
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Liquid-phase hydrogenation of carbon monoxide to methanol using a recyclable manganese-based catalytic system†
Sebastian
Stahl
ab,
Niklas
Wessel
ab,
Andreas J.
Vorholt
 *a and
Walter
Leitner
*ab
*a and
Walter
Leitner
*ab
aMax Planck Institute for Chemical Energy Conversion, Stiftstraße 34–36, 45470 Mülheim an der Ruhr, Germany. E-mail: andreas-j.vorholt@cec.mpg.de; walter.leitner@cec.mpg.de
bInstitute for Technical and Macromolecular Chemistry, RWTH Aachen University, Worringerweg 2, 52074 Aachen, Germany
First published on 7th June 2024
Abstract
A simple and recyclable homogeneous catalytic system for the hydrogenation of carbon monoxide to methanol was established. The reaction is catalyzed by a molecular manganese complex using a high-boiling alcohol as the solvent for catalyst immobilization. The CO hydrogenation is assisted by the product itself and the solvent through the formation of a methyl or dodecyl formate ester intermediate mediated by catalytic amounts of NaOMe as the base. This allows the catalytic formation of methanol in alcohols combined with facile product separation and catalyst recycling via distillation. Initial turnover frequencies (TOF) of 2250 h−1 were reached under optimized conditions in 1-dodecanol/methanol as the reaction medium (T = 160 °C, p(H2/CO) = 80/10 bar). The performance was stabilized in batch-wise recycling over 6 runs achieving a total turnover number (TTON) of >12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 corresponding to an enhancement of more than five times compared to single batch operation under identical conditions. Minimal leaching of the components of the organometallic catalyst was observed during distillative product separation and catalyst activity could be fully restored by re-addition of the base NaOMe.
000 corresponding to an enhancement of more than five times compared to single batch operation under identical conditions. Minimal leaching of the components of the organometallic catalyst was observed during distillative product separation and catalyst activity could be fully restored by re-addition of the base NaOMe.
Introduction
Methanol is of central importance for the future “defossilized” chemical supply chain.1,2 Already today methanol is one of the largest volume basic chemicals and an important C1 building block for numerous organic transformations such as the production of formaldehyde or acetic acid.3–5 In addition, H3COH can be viewed as “liquid” synthesis gas providing H2/CO (2:1) upon catalytic dehydrogenation as high energy and valuable industrial building blocks.6 Methanol to olefins (MTO) or gasoline (MTG) and related technologies enable the production of aliphatic and aromatic hydrocarbons as a sustainable substitute for fossil resources when employing “green” methanol.7,8 Methanol itself holds great potential as a substitute fuel in hard-to-electrify sectors such as heavy-duty transportation and the shipping industry.9,10 Additionally, its properties (12.6 wt% hydrogen, liquid at room temperature) make methanol a promising candidate for a key pivot point in a hydrogen economy.11,12
While the heterogeneously catalysed gas phase process from syngas to methanol operating at high temperatures (>200 °C) is well-established.13–15 Typically, the gas mixture contains certain amounts of CO2 which is the more active C1 compound over standard catalysts involving water–gas-shift equilibria as part of the reaction network. In contrast, the use of organometallic complexes in homogeneous catalysis for the transformation of H2/CO to methanol is far less explored. Possible lower reaction temperatures and operation in liquid phase can be expected to result in higher one-through conversions of CO due to favourable thermodynamics, making such systems particularly attractive for the integration with decentralized renewable carbon and energy supply. The direct hydrogenation of CO has been proven challenging, however, requiring harsh conditions even for only small turnover numbers.16–23 This is mainly due to the high binding affinity of CO with organometallic complexes and the high endothermicity of its migratory insertion into metal-hydride bonds.24–26 The groups of Prakash27 and Beller28 aimed to evade this limitation via the use of amines as co-reagents to form formamides as reactive intermediates. While marking a major advance in the productivity of CO hydrogenation under mild conditions, these systems suffer from incomplete formamide conversion and trace amounts of N-methylated side-product formation.
In this context, our group developed a homogeneous catalytic approach for the alcohol-assisted hydrogenation of CO to methanol.29 The CO is activated as formate esters in situ in the presence of the alcohol co-reagent using a catalytic amount of NaOtBu as the base. The formate group is then hydrogenated in two consecutive steps to methanol liberating the alcohol to act as a co-reagent again (Scheme 1), which is based on the seminal work by Milstein,30,31 Sanford32,33 and others.34,35 In particular, it was demonstrated that the reaction can also be performed with the product itself, methanol, as the alcohol component, opening the possibility for catalytic “breeding” of methanol from H2/CO. Using a manganese pincer-complex comprising the MACHO-type ligand as catalyst (Mn-1), a TON of 4023 and a turnover frequency (TOF) of 857 h−1 with a methanol selectivity >99% was reached at 150 °C and 55 bar with ethanol as the alcohol component. During the preparation of the present article, the Beller group reported the utilization of a novel PNP ligand design (Mn-2) increasing the TOF to >1600 h−1 (Fig. 1).36
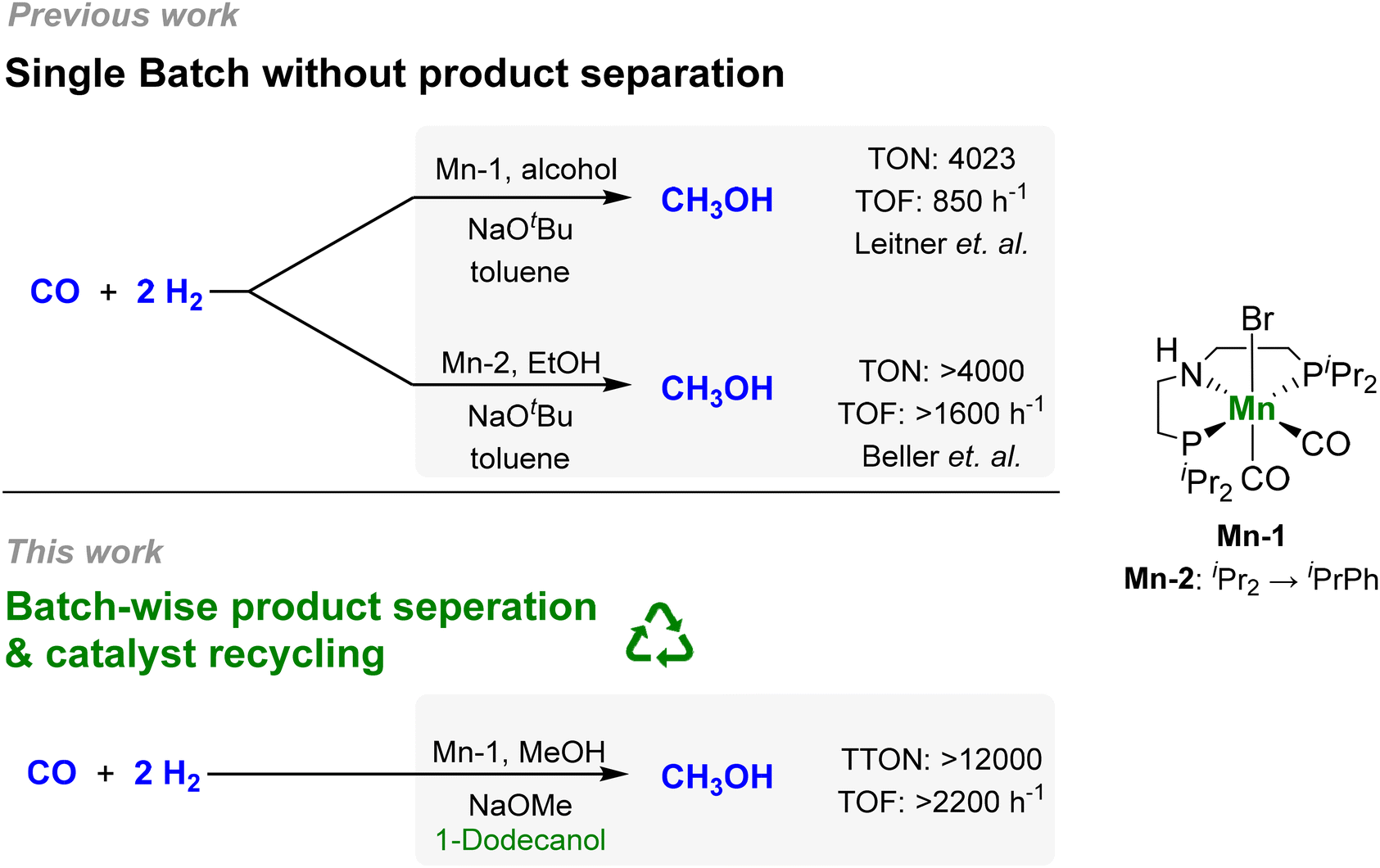 | ||
| Fig. 1 Development of the alcohol-assisted carbon monoxide hydrogenation with molecular pincer catalysts.29,36 | ||
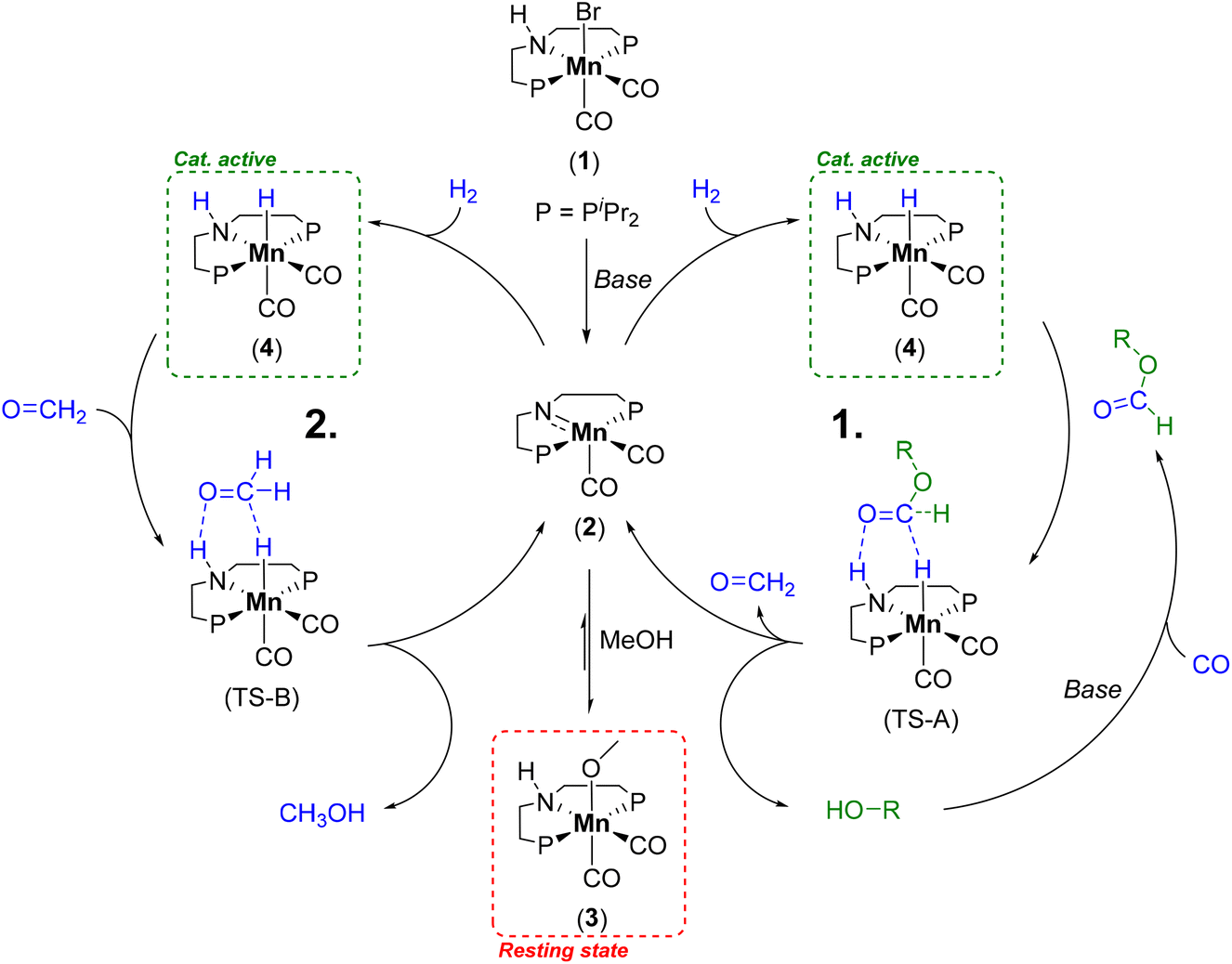 | ||
| Scheme 1 Catalytic cycle for the alcohol mediated hydrogenation of CO by Mn-1 to methanol illustrating the formate ester hydrogenation to formaldehyde (cycle 1) and the consecutive hydrogenation to methanol (cycle 2).29 | ||
Based on these recent developments in Mn-catalysed liquid phase CO hydrogenation, a first attempt to demonstrate the homogeneously catalyzed methanol synthesis in combination with syngas production at a pilot facility was recently implemented.37 A major challenge for homogeneous catalysis is, however, the inherent challenge to separate and recycle the organometallic active species from the product. In the present study, we therefore aimed to develop an optimized catalytic system integrating the alcohol-assisted CO hydrogenation with facile product separation for the synthesis of methanol and recycling of the organometallic catalyst.
Results and discussion
Due to the low boiling point of methanol and the high selectivity of the reaction, we decided on a recycling strategy via distillation. For an initial screening of possible high boiling solvents allowing for separation of catalytically produced methanol, we used the defined molecular complex Mn-MACHO-iPr (Mn-1) as pre-catalyst which is air-stable in its solid form. NaOtBu was used as a base to activate the precatalyst and the CO in the presence of pre-charged methanol via methyl formate as the intermediate (Scheme 1). With the solvent screening, we aim to increase the boiling point (BP) difference between solvent and product, to avoid azeotrope formation as in the reported toluene system,29 and to ideally use green solvents.38For this screening 5 μmol of Mn-1 with 10 eq. of NaOtBu was employed in 1.5 ml of a mixture of the solvent with methanol at a temperature of 150 °C and a pressure of 10 bar of CO and 50 bar of H2 for 4 h. The previously used solvent toluene as the reference system gave high TONs, as expected, but product isolation was not possible due to azeotrope formation (Table 1 entry 1). Product inhibition effects were found to impede the utilization of methanol itself as the solvent (TON of 64, entry 2). This most likely reflects that the heterolytic hydrogen cleavage to form the catalytically active complex (4) competes with the methanolate complex (3) (Scheme 1).39
| Entry | Solvent | MeOH wt% (%) | n (Methyl formate) (mmol) | n (Dodecyl formate) (mmol) | n (MeOH) (mmol) | S to MeOH (%) | TON (MeOH) |
|---|---|---|---|---|---|---|---|
| Conditions: CO (10 bar), H2 (50 bar), Mn-1 (5 μmol), NaOtBu (50 μmol), solvent + methanol (1.5 mL), 150 °C, 4 h, 20 mL autoclave. Selectivities calculated as molar fraction of total products detected (methanol, methyl and dodecyl formate).a Azeotrope formation with methanol.b Octyl formate. | |||||||
| 1 | Toluenea | 33 | 0.23 | — | 7.28 | 97 | 1289 |
| 2 | Methanol | 100 | 0.99 | — | 0.37 | 27 | 64 |
| 3 | 1-Dodecanol | 50 | 0.63 | 0.09 | 2.87 | 80 | 567 |
| 4 | 1-Dodecanol | 33 | 0.31 | 0.06 | 4.73 | 93 | 869 |
| 5 | 1-Dodecanol | 25 | 0.21 | 0.06 | 6.48 | 96 | 1189 |
| 6 | 1-Octanol | 33 | 0.34 | 0.11b | 4.51 | 91 | 827 |
Polar high boiling (>200 °C) solvents like N-Methyl-2-pyrrolidon (NMP) or γ-valerolacton showed no activity (ESI, Table S1, entries 1 and 2†). We then turned our attention to long-chain alcohols as they can also act as CO activators in the presence of base and fulfils all desired criteria. Indeed, promising TONs for methanol formation were observed with formate esters of methanol and the solvents as the only detectable side-products (entries 3–6). The performance in terms of TONs in these solvents could be further enhanced by decreasing the initial methanol content in the solvent mixture. With 1-dodecanol (BP 259 °C) containing initially 25% MeOH a TON of 1189 was finally reached, which is comparable to the benchmark system in toluene (entries 1 and 5). While similar productivity was reached in 1-octanol (entry 6), its lower boiling point (BP 195 °C) makes it less promising for separation and product isolation. NMR spectra and GC-MS analysis of the solution after the catalytic reaction confirmed that the reaction system does not form any side-products and only consists of the initially charged components when long-chain alcohols are used as the solvent.
With a solvent system combining high productivity and potential for recycling in hand (25% methanol in 1-dodecanol) we aimed to understand the influence of different reaction parameters to increase the TON values of the system. Variation of the phosphine substituents at the MACHO ligand of catalyst Mn-1 (Fig. 1) showed slightly decreased reactivity for the cyclohexyl (TON 1034) and no reactivity for the tert-butyl derivative (ESI, Table S1, entries 3 and 4†). Presumably, the increased steric demand compared to Mn-1 is impeding hydride transfer to the formate ester intermediate and thus reducing catalytic activity. We significantly simplified the system by exchanging the previously used base NaOtBu with NaOMe, as the corresponding base of the product, which both show similar performance. A minimum base amount of 10 eq. relative to the Mn-1 was found necessary to achieve reactivity under these conditions (Table 2, entries 1–5). A higher base concentration improves the reaction to a maximum TON of 3411 at a base/Mn ratio of 75:1. Computational analysis of the mechanism indicated the first hydrogenation of methyl formate to formaldehyde as the rate determining step.36 Thus, it can be assumed that a higher base amount is increasing the formate concentration which accelerates this step in the catalytic cycle. However, very high base concentrations were found detrimental, presumably due to catalyst deactivation. Selectivity to methanol was above 90% for all reactions and only unreacted formate ester intermediates were detected as side-products.
| Entry | H2/CO Ratio | Base equivalents | n (Methyl formate) (mmol) | n (Dodecyl formate) (mmol) | n (MeOH) (mmol) | S to MeOH (%) | TON (MeOH) |
|---|---|---|---|---|---|---|---|
| Conditions: CO + H2 (60 bar total), Mn-1 (2 μmol), NaOMe, 25% Methanol in 1-Dodecanol (1.5 ml), 150 °C, 5 h, 20 mL autoclave. Selectivities calculated as molar fraction of total products detected (methanol, methyl and dodecyl formate).a 90 bar total pressure. | |||||||
| 1 | 5:1 |
10 | 0.25 | 0.10 | 3.33 | 90 | 1665 |
| 2 | 5:1 |
25 | 0.24 | 0.08 | 4.66 | 94 | 2360 |
| 3 | 5:1 |
50 | 0.24 | 0.04 | 5.49 | 95 | 2745 |
| 4 | 5:1 |
75 | 0.28 | 0.09 | 6.48 | 95 | 3411 |
| 5 | 5:1 |
100 | 0.29 | 0.10 | 5.30 | 93 | 2522 |
| 6 | 2:1 |
25 | 0.63 | 0.35 | 1.50 | 61 | 712 |
| 7 | 3:1 |
25 | 0.44 | 0.21 | 3.08 | 82 | 1464 |
| 8 | 7:1 |
25 | 0.05 | 0.02 | 5.82 | 99 | 2844 |
| 9a | 8:1 |
25 | 0.07 | 0.03 | 8.06 | 99 | 3930 |
Subsequently, the influence of the partial pressures of the reactive gases was investigated (Table 2, entries 6–9). While a higher CO partial pressure would seem beneficial for a higher formate ester intermediate concentration, CO as a strong binding ligand can also inhibit the hydrogenation activity of the catalyst. When keeping the total pressure constant, a higher H2/CO ratio above the stochiometric ratio of 2:1 leads indeed to an increase of the TON. A further improvement was achieved by simultaneously increasing the H2 partial pressure (H2/CO = 8:1) and the total pressure to 90 bar reaching a TON of 3930 within 5 h.
Varying the reaction temperature (Fig. 2) confirms the formate ester hydrogenation as the overall rate determining transformation. While the formation of the formate esters already takes place at lower temperatures, a minimum of 130 °C is necessary to produce methanol. A temperature of 160 °C provided the optimal balance between reaction rate and catalyst stability. At temperatures above 160 °C deactivation of the catalyst starts to set in as indicated by a color change from a clear, slightly yellow solution to a cloudy brown mixture and supported by NMR experiments (see ESI† for details). The highest TON of 4555 was achieved after 20 h at 160 °C corresponding to almost full CO conversion, derived from the theoretical yield calculated via the ideal gas equation (ESI, Table S3†), at 99% methanol selectivity.
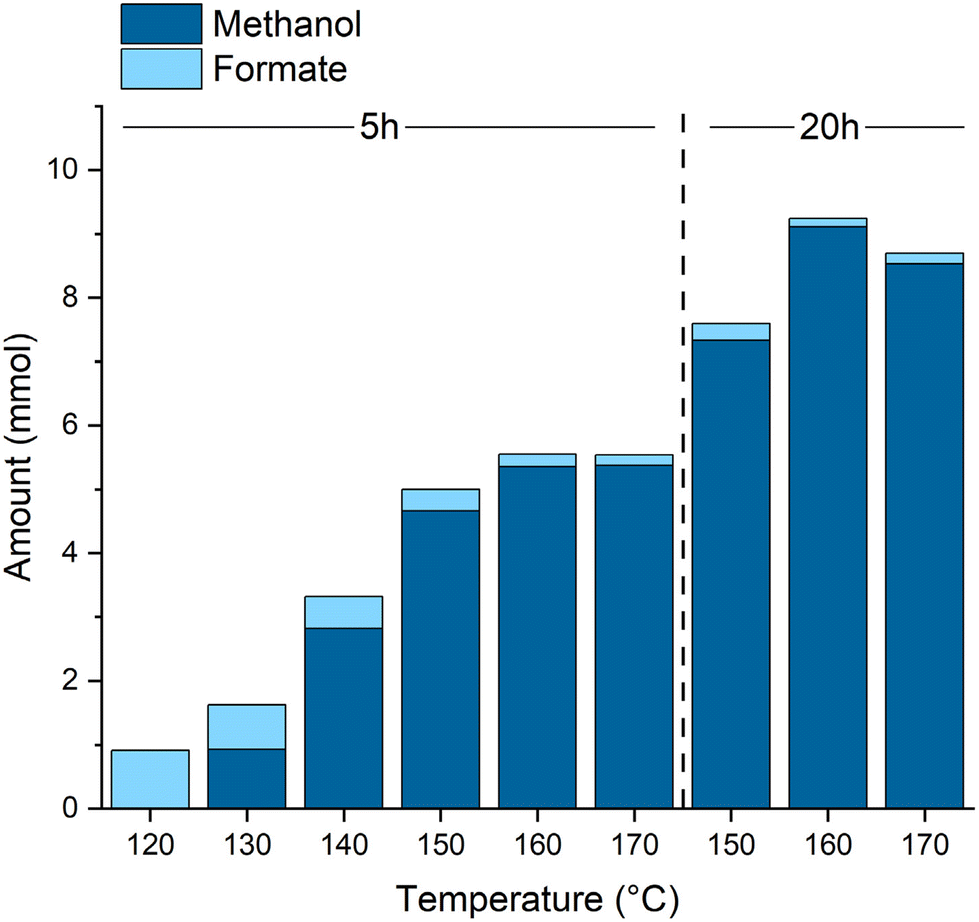 | ||
| Fig. 2 Amount of Methanol and Formate ester (total of methyl- and dodecylformate) at various temperatures and reaction times. Conditions: CO (10 bar), H2 (50 bar), Mn-1 (2 μmol), NaOMe (50 μmol), 25% methanol in 1-dodecanol (1.5 ml), T, t, 20 mL autoclave. | ||
A concentration/time profile under the typcial conditions was obtained by performing the reaction in a 280 ml reactor equipped with on-line NMR and IR analytics. In Fig. 3 the course of the concentration of methanol and the two formate ester intermediates are illustrated as monitored by the indicated NMR signals. Both formate ester concentrations can also be followed via IR (ESI, Fig. S3 and 4†). After pressurization, the methyl formate ester is formed rapidly within minutes, while the dodecyl formate takes around 5 h to reach its highest concentration. Consequently, besides different pKa values, both alcohols get deprotonated and form formate ester intermediates in the course of the reaction. The rate of methanol formation is highest in the beginning while the highest total formate concentration (methyl and dodecyl formate) is present. The total formate ester concentration decreases in line with the recorded pressure drop (ESI, Fig. S5†) supporting that the concentration of formate esters depends on the partial pressure of carbon monoxide. The methyl formate ester appears to react preferentially to methanol being consumed completely after a reaction time of 10 h. However, the reaction is still proceeding after this point suggesting that the dodecyl formate ester can also act as a reactive intermediate, albeit apparently at a lower rate.
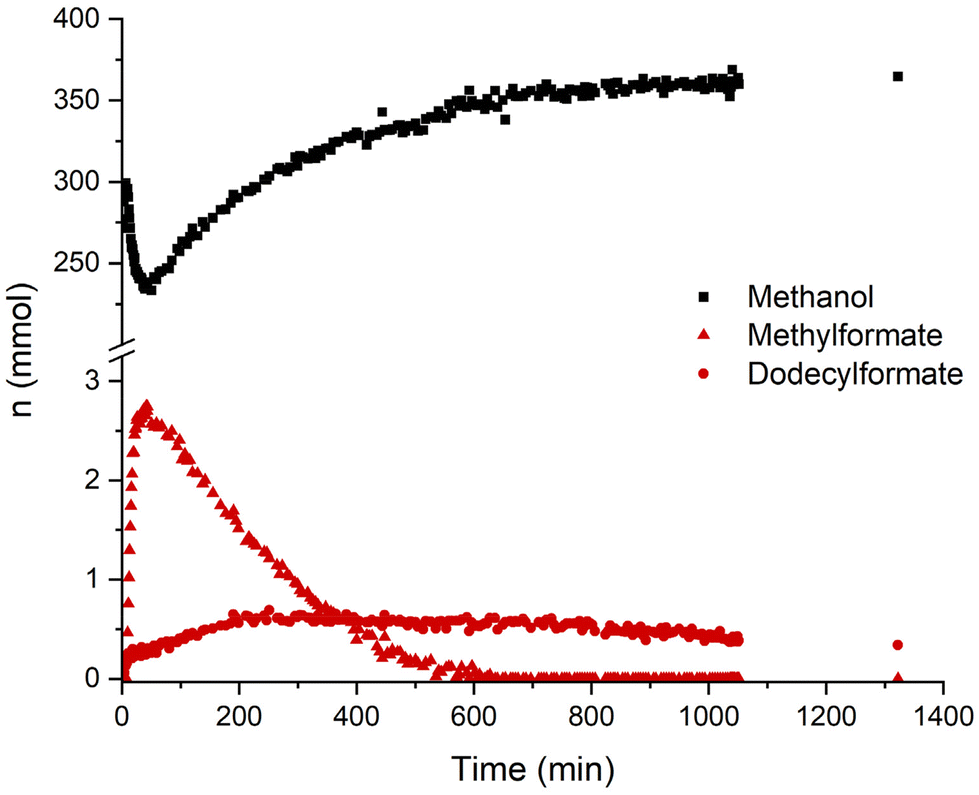 | ||
| Fig. 3 Product and intermediate time profile based 1H NMR measurements with internal standard THF. Conditions: CO (8 bar), H2 (32 bar), Mn-1 (48 μmol), NaOMe (2.4 mmol), 25% methanol in 1-dodecanol (25 ml), 150 °C, 280 mL reactor. δ = 3.33 (s, 3 H, H3C-OH (methanol)), 8.02 (s, 1 H, OOCH (methyl formate)), 8.53 (m, 1 H, OOCH (dodecyl formate)) ppm. | ||
Consequently, two parallel pathways for the hydrogenation of carbon monoxide exist when employing long-chain alcohols as the solvent in this reaction (Scheme 2). While the initial loading with methanol is beneficial for a rapid start-up of the reaction, the process can be performed also without the need for pre-charged methanol.
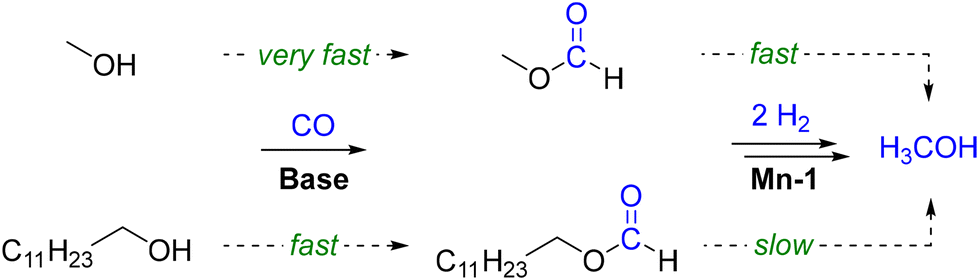 | ||
| Scheme 2 Parallel pathways for activation of carbon monoxide with methanol or 1-dodecanol via their respective formate esters and the consecutive hydrogenations to methanol. | ||
We then combined all previous optimizations to maximize the productivity and activity of the catalytic system (Table 3). Using the standard pre-charged 25% methanol concentration, the activity reached a TOFini of 2025 h−1 within the first hour and a TON of 4909 corresponding to nearly full conversion at high selectivity after only 4 h. Initiating the reaction with lower or even no methanol lead to similar performance, albeit a small amount of methanol as “starter” proves beneficial, as expected. As the CO is consumed nearly completely under these conditions, the TON could be increased to 7139 by increasing the CO partial pressure to 15 bar while maintaining a H2/CO ratio of 5:1.
| Entry | CO (bar) | H2 (bar) | MeOH wt% (%) | S (%) to MeOH | TOFini (MeOH) (h−1) | TON (MeOH) |
|---|---|---|---|---|---|---|
| Conditions: CO, H2, Mn-1 (2 μmol), NaOMe (150 μmol, 75 eq.), 25%, 10% or 0% Methanol in 1-Dodecanol (1.5 ml), 160 °C, 4 h, 20 mL autoclave. Selectivities calculated as molar fraction of total products detected (methanol, methyl and dodecyl formate). | ||||||
| 1 | 10 | 80 | 25 | 99 | 2025 | 4909 |
| 2 | 10 | 80 | 10 | >99 | 2250 | 5080 |
| 3 | 10 | 80 | 0 | 99 | 1895 | 4742 |
| 4 | 15 | 75 | 0 | >99 | n. d. | 7139 |
Finally, we turned our attention to the recyclability of the system. In order to produce sufficient quantities for a reproducible product separation via distillation, the system was scaled up to a 300 ml autoclave. The Mn-1 concentration was kept at 1.3 × 10−3 M at a NaOMe/Mn ratio of 50:1. 70 ml of 1-dodecanol was used as the solvent without pre-charged methanol to ensure that all distilled product would come from the catalytic reaction. An initial pressure of 120 bar at room temperature (H2/CO = 5:1) together with a reaction temperature of 150 °C was chosen. Due to the geometry of the reactor, the gas/liquid-phase ratio is lower (∼3.6/1) compared to the autoclave setup (∼14.5/1) thus, despite the doubled CO pressure, reducing the maximum possible TON per run by approx. 50%. Even though the reaction can be performed at significantly lower pressures, the high pressure is necessary in this setup to produce sufficient quantities of the product methanol for a reliable collection and quantification. After cooling down the reactor, the excess pressure was released. The product separation was achieved via a vacuum distillation directly from the reactor at 70 °C with the product collected in a cooling trap. The isolated product was analyzed by NMR and catalyst leaching was quantified by X-ray fluorescence (XRF) measurements.
Methanol was detected as the only product in the distillate after the first run (ESI, Fig. S7†). The amount of methanol collected (201–213 mmol, ESI, Table S5†) correlates well with the amount of CO in the reactor estimated from the ideal gas law (205 mmol, ESI, Table S3†) indicating full conversion as expected under these conditions and nearly quantitative yield even in the simple and non-optimized set-up. The reaction was repeated in the same reactor by simply re-pressurizing and re-adjusting the temperature again for 4 h. Following the batch-wise operation, the performance remained constant for 4 consecutive runs with only small fluctuations that can be attributed to manual handling errors at high yields (Fig. 4). In run 5, however, the isolated methanol quantity was significantly lower reflecting lower conversion as also indicated by a reduced pressure drop (ESI, Table S5†). However, the addition of another batch of NaOMe between run 5 and 6 restored the catalytic performance back to previous levels. This base deactivation can possibly be attributed to (moisture) impurities entering the catalytic system through manual operation between runs. A continuous operation of the product separation and catalyst recycling in a closed system will presumably suppress this deactivation and give more insights into other possible deactivation modes. Analyzing the contamination of the isolated methanol with phosphorous and manganese confirmed a minor loss of 1.83% of the employed ligand over the 6 recycling runs, while manganese leaching in each run was below the detection limit (detailed leaching per run can be found in the ESI, Table S5†). The ligand leaching is presumably attributed to the non-optimized distillation conditions and can be lowered with a more controlled distillation setup.
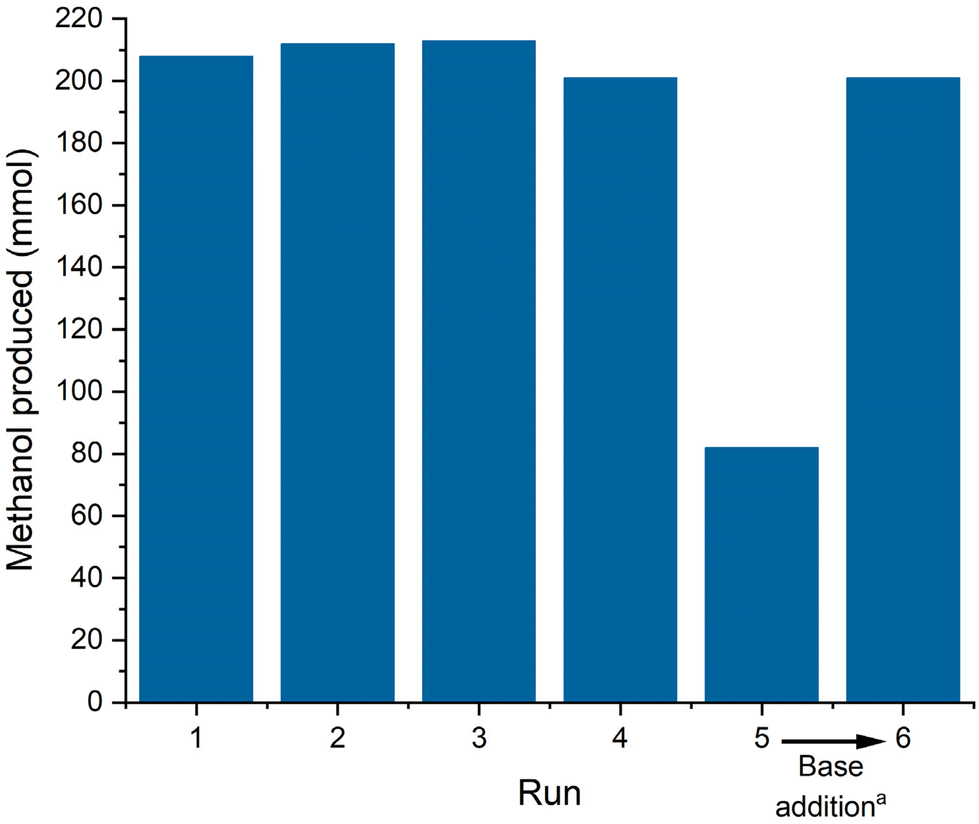 | ||
| Fig. 4 Amount of methanol produced and theoretical yield in the catalyst recycling experiment in the hydrogenation of CO. Conditions: CO (20 bar), H2 (100 bar), Mn-1 (92 μmol), NaOMe (4.65 mmol, 50 eq.), 1-dodecanol (70 ml), 150 °C, 4 h, 300 mL reactor. (a) Addition of NaOMe (4.65 mmol) in 1-dodecanol (10 ml). | ||
Over the 6 runs of repetitive batch operation, a TTON of 12.138 was achieved, marking the highest reported productivity for the homogenous carbon monoxide hydrogenation to methanol.
Conclusion
In conclusion, we have developed a simple and recyclable system for the alcohol-assisted homogeneously catalyzed formation of methanol from carbon monoxide and hydrogen. The use of sodium methanolate as the base in long-chain alcohols, in the case of 1-dodecanol, enables the activation of CO via the formation of formate esters allowing initiation of the catalytic hydrogenation by a manganese pincer complex such as Mn-1. The difference in volatility of the low-boiling product methanol and all other components of the catalytic systems allows simple product isolation by flash distillation directly from the reactor. Re-using the catalyst solution in a batch-wise recycling was demonstrated for six consecutive runs yielding a TTON of over 12000 corresponding to a more than five times enhancement over the TON in single batch mode under identical conditions. This defines the highest productivity for methanol formation from H2/CO in terms of turnover per metal reported today. Notably, the demonstrated stability of the organometallic catalyst beyond the actual series of experiments makes this system very attractive for continuous operation as the next phase of development.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy – Cluster of Excellence 2186 “The Fuel Science Center” ID: 390919832 at RWTH Aachen and the Max-Planck Society for financing this research. In addition, we would like to thank Rucha Shirish Medhekar for her assistance in the lab concerning this work. Open Access funding provided by the Max Planck SocietyReferences
- S. Simon Araya, V. Liso, X. Cui, N. Li, J. Zhu, S. L. Sahlin, S. H. Jensen, M. P. Nielsen and S. K. Kær, Energies, 2020, 13, 596 CrossRef.
- A. Sonthalia, N. Kumar, M. Tomar, V. Edwin Geo, S. Thiyagarajan and A. Pugazhendhi, Clean Technol. Environ. Policy, 2023, 25, 551–575 Search PubMed.
- K. Natte, H. Neumann, M. Beller and R. V. Jagadeesh, Angew. Chem., Int. Ed., 2017, 56, 6384–6394 CrossRef CAS PubMed.
- G. A. Olah, A. Goeppert and G. K. S. Prakash, Beyond Oil and Gas: The Methanol Economy, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2018 Search PubMed.
- M. Bertau, H. Offermanns, L. Plass, F. Schmidt and H.-J. Wernicke, Methanol: The Basic Chemical and Energy Feedstock of the Future, Springer-Verlag, Berlin, Heidelberg, 2014 Search PubMed.
- A. Kaithal, B. Chatterjee, C. Werlé and W. Leitner, Angew. Chem., Int. Ed., 2021, 60, 26500–26505 CrossRef CAS PubMed.
- V. Dieterich, A. Buttler, A. Hanel, H. Spliethoff and S. Fendt, Energy Environ. Sci., 2020, 13, 3207–3252 RSC.
- M. Yang, D. Fan, Y. Wei, P. Tian and Z. Liu, Adv. Mater., 2019, 31, 1902181 CrossRef CAS PubMed.
- N. R. Ammar, Transp. Res. D Trans. Environ., 2019, 69, 66–76 CrossRef.
- M. Svanberg, J. Ellis, J. Lundgren and I. Landälv, Renewable Sustainable Energy Rev., 2018, 94, 1217–1228 CrossRef CAS.
- M. Nielsen, E. Alberico, W. Baumann, H.-J. Drexler, H. Junge, S. Gladiali and M. Beller, Nature, 2013, 495, 85–89 CrossRef CAS PubMed.
- H. V. M. Hamelers, O. Schaetzle, J. M. Paz-García, P. M. Biesheuvel and C. J. N. Buisman, Environ. Sci. Technol. Lett., 2014, 1, 31–35 CrossRef CAS.
- M. Behrens, F. Studt, I. Kasatkin, S. Kühl, M. Hävecker, F. Abild-Pedersen, S. Zander, F. Girgsdies, P. Kurr, B.-L. Kniep, M. Tovar, R. W. Fischer, J. K. Nørskov and R. Schlögl, Science, 2012, 336, 893–897 CrossRef CAS PubMed.
- L. C. Grabow and M. Mavrikakis, ACS Catal., 2011, 1, 365–384 CrossRef CAS.
- K. C. Waugh, Catal. Lett., 2012, 142, 1153–1166 CrossRef CAS.
- B. D. Dombek, J. Am. Chem. Soc., 1980, 102, 6855–6857 CrossRef CAS.
- B. D. Dombek, J. Am. Chem. Soc., 1981, 103, 6508–6510 CrossRef CAS.
- B. Li and K.-J. Jens, Ind. Eng. Chem. Res., 2014, 53, 1735–1740 CrossRef CAS.
- B. Li and K. J. Jens, Top. Catal., 2013, 56, 725–729 CrossRef CAS.
- K. Li and D. Jiang, J. Mol. Catal. A: Chem., 1999, 147, 125–130 CrossRef CAS.
- D. Mahajan, V. Krisdhasima and R. D. Sproull, Can. J. Chem., 2001, 79, 848–853 CrossRef CAS.
- M. Marchionna, L. Basini, A. Aragno, M. Lami and F. Ancillotti, J. Mol. Catal., 1992, 75, 147–151 CrossRef CAS.
- S. Ohyama, Appl. Catal., A, 1999, 180, 217–225 CrossRef CAS.
- H. Berke and R. Hoffmann, J. Am. Chem. Soc., 1978, 100, 7224–7236 CrossRef CAS.
- M. M. Deegan and J. C. Peters, J. Am. Chem. Soc., 2017, 139, 2561–2564 CrossRef CAS PubMed.
- T. Ziegler, L. Versluis and V. Tschinke, J. Am. Chem. Soc., 1986, 108, 612–617 CrossRef CAS.
- S. Kar, A. Goeppert and G. K. S. Prakash, J. Am. Chem. Soc., 2019, 141, 12518–12521 CrossRef CAS PubMed.
- P. Ryabchuk, K. Stier, K. Junge, M. P. Checinski and M. Beller, J. Am. Chem. Soc., 2019, 141, 16923–16929 CrossRef CAS PubMed.
- A. Kaithal, C. Werlé and W. Leitner, JACS Au, 2021, 1, 130–136 CrossRef CAS PubMed.
- E. Balaraman, C. Gunanathan, J. Zhang, L. J. W. Shimon and D. Milstein, Nat. Chem., 2011, 3, 609–614 CrossRef CAS PubMed.
- J. R. Khusnutdinova, J. A. Garg and D. Milstein, ACS Catal., 2015, 5, 2416–2422 CrossRef CAS.
- N. M. Rezayee, C. A. Huff and M. S. Sanford, J. Am. Chem. Soc., 2015, 137, 1028–1031 CrossRef CAS PubMed.
- C. A. Huff and M. S. Sanford, J. Am. Chem. Soc., 2011, 133, 18122–18125 CrossRef CAS PubMed.
- W.-Y. Chu, Z. Culakova, B. T. Wang and K. I. Goldberg, ACS Catal., 2019, 9, 9317–9326 CrossRef CAS.
- S. Kar, A. Goeppert, J. Kothandaraman and G. K. S. Prakash, ACS Catal., 2017, 7, 6347–6351 CrossRef CAS.
- G. Neitzel, R. Razzaq, A. Spannenberg, K. Stier, M. Checinski, R. Jackstell and M. Beller, ChemCatChem, 2023, e202301053 Search PubMed.
- C1 Green Chemicals AG, DBI-Gastechnologisches Institut gGmbH Freiberg, Fraunhofer UMSICHT, Fraunhofer IWES, TU Berlin, “Leuna 100”, to find at https://www.leuna100.de/de, 2023.
- F. P. Byrne, S. Jin, G. Paggiola, T. H. M. Petchey, J. H. Clark, T. J. Farmer, A. J. Hunt, C. Robert McElroy and J. Sherwood, Sustain. Chem. Process., 2016, 4, 7 CrossRef.
- D. A. Kuß, M. Hölscher and W. Leitner, ChemCatChem, 2021, 13, 3319–3323 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Additional graphics, detailed experimental procedures and analytical data. See DOI: https://doi.org/10.1039/d4gc01050g |
| This journal is © The Royal Society of Chemistry 2024 |