instead of their typical open-chain value of 104.9°. Additionally, the three biphenyl residues have an inter-ring torsion angle within a range of 34–38°, despite usually having a coplanar conformation.51
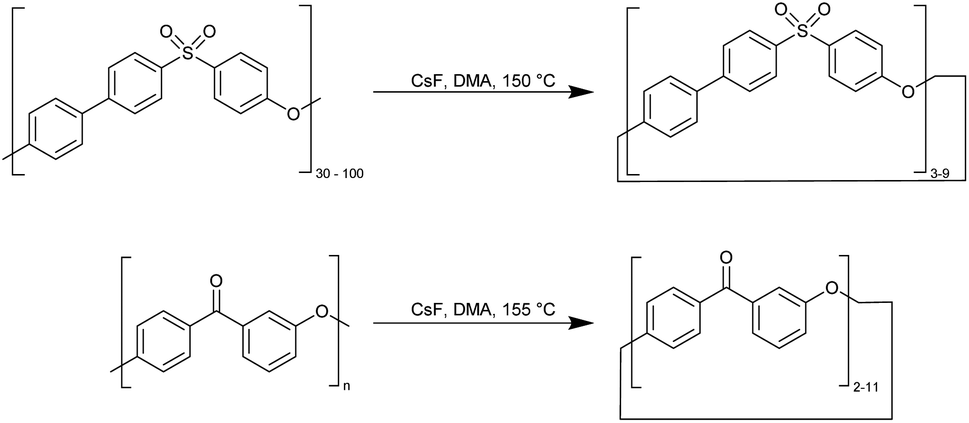 | ||
| Fig. 7 Synthesis of macrocycles by depolymerisation of aromatic polyether sulfone and polyether ketones.51 | ||
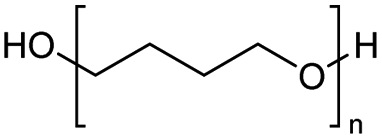
The reverse depolymerization of polytetrahydrofuran (PTHF) back to its monomer tetrahydrofuran (THF) is a widespread subject in C–O ring closure depolymerization. This reversible process is primarily controlled by temperature and monomer concentration, enabled by the low ring strain of THF.52 It has been reported that PTHF can be depolymerized into THF at temperatures above 160 °C.27,28,53,54 The depolymerization of PTHF is achieved through a C–O bond ring closing metathesis reaction, which can be catalysed using iron(III)-chloride (FeCl3) and zinc(II)-triflate (Zn(OTf)3) as catalyst precursors. The reaction takes place within the temperature range of 120–160 °C and 160–180 °C. Due to the equilibrium reaction, the formed THF must be continuously removed from the system via distillation.27,28
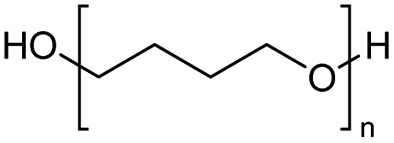
Out of various iron precursors, only FeCl3 works as a catalyst precursor for PTHF depolymerization, while Fe3(CO)9, FeCl2·4H2O, Fe(OAc)2, and Fe(acac)2 do not. Similarly, zinc chloride (ZnCl2), Zn(OAc)2, Zn(acac)2, and ZnO are not effective. It was found that the catalyst activity decreased over time when a new portion of PTHF was added to the reaction within a certain time span.27 The depolymerization of PTHF catalyzed by triflic acid (TfOH) showed comparable reaction performance, indicating the probable formation of TfOH during catalysis with Zn(OTf)3. In this study, no thermal decomposition was observed at 160–180 °C.27,28 This method is not effective for depolymerizing PEG and PPG into their original monomers. The high ring tension of the ethylene and propylene oxide may be a contributing factor.28,54
Wang et al. compared the use of heteropolyacids (HPA) to concentrated sulfuric acid (H2SO4) and previously published FeCl3 for the ring-closing depolymerization of PTHF. The resulting yield was evaluated, and HPA demonstrated better reusability.54 The HPAs, including silicomolybdic acid (H3W12PO40), silicotungstic acid (H3Mo12PO40), phosphomolybdic acid (H4SiW12O40), and phosphotungstic acid (H4SiMo12O40), were utilized. These HPAs, along with H2SO4, resulted in yields exceeding 95% (90% with FeCl3). They were reused ten times with a slightly decreased final yield of over 80%, while FeCl3 and H2SO4 could only be reused four to five times with a significantly decreased final yield of 40–50%.
For the use of a Brønsted acid, the mechanism shown in Fig. 8 has been proposed.54 The terminal hydroxyl group is protonated to form an oxonium cation, and the terminal carbon is then attacked by the oxygen of the next chain unit in an SN2 manner. Water acts as a leaving group, forming a tetrahydrofuranyl cation. This cation also acts as a leaving group when the adjacent oxygen unit undergoes nucleophilic attack at the terminal carbon. It is possible that this mechanism may also be at work with Lewis acids. The depolymerization of PTHF is affected by the end group, as for PTHF containing an acetyl end group instead of a hydroxyl group it does not occur.54
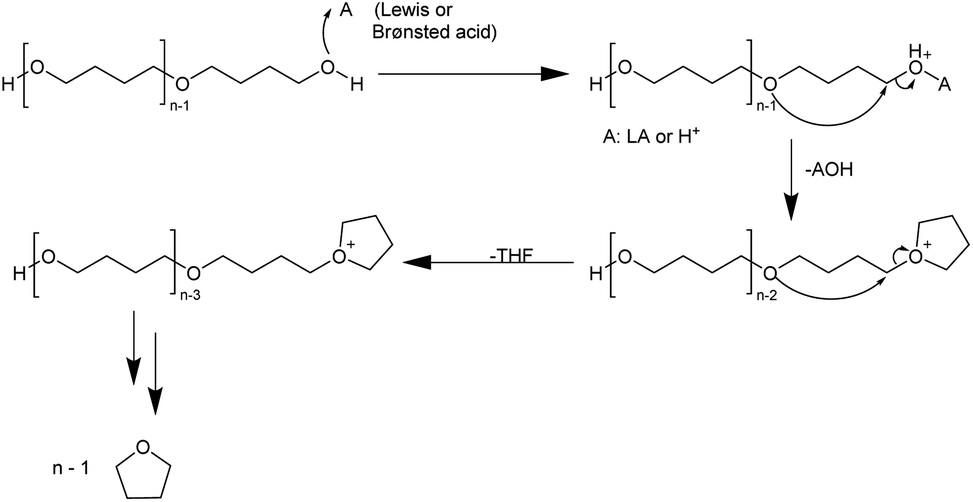 | ||
| Fig. 8 Proposed mechanism for the ring-closing depolymerization of PTHF with acidic catalysts.54 | ||
Dioxanes can be synthesised from oligomeric ethylene glycols, PEG and PPG using silicon-based catalysts.55–57 Jutzi et al. used the low valent half-sandwich complex pentamethylcyclopentadienyl silicon(II) cation Cp*Si+ shown in Fig. 9.55 In that study, oligomeric and polymeric ethylene glycol dimethylethers with up to 10 subunits of poly(ethylene glycol) dimethylether were also depolymerised to dioxane by Cp*Si+. Complete conversion of dimethoxyethane (DME) was achieved at room temperature within 15 days. A catalytic cycle for the DME conversion was proposed (Fig. 9). The open coordination sphere at the silicon atom allows two DME molecules to be coordinated to the Si centre via up to four oxygen atoms. These weak Si–O interactions are of low strength and therefore flexible, but strong enough to induce rearrangements of σ bonds and lone pair electrons, leading to the formation of diglyme and dimethylether. The C–O ring closure of diglyme to dioxane is similar. Due to the chelating effect, DME is more likely to be coordinated to Cp*Si+ than dioxane or dimethylether.55
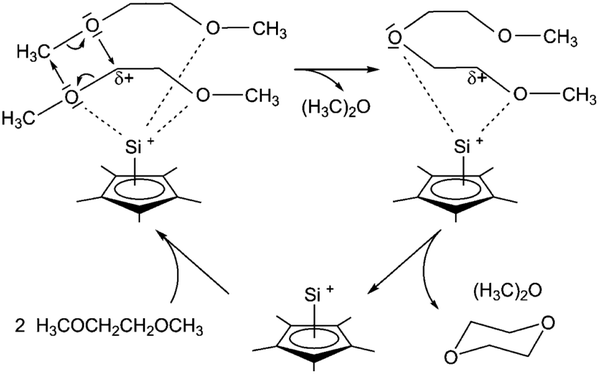 | ||
| Fig. 9 Proposed reaction mechanism for the production of dioxane from dimethyl ether, catalysed by Cp*Si+. (reprinted with permission from Wiley, ©2011).55 | ||
Greb et al. recently reported the depolymerisation of 18-crown-6, PEG and PPG via C–O bond ring closure metathesis using a silicon-based Lewis superacid, bis(perchlorocatecholato)silane, resulting in the formation of 1,4-dioxane and 2,5-dimethyl-1,4-dioxane. Yields of 91% and 25% were obtained for the depolymerisation of PEG and PPG, respectively. It was suggested that C–O cross metathesis can occur as a side reaction. This was further demonstrated by the reaction of dibutyl and dioctyl ether to butyl octyl ether.56 Calculations were carried out with 18-crown-6 as a theoretical approach to the study of polyether depolymerisation is limited by its size and conformational flexibility. Four phases of the reaction were identified (Fig. 10). First, substrate binding, formation of a zwitterion, formation of dioxane via a SN2 cascade and release of the product.56
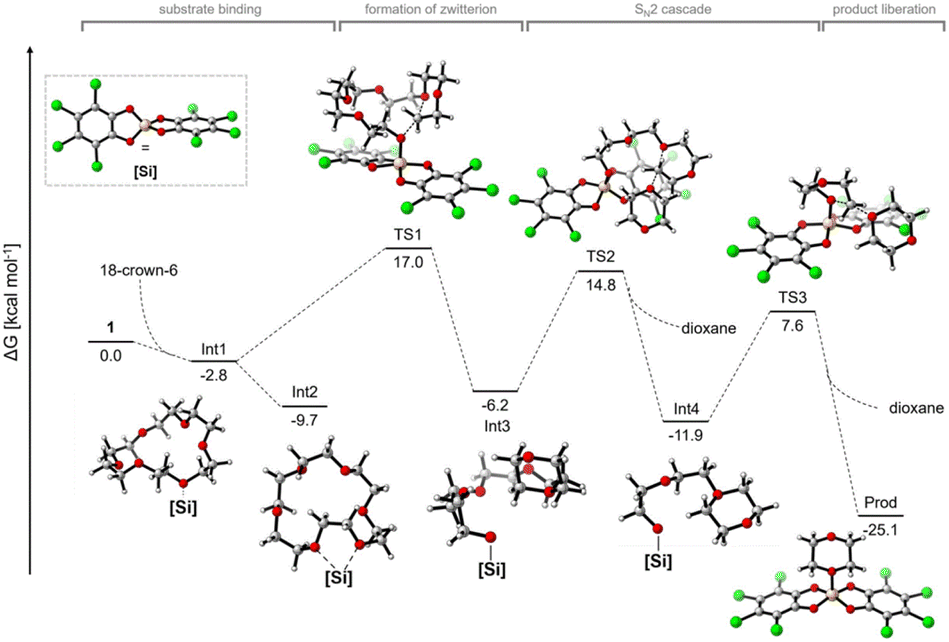 | ||
| Fig. 10 Computational reaction profile of 18-crown-6 degradation by bis(perchlorocatecholato)silane with possible intermediates and transition states (reprinted with permission from Wiley, ©2022).56 | ||
Recently, Inuoe et al. published an acetonitrile adduct of a super Lewis acid silane with perfluorinated pinacolato groups Si(pinF)2 catalysing the depolymerisation of oligomers 1,5-dimethoxypentane (1,5-DMP), diglyme, tetraglyme and poly(ethylene glycol)dimethyl ether (PEG-DME) with excellent yields of 87–97%.57 However, the longer the depolymerised oligomer or polymer, the higher the amounts of catalyst required to achieve high yields, resulting in a lower TON, indicating catalyst deactivation. The proposed mechanism starts with the mono coordination of an ether substrate (Fig. 11). Coordination of a substrate to the Si centre results in shielding of a second coordination site by the perfluorinated pinacolato groups. Coordination is followed by intramolecular SN2 attack of the adjacent uncoordinated oxygen atom on the activated carbon of the C–O bond, resulting in cleavage of the C–O bond, producing an oxonium cation and a pentavalent silicate anion. The methoxy group of the anion then attacks the primary carbon centre in the methyl moiety of the cation, releasing 1,4-dioxane and dimethyl ether in the case of diglyme. In the case of oligoethers, the methoxy group anions attack the kinetically less favoured secondary carbon centre, reducing the overall reaction rate. Consequently, the accumulation of the highly electrophilic oxonium species increases the possibility of side reactions. In addition, the oxonium species is susceptible to nucleophilic attack, so an attack on another secondary atom of the oxonium species would lead to irreversible catalyst deactivation.57
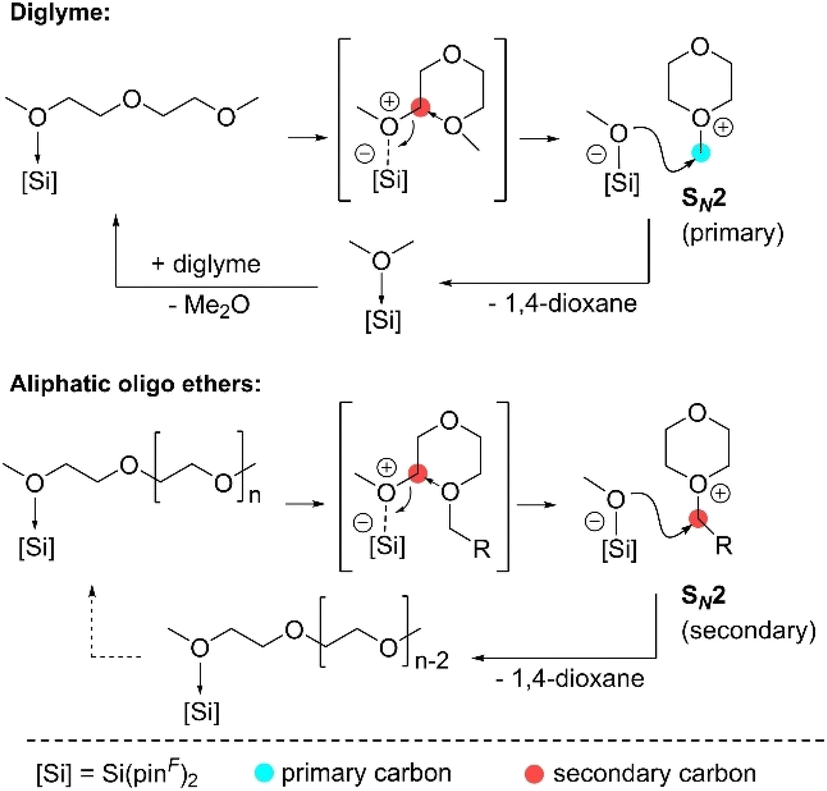 | ||
| Fig. 11 Proposed Si(pinF)2-catalysed ring-closing metathesis of diglyme and oligo(ethylene glycol) (reprinted with permission from Wiley, ©2022).57 | ||
|
|
|||
|---|---|---|---|
| Entry | Additive | Major products | Yield (%) |
| a Reaction conditions: 50 mg Ti4+-mont, 0.3 mmol PTHF (based on polymer subunit), 8 mL aromatic compound, 160 °C, 12 h. b Reaction conditions: 50 mg Ti4+-mont, 0.6 mmol PTHF (based on polymer subunit), 0.5 mmol aniline, 180 °C, 48 h. | |||
| 1a |
|
|
75 |
| 2a |
|
|
65 (3:2) |
| 3a |
|
|
65 (1.1) |
| 4a |
|
|
64 |
| 5a |
|
|
63 (2:7) |
| 6b |
|
|
36 |
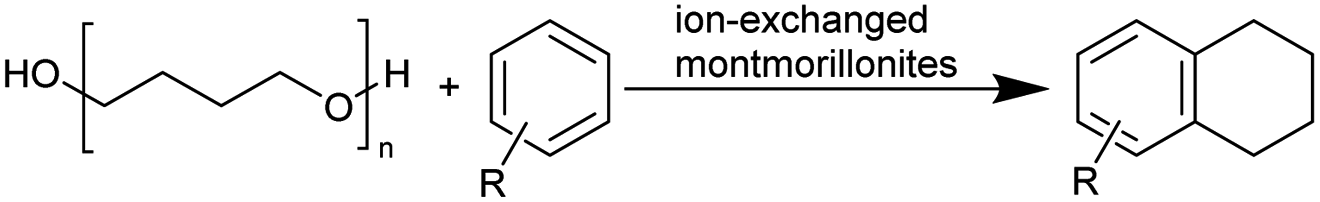
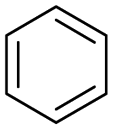
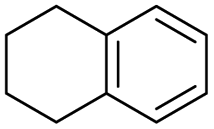
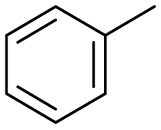
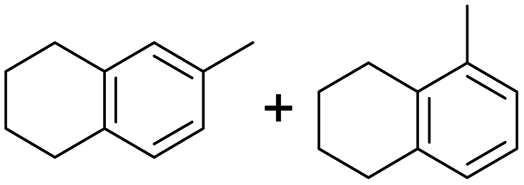
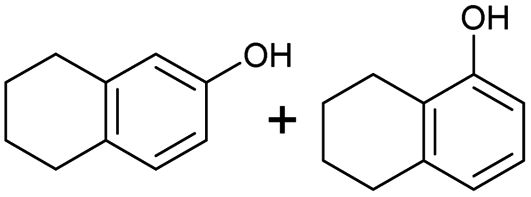
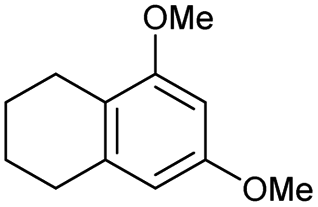
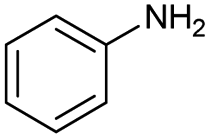
The interlayer distance of the Ti4+ mont of 2.8 Å is broadened to 4.5 Å when treated with PTHF and returns to its original distance after the d-polymerisation process. The extended interlayer of the Ti4+-mont with intercalated PTHF is significantly smaller than the extended interlayer (12 Å) of the H-mont with intercalated PEG. Interestingly, the active centre is not the Ti4+ itself, but catalytically active Brønsted acid sites within the interlayer due to the suppressed reaction of the Ti4+-mont upon introduction of 2,6-lutidine. A Friedel–Crafts alkylation mechanism is proposed. After activation of the C–O bond at the Brønsted acid site, a PTHF fragment with a carbocation is formed. This carbocation attacks the aromatic compound, followed by intramolecular alkylation at the acid site to produce the corresponding tetralin.36
2.5. Depolymerisation of aromatic polyethers and epoxy resins
Depolymerization of polyethers with aromatic subunits can lead to the production of valuable aromatic molecules. Wang and colleagues studied the depolymerisation of poly(p-phenylene oxide) (PPO) and PPO/polystyrene blends (MPPO) over Ru@Nb2O5 catalysts via hydrogenolysis to a mixture or arenes under harsh reaction conditions as shown in Fig. 12.38 The composition of the product mixture as a function of the loading and support of the Ru catalyst has been extensively studied by the authors. The product mixture consists of intermediates from the hydrogenolysis steps. From these products, 3,5-dimethylphenol (3,5-DMP) is important in various industries, including pharma (for antibiotics and vitamin E), food and agriculture.38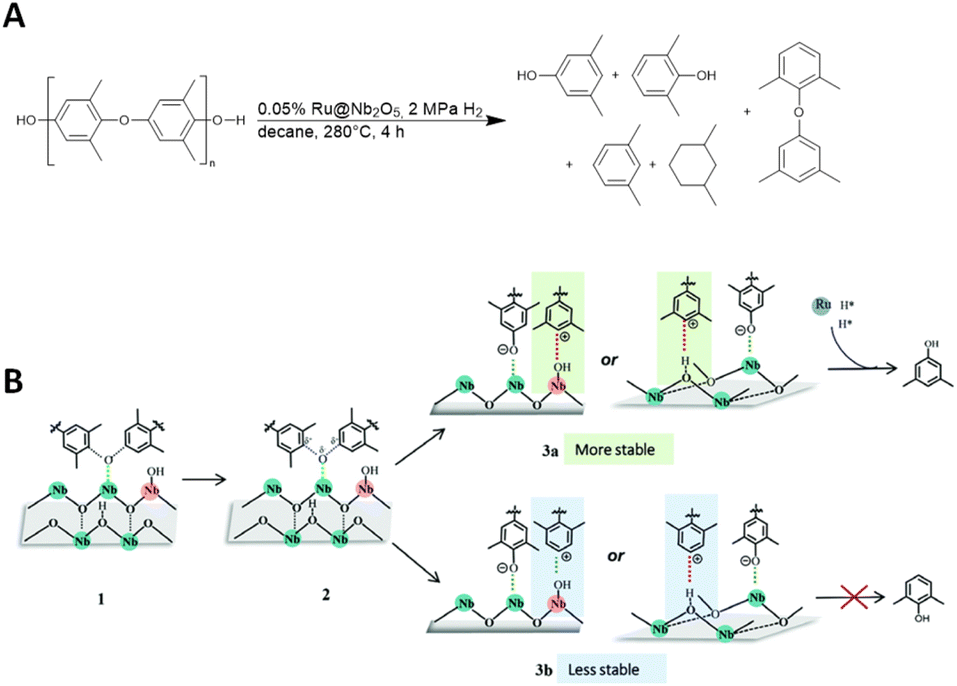 | ||
| Fig. 12 (A) of PPO via hydrogenolysis in the presence of a supported ruthenium catalyst (Ru@Nb2O5). (B) Proposed reaction mechanism (adapted with permission from The Royal society of Chemistry, ©2021).38 | ||
The size of the Ru particles determines the product selectivity. When using high-loaded (2%) Ru@Nb2O5, PPO is completely depolymerized and undergoes complete hydrogenolysis to m-xylene, as well as hydrogenation to 1,3-dimethylcyclohexane as the main product. Conversely, lower-loaded (0.5%) Ru@Nb2O5 results in a mixture of 1,3-dimethylcyclohexane and 3,5-DMP. A significant portion of the low yield observed when using pure Nb2O5 was attributed to the presence of 3,5-DMP, along with smaller quantities of 2,6-dimethylphenol (2,6-DMP) and PPO residues. However, by depolymerisation of PPO with exceptionally low loading (0.05%) Ru@Nb2O5 under given reaction conditions, a yield of 60% and the highest selectivity for 3,5-DMP (∼95%) can be achieved.38
Comparing Nb2O5 with other basic and acid catalyst supports, such as TiO2, MgAl2O4, and hydroxyapatite, demonstrates its significant role in product selectivity and its involvement in the reaction mechanism. CO2-TPD (temperature programmed desorption) confirmed the presence of basic sites in MgAl2O4 and hydroxyapatite, which decompose PPO mainly to 2,6-DMP. A significantly higher amount of Brønsted acid sites were found in Nb2O5 than in TiO2. On the basis of these results, the authors concluded that the acid catalyst supports preferably cleaved the C(ortho)–O linkage, whereas the basic catalyst supports favoured the C(meta)–O bond-cleavage. The mechanism is also influenced by the Brønsted acid sites, which cleave the C(ortho)–O bond and stabilize the intermediates (Fig. 12B). The proposed mechanism begins with PPO adsorption (1) and activation of the C–O bond, resulting in an oxygen anion (2) and two carbocation intermediates. The intermediates are anchored by the Brønsted acid sites, with anchoring at the ortho-position of the arene relative to the methyl groups (3a) being more stable than at the meta-position. The methyl groups donate electrons to the carbocation in the ortho-position via the σ–π-hyperconjugation effect, which lowers the kinetic barrier for the C(ortho)–O cleavage. The weakened C–O bond is attacked by the hydrides on the Ru cluster, resulting in the formation of 3,5-DMP.38
Other studies have shown that small particles such as Ru and Pt with low loadings result in small metal clusters which limit the surface area and thus limit the adsorption and hydrogenation of aromatic rings, which is consistent with the complete hydrogenolysis and hydrogenation of PPO with highly loaded (2%) Ru@Nb2O5. The limited adsorption sites are also attributed to the restriction of activated hydrogen supply, which prevents the hydrogenolysis of the hydroxy group. The cleavage of the C–O bond at the ortho position is preferred over the meta position relative to the methyl residues, resulting in 3,5-DMP. This preference is due to the larger dissociation energy of the C(ortho)–O bond (305.2 kJ mol−1) compared to the C(meta)–O bond (305.2 kJ mol−1), as calculated.58,59
Nakajima's group recently reported the depolymerisation of polyetheretherketone (PEEK) using sulphur nucleophiles to dithiofunctionalised benzophenone derivatives. This was achieved without a catalyst and resulted in the generation of valuable chemicals. Table 7 shows a wide range of substrates explored using 2-phenylethanol and various alkyl halides. The depolymerization process allows for the introduction of reactive functional groups to the alkyl halide. This method is applicable for PEEK in the form of powder, pellets, or film.39
|
|
|||||
|---|---|---|---|---|---|
| Entry | R | X | T (°C) | Time (h) | Yield (%) |
| a Reaction conditions: PEEK powder (0.3 mmol relative to the molecular weight of the monomer), PhCH2CH2SH (1.2 mmol), NaOt-Bu (0.9 mmol), DMAc (0.6 mL), R–X (0.9 mmol). | |||||
| 1 | Me | I | 100 | 1 | 93 |
| 2 |
|
Br | 100 | 2 | 77 |
| 3 |
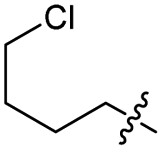
|
Cl | 80 | 23 | 67 |
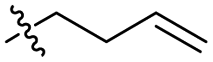
| 4 |
|
Br | 100 | 2 | 97 |
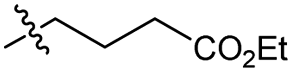
| 5 |
|
Br | 100 | 2 | 89 |
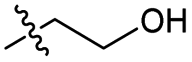
| 6 |
|
Br | 100 | 2 | 83 |
| 7 |
|
Br | r.t. | 20 | 53 |
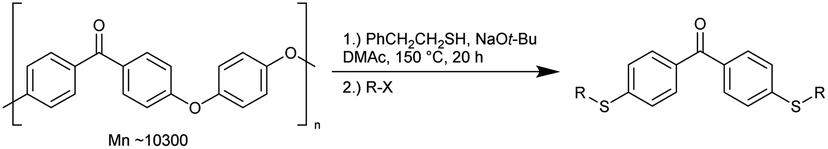
Epoxy resins typically contain bisphenol A (BPA) and are not biodegradable. Incineration of these resins results in the emission of toxic substances. Due to the health risks associated with these emissions, substitutes are currently under investigation.60 However, methods are needed for a safe disposal of the remaining end-of-use composites. Skrydstrup et al. have recently reported the catalysed depolymerisation of epoxy resins and their fibre composites to recover (BPA) derivatives. The homogeneous Triphos-Ru-TMM catalyst complex (Fig. 13) was used for C–O bond cleavage in both BPA and non-BPA model compounds and in selected commercial products. The Airstone 760/766H commercial resin, used in the construction of wind turbine blades, along with UHU two-component glue for handicrafts, Roizefar epoxy resin, and an epoxy resin for maritime engineering were successfully depolymerized. The reaction protocol was successfully applied to fibre-reinforced epoxy composite samples, such as those from laminates and wind turbine blades. Longer reaction times of up to three days were required for complete disassembly. The glass and carbon fibres were recovered to a high level of quality.37
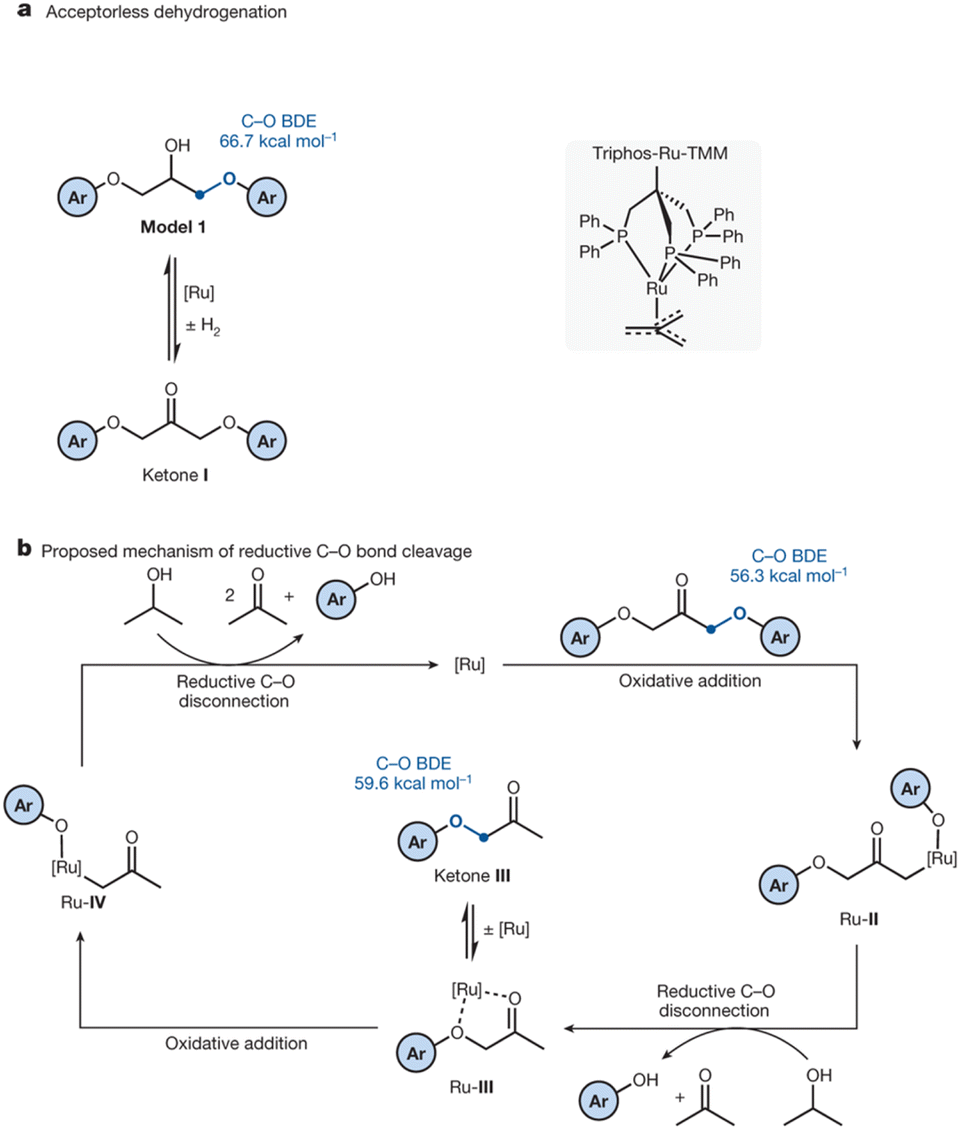 | ||
| Fig. 13 (a) Concept scheme for acceptorless hydrogenation. (b) Proposed reaction mechanism for the Ru-catalysed C–O disconnection in epoxy resins and composites (adapted with permission from Springer Nature under Creative Commons licence, 2023).38 | ||
The authors propose a depolymerization mechanism based on their observations from the model reactions on acceptorless hydrogenations. The mechanism relies on the cleavage of C–O bonds, as shown in Fig. 13. The alcohol group is dehydrogenated to a ketone by the Ru-catalyst, as evidenced by the inactivity of the ether-substituted models. The C–O single bond of the ketone's β-carbon is then susceptible to cleavage via oxidative addition, generating intermediate Ru-II. Isopropanol acts as an additional hydrogen source via transfer hydrogenation, reducing Ru-II to a low-valent R-III, and producing a phenol derivative. Subsequently, a second oxidative addition step of Ru-III initiates a reduction cascade, resulting in the cleavage of the remaining polymeric substrate to yield the corresponding phenol derivatives and acetone.37
3 Depolymerization of polysiloxanes
Polyorganosiloxanes, also known as silicones, are hybrid organic/inorganic polymers. Their chains are characterized by a backbone of alternating silicon and oxygen atoms, which confers them with high flexibility and chemical stability. They display unique properties that are different from, or complementary to, those of common organic polymers.61 The construction, automotive, electronics, medical, personal care and consumer goods industries use them in a variety of applications. These include their use as insulators, dispersants, defoamers, release agents, implants, matrices in drug delivery systems, adhesives, and sealants.13,62–64The main organosilicon polymers are poly(dimethylsiloxane)s (PDMS). They are mainly produced from cyclosiloxane monomers, such as hexamethylcyclotrisiloxane (D3) and octamethylcyclotetrasiloxane (D4), through ionic ring-opening polymerization, catalysed by acids or bases.65–67 Studies have shown that for polymerization to occur, cyclic monomers and linear polymers must reach a thermodynamic equilibrium. Thus, PDMS can be depolymerized into cyclosiloxane monomers by continuously removing them from the equilibrium under acid or base catalysis.68–74
Individual PDMS chains can be joint into a three-dimensional network, creating an elastomeric material that resists shear, tension, and compression. This process is known as curing or vulcanization in silicone chemistry, possibly in tribute to Charles Goodyear's accidental discovery in 1839. Therefore, it is common to refer to these networks as silicone rubbers.75,76
Recycling vulcanized silicone rubber wastes to its monomers is challenging due to the slow rate of depolymerization caused by the network structure, even with the use of catalysts. To accelerate the process, it is common to conduct depolymerization in an inert gas environment at a temperature of 300 °C or higher.77–79 Surface etching with acid, base, and fluoride are alternative methods for depolymerizing silicone elastomers.80
This subsection presents the most representative methods for the chemical depolymerization of linear PDMS and silicone rubbers. These methods use either stoichiometric depolymerization reagents or catalytic technologies. The underlying reaction mechanisms and industrial applications have been included. This review will not discuss the historically relevant and widely used methods for thermal degradation of silicones. Interested readers can find abundant information on these approaches elsewhere.81–86 Recently, Rupasinghe and Furgal published an interesting overview on the various available silicone depolymerization methods sorted out by depolymerization temperature and mechanism.87
3.1. Acid catalysed depolymerization
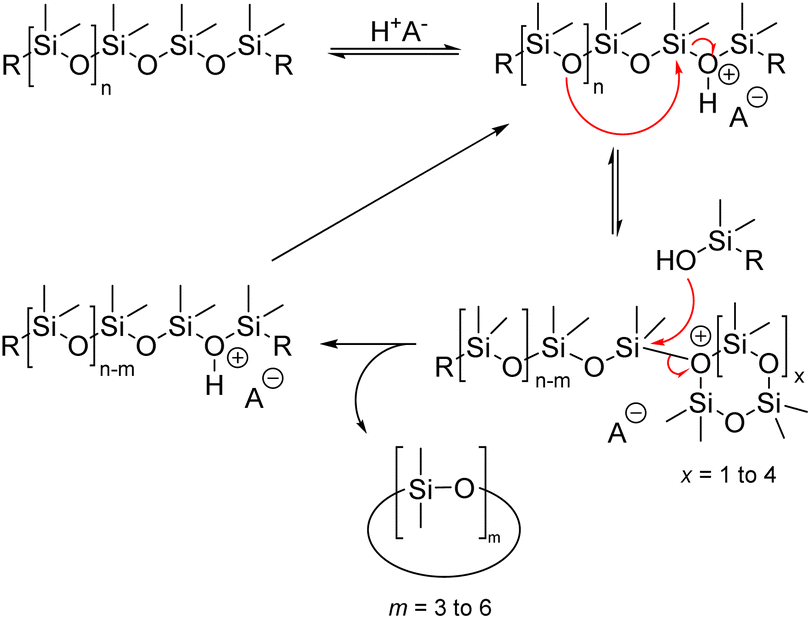 | ||
| Fig. 14 Reaction mechanism for acid-catalysed depolymerisation of siloxanes. | ||
Siloxane depolymerization through acid catalysis occurs via an equilibrium between the initial siloxane and cyclic siloxane product. The acidic proton coordinates with the siloxane oxygen, leading to the disruption of the siloxane bond and the formation of a silanol group, silyl halide, and silyl ester group. Strong protonic acids seem to utilize silanol end groups as active intermediates in the polymerization and rearrangement of diorganosiloxanes. The rate at which equilibrium is reached and the products produced depend on the concentrations of silanol and silyl ester (or halide) end groups in the siloxane phase.66
In 1956, Hyde et al. at the Dow Corning Corp. introduced a method to depolymerize silicone polymers, such as filled and vulcanized siloxane elastomers, using anhydrous hydrogen chloride in various hydrocarbons, ranging from aliphatic, aromatic, or chlorinated solvents.91
In 1973, Miller et al. patented a distillation process for the preparation of cyclic polysiloxanes, using clay treated with sulphuric acid at 300 °C to successfully produce cyclic products. However, the prolonged exposure of the reactant to acidity and heat, large amounts of evolved hydrogen gas, and the low yield of cycles made the process commercially impractical.92
Burkhardt and colleagues presented a technique for producing cyclic siloxanes by means of acid-catalysed depolymerisation of linear siloxanes. Linear poly(dimethylsiloxanes) were treated with 50–85% aq. sulfuric acid for 1.5–6 hours at 130–150 °C. The siloxane cycle and water were recovered by distillation, yielding 89%. This method can have a significant impact even on a smaller scale. However, due to the corrosive nature of sulfuric acid and the inherent risk of explosion when large amounts of highly concentrated sulfuric acid come into contact with hydrosiloxanes, the industrial-scale synthesis of cyclic siloxane is still not feasible.93 Later in 1994, the same group reported another acid-catalysed depolymerisation of silicone elastomers using sulfuric acid and aluminosilicate.94
Similarly, in 1990, Crivello patented the high temperature, acid catalysed depolymerisation of polyalkylhydrosiloxanes using a fixed acid bed at reduced pressure. The use of an acidic zeolite catalyst is described for the production of cyclic siloxanes from linear polysiloxanes.95 The method is based on incrementally adding a reactant to a hot acid catalyst at temperatures ranging from 200 to 800 °C under reduced pressure. Volatile cyclic compounds were recovered through condensation. However, the possibility of linear hydrosiloxane build-up in contact with heat and acidity still exists, despite the semi-batch nature of this procedure. As the reaction progresses, high molecular weight non-volatile species tend to stick to the catalyst surface, forming a gel that deactivates the catalyst. This results in hydrogen production and leads to the accumulation of linear hydrosiloxanes. To avoid an ineffective and expensive use of the reactor volume and catalyst active surface area, it is important to expose the reactants to the whole catalyst bed. The method's versatility in producing variable amounts of distinct cyclic species is limited by the set residence period at the catalyst.95
The reuse of waste silicone by dissolving it in a suitable solvent is the subject of a US patent. The waste silicone is converted into cyclic dimethylcyclosiloxanes, such as D3, D4, and D5, using a two-step acid/base catalysed scission process. The waste silicone used is of high molecular weight, including liquids and elastomeric solid silicone polymer products with short alkyl groups, such as methyl groups. A high-boiling organic solvent containing sulphuric acid can be used to heat silicone polymers until they dissolve, thereby cracking high molecular weight silicone polymers. The polymers are heated until they dissolve, then the mixture is made basic, and the resulting cyclic siloxanes are separated by distillation. Filtration is the only way to remove the solvent from semi-volatile by-products and silicone polymer components such as fillers. However, the finely divided nature of the fillers used in silicone formulations clogs the filters. Unfortunately, it is not economically viable to re-use the filtered fillers. Additionally, the solvent has a short shelf life and requires periodic replacement.96
Katsuragi and Mori developed a method to remove vulcanized silicone resins from non-sensitive, hard supports. They used a mixture of solvents, including chlorinated aliphatic hydrocarbons, aliphatic hydrocarbons, and monocyclic aromatics, as well as methyl ethyl ketone, acetone, cyclohexanone, acetic acid/n-butyl acetate, or a mixture of these solvents with tetrahydrofuran and dodecylbenzenesulfonic acid. However, the process of desiliconisation mentioned above focuses on the removal of the silicone component from the surface. It does not provide an up-cycling process to convert this silicone waste into a value-added product.97 Very recently, Knott and colleagues at Evonik patented a similar approach to perform a series of acid catalysed depolymerisations of silicone waste (silicone oil, elastomer, rubber, adhesive, sealant, sheet like silicone coated material) to acetoxysilane using acetic anhydride, acetic acid and one Brønsted acid within the temperature range of 50 to 200 °C with and without solvent.98–101
In 1992, Humphrey and colleagues introduced a method to partially depolymerize silicone elastomers into a viscous liquid silicone product. They treated milled silicone rubber with anhydrous hydrogen chloride at room temperature for several hours to obtain liquid silicone. The patent suggests that silicone rubber can be depolymerized using anhydrous halogen acid or certain easily hydrolysable halides to generate hydrochloric acid, which is a useful catalyst.102
In 1995, Wilford and colleagues proposed a method to break down silicones into shorter chains using dodecylbenzenesulphonic acid as a depolymerization agent in a hydrocarbon solvent with a chain length between C9 and C15.103 Shin Etsu Chemical Co Ltd later patented a similar approach to decompose waste silicone rubber using acid catalysed depolymerisation. An organic sulfonic acid was used as a catalyst for the depolymerisation in a non-aqueous solvent.104 More recently, Wu and Tang patented very similar process, where silicone waste is depolymerised into cyclosiloxanes by treatment with concentrated sulfuric acid and dodecylbenzenesulfonic acid at temperatures between 100 and 250 °C in vacuuo.105,106 Using slightly different processes, Chen and Zhao disclosed the chemical degradation of silicones in acidic media containing formamide or dimethylformamide, and a mixture of sulfonic and sulfuric acid at 200–280 °C.107,108
In 1996, Knies developed an inexpensive method of depolymerizing addition-cured silicones into volatile siloxanes and fillers using a small amount of acid. The process consists of three steps: first, a solvent-based solution of strong Brønsted acid is introduced into the silicone rubber vulcanizate; second, the solvent is removed; and finally, the silicone rubber vulcanisate is heated to a temperature of at least 350 °C. For this process, it is preferable to use non-polar solvents with boiling points up to 100 °C at 0.1 MPa, whether halogenated or non-halogenated.109
In 1996, Kostas proposed a method to produce cyclosiloxanes from various linear polyorganosiloxanes and siloxane copolymers. The process involves continuously passing silicone feedstock through a reactor containing an acidic catalyst under an inert atmosphere with a pressure of more than 14.7 psi and a temperature ranging from room temperature to 800 °C. Zeolite catalysts, such as HY type and zeolite molecular sieve with a pore size of 5–20 Å, silica alumina, titano silica alumina, hetero polyacid, supported mineral acid catalyst, and ion exchange resin catalyst, are preferred for their acidity. This method has the main advantage of not requiring a solvent for the recovery of this cyclic siloxane.110
In 1996, Hirose and colleagues introduced a halogen-free method for decomposing silicones into alkoxysilanes at room temperature. The silicone polymers are decomposed using an acidic catalyst, orthoester, and active hydrogen groups, such as sulphuric acid, methyl orthoester, and methanol. This method can be used for recycling of silicone rubber to alkoxysilanes, and for surface modification of silicone coated substrates by partially decomposing and removing the polysiloxane film from the substrate with high yield and without the formation of undesired side products.111
In 1997, Tsuji et al. proposed a method to produce alkoxysilane monomers in a cost-effective and safe manner. This was achieved by quantitatively decomposing a range of silicone products, including silicone oils, rubber, resin, elastomers, and varnish, under mild conditions. Waste silicone is treated in the presence of orthocarboxylic ester, specifically trimethyl orthoformate, and a proton donor such as alcohol or water. The alkoxysilane monomer or oligomer is hydrolysed and the resulting hydrolysate is condensed. However, this method produces an unwanted hydrolysis product of orthocarboxylic ester, which requires further purification. As a result, this process has not been practical on an industrial scale.112 Kawamoto et al. proposed an improved method to prevent the unwanted production of side products and produce pure monomers or oligomers. Under reflux conditions at 90 °C for 4 hours, silicone scrap is decomposed using dimethyl carbonate, an alcohol and an acid catalyst such as sulphuric acid, hydrochloric acid, nitric acid, fuming sulphuric acid or acetic acid, etc.113
In 2004, Oishi developed a method to recover silicone oil or grease from used silicone rubber by acid catalysed depolymerisation. The reaction is performed between 70 and 170 °C with various acidic catalysts, such as concentrated inorganic acids like sulphuric acid, nitric acid, and hydrochloric acid, as well as organic methyl sulfonic acid, phenyl sulfonic acid, and triflic acid in toluene or xylene as a solvent.114
3.2. Base catalysed depolymerization
Already in 1946, Hyde and co-workers described a process for the synthesis of highly purified cyclic dimethylsiloxanes with 91% yield from liquid polydimethylsiloxane using powdered sodium hydroxide at high temperatures between 230 and 240 °C. Strong bases such as sodium hydroxide, potassium hydroxide, or lithium hydroxide, as well as alkaline earth metal hydroxides and alkali metal fluorides, or mixtures of these hydroxides and fluorides, were used.116 This work was soon followed by Kantor, who used quaternary ammonium bases and quaternary phosphonium bases as initiators for the depolymerisation of polydiorganosiloxanes.117
In 1953, Hurd et al. proposed a mechanism for the rearrangement of organopolysiloxanes using a strong base. The mechanism involves two main steps. In step 1, the catalyst molecule is added to the siloxane system and in step 2, the catalyst molecule, or a part of it, is continuously involved in the rearrangement of the siloxane bonds (Fig. 15). It is well known that the silicon atom is a relatively good electron acceptor. It can coordinate with electron donor groups such as alkali metal hydroxides or fluorides to form corresponding potassium silanolate or fluorosilicate ions.65 When a hydroxyl group-containing base is introduced into the system, the oxygen atom in the hydroxyl group coordinates with the silicon atom in the siloxane chain, leading to successive chain-breaking rearrangements of the activated complex. Subsequently, these active intermediate coordination complexes undergo further rearrangement, such as chain scission and backbiting, resulting in the formation of a mixture of cyclic and linear siloxanes, depending on the chosen starting material.80
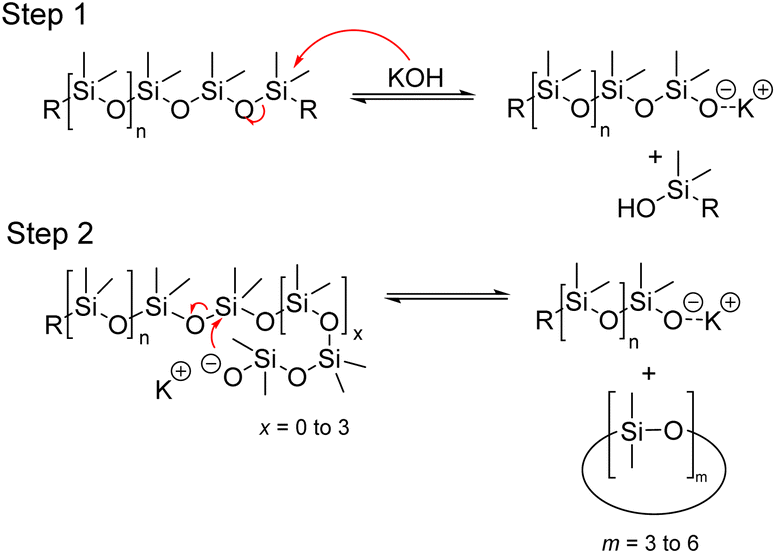 | ||
| Fig. 15 General mechanism of base-catalysed depolymerization of polysiloxanes. | ||
:1 and 0.007–0.073:1, respectively, at a temperature of 150–200 °C. To simplify the process, the reaction was carried out at atmospheric pressure. The simultaneous passing of water vapour and an inert gas improves the quality of the target product.71
In 1997, Allandrieu described a kinetically favourable method for the selective depolymerisation of polydiorganisiloxane (PDOS) into high purity D4 and D5 in good yields. The depolymerization is conducted at temperatures between 110 and 150 °C using hydroxide of rubidium, caesium or quaternary phosphonium hydroxides.118
In 1999, Yang et al. introduced a process for effectively decomposing siloxane products, such as silicone rubber, resin, and silica, using an alkali reagent in secondary and tertiary aliphatic alcohols. The base is a metal hydroxide or alkoxide that is present in an amount of 5–40% by weight with respect to polyorganosiloxane. The reaction was carried out either at atmospheric pressure or at a pressurised reactor varying between 20 and 100 °C within a time frame of one hour to three days. The pressure, temperature, and time required for the reaction varied depending on the starting material to be recovered.119
Mukherjee et al. described in 2001 an efficient process for recovering cyclosiloxane from silicone waste. The process involves a four-step reaction to recover cyclic siloxane from silicone scrap. First, the silicone is dissolved in octyl alcohol with linear alkylbenzene sulfonic acid (LABS) at 140 °C for 4 hours. In the second step, the reaction mixture was heated at 140 °C and 4 bar pressure after adding 50% aqueous NaOH. After 1 hour, the system was cooled down and the sodium silicate was separated from the mixture. To remove the octyl alcohol, the neutralised liquid mixture was maintained at 130 °C under vacuum.120
In 2002, Oku et al. reported an effective method for depolymerising PDMS using potassium hydroxide and buffering acids such as KH2PO4 and KCOOC6H4COOH to produce monomeric cyclic siloxanes such as D3 and D4. This method not only depolymerises silicone oil but also efficiently depolymerises vulcanised PDMS into a mixture of cyclosiloxane monomers with an 85% yield.121 Later the group depolymerised high-temperature vulcanised silicone rubbers containing fillers using KOH catalyst in a mixture of hexane, diethylamine and methanol. They not only separate cyclic siloxane with a high yield but also recover the filler present in the silicone rubber. However, the catalyst used in the reaction is present in the fillers as potassium silanolate, which does not allow the filler to be reused for reproducing corresponding rubber with good thermal stability.122 Later on, they a complex catalytic system was developed to recover both the cycles and the fillers contaminated by alkali metal cations. To achieve this, an organic base catalyst, tetramethylammonium hydroxide, was used in a mixture of diethylamine and hexane under reflux conditions.123
In 2008, Sik proposed a process for regenerating silicone oil from silicone waste. The process involves treating waste siloxanes with alkali metal hydroxide, such as sodium or potassium hydroxide, and to remove hydrosilane and trihydroxysilane impurities, which cause gelation. The process also involves treating these impurities with an oxidant, such as NaOCl or CaOCl2, to convert them into siloxanes.124
In 2009, Eyster et al. introduced a solvent-free method for recovering the monomer or cyclic siloxane from filled silicone material. They used a special reactor with a heated side wall, an agitator, and an additional heating setup, keeping the temperature below 200 °C and the pressure at 15 mmHg with potassium silanolate as a catalyst. This method is more environmentally friendly and cost-effective than traditional solvent-based methods.125
In 2010, Zhao presented a three-step method for producing silicone rubber from waste silicone rubber. Firstly, discarded silicone rubber was catalytically decomposed with a basic catalyst, decolourised with activated carbon filters and dimethylsilane was produced. Secondly, silicone rubber was produced using the recovered dimethylsilane and tetramethyl-oxyammonia.126
In 2022, Xu introduced a method for cracking silicones using alkali metal hydroxide and Schiff base metal complex, which has potential applications in the field of silicone production. The method mainly employs 5–8% of sodium or potassium hydroxide with 0.2–0.5% Schiff base metal complex, preferably copper, in a high boiling inert solvent such as tetraethylene glycol dimethyl ether or phenyl ether at 140 °C to recover cyclic siloxanes.127
 | ||
| Fig. 16 Cleavage of Si–O bond by an amine. | ||
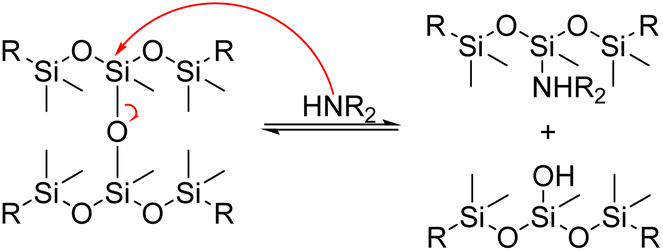
In 1978, Schatz and colleagues patented a method for reclaiming siloxane formulations containing fillers using an amine. This technique involves dissolving siloxane polymers and mixtures of them in a butyl amine solution. Quaternary ammonium and phosphonium bases may also be added, along with 1 to 10% by weight of alkali metal hydroxide. The solution is then heated to the boiling point of the amine while being stirred. Distillation is used to extract the amine and remove low molecular weight products from the mixture. The resulting monomers can then be used as raw materials to create polysiloxanes. Additionally, fillers from the original mixture can be retrieved and reused.133
Ionic potassium hydroxide is only sparingly soluble in amines, and understanding the dynamic study of the splitting reaction can be tedious. It took hours or days for the complete solubilization of PDMS in primary and secondary amines. To avoid this situation, Chang et al. prepared a homogeneous catalytic solution of potassium hydroxide in ethanol, where ethanol acts as a second nucleophile to enhance the rate of aminolysis. This method is potentially important from a commercial standpoint.134
The group later used different solvents to swell the silicone matrix, reducing the internal resistance of diffusion of the nucleophilic agents. The results were used to model the effect of the solvent on product distribution and the nucleophilic cleavage, aminolysis and alcoholysis reactions.135 In 2005, a mild catalyst for nucleophilic cleavage of crosslinked silicones into cyclic products in polar solvents was developed by preparing a homogeneous solution of potassium hydroxide in diethylamine (DEA) and ethanol. The silicone rubber was then swelled overnight in toluene, tetrahydrofuran or DEA in a glass reactor. The mixture was then agitated at room temperature to dissolve the silicone rubber in the system. Finally, during the filtration and washing process, the filler and cyclic siloxanes are recovered as a recycled product.136
In 1998, Cho and colleagues expanded the range of depolymerization by converting industrial silicone waste, such as oil, grease, gel, resin, and rubber, into value-added cyclic siloxane, disubstituted alkoxy silane, and filler. These methods use catalysts such as alkali metal alcoholate or quaternary ammonium alcoholate, alone or in combination with alkoxysilane, at temperatures below 300 °C under reflux and pressure.137
Sobota and colleagues recently reported an efficient method for degrading hard silicone rubber waste through solvothermal alcoholysis with fatty alcohols, with and without a catalyst. The most effective results were achieved using alkali-metal aryloxides (of Mg, Zn, Li, K and Na) supported by a methylsalicylate ligand. This process allowed the authors to recover a variety of liquid alkoxysiloxane derivatives (R(OSiMe2)xOR) in high yields.138
Phosphazene bases, which are a combination of nitrogen and phosphorus, have gained significant attention in organic synthesis.139 In 2008, Hupfiled patented a method for producing silicones that addresses some of the aforementioned issues. Polysiloxane and a high boiling end blocker were treated with phosphazene or carborane acid catalyst to continuously form cyclic polysiloxanes at reduced temperatures. The reaction mixtures were less corrosive and produced good yields with low levels of catalyst.140
Phosphazene bases react with water to form hydroxyl ion salts with uncoordinated phosphazenium cations. In 2023, Khedaioui et al. used P4-tBu at 90 °C for the conversion of thermoset silicone networks into liquid siloxanes, taking advantage of this specific property. In this process, the phosphazenium hydroxide is formed by deprotonation of adventitious moisture by the highly reactive P4-tBu (Fig. 17).141
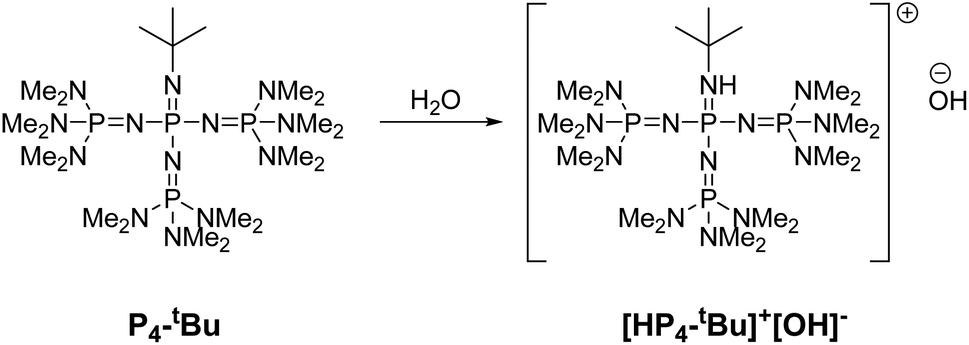 | ||
| Fig. 17 Activation of P4-tBu by the adventitious presence of water in silicones. | ||
Weitkamp et al. reported the synthesis of a phosphazenium hydroxide salt (2, Fig. 18). This compound exhibits notable reactivity due to the weak coordination nature of the phosphazenium ion.142,143 Later, they reported the first silanol–silanolate anion generated in situ by the hydroxide anion in the presence of a phosphazenium cation (Fig. 18B). The salt (4, Fig. 18) enables the rapid depolymerization of polydimethylsiloxanes into cyclic siloxanes without the need for solvents. At room temperature, PDMS was readily decomposed into a mixture of D4 (82%), D5 (8%), and traces of D3 (2%) using only a concentration of 0.1 mol% of catalyst.144
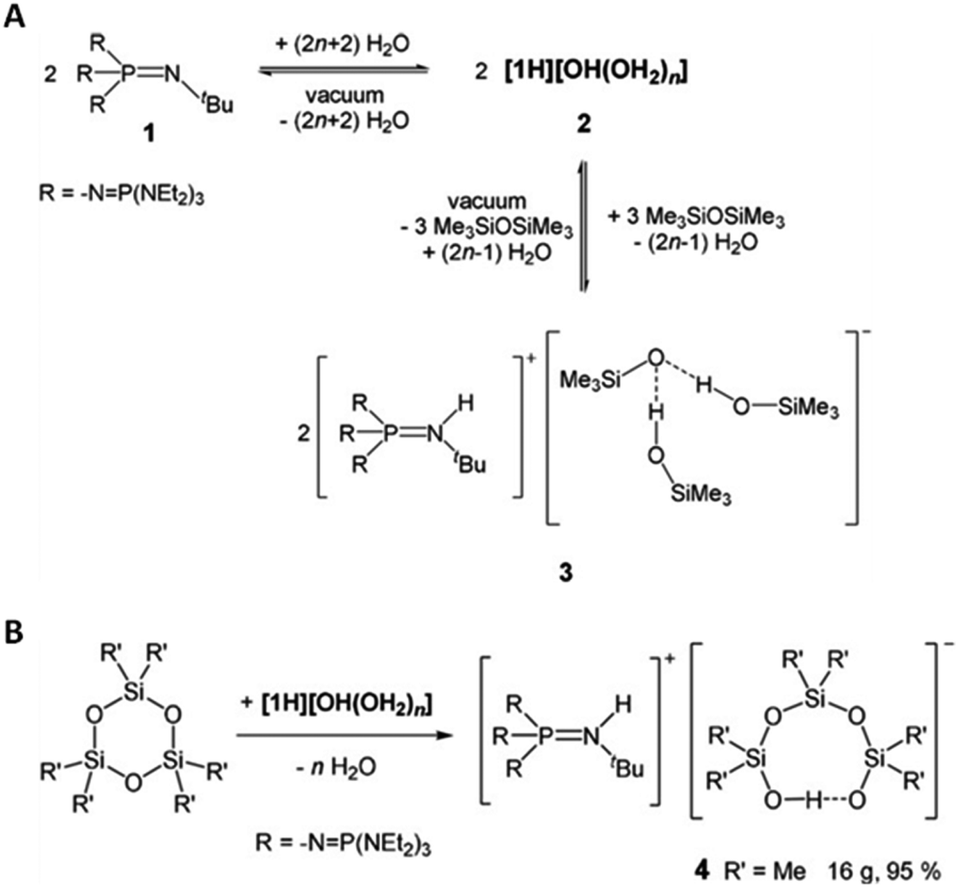 | ||
| Fig. 18 (A) Equilibrium reaction of phosphazene 1, hydroxide salt 2, and silanol–silanolate salt 3; 9 (B) reaction of cyclotrisiloxanes with in situ generated phosphazenium hydroxide. Reprinted with permission from Wiley under Creative Commons License, 2019.144 | ||
In 2023, Vu et al. chemically recycled used silicones into cyclic monomers using a polydentate crown ether and potassium silanolate catalyst. The reaction was performed at a wide range of temperatures, from 60–170 °C, resulting in a 99% yield of cyclic siloxane. The catalyst was recycled without any loss of activity over five runs. The mechanism for silicone depolymerization was explained through base-catalysed depolymerization with a polydentate ligand crown ether (Fig. 19). The dissociation of the ligand into an ion pair enhances the nucleophilicity of the counter anions, improving the kinetics of silicone depolymerization. The potassium cation in the crown ether behaves as a mild Lewis acid, activating the Si–O bond to facilitate depolymerization. Furthermore, this combination may restrict the demethylation pathway, which involves the K+ cation as its primary intermediate.145
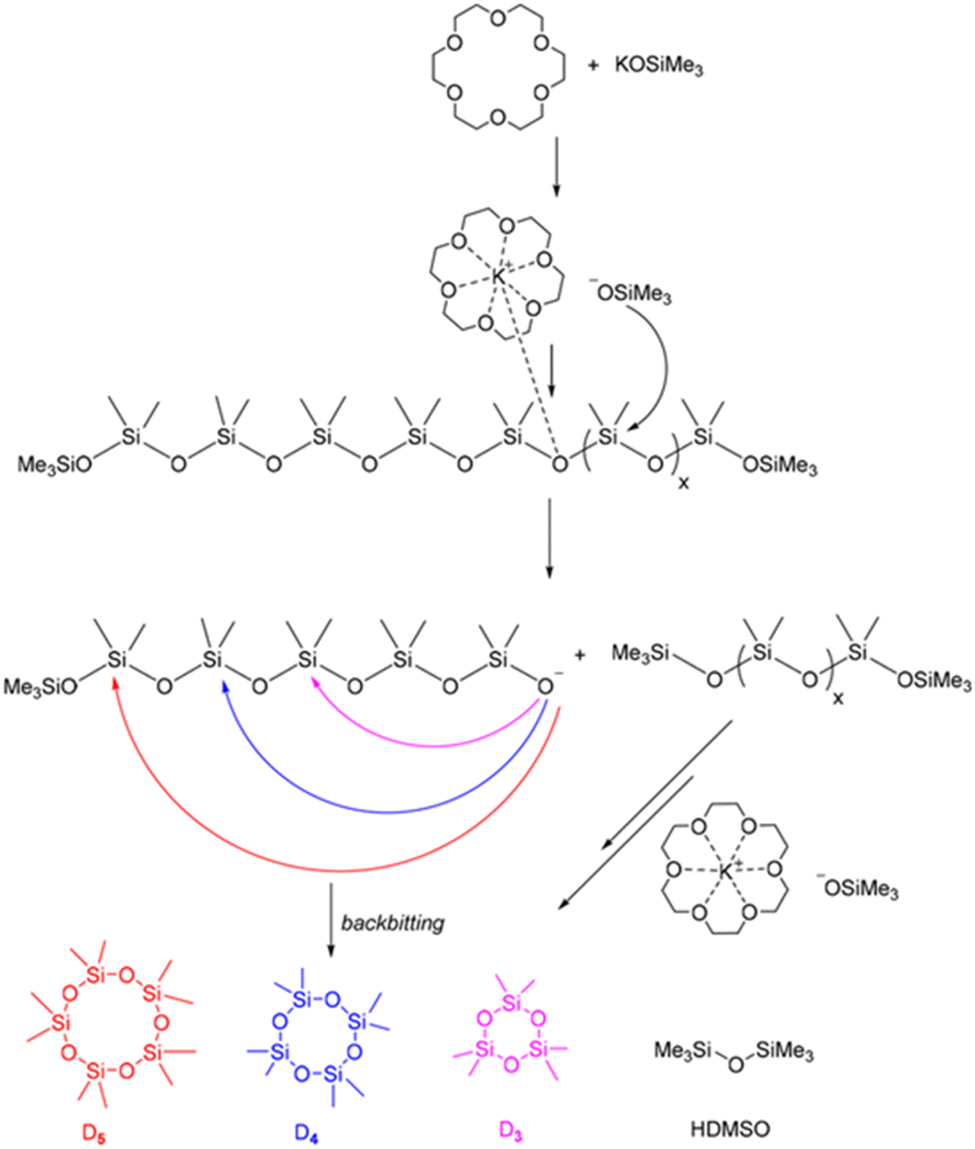 | ||
| Fig. 19 Proposed mechanism for the polydentate ligand–silanolate catalysed depolymerization of silicones. Reprinted with permission from The Royal society of Chemistry under Creative Commons License, 2023.145 | ||
Torgunrud et al. introduced an entropy-driven depolymerisation of polydimethylsiloxane. Various silicone acetal cycles were produced with good yield by depolymerising PDMS with different alcohols and glycols in the presence of a base. The authors mainly use monoalcohols such as methanol and isopropanol, as well as different glycols. They proposed a mechanism for the base-catalysed depolymerisation of PDMS with different end groups in hexylene glycol (Fig. 20). In the absence of a catalyst, hexylene glycol (HG) is a weak nucleophile, but when it is deprotonated with a base, it becomes an excellent nucleophile. To create a pentavalent silicate anion, a nucleophile can attack any silicon at either the chain ends or the main chain. The next step involves a second heterolytic Si–O bond cleavage, followed by proton transfer, intramolecular nucleophilic attack and heterolytic Si–O bond cleavage (leaving group loss). Distillation is necessary to remove the product from the process and prevent repolymerization, as each phase of the mechanism is in equilibrium.146
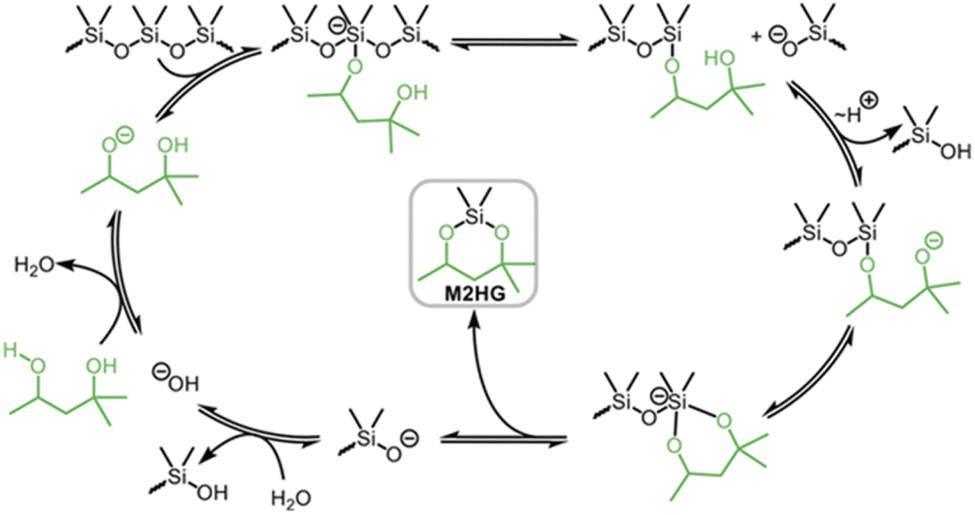 | ||
| Fig. 20 Postulated reaction mechanism for the base-catalyzed depolymerization of PDMS with hexylene glycol. Reprinted with permission from The American Chemical Society, © 2023.146 | ||
3.3. Depolymerization with dimethyl carbonate and fluorides
Several reagents, such as alcohol,147–149 thionyl chloride,150–153 hydrogen chloride,154,155 and amines,128–132,134 have been reported for the depolymerization of silicone. Although alcohol is the most commonly used reagent, it is important to note that depolymerization with alcohol is an equilibrium reaction, and therefore, it is necessary to remove water as a byproduct during the reaction. Conventional methods for producing regenerated silicone oils from waste silicon compounds involve the use of alkaline catalysts, such as caustic soda (sodium hydroxide) or caustic gallium (potassium hydroxide). Regenerated silicone oil is produced by distilling monomeric silane (dimethylcyclic silane) through high-temperature heat treatment cracking in an inert, high-boiling-point solvent under polymerization or polycondensation reaction with the aid of an alkali or acidic catalyst. However, the conventional method of breaking down, distilling, and recovering cyclic silane, followed by synthesizing oil from the waste silane hydrolysate, cannot be used because the reactants tend to gel during the cracking process.The use of alkali metal hydroxides as initiators has not been entirely successful, particularly on an industrial scale. Therefore, there was a need to discover a new, non-halide, non-toxic reagent that does not produce water during the depolymerization reaction. In their report, Zhurkina et al. found that ethyl orthoformate, a non-halogen reagent, can cleave Si–O bonds into D4 using strong acid without the formation of water, alas with low yields.156
In 2001, Suzuki and colleagues reported the complete degradation of hexamethyldisiloxane and D3 using dimethyl carbonate (DMC) over an alumina-supported KF catalyst, resulting in 85% methoxytrimethylsilane and 94% dimethoxydimethylsilane. The addition of a small amount of methanol to the deoligomerization reaction increased the yield of methoxytrimethylsilane to 98%. Importantly, the use of a solid catalyst makes it possible to avoid the disposal of acidic waste.157 Deoligomerization of larger cycloorganosiloxanes, such as D4 and D5 was later achieved with the same catalytic system.158 They later reported that depolymerization of silicone polymers with DMC was carried out using a catalyst other than strong acid. In this case, basic silicone products such as silicone oil and silicone rubber were depolymerized at 180 °C for 15 hours using DMC, sodium chloride, and methanol. The main advantage of this method is that the reagents used for depolymerization are non-corrosive, less toxic, readily available, and inexpensive. The siloxane bond is cleaved by the attack of methanol, and DMC acts as a scavenger of the byproduct, i.e. water.159
The proposed mechanism for this process using potassium fluoride supported on alumina begins with the activation of methylcyclooligosiloxane on the catalyst surface (Fig. 21). The catalyst partially breaks the siloxane bond of methylcyclooligosiloxane, resulting in the creation of methoxy-terminated linear siloxane and carbon dioxide. This linear siloxane then reacts with DMC, like the ring opening reaction of cyclooligosiloxane with DMC. Ultimately, cyclooligosiloxane is transformed into dimethoxydimethylsilane monomer. The cleavage of the siloxane bond in cyclooligosiloxanes or linear siloxanes is the rate-determining step (RDS).158
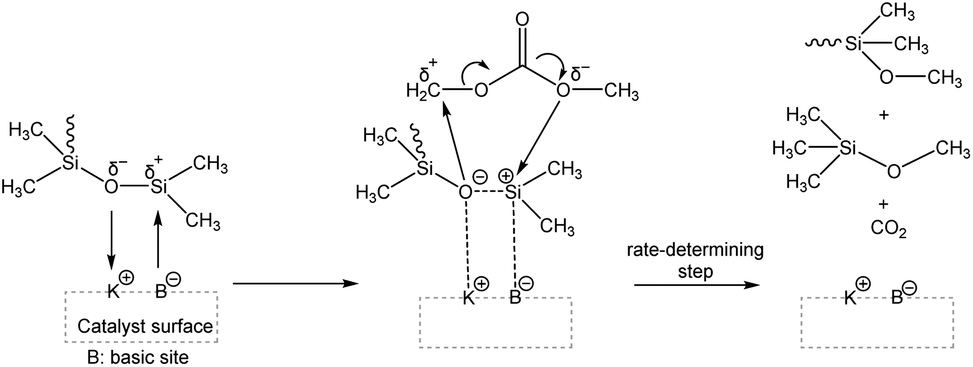 | ||
| Fig. 21 Speculated mechanism for the deoligomerization of linear siloxane in the presence of DMC on a supported catalyst.158 | ||
In 2000, Takeshi reported the applicability of industrial-scale silicone depolymerisation using alkylcarbonate. Silicone products such as silicone oil, silicone grease, silicone rubber, liquid silicone rubber, silicone sealant, silicone elastomer, silicone resin, silicone varnish, siloxane bond-containing silane coupling agent, and silicone–organic polymer copolymer can be recycled using dimethylcarbonate and an active hydrogen group-containing compound like methanol in the presence of acidic catalysts such as sulfuric acid, trifluoromethanesulfonic acid, or p-toluenesulfonic acid.160
In 1989, Schiem and his colleagues were the first to observe the catalytic activity of Boron trifluoride on silicone degradation.161 They used BF3–dioxane adduct to cleave siloxane and form fluorosilane. This reactivity was exploited by Enthaler et al., who were able to cleave the Si–O bond in siloxane and the Si–N bond in silazene and convert them to Si–F to give difluorodimethylsilane and 1,3-difluoro-1,1,3,3-tetramethyldisiloxane. The reaction was achieved with boron trifluoride diethyletherate as a catalyst under solventless conditions at 100 °C.162 They reported the degradation of short-chain silicone, silicone oil, and crosslinked silicones into their corresponding fluorinated products. The catalyst was used to convert poly(methylhydrosiloxane) into methyltrifluorosilane and difluoromethylsilane.163–165
The authors propose that the Lewis acid boron trifluoride activates a Si–O bond of the silicone by coordinating the oxygen to the BF3 (Fig. 22). This cleaves the Si–O bond, forming a [Si–F] moiety B and a [SiOBF2] moiety C. The [Si–F] moiety B can then react with boron trifluoride to cleave the Si–O bond of the [SiF] moiety B, forming difluoromethyl silane 3 (where X = H). This compound is removed from the reaction mixture by distillation, leaving a [SiOBF2] moiety. The [FSiOBF2] unit D can be formed by breaking the Si–O bond adjacent to the fluorine atom. This unit can then react with boron trifluoride to produce 3, which is the final product of the depolymerization process. To obtain trifluoromethylsilane 4 (for X = OMe, OSiR3), similar depolymerization reactions are necessary, but an additional step is required to convert the third Si–O bond.165
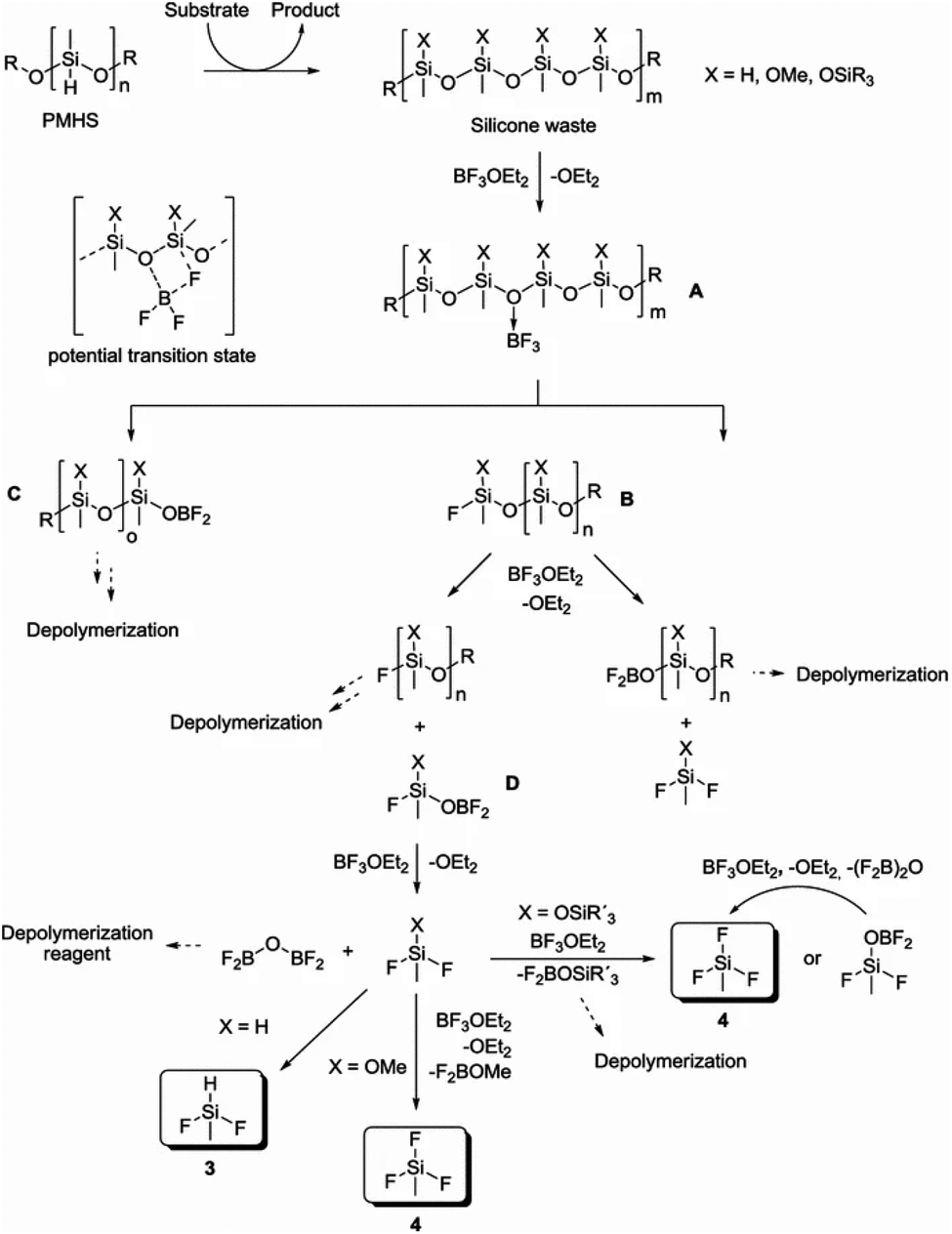 | ||
| Fig. 22 Proposed mechanism for the depolymerization of silicones catalyzed by boron trifluoride. Reprinted with permission of Springer Nature, © 2015.165 | ||
Furgal and Rupasinghe recently investigated an effective method for depolymerizing silicone-based polymers, elastomers, and resins at room temperature using catalytic amounts of tetrabutylammonium fluoride (TBAF) and highly swelling organic solvents such as THF.166,167 They successfully depolymerized common silicone samples such as PDMS and PMPS. The researchers then applied the same method to commercial polymers, such as cured Ace brand 100% silicone caulk, SYLGARD 184, Ecoflex 00-30, Smooth-Sil 950, Dragon Skin 10 FAST, and ELASTOSIL 3003. The results showed that the method was effective even with different additives and morphologies. The authors propose a reaction mechanism similar to the one reported by Laine for the equilibration of silsesquioxane cages using TBAF.168 In the key step, fluoride activates a silicone atom either at the chain-end or in the middle, followed by back-biting to produce cyclic siloxanes (Fig. 23).167
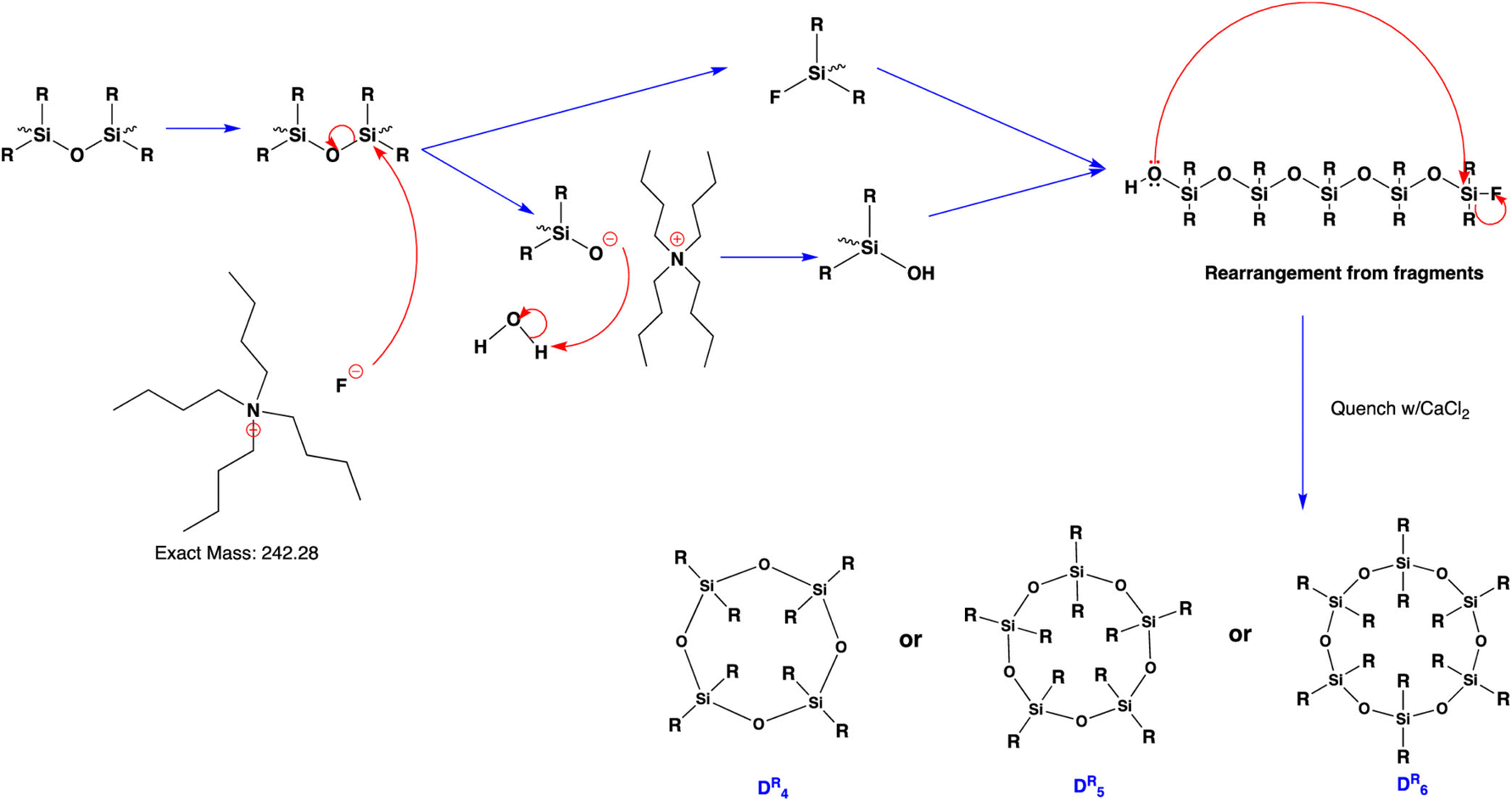 | ||
| Fig. 23 Reaction mechanism for the depolymerization of PDMS catalysed by tetrabutylammonium fluoride. (Reprinted with permission of the American Chemical society, © 2021).167 | ||
3.4. Metal-catalysed processes
Enthaler et al. introduced a series of catalysts for the depolymerisation of hydroxy-terminated silicones into low molecular weight dihalomethylsilane, 1,3-difluoro-1,1,3,3-tetramethyldisiloxane, dichlorodimethylsilane, dimethoxydimethylsilane, and diacetoxydimethylsilane using a cheap iron catalyst and a halogen source.169–172 Carboxylic acid chlorides or anhydrides, potassium fluoride or alcohols are used as depolymerization agents along with the iron salt. The authors proposed that the activation of a Si–O bond of PDMS by FeCl3 is the key step in the depolymerisation reaction when using an acyl anhydride and potassium fluoride (Fig. 24). This generates an acyl fluoride, which cleaves the Si–O bond to form a lower molecular weight Si–F moiety (A) and a SiOC(O)Ph moiety (B). A molecule of A is reacted with another molecule of acyl fluoride in the presence of a catalyst to cleave the Si–O bond adjacent to the fluorine atom. This forms difluorodimethylsilane (3) and a SiOC(O)Ph moiety. The difluorodimethylsilane (3) is then removed by distillation. Alternatively, the Si–O bond adjacent to the fluorine atom can be cleaved to form C. C can then react with 2 to form 3 and an anhydride. By repeating the described reaction sequences, depolymerization can be achieved, resulting in 3 as the end product.170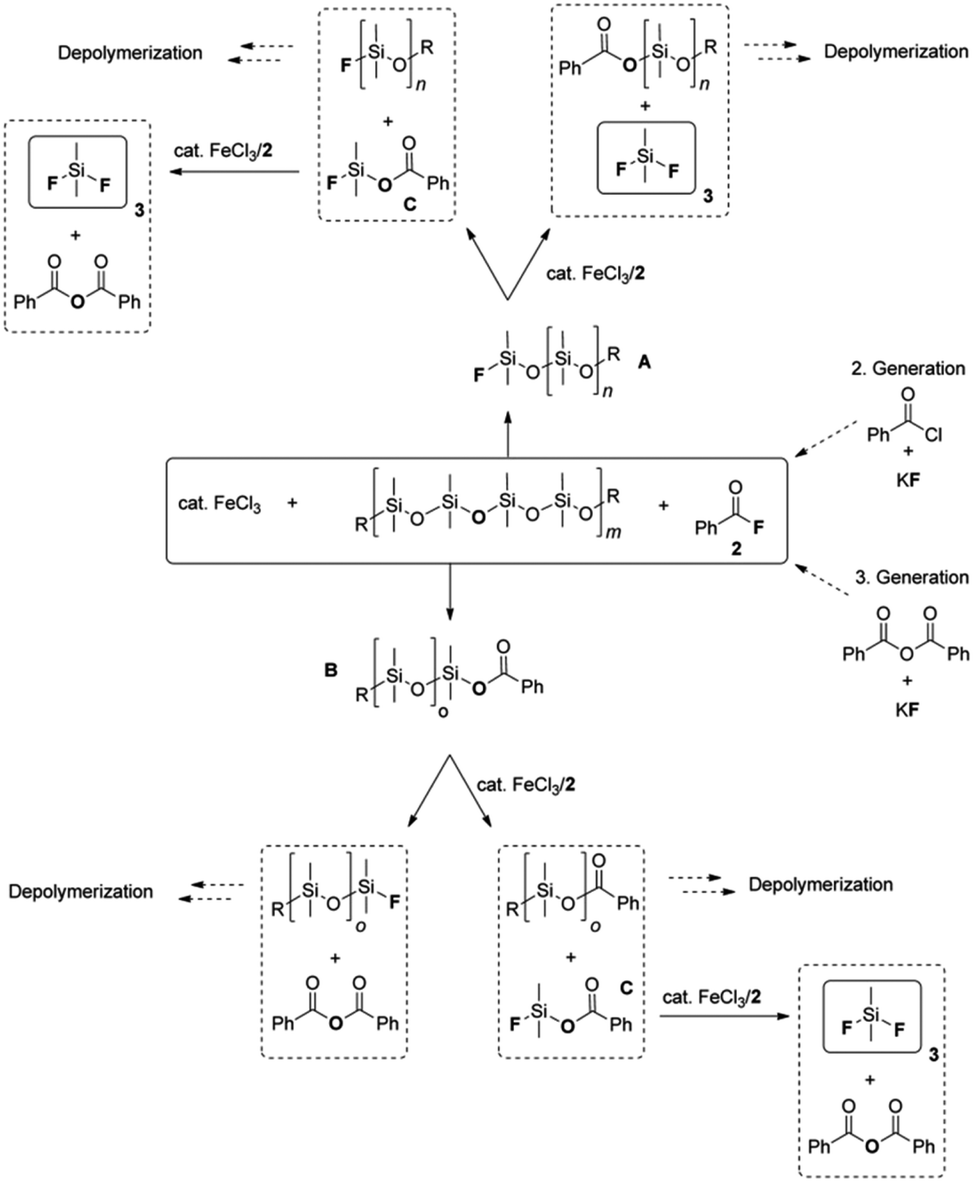 | ||
| Fig. 24 Proposed mechanism for the depolymerization of PDMS catalysed by iron salts. Reprinted with permission of Wiley, © 2014.170 | ||
Using zinc(II) halides and zinc(II) acetate, Daeschlein et al. selectively cleaved poorly reactive Si–O–Si bonds in functionalised disiloxanes to form zwitterionic, water-stable molecular zinc silanolates. X-ray crystallography studies confirmed the formation of zwitterionic species with a positive charge on the nitrogen and a negative charge on the oxygen. This makes the species stable in the presence of water.173,174
More recently, the Enthaler's group developed a new protocol for depolymerizing poly(siloxanes) using benzoyl fluoride as the depolymerizing reagent and a catalytic zinc salt to produce difluorodimethylsilane and 1,3-difluoro-1,1,3,3,tetramethyldisiloxane.175
Aluminium chloride and trimethylaluminum have also been used to promote thermal depolymerisation of silicone material. Alexander et al. disclosed a method to depolymerize silicone grease at 120 °C using AlCl3 with a Si:Al concentration of about 1:1. Their study revealed the formation of aluminium siloxide along with cyclic products.176 Barron and Apblett confirmed the formation of dimeric aluminium siloxides through the cleavage of silicones at 175 °C using trimethylaluminum.177 Mulhaupt and colleagues (104) obtained similar results using crystallographic methods with trimethylaluminium at 150 °C.178
4 Conclusions
Many commendable efforts have been made, both in academia and industry, to develop efficient processes for the chemical degradation of polyether and polysiloxane waste to recover valuable molecules and monomers. However, many obstacles remain, as most of the processes presented here are still far from large-scale implementation. Efficient separation of mixed plastic waste streams, the presence of contaminants such as additives, plasticisers and fillers, high energy requirements and process complexity are the main challenges.In addition, especially in the industrial setting, most developments have been directed towards process optimisation, with only marginal efforts devoted to understanding the chemical principles behind the working reactions and mechanistic understanding at the molecular level, as noted elsewhere.7 This has set back the progress of the technology enormously. Therefore, better communication between academic and industrial catalysis scientists is needed.
Even more worrying is the complete disregard for sustainability metrics in the design and implementation of the proposed chemical recycling technologies. All process conditions, solvents, energy and water use, and transport and distribution of waste and products need to be integrated into a life cycle assessment. Otherwise, the potentially useful technologies described here (and elsewhere) will remain wishful thinking and their real benefits and drawbacks will not be properly identified.
As polyethers and silicones (as well as other classes of synthetic polymers) have become irreplaceable in many consumer products and technological applications, it is realistic to say that they are here to stay. Therefore, it is only through the proper integration of waste streams into production chains, the systematic replacement of landfill and mechanical recycling by chemical and catalytic technologies, as well as widespread awareness of waste management, that the negative footprint left by the plastics era can be mitigated and the goal of a circular economy can be realised.179
To achieve this goal, it would be optimal for closed-loop recycling technologies to be integrated into all polymer waste treatment facilities. Nevertheless, chemical recycling methodologies are not always de facto capable of producing polymerizable monomers (as they are not typically designed to do so). In the case of polyether, its degradation can result in the formation of unstrained 5- or 6-membered rings, which may be subjected to ring-opening polymerisation. However, the recovery of strained epoxides from glycol esters remains a significant challenge. Polysiloxanes offer a more favourable outcome in this regard, as their depolymerisation typically results in mixtures of cyclic polysiloxanes that can be directly employed for de novo polymerisation processes.
Author contributions
E. M. conceptualized and supervised the project. T. T. and S. H. made the literature survey and wrote various portions of the manuscript. E. M. co-wrote the manuscript and edited its final version.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors express their gratitude to Felix Geitner, Andrea Gutacker, Jan-Erik Damke, Niklas Schafhausen, and Udo Kragl for their valuable assistance and insightful discussions.Notes and references
- L. H. Baekeland, Method of making insoluble products of phenol and formaldehyde, US Pat. US942699A, 1909.
- A. J. Ihde, The Development of Modern Chemistry, Dover Publications, Inc., New York, 1984, ch. 26, pp. 711–719 Search PubMed.
- K. L. Law and R. Narayan, Nat. Rev. Mater., 2022, 7, 104–116 CrossRef CAS.
- Q. Hou, M. Zhen, H. Qian, Y. Nie, X. Bai, T. Xia, M. Laiq Ur Rehman, Q. Li and M. Ju, Cell Rep. Phys. Sci., 2021, 2, 100514 CrossRef CAS.
- J. A. Ivar do Sul and M. Labrenz, in Handbook of Microplastics in the Environment, ed. T. Rocha-Santos, M. Costa and C. Mouneyrac, Springer International Publishing, Cham, 2020, pp. 1–16, DOI:10.1007/978-3-030-10618-8_25-2.
- I. Dimante-Deimantovica, S. Saarni, M. Barone, N. Buhhalko, N. Stivrins, N. Suhareva, W. Tylmann, A. Vianello and J. Vollertsen, Sci. Adv., 2024, 10, eadi8136 CrossRef CAS PubMed.
- A. J. Martín, C. Mondelli, S. D. Jaydev and J. Pérez-Ramírez, Chem, 2021, 7, 1487–1533 Search PubMed.
- M. Burelo, A. Martínez, J. D. Hernández-Varela, T. Stringer, M. Ramírez-Melgarejo, A. Y. Yau, G. Luna-Bárcenas and C. D. Treviño-Quintanilla, Molecules, 2024, 29, 387 CrossRef CAS PubMed.
- B. Liu, Z. Westman, K. Richardson, D. Lim, A. L. Stottlemyer, T. Farmer, P. Gillis, V. Vlcek, P. Christopher and M. M. Abu-Omar, ACS Sustain. Chem. Eng., 2023, 11, 6114–6128 CrossRef CAS.
- A. Kemona and M. Piotrowska, Polymers, 2020, 12, 1752 CrossRef CAS PubMed.
- M. B. Johansen, B. S. Donslund, E. Larsen, M. B. Olsen, J. A. L. Pedersen, M. Boye, J. K. C. Smedsgård, R. Heck, S. K. Kristensen and T. Skrydstrup, ACS Sustain. Chem. Eng., 2023, 11, 10737–10745 CrossRef CAS.
- M. Grdadolnik, A. Drinčić, A. Oreški, O. C. Onder, P. Utroša, D. Pahovnik and E. Žagar, ACS Sustain. Chem. Eng., 2022, 10, 1323–1332 CrossRef CAS PubMed.
- T. Köhler, A. Gutacker and E. Mejía, Org. Chem. Front., 2020, 7, 4108–4120 RSC.
- R. Müller, J. Chem. Educ., 1965, 42, 41 CrossRef.
- E. G. Rochow, Organosilicon halides, US Pat. US2380995, General Electric Co., 1945.
- E. G. Rochow, J. Am. Chem. Soc., 1945, 67, 963–965 CrossRef CAS.
- Y. Zhang, J. Li, H. Liu, Y. Ji, Z. Zhong and F. Su, ChemCatChem, 2019, 11, 2757–2779 CrossRef CAS.
- M. G. Voronkov, Glass Phys. Chem., 2009, 35, 231–236 CrossRef CAS.
- D. Pakuła, B. Marciniec and R. E. Przekop, AppliedChem, 2023, 3, 89–109 CrossRef.
- D.-G. Yu, B.-J. Li and Z.-J. Shi, Acc. Chem. Res., 2010, 43, 1486–1495 CrossRef CAS PubMed.
- R. Parthasarathi, R. A. Romero, A. Redondo and S. Gnanakaran, J. Phys. Chem. Lett., 2011, 2, 2660–2666 CrossRef CAS.
- L. Pietrelli, S. Ferro, A. P. Reverberi and M. Vocciante, Chemosphere, 2021, 273, 129725 CrossRef CAS PubMed.
- N. Jonkers and P. de Voogt, in Comprehensive Analytical Chemistry, ed. T. P. Knepper, D. Barcelo and W. P. de Voogt, Elsevier, 2003, vol. 40, pp. 719–747 Search PubMed.
- J. Liu, L. Zhang, G. Lu, R. Jiang, Z. Yan and Y. Li, Ecotoxicol. Environ. Saf., 2021, 208, 111481 CrossRef CAS PubMed.
- J. R. Rochester, Reprod. Toxicol., 2013, 42, 132–155 CrossRef CAS PubMed.
- L. S. Yang and D. A. Macarevich, Preparation of glycol diesters from polyethers, US Pat. US5254723A, ARCO Cemichal Technology, 1993.
- S. Enthaler and A. Trautner, ChemSusChem, 2013, 6, 1334–1336 CrossRef CAS PubMed.
- S. Enthaler, J. Appl. Polym. Sci., 2014, 131, 39791 CrossRef.
- S. Enthaler and M. Weidauer, Chem.–Eur. J., 2012, 18, 1910–1913 CrossRef CAS PubMed.
- S. Enthaler, Catal. Lett., 2014, 144, 850–859 CrossRef CAS.
- S. Enthaler and M. Weidauer, ChemSusChem, 2012, 5, 1195–1198 CrossRef CAS PubMed.
- S. Enthaler, Eur. J. Lipid Sci. Technol., 2012, 115, 239–245 CrossRef.
- Z. Maeno, S. Yamada, T. Mitsudome, T. Mizugaki and K. Jitsukawa, ChemistrySelect, 2016, 1, 201–204 CrossRef CAS.
- Z. Maeno, S. Yamada, T. Mitsudome, T. Mizugaki and K. Jitsukawa, Green Chem., 2017, 19, 2612–2619 RSC.
- Z. Maeno, K. Midogochi, T. Mitsudome, T. Mizugaki and K. Jitsukawa, Tetrahedron Lett., 2018, 59, 832–835 CrossRef CAS.
- Z. Maeno, H. Torii, S. Yamada, T. Mitsudome, T. Mizugaki and K. Jitsukawa, RSC Adv., 2016, 6, 89231–89233 RSC.
- A. Ahrens, A. Bonde, H. Sun, N. K. Wittig, H. C. D. Hammershoj, G. M. F. Batista, A. Sommerfeldt, S. Frolich, H. Birkedal and T. Skrydstrup, Nature, 2023, 617, 730–737 CrossRef CAS PubMed.
- B. Feng, Y. Jing, Y. Guo, X. Liu and Y. Wang, Green Chem., 2021, 23, 9640–9645 RSC.
- Y. Minami, N. Matsuyama, Y. Takeichi, R. Watanabe, S. Mathew and Y. Nakajima, Commun. Chem., 2023, 6, 14 CrossRef CAS PubMed.
- E. Palazzi, C. Caviglione, A. P. Reverberi and B. Fabiano, Process Saf. Environ. Prot., 2017, 110, 89–101 CrossRef CAS.
- J. F. Rabek, J. Lucki, B. J. Qu and W. F. Shi, Macromolecules, 1991, 24, 836–843 CrossRef CAS.
- K. Motokura, N. Nakagiri, T. Mizugaki, K. Ebitani and K. Kaneda, J. Org. Chem., 2007, 72, 6006–6015 CrossRef CAS PubMed.
- T. Mitsudome, T. Matsuno, S. Sueoka, T. Mizugaki, K. Jitsukawa and K. Kaneda, Green Chem., 2012, 14, 610–613 RSC.
- T. Mizugaki, R. Arundhathi, T. Mitsudome, K. Jitsukawa and K. Kaneda, ACS Sustain. Chem. Eng., 2014, 2, 574–578 CrossRef CAS.
- C. Sanchez, B. Julián, P. Belleville and M. Popall, J. Mater. Chem., 2005, 15, 3559–3592 RSC.
- E. Finocchio, I. Baccini, C. Cristiani, G. Dotelli, P. Gallo Stampino and L. Zampori, J. Phys. Chem. A, 2011, 115, 7484–7493 CrossRef CAS PubMed.
- Y. Deng, J. B. Dixon and G. N. White, Colloid Polym. Sci., 2005, 284, 347–356 CrossRef.
- J. Yang, P. S. White and M. Brookhart, J. Am. Chem. Soc., 2008, 130, 17509–17518 CrossRef CAS PubMed.
- L. Monsigny, J.-C. Berthet and T. Cantat, ACS Sustain. Chem. Eng., 2018, 6, 10481–10488 CrossRef CAS.
- E. Feghali and T. Cantat, ChemSusChem, 2015, 8, 980–984 CrossRef CAS PubMed.
- A. Ben-Haida, I. Baxter, H. M. Colquhoun, P. Hodge, F. H. Kohnke and D. J. Williams, Chem. Commun., 1997, 1533–1534, 10.1039/A703854B.
- J. S. Hrkach and K. Matyjaszewski, Macromolecules, 1990, 23, 4042–4046 CrossRef CAS.
- X. Zhang, Y. Sun, C. Zhang and X. Zhang, CCS Chem., 2022, 1–9, DOI:10.31635/ccschem.022.202202072.
- Y. Wang, Y. Hou and H. Song, Polym. Degrad. Stab., 2017, 144, 17–23 CrossRef CAS.
- K. Leszczyńska, A. Mix, R. J. F. Berger, B. Rummel, B. Neumann, H.-G. Stammler and P. Jutzi, Angew. Chem., Int. Ed., 2011, 50, 6843–6846 CrossRef PubMed.
- N. Ansmann, T. Thorwart and L. Greb, Angew. Chem., Int. Ed., 2022, 61, e202210132 CrossRef CAS PubMed.
- F. Tschernuth, L. Bichlmaier and S. Inoue, ChemCatChem, 2023, 15, e202300281 CrossRef CAS.
- Y. Jing, L. Dong, Y. Guo, X. Liu and Y. Wang, ChemSusChem, 2020, 13, 4181–4198 CrossRef CAS PubMed.
- U. Sanyal, Y. Song, N. Singh, J. L. Fulton, J. Herranz, A. Jentys, O. Y. Gutiérrez and J. A. Lercher, ChemCatChem, 2018, 11, 575–582 CrossRef.
- K. R. Mulcahy, A. F. R. Kilpatrick, G. D. J. Harper, A. Walton and A. P. Abbott, Green Chem., 2022, 24, 36–61 RSC.
- J. E. Mark, R. West and H. R. Allcock, in Inorganic Polymers, Oxford University Press, New York, 2nd edn, 2005, pp. 154–199 Search PubMed.
- R. N. Meals, Ann. N. Y. Acad. Sci., 1965, 125, 137–146 CrossRef CAS PubMed.
- S. Geier, H. Schmitz, U. Göschel, P. Eyerer, A. Ostrowicki, N. Woicke, C. Ulrich, W. Lutz, J. Eschl, G. Rüb, M. Keuerleber, J. Diemert, J. Hauk, A. Stieneker, J. Woidasky, I. Fischer, K. Kretschmer, L. Ober, C. Kohlert, S. Ganslmeier, C. Schlade, H. Schüle, K. Kurz, K. U. Tönnes, P. Elsner, R. Protte, D. Liebing, A. Rodríguez, S. R. Raisch, H.-J. Dern, S. Schlünken, R. Bräuning and A. König, in Kunststoffe: Eigenschaften und Anwendungen, ed. P. Elsner, P. Eyerer and T. Hirth, Springer, Berlin, Heidelberg, 2012, ch. 2, pp. 115–1201, DOI:10.1007/978-3-642-16173-5_2.
- A. Tomanek, Silicones & Industry: A Compendium for Practical Use, Instruction and Reference, Wacker-Chemie, 1991 Search PubMed.
- D. T. Hurd, R. C. Osthoff and M. L. Corrin, J. Am. Chem. Soc., 1954, 76, 249–252 CrossRef CAS.
- D. T. Hurd, J. Am. Chem. Soc., 1955, 77, 2998–3001 CrossRef CAS.
- J. F. Hyde, Polymerization of siloxanes, US Pat. US2490357A, Corning Inc, 1949.
- S. W. Kantor, W. T. Grubb and R. C. Osthoff, J. Am. Chem. Soc., 1954, 76, 5190–5197 CrossRef CAS.
- M. Mazurek and J. Chojnowski, Makromol. Chem., 1977, 178, 1005–1017 CrossRef CAS.
- H. R. Allok, J. Macromol. Sci., Part A: Pure Appl.Chem., 1970, 4(2), 149–189 CrossRef.
- A. S. Shapatin, E. A. Simanenko, G. Y. Zhigalin, A. G. Trufanov and I. M. Golyaeva, Process for producing organocyclosiloxanes, SU939445A1, Predpriyatie P/Y G-4236, 1982.
- T. D. Wilford, Depolymerization agents and depolymerization processes for silicones, DE4300168A1, Penn White Ltd, 1994.
- C. Allandrieu and D. Cardinaud, Process for the production of cyclosiloxanes by depolymerization of polysiloxanes, DE19619002A1, Rhodia Chimie SAS, 1996.
- J. S. Razzano, Cracking of fluorosilicones to produce 1,3,5-tris (3,3,3-trifluoropropyl)-1,3,5-trimethylcyclotrisiloxane, US Pat. US6037486, General Electric Company, 2000.
- H.-H. Moretto, M. Schulze and G. Wagner, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2000, DOI:10.1002/14356007.a24_057.
- D. Wang, J. Klein and E. Mejía, Chem.–Asian J., 2017, 12, 1180–1197 CrossRef CAS PubMed.
- F.-H. Kreuzer and H. Gebauer, Process for the preparation of hexamethyl cyclotrisiloxane and an application of the cyclotrisiloxane so prepared, DE3366928D1, Consortium fuer Elektrochemische Industrie GmbH, 1984.
- W. Knies, G. Vogl and V. Frey, Verfahren zur Pyrolyse von Silikonkautschukvulkanisaten, DE4126319 A1, Wacker Chemie AG, 1993.
- T. Bunce and A. E. Surgenor, Process for making cyclic organotrisiloxanes, GB2331992A, Dow Corning Corp, 1999.
- M. A. Brook, S. Zhao, L. Liu and Y. Chen, Can. J. Chem., 2012, 90, 153–160 CrossRef CAS.
- N. Grassie and I. G. Macfarlane, Eur. Polym. J., 1978, 14, 875–884 CrossRef CAS.
- N. Grassie, I. G. Macfarlane and K. F. Francey, Eur. Polym. J., 1979, 15, 415–422 CrossRef CAS.
- N. Grassie and K. F. Francey, Polym. Degrad. Stab., 1980, 2, 53–66 CrossRef CAS.
- T. H. Thomas and T. C. Kendrick, J. Polym. Sci., Part A-2, 1970, 8, 1823–1830 CrossRef CAS.
- G. Camino, S. M. Lomakin and M. Lazzari, Polymer, 2001, 42, 2395–2402 CrossRef CAS.
- G. Camino, S. M. Lomakin and M. Lageard, Polymer, 2002, 43, 2011–2015 CrossRef CAS.
- B. Rupasinghe and J. C. Furgal, Polym. Int., 2022, 71, 521–531 CrossRef CAS.
- R. H. Krieble and J. R. Elliott, J. Am. Chem. Soc., 1946, 68, 2291–2294 CrossRef CAS.
- R. Müller, R. Köhne and S. Sliwinski, J. Prakt. Chem., 1959, 9, 63–70 CrossRef.
- S. W. Kantor, W. T. Grubb and R. C. Osthoff, J. Am. Chem. Soc., 1954, 76, 5190–5197 CrossRef CAS.
- J. F. Hyde and G. J. Kookootsedes, Process for the recovery of organosiloxane polymers from silicone rubber material, DE954455C, Dow Corning Corp, 1956.
- C. J. Miller and W. P. Ryan, Cyclopolysiloxanes containing silanic hydrogen, US Pat. US3714213, General Electric Co., 1973.
- J. Burkhardt and E. Louis, Process for preparing cyclic dimethylpolysiloxanes, US Pat. US4276425A, Wacker Chemie AG, 1981.
- J. Burkhardt and A. B. A. Pressl, Process for the production of methylpolysiloxanes, DE4235877A1, Wacker Chemie AG, 1994.
- J. V. Crivello and J. L. Lee, Method for making cyclic poly(siloxanes), US Pat. US4895967, General Electric Co., 1990.
- T. W. Greenlee, Process for recycling silicone scrap and its products, US Pat. US5110972, Tremco Inc., 1992.
- H. Katsuragi and K. Mori, Solution for dissolving and removing cured material of siloxane resin, JPH04318075A, Kanto Chemical Co Inc, 1992.
- W. Knott, H. Dudzik and D. Schaefer, Upcycling process for processing silicone wastes, US Pat. US20220119617A1, Evonik Operations GmbH, 2022.
- W. Knott, H. Dudzik and D. Schaefer, Process for recycling silicones, US Pat. US11286366B2, Evonik Operations GmbH, 2022.
- W. Knott, H. Dudzik and D. Schaefer, Upcycling method for processing silicone wastes, EP3985055, Evonik Operations GmbH, 2022.
- W. Knott and H. Dudzik, Process for producing endcapped, liquid siloxanes from silicone wastes, US Pat. US20220348721A1, Evonik Operations GmbH, 2022.
- B. J. Humphrey and H. H. Wasserman, Liquefaction of silicone rubber gum and products thereof, US Pat. US2673843A, Connecticut Hard Rubber Co, 1954.
- T. D. Wilford, Depolymerization agent and method of depolymerizing silicones, EP0678110A1, Penn White Ltd, 1995.
- E. Tabei and M. Fukushima, Processing method for reuse of waste silicone hardened matter, JP2002348407A, Shin Etsu Chemical Co Ltd, 2002.
- S. Wu, Process for preparing cyclic siloxane by using silicone rubber leftover materials, CN100427491C, Wu, Shiwei, 2008.
- L. G. Dai and L. Tang, Silicon rubber waste cracking, recycling and reusing method, CN109233285A, Yangzhong Huifeng Packing Co Ltd, 2019.
- J. Chen, Technology for preparing dimethylcyclosiloxane by performing acid-method inner depolymerization on organosilicon waste, CN103936784A, Dongzhi Oasis Environmental Protection Chemical Co Ltd, 2014.
- J. Zhao and M. Wang, Method for producing organosilicone monomer from waste silicone rubber, CN106633177A, Yangzhou Hongyuan Chemical Industry New Material Co Ltd, 2017.
- W. Knies, G. Vogl and W. Guske, Depolymerisation of addn.-cured silicone rubber vulcanisate at low cost, DE19502393A1, Wacker Chemie AG, 1996.
- J. N. Kostas, Process for preparing cyclic polysiloxanes from linear polysiloxanes, US Pat. US5491249, Hercules Inc., 1996.
- T. Hirose, R. Tsuji and K. Ouchi, Method for decomposing polysiloxane, US Pat. US5504235A, Kaneka Corp, 1996.
- R. Tsuji, Recycling of silicone compound, JPH09176364A, Kanegafuchi Chemical Industry Co Ltd, 1997.
- T. Kawamoto, Process for recycling silicone compounds, US Pat. US6172253B1, Yazaki Corp, 2001.
- F. Oishi, M. Fukawa and F. Niiguni, Method for regenerating silicone oil/silicone grease from used silicone rubber, JP2005307119A, SHINKO GIKEN KK, 2005.
- J. F. Hyde, Polymerization of organosiloxanes, US Pat. US2634284A, Dow Corning Corp, 1953.
- M. J. Hunter, J. F. Hyde, E. L. Warrick and H. J. Fletcher, J. Am. Chem. Soc., 1946, 68, 667–672 CrossRef CAS PubMed.
- A. R. Gilbert and S. W. Kantor, J. Polym. Sci., 1959, 40, 35–58 CrossRef CAS.
- C. Allandrieu and D. Cardinaud, Process for the manufacture of cyclosiloxanes by depolymerization of polysiloxanes, US Pat. US5670689A, Rhodia Chimie SAS, 1997.
- J.-K. Yang, J.-R. Han, J.-B. Shin and J.-J. Hong, Process for decomposing siloxane bond-containing compound, US Pat. US5892087A, 1999.
- S. R. Mukherjee and A. K. Paul, Process for the recovery of methyl polysiloxanes in the form of cyclosiloxanes, US Pat. US6187827B1, Soumitra Ranjan Mukherjee, 2001.
- A. Oku, W. Huang and Y. Ikeda, Polymer, 2002, 43, 7289–7293 CrossRef CAS.
- W. Huang, Y. Ikeda and A. Oku, Polymer, 2002, 43, 7295–7300 CrossRef CAS.
- Y. Ikeda, W. Huang and A. Oku, Green Chem., 2003, 5, 508–511 RSC.
- G. G. Sik, Reclaimed silicone oil using waste silane hydrolysate, process for preparing them same and uses of the same, KR100953734B1, Silan Tech Corp Co Ltd, 2010.
- P. Eyster, J. Bratina, T. Roberts and C. Peregrine, Method of processing silicone wastes, US Pat. US20090281202A1, Heritage Research Group LLC, 2009.
- G. Zhao, Process for producing silicon rubber by using waste silicon rubber materials, CN101649050A, Yangzhou Hongyuan Chemical Industry New Material Co Ltd, 2010.
- Y. Xu, J. Jinbo, Z. Rong, L. Fei and H. Hengchao, High-efficiency cracking recovery method for silicone rubber waste, CN111040243B, Guangzhou Baiyun Chemical Industry Co Ltd, 2020.
- Y. C. Hsiao, L. W. Hill and S. P. Pappas, J. Appl. Polym. Sci., 1975, 19, 2817–2820 CrossRef.
- S. P. Pappas and R. L. Just, J. Polym. Sci., Polym. Chem. Ed., 1980, 18, 383–761 CrossRef.
- K. H. Schimmel, Acta Polym., 1987, 38, 495–498 CrossRef CAS.
- K. H. Schimmel and J. Schulz, Acta Polym., 1987, 38, 536–538 CrossRef CAS.
- K. H. Schimmel, E. Schröder, J. Schulz and T. Souvimonh, Acta Polym., 1988, 39, 310–314 CrossRef CAS.
- M. Heidingfeldova, P. Hron and M. Schatz, Reclamation of siloxanes, GB2015550A, Vysoka Skola Chemicko Technologicka V Praze, 1979.
- C. L. Chang, H. S. Lee and C. K. Chen, Polym. Degrad. Stab., 1999, 65, 1–4 CrossRef CAS.
- C. L. Chang and Y. K. Lin, Polym. Degrad. Stab., 2005, 87, 207–211 CrossRef CAS.
- C.-L. Chang, H. S.-J. Lee and C.-K. Chen, J. Polym. Res., 2005, 12, 433–438 CrossRef CAS.
- T. Cho, Y. Ohta, T. Cho, T. Yamashita, N. Ohkawa and M. Nishida, Process for recovering organoalkoxysilane from polyorganosiloxane, US Pat. US5783609A, Tama Chemicals Co Ltd, Toshiba Silicone Co Ltd, 1998.
- R. Petrus, J. Utko, R. Gniłka, M. G. Fleszar, T. Lis and P. Sobota, Macromolecules, 2021, 54, 2449–2465 CrossRef CAS.
- S. Boileau and N. Illy, Prog. Polym. Sci., 2011, 36, 1132–1151 CrossRef CAS.
- P. C. Hupfield, R. A. Kneale, D. C. Miller, S. O'Hare, A. Surgenor and R. G. Taylor, Method for the Production of Cyclic Polysiloxanes, US Pat., US20100179289A1, 2008 Search PubMed.
- D. Z. Khedaioui, C. Tribout, J. Bratasanu, F. D'Agosto, C. Boisson and D. Montarnal, Angew. Chem., Int. Ed., 2023, 62, e202300225 CrossRef CAS PubMed.
- R. F. Weitkamp, B. Neumann, H.-G. Stammler and B. Hoge, Angew. Chem., Int. Ed., 2019, 58, 14633–14638 CrossRef CAS PubMed.
- R. F. Weitkamp, B. Neumann, H.-G. Stammler and B. Hoge, Angew. Chem., Int. Ed., 2019, 131, 14775–14780 CrossRef.
- R. F. Weitkamp, B. Neumann, H.-G. Stammler and B. Hoge, Angew. Chem., Int. Ed., 2020, 59, 5494–5499 CrossRef CAS PubMed.
- N. D. Vu, A. Boulègue-Mondière, N. Durand, J. Raynaud and V. Monteil, Green Chem., 2023, 25, 3869–3877 RSC.
- J. L. Torgunrud, A. M. Reverón Pérez, E. B. Spitzberg and S. A. Miller, Macromolecules, 2023, 56, 3668–3678 CrossRef CAS.
- M. G. Vornokov and Z. I. Shabarova, J. Gen. Chem. USSR, 1959, 29, 1501 Search PubMed.
- D. L. Bailey and F. M. O'connor, Process for producing alkoxysilicon compounds, US Pat. US2881199A, Union Carbide Corp, 1959.
- R. Tsuji, K. Ouchi and T. Hirose, Production of alkoxysilane, JPH07102067A, Kanegafuchi Chemical Industry Co Ltd, 1995.
- R. N. Lewis, Preparation of organo-substituted halogenosilanes, US Pat. US2500761A, General Electric Co, 1950.
- E. P. Micheev and G. N. Malnova, J. Prakt. Chem., 1964, 23, 206–207 CrossRef CAS.
- M. Lefort, Preparation of chlorosilanes from disiloxanes, US Pat. US3689519A, Rhone Poulenc SA, 1972.
- M. Takeuchi, T. Ishihara, T. Kubota and M. Endo, Production of organomonochlorosilatne, JPH05222062A, Shin Etsu Chemical Co Ltd, 1993.
- J. F. Hyde, Method of preparation of trimethylchlorosilane, US Pat. US2615034A, Dow Corning Corp, 1952.
- H.-J. Kotzsch and H.-J. Vahlensieck, Method of cleaving organosiloxanes, US Pat. US4417067A, Dynamit Nobel AG, 1983.
- I. P. Zhurkina, E. P. Nedogrei, R. S. Musavirov and D. L. Rakhmankulov, J. Gen. Chem. USSR, 1989, 59, 1066 Search PubMed.
- M. Okamoto, K. Miyazaki, A. Kado and E. Suzuki, Chem. Commun., 2001, 1838–1839, 10.1039/b104371b.
- M. Okamoto, K. Miyazaki, A. Kado, S. Suzuki and E. Suzuki, Catal. Lett., 2003, 88, 115–118 CrossRef CAS.
- M. Okamoto, S. Suzuki and E. Suzuki, Appl. Catal., A, 2004, 261, 239–245 CrossRef CAS.
- T. Kawamoto, Reutilization of silicone compound as resource, JP2000169484A, Yazaki Corp, 2000.
- U. Scheim, A. Porzel and K. Rühlmann, J. Organomet. Chem., 1988, 354, 31–37 CrossRef CAS.
- P. Döhlert, J. Pfrommer and S. Enthaler, Phosphorus, Sulfur, and Silicon and the Related Elements, 2016, vol. 191, pp. 1189–1193 Search PubMed.
- P. Döhlert, J. Pfrommer and S. Enthaler, ACS Sustain. Chem. Eng., 2015, 3, 163–169 CrossRef.
- P. Doehlert and S. Enthaler, J. Appl. Polym. Sci., 2015, 132, 42814 CrossRef.
- P. Döhlert and S. Enthaler, Catal. Lett., 2016, 146, 345–352 CrossRef.
- D. J. Krug, M. Z. Asuncion and R. M. Laine, ACS Omega, 2019, 4, 3782–3789 CrossRef CAS PubMed.
- B. Rupasinghe and J. C. Furgal, ACS Appl. Polym. Mater., 2021, 3, 1828–1839 CrossRef CAS.
- J. C. Furgal, T. Goodson Iii and R. M. Laine, Dalton Trans., 2016, 45, 1025–1039 RSC.
- S. Enthaler, J. Appl. Polym. Sci., 2015, 132, 41281–41288 CrossRef.
- S. Enthaler and R. Kretschmer, ChemSusChem, 2014, 7, 2030–2036 CrossRef CAS PubMed.
- M. Weidauer, B. Heyder, D. Woelki, M. Tschiersch, A. Köhler-Krützfeldt and S. Enthaler, Resour.-Effic. Technol., 2015, 1, 73–79 Search PubMed.
- M. Weidauer, B. Heyder, D. Woelki, M. Tschiersch, A. Koehler-Kruetzfeldt and S. Enthaler, Eur. J. Lipid Sci. Technol., 2015, 117, 778–785 CrossRef CAS.
- C. Daeschlein and C. Strohmann, Z. Naturforsch., B: J. Chem. Sci., 2009, 64b, 1558–1566 CrossRef.
- C. Daeschlein, J. O. Bauer and C. Strohmann, Angew. Chem., Int. Ed., 2009, 48, 8218–8221 CrossRef.
- S. Enthaler, Angew. Chem., Int. Ed., 2014, 53, 2716–2721 CrossRef CAS PubMed.
- M. R. Alexander, F. S. Mair, R. G. Pritchard and J. E. Warren, Appl. Organomet. Chem., 2003, 17, 730–734 CrossRef CAS.
- A. W. Apblett and A. R. Barron, Organometallics, 1990, 9, 2137–2141 CrossRef CAS.
- R. Mulhaupt, J. Calabrese and S. D. Ittel, Organometallics, 1991, 10, 3403–3406 CrossRef CAS.
- F. Vidal, E. R. van der Marel, R. W. F. Kerr, C. McElroy, N. Schroeder, C. Mitchell, G. Rosetto, T. T. D. Chen, R. M. Bailey, C. Hepburn, C. Redgwell and C. K. Williams, Nature, 2024, 626, 45–57 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |