
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnraveling the molecular and growth mechanism of colloidal black In2O3−x†
Cameron
Armstrong
 ,
Kayla
Otero
and
Emil A.
Hernandez-Pagan
*
,
Kayla
Otero
and
Emil A.
Hernandez-Pagan
*
Department of Chemistry and Biochemistry, University of Delaware, Newark, DE 19716, USA. E-mail: emilhp@udel.edu
First published on 19th April 2024
Abstract
Black metal oxides with varying concentrations of O-vacancies display enhanced optical and catalytic properties. However, direct solution syntheses of this class of materials have been limited despite being highly advantageous given the different synthetic handles that can be leveraged towards control of the targeted material. Herein, we present an alternate colloidal synthesis of black In2O3−x nanoparticles from the simple reaction between In(acac)3 and oleyl alcohol. Growth studies by PXRD, TEM, and STEM-EDS coupled to mechanistic insights from 1H, 13C NMR revealed the particles form via two paths, one of which involves In0. We also show that variations in the synthesis atmosphere, ligand environment, and indium precursor can inhibit formation of the black In2O3−x. The optical spectrum for the black nanoparticles displayed a significant redshift when compared to pristine In2O3, consistent with the presence of O-vacancies. Raman spectra and surface analysis also supported the presence of surface oxygen vacancies in the as-synthesized black In2O3−x.
Introduction
Materials to be considered for photo(electrochemical)chemical applications must meet several criteria: be relatively abundant, have band edges that straddle the redox potentials of the desired reaction, and should be capable of photoexcitation from a wide range of photon energies.1 Wide band gap metal oxide semiconductors are compelling targets for such applications due to their chemical and thermal stability, with valence/conduction bands which can straddle a large range of redox potentials.2 However, due to the wide band gap, many oxides fall short when considering the window of usable photons. For example anatase TiO2, perhaps the most studied photo(electro) active catalysts, has a band gap (3.2 eV) that limits absorption of photons to the UV region of the solar spectrum.3,4 Great efforts have been focused on expanding the functional spectral range into the visible and near-IR (NIR) regions of the solar spectrum due to the higher photon flux.5 Band gap engineering through the incorporation of dopants is the most common technique for modifying the optical absorption range of semiconductors through the formation of donor or acceptor states within the band gap.6–9 In some notable cases a reduction of the overall band gap can also be achieved.10,11 However, doping can also lead to undesirable properties. Aliovalent dopants typically illicit a greater increase in the photon wavelengths range absorbed but result in higher concentrations of defects.4,12 These defects can act as deleterious trap states preventing photogenerated charge carriers from driving the desired chemical reaction.4 Conversely, isovalent dopants incorporate better into the host material resulting in fewer defects but a lesser change in optical properties when compared to the aliovalent species.6,13,14 Dopants in general can also contribute to reduced carrier lifetime due to enhanced recombination as is the case for N-doped TiO2.15 Alternatively, the intentional engineering of defect states in metal oxides in the form of oxygen vacancies has been shown to be a promising alternative to doping.Oxygen vacant metal oxides have a non-stoichiometric oxygen composition. The non-stoichiometry or oxygen deficiency can arise from “self-doping”. This effect is a result of aliovalent constituent metal species, resulting in the elimination of oxygen as a means of maintaining charge neutrality.16–22 Additionally, the elimination of oxygen results in discrete regions of disorder (amorphization) in the lattice.16,21,23 While oxygen vacancies and their effects have been known for some time, one of the most notable reports on the topic came from Chen and co-workers in 2011 through the controlled hydrogenation of TiO2 nanoparticles.24 The hydrogenated particles shifted from white to black exhibiting an extreme extension of absorbance into NIR region. This resulted in a paradigm shift in the way researchers approached the development of visible light absorbing photo(electro)catalysts.
In addition to TiO2, metal oxides such as In2O3, CeO2, ZrO2 and WO3 have all displayed an extended absorption range, as well as other distinct properties through oxygen vacancy control.14,16,25–27 Ozin and co-workers evaluated black In2O3 under photothermal conditions which displayed a relative fivefold increase in the production of CO from CO2 hydrogenation under illuminated and varying temperature conditions when compared with pristine In2O3.16,28 Tungsten oxides, having a slew of sub-stoichiometric compositions (WO2.72, WO2.8, WO2.83, and WO2.9) display surface plasmon resonance (SPR) extending absorbance into the NIR. Additionally, Xie and co-workers reported extended catalytic activity in reduced WO3 through the continued degradation of formaldehyde which continued 5 h post-illumination.27,29,30
The formation of oxygen vacancies has been predominately achieved though post-synthetic methods including, plasma treatment, laser irradiation, and electrochemically.31–33 However, the most common method has been via hydrogenation.3,4,16,18,20,34–36 While effective, this approach requires high temperatures (i.e., energy demand) and a hydrogen atmosphere which albeit not uncommon can present safety concerns. The elevated temperatures can also lead to sintering which decreases the surface area, an important property for catalytic applications.37
Solution based synthesis offers an alternative to these harsh conditions but only a few have been reported. Fang and co-workers produced black TiO2 post-synthesis via the generation of in situ H2 from the addition of sodium borohydride to an aqueous particle suspension.25 Similar results were reported by Jung and co-workers following an analogous approach that used 2-methylimidazole under aqueous conditions as well.38 Alternatively, the direct synthesis of materials with oxygen vacancies using precursor chemistry offers a more facile approach, eliminating the need for particle formation followed by post-treatment. Several methods have been reported to prepare WO3−x using precursors with readily oxidizable W4+ or W5+.27,29,39 Guo and co-workers developed a solvothermal method based on cerium(III) acetylacetonate which resulted in reduced CeO2 quantum dots at temperatures as low as 120 °C.20 Recently, Milliron and co-workers reported a colloidal synthesis to produce oxygen deficient In2O3 through the presence of spectator cations which was extended to CdO and γ-Fe3O4.26
Herein, we report on an alternate non-aqueous colloidal synthesis of black, oxygen deficient In2O3−x nanoparticles. The synthesis is based on the reaction between indium(III) acetylacetonate (acac) and oleyl alcohol. Control experiments and in-depth analysis of the reaction using NMR revealed the importance of the synthesis atmosphere, the ligand environment, and the acac precursor in the formation of In2O3−x. PXRD and STEM-EDS elemental mapping provided unique insight into the growth mechanism. Comparing optical and vibrational studies to previous literature reports we find good agreement with the presence of oxygen vacancies. XPS analysis and gas adsorption measurements further validate and provided quantification of the surface vacancies. These findings highlight the importance of understanding reaction mechanics and nanoparticle growth, to be used to develop broadly absorbing photoactive materials through highly versatile colloidal means.
Experimental section
Chemicals & methods
Oleyl alcohol (OLOH, 80–85%) was purchased from Thermo Scientific. Indium acetylacetonate (In(acac)3, 98%) and Indium acetate (In(OAc)3, 99.99%) were purchased from Strem. Oleic Acid (OLAC, 90%), oleylamine (OLAM, 70%), deuterated chloroform (CDCl3, 99.8%), and barium sulfate (BaSO4, 98%) were purchased from Sigma. Tri-n-octylamine (TOA, 97%), hexanes, ethanol, and isopropanol were obtained from Fischer. Dioctylamine (DOA, 96%) and 4-ocytlaniline (95%) were acquired from J&K. All chemicals were used as received without further purification. All synthesis were carried out in 3-neck 50 mL round bottom flasks loaded with a magnetic stir bar and a condenser connected to the middle neck with rubber septa on the side necks. One septum was perforated, and a k-type thermocouple connected to a Gemini temperature controller (J-Kem Scientific) which controlled the Glass-Col heating mantles. Air free synthesis was carried out using standard Schlenk line techniques under a nitrogen atmosphere. Open air reactions used the same glassware but omitted the connection to the Schlenk line. Reactions under air flow were carried using a 3-inch 18-gauge needle through one of the septa while the other side neck was fitted with a stopcock adapter. The air was passed through a Drierite laboratory gas drying unit prior to reaching the reaction flask.Safety considerations
Care should be taken when using a Schlenk line to ensure over pressurization in a closed system does not occur. Constant temperature monitoring and care should be utilized when conducting reactions at elevated temperatures. When the heating mantles are removed from the flask, heating must first be turned off and extreme caution should be used when removing and placing the mantle. When working with a furnace, heat resistant gloves and tongs should be used when the furnace is in operation.Synthesis of black In2O3 nanoparticles
The procedure to prepare black In2O3−x nanoparticles was adapted from the literature.39 In a typical synthesis, 1.0 mmol of In(acac)3 was dissolved in 10.0 mL of OLOH and 2.0 mL of DOA. The reaction was degassed under vacuum at 120 °C for 30 min while stirring at 700 rpm. The reaction was then placed under nitrogen blanket and heated to 270 °C where the particles aged for 1 h. Upon completion the heating mantle was turned off and carefully removed allowing the flask to cool naturally to room temperature. The particles were washed three times by precipitating with isopropanol, centrifugation at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 2 min, and resuspension in toluene. The washed particles were stored in 10.0 mL of toluene. Alterations to this procedure will be discussed and noted in the following sections.
000 rpm for 2 min, and resuspension in toluene. The washed particles were stored in 10.0 mL of toluene. Alterations to this procedure will be discussed and noted in the following sections.
Supernatant collection and analysis
The collection of the reaction supernatant was carried out using a modified method to the one mentioned above. In a typical synthesis 1.0 mmol of In(acac)3 was mixed with 2.0 mL OLOH (∼6 mmol) in a 15 mL three-neck round-bottom flask with a magnetic stir bar, condenser in the center neck, thermocouple, and two rubber septa on the side necks with stirring set to 700 rpm. The reaction was degassed for 30 min at 120 °C and then heated to 270 °C. Upon completion, the reaction was cooled naturally to room temperature. The reaction was then transferred to a 50 mL centrifuge tube without the aid of additional solvents and centrifuged at 10000 rpm for 2 min to remove any insoluble material. The remaining supernatant was transferred with a glass pipette to 4 mL septum-capped vial for storage. Reactions intended for analysis at various stages of the reaction were heated to the desired point then cooled naturally to room temperature followed by the same workup used above. NMR samples were prepared using 100 μL of sample in 700 μL of CDCl3.
Synthesis of pristine control In2O3 nanoparticles
Preparation of pristine In2O3 nanoparticles of large (∼120 nm) and small (∼9 nm) size were prepared using two different literature procedures.6,40 In a typical synthesis of 120 nm In2O3 nanoparticles, 1.0 mmol of In(acac)3 was dissolved in 3.5 mL of TOA, 1.0 mL of OLAC, and 0.5 mL of OLAM. The reaction was degassed under vacuum at 120 °C for 15 min while stirring at 700 rpm. The reaction was then placed under nitrogen blanket and heated quickly to 320 °C and aged for 5 min. Upon completion, the heating mantle was removed, and the flask was quickly cooled to room temperature by passing an air stream on the flask. The particles were washed three times by precipitation with isopropanol, centrifugation at 10000 rpm for 2 min, and resuspension in toluene. The washed particles were stored in 10.0 mL of toluene.
In a typical synthesis of 9 nm In2O3 nanoparticles, a precursor solution of 1.0 mmol of In(acac)3 was dissolved in 2.0 mL of OLAC in a 20 mL scintillation vial. The vial was placed on a hot plate in an Al foil jacket with stirring set to 700 rpm and was degassed for 30 min at 120 °C. The vial was then placed under N2 blanket and heated to 150 °C for 30 min. Concurrently, 10.0 mL of OLOH were degassed for 1 h at 120 °C in N2 flow configuration with stirring set to 700 rpm. The reaction was then switched to nitrogen flow and heated to 290 °C. The precursor solution was then loaded into a 5 mL syringe and injected via syringe pump at a rate of 3.5 mL min−1. Upon completion of the injection the reaction was aged for 5 min. The heating mantle was removed, and the flask was cooled naturally to room temperature. The particles were washed three times by precipitating with ethanol and acetone (50/50 vol.), centrifugation at 10000 rpm for 2 min, and resuspension in hexanes. The washed particles were stored in 10.0 mL of hexane.
Oxidative annealing
A suspension of black particles was transferred to a 20 mL septa-cap vial and residual solvent was removed under vacuum on a Schlenk line. Air annealing was carried out in 20 mL vial (without the cap) placed in a Barnstead muffle furnace. Samples were heated to 400 °C for 24 h in air and allowed to cool naturally to room temperature.Solid state materials characterization
Material characterization was conducted via powder X-ray diffraction (PXRD), high-resolution transmission electron microscopy (HR-TEM), selected area electron diffraction (SAED), X-ray photoelectron spectroscopy (XPS), UV-vis-NIR spectroscopy, Raman spectroscopy. High-resolution TEM and SAED were performed on a Talos F200C microscope at an operating voltage of 200 kV. STEM-EDS mapping images were collected on a Tecnai Osiris TEM/STEM at an operating voltage of 200 kV equipped with a SuperX™ quad EDS detection system. Drift-corrected maps used a spot size of 4 with a beam current of ∼1 nA. Maps were rendered using Bruker Espirit version 1.9. TEM samples were prepared via drop-cast technique onto 400 mesh Cu grids with carbon type-B support films (Ted Pella). X-ray diffraction patterns were collected using a Bruker D2 phaser and a Bruker D8 Discover (for the Rietlved refinement) X-ray diffractometers both with LynxEye position-sensitive detectors and Cu Kα1 X-ray source (λ = 1.54 Å). Rietveld refinement was performed using Profex. XPS was conducted on a Thermo K-Alpha with a monochromatic Al Kα X-ray source with a spot size of 400 μm, 20 scan accumulation, pass energy of 40 eV, dwell time of 50 ms, and energy step of 0.1 eV. Survey scans were collected with 10 scan accumulation, pass energy of 200 eV, dwell time of 10 ms, and energy step of 0.1 eV. XPS fitting was performed using CasaXPS. UV-vis-NIR reflectance spectra were collected with a Jasco V-770 with an integrating sphere. Samples were prepared by mixing 2% sample with 98% BaSO4 (w/w%) and deposited on a glass slide. Raman spectra were collected with a HORIBA LabRAMM HR Evolution Nano with a 532 nm excitation laser at 5% intensity (∼5 mW), 1800 g mm−1 grating, and 100× optical focus; calibrated using a Si wafer (520.7 cm−1). N2O titration was performed on Michromeritics AutoChem II connected to a Pfeiffer Vacuum Omnistar GSD301 quadrupole mass spectrometer with a secondary electron multiplier. 120–150 mg of dry nanoparticles were loaded into a U-shaped quartz flow tube with the particles supported between quartz wool. Samples were purged with pure Ar for 5 min and heated to a constant 35 °C. Pure N2O doses were delivered using a 0.5 mL sampling loop until complete saturation with 10 pulses. The mass spectrometer was calibrated using pure N2O pulses at the end of each experiment. To remove surface coordinated species samples were hydrogen pre-treated by heating to 270 °C (10 °C min−1) for 60 min in 10% H2/Ar flowed at 20 mL min−1. Calculation of surface vacancy density was adapted from Kameyama et al. and Rosowski et al.18,41 A 44 m/z (N2O) was used for quantification. Assuming a 1:1 N2O:O-vacancy ratio, the N2O consumption was calculated by numerically integrating the dosing peaks and multiplying each by the N2O calibration factor of 1.687 × 109 μmol per unit area to obtain the μmol's of N2O measured after each dose. The total moles of N2O consumed was calculated by assuming the final 3 titrations experienced no N2O consumption and multiplying the average of those 3 titrations by 10. Setting that value to the total amount of N2O dosed (μmol) and subtracting the sum of the first 7 titrations from the calculated total dose equals the N2O consumed (μmol). Dividing the consumed N2O by the mass of nanoparticles used gives the density of vacancies (μmol g−1).
Results and discussion
Synthesis and mechanism
The colloidal synthesis of black In2O3 was adapted from a modified established literature procedure for pristine In2O3.42 This procedure entailed the decomposition of an In salt (In(acac)3) in the presence of oleyl alcohol at 320 °C for 2 h under N2 blanket. In our case, the color of the reaction progressed from transparent yellow to black. This observation was unexpected given that pristine In2O3 is off-white due to its wide bandgap (∼3.75 eV direct).43,44 The PXRD pattern of the obtained black product showed sharp reflections consistent with the bixbyite In2O3 simulated pattern seen in Fig. 1a, and validated through SAED (Fig. S1†). This result suggested the formation of In2O3−x with O-vacancies. High-resolution TEM imaging revealed lattice fringes with a spacing of 4.18 Å, consistent with the (211) plane (Fig. S2†). TEM analysis shown in Fig. 1b revealed the particles are highly agglomerated with a nonuniform morphology.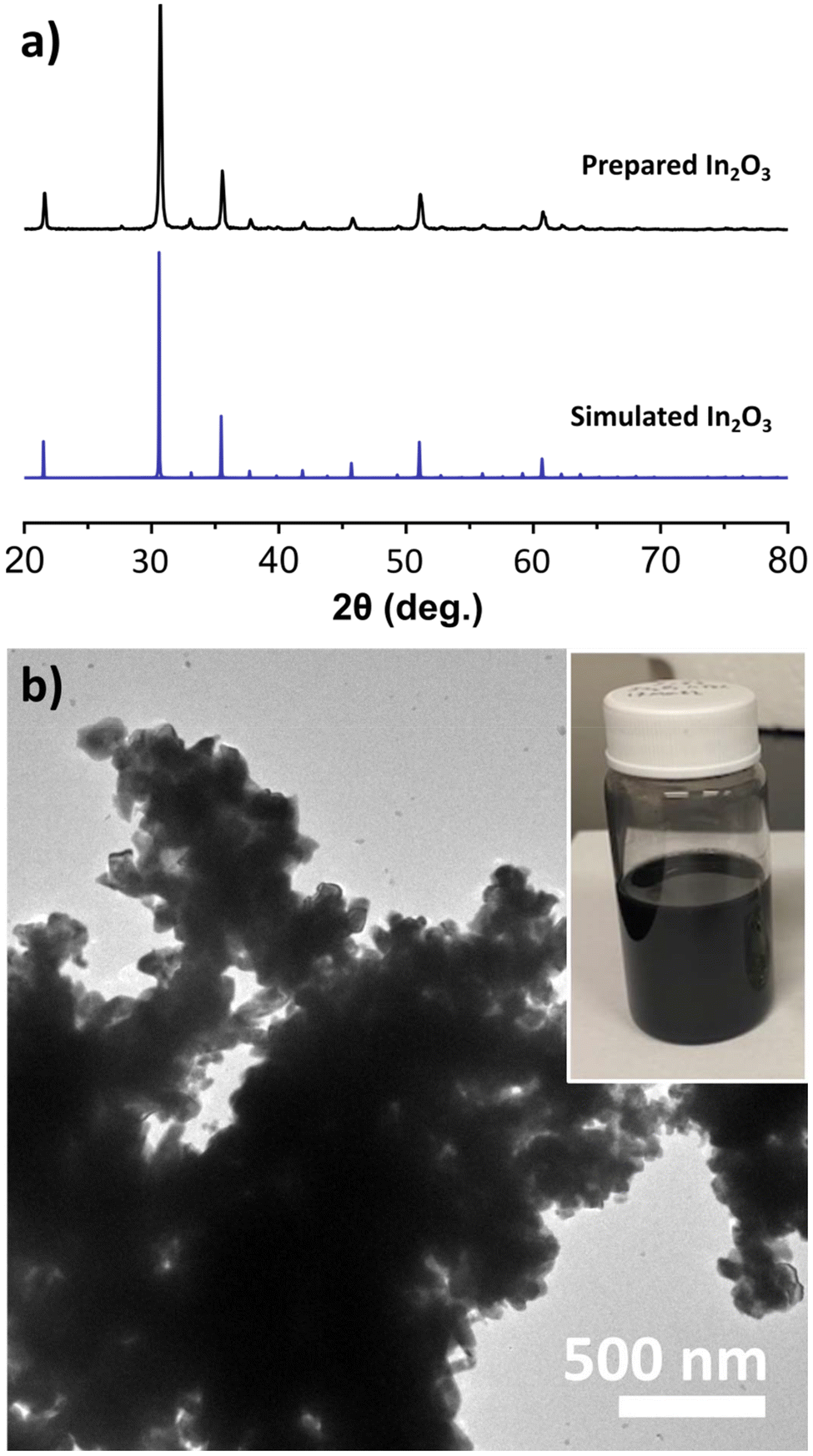 | ||
| Fig. 1 (a) PXRD of the obtained black In2O3−x compared to the simulated pattern for bixbyite In2O3. (b) TEM image of the black In2O3−x showing dense agglomerations. The inset shows a picture of the particles dispersed in toluene. | ||
In efforts to improve the morphology (size/shape) we began by varying reaction conditions starting with the solvent volume. During these studies we observed that decreasing the volume of the reaction from 25 mL to 12 mL enabled more mild reactions conditions with a more abrupt appearance of the black color at ∼210 °C and the reaction temperature lowered from 320 °C to 270 °C. While the crystal domains seemed to decrease in size as evident by a slight widening of the PXRD reflections, particles remained heavily agglomerated (Fig. S3†). We then explored the addition of different ligands. Introducing DOA or TOA had a slight improvement in shape and resulted in smaller agglomerations. Conversely, addition of OLAM or OLAC yielded small, off-white nanoparticles (Fig. S4 and S5†). The production of white nanoparticles when using OLAM and OLAC was surprising and will be discussed below. Due to the improvement of morphology when employing the more substituted amines, we opted to utilize DOA in our finalized procedure.
We then focused our efforts on interrogating early stages of the reaction to gain insights on the growth process of the black In2O3−x. A series of aliquots taken at different time intervals during the reaction were analyzed using PXRD, TEM and STEM-EDS imaging/elemental mapping. For simplicity, Fig. 2 contains data related to the first aliquot and the final product. At time 0 min, defined as the onset of black coloration around 210 °C, the PXRD pattern revealed the unexpected presence of In2O3 and indium metal. TEM imaging, seen in Fig. S6,† showed two distinct particle populations with lattice fringes corresponding to highly polycrystalline In2O3−x and large In particles, in agreement with the PXRD including the relative broadness of the reflections (e.g., In (101) vs. In2O3 (222)).
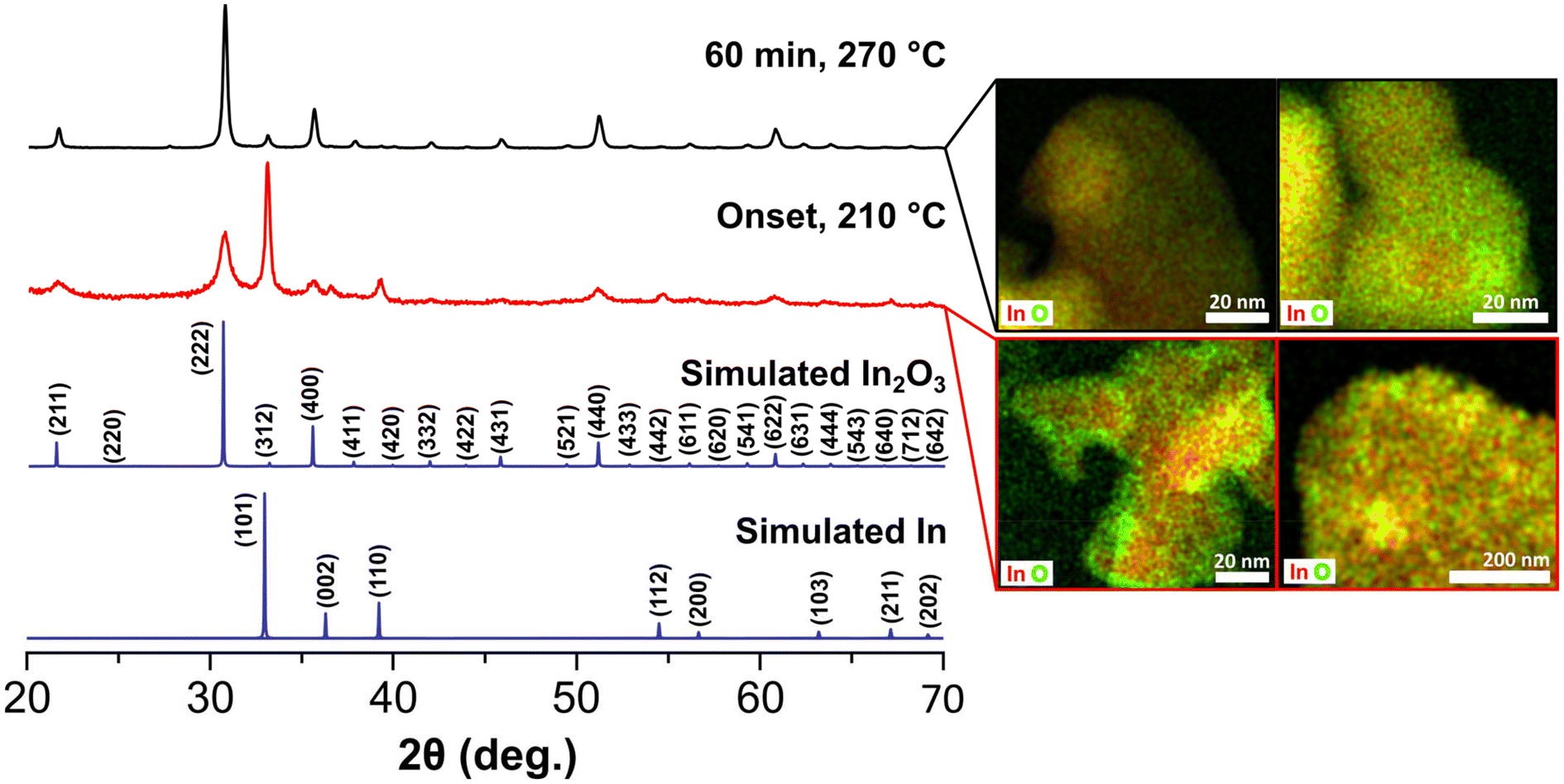 | ||
| Fig. 2 PXRD powder patterns and corresponding STEM-EDS elemental mapping images of the particles obtained from an aliquot taken at the onset of the color change (210 °C) and the final reaction product (60 min, 270 °C). The top-left map was taken from a region with not core component while the top-right was taken from a particle displaying the core–shell morphology. The bottom-left was taken from one of the larger agglomerations showing high contrast while the bottom-right was taken from a region of smaller highly agglomerated lighter contrast. | ||
STEM-EDS elemental mapping (Fig. 2) validated these observations and further showed the presence of an oxide shell on the In particles. Analysis of the aliquots taken as the reaction reached the final temperature (270 °C) showed improved crystallinity of the In2O3−x and a gradual decrease of the reflections corresponding to In (Fig. S7†). After 1 h at 270 °C, the observed diffraction pattern is predominately that of In2O3 with perhaps a minor contribution of In, convoluted by overlapping of the In2O3 (312) and In (101) reflections (Fig. 2). STEM-EDS elemental mapping revealed the In particles convert to In2O3−x with some containing an In rich core as seen in Fig. 2. Line-scans of the In rich core shows these are not pure metal but rather highly O-deficient regions (Fig. S8†). It should be noted that we did not observe any hollow structures, indicating the Kirkendall effect was not at play. Taken together, the results from the aliquot study suggest a poorly oxidative reaction environment, favoring the reduction of the In(acac)3 precursor to In0 and oxygen deficient In2O3−x. A poorly oxidative reaction environment was also recently reported by Robinson et al. in the synthesis of CoMn2O4 spinels.45
To complement the TEM aliquot studies, we performed Rietveld refinement on the onset and final reaction samples. This method provides the relative percentages of a mixture and stoichiometric information. Fig. S9† shows the refined patterns with tabulated results seen in Table S1.† Analysis of the onset pattern reveals the relative percentage of In metal is ∼37%, which decreases to ∼4% in the final particles. By refining the oxygen occupancy in the onset and final samples, both display an oxygen deficiency of approximately 10% supporting the hypothesis that the oxide is oxygen deficient from an early stage. As control, we performed the same analysis for 120 nm pristine In2O3 control sample (vide infra) which showed no presence of In metal and a stoichiometric composition, validating our refinement model.
In order to probe the reaction environment, we performed the synthesis under altered conditions. First, the synthesis was done under ambient atmosphere by leaving the condenser open. The reaction proceeded similarly with formation of the black In2O3−x product (Fig. S10†). In contrast, syntheses carried out saturating the flask headspace with air flow or bubbling air directly into the solution yielded off-white, pristine In2O3 (Fig. S10†). As expected, the increased supply of air creates a more oxidizing environment that prevents the reduction of In3+ and inhibits the formation of the black oxide, while the open condenser setup most likely does not allow for a significant mixing of the ambient air during the reaction. It should be noted that exposure to air during the synthesis can be used to provide some tunability of the stoichiometry (vide infra), albeit not full control. Aside from the reaction atmosphere, other factors that play a role in the formation of the black In2O3−x were the ligand environment and the metal precursor. As mentioned earlier, addition of OLAM or OLAC to the reaction under a N2 atmosphere yielded off-white, pristine In2O3 nanoparticles (Fig. S5 and S4†). When In(acac)3 is replaced with In(OAc)3 under otherwise identical conditions (DOA, OLOH, N2 atmosphere at 270 °C) the obtained product was also pristine In2O3 as seen in Fig. S5.†
To gain a more mechanistic understanding of the different reaction outcomes we proceeded to analyze the supernatant of a simplified synthesis that yielded black In2O3−x. Here, 2.0 mL of OLOH were combined with 1.0 mmol of precursor and aliquots were taken at different times and temperatures. This volume was selected as it was the smallest working volume that allowed for 1.0 mmol of precursor to dissolve and react while preventing the potential for signal saturation from unreacted OLOH. The initial focus was the formation of the indium metal.
Given the poorly oxidative reaction environment (demonstrated by the control experiments) the formation of indium metal nanoparticles can be expected, as alcohols are known reducing agents.46–49 However, we wondered whether the alcohol was the active reducing agent or if it first formed an aldehyde. Fig. 3 shows the 1H NMR comparisons of stock OLOH, the degassed reaction supernatant (120 °C, 30 min), the supernatant early in the black color appearance (235 °C), and the full reaction (270 °C, 60 min) (13C NMR spectra Fig. S11†). Observing the region between 9–10 ppm in the 1H NMR spectrum of the full reaction a very small triplet of doublets appears at 9.75 ppm. Due to the very low intensity we failed to see any coupling in the 2D techniques, however when comparing the shift to the spectrum of dodecyl aldehyde (Fig. S12†), the observed triplet is consistent with the analog. Additionally, the splitting is consistent with protons split by alkyl free rotation (∼8 Hz) then split to a smaller extent by the aldehyde rotation (∼2 Hz). It should be noted that the aldehyde triplet appears in the stock OLOH prior to any use, with minimal change in intensity after heating the degassed OLOH alone under the reaction conditions (Fig. S13†). Conversely, an increase in peak intensity was observed in the supernatant of the final reaction spectrum, indicating oxidation of the alcohol as it reduces the indium precursor.
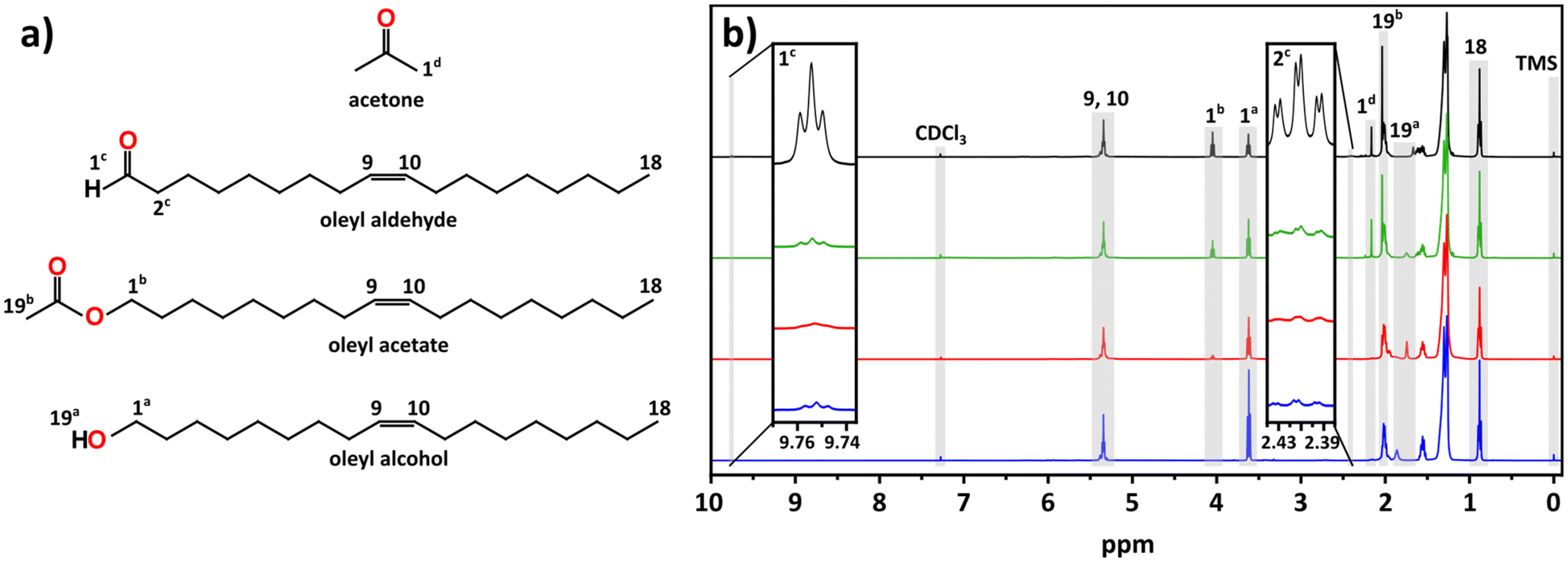 | ||
| Fig. 3 (a) Structure and key protons for the products observed in the NMR analysis of the supernatant for aliquots and the end of the reaction. (b) Stacked NMR progression of OLOH (bottom, blue) and supernatant aliquots taken after degassing with In(acac)3 (middle bottom, red), the nucleation onset (middle top, green), and the end of the reaction (top, black). The key features are highlighted in gray with notation corresponding to the molecular counterpart in part (a). | ||
It should be noted that the amount of aldehyde observed in the final product is very minor compared to the relative amount of metal observed throughout the reaction. We would expect the possibility of the aldehyde also serving as reducing agent, further oxidizing to the carboxylic acid. However, we fail to see any of the features associated with such a species. The work done by Lee and co-workers presents one possible explanation for the low concentration of aldehyde. Their work showed the catalytic reduction of an aldehyde to an alcohol with In(iOPr)3 as catalyst following a Meerwin–Pondorf–Verley type mechanism.50 At the elevated temperatures in our procedure, it is very possible an equilibrium exists between the acac and alcohol to form acetylacetone and an oleyl alkoxide. From this we speculated that the aldehyde in our reaction was being catalytically converted back to OLOH. To test this hypothesis, we carried out a 2 mL volume reaction spiked with 1.0 mmol of dodecanal. Comparing the 1H NMR of the mix of OLOH and aldehyde to the supernatant of a completed reaction we observed a drastic decrease in the aldehyde proton signal in the final product (Fig. S14†). We failed to observe any evidence of a carboxylic acid forming suggesting the low amount of aldehyde in the final aliquot does not reflect the true amount generated due to a complex underlying process. Based on these results we propose reaction path 1 illustrated in Scheme 1. At the onset of nucleation/observance of the black color, part of the precursor is rapidly reduced from In3+ to In0 through oxidation of OLOH to oleyl aldehyde and possible conversion of the aldehyde back to the alcohol. With a better understanding of the molecular path that yields the In0 particles, we turned to the oxide formation.
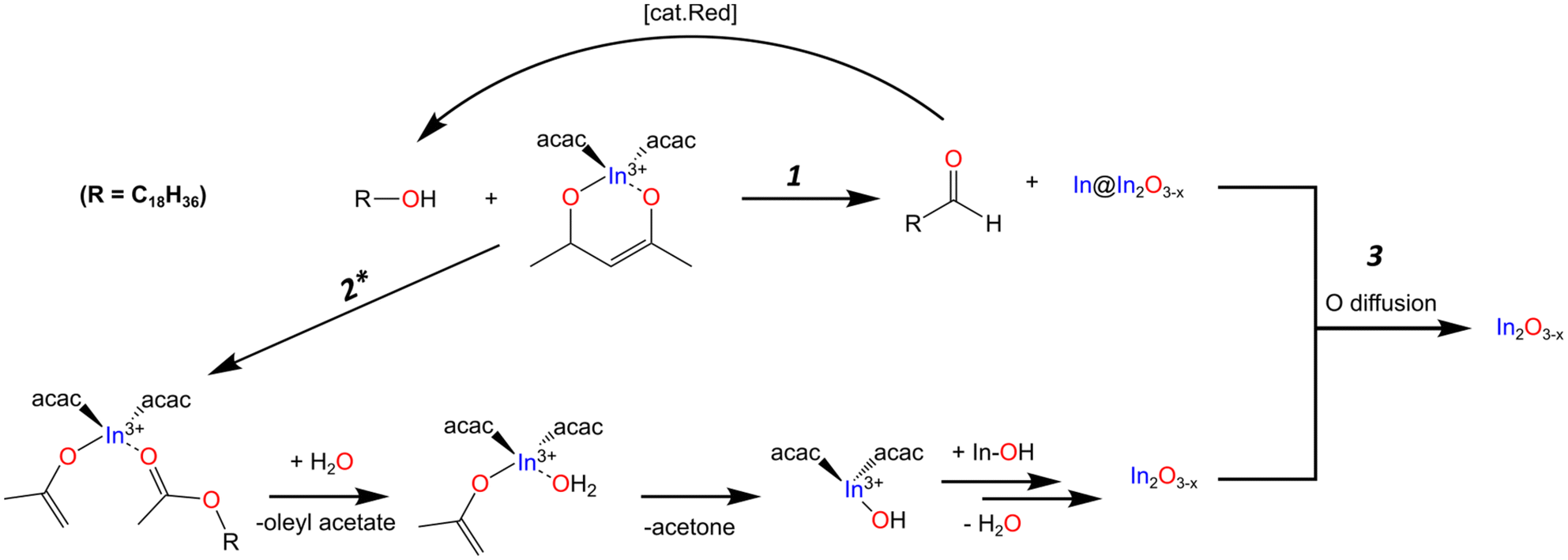 | ||
| Scheme 1 Proposed mechanistic pathway based on the NMR, PXRD, and TEM/STEM-EDS mapping analysis of the reaction progression. Path 1 shows the formation of the metal particles with an oxide shell In@In2Ox−3 structure and subsequent reduction of the aldehyde back to the alcohol. Path 2* shows the formation of the oxide through the most likely organic pathway. The star denotes the path may experience interference from the formation of the aldehyde as the reaction progresses to form the oxygen deficient oxide. Path 1 and 2* then converge forming path 3 to continue oxide growth through OR with simultaneous inward diffusion of oxygen to convert the metal to the highly oxygen deficient/In rich oxide. | ||
From the comparison of the 1H NMR spectra shown in Fig. 3, we can see distinct changes throughout the progression of the reaction between 1.00–4.25 ppm. The most notable change is the appearance of an additional triplet centered at 4.05 ppm, downfield of a triplet associated with the alcohol 1a protons (Fig. 3a). From its chemical shift in the 13C NMR, the appearance of a new carbon signal at 171.21 ppm, we identified this feature as an ester (Fig. S11†). The intense singlet at 2.04 ppm is coupled to a carbon at 20.95 ppm in the heteronuclear single quantum coherence (HSQC) spectrum (Fig. S15†), both of which display the appropriate chemical shift for an acetate ester with the appropriate phasing displaying CH or CH3 characteristic. The heteronuclear multiple bond correlation (HMBC) spectrum (Fig. S16†) displays coupling to the ester carbonyl, allowing us to assign this as a terminal methyl group adjacent to the ester carbonyl. Presence of an ester was validated by FTIR (Fig. S17†) through the appearance of peaks at 1743 cm−1 and 1237 cm−1 which can be assigned to the ester's C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–O stretches, respectively; the latter distinguishable from the alcohol C–O stretch observed at 1051 cm−1.
O and C–O stretches, respectively; the latter distinguishable from the alcohol C–O stretch observed at 1051 cm−1.
The other notable feature in the full reaction 1H NMR is the appearance of a small singlet at 2.16 ppm. This signal couples to a small feature in the 13C NMR at 30.84 ppm seen in the 1H NMR and HSQC spectra with CH/CH3 phasing (Fig. 3 and S15†). The appearance of the expected ketone chemical shift in the 13C NMR is not observed, most likely due to a lower concentration of acetone combined with the low sensitivities of carbonyl groups in 13C NMR.51 However, in the HMBC spectrum the singlet at 2.16 ppm displays coupling to a carbon at ∼207 ppm (Fig. S16†). These shifts are consistent with acetone in CDCl3.52
The appearance of an ester and acetone are two of the expected products seen in the literature for the reaction between a metal acac and a primary alcohol.46,48,53 Based on these observations and the lack of features indicative of an ether, the oxide formation mechanism most likely follows the path reported by Niederberger and co-workers as laid out in path 2 of Scheme 1. The alcohol performs a nucleophilic attack on the acac carbonyl while an enolate and ester ligand coordinate to the In center. The ester is released and replaced with water. The water then undergoes proton transfer with the enolate generating acetone and a hydroxyl ligand. The L2In-OH species can then condense to form the oxygen deficient In2O3−x (Scheme 1). When DOA is present, it can also coordinate to the In center. With this mechanism in mind, we provide possible explanations for the different results observed when OLAM, OLAC, or In(OAc)3 were used in the reaction.
The addition of OLAM producing pristine particles and no indium metal (although its role as reducing agent is well documented), suggests it follows a much faster amidation analogue of path 2.45,48,54 Hutchison and co-workers assessed the use of primary amine vs. primary alcohol in the preparation of In2O3 and found that primary amines reacted much faster than the alcohol.54 To validate this report, we performed the same 2 mL volume test by using OLAM instead of OLOH; analyzing aliquots at the same interval with NMR as the OLOH trials seen in Fig. S18.† We tracked the diminishing of the amine adjacent protons at ∼2.66 ppm and the increase of the amide proton signal at ∼3.21 ppm. Forming ratios of amide:amine and ester:alcohol from the integrated proton signals (Table S2†), it can be seen that the amide quantity very quickly over takes the amine by approximate 2.5. In contrast, the ester never overtakes the alcohol amount in the same timeframe, validating the greater reactivity of the amine. To probe the effects of sterics, we replaced the 2 mL of DOA in the reaction with 2 mL of 4-octylaniline. The aniline derivative is a weaker primary amine with sterics between that of OLAM and DOA. The reaction initially turned black at 235 °C which subsequently lightened in color after ∼20 min at 270 °C. The PXRD in Fig. S19† shows a similar pattern to the black oxide pattern with a wider base suggesting increased polydispersity. TEM analysis seen in Fig. S19† shows a mix of large particles similar with the black particles’ morphology and small particles consistent with the pristine ones obtained when using OLAM. This result suggests that aniline initially reacts slow enough to allow for the indium metal and oxygen deficient particles to form but at the final reaction temperature the amidation pathway begins to take over. When OLAC is present in the reaction we posit a similar rate effect as that of the OLAM takes place. The presence of OLAC in the reaction typically results in the in situ formation of a metal oleate that serves as precursor.6,26 The reaction of carboxylates with an alcohol to produce the metal oxide proceeds via a simpler esterification pathway that does not require C–C bond breaking. This simpler pathway would also explain the observed difference in reactivity between In(acac)3 and In(OAc)3 as the acac involves C–C bond breaking while the acetate does not. Performing a similar small volume experiment we added 1 mL (∼3 mmol) of OLAC to the 2 mL of OLOH; a stoichiometric amount relative to the acac ligands. The 1HNMR analysis seen in Fig. S18† reveals an approximate 3 times increase in the ester:alcohol ratio, supporting our hypothesis that a faster condensation reaction occurs between the carboxylate when compared to the acac. Therefore, it is likely a synergistic effect between the acac and primary alcohol which allows for the conditions necessary to form the oxygen vacant oxide.
As illustrated in Scheme 1, the proposed overarching mechanism is as follows. At the visual onset of the black color change the reduction of In3+ to In0 through the oxidation of OLOH to oleyl aldehyde occurs rapidly (path 1) with concurrent formation of oxide particles containing oxygen vacancies (path 2). As the reaction is held at 270 °C for 1 h, the two paths converge (path 3). The metal particles get oxidized to form oxygen deficient In2O3−x with some particles containing an InyOz core while the In2O3−x formed through path 2 becomes more crystalline. The fact that in the standard reaction, all aliquots analyzed were black with their PXRD patterns containing reflections for the oxide is a good indication of oxygen deficient particles. In control reactions where pristine In2O3 particles formed concurrently with In0, the solution color was gray. To further validate that the obtained particles were rich in oxygen vacancies we performed a range of characterization techniques as discussed in the next sections.
Optical and vibrational characterization
A well-documented signature of metal oxides with oxygen vacancies is the emergence of distinct optical properties. Therefore, we performed diffuse reflectance measurements on the black In2O3−x nanoparticles. As control, we measured the spectra of 9 nm and 120 nm pristine, off-white In2O3 nanoparticles synthesized as reported by the Hutchison and Milliron groups, respectively (see Fig. S20 and S21† for PXRD and TEM).6,40 These two sizes allowed us to rule out any potential size induced effects. We also compared the spectrum for a batch of black In2O3−x nanoparticles that were annealed under an air atmosphere in a tube furnace at 400 °C for 24 h as a means to eliminate oxygen vacancies. Fig. 4 shows Kubelka–Munk curves obtained from the diffuse reflectance spectra of the four samples: black In2O3−x, annealed, “9 nm” and “120 nm”. The three control samples have very similar curve profiles with an onset of absorption around 3.6–3.7 eV, as expected based on the wide band gap of pristine In2O3. In contrast, the curve for the black In2O3−x particles displayed a significant red shift in the absorbance range extending into the NIR region. The observed enhanced absorption range is in agreement with literature reports on black In2O3−x, including ones made through other routes.16,26,28,55 Furthermore, some degree of tunability can be achieved by introducing dry air during the reaction after the particles are formed. These particles (PXRD Fig. S22†) showed optical properties between the black In2O3−x and pristine controls These results support the as-synthesized black particles poses oxygen vacancies which are eliminated when annealed in air post-synthesis, or have a degree of attenuation when subjecting the reaction to an oxidating environment during the reaction. Comparison of TEM images in Fig. S23† of the as-synthesized and post annealed particles confirms no significant size or shape differences, ruling out any potential morphological contribution to the observed differences in optical properties.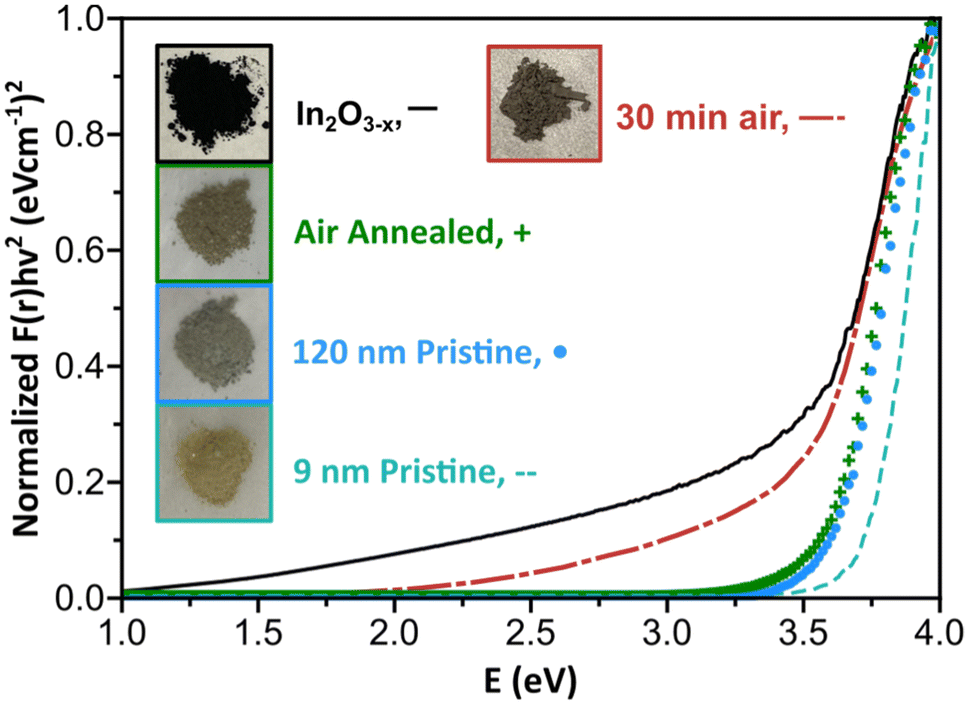 | ||
| Fig. 4 Kubelka–Munk plot of the black In2O3−x, 30 minutes of air flow, air annealed, 120 nm, and 9 nm nanoparticles using r = ½ for the direct transition. Inset shows dried powders of each of the measured nanoparticles. | ||
Aside from the enhanced optical properties, an additional signature of oxygen vacancies is a significantly altered Raman spectrum. The Millirion group and several other groups have attributed broadened and reduced Raman modes observed in black In2O3−x and other oxygen deficient oxides to phonon confinement.3,26,30,55,56 The phonon confinement arises from oxygen vacancies forming discrete disordered regions as opposed to being randomly distributed throughout the lattice. As a result, the long-range ordering in the lattice is disrupted quenching/broadening many of the expected modes. Such disordered regions have been observed through HR-TEM by the Ozin group and others.16,57,58Fig. 5 displays the Raman spectra for the black In2O3−x and the three control samples (annealed, “9 nm” and “120 nm”). Each control displays defined modes at ∼130, 305, 365, 494, and 627 cm−1, while the black In2O3−x only displays one easily identifiable peak at 127.8 cm−1 with the remaining four modes significantly diminished/broadened or absent. The high degree of crystallinity of the black In2O3−x (as evidenced by PXRD, HR-TEM, and SAED analysis), supports that the less defined spectrum is not due to discrete disordered regions rather than an overall amorphization. Comparing these results with the work of Milliron and coworkers on colloidal black In2O3, we can see a striking similarity between their most oxygen deficient Raman spectrum and our own.26 We also observed a slight red shift in the spectrum for black In2O3−x which has been attributed to laser induced heating of the dark material during measurements.26,55
 | ||
| Fig. 5 Raman spectra of the black In2O3−x, air annealed, 120 nm, and 9 nm nanoparticles. The observed Raman modes are labeled with their respective wavenumbers with smaller modes in the 120 nm and air annealed nanoparticles denoted by `. The vertical line crosses the peak at ∼130 cm−1 and shows a red-shift in the spectrum for the black In2O3−x sample. The asterisks denote signal from the room lighting. | ||
Surface analysis
Given the observation of indium metal during early stages of the reaction, XPS was employed to determine if any metal remained in the final product (within the 5–10 nm penetration depth limit of this technique). Fig. 6 shows the In 3d spectra for the black In2O3−x nanoparticles, and four controls (annealed, old In2O3−x, “9 nm” and “120 nm”) with a comparison to bulk indium metal seen in Fig. S25.† The overlay of each of the oxide In 3d spectra have nearly the same peak position (∼443.9 eV, 3d5/2 and ∼451.5 eV) with spin–orbit coupling of 7.6 eV. Peak position deviations fell within the C 1s calibration uncertainty of ±0.2 eV and were consistent with negligible deviations observed for the O 1s peaks.59 From the spectrum of the bulk indium metal sample, we see two discrete peaks superimposed upon one another. The lower binding energy peak and narrower line shape is due to the 0 valent oxidation state while the higher binding energy peak corresponds to surface oxidation. We can further differentiate the metal due to its plasmon loss feature at 454.9 eV.60 As an additional check for surface indium metal, we collected the InMNN Auger spectrum. Auger signals are much more sensitive to surface environments and can have drastically different line shapes, as is the case of In2O3 and In.61–63 Comparing the same five samples from above, we see in Fig. S25† a consistent line shape between all oxide samples. In contrast, while the Auger line shape for the metal sample was similar to the oxides it also displayed small features at 403.2 eV and 410.9 eV belonging to the metal. From the In 3d and InMNN Auger spectra we can reasonably conclude that any surface metal, if present, is in concentrations below the detection limit of XPS.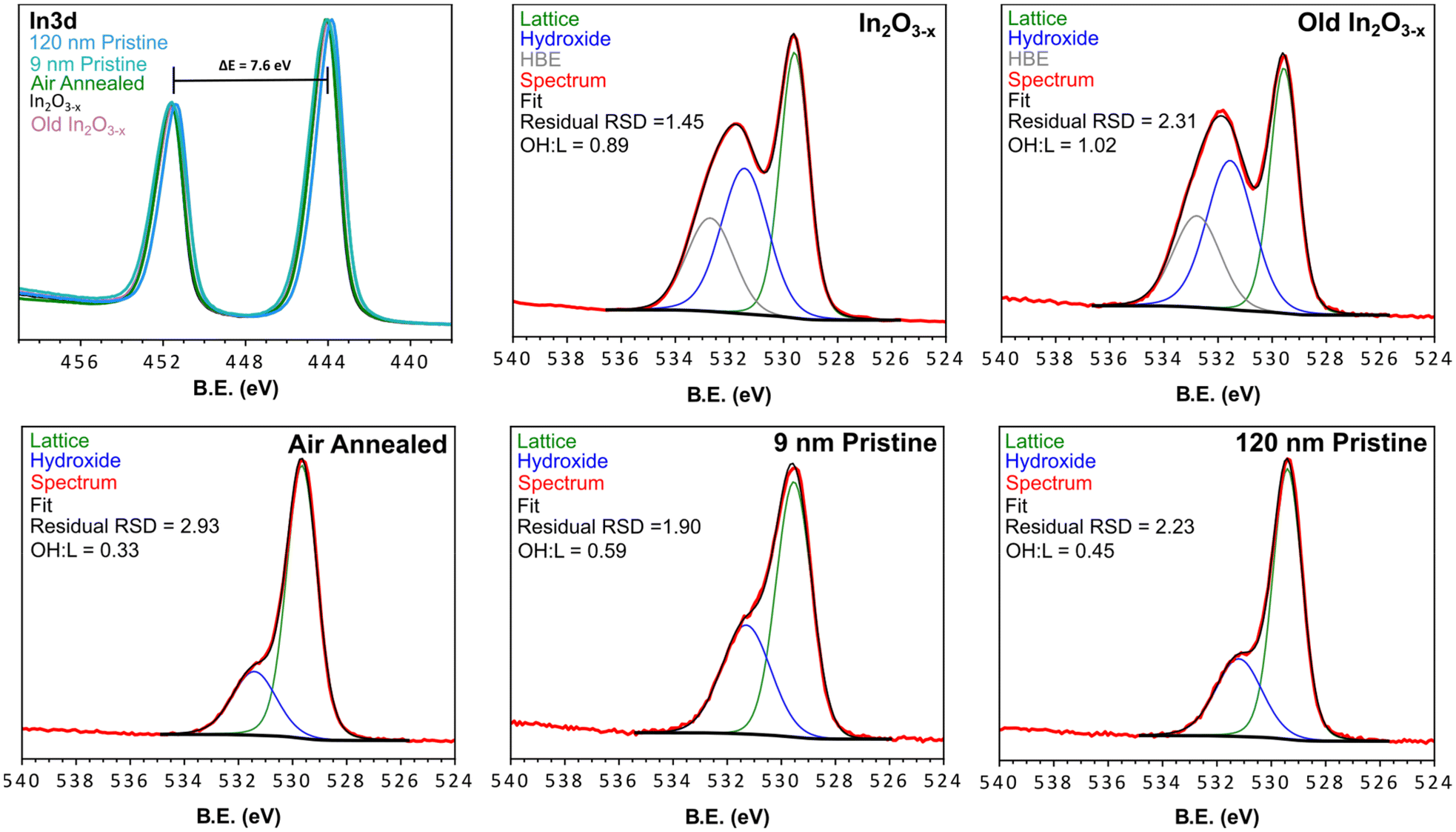 | ||
| Fig. 6 XPS spectra of the black In2O3−x, aged black In2O3−x, air annealed, 9 nm pristine, 120 nm pristine, and commercial samples. Each spectrum was fit with lattice and surface hydroxyl groups, while the black In2O3−x samples displayed an additional HBE feature. The Survey scans of each material are found in Fig. S24† and peak fitting parameters for the O 1s fitting are found in Table S3.† | ||
The O 1s XPS spectrum is commonly reported as tool to characterize and/or quantify oxygen vacancies in metal oxides. The rationale being that the presence of different oxidation states and/or coordination environment in the host metal centers, results in a shift in binding energy of the surrounding oxygen, or the appearance of new peaks. Upon inspection of the O 1s spectra in Fig. 6, a distinct difference is observed in the In2O3−x samples. The shoulder a few eV higher than the strongest peak displays greater broadening and intensity relative to the non-black control samples. By fitting the O 1s spectra of the black and control materials we can relate the areas to approximate atomic % of oxygen species at the surface and up to ∼10 nm within the particles (Fig. 6). From the peak fitting we observed 2 distinct regions corresponding to lattice oxygen of the metal oxide (∼529.5 eV), and surface coordinated hydroxyl groups (∼531.3 eV).64,65 Additionally, we observed a third feature at higher binding energy (∼532.7 eV, further referred to as HBE) of an unknown identity absent in the controls. In other reports, this feature has been assigned to loosely bound water, however this is unlikely under the ultra-high vacuum conditions the measurements are conducted under.
Upon inspection of the spectrum for the black In2O3−x, one can observe a much higher intensity with a larger peak-fit area relating to surface hydroxyl groups. Since the peak area is proportional to concentration, this observation indicates the black In2O3−x contains a higher concentration of surface hydroxyl groups relative to the controls. This result is consistent with literature reports that have shown an increased concentration of surface hydroxyl groups is associated with oxygen deficient metal oxides.28,66 In fact, the majority of oxygen vacant materials display this increase in surface coordinated oxygen containing species.16,26,28,35,66–68 While each of the analyzed materials displayed the expected hydroxyl peak, a comparison of the hydroxyl:lattice (OH:L) ratios (Fig. 6) showed the black In2O3−x displayed almost double the amount of surface hydroxyl groups compared to the control samples. Most notably, the comparison between the black and 9 nm pristine In2O3 eliminates the possibility of a size/surface area component as the larger black In2O3−x far exceed the 9 nm particles in terms of OH:LO ratio; as well as the 9 nm particles experiencing a more complete sampling due to their size. The increase in hydroxyl signal in the XPS fit is consistent with that reported by Milliron and Ozin for black and pristine In2O3.16,26 Furthermore, the appearance of the HBE feature in only the black indium oxide may be a further signature of additional oxygen species quenching the surface vacant sites. The stability of the particles was assessed by XPS analysis of old (>3 months) In2O3−x samples stored in toluene under ambient conditions. The spectrum for this sample (Fig. 6, top right corner) on par with the freshly prepared In2O3−x, indicating a consistent surface environment and long-term stability.
To complement the XPS analysis, we performed gas chemisorption measurement. Rosowski and co-workers utilized a creative approach using constant flow N2O chemisorption to quantify surface vacancies in hydrogenated In2O3 and ZrO2.18 The mechanism of N2O chemisorption has been described as the insertion of oxygen into a surface vacant oxygen site, followed by the release of diatomic nitrogen. As such, it is possible to monitor the consumption of N2O as a means to detect and quantify oxygen vacancies. As discussed above, oxygen vacant sites are higher in energy, making them susceptible to quenching. Prior to the measurements, a mild hydrogen pre-treatment was performed, as reported by Rosowski to strip the nanoparticles of any ligands utilized in the synthesis and surface quenching species.18 Performing the titration following the pre-treatment we observed N2O consumption/evolution in all samples as seen in Fig. S26.† From these measurements, we were able to calculate a surface vacancy density for the 9 nm pristine, 120 nm pristine, and black In2O3−x to be 205.6, 131.3, and 230.8 μmol g−1, respectively. Thus, the black In2O3−x displayed the highest degree of consumption, indicating a higher concentration of vacancies. We acknowledge that the hydrogenation pre-treatment could induce vacancies in all samples. However, from the H2 consumption plots (Fig. S26†) we see a very consistent trend in uptake, suggesting similar responses by each of the trials. Holding all things equal, it would be expected that the higher surface area of the 9 nm pristine In2O3 would have the highest vacancy density, while the 120 nm and black particles would have similar values. Therefore, the larger response of the black In2O3−x can be attributed to the pre-existing vacancies and not from the hydrogenation pre-treatment.
Conclusions
In this report, the molecular and growth mechanism that led to the formation of black In2O3−x nanoparticles in a simple one-pot heat-up synthesis were studied via time- and temperature-dependent analysis of the reaction. Two concurrent/convergent paths were uncovered that involved formation of In0 particles that would then oxidize and direct formation of In2O3−x. The synthesis atmosphere, ligand environment, and In precursor were key factors in obtaining the black In2O3−x nanoparticles. Optical absorption measurements revealed the synthesized material had an absorption range that extended into the NIR as expected for a wide band gap material with high oxygen vacancies. The presence of oxygen vacancies was further validated through Raman spectroscopy, XPS, and gas adsorption measurements. Ongoing work is focused on incorporation of dopants into this reaction to further tune the optical properties, as well as evaluating the photo(electro)catalytic properties of these nanoparticles. Additionally, we strive to perform in situ PXRD and TEM experiments in future work to gain further insight into the nucleation and growth of this material. We believe the results presented here could be insightful towards the synthetic control of oxygen vacancies in other metal oxides.Author contributions
C. A. co-developed experimental methodology, performed synthesis, characterization/data analysis, wrote initial draft, worked on revisions/edits, and prepared figures for the manuscript. K. O. aided in the reproduction of experiments, collection of samples for analysis, aided in NMR data collection, and revised the manuscript. E. A. H-P. co-designed experimental methodology, supervised/advised and revised/edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Brandon Vance for his help in our collection of N2O chemisorption data, Dr Sviatoslav Baranets for his help with the thermal annealing, Danielle Gendler for aiding in the collection of FTIR and NMR data, Ashkan Yazdanshenas for aiding in NMR data collection. Financial support for this work was provided by University of Delaware start-up funds and a Delaware COBRE seed grant supported by the National Institute of General Medical Sciences (NIGMS 1 P20 GM104316-01A1). K. O. thanks the GEM Fellowship Program for support. XPS analysis was performed with the instrument sponsored by the National Science Foundation under grant no. CHE-1428149. Raman analysis was performed with the instrument sponsored by the National Science Foundation under grant no. CHE-1828325. NMR facilities are supported through NIH P20GM104316. HRTEM and STEM-EDS was conducted at the Vanderbilt Institute of Nanoscale Science and Engineering. We also thank the reviewers for their comments and suggested experiments.References
- L. Zhang, J. Ran, S.-Z. Qiao and M. Jaroniec, Chem. Soc. Rev., 2019, 48, 5184–5206 RSC.
- M. S. S. Danish, L. L. Estrella, I. M. A. Alemaida, A. Lisin, N. Moiseev, M. Ahmadi, M. Nazari, M. Wali, H. Zaheb and T. Senjyu, Metals, 2021, 11, 80 CrossRef CAS.
- A. Naldoni, M. Allieta, S. Santangelo, M. Marelli, F. Fabbri, S. Cappelli, C. L. Bianchi, R. Psaro and V. Dal Santo, J. Am. Chem. Soc., 2012, 134, 7600–7603 CrossRef CAS PubMed.
- Á. Balog, G. F. Samu, S. Pető and C. Janáky, ACS Mater. Au, 2021, 1, 157–168 CrossRef PubMed.
- Y. Sang, H. Liu and A. Umar, ChemCatChem, 2015, 7, 559–573 CrossRef CAS.
- A. W. Jansons, K. M. Koskela, B. M. Crockett and J. E. Hutchison, Chem. Mater., 2017, 29, 8167–8176 CrossRef CAS.
- C. Di Valentin, G. Pacchioni and A. Selloni, J. Phys. Chem. C, 2009, 113, 20543–20552 CrossRef CAS.
- K. R. Reyes-Gil, E. A. Reyes-García and D. Raftery, J. Phys. Chem. C, 2007, 111, 14579–14588 CrossRef CAS.
- R. Medhi, M. D. Marquez and T. R. Lee, ACS Appl. Nano Mater., 2020, 3, 6156–6185 CrossRef CAS.
- Y. Liu, W. Wang, X. Xu, J.-P. M. Veder and Z. Shao, J. Mater. Chem. A, 2019, 7, 7280–7300 RSC.
- H. Sun, W. Fan, Y. Li, X. Cheng, P. Li and X. Zhao, J. Phys. Chem. C, 2010, 114, 3028–3036 CrossRef CAS.
- N. Sareecha, W. A. Shah, M. L. Mirza, A. Maqsood and M. S. Awan, Phys. B, 2018, 530, 283–289 CrossRef CAS.
- R. Jaiswal, J. Bharambe, N. Patel, A. Dashora, D. C. Kothari and A. Miotello, Appl. Catal., B, 2015, 168–169, 333–341 CrossRef CAS.
- F. Qi, Z. Yang, J. Zhang, Y. Wang, Q. Qiu and H. Li, ACS Appl. Mater. Interfaces, 2020, 12, 55417–55425 CrossRef CAS PubMed.
- K. Yamanaka and T. Morikawa, J. Phys. Chem. C, 2012, 116, 1286–1292 CrossRef CAS.
- L. Wang, Y. Dong, T. Yan, Z. Hu, A. A. Jelle, D. M. Meira, P. N. Duchesne, J. Y. Y. Loh, C. Qiu, E. E. Storey, Y. Xu, W. Sun, M. Ghoussoub, N. P. Kherani, A. S. Helmy and G. A. Ozin, Nat. Commun., 2020, 11, 2432 CrossRef CAS PubMed.
- S. Arooj, T. Xu, X. Hou, Y. Wang, J. Tong, R. Chu and B. Liu, RSC Adv., 2018, 8, 11828–11833 RSC.
- R. Baumgarten, R. Naumann d'Alnoncourt, S. Lohr, E. Gioria, E. Frei, E. Fako, S. De, C. Boscagli, M. Drieß, S. Schunk and F. Rosowski, Chem. Ing. Tech., 2022, 94, 1765–1775 CrossRef CAS.
- L. Liu, Y. Jiang, H. Zhao, J. Chen, J. Cheng, K. Yang and Y. Li, ACS Catal., 2016, 6, 1097–1108 CrossRef CAS.
- Z.-Q. Wang, M.-J. Zhang, X.-B. Hu, V. P. Dravid, Z.-N. Xu and G.-C. Guo, Chem. Commun., 2020, 56, 403–406 RSC.
- D. B. Buchholz, Q. Ma, D. Alducin, A. Ponce, M. Jose-Yacaman, R. Khanal, J. E. Medvedeva and R. P. H. Chang, Chem. Mater., 2014, 26, 5401–5411 CrossRef CAS PubMed.
- L. Liu, C.-C. He, J. Zeng, Y.-H. Peng, W.-Y. Chen, Y.-J. Zhao and X.-B. Yang, J. Phys. Chem. C, 2021, 125, 7077–7085 CrossRef CAS.
- H. H. Pham and L.-W. Wang, Phys. Chem. Chem. Phys., 2014, 17, 541–550 RSC.
- X. Chen, L. Liu, P. Y. Yu and S. S. Mao, Science, 2011, 331, 746–750 CrossRef CAS PubMed.
- W. Fang, M. Xing and J. Zhang, Appl. Catal., B, 2014, 160–161, 240–246 CrossRef CAS.
- K. Kim, J. Yu, J. Noh, L. C. Reimnitz, M. Chang, D. R. Gamelin, B. A. Korgel, G. S. Hwang and D. J. Milliron, J. Am. Chem. Soc., 2022, 144, 22941–22949 CrossRef CAS PubMed.
- K. Manthiram and A. P. Alivisatos, J. Am. Chem. Soc., 2012, 134, 3995–3998 CrossRef CAS PubMed.
- Z. Zhang, C. Mao, D. M. Meira, P. N. Duchesne, A. A. Tountas, Z. Li, C. Qiu, S. Tang, R. Song, X. Ding, J. Sun, J. Yu, J. Y. Howe, W. Tu, L. Wang and G. A. Ozin, Nat. Commun., 2022, 13, 1512 CrossRef CAS PubMed.
- T. M. Mattox, A. Bergerud, A. Agrawal and D. J. Milliron, Chem. Mater., 2014, 26, 1779–1784 CrossRef CAS.
- J. Li, Y. Liu, Z. Zhu, G. Zhang, T. Zou, Z. Zou, S. Zhang, D. Zeng and C. Xie, Sci. Rep., 2013, 3, 2409 CrossRef PubMed.
- H. Han, S. Jin, S. Park, Y. Kim, D. Jang, M. H. Seo and W. B. Kim, Nano Energy, 2021, 79, 105492 CrossRef CAS.
- S. Sharma Kanakkillam, B. Krishnan, S. Sepulveda Guzman, J. Amilcar Aguilar Martinez, D. Avellaneda Avellaneda and S. Shaji, Appl. Surf. Sci., 2021, 567, 150858 CrossRef CAS.
- X. He, H. Guo, T. Liao, Y. Pu, L. Lai, Z. Wang and H. Tang, Nanoscale, 2021, 13, 16307–16315 RSC.
- F. Gu, C. Li, D. Han and Z. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 933–942 CrossRef CAS PubMed.
- A. Sinhamahapatra, J.-P. Jeon, J. Kang, B. Han and J.-S. Yu, Sci. Rep., 2016, 6, 27218 CrossRef CAS PubMed.
- W. Wei, Z. Wei, R. Li, Z. Li, R. Shi, S. Ouyang, Y. Qi, D. L. Philips and H. Yuan, Nat. Commun., 2022, 13, 3199 CrossRef PubMed.
- A. Solieman, J. Sol-Gel Sci. Technol., 2011, 60, 48 CrossRef CAS.
- D. Seok, M. Wu, K. B. Shim, Y. Kang and H.-K. Jung, Nanotechnology, 2016, 27, 435401 CrossRef PubMed.
- J. Seo, Y. Jun, S. J. Ko and J. Cheon, J. Phys. Chem. B, 2005, 109, 5389–5391 CrossRef CAS PubMed.
- S. H. Cho, S. Ghosh, Z. J. Berkson, J. A. Hachtel, J. Shi, X. Zhao, L. C. Reimnitz, C. J. Dahlman, Y. Ho, A. Yang, Y. Liu, J.-C. Idrobo, B. F. Chmelka and D. J. Milliron, Chem. Mater., 2019, 31, 2661–2676 CrossRef CAS.
- S. Tada, M. Yokoyama, R. Kikuchi, T. Haneda and H. Kameyama, J. Phys. Chem. C, 2013, 117, 14652–14658 CrossRef CAS.
- D. Caruntu, K. Yao, Z. Zhang, T. Austin, W. Zhou and C. J. O'Connor, J. Phys. Chem. C, 2010, 114, 4875–4886 CrossRef CAS.
- T. Suzuki, H. Watanabe, T. Ueno, Y. Oaki and H. Imai, Langmuir, 2017, 33, 3014–3017 CrossRef CAS PubMed.
- P. Erhart, A. Klein, R. G. Egdell and K. Albe, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 153205 CrossRef.
- J. L. Rowell, Y. Jia, Z. Shi, A. Molina Villarino, M. Kang, D. Yoon, K. Z. Jiang, H. D. Abruña, D. A. Muller and R. D. Robinson, J. Am. Chem. Soc., 2023, 145, 17406–17419 CrossRef CAS PubMed.
- M. Staniuk, D. Zindel, W. van Beek, O. Hirsch, N. Kränzlin, M. Niederberger and D. Koziej, CrystEngComm, 2015, 17, 6962–6971 RSC.
- M. Niederberger, Acc. Chem. Res., 2007, 40, 793–800 CrossRef CAS PubMed.
- M. Niederberger and G. Garnweitner, Chem. – Eur. J., 2006, 12, 7282–7302 CrossRef CAS PubMed.
- M. E. F. Brollo, R. López-Ruiz, D. Muraca, S. J. A. Figueroa, K. R. Pirota and M. Knobel, Sci. Rep., 2014, 4, 6839 CrossRef CAS PubMed.
- J. Lee, T. Ryu, S. Park and P. H. Lee, J. Org. Chem., 2012, 77, 4821–4825 CrossRef CAS PubMed.
- J. Joseph, C. Baker, S. Mukkamala, S. H. Beis, M. C. Wheeler, W. J. DeSisto, B. L. Jensen and B. G. Frederick, Energy Fuels, 2010, 24, 5153–5162 CrossRef CAS.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- L. Zhang, G. Garnweitner, I. Djerdj, M. Antonietti and M. Niederberger, Chem. – Asian J., 2008, 3, 746–752 CrossRef PubMed.
- T. A. Knecht and J. E. Hutchison, Chem. Mater., 2023, 35, 3151–3161 CrossRef CAS.
- M. Ziemba, L. Schumacher and C. Hess, J. Phys. Chem. Lett., 2021, 12, 3749–3754 CrossRef CAS PubMed.
- A. K. Arora, M. Rajalakshmi, T. R. Ravindran and V. Sivasubramanian, J. Raman Spectrosc., 2007, 38, 604–617 CrossRef CAS.
- H. Zhang, B. M. May, F. Omenya, M. S. Whittingham, J. Cabana and G. Zhou, Chem. Mater., 2019, 31, 7790–7798 CrossRef CAS.
- Z. Sadighi, J. Huang, L. Qin, S. Yao, J. Cui and J.-K. Kim, J. Power Sources, 2017, 365, 134–147 CrossRef CAS.
- T. R. Gengenbach, G. H. Major, M. R. Linford and C. D. Easton, J. Vac. Sci. Technol., A, 2021, 39, 013204 CrossRef CAS.
- Z. M. Detweiler, S. M. Wulfsberg, M. G. Frith, A. B. Bocarsly and S. L. Bernasek, Surf. Sci., 2016, 648, 188–195 CrossRef CAS.
- M. C. Biesinger, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Phys. Chem. Chem. Phys., 2012, 14, 2434–2442 RSC.
- A. W. C. Lin, N. R. Armstrong and T. Kuwana, Anal. Chem., 1977, 49, 1228–1235 CrossRef CAS.
- A. J. Barlow, N. Sano, B. J. Murdoch, J. F. Portoles, P. J. Pigram and P. J. Cumpson, Appl. Surf. Sci., 2018, 459, 678–685 CrossRef CAS.
- H. Idriss, Surf. Sci., 2021, 712, 121894 CrossRef CAS.
- J. Y. Y. Loh and N. P. Kherani, Molecules, 2019, 24, 3818 CrossRef CAS PubMed.
- G. Zhan, J. Li, Y. Hu, S. Zhao, S. Cao, F. Jia and L. Zhang, Environ. Sci.: Nano, 2020, 7, 1454–1463 RSC.
- S.-S. Li and Y.-K. Su, RSC Adv., 2019, 9, 2941–2947 RSC.
- J. Gan, X. Lu, J. Wu, S. Xie, T. Zhai, M. Yu, Z. Zhang, Y. Mao, S. C. I. Wang, Y. Shen and Y. Tong, Sci. Rep., 2013, 3, 1021 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: SAED, PXRD, NMR, FTIR, XPS, gas adsorption measurements. See DOI: https://doi.org/10.1039/d3nr05035a |
| This journal is © The Royal Society of Chemistry 2024 |