
Facile synthesis of asymmetric molecular brushes with triple side chains using a multivalent monomer strategy†
Shiya
Cao‡
,
Wenyi
Liu‡
,
Bicai
Yang
,
Yuan
Zheng
,
Shaoliang
Lin
 * and
Binbin
Xu
*
* and
Binbin
Xu
*
Shanghai Key Laboratory of Advanced Polymeric Materials, Key Laboratory for Ultrafine Materials of Ministry of Education, School of Materials Science and Engineering, East China University of Science and Technology, 130 Meilong Road, Shanghai 200237, People's Republic of China. E-mail: slin@ecust.edu.cn; binbinxu@ecust.edu.cn
First published on 8th August 2024
Abstract
Heterografted molecular brushes (HMBs) bearing two or more types of side chains have attracted tremendous attention because of their asymmetric structures and multiple functionalities. However, the controlled synthesis of HMBs with three types of side chains remains a challenge. The structure–property–application correlations of HMBs with three types of side chains are still not clear. Herein, based on the multivalent monomer strategy, we report the synthesis of well-defined asymmetric HMBs comprising three different side chains by sequential reversible addition–fragmentation chain transfer (RAFT) polymerization, atom transfer radical polymerization (ATRP), thiol–epoxy coupling reaction, and ring-opening polymerization (ROP). PA-g-PAzo/PEG/PLA with a polyacrylate (PA) backbone, hydrophobic poly(azobenzene-methacrylate) (PAzo) brush, hydrophilic polyethylene glycol (PEG) brush, and hydrophobic polylactide (PLA) brush was synthesized and characterized. The self-assembly behavior of PA-g-PAzo/PEG/PLA with a short backbone and relatively strong intermolecular association in solutions was investigated. Well-defined platelets with tunable morphologies were constructed. Subsequently, PA-g-PFA/PEG/PLA with a fluorophilic poly(pentafluoropropyl acrylate) (PFA) brush, hydrophilic PEG brush, and lipophilic PLA brush was synthesized and further used as an efficient surfactant for the stabilization of different emulsions. Our study offers a platform for exploring the unique properties of asymmetric HMBs.
Introduction
The synthesis of complex polymers with different topological structures and diverse functionalities is of great importance to establish architecture–property relationships and to create advanced polymeric materials with desirable properties.1–5 Aside from the more established linear polymers, molecular brushes (MBs), as one of the most accessible complex polymers, have garnered particularly extensive interest.6–9 MBs are a special class of branched copolymers with dense side chains covalently grafted onto a linear polymeric backbone. Compared to linear copolymers, more variables of MBs, particularly the tunable degree of polymerization (DP) of the backbone and side chains and the grafting density, provide wide opportunities to control molecular conformation and features.10–12 In addition, due to the steric repulsion between dense side chains, MBs exhibit a variety of unique properties (e.g., extended backbone, low entanglement of side chains, distinct phase-separated nanostructures, and unusual rheological and mechanical performance), which have empowered various applications, such as drug delivery, sensors, photonic crystals, supersoft elastomers, and anti-fouling materials.13–16With the development of controlled/living radical polymerization methods and highly efficient click reactions,17–19 pathways have been created that allow access to MBs with diverse structures.20 Recently, heterografted molecular brushes (HMBs) whose backbones are simultaneously grafted with two or more types of side chains have attracted significant attention because of their asymmetric structures and multiple functionalities.21–24 When endowed with an amphiphilic character, HMBs can act as efficient surfactants in biphasic systems and solute stabilizers and yield intriguing nanostructures.25,26 For instance, Cheng et al. synthesized amphiphilic HMBs PNB-g-PEG/PLA with each graft site bearing a hydrophilic poly(ethylene glycol) (PEG) graft and a hydrophobic polylactide (PLA) graft. PNB-g-PEG/PLA represented a new type of giant surfactant and could help stabilize miniemulsions by selective interactions of the heterografts with different phases.27 Herrera-Alonso et al. reported the preparation of amphiphilic HMBs PGMA-g-PEG/PLA with a poly(glycidyl methacrylate) (PGMA) backbone and PEG and PLA side chains. PGMA721-g-PEG45/PLA15 nanoparticles achieved high solute encapsulation capacity.28 Moreover, driven by hydrophobic interactions, PGMA-g-PEG/PLA with a short backbone and side chains self-assembled into well-defined compartmentalized spherical nanoparticles, long rods, and toroids.29 However, the controlled synthesis of HMBs with three types of side chains remains a challenge. The structure–property–application correlations of HMBs with three types of side chains are still not clear enough. Thus, it is highly desirable to develop a systematic method to synthesize HMBs with three types of side chains.
There are three grafting approaches for the synthesis of MBs: grafting-through,30,31 grafting-to,32,33 and grafting-from.34,35 The grafting-through method allows direct polymerization of macromonomers, which leads to the creation of MBs with a high density of side chains. However, this method is restricted to polynorbornene backbones obtained via ring-opening metathesis polymerization (ROMP). The grafting-to method involves coupling between reactive side chains and active sites on the backbone. The backbone and side chains can be produced separately with controlled structures and compositions. Nevertheless, the grafting density of side chains is often limited due to the steric hindrance. Gao and Matyjaszewski showed that this limitation could be overcome by the combination of thin linear poly(ethylene glycol)-N3 (PEG-N3) side chains and click reaction.36 The grafting-from approach introduces the side chains by initiating monomers from the active sites on the backbone. The steric crowding is maximally minimized during the graft-from process. To overcome the synthetic hurdle of HMBs, we have designed a functional monomer Br-acrylate-alkyne that contains an alkynyl group for click reaction and a 2-bromopropionate initiating group for atom transfer radical polymerization (ATRP). The combination of grafting-from and grafting-to methods based on the multivalent monomer strategy allows for the precise fabrication of HMBs with double-brushes.37–39 Thus, it is envisaged that the multivalent monomer strategy can be extended to the synthesis of HMBs with three types of side chains.
Herein, we report the synthesis of well-defined asymmetric HMBs comprising three different side chains based on a Br-acrylate-epoxide multivalent monomer by sequential reversible addition–fragmentation chain transfer (RAFT) polymerization, ATRP, thiol–epoxy coupling reaction, and ring-opening polymerization (ROP). The Br-acrylate-epoxide monomer combines an acrylate skeleton for RAFT polymerization, a 2-bromopropionate group for ATRP grafting reaction, an epoxy group for thiol–epoxy coupling reaction, and a potential hydroxy group for ROP grafting reaction (Scheme 1). PA-g-PAzo/PEG/PLA with a polyacrylate (PA) backbone, hydrophobic poly(azobenzene-methacrylate) (PAzo) brush, hydrophilic polyethylene glycol (PEG) brush, and hydrophobic polylactide (PLA) brush was synthesized and characterized. The self-assembly behavior of PA-g-PAzo/PEG/PLA with a short backbone and relatively strong intermolecular association (π–π interaction of PAzo brushes) in solutions was investigated. Subsequently, PA-g-PFA/PEG/PLA with a PA backbone, fluorophilic poly(pentafluoropropyl acrylate) (PFA) brush, hydrophilic PEG brush, and lipophilic PLA brush was synthesized and further used as a multifunctional surfactant for the stabilization of emulsions. The emulsifying performance of PA-g-PFA/PEG/PLA was compared with surfactants formed from the linear analogues. Our study presents an efficient platform for the facile synthesis of asymmetric HMBs with structural and functional control.
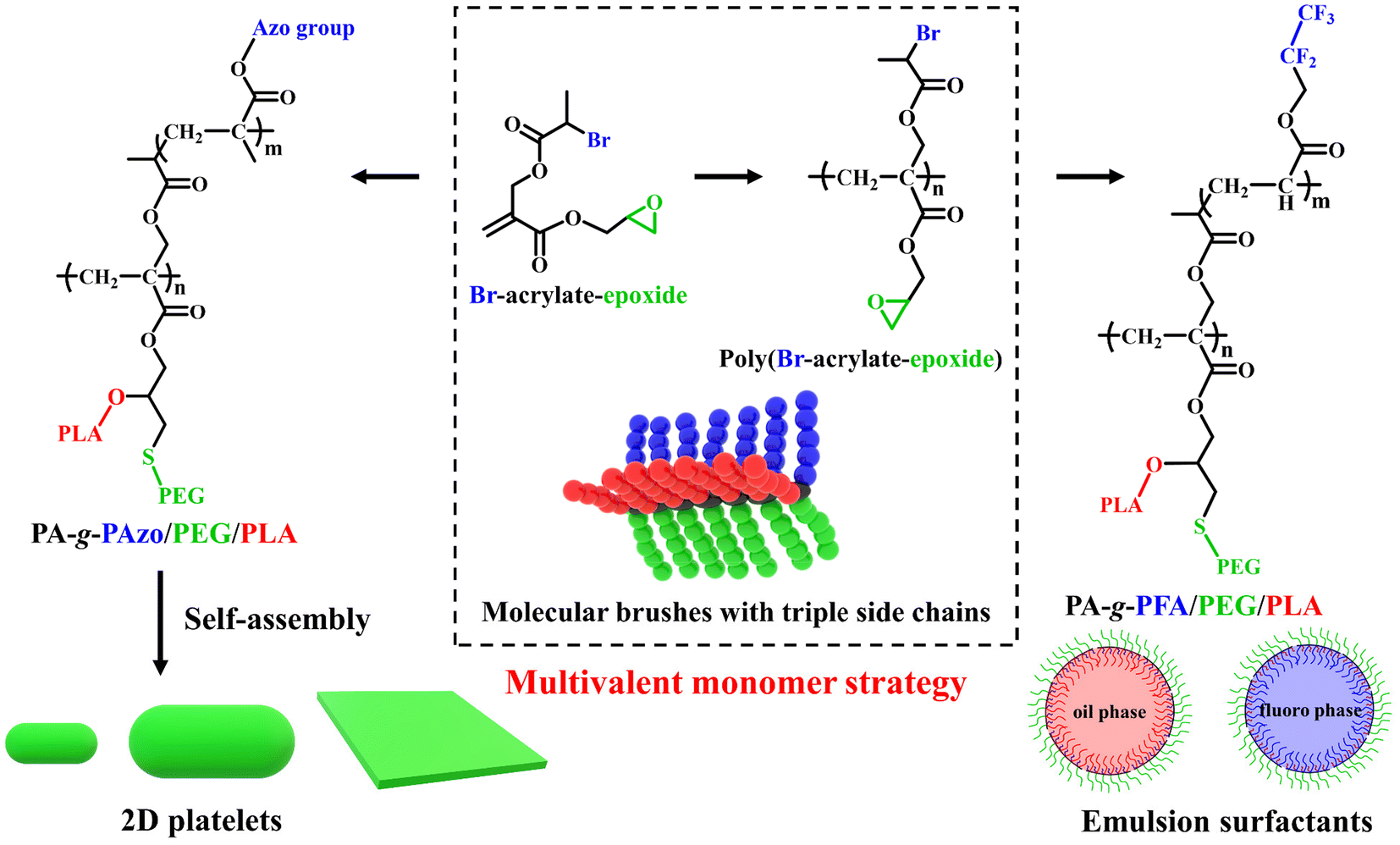 | ||
| Scheme 1 Chemical structures of the multivalent monomer Br-acrylate-epoxide, the macro-agent poly(Br-acrylate-epoxide), and the asymmetric molecular brushes with triple side chains PA-g-PAzo/PEG/PLA and PA-g-PFA/PEG/PLA. Schematic representations of the self-assembly behavior of PA-g-PAzo/PEG/PLA and the surfactant properties of PA-g-PFA/PEG/PLA. | ||
Materials and methods
Materials
2,2,3,3,3-Pentafluoropropyl acrylate (FA, Aladdin, 98%) was passed through a basic alumina column to remove the stabilizer and distilled under reduced pressure from CaH2 prior to use. Lactide (Aldrich, 99%) was distilled under reduced pressure from CaH2 prior to use. 2,2′-Azobis(isobutyronitrile) (AIBN, Aldrich, 98%) was recrystallized from anhydrous ethanol twice. Copper(I) bromide (CuBr, Aldrich, 98%) was purified by stirring overnight over CH3COOH at room temperature, followed by washing with ethanol, diethyl ether, and acetone prior to drying at 40 °C in vacuo for one day. Tetrahydrofuran (THF, Aldrich, 99.9%), toluene (Aldrich, 99.8%), and dichloromethane (DCM) were dried over CaH2 and distilled from sodium and benzophenone under N2 prior to use. Glycidol, poly(ethylene glycol) methyl ether thiol (PEG-SH, Mn ≈ 2000 g mol−1, Aldrich, 99%), 2,2′-bipyridyl (Macklin, 99%), and 1,1,4,7,10,10-hexamethyl-triethylenetetramine (HMTETA, Aldrich, 97%) were used as received.Synthesis of the Br-acrylate-epoxide functional monomer
tBA-Br (3 g, 10.23 mmol) and dry CH2Cl2 (100 mL) were added to a 250 mL round bottom flask. The solution was stirred at 0 °C for 30 min followed by adding TFA (35 g, 307 mmol) and the reaction mixture was warmed to 25 °C. After stirring at room temperature for 4 h, the solution was concentrated and a viscous solid was obtained after drying in vacuo. The crude product of 2-((2-bromopropanoyloxy)methyl) acrylic acid was used without further purification.2-((2-Bromopropanoyloxy)methyl) acrylic acid (2.97 g, 12.5 mmol), glycidol (1.86 g, 25.08 mmol), DMAP (0.15 g, 1.25 mmol), and 20 mL of DCM were first added to a 100 mL Schlenk flask (flame-dried under vacuum prior to use) sealed with a rubber septum and kept under N2. The reaction mixture was stirred at 0 °C for 20 min. A solution of DCC (5.17 g, 25.08 mmol) in 20 mL of DCM was added. The reaction mixture was warmed to 25 °C with stirring for 14 h. The reaction was quenched by adding NaCl aqueous solution. The aqueous phase was extracted with CH2Cl2, and all organic layers were merged. The combined organic extracts were washed with brine three times, dried over MgSO4, and concentrated. The residue was purified by silica column chromatography (eluent: ethyl acetate/hexane, v![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :v = 1:10) to afford Br-acrylate-epoxide. The 1H NMR, 13C NMR, and HRMS spectra of Br-acrylate-epoxide are shown in Fig. 1a, Fig. S1 and S2,† respectively. 1H NMR (CDCl3): δ (ppm): 1.87 (d, 3H, CH3CH), 2.66, 2.85 (dd, 2H, O–CH2–CH), 3.25 (m, 1H, –OCH–), 4.03, 4.51 (dd, 2H, –OCH2–), 4.41 (q, 1H, CHBr), 4.92 (ABq, 2H, CH2OCO), 5.97, 6.45 (dd, 2H, CH2
:v = 1:10) to afford Br-acrylate-epoxide. The 1H NMR, 13C NMR, and HRMS spectra of Br-acrylate-epoxide are shown in Fig. 1a, Fig. S1 and S2,† respectively. 1H NMR (CDCl3): δ (ppm): 1.87 (d, 3H, CH3CH), 2.66, 2.85 (dd, 2H, O–CH2–CH), 3.25 (m, 1H, –OCH–), 4.03, 4.51 (dd, 2H, –OCH2–), 4.41 (q, 1H, CHBr), 4.92 (ABq, 2H, CH2OCO), 5.97, 6.45 (dd, 2H, CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C). 13C NMR (125 MHz, CDCl3): δ (ppm): 22.3 (CH3CHBr), 39.8 (CH3CHBr), 44.7 (CH2CHOCH2), 49.3 (CH2CHOCH2), 63.6 (CH2CCH2O2C), 65.6 (CH2CCO2CH2), 129.7 (CH2C), 134.3 (CH2C), 164.7 (CH2CCO2CH2), 169.7 (CH2CCH2O2C). HRMS (m/z): calcd for C10H14BrO5 [M + H]+: 293.0025, found: 293.0027.
C). 13C NMR (125 MHz, CDCl3): δ (ppm): 22.3 (CH3CHBr), 39.8 (CH3CHBr), 44.7 (CH2CHOCH2), 49.3 (CH2CHOCH2), 63.6 (CH2CCH2O2C), 65.6 (CH2CCO2CH2), 129.7 (CH2C), 134.3 (CH2C), 164.7 (CH2CCO2CH2), 169.7 (CH2CCH2O2C). HRMS (m/z): calcd for C10H14BrO5 [M + H]+: 293.0025, found: 293.0027.
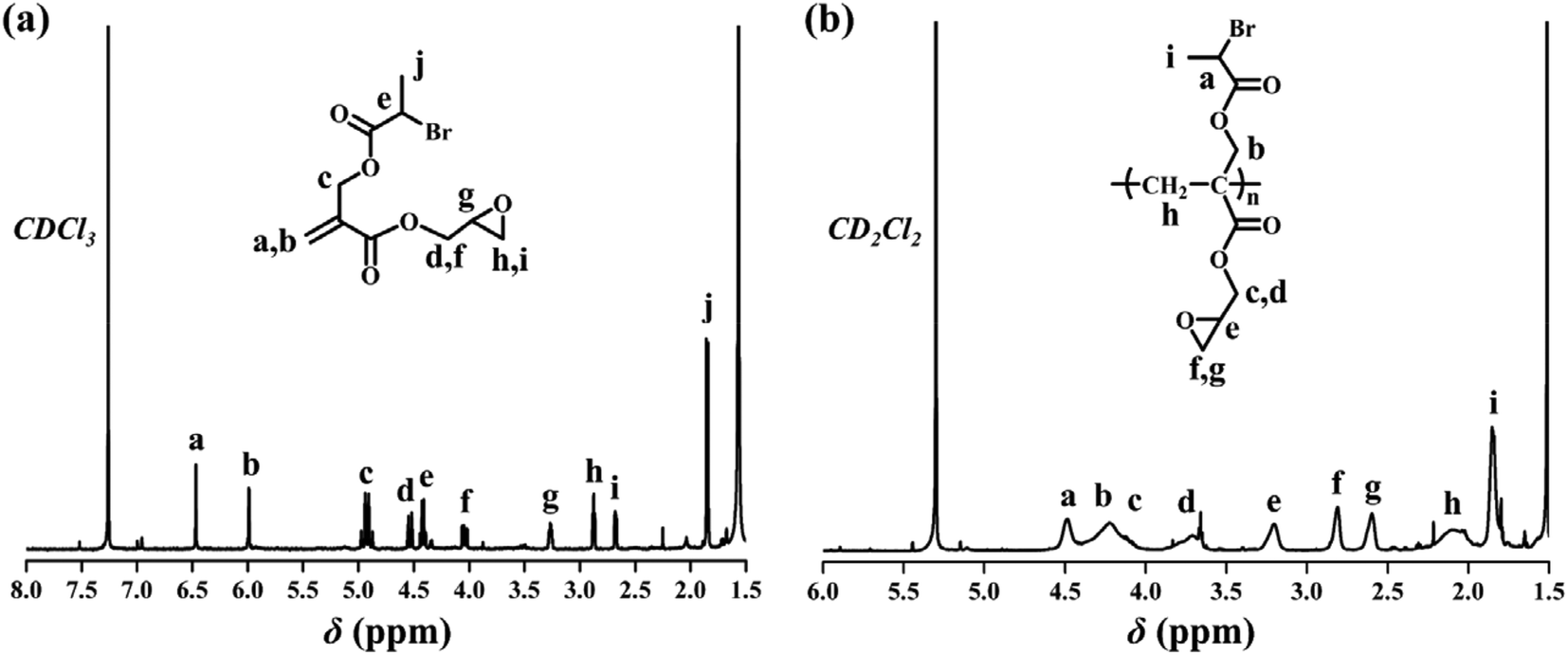 | ||
| Fig. 1 1H NMR spectra of (a) the Br-acrylate-epoxide functional monomer and (b) the poly(Br-acrylate-epoxide) macro-agent. | ||
RAFT homopolymerization of the Br-acrylate-epoxide monomer
CDB (14.0 mg, 0.051 mmol) and AIBN (3.0 mg, 0.017 mmol) were first added to a 10 mL Schlenk flask (flame-dried under vacuum prior to use) sealed with a rubber septum for degassing and kept under N2. Next, Br-acrylate-epoxide (0.88 g, 3.0 mmol) and dry toluene (1.0 mL) were added via a gastight syringe. The flask was degassed by three cycles of freeze–pump–thaw followed by immersing the flask in an oil bath set at 65 °C. The polymerization was terminated by immersing the flask in liquid N2 after 16 h. The solution was precipitated into cold n-hexane. The crude product was purified by repeated dissolution and precipitation followed by drying in vacuo overnight to give 0.47 g of poly(Br-acrylate-epoxide) functional macro-agent. The 1H NMR spectrum of poly(Br-acrylate-epoxide) is shown in Fig. 1b. According to the integration area of the peaks between 7.00 ppm and 8.00 ppm (CTA end group) and the peak at 3.20 ppm (–CO2CH in the Br-acrylate-epoxide repeat unit) in the 1H NMR spectrum, the average degree of polymerization of the poly(Br-acrylate-epoxide) backbone (n) was calculated to be 35. GPC: Mn = 9000 g mol−1, Mw/Mn = 1.19. 1H NMR (CD2Cl2): δ (ppm): 1.84 (3H, CH3CHBr), 2.03 (2H, CH2C), 2.59, 2.81 (2H, OCH2CH), 3.20 (1H, OCH2CH), 3.61–4.45 (2H, COOCH2CH; 2H, CH2CCH2O2C), 4.49 (1H, CH3CHBr).Synthesis of PA-g-PAzo/PEG/PLA heterografted molecular brushes
CuBr (14 mg, 0.10 mmol), Azo monomer (2.16 g, 5.11 mmol), and poly(Br-acrylate-epoxide) (14 mg, 0.05 mmol ATRP initiating group) were first added to a 50 mL Schlenk flask (flame-dried under vacuum prior to use) sealed with a rubber septum for degassing and kept under N2. Next, HMTETA (36 μL, 0.10 mmol) and 1,4-dioxane (15 mL) were charged via a gastight syringe. The flask was degassed by three cycles of freeze–pump–thaw followed by immersing the flask in an oil bath set at 80 °C. The polymerization and coupling reaction lasted 7 h and were terminated by immersing the flask in liquid N2. The mixture was diluted with THF and passed through an alumina column to remove the residual copper catalyst. The solution was concentrated and precipitated into a mixture of hexane/diethyl ether (v:v = 1:1). After repeated purification by dissolving in THF and precipitating in hexane/diethyl ether three times to completely remove the unreacted Azo monomer, 0.32 g of PA-g-PAzo was obtained after drying in vacuo overnight. GPC: Mn = 66400 g mol−1, Mw/Mn = 1.26. 1H NMR (CD2Cl2): δ (ppm): 0.88 (CH2CH2CH2CH3, CH3CHCO2, CH3CCO2), 1.35, 1.61, 1.73 (CH2CH2CH2CH3, CH2CCO2, OCH2CH2CH2CH2CH2CH2O), 2.61 (CH2CH2CH2CH3), 3.57–4.02 (OCH2CH2CH2CH2CH2CH2O, CH2CCO2CH2, CH2CCH2OCO), 6.85, 7.20, 7.75 (C6H4N2C6H4).
To a stirred and ice-cold solution of PA-g-PAzo (0.11 g, 0.03 mmol of epoxy group) and PEG-SH (0.18 g, 0.09 mmol) in THF (9 mL), LiOH (1.0 mg, 0.04 mmol) in water (0.5 mL) was added slowly under an argon atmosphere. The reaction mixture was stirred for 30 h at ambient temperature. THF was evaporated, the resulting solid was dried, and the crude polymer was dissolved in DCM and washed with water. The organic layer was dried, concentrated, and precipitated three times in methanol. In addition, the reaction mixture could also be dialyzed against methanol using a dialysis membrane (MW cut-off = 2.0 kDa) to remove excess PEG-SH until the dialysate did not show any detectable PEG-SH. The precipitate was collected and dried under vacuum to give 0.11 g of PA-g-PAzo/PEG. GPC: Mn = 109700 g mol−1, Mw/Mn = 1.27. 1H NMR (CD2Cl2): δ (ppm): 0.88 (CH2CH2CH2CH3, CH3CHCO2, CH3CCO2), 1.35, 1.61, 1.73 (CH2CH2CH2CH3, CH2CCO2, OCH2CH2CH2CH2 CH2CH2O), 2.61 (CH2CH2CH2CH3), 3.30 (OCH3), 3.57 (OCH2CH2), 3.63–4.02 (OCH2CH2CH2CH2CH2CH2O, CH2CCO2CH2, CH2CCH2OCO), 6.85, 7.20, 7.75 (C6H4N2C6H4).
PA-g-PAzo/PEG (20 mg, 0.003 mmol) and lactide (40 mg, 0.28 mmol) were loaded into a round-bottom flask, and placed under high vacuum at 35 °C for ∼5 h. After backfilling with argon, anhydrous DCM (1 mL) was added to dissolve the reagents. DBU (0.5 mg, 0.003 mmol) was then injected and the reaction was allowed to proceed for 1 h under argon at room temperature. The polymerization was quenched. DCM was removed under vacuum and the polymer was re-dissolved in THF, followed by precipitation into methanol. The precipitate was collected and dried under vacuum to give 0.05 g of PA-g-PAzo/PEG/PLA. GPC: Mn = 147600 g mol−1, Mw/Mn = 1.30. 1H NMR (CD2Cl2): δ (ppm): 0.88 (CH2CH2CH2CH3, CH3CHCO2, CH3CCO2), 1.35, 1.61, 1.73 (CH2CH2CH2CH3, CH2CCO2, OCH2CH2CH2CH2 CH2CH2O, OCHCH3), 2.61 (CH2CH2CH2CH3), 3.30 (OCH3), 3.57 (OCH2CH2), 3.63–4.02 (OCH2CH2CH2CH2 CH2CH2O, CH2CCO2 CH2, CH2CCH2OCO), 5.15 (OCHCH3), 6.85, 7.20, 7.75 (C6H4N2C6H4).
Results and discussion
Synthetic route for asymmetric HMBs with three types of side chains
In traditional grafting-from and grafting-to processes, less efficient transformations of polymeric functional groups are frequently used.40,41 By employing the multivalent monomer strategy, we can prepare asymmetric HMBs with three types of side chains without the need for unnecessary polymeric functionality transformations. In this study, a special functional monomer, Br-acrylate-epoxide, was synthesized in the first step (Scheme 1). Then, the poly(Br-acrylate-epoxide) macro-agent was prepared by RAFT homopolymerization of Br-acrylate-epoxide, which bears a Br-containing ATRP initiating group and an epoxy group. Two independent side chains could be attached to the backbone by sequential ATRP and thiol–epoxy coupling reactions. The thiol–epoxy coupling reaction also produced reactive hydroxyl groups on the backbone. Then, the third side chain could be obtained by ROP of LA initiated by the hydroxyl group.Synthesis of the Br-acrylate-epoxide monomer and poly(Br-acrylate-epoxide) macro-agent
The multivalent monomer Br-acrylate-epoxide was synthesized in four steps (Scheme 2). Based on commercially available tert-butyl acrylate (tBA), the intermediate of tBA-OH was synthesized via the Baylis–Hillman reaction.42 Next, the 2-bromopropionate group was introduced through an esterification reaction. The tert-butoxycarbonyl group was then hydrolyzed to the carboxyl group using CF3COOH, followed by an esterification reaction with glycidol to yield the Br-acrylate-epoxide monomer. The chemical structure of Br-acrylate-epoxide was confirmed using 1H NMR, 13C NMR, and high-resolution mass spectrometry (Fig. 1a, S1, and S2†). The resonance signals of the double bond appeared at 5.97 and 6.45 ppm. The peaks located at 4.41 and 3.25 ppm belonged to CHBr and –OCH–, respectively, which demonstrated the successful introduction of the 2-bromopropionate and epoxy groups. | ||
| Scheme 2 Synthesis of (a) the Br-acrylate-epoxide functional monomer and (b) the poly(Br-acrylate-epoxide) macro-agent. | ||
RAFT homopolymerization of Br-acrylate-epoxide was carried out in toluene at 65 °C using AIBN as the initiator, while cumyl dithiobenzoate (CDB) was used as the chain transfer agent. Well-defined poly(Br-acrylate-epoxide) was obtained with a narrow molecular weight distribution (Mn = 9000 g mol−1, Đ = 1.19). The 1H NMR spectrum of the poly(Br-acrylate-epoxide) macro-agent is shown in Fig. 1b. The appearance of CHBr (peak a, 4.48 ppm) and –OCH– (peak e, 3.20 ppm) resonance signals demonstrated the presence of the ATRP initiating group and the epoxy group after RAFT homopolymerization. The average degree of polymerization of the poly(Br-acrylate-epoxide) backbone (n) was calculated to be 35 by 1H NMR.
Preparation of PA-g-PAzo/PEG/PLA and PA-g-PFA/PEG/PLA asymmetric HMBs
In our polymer design, the ATRP grafting reaction was performed to introduce the primary side chain in the asymmetric HMBs. To begin, we first prepared an azobenzene-methacrylate monomer (Azo, 6-(4-butyl-4′-oxyazobenzene) hexyl methacrylate). ATRP of the Azo monomer was directly initiated by the poly(Br-acrylate-epoxide) macro-agent to afford PA-g-PAzo via the grafting-from strategy (Scheme 3a). The reaction was carried out at 80 °C in 1,4-dioxane, with feeding ratios of [monomer]/[initiator]/[CuBr]/[HMTETA] = 100/1/2/2. Both a high feeding ratio of the monomer to the ATRP initiating group and a low conversion of the monomer were employed to suppress possible chain coupling or crosslinking.43,44 PA-g-PAzo was characterized by 1H NMR. As shown in Fig. 2a, typical peaks corresponding to the PAzo brush appeared. The peaks at 6.85, 7.20, and 7.75 ppm are assigned to the aromatic group in the PAzo brush. The peak at 2.61 ppm corresponds to CH2CH2CH2CH3. The mean number of repeat units (m) of the PAzo brush was calculated as 10. The second side chain was then grafted onto the backbone by a thiol–epoxy coupling reaction. PEG-SH (Mn ≈ 2000 g mol−1) was used to guarantee the grafting density because of its “thinner” structure and low steric congestion.36 By using LiOH as the catalyst and THF/H2O as the solvent, the coupling reaction between PEG-SH and PA-g-PAzo was conducted at a feeding ratio of [thiol]/[epoxy] = 3/1. PA-g-PAzo/PEG was obtained with a high grafting efficiency (>90%) of the PEG side chain. The 1H NMR spectrum of PA-g-PAzo/PEG is shown in Fig. 2b and proton resonance signals of the PEG side chain (3.30, 3.57 ppm) appeared in the spectrum. In addition, the GPC trace after the thiol–epoxy coupling reaction shifted toward the high molecular weight region (Fig. 2d). This evidence in combination with the proton resonance signals at 6.85, 7.20, and 7.75 ppm (the aromatic group of Azo) confirmed the formation of PA-g-PAzo/PEG without affecting the polymeric skeleton and PAzo side chain.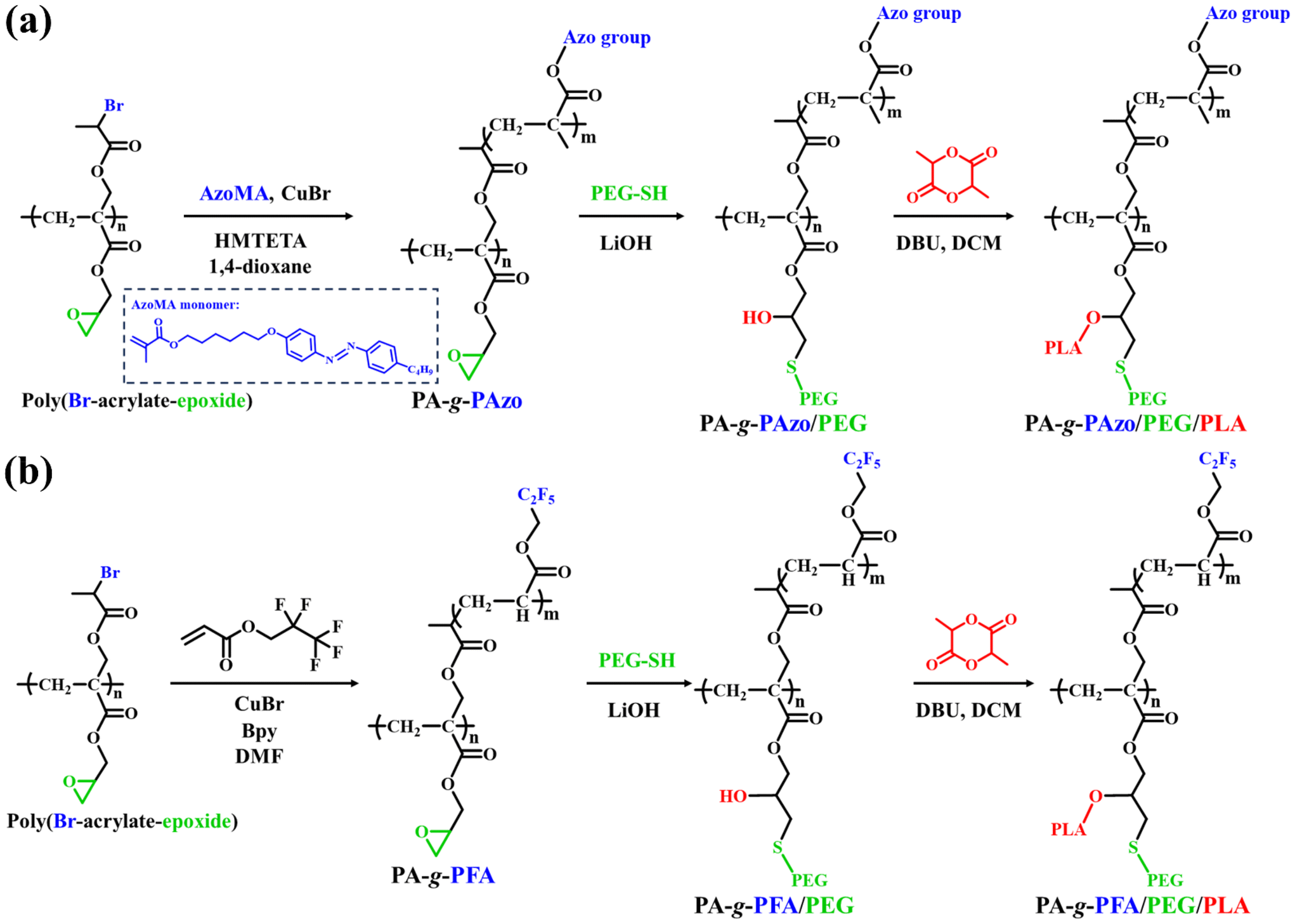 | ||
| Scheme 3 Synthesis of (a) PA-g-PAzo/PEG/PLA and (b) PA-g-PFA/PEG/PLA. | ||
 | ||
| Fig. 2 1H NMR spectra of (a) PA-g-PAzo, (b) PA-g-PAzo/PEG, and (c) PA-g-PAzo/PEG/PLA. (d) GPC curves of PA-g-PAzo, PA-g-PAzo/PEG, and PA-g-PAzo/PEG/PLA. | ||
Next, catalyzed by 1,8-diazabicyclo[5.4.0]-7-undecene (DBU), the ROP reaction of LA using the hydroxyl group of PA-g-PAzo as an initiator was conducted in CH2Cl2 at 35 °C. PA-g-PAzo/PEG/PLA with well-defined PAzo, PEG, and PLA brushes was obtained. The structure of PA-g-PAzo/PEG/PLA was characterized by 1H NMR (Fig. 2c). The resonance signal located at 5.15 ppm was attributed to OCHCH3 in the PLA side chains. The existence of peaks at 6.85, 7.20, and 7.75 ppm (C6H4N2C6H4 of Azo), and 3.57 ppm (OCH2CH2 of PEG) indicates the presence of PAzo and PEG brushes. The average degree of polymerization of PLA obtained by 1H NMR was calculated as 18. The GPC curve of PA-g-PAzo/PEG/PLA shows a narrow and monomodal peak (Fig. 2d) in the higher molecular weight region compared to the corresponding PA-g-PAzo/PEG. The dispersities of PA-g-PAzo, PA-g-PAzo/PEG, and PA-g-PAzo/PEG/PLA are all below 1.30 (Fig. 2d), indicating good control of the whole synthetic system. Thus, these results clearly demonstrated the successful synthesis of well-defined PA-g-PAzo/PEG/PLA HMBs.
Subsequently, we prepared PA-g-PFA/PEG/PLA HMBs under similar conditions. Here, pentafluoropropyl acrylate (FA) is a fluorophilic monomer (Scheme 3b). The details about the synthetic process of PA-g-PFA/PEG/PLA are provided in the ESI.† The chemical structure of PA-g-PFA/PEG/PLA was confirmed by 1H NMR (Fig. S3†). All proton resonance signals of the PFA, PEG, and PLA side chains were observed. In addition, it can be seen in Fig. S3d† that the typical 19F signals corresponding to the PFA side chain appeared. All synthesized PA-g-PFA, PA-g-PFA/PEG, and PA-g-PFA/PEG/PLA showed monomodal GPC elution curves and low dispersities (Mn = 46100 g mol−1, Đ = 1.29; Mn = 79300 g mol−1, Đ = 1.31; Mn = 108300 g mol−1, Đ = 1.35). These results affirmed the successful preparation of PA-g-PFA/PEG/PLA HMBs with PFA, PEG, and PLA brushes.
Self-assembly of amphiphilic PA-g-PAzo/PEG/PLA
Two-dimensional (2D) platelet structures have gained tremendous interest due to their wide applications in optical and electrical devices.45–47 However, the preparation of morphologically tunable 2D platelets through block copolymer self-assembly remains a challenge. 2D platelet structures are hardly created due to the existence of lateral solvophobic edges with high free energy. As a representative equilibrium morphology, a vesicle is essentially a closed 2D molecular membrane. Thus, 2D platelets can be fabricated when the molecular membrane is difficult to bend.48 On the other hand, MBs may form 2D structures by orderly packing cylindrical molecular chains.49 We propose that HMBs can easily form well-defined 2D structures because their rigid structure and hetero-brushes limit the bending of 2D assemblies to form other assemblies.Previous studies have demonstrated that the planar π-conjugated structure of azobenzene is advantageous for enhancing molecular rigidity and π–π stacking, which can increase the 2D platelet assembling tendency of azobenzene-containing polymers.50 In the present study, the self-assembly behavior of PA-g-PAzo/PEG/PLA with a relatively short backbone and PAzo brush was investigated. The intermolecular association of PA-g-PAzo/PEG/PLA occurred in a CH3OH/THF mixed solution to form well-defined 2D platelets. The self-assembly of PA-g-PAzo/PEG/PLA was performed through a “heating–cooling–aging” process. After the PA-g-PAzo/PEG/PLA solution (Vmethanol:VTHF = 92:8) was subjected to the process of heating to 68 °C, cooling to 25 °C, and finally aging at 10 °C, the assemblies were examined by transmission electron microscopy (TEM). As shown in Fig. 3, 2D platelets were obtained with lateral sizes ranging from several tens to thousands of nanometers. The size of the resulting platelets could be tailored by controlling the polymer concentration.
 | ||
| Fig. 3 TEM images of 2D platelets formed by PA-g-PAzo/PEG/PLA in a mixture of CH3OH and THF (Vmethanol:VTHF = 92:8) with concentrations of (a, b) 0.015 mg mL−1, platelet-1, (c, d) 0.02 mg mL−1, platelet-2, and (e, f) 0.04 mg mL−1, platelet-3. (g) Statistical analysis of the length distribution of the platelets. | ||
Fig. 3a and b taken from the samples with a concentration of 0.015 mg mL−1 show that platelets with a number-average length of 45 nm were formed (defined as platelet-1, Fig. 3g). The platelets in a 0.02 mg mL−1 solution grew to a larger size (defined as platelet-2, DLlength = 263 nm, Fig. 3c, d and g). As the concentration was increased to 0.04 mg mL−1, the length of the formed platelets further increased to 1343 nm (defined as platelet-3, Fig. 3e–g). The details of the size comparison between different platelets are presented in Fig. 3g. TEM images of the typical intermediate structures during the formation of the platelets are provided in Fig. S9.† We proposed that the platelets were composed of several interconnected PA-g-PAzo/PEG/PLA sandwich-like layers (Scheme S4†). The middle layer is a solvophobic layer formed by PAzo and PLA brushes, while the two outer layers are PEG brushes. The formation of the sandwich-like layers was driven by both solvophobic interaction and π–π interaction of PAzo brushes. Previous studies explained the formation of the multi-layered platelets due to the crystallization of PEG chains. Below the crystallization temperature, the PEG chains can crystallize and thus form a compact state, which drives the association of monolayers.51,52
To verify the molecular restacking of polymers in the formation of platelets, PA-g-PAzo/PEG/PLA was examined by UV-vis spectroscopy. The absorption of Azo groups in PA-g-PAzo/PEG/PLA appeared at 355 nm in THF and shifted to 360 nm when it was assembled in a CH3OH/THF mixed solution (Fig. S6†). Compared with PA-g-PAzo/PEG/PLA in the THF solution, where the polymer molecules dissolved completely, PA-g-PAzo/PEG/PLA displayed red-shifted peaks in the CH3OH/THF solution. Such red-shifted absorption signified more closely packed J-type stacking during the formation of the 2D platelets due to the π–π interaction of PAzo brushes. On the other hand, the shapes and sizes of the platelets were also influenced by the ratio of CH3OH/THF (solvent polarity). As shown in Fig. 4a, platelets with a number-average length of 193 nm were formed in a 0.01 mg mL−1 solution of PA-g-PAzo/PEG/PLA in CH3OH/THF (95/5). The length of the platelets formed in 0.05 mg mL−1 solution was 3520 nm (Fig. 4c and d).
 | ||
| Fig. 4 TEM images of 2D platelets formed by PA-g-PAzo/PEG/PLA in a mixture of CH3OH and THF (Vmethanol:VTHF = 95:5) with concentrations of (a) 0.01 mg mL−1 and (b) 0.05 mg mL−1. | ||
Emulsifying performance of PA-g-PFA/PEG/PLA
Previous studies suggested that molecular brushes have a lower tendency for intermolecular self-assembly than the corresponding linear copolymers, especially for the HMBs that lack interchain forces (π–π interaction, hydrogen bonds, etc.).27 Thus, HMBs may have facilitated interactions with solvents for enhanced performance as emulsifiers.53 In the present study, special HMBs with fluorophilic PFA, lipophilic PLA, and hydrophilic PEG side chains, designated as PA-g-PFA/PEG/PLA, were used as surfactants to stabilize emulsion interfaces. As a proof of concept, we selected two oil phases: lipophilic toluene and fluorophilic hexafluorobenzene. Both highly stable toluene-in-water and hexafluorobenzene-in-water emulsions could be gained by adopting PA-g-PFA/PEG/PLA HMBs as surfactants.The toluene-in-water emulsion formed by PA-g-PFA/PEG/PLA is shown in Fig. 5. As a control experiment, the performance of the corresponding PEG-b-PLA as a surfactant to stabilize the water–toluene interface was also investigated. Optical microscopy (OM) and DLS were used to study the structure and size of oil droplets in the emulsions. The average sizes of emulsion droplets formed by PEG-b-PLA and PA-g-PFA/PEG/PLA were ca. 4 μm and 5 μm, respectively (Fig. 5a, b and S11†). On the other hand, hexafluorobenzene-in-water emulsions formed by PA-g-PFA/PEG/PLA are shown in Fig. 6. The droplet diameters of the emulsions were ca. 3.5 μm for the PEG-b-PFA surfactant and ca. 3 μm for the PA-g-PFA/PEG/PLA surfactant (Fig. 6a, b and S12†). For both PA-g-PFA/PEG/PLA and PEG-b-PFA-based emulsions, the oil droplets showed relatively narrow size distributions.
 | ||
| Fig. 5 Optical microscopy images of the emulsions prepared using toluene as the oil phase: PEG-b-PLA as the surfactant after storing for (a) 24 h and (c) 7 d, and PA-g-PFA/PEG/PLA as the surfactant after storing for (b) 24 h and (d) 7 d. | ||
 | ||
| Fig. 6 Optical microscopy images of the emulsions prepared using hexafluorobenzene as the oil phase: PEG-b-PFA as the surfactant after storing for (a) 24 h and (c) 7 d, and PA-g-PFA/PEG/PLA as the surfactant after storing for (b) 24 h and (d) 7 d. | ||
In order to investigate the emulsion stability, the diameters of oil droplets in the emulsions were monitored by OM. Compared with PEG-b-PLA, PA-g-PFA/PEG/PLA resulted in a much more stable emulsion. For the emulsion using PEG-b-PLA as the surfactant, the size of oil droplets increased by 80% from 24 h to 7 d (Fig. 5c). The emulsion using PA-g-PFA/PEG/PLA as the surfactant showed only 10% increase in diameter during the same period (Fig. 5d). The hexafluorobenzene-in-water emulsion using PA-g-PFA/PEG/PLA as the surfactant also showed high stability. The droplet diameter increased by 200% for the PEG-b-PFA-based system (Fig. 6c), but the variation in droplet diameter was only 20% for the PA-g-PFA/PEG/PLA-based system (Fig. 6d). Evidently, the emulsifying performance of PA-g-PFA/PEG/PLA was significantly better than that of its corresponding linear copolymers.
Conclusions
In conclusion, we presented a feasible approach to synthesize well-defined asymmetric HMBs comprising three different side chains based on a multivalent monomer Br-acrylate-epoxide by sequential RAFT polymerization, ATRP, thiol–epoxy coupling reaction, and ROP. PA-g-PAzo/PEG/PLA with a short backbone and relatively strong intermolecular association was designed to promote the formation of 2D platelet structures. Through a heating–cooling–aging process, well-defined platelets with tunable morphologies were constructed by varying solvent polarity and concentration. Besides, PA-g-PFA/PEG/PLA with fluorophilic PFA, hydrophilic PEG, and lipophilic PLA brushes was synthesized and further used as an efficient surfactant for the stabilization of different emulsions. The emulsifying performance of PA-g-PFA/PEG/PLA was significantly better than that of its corresponding linear copolymers. Overall, this study offers an efficient platform for the facile synthesis of asymmetric HMBs with structural and functional control.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful for financial support from the National Natural Science Foundation of China (52273008, 52325308, 52073092), the Shanghai Scientific and Technological Innovation Project (22ZR1479300), and the Shanghai Rising-Star Program (23QA1402500).References
- J.-F. Lutz, J.-M. Lehn, E. W. Meijer and K. Matyjaszewski, Nat. Rev. Mater., 2016, 1, 16024 CrossRef CAS.
- K. Matyjaszewski, Science, 2011, 333, 1104–1105 CrossRef CAS PubMed.
- G. Polymeropoulos, G. Zapsas, K. Ntetsikas, P. Bilalis, Y. Gnanou and N. Hadjichristidis, Macromolecules, 2017, 50, 1253–1290 CrossRef CAS.
- A. Gregory and M. H. Stenzel, Prog. Polym. Sci., 2012, 37, 38–105 CrossRef CAS.
- H. Bayraktutan, R. J. Kopiasz, A. Elsherbeny, M. Martinez Espuga, N. Gumus, U. C. Oz, K. Polra, P. F. McKay, R. J. Shattock, P. Ordóñez-Morán, A. Mata, C. Alexander and P. Gurnani, Polym. Chem., 2024, 15, 1862–1876 RSC.
- J. Rzayev, ACS Macro Lett., 2012, 1, 1146–1149 CrossRef CAS PubMed.
- G. Xie, M. R. Martinez, M. Olszewski, S. S. Sheiko and K. Matyjaszewski, Biomacromolecules, 2018, 20, 27–54 CrossRef.
- H.-i. Lee, J. Pietrasik, S. S. Sheiko and K. Matyjaszewski, Prog. Polym. Sci., 2010, 35, 24–44 CrossRef CAS.
- M. Müllner, K. Yang, A. Kaur and E. J. New, Polym. Chem., 2018, 9, 3461–3465 RSC.
- R. Verduzco, X. Li, S. L. Pesek and G. E. Stein, Chem. Soc. Rev., 2015, 44, 2405–2420 RSC.
- H. Mei, T. S. Laws, J. P. Mahalik, J. Li, A. H. Mah, T. Terlier, P. Bonnesen, D. Uhrig, R. Kumar, G. E. Stein and R. Verduzco, Macromolecules, 2019, 52, 8910–8922 CrossRef CAS.
- X. Fu, Z.-H. Guo, A. N. Le, J. Lei and M. Zhong, Polym. Chem., 2020, 11, 270–274 RSC.
- B. Saha, N. Choudhury, S. Seal, B. Ruidas and P. De, Biomacromolecules, 2019, 20, 546–557 CrossRef CAS PubMed.
- W. F. M. Daniel, J. Burdyńska, M. Vatankhah-Varnoosfaderani, K. Matyjaszewski, J. Paturej, M. Rubinstein, A. V. Dobrynin and S. S. Sheiko, Nat. Mater., 2016, 15, 183–189 CrossRef CAS PubMed.
- T. Deng, B. Xu, L. Zhang and C. Li, Macromolecules, 2022, 55, 10703–10712 CrossRef CAS.
- C. M. Tonge and Z. M. Hudson, J. Am. Chem. Soc., 2019, 141, 13970–13976 CrossRef CAS.
- D. Zhou, L.-W. Zhu, B.-H. Wu, Z.-K. Xu and L.-S. Wan, Polym. Chem., 2022, 13, 300–358 RSC.
- Y. Zhao, H. Wu, Y. Zhang, X. Wang, B. Yang, Q. Zhang, X. Ren, C. Fu, Y. Wei, Z. Wang, Y. Wang and L. Tao, ACS Macro Lett., 2015, 4, 843–847 CrossRef CAS PubMed.
- Y. Li, H. T. T. Duong, M. W. Jones, J. S. Basuki, J. Hu, C. Boyer and T. P. Davis, ACS Macro Lett., 2013, 2, 912–917 CrossRef CAS PubMed.
- C. Feng and X. Huang, Acc. Chem. Res., 2018, 51, 2314–2323 CrossRef CAS PubMed.
- A. N. Le, R. Liang and M. Zhong, Chem. – Eur. J., 2019, 25, 8177–8189 CrossRef CAS PubMed.
- K. Kawamoto, M. Zhong, K. R. Gadelrab, L.-C. Cheng, C. A. Ross, A. Alexander-Katz and J. A. Johnson, J. Am. Chem. Soc., 2016, 138, 11501–11504 CrossRef CAS PubMed.
- M. Tong, X. An, W. Pan, H. Liu and Y. Zhao, Polym. Chem., 2016, 7, 2209–2221 RSC.
- B. Wang, Y. Mi, B. Wang, L. Pan, Y. Li and D.-P. Song, Macromolecules, 2024, 57, 1770–1778 CrossRef CAS.
- Y. Liu, Y. Zhang, Z. Wang, J. Wang, K. Wei, G. Chen and M. Jiang, J. Am. Chem. Soc., 2016, 138, 12387–12394 CrossRef CAS PubMed.
- Y. Li, E. Themistou, J. Zou, B. P. Das, M. Tsianou and C. Cheng, ACS Macro Lett., 2012, 1, 52–56 CrossRef CAS PubMed.
- Y. Li, J. Zou, B. P. Das, M. Tsianou and C. Cheng, Macromolecules, 2012, 45, 4623–4629 CrossRef CAS.
- H. Luo, M. Szymusiak, E. A. Garcia, L. L. Lock, H. Cui, Y. Liu and M. Herrera-Alonso, Macromolecules, 2017, 50, 2201–2206 CrossRef CAS.
- H. Luo, J. L. Santos and M. Herrera-Alonso, Chem. Commun., 2014, 50, 536–538 RSC.
- H. Ma and K. T. Kim, Macromolecules, 2020, 53, 711–718 CrossRef CAS.
- Z. Dong, W. Huang, X. Liu, F. Yu, C. Long, S. Feng, L. Luo and Z.-R. Chen, Macromolecules, 2021, 54, 9385–9392 CrossRef CAS.
- Y. Chen, Z. Sun, H. Li, Y. Dai, Z. Hu, H. Huang, Y. Shi, Y. Li and Y. Chen, ACS Macro Lett., 2019, 8, 749–753 CrossRef CAS.
- G. Wu, S.-C. Chen, Q. Zhan and Y.-Z. Wang, Macromolecules, 2011, 44, 999–1008 CrossRef CAS.
- R. Chen, X. Jiang, G. Lu, W. Liu, W. Jin, G. Tian and X. Huang, Polym. Chem., 2022, 13, 2791–2802 RSC.
- Y. Yao, L. Zhang, S. Zhang, X. Huang, C. Feng, S. Lin and B. Xu, Langmuir, 2023, 39, 18880–18888 CrossRef CAS.
- H. Gao and K. Matyjaszewski, J. Am. Chem. Soc., 2007, 129, 6633–6639 CrossRef CAS.
- B. Xu, C. Feng and X. Huang, Nat. Commun., 2017, 8, 333 CrossRef PubMed.
- B. Xu, C. Feng, Y. Lv, S. Lin, G. Lu and X. Huang, ACS Appl. Mater. Interfaces, 2020, 12, 1588–1596 CrossRef CAS PubMed.
- B. Xu, H. Qian and S. Lin, ACS Macro Lett., 2020, 9, 404–409 CrossRef CAS PubMed.
- D. Wu, Y. Yang, X. Cheng, L. Liu, J. Tian and H. Zhao, Macromolecules, 2006, 39, 7513–7519 CrossRef CAS.
- W. Gan, Y. Shi, B. Jing, X. Cao, Y. Zhu and H. Gao, Macromolecules, 2017, 50, 215–222 CrossRef CAS.
- Y. Zhang, Z. Shen, D. Yang, C. Feng, J. Hu, G. Lu and X. Huang, Macromolecules, 2010, 43, 117–125 CrossRef CAS.
- Z. Shen, Y. Chen, E. Barriau and H. Frey, Macromol. Chem. Phys., 2006, 207, 57–64 CrossRef CAS.
- C. Liu, Y. Zhang and J. Huang, Macromolecules, 2008, 41, 325–331 CrossRef CAS.
- H. Duan, P. Lyu, J. Liu, Y. Zhao and Y. Xu, ACS Nano, 2019, 13, 2473–2480 CAS.
- L. Han, M. Wang, X. Jia, W. Chen, H. Qian and F. He, Nat. Commun., 2018, 9, 865 CrossRef PubMed.
- D. Jang, J.-M. Heo, F. Jannah, M. I. Khazi, Y. J. Son, J. Noh, H. An, S. M. Park, D. K. Yoon, N. N. Kadamannil, R. Jelinek and J.-M. Kim, Angew. Chem., Int. Ed., 2022, 61, e202211465 CrossRef CAS.
- B. Ni, M. Huang, Z. Chen, Y. Chen, C.-H. Hsu, Y. Li, D. Pochan, W.-B. Zhang, S. Z. D. Cheng and X.-H. Dong, J. Am. Chem. Soc., 2015, 137, 1392–1395 CrossRef CAS PubMed.
- Y. Shi, W. Zhu, D. Yao, M. Long, B. Peng, K. Zhang and Y. Chen, ACS Macro Lett., 2014, 3, 70–73 CrossRef CAS PubMed.
- Z. Li and Z. Lin, ACS Nano, 2021, 15, 3152–3160 CrossRef CAS PubMed.
- D. Wu, F. Xu, Y. Huang, C. Chen, C. Yu, X. Feng, D. Yan and Y. Mai, Macromolecules, 2018, 51, 161–172 CrossRef CAS.
- Y. Huang, Y. Mai, X. Yang, U. Beser, J. Liu, F. Zhang, D. Yan, K. Müllen and X. Feng, J. Am. Chem. Soc., 2015, 137, 11602–11605 CrossRef CAS PubMed.
- Y. Gao, X. Wu, Z. Xiang and C. Qi, Langmuir, 2021, 37, 2376–2385 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Materials, measurements, experimental details of the synthetic route of polymers, preparation and characterization of assemblies. See DOI: https://doi.org/10.1039/d4py00656a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |