
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceResearch progress and perspectives on photocatalysts based on the lead-free double halide perovskite
Do Yeon
Heo
,
Mahider Asmare
Tekalgne
and
Soo Young
Kim
 *
*
Department of Materials Science and Engineering Korea University, Seoul 02841, Republic of Korea. E-mail: sooyoungkim@korea.ac.kr
First published on 23rd October 2023
Abstract
Photocatalytic technology stands as a promising solution to address the current energy and environmental challenges. Halide perovskites, particularly lead-free double halide perovskites, have garnered recognition as next-generation photocatalysts due to their adjustable bandgap, low binding energy, broad visible light absorption range, and efficient charge carrier transfer. In this review, we explore the utilization of lead-free double halide perovskites characterized by their non-toxic attributes and diverse chemical compositions and properties as photocatalysts for both hydrogen production and carbon dioxide reduction. We commence by presenting an overview of lead-free double halide perovskites, followed by a comprehensive analysis of recent research outcomes pertaining to their application as photocatalysts for hydrogen production and carbon dioxide reduction. Lastly, we discuss the challenges and prospects associated with lead-free double halide perovskite photocatalysts. This review is anticipated to serve as a valuable reference for the development of lead-free double halide perovskite-based photocatalysts, addressing critical aspects in the pursuit of achieving high-efficiency hydrogen generation and carbon dioxide reduction, crucial for our future energy and environmental needs.
Broader contextIn the ever-pressing quest for sustainable energy solutions and environmentally conscious technologies, photocatalytic advancements play a pivotal role. This review paper sheds light on a compelling avenue of research that showcases lead-free double halide perovskites as catalysts of substantial promise. Their ability to fine-tune bandgaps, their low binding energy, broad light absorption range, and effective charge transfer properties make them a new beacon in the realm of energy and environmental catalysis. The investigations discussed herein not only expand our comprehension of semiconductor materials but also hold immense potential for real-world applications. In particular, their application in the simultaneous production of hydrogen and the reduction of carbon dioxide marks a significant step towards sustainable energy solutions and mitigation of climate change. This work exemplifies the impact of materials science on critical energy and environmental challenges and paves the way for greener, more efficient future technologies. The findings underscore the importance of innovative, non-toxic materials in addressing our contemporary global challenges and herald a promising era for energy and environmental catalysis. |
1. Introduction
The Earth's temperature has increased by more than 1.2 °C compared to pre-industrial levels. If current trends persist, the Paris Agreement's target of limiting global warming to 1.5 °C is expected to be reached by 2035.1 The primary driver of global warming is the presence of greenhouse gases, which traps infrared radiation while allowing visible light to pass through. This imbalance in energy absorption and emission results in a net increase in Earth's temperature, leading to a range of severe consequences, including floods, heatwaves, cold spells, reduced solar reflectance, and rising sea temperatures.Among greenhouse gases, carbon dioxide (CO2) emissions from fossil fuels, the predominant source of energy, account for a staggering 76% of the total volume. Consequently, there is an urgent need to reduce greenhouse gas concentrations and develop sustainable energy technologies as viable alternatives. Solar energy, as the most abundant renewable energy source, holds immense promise. Harnessing the solar energy incident on Earth for various applications is crucial, with the potential to meet global energy demands within a relatively short timeframe. While solar energy technologies have primarily focused on electricity generation, a novel approach exists, leveraging solar energy to drive or initiate chemical reactions—a field known as photocatalysis. Photocatalysis finds applications in diverse areas, including hydrogen (H2) production, CO2 conversion, and pollution abatement, offering significant potential to address our current energy and environmental crises.2
The concept of photocatalysis was first introduced by Fujishima and Honda in 1972, utilizing titanium dioxide as a photocatalyst to demonstrate hydrogen production through solar-driven water splitting.3 This groundbreaking discovery sparked extensive research into solar energy conversion using photocatalysts. Numerous materials have been explored for photocatalysis, encompassing metal oxides,4–8 metal nitrides,9,10 metal sulfides,11–13 metal–organic frameworks,14–17 covalent organic frameworks,18,19 and metal halides.20 However, many of these materials present limitations such as inefficient utilization of visible light, rapid recombination of charge carriers, and instability. Additionally, some photocatalyst components may not align with environmental sustainability goals.21,22 Nonetheless, halide perovskites have exhibited remarkable photocatalytic performance owing to their strong response to visible light, prolonged charge carrier lifetimes, high light absorption coefficients, and minimal non-radiative carrier recombination.23 Although lead-based perovskites were initially employed as photocatalysts,24 their commercial viability is hampered by their solubility in water, resulting in the release of toxic lead. Consequently, the development of lead-free halide perovskite materials has emerged as a critical objective.
Lead-free halide perovskites have garnered considerable attention across a range of fields, including perovskite solar cells,25–29 light-emitting diodes,30–33 X-ray detectors,34,35 memristors,36,37 and more. The substitution of lead with alkali metals in halide perovskites leads to an increase in the electronic bandgap,38,39 which significantly influences photocatalytic performance. For efficient solar energy utilization, extending light absorption into the visible spectrum is essential, necessitating a photocatalyst with a bandgap energy less than 3 eV.40 Moreover, semiconductor photocatalysts designed for water splitting require a minimum bandgap of 1.23 eV, and research has demonstrated that photocatalysts with bandgaps exceeding 2 eV exhibit superior performance.41 Double perovskites, characterized by their diverse chemical compositions and properties relative to traditional perovskite structures, offer tunable bandgaps that can enable effective photocatalytic properties.
In this review, we aim to provide insights into recent research and future prospects of lead-free double perovskites as photocatalysts in environmentally sustainable industries, with a specific focus on H2 production and CO2 reduction. Our discussion will commence with an exploration of the theoretical foundations and synthesis methodologies associated with lead-free double perovskites and photocatalysts. Subsequently, we will introduce recent research endeavors pertaining to lead-free double perovskite photocatalysts in the context of H2 production and CO2 reduction, culminating in a discussion of the challenges and prospects shaping the future of this dynamic field.
2. Double halide perovskite
2.1. Crystal structure
The traditional crystal structure of perovskites adopts a cubic structure denoted as ABX3. In contrast, double perovskites share similarities with the conventional perovskite structure but are composed of trivalent cations (B3+) and monovalent cations (B+) instead of the typical divalent cation (B2+), as shown in Table 1. The chemical formula for double perovskite is represented as A2B+B3+X6. The structure of double perovskites forms a cubic lattice. Within the A site, there are two different cations, while the B site contains two distinct anions. This arrangement results in alternating cations and anions at the corners of the cubes. In Fig. 1, it can be observed that A+ ions are coordinated with 12 X− ions, whereas B+ and B3+ ions are coordinated with 6 X− ions, forming a network structure known as rock-salt ordering.42,43 Consequently, the unit cell can be understood as a doubled cubic perovskite, essentially a double perovskite structure.41,44| A | CH3NH3+, CH(NH2)2+, Cs+etc. |
| B2+ | Pb2+, Sn2+etc. |
| B3+ | Bi3+, Sb3+, In3+etc. |
| B+ | Ag+, Cu+, Na+etc. |
| X | I−, Br−, Cl− |
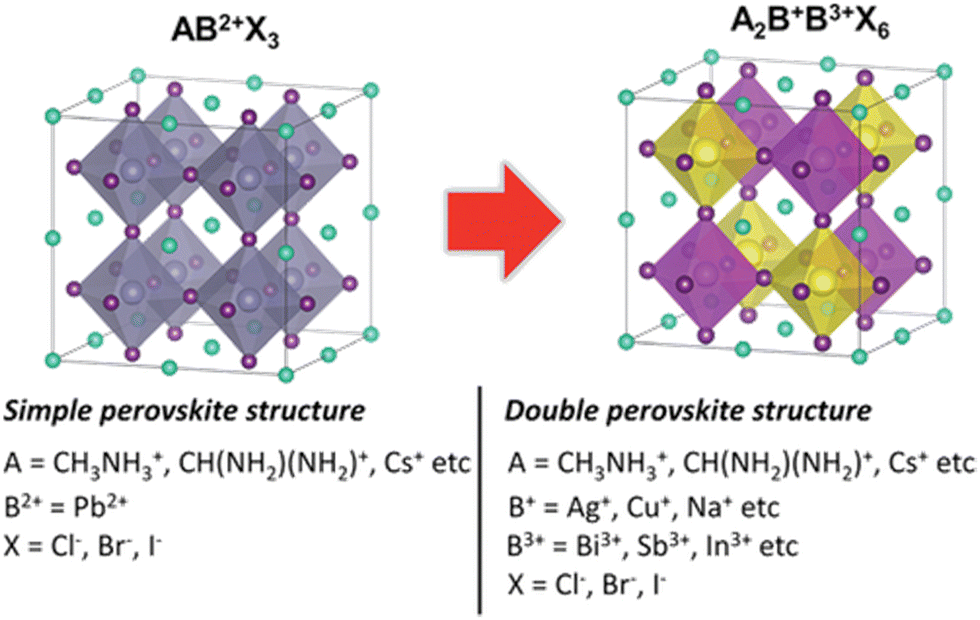 | ||
| Fig. 1 Illustration of structural contrasts between simple (AB2+X3) and double perovskites (A2B+B3+X6). Visualization of A+, B2+, B+, B3+, and X− ions depicted as cyan, gray, yellow, pink, and purple spheres, respectively.43 Reproduced with permission from ref. 43 © 2019 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
The structural stability of a double perovskite can be determined through the octahedral (μ) and Goldschmidt (t) tolerance factors. μ evaluates the stability of BX6 octahedra, while t indicates the degree of distortion in the perovskite structure. These factors can be calculated using the following formulas:45
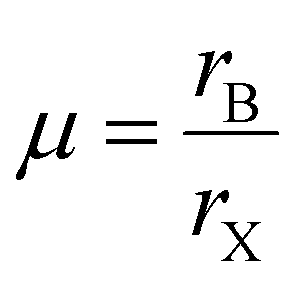
Here, rA, rB, and rX represent the radii of the respective ions in the material. Both t and μ can be adjusted by substituting ions with different-sized elements. For perovskites to form a stable structure, it is generally accepted that 0.75 < t < 1 and μ > 0.41. Particularly, cubic structures within the range 0.9 ≤ t ≤ 1.0 are considered the most stable perovskite structures. Furthermore, stable tetragonal structures can be achieved within the range 0.44 < μ < 0.9.46 Thus, ion substitutions favoring the formation of cubic structures represent one of the best routes to realize stable perovskite structures. However, only specific metal ions that simultaneously satisfy both criteria can create such structures.
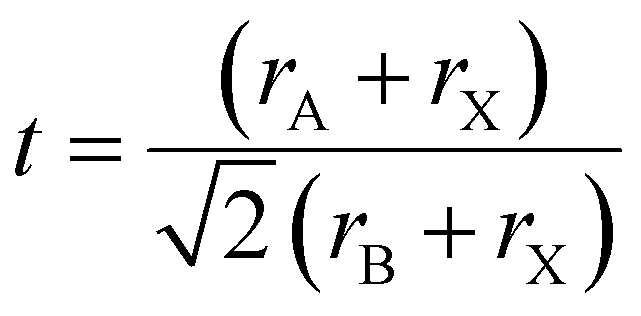
2.2. Synthesis protocol
The general steps of the hot injection method are as follows (see Fig. 2(a)):47
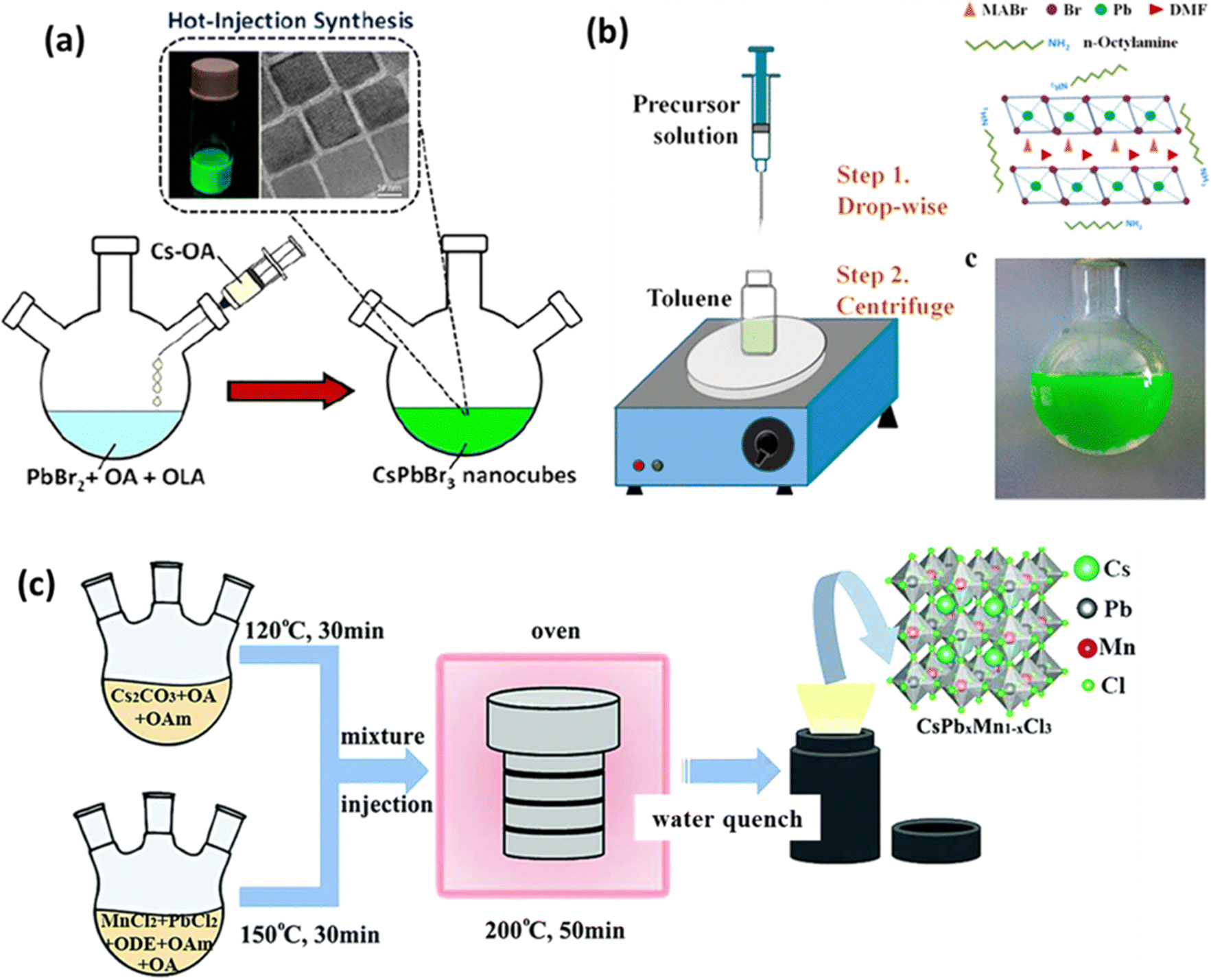 | ||
| Fig. 2 Diagram depicting perovskite synthesis techniques: (a) hot injection,47 (b) LARP,50 and (c) solvothermal method.53 Reproduced with permission from ref. 47 Copyright © 2022 American Chemistry Society. Reproduced with permission from ref. 50 Copyright © 2015 American Chemistry Society. Reproduced with permission from ref. 53 Copyright © 2018 Royal Society of Chemistry. | ||
1. Mix reactants in the required proportions in a solvent, creating the reaction mixture to control the reaction environment and promote the reaction.
2. Place the synthesis reaction vessel into a heat conversion device and heat it to a high temperature.
3. Once the reaction is complete, cool and separate the components.
4. Harvest and refine the perovskite powder produced in the reaction vessel, removing any by-products or remaining residues to obtain pure perovskite.
By adjusting the elemental composition of the reactants, the type of ligand used, and the reaction temperature, one can modify the shape and size of the resulting nanoparticles. The hot injection method is a convenient way to produce nanoparticles with well-dispersed cubic CsPbBr3 nanocubes, as demonstrated by Vighnesh et al.47 They outlined a comprehensive hot injection protocol for synthesizing CsPbBr3 nanocrystals, considering both intrinsic and extrinsic factors affecting reproducibility. The protocol covered aspects such as precursor solution preparation, ligands, the roles of temperature and solvents, nanocrystal refinement, and storage. Moreover, the hot injection method has also been used for synthesizing double perovskites. Cs2Na1−xAgxBiCl6 nanocrystals were synthesized through a hot injection method by reacting metal carboxylates and carbonates with benzoyl chloride to induce nucleation and growth.48
Liu et al. successfully synthesized Cs4CuIn2Cl12 double perovskites using a moisture-assisted hot injection method, overcoming the challenges posed by moisture, which is known to be detrimental in nanocrystal synthesis.49 They demonstrated that the presence of water molecules during Cs4CuIn2Cl12 synthesis improves photoluminescence quantum yields, induces morphological transformations from 3D nanocubes to 2D nanoplatelets, and triggers photocarrier trapping. The hot injection method offers ease of modification in the synthesis process, making it a widely used approach in perovskite synthesis.
1. Prepare a precursor solution of perovskite by dissolving it in a solvent.
2. Dissolve ligand molecules in the solvent and add them to the precursor solution. Ligands play a crucial role in controlling the growth and morphology of nanoparticles.
3. Heat the ligand-treated solution in a reaction vessel using an appropriate heat source, initiating the formation of ligand-perovskite complexes.
4. Upon completion of the reaction, cool the solution to promote nanoparticle formation.
5. Separate and purify the formed perovskite nanoparticles. During this step, any by-products or remaining impurities are removed to obtain pure perovskite nanoparticles.
LARP allows for the synthesis of perovskite nanoparticles at room temperature without the need for high-temperature reactions. However, maintaining purity during synthesis can be challenging due to the rapid recrystallization rate associated with this method.
Yoo et al. investigated the impact of carboxylic acid and carboxylamine ligands on the size, shape, and crystal structure of CsPbBr3 nanocrystals synthesized using LARP.51 They found that changes in carboxylic acid length primarily affected the shape and crystal structure of the perovskite nanocrystals, while increasing the carboxylamine ligand length led to a decrease in nanoparticle size, with no significant impact on shape. Additionally, they demonstrated that carboxylic acid ligands directly bond to the perovskite nanocrystals, whereas carboxylamine ligands either directly or indirectly interact with the perovskite nanocrystals through amine ligands. LARP has also been used for synthesizing Cs2AgSbCl6 nanocrystals, which are employed as photocatalysts.52 By controlling the surface ligand density through α-alkylation of aldehydes, high alkylation yields were achieved.
1. Dissolve metal precursors and halide compounds in an initial solvent. The reason for heating the initial reaction medium to temperatures exceeding 100 °C in the solvothermal method is to ensure the necessary reaction rate and thermodynamic stability for synthesizing complex compounds like double perovskites. At this stage, ligand molecules are added to facilitate interaction with metal precursors.
2. Use the solvent within a reactor to create high-temperature and high-pressure conditions, initiating chemical reactions leading to the formation of perovskite nanoparticles. Ligands can influence the size and shape of nanoparticles during this process.
3. Upon completion of the reaction, cool the mixture, promoting nanoparticle formation.
4. Separate and refine the formed perovskite nanoparticles.
Solvothermal reactions offer control over the resulting product's characteristics based on reactor design, parameter settings, and the choice of ligands. Ligands play a crucial role in adjusting nanoparticle size, shape, dispersion, and other properties. Compared to the hot injection method, which can be challenging for heat-sensitive materials, the solvothermal method allows for chemical reactions at high temperatures and pressures using solvents, making it suitable for a wide range of material syntheses. Chen et al. synthesized Mn-doped CsPbCl3 using both the hot injection and solvothermal methods, demonstrating that the nanocrystals produced by the solvothermal method exhibited better stability.54 They explained that the solvothermal method can provide more energy due to its operation at high temperature and pressure, enhancing the binding of Mn2+ within the ligand field of CsPbCl3.
As an application of the solvothermal method, there is the microwave-assisted solvothermal method. This technique employs microwave energy to synthesize nanoparticles under high-temperature and high-pressure conditions. Microwave energy is delivered in the form of electromagnetic waves and rapidly transfers heat throughout the material, significantly increasing reaction rates and allowing for the synthesis of smaller nanoparticles. Cs2AgBiBr6 nanocrystals were synthesized using the microwave-assisted solvothermal method in combination with the solvent IPA (isopropanol), which efficiently absorbs microwave energy.55 The Cs2AgBiBr6 nanoparticles synthesized by Slavney et al. in a simple and fast (2 min synthesis) method had a long carrier lifetime of 921 ns, comparable to the single crystal form (668 ns).56
2.3. Lead-free double halide perovskite (LFDHP) and their stability
Perovskites with the APbX3 structure have been extensively researched due to their high efficiency in various applications such as photovoltaics and LEDs. However, the use of Pb compounds has been globally restricted or prohibited since the 2000s due to the risk of lead poisoning upon human exposure.Therefore, research on lead-free perovskites is essential for the commercialization of perovskite applications. Substituting Pb2+ with 2+ cations like Sn2+ and Ge2+ was attempted, but these alternatives suffer from instability issues due to their unstable oxidation states.57–59 Consequently, research turned to partial doping of lead, although this approach does not provide a fundamental solution to lead toxicity.60–62 Thus, there is an urgent need to explore new materials to address toxicity and stability issues, with lead-free double halide perovskites emerging as a potential solution.
As previously mentioned, stable perovskites are formed when 0.9 ≤ t ≤ 1.0 and 0.44 < μ < 0.9. To assess the thermodynamic stability of A2B+B3+X6 double perovskites, the decomposition energy for possible decomposition pathways is calculated. Typically, halide perovskites are synthesized through reverse reactions, and the decomposition enthalpy can be calculated using the following equation:63
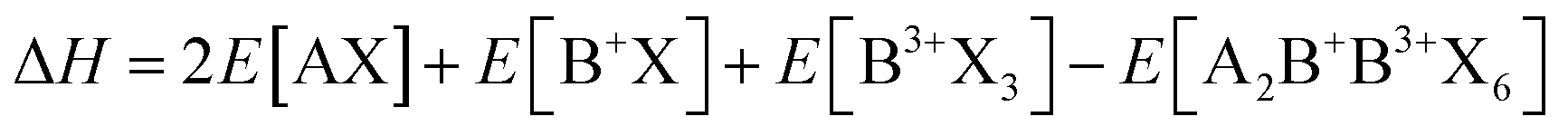
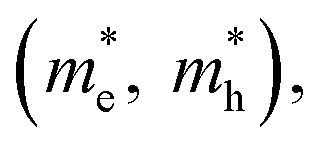 and exciton binding energy (ΔEb), as summarized in Fig. 3(a).63 Zhao et al. systematically compiled this data through first-principles calculations.
and exciton binding energy (ΔEb), as summarized in Fig. 3(a).63 Zhao et al. systematically compiled this data through first-principles calculations.
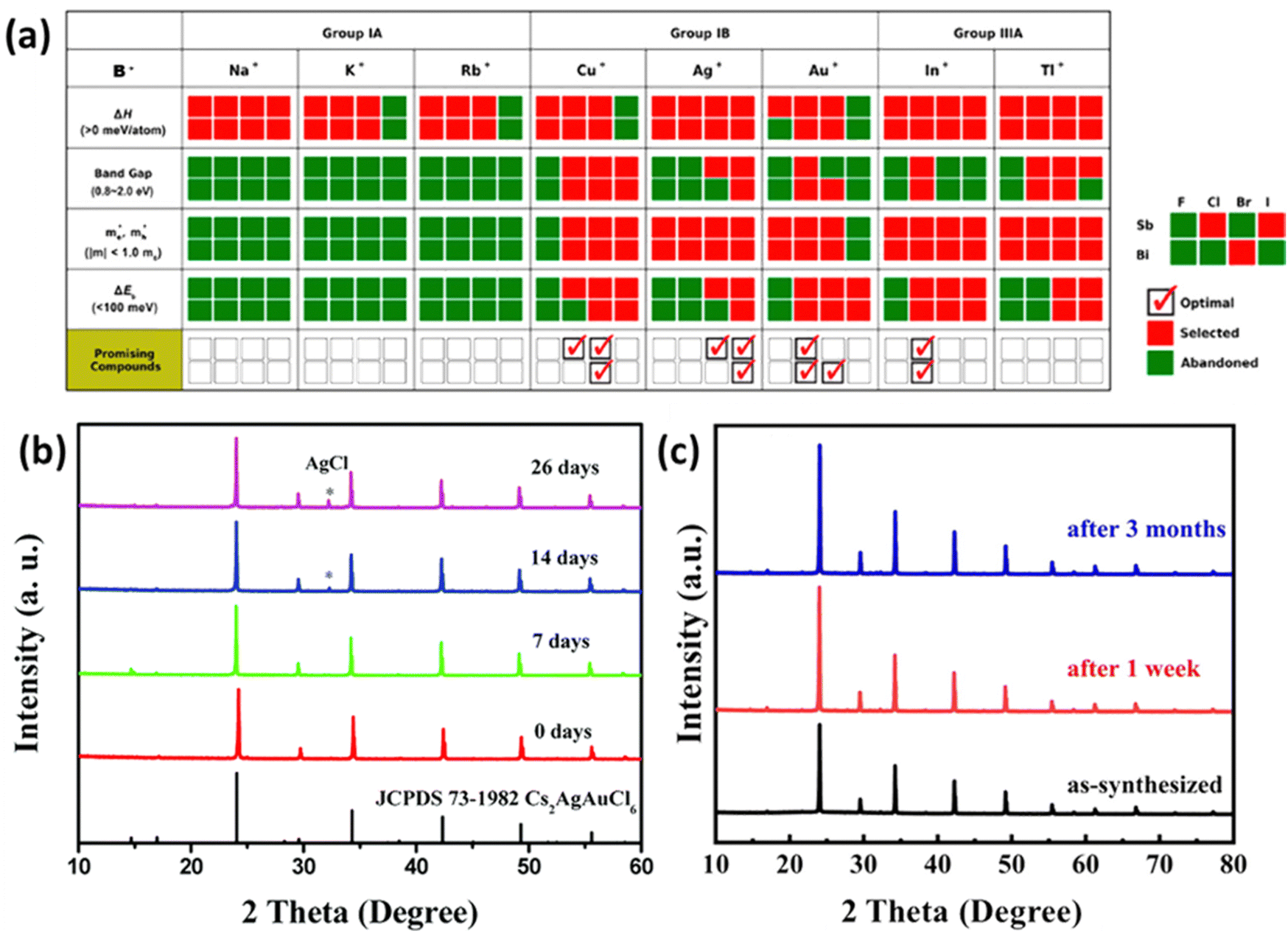 | ||
Fig. 3 (a) Stepwise materials evaluation process for photovoltaic performance, considering parameters such as decomposition enthalpy (ΔH), band gap, effective carrier masses 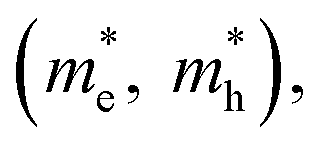 , and exciton binding energy (ΔEb). Materials successfully passing the screening are indicated by red squares (Selected), while those not meeting the criteria are marked with green squares (Abandoned). The optimal environmentally friendly A2M+M3+XVII6 perovskites meeting all criteria are denoted by red checkmarks.63 Reproduced with permission from ref. 63 Copyright © 2017 American Chemistry Society. (b) XRD of Cs2AgInCl6 after 7 days, 14 days and 26 days of exposure to light and moisture conditions.72 Reproduced with permission from ref. 72 Copyright © 2017 RSC. (c) XRD patterns of Cs2InAgCl6 after storage for one week and three months under ambient conditions.73 Reproduced with permission from ref. 73 Copyright © 2021 Elsevier Inc. All rights reserved. , and exciton binding energy (ΔEb). Materials successfully passing the screening are indicated by red squares (Selected), while those not meeting the criteria are marked with green squares (Abandoned). The optimal environmentally friendly A2M+M3+XVII6 perovskites meeting all criteria are denoted by red checkmarks.63 Reproduced with permission from ref. 63 Copyright © 2017 American Chemistry Society. (b) XRD of Cs2AgInCl6 after 7 days, 14 days and 26 days of exposure to light and moisture conditions.72 Reproduced with permission from ref. 72 Copyright © 2017 RSC. (c) XRD patterns of Cs2InAgCl6 after storage for one week and three months under ambient conditions.73 Reproduced with permission from ref. 73 Copyright © 2021 Elsevier Inc. All rights reserved. | ||
One persistent challenge with perovskites is their vulnerability to light and moisture. In particular, Methylammonium lead iodide (MAPbI3) undergoes accelerated degradation to PbI2 when exposed to light or moisture, resulting in a severe drop in performance.69 In contrast, halide double perovskites have been shown to be stable under ambient conditions through extensive research. Cs2AgBiBr6, known for its high stability, has been investigated in various fields. Films of Cs2AgBiBr6 used in photodetectors maintained excellent intrinsic and environmental stability for up to two weeks.70 These films exhibited stability in thermal tests within a temperature range of 20–120 °C and continuous ultraviolet irradiation tests. Cs2AgBiBr6 films designed for neuromorphic computing retained stable operation even after 20 days of storage in ambient conditions (approximately 40–50% humidity and around 25 °C).71 X-ray diffraction (XRD) confirmed the maintenance of stable forms in Cs2AgInCl6 synthesized by Zhou even after 26 days of exposure to light and humidity as shown in Fig. 3(b).72 Another study found that Cs2AgInCl6 remained stable in an air ambient environment for three months as shown in Fig. 3(c).73
Mechanical stability, which relates to a material's ability to restore its structure under applied distortion pressure, is also crucial. Many systems with the chemical structure of A2B+B3+X6 double perovskites are known to possess mechanical stability. Traditional mechanical stability for cubic phase crystals under equilibrium conditions requires satisfying the following elastic conditions:74
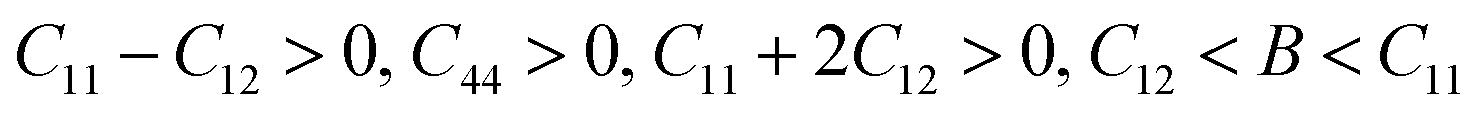
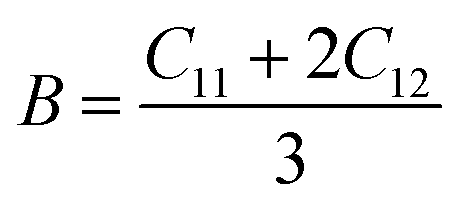
Here, C11, C12, and C44 represent elastic constants, B represents bulk modulus, and G denotes shear modulus. Lower values of elastic constants indicate softer materials. Lower values of B and C11 imply weaker resistance to volume changes caused by applied pressure. Cs2AgFeCl6, for instance, exhibits low B values, indicating volume deformation under external stress.75 Furthermore, the ratio of shear elastic modulus to bulk elastic modulus classifies Cs2AgBiX6 (X = Br, Cl) as ductile materials.76
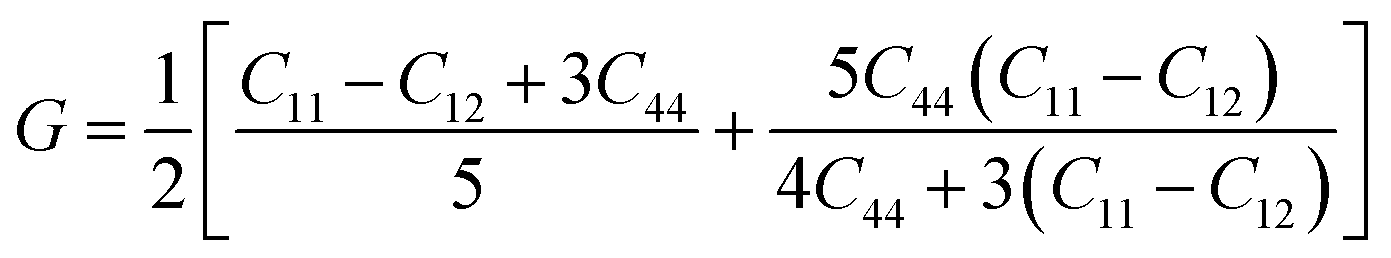
3. Photocatalytic application
3.1. The principle of photocatalysts
As depicted in Fig. 4(a), visible and infrared light account for over 90% of the total solar energy.77 To harness solar energy efficiently, the development of narrow bandgap photocatalysts is essential. In particular, the valence band maximum (VBM) position should be more positive than the potential of the oxidative half-reaction, while the conduction band minimum (CBM) position should be more negative than the potential of the reductive half-reaction. Due to the advantages of bandgap and band position adjustments, double-halide perovskites are considered promising candidates for photocatalysis. In semiconductor-based photocatalytic reactions, the role of the photocatalyst is to absorb light, generate photo-induced electron–hole pairs, and transport charge carriers to the surface to induce oxidative–reductive reactions. Photocatalysts typically have active sites on their surfaces to capture charges and activate reactants. The fundamental steps of a photocatalytic process can be seen in Fig. 4(b).78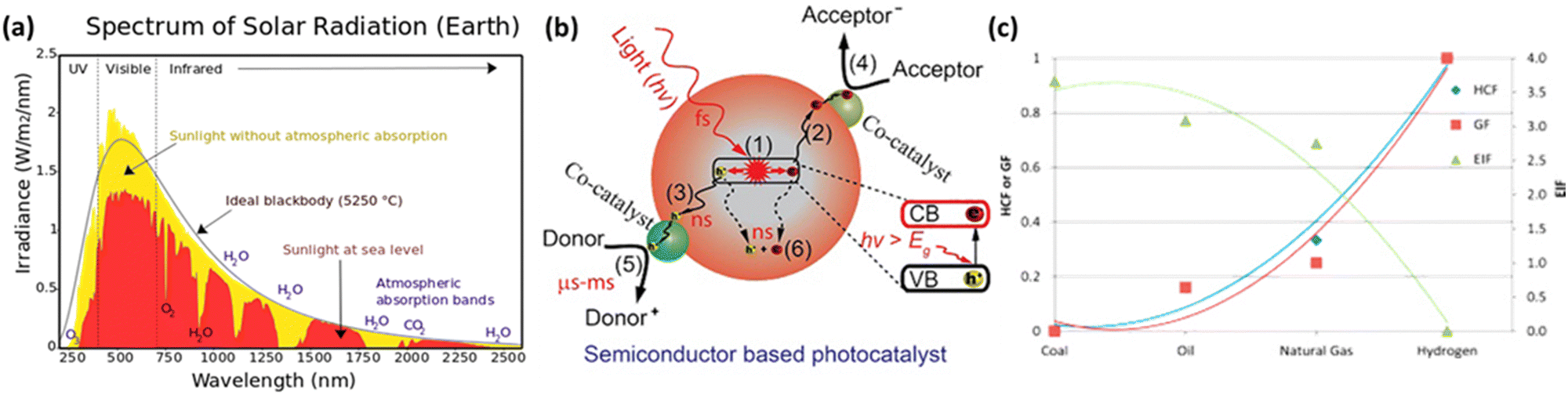 | ||
| Fig. 4 (a) Illustration of the solar spectrum and absorption by atmospheric gases in the wavelength range of 240 nm to 2.5 μm.77 Reproduced with permission from ref. 77 Copyright via Wikimedia Commons. (b) Fundamental operating mechanisms of semiconductor-based photocatalysts.78 Reproduced with permission from ref. 78 Copyright © 2022 Tsinghua University Press Ltd. (c) Comparison of Hydrogen Combustion Efficiency (HCF), Greenhouse Gas Footprint (GF), and Energy Intensity Factor (EIF) between hydrogen and other fossil fuels.81 Reproduced with permission from ref. 81 Copyright © 2014 Hydrogen Energy Publications, LLC. Published by Elsevier Ltd. All rights reserved. | ||
(1) Photogenerated electrons jump from the valence band (VB) to the conduction band (CB) after accepting holes. This process occurs rapidly at the femtosecond level (∼10−15 seconds).
(2 and 3) Subsequently, the electrons and holes migrate to the photocatalyst surface. Charge diffusion during this step is swift, occurring at the nanosecond level (∼10−9 seconds).
(4 and 5) Electrons and holes approaching the surface are utilized by catalytic active sites to initiate the desired oxidative–reductive reactions. This is a relatively slow process occurring at the microsecond to millisecond range (10−6 to 10−3 seconds).
(6) Electron–hole pairs can recombine with each other, generating energy in the form of fluorescence or heat within nanoseconds.78,79
3.2. Photocatalysis for H2 production
Hydrogen is gaining attention as a promising renewable fuel source because it offers energy without carbon emissions. Unlike conventional electricity, hydrogen fuel can utilize storage and transportation infrastructure, and it can be stored for extended periods. Hydrogen can be stored as a gas, liquid, or absorbed in metal hydrides. Additionally, the production of hydrogen benefits from an abundant water supply, and there are no exhaust emissions during the production process. As shown in Fig. 4(c), as the hydrogen content factor (HCF) increases, the greenization factor (GF) rises, while the environmental impact factor (EIF) decreases.80,81 EIFmax is considered for coal. In other words, hydrogen is expected to play a crucial role in reducing carbon emissions.Hydrogen can be produced from various forms of energy sources, including electricity, heat, light, and biochemical processes. Among these, hydrogen production using photocatalysis converts photon energy into chemical energy (hydrogen). To choose the optimal photocatalyst for hydrogen production, the rapid generation and separation of electron–hole pairs are essential. Semiconductors and metal oxides have been extensively studied as photocatalysts. Semiconductors with narrower bandgaps can harvest more visible light, enabling them to utilize solar energy more effectively and achieve higher solar-hydrogen efficiency. However, the bandgap of semiconductors should theoretically be greater than 1.23 eV, and practical consideration of overpotential is required for actual oxidative–reductive reactions. Photoreduction and photooxidation reactions are expressed as follows:80
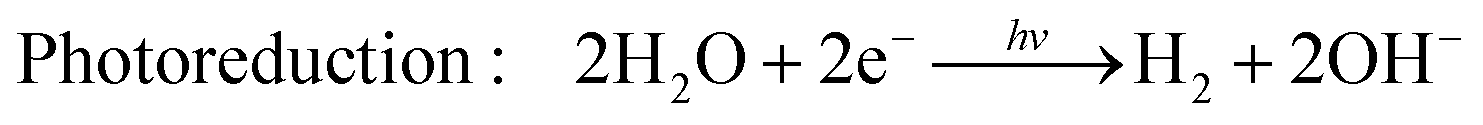
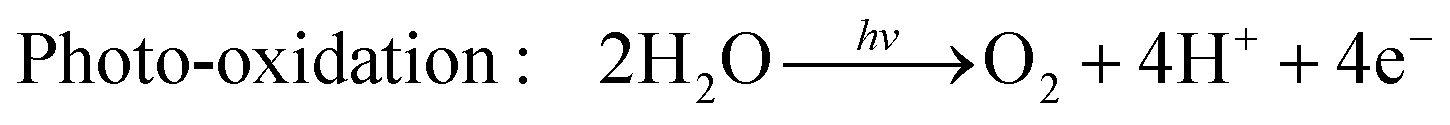
3.3. Photocatalysis for CO2 reduction
Converting CO2 into CO and hydrocarbons requires a substantial amount of energy. Therefore, photocatalytic CO2 conversion using solar energy is a promising technology for reducing CO2 emissions. While photocatalytic CO2 reduction technology offers the advantage of reducing greenhouse gas concentrations and producing available energy sources, it faces the challenge of relatively low overall efficiency. The CO2 conversion process unfolds as follows:821. CO2 molecules adsorb onto the photocatalyst's surface.
2. The photocatalyst absorbs photons with energy greater than its bandgap, generating electron–hole pairs in the conduction and valence bands.
3. The photo-generated electrons and holes then move to the surface of the photocatalyst.
4. The electrons react with adsorbed CO2 on the surface to produce CO and hydrocarbons.
5. Finally, the generated products (CO and hydrocarbons) are desorbed from the surface.
Through this conversion technology, both C1 and C2+ products can be produced.83 Notable C1 products include CO, CH4, HCOOH, and CH3OH, while C2+ products include C2H4, C2H5OH, C2H6, propanol, and acetone. C2+ products have broader applications and higher added value compared to C1 products. They boast high energy density and play a significant role in the chemical industry. Specifically, C2H4 is a primary component used in various plastic and cosmetic productions, while C2H5OH serves as alcohol beverages, pharmaceutical preservatives, and fuel. Given their higher value and derivation from C1 products, proposing the production of C2+ through a single CO2 reduction process is suggested.
To enhance selectivity for C2+ products, it is essential to suppress the ratio of C1 products and inhibit the hydrogen evolution reaction pathway. The preference for reaction pathways leading to desired products depends on the adsorption energies of intermediates at the active sites of the catalyst surface. Therefore, research on efficient photocatalysts for CO2 conversion is required. Table 2 presents common chemical reaction equations and associated potentials for CO2 reduction.84 For instance, when converting CO2 into CO, the photocatalyst's CBM and VBM should be less than −0.53 V and greater than +0.81 V, respectively. This implies that the photocatalyst's bandgap should be greater than 1.34 eV.
| Reaction equations | E 0 (V vs. NHE) | Main products |
|---|---|---|
| CO2 + 2H+ + 2e− → HCOOH | −0.61 | Formic acid |
| CO2 + 2H+ + 2e− → CO + 2H2O | −0.53 | Carbon monoxide |
| CO2 + 4H+ + 4e− → HCHO + H2O | −0.48 | Formaldehyde |
| CO2 + 6H+ + 6e− → CH3OH + H2O | −0.38 | Methanol |
| CO2 + 8H+ + 8e− → CH4 + 2H2O | −0.24 | Methane |
| 2CO2 + 8H+ + 8e− → CH3COOH + 2H2O | −0.31 | Acetic acid |
| 2CO2 + 14H+ + 14e− → C2H6 + 4H2O | −0.51 | Ethane |
4. LFDHP photocatalytic for H2 production
To utilize perovskite photocatalysts effectively, the bandgap needs to be reduced, and they should possess a broad absorption spectrum. Achieving this involves adopting 1+ and 3+ cations and arranging them orderly on the B-site to form double perovskites, as mentioned earlier. Let's delve into the research results on H2 production using LFDHP photocatalysts.4.1. Doping strategies for improved photocatalysis
Metal halide perovskites are ionic substances, making it challenging to use them directly as photocatalysts for water splitting. To overcome this issue, a concept called HX splitting was proposed, involving the use of saturated hydrohalic acid (HX) solutions. Saturated HX solution contributes to improving the activity of photocatalysts. HX splitting is employed by photocatalysts to absorb photon energy and facilitate chemical reactions, impacting charge transfer processes. Furthermore, it affects the chemical reactions occurring at the active sites on the surface of the photocatalyst, accelerating and enhancing the efficiency of the reactions. In other words, the use of HX enhances the activity of the photocatalyst, leading to increased efficiency in chemical reactions. Li et al. combined Ag atoms with Cs3Br2Br9 to induce the transformation into the double perovskite structure Cs2AgBiBr6.85 They found that Ag atoms occupying vacancies eliminate the distortion of the BiBr6 octahedra and remove strong localization of electron–hole pairs. As a result, Cs2AgBiBr6 exhibited lower exciton binding energy, higher carrier mobility, and longer lifetime, leading to a significant enhancement in photocatalytic performance (Fig. 5(a)). Additionally, when electron acceptor H3PO2 was introduced into HBr solution, the photocatalytic activity of Cs2AgBiBr6 was further improved, reaching 54.7 μmol g−1 h−1, as seen in Fig. 5(b). This demonstrates that simple doping methods can enhance the photocatalytic properties of perovskites. Chen et al. synthesized Cs3Bi2xSb2−2xI9 by doping Sb into Cs3Bi2I9.86 Sb doping effectively reduced the contribution of Bi3+ to the conduction band and mitigated the influence of Bi vacancies on the band structure (Fig. 5(c)). The introduction of Sb, with a smaller ionic radius than Bi, into Cs3Bi2I9 has been found to reduce the contribution of Bi metal ions to the conduction band, thereby mitigating the impact of defects and decreasing the intermediate gap state. Cs3Bi2I9 exhibited a photocatalytic activity of 1.12 μmol h−1 for HI solution, but after Sb doping, this activity significantly increased to 78.6 μmol h−1, as shown in Fig. 5(d).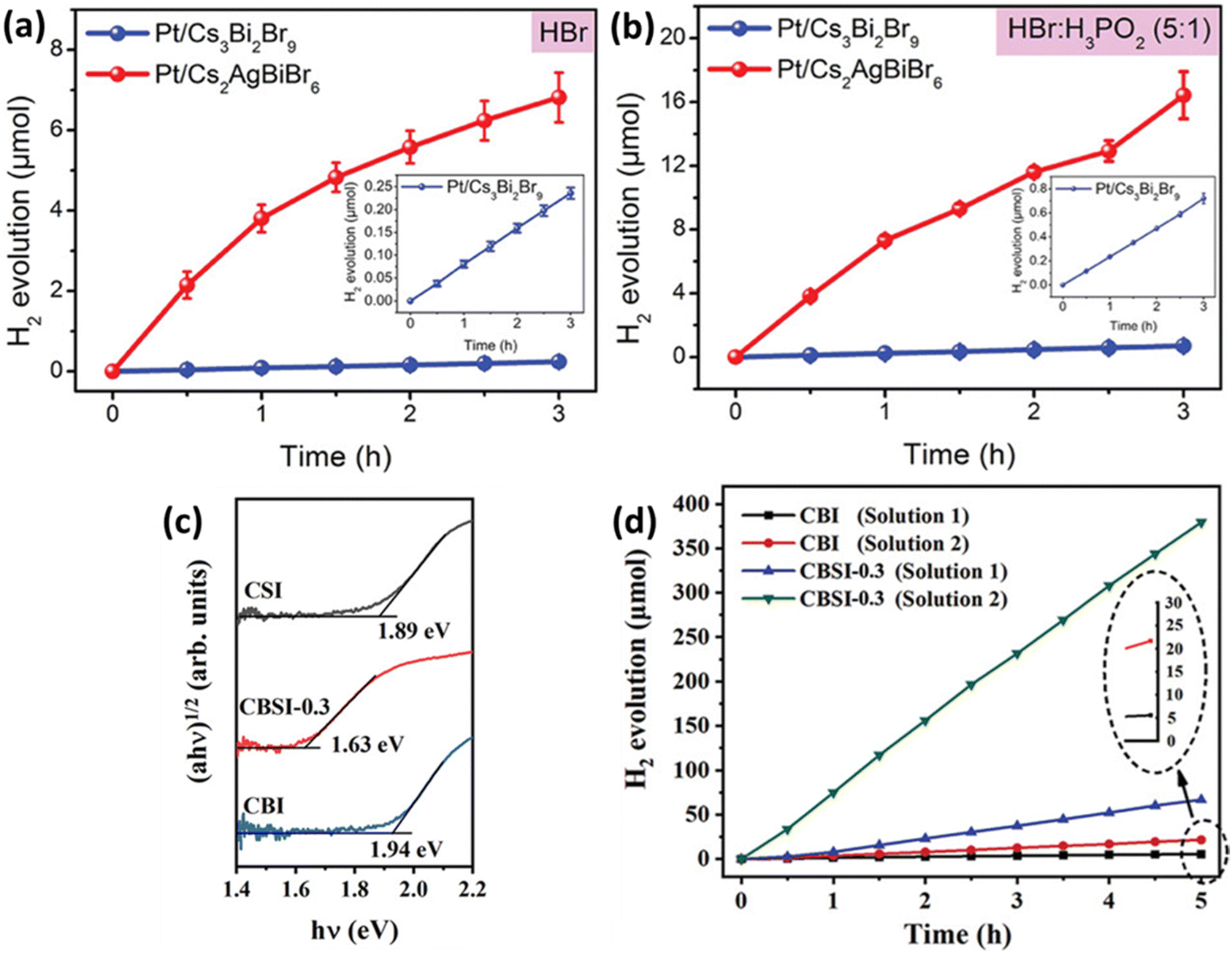 | ||
| Fig. 5 Hydrogen evolution performance of Pt/Cs3Bi2Br9 and Pt/Cs2AgBiBr6 in aqueous solutions of (a) HBr and (b) HBr/H3PO2.85 Reproduced with permission from ref. 85 Copyright © 2020 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. (c) Optical absorption spectra of Cs3Bi2I9 (CBI), Cs3Sb2I9 (CSI), and Cs3Bi2xSb2−2xI9 (CBSI-0.3). (d) Comparative analysis of the hydrogen evolution activity between CBI and CBSI-0.3 in Solution 1 and 2.86 Reproduced with permission from ref. 86 Copyright © 2020 Wiley-VCH GmbH. | ||
Surface defects in Bi-based materials have been reported to enhance surface charge separation and improve photocatalytic performance.87,88 He et al. formed defect-rich Cs2AgBiBr6 by exposing Cs2AgBiBr6 to visible light, demonstrating that the formation of surface defects optimizes local atomic arrangements, electronic structures, and provides active sites for trapping electrons, leading to higher charge separation and increased photocatalytic efficiency.89 The defect-rich Cs2AgBiBr6 exhibited powerful photocatalytic hydrogen evolution, reaching 4.06 μmol g−1 within 10 hours, about 5.27 times better than pristine Cs2AgBiBr6 (0.77 μmol g−1 within 10 hours), with sustained performance over 80 hours of photocatalytic reaction.
4.2. Composite strategies with 2D materials
Ongoing research aims to enhance the performance of LFDHP by combining them with 2D conductive materials. Reduced graphene oxide (RGO) is known for its excellent electron transfer properties and chemical stability. Wang et al. produced Cs2AgBiBr6/RGO composites through photoreduction and confirmed their photocatalytic activity.90 Photogenerated electrons in Cs2AgBiBr6 migrate to the conductive RGO, where they reduce H+ to form H2, while holes oxidize Br− to Br3− on Cs2AgBiBr6 particles. The introduction of RGO significantly increased the H2 production rate to ∼80 times that of Cs2AgBiBr6 in HBr/H3PO2 solution and maintained high activity continuously for 120 hours. Another approach involves introducing nitrogen-doped carbon (N–C) into LFDHP to enhance charge transfer.91 Cs2AgBiBr6/N–C demonstrated a high hydrogen evolution rate of 380 μmol g−1 h−1 under visible light irradiation (λ ≥ 420 nm), which was 19 times higher than pure Cs2AgBiBr6 (Fig. 6(a)). High-surface-area N–C prevents perovskite aggregation and provides more pathways for the movement of photogenerated carriers. Nitrogen defects promote photogenerated electron transfer, spatially separating electrons and holes within perovskites, significantly improving photocatalytic performance.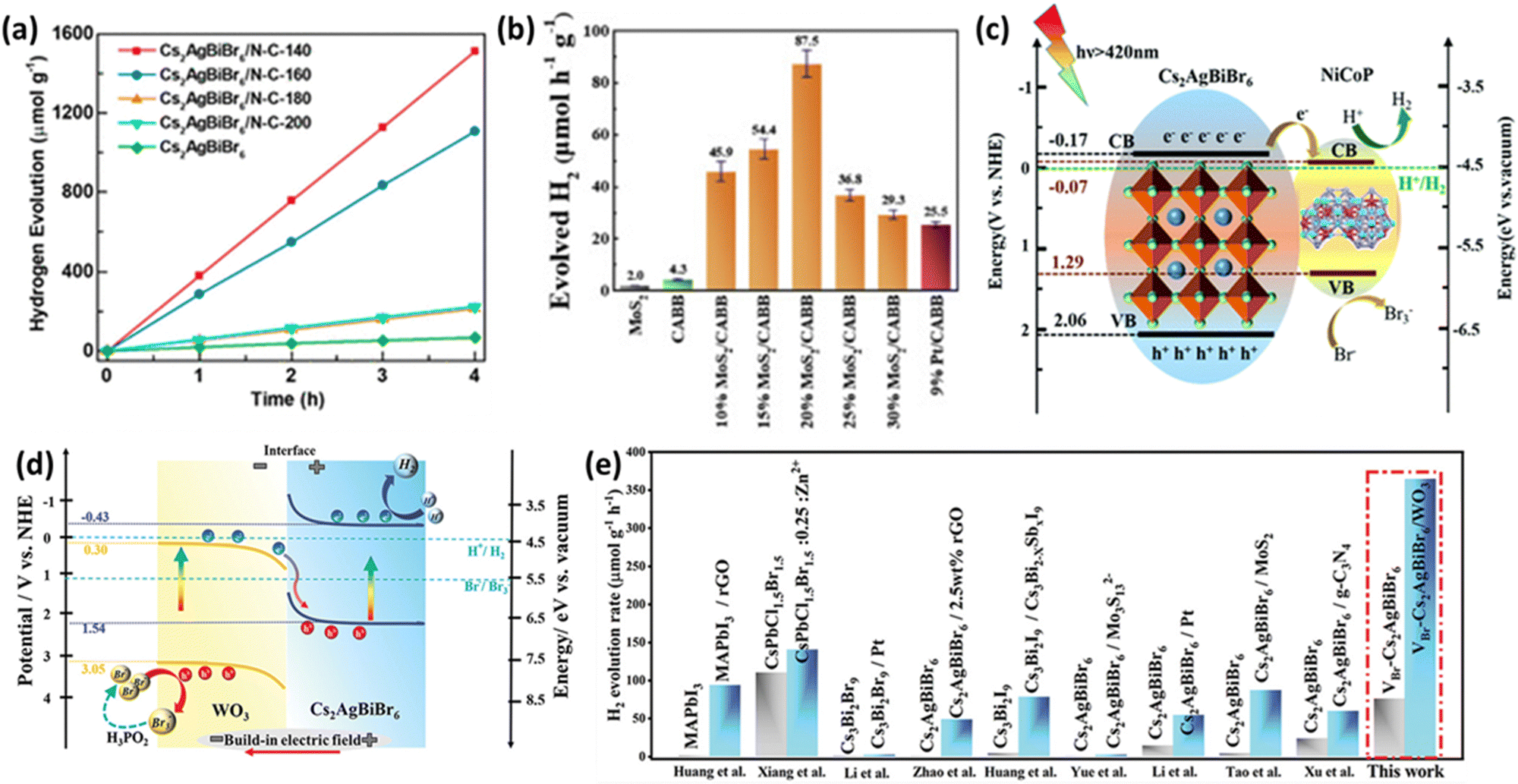 | ||
| Fig. 6 (a) Hydrogen production rate from HBr aqueous solution using Cs2AgBiBr6 and Cs2AgBiBr6/N–C–Th composites under visible light with a cutoff filter (λ ≥ 420 nm), and stability assessment.91 Reproduced with permission from ref. 91 Copyright © 2021, American Chemistry Society. (b) Photocatalytic hydrogen evolution rates for MoS2, CABB, MoS2/CABB, and Pt/CABB under visible light illumination (300 mW cm−2, λ ≥ 420 nm).93 Reproduced with permission from ref. 93 Copyright © 2021, American Chemical Society. (c) Mechanism for photocatalytic hydrogen generation over NiCoP (NCP)/Cs2AgBiBr6 (CABB).94 Reproduced with permission from ref. 94 Copyright © 2022 Royal Society of Chemistry. (d) Schematic representation of charge transfer and dynamic equilibrium processes. (e) Comparative analysis of hydrogen evolution performance between previously reported perovskite-based catalysts and VBr-Cs2AgBiBr6/WO3.98 Reproduced with permission from ref. 98 Copyright © 2023 Wiley-VCH GmbH. | ||
Research also explores the use of LFDHP combined with transition metal dichalcogenides (TMDs) to achieve high hydrogen production rates and long-term stability. TMDs, such as MoS2, have been widely used as photocatalysts for hydrogen evolution reactions (HER) due to their similar hydrogen binding energy to noble metal catalysts and excellent stability in HBr solutions.92 Zhang et al. proposed a MoS2/Cs2AgBiBr6 (MoS2/CABB) composite photocatalyst that exhibited an HER rate of 87.5 μmol h−1 g−1, which was about 20 times higher than Cs2AgBiBr6 (CABB) and even outperformed Pt/Cs2AgBiBr6 (Pt/CABB) as shown in Fig. 6(b).93 The composite maintained excellent photocatalytic stability during a discontinuous 500 hour test over 40 days.
Auxiliary catalysts are used to facilitate electron transfer and electron–hole pair separation. Transition metal phosphides (TMPs) are gaining attention as alternative catalysts that can replace noble metals. Huang et al. synthesized the NiCoP/Cs2AgBiBr6 (NCP/CABB) composite through electrostatic binding.94 The optimized NCP/CABB composite achieved a high H2 generation rate of 373.16 μmol g−1 h−1, which was about 88 times higher than pure CABB (4.23 μmol g−1 h−1) and significantly superior to CABB/5% Pt (7.78 μmol g−1 h−1). As shown in Fig. 6(c), the mechanism involves easy activation of electrons and holes under visible light, where electrons transfer easily from CABB's valence band to NCP's conduction band, facilitating the H2 production reaction. The change in interfacial energy positions during electron transfer resulted in band bending at the interface. The introduction of highly conductive TMP nanoparticles enhanced photogenerated charge separation and movement, significantly improving photocatalyst performance.
4.3. Heterojunction approaches for enhanced photocatalysis
Recent research has revealed that a step-scheme (S-scheme) heterojunction composed of n-type reduction photocatalysts and n-type oxidation photocatalysts with staggered band alignment has a strong oxidation–reduction potential.95–97 Cs2AgBiBr6 rich in Br-vacancies combined with WO3 nanorods in an S-scheme heterojunction exhibited excellent photocatalytic properties.98 Abundant Br-vacancies provided effective active sites, while the S-scheme heterojunction ensured efficient charge transfer at the interface (Fig. 6(d)). The optimized VBr-Cs2AgBiBr6/WO3 S-scheme photocatalyst demonstrated a high hydrogen evolution rate of 364.89 μmol g−1 h−1, surpassing other perovskite-based catalysts (Fig. 6(e)). These findings highlight the promising potential of perovskite photocatalysts, especially LFDHP, in various applications with enhanced performance through doping, defect engineering, and composite strategies.5. LFDHP photocatalytic for CO2 reduction
In this section, we delve into the promising applications of LFDHP as photocatalysts for converting CO2 into CO and hydrocarbons, highlighting their potential in addressing environmental challenges and clean energy generation.5.1. LFDHP as photocatalysts
Zhou et al. synthesized Cs2AgBiBr6 nanocrystals using a simple hot injection method in Fig. 7(a) and used them as catalysts for CO2 reduction.99 They achieved a total electron consumption of 105 μmol g−1 under AM 1.5G illumination, demonstrating its significant potential as a photocatalyst (Fig. 7(b)). It also exhibited excellent stability in nonpolar solvents, 55% relative humidity, and thermal and light stability tests. Additionally, the performance of LFDHP photocatalysts was found to vary significantly depending on their morphology. Cs2AgBiX6 synthesized in a 2D nanoplatelet form outperformed nanocubes in CO2 reduction, attributed to enhanced charge transport and longer diffusion lengths within nanoplatelets.100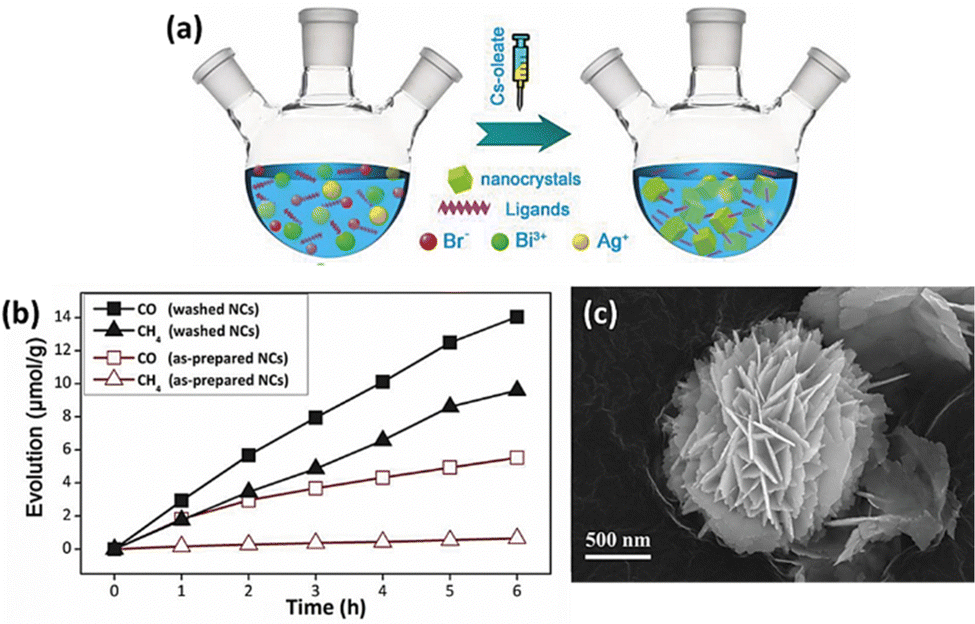 | ||
| Fig. 7 (a) Diagram depicting the solution-phase synthesis of Cs2AgBiBr6 NCs using the hot-injection route. (b) Evolution of CO and CH4 over as-prepared and washed Cs2AgBiBr6 NCs over time.99 Reproduced with permission from ref. 99 Copyright © 2018 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. (c) FE-SEM (Field Emission Scanning Electron Microscopy) image of Cs2NaBiCl6.102 Reproduced with permission from ref. 102 Copyright © 2022 Wiley-VCH GmbH. | ||
Reducing the size of a material leads to an increase in its surface area. This, in turn, creates more active sites and shortens the distance for charges (electrons and holes) to move across the surface. As a result, the likelihood of electrons and holes recombining decreases.101 This phenomenon enhances the activity of the photocatalyst. Pi et al. introduced the 3D hierarchical Cs2NaBiCl6 photocatalyst with abundant chlorine vacancies.102Fig. 7(c) depicts numerous 3D hierarchical microspheres assembled from nano flakes. Chlorine vacancies suppressed electron–hole recombination, enhanced CO2 adsorption, and reduced the reaction energy barrier for CO2 to CO conversion. This defect-engineered perovskite photocatalyst demonstrated a substantial electron consumption of 412.26 μmol g−1 during a 6 hour test.
5.2. Combining LFDHP with other materials
Combining LFDHP with other materials can enhance CO2 reduction performance. Chen et al. synthesized Cs2AgInCl6 quantum dots via a hot injection method and controlled the Cl source to create composites with varying Ag loadings.103 These composites exhibited improved CO2 reduction efficiency through the surface plasmon resonance effect of Ag nanoparticles, achieving maximum yields of 26.4 μmol g−1 for CO and 28.9 μmol g−1 for CH4 during CO2 reduction. Furthermore, the study explored the CO2 reduction capabilities of a 2D Cs2AgBiBr6 nanoflake-Cu-RGO heterostructure without organic solvents, utilizing H2O vapor as a proton source.104 The results showed an overall enhancement in photocatalytic performance, particularly in CH4 selectivity as shown in Fig. 8(a) and (b).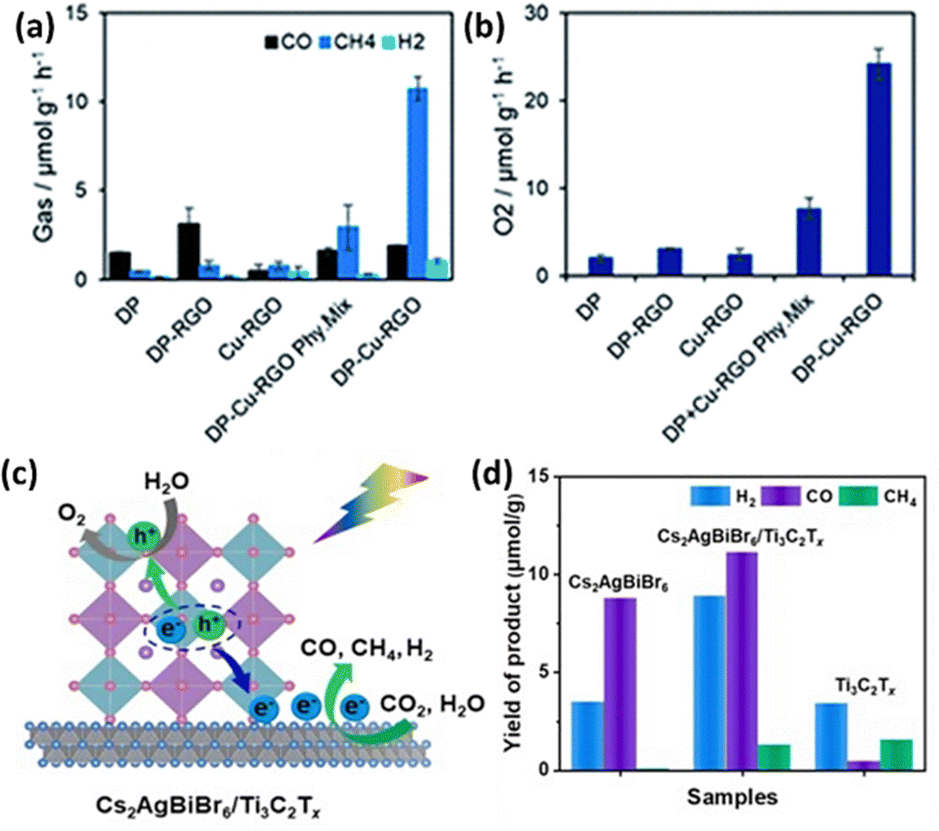 | ||
| Fig. 8 (a) Production rates of CO, CH4, H2, and (b) O2 from the photocatalytic reduction of CO2 using water vapor as a proton source on various photocatalysts under simulated solar light for 4 hours.104 Reproduced with permission from ref. 104 Copyright © 2022 Wiley-VCH GmbH. (c) Diagram illustrating the mechanism of photocatalytic CO2 reduction by Cs2AgBiBr6/Ti3C2Tx heterostructures under visible light irradiation. (d) Comparison of the photocatalytic activity between Cs2AgBiBr6, Ti3C2Tx, and Cs2AgBiBr6/Ti3C2Tx heterostructures for CO2 reduction.110 Reproduced with permission from ref. 110 Copyright © 2022 Elsevier B.V. All rights reserved. | ||
In a prior study, a 2D Bismuth monolayer exhibited a remarkable electrocatalytic efficiency in producing formate (HCOO−) during CO2 reduction reactions.105,106 Building upon this foundational research, Sena et al. assembled Cs2AgBiBr6 onto this bismuth monolayer, creating a composite material.107 This composite photocatalyst significantly improved charge separation and reduced the rate of electron–hole recombination, thereby enhancing the production and selectivity of the eight-electron CH4 pathway. The Cs2AgBiBr6/Bi monolayer composite displayed outstanding photocatalytic activity, with rates of 1.49(±0.16) μmol g−1 h−1 for CH4, 0.67(±0.14) μmol g−1 h−1 for CO, and 0.75(±0.20) μmol g−1 h−1 for H2, along with a CH4 selectivity of 81(±1)% based on one solar electron standard.
Among 2D materials, MXenes, with the general chemical formula Mn+1XnTx (M = early transition metal, X = carbon and/or nitrogen, Tx = surface group), exhibit excellent conductivity, high electrical capacity, a high elastic modulus, and an adjustable bandgap.108 However, 2D nanosheets with atomic thickness often have limited light absorption capabilities.109 As shown in Fig. 8(c), Zhang et al. proposed a self-assembled heterostructure of Cs2AgBiBr6 nanocrystals on MXene nanosheets' surfaces through mutual electrostatic interactions.110 These MXene nanosheets effectively facilitated the formation of free charge carriers in Cs2AgBiBr6 nanocrystals by reducing the exciton binding energy. Consequently, this resulted in a high photocurrent consumption rate of 50.6 μmol g−1 h−1 during the photocatalytic reduction of CO2 (Fig. 8(d)).
5.3. Z-Scheme systems for enhanced photocatalysis
In the pursuit of developing efficient photocatalysts, researchers have introduced a concept known as Z-scheme heterojunctions. These Z-scheme systems offer the advantage of spatially separating photoinduced charge carriers, enabling two semiconductors to simultaneously harness solar energy.111 When light is incident upon them, electrons generated in the conduction band of the oxidative semiconductor are transferred to the valence band of the reductive semiconductor, where they recombine.112Mahmoud et al. proposed a Z-scheme heterostructure involving Cs2AgBiBr6 (CABB) and Sr2FeNbO6 (SFNO).113 The energy band gaps of CABB and SFNO were determined to be 2.17 eV and 2.04 eV, respectively, with conduction band minima (CBM) located at −0.57 V and 0.10 V (relative to the NHE at pH = 7), as depicted in Fig. 9(a). This configuration facilitates the efficient transfer of electrons from SFNO's CBM to CABB's VBM when exposed to light. As observed in the time-resolved PL decay shown in Fig. 9(b), this heterostructure promotes effective spatial charge separation. The CABB/SFNO Z-scheme photocatalyst exhibited remarkable CO and CH4 production rates of 50.00 and 8.12 μmol g−1 h−1, respectively, as illustrated in Fig. 9(c). This enhanced performance is attributed to the unique Z-scheme heterojunction, which effectively separates charge carriers in space. Research utilizing the Z-scheme approach is gaining attention due to its simplicity in synthesizing heterostructures. Wu et al. used a simple hot injection method to grow CABB nanocrystals in situ on the surface of Bi2WO6 (BWO) nanosheets.114 As shown in Fig. 9(d), the resulting CABB/BWO heterojunction, thanks to the presence of spatial charge separation characteristic of a Z-scheme, exhibited high photocatalytic activity with a substantial electron consumption rate of 87.66 μmol g−1 h−1 under simulated sunlight conditions (AM 1.5G).
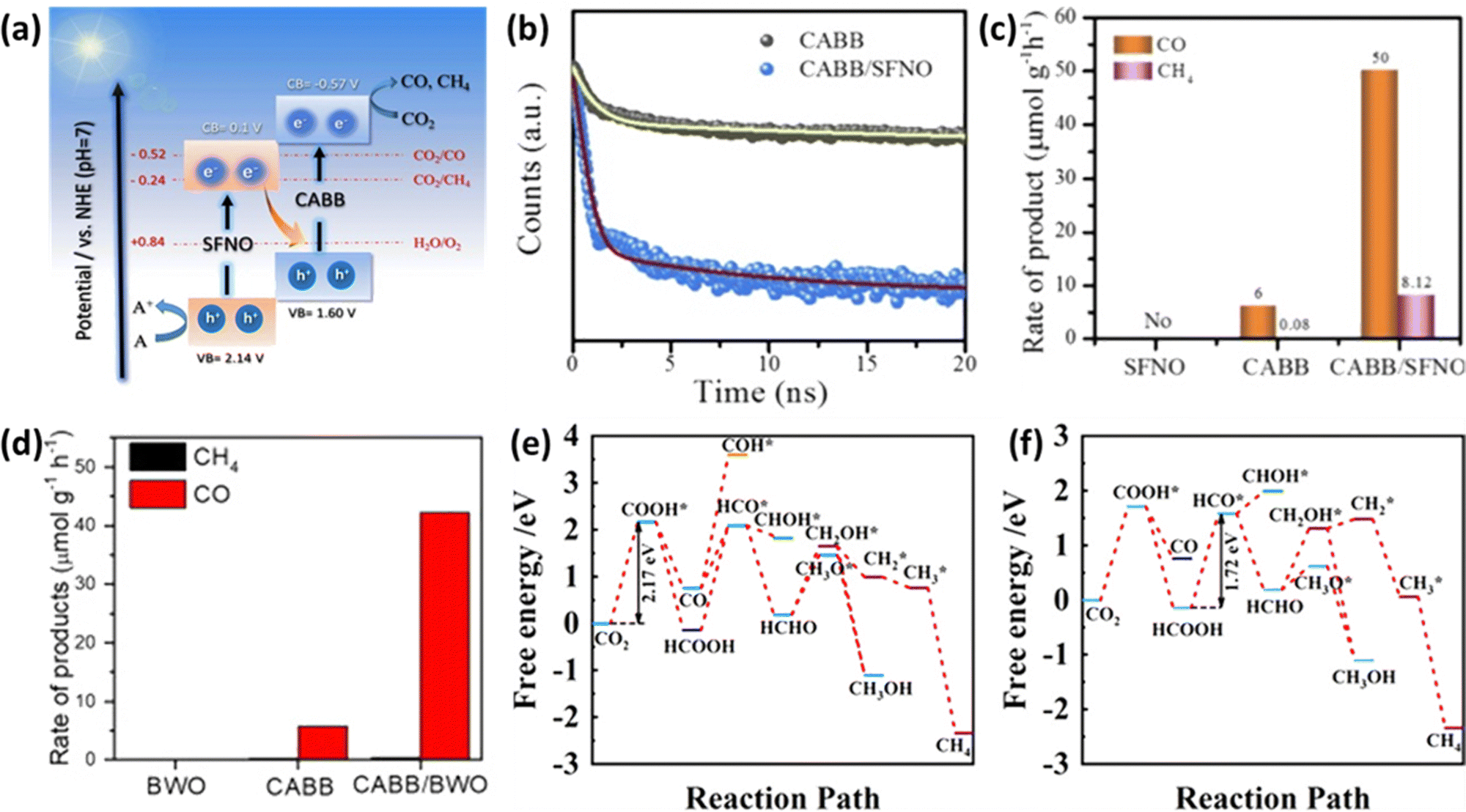 | ||
| Fig. 9 (a) Illustration of the potential reaction mechanism of the CABB/SFNO Z-scheme heterojunction. (b) Steady-state PL (photoluminescence) and TRPL (Time-Resolved Photoluminescence) decay spectra of CABB and CABB/SFNO heterojunction. (c) Photocatalytic activity test comparing SFNO, CABB, and CABB/SFNO heterojunction.113 Reproduced with permission from ref. 113 Copyright © 2022 Elsevier B.V. All rights reserved. (d) Photocatalytic activity test comparing CABB, BWO, and CABB/BWO.114 Reproduced with permission from ref. 114 Copyright © 2022 Elsevier Inc. All rights reserved. (e) Free energy diagram depicting possible reaction pathways for CO2 reduction on the (CH3NH3)2AgBiI6 (010) surface. (f) Free energy diagram illustrating possible reaction pathways for CO2 reduction on the SnS/(CH3NH3)2AgBiI6 heterostructure.117 Reproduced with permission from ref. 117 Copyright Copyright © 2022, American Chemical Society. | ||
Continuing research endeavors involve the integration of 2D materials with perovskites within Z-scheme systems to enhance CO2 reduction performance. For example, the 2D organic semiconductor g-C3N4, which previously exhibited unsatisfactory photocatalytic activity for CO2 reduction due to rapid carrier recombination and inefficient utilization of visible light,115 has been combined with CABB in a Z-scheme configuration.116 This innovative combination capitalizes on g-C3N4's CBM reduction ability and CABB's VBM oxidation capability, resulting in impressive CH4 production with high selectivity. Furthermore, research by Li et al. has elucidated the Z-scheme photocatalytic mechanism of SnS/(CH3NH3)2AgBiI6, analyzing band energy positions and interfacial charge transfer.117 The interaction at the interface between the (CH3NH3)2AgBiI6 (010) surface and SnS monolayers induces electron flow from SnS to the (CH3NH3)2AgBiI6 (010) surface. As seen in Fig. 9(e) and (f), Calculations of Gibbs free energy changes (ΔG) in comparison between the (CH3NH3)2AgBiI6 surface and SnS/(CH3NH3)2AgBiI6 revealed a decrease from 2.17 to 1.72 eV. This outcome signifies that SnS/(CH3NH3)2AgBiI6 enhances the photocatalytic activity for CO2 reduction.
6. Summary and prospects
LFDHP has garnered significant attention in the fields of photocatalytic hydrogen production and CO2 reduction due to its exceptional optical properties, adjustable bandgap, superior charge carrier transport capabilities, and abundant active sites. This review provides an overview of LFDHP, its synthesis methods, and recent research on its applications in hydrogen production and CO2 reduction over the past few years. It was summarized in Fig. 10. It serves as a valuable reference for the development of new LFDHP photocatalysts with high efficiency and selectivity for hydrogen production and CO2 conversion. While LFDHP photocatalysts for hydrogen production and CO2 reduction have achieved many successes in recent years, there is still much room for improvement, which can be summarized in terms of material aspects, hydrogen production catalysis, CO2 reduction catalysis, and theoretical considerations. | ||
| Fig. 10 Visual overview of review content. | ||
6.1. Material aspects
Currently, LFDHP photocatalysts primarily focus on the BiAg-based perovskite structure type. These photocatalysts possess an appropriate bandgap and band structure but suffer from unsatisfactory performance due to undesired electron–hole recombination. To address this issue, it is possible to adjust the electron band structure through doping or substituting elements, thereby enhancing visible light utilization efficiency and oxidative reduction capacity. Another approach to suppress carrier recombination in bulk photocatalysts is by reducing the size of perovskite powder to nanocrystals or quantum dots. Heterojunction photocatalysts offer a method to improve the spatial separation of photogenerated charge carriers, resulting in high-efficiency photocatalytic performance. Loading cocatalysts can inhibit charge carrier recombination and increase active sites, further improving photocatalytic activity.6.2. Aspects of hydrogen production photocatalysis
Due to LFDHP's sensitivity to moisture, conducting photocatalytic reactions in halogen acid solutions has proven effective for hydrogen generation. However, it's essential to note that this does not imply complete safety of LFDHP in halogen acids. Thus, researching excellent sealing techniques to restrict contact between LFDHP and polar solvents is crucial. The sealing material must be transparent enough to ensure sufficient light absorption by LFDHP and possess excellent conductivity for effective carrier extraction. Commonly used sealing materials include transparent resins (epoxy) and conductive carbon paste. Some studies encapsulate LFDHP within a Photovoltaic-photoelectrochemical cell (PV-PEC) to protect perovskite from polar solvents. This system gains attention as it increases the oxidation–reduction capacity of the PEC cell, enabling higher voltages in series due to the presence of solar components. Additionally, research on LFDHP with excellent water stability for direct hydrogen production in water is gradually increasing. Alongside improving water stability, researching LFDHP with high photocatalytic activity will be a crucial breakthrough in future studies.6.3. Aspects of CO2 reduction
Research on the stability of LFDHP for photocatalytic CO2 reduction, charge carrier mobility, electron–hole recombination, and catalyst activity mechanisms is essential. Furthermore, studying the impact of photon energy, irradiation time, catalytic reaction, reaction medium, and O2 production on LFDHP's stability and photocatalytic performance is necessary. Doping is one method to enhance the CO2 reduction performance of photocatalysts by partially replacing some atoms in the perovskite composition. This approach allows for bandgap adjustment to increase light absorption efficiency and control the CBM. Heterojunctions with superior optical properties, such as 2D semiconductors, can facilitate the separation of photogenerated electron–hole pairs. Another effective approach is immobilizing metal nanoparticles on the surface of LFDHP, forming Schottky heterojunctions.6.4. Theoretical aspects
To provide guidelines for improving photocatalytic efficiency, detailed information about the photocatalytic process, especially the reaction pathways and kinetics of photogenerated charge carriers in perovskite materials, is needed. Therefore, a comprehensive theoretical model is essential to interpret the role of perovskite materials in photocatalytic oxidation–reduction reactions. Theoretical approaches like Density Functional Theory (DFT) can help understand existing research and provide guidance for developing more efficient photocatalysts for oxidation–reduction reactions. In addition to theoretical calculations, on-site characterization techniques can support mechanism studies.118 For example, in situ X-ray photoelectron spectroscopy (XPS) can investigate electron transfer pathways by observing changes in binding energy. In situ X-ray absorption spectroscopy (XAS) can study the evolution of electronic structure on the catalyst's surface and monitor dynamic changes in the state of metal atoms, identifying active sites. Time-resolved ultrafast spectroscopy can provide a comprehensive and reliable understanding of charge carrier behaviour in LFDHP. Continued exploration in these promising areas is expected to lead to significant improvements in the efficiency and stability of LFDHP hydrogen production and CO2 reduction photocatalysts, making practical applications possible in the near future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the National Research Foundation of Korea (NRF) (2022M3H4A1A01012712, 2022M3H4A1A04096380).Notes and references
- Global temperature trend monitor, https://cds.climate.copernicus.eu/cdsapp#!/software/app-c3s-global-temperature-trend-monitor?tab=app, (accessed 11 July 2023).
- J. Luo, W. Zhang, H. Yang, Q. Fan, F. Xiong, S. Liu, D. S. Li and B. Liu, EcoMat, 2021, 3, e12079 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS.
- L. Wang, J. Zhang, Y. Zhang, H. Yu, Y. Qu and J. Yu, Small, 2022, 18, 2104561 CrossRef CAS.
- W. Jiang, H. Loh, B. Q. L. Low, H. Zhu, J. Low, J. Z. X. Heng, K. Y. Tang, Z. Li, X. J. Loh, E. Ye and Y. Xiong, Appl. Catal., B, 2023, 321, 122079 CrossRef CAS.
- C. Karthikeyan, P. Arunachalam, K. Ramachandran, A. M. Al-Mayouf and S. Karuppuchamy, J. Alloys Compd., 2020, 828, 154281 CrossRef CAS.
- H. H. Do, D. L. T. Nguyen, X. C. Nguyen, T. H. Le, T. P. Nguyen, Q. T. Trinh, S. H. Ahn, D. V. N. Vo, S. Y. Kim and Q. V. Le, Arabian J. Chem., 2020, 13(2), 3653–3671 CrossRef CAS.
- T. P. Nguyen, D. L. T. Nguyen, V. H. Nguyen, T. H. Le, D. V. N. Vo, Q. T. Trinh, S. R. Bae, S. Y. Chae, S. Y. Kim and Q. Van Le, Nanomaterials, 2020, 10 Search PubMed.
- Z. Cheng, W. Qi, C. Heng Pang, T. Thomas, T. Wu, S. Liu, M. Yang, Z. Cheng, W. Qi, S. Liu, M. Yang, C. H. Pang, T. Wu and T. Thomas, Adv. Funct. Mater., 2021, 31, 2100553 CrossRef CAS.
- H. S. Gujral, G. Singh, A. V. Baskar, X. Guan, X. Geng, A. V. Kotkondawar, S. Rayalu, P. Kumar, A. Karakoti and A. Vinu, Sci. Technol. Adv. Mater., 2022, 23, 76–119 CrossRef CAS PubMed.
- S. L. Lee and C.-J. Chang, Catalysts, 2019, 9, 457 CrossRef CAS.
- S. Zhang, X. Ou, Q. Xiang, S. Carabineiro, J. Fan and K. Lv, Chemosphere, 2022, 303, 135085 CrossRef CAS.
- Q. Zhu, Q. Xu, M. Du, X. Zeng, G. Zhong, B. Qiu, J. Zhang, Q. H. Zhu, Q. Xu, X. F. Zeng, G. F. Zhong, M. M. Du, B. C. Qiu and J. L. Zhang, Adv. Mater., 2022, 34, 2202929 CrossRef CAS.
- S. Tasleem, M. Tahir and W. A. Khalifa, Int. J. Hydrogen Energy, 2021, 46, 14148–14189 CrossRef CAS.
- S. Gautam, H. Agrawal, M. Thakur, A. Akbari, H. Sharda, R. Kaur and M. Amini, J. Environ. Chem. Eng., 2020, 8, 103726 CrossRef CAS.
- J. Chen, R. Abazari, K. A. Adegoke, N. W. Maxakato, O. S. Bello, M. Tahir, S. Tasleem, S. Sanati, A. M. Kirillov and Y. Zhou, Coord. Chem. Rev., 2022, 469, 214664 CrossRef CAS.
- S. A. Younis, E. E. Kwon, M. Qasim, K. H. Kim, T. Kim, D. Kukkar, X. Dou and I. Ali, Prog. Energy Combust. Sci., 2020, 81, 100870 CrossRef.
- J. You, Y. Zhao, L. Wang and W. Bao, J. Cleaner Prod., 2021, 291, 125822 CrossRef CAS.
- H. Wang, H. Wang, Z. Wang, L. Tang, G. Zeng, P. Xu, M. Chen, T. Xiong, C. Zhou, X. Li, D. Huang, Y. Zhu, Z. Wang and J. Tang, Chem. Soc. Rev., 2020, 49, 4135–4165 RSC.
- Y. Zhou, F. Lu, T. Fang, D. Gu, X. Feng, T. Song and W. Liu, J. Alloys Compd., 2022, 911, 165062 CrossRef CAS.
- Y. Tang, C. H. Mak, G. Jia, K. C. Cheng, J. J. Kai, C. W. Hsieh, F. Meng, W. Niu, F. F. Li, H. H. Shen, X. Zhu, H. M. Chen and H. Y. Hsu, J. Mater. Chem. A, 2022, 10, 12296–12316 RSC.
- V. H. Nguyen, B. S. Nguyen, Z. Jin, M. Shokouhimehr, H. W. Jang, C. Hu, P. Singh, P. Raizada, W. Peng, S. Shiung Lam, C. Xia, C. C. Nguyen, S. Y. Kim and Q. Van Le, Chem. Eng. J., 2020, 402 Search PubMed.
- B. M. Bresolin, Y. Park and D. W. Bahnemann, Catalysts, 2020, 10, 709 CrossRef CAS.
- X. Qian, Z. Chen, X. Yang, W. Zhao, C. Liu, T. Sun, D. Zhou, Q. Yang, G. Wei and M. Fan, J. Cleaner Prod., 2020, 249, 119335 CrossRef CAS.
- D. Y. Heo, T. H. Lee, A. Iwan, L. Kavan, M. Omatova, E. Majkova, K. Kamarás, H. W. Jang and S. Y. Kim, J. Power Sources, 2020, 458, 228067 CrossRef CAS.
- E. Aktas, N. Rajamanickam, J. Pascual, S. Hu, M. H. Aldamasy, D. Di Girolamo, W. Li, G. Nasti, E. Martínez-Ferrero, A. Wakamiya, E. Palomares and A. Abate, Commun. Mater., 2022, 3, 1–14 CrossRef.
- P. K. Kung, M. H. Li, P. Y. Lin, J. Y. Jhang, M. Pantaler, D. C. Lupascu, G. Grancini and P. Chen, Solar RRL, 2020, 4, 1900306 CrossRef CAS.
- W. Cao, Z. Hu, Z. Lin, X. Guo, J. Su, J. Chang and Y. Hao, J. Energy Chem., 2022, 68, 420–438 CrossRef CAS.
- M. Ikram, R. Malik, R. Raees, M. Imran, F. Wang, S. Ali, M. Khan, Q. Khan and M. Maqbool, Sustain. Energy Technol. Assess., 2022, 53, 102433 Search PubMed.
- E. Aktas, N. Rajamanickam, J. Pascual, S. Hu, M. H. Aldamasy, D. Di Girolamo, W. Li, G. Nasti, E. Martínez-Ferrero, A. Wakamiya, E. Palomares and A. Abate, Commun. Mater., 2022, 3, 1–14 CrossRef.
- J. Lu, C. Yan, W. Feng, X. Guan, K. Lin and Z. Wei, EcoMat, 2021, 3, e12082 CrossRef CAS.
- S. Z. Wang, J. T. Wang, Y. H. Lou, Y. H. Zhou and Z. K. Wang, Adv. Mater. Interfaces, 2022, 9, 2200772 CrossRef CAS.
- M. J. Seol, J. W. Han, S. H. Hwang and S. Y. Kim, J. Korean Institute Metals Mater., 2022, 60 Search PubMed.
- Y.-T. Huang, S. R. Kavanagh, D. O. Scanlon, G. Kaur, A. Shukla, K. Justice Babu, M. Nath Tripathi, A. Saha, S. Singh, J. Ghosh, P. J. Sellin and P. K. Giri, Nanotechnology, 2022, 33, 312001 CrossRef.
- T. Jiang, W. Ma, H. Zhang, Y. Tian, G. Lin, W. Xiao, X. Yu, J. Qiu, X. Xu, Y. Yang, D. Ju, T. Jiang, W. Ma, Y. Tian, W. Xiao, Y. Yang, H. Zhang, X. Yu, J. Qiu, X. Xu, G. Lin and D. Ju, Adv. Funct. Mater., 2021, 31, 2009973 CrossRef CAS.
- G. S. H. Thien, M. Ab Rahman, B. K. Yap, N. M. L. Tan, Z. He, P. L. Low, N. K. Devaraj, A. F. Ahmad Osman, Y. K. Sin and K. Y. Chan, ACS Omega, 2022, 7, 39472–39481 CrossRef CAS PubMed.
- Y. Fang, S. Zhai, L. Chu and J. Zhong, ACS Appl. Mater. Interfaces, 2021, 13, 17141–17157 CrossRef CAS PubMed.
- W. J. Jang, H. W. Jang and S. Y. Kim, Small Methods, 2023, 2300207 CrossRef.
- U. Farooq, M. Ishaq, U. A. Shah, S. Chen, Z. H. Zheng, M. Azam, Z. H. Su, R. Tang, P. Fan, Y. Bai and G. X. Liang, Nano Energy, 2022, 92, 106710 CrossRef CAS.
- I. Dincer and C. Zamfirescu, Sustainable Hydrogen Production, 2016, pp. 309–391 Search PubMed.
- C. Acar, I. Dincer and C. Zamfirescu, Int. J. Energy Res., 2014, 38, 1903–1920 CrossRef CAS.
- P. Zhang, J. Yang and S. H. Wei, J. Mater. Chem. A, 2018, 6(4), 1809–1815 RSC.
- F. Igbari, Z. K. Wang and L. S. Liao, Adv. Energy Mater., 2019, 9, 1803150 CrossRef.
- F. Giustino and H. J. Snaith, ACS Energy Lett., 2016, 1, 1233–1240 CrossRef CAS.
- G. Li, K. Chen, Y. Cui, Y. Zhang, Y. Tian, B. Tian, Y. Hao, Y. Wu and H. Zhang, Adv. Opt. Mater., 2020, 8, 1902012 CrossRef CAS.
- W. Travis, E. N. K. Glover, H. Bronstein, D. O. Scanlon and R. G. Palgrave, Chem. Sci., 2016, 7, 4548–4556 RSC.
- K. Vighnesh, S. Wang, H. Liu and A. L. Rogach, ACS Nano, 2022, 16, 19618–19625 CrossRef CAS.
- D. Zhu, J. Zito, V. Pinchetti, Z. Dang, A. Olivati, L. Pasquale, A. Tang, M. L. Zaffalon, F. Meinardi, I. Infante, L. De Trizio, L. Manna and S. Brovelli, ACS Energy Lett., 2020, 5, 1840–1847 CrossRef CAS.
- M. Liu, S. K. Matta, H. Ali-Löytty, A. Matuhina, G. K. Grandhi, K. Lahtonen, S. P. Russo and P. Vivo, Nano Lett., 2022, 22, 311–318 CrossRef CAS.
- F. Zhang, H. Zhong, C. Chen, X. G. Wu, X. Hu, H. Huang, J. Han, B. Zou and Y. Dong, ACS Nano, 2015, 9, 4533–4542 CrossRef CAS PubMed.
- Y. T. Yoo, D. Y. Heo, S. R. Bae, J. Park, T. W. Lee, H. W. Jang, S. H. Ahn and S. Y. Kim, Small Methods, 2021, 5, 2100054 CrossRef CAS.
- Y. Guo, Y. Lou, J. Chen and Y. Zhao, ChemSusChem, 2022, 15, e202102334 CrossRef CAS PubMed.
- D. Chen, G. Fang, X. Chen, L. Lei, J. Zhong, Q. Mao, S. Zhou and J. Li, J. Mater. Chem. C, 2018, 6, 8990–8998 RSC.
- D. Chen, G. Fang, X. Chen, L. Lei, J. Zhong, Q. Mao, S. Zhou and J. Li, J. Mater. Chem. C, 2018, 6, 8990–8998 RSC.
- Y. J. Low, J. Y. C. Liew, M. A. Kamarudin, H. N. Lim, F. D. Muhammad, K. P. Lim, M. H. M. Zaid, T. F. Choo, H. K. Lee, Y. W. Fen, S. Hayase and Z. A. Talib, Mater. Today Chem., 2023, 29, 101477 CrossRef CAS.
- A. H. Slavney, T. Hu, A. M. Lindenberg and H. I. Karunadasa, J. Am. Chem. Soc., 2016, 138, 2138–2141 CrossRef CAS PubMed.
- H. Dong, C. Ran, W. Gao, N. Sun, X. Liu, Y. Xia, Y. Chen and W. Huang, Adv. Energy Mater., 2022, 12, 2102213 CrossRef CAS.
- A. Aftab and M. I. Ahmad, Sol. Energy, 2021, 216, 26–47 CrossRef CAS.
- R. Chiara, M. Morana and L. Malavasi, ChemPlusChem, 2021, 86, 879–888 CrossRef CAS PubMed.
- D. Y. Heo, T. H. Lee, A. Iwan, L. Kavan, M. Omatova, E. Majkova, K. Kamarás, H. W. Jang and S. Y. Kim, J. Power Sources, 2020, 458, 228067 CrossRef CAS.
- A. Yadegarifard, H. Lee, H. J. Seok, I. Kim, B. K. Ju, H. K. Kim and D. K. Lee, Nano Energy, 2023, 112, 108481 CrossRef CAS.
- F. Yang, D. Hirotani, G. Kapil, M. A. Kamarudin, C. H. Ng, Y. Zhang, I. Shen, S. Hayase and Y. H. Zhang, Angew. Chem., 2018, 130, 12927–12931 CrossRef.
- X. G. Zhao, J. H. Yang, Y. Fu, D. Yang, Q. Xu, L. Yu, S. H. Wei and L. Zhang, J. Am. Chem. Soc., 2017, 139, 2630–2638 CrossRef CAS PubMed.
- S. Chand Yadav, A. Srivastava, V. Manjunath, A. Kanwade, R. S. Devan and P. M. Shirage, Mater. Today Phys., 2022, 26, 100731 CrossRef CAS.
- P. Barbier, M. Drache, G. Mairesse and J. Ravez, J. Solid State Chem., 1982, 42, 130–135 CrossRef CAS.
- W. Urland, Chem. Phys. Lett., 1981, 83, 116–119 CrossRef CAS.
- E. T. McClure, M. R. Ball, W. Windl and P. M. Woodward, Chem. Mater., 2016, 28, 1348–1354 CrossRef CAS.
- A. H. Slavney, T. Hu, A. M. Lindenberg and H. I. Karunadasa, J. Am. Chem. Soc., 2016, 138, 2138–2141 CrossRef CAS.
- M. Shirayama, M. Kato, T. Miyadera, T. Sugita, T. Fujiseki, S. Hara, H. Kadowaki, D. Murata, M. Chikamatsu and H. Fujiwara, J. Appl. Phys., 2016, 119, 115501 CrossRef.
- L. Z. Lei, Z. F. Shi, Y. Li, Z. Z. Ma, F. Zhang, T. T. Xu, Y. T. Tian, D. Wu, X. J. Li and G. T. Du, J. Mater. Chem. C, 2018, 6, 7982–7988 RSC.
- J. Lao, W. Xu, C. Jiang, N. Zhong, B. Tian, H. Lin, C. Luo, J. Travas-Sejdic, H. Peng and C. G. Duan, J. Mater. Chem. C, 2021, 9, 5706–5712 RSC.
- J. Zhou, Z. Xia, M. S. Molokeev, X. Zhang, D. Peng and Q. Liu, J. Mater. Chem. A, 2017, 5, 15031–15037 RSC.
- K. Li, S. Li, W. Zhang, Z. Shi, D. Wu, X. Chen, P. Lin, Y. Tian and X. Li, J. Colloid Interface Sci., 2021, 596, 376–383 CrossRef CAS PubMed.
- J. Wang, S. Yip, S. R. Phillpot and D. Wolf, Phys. Rev. Lett., 1993, 71, 4182–4185 CrossRef CAS.
- K. Radja, B. L. Farah, A. Ibrahim, D. Lamia, I. Fatima, B. Nabil, A. Mohamed, Y. Al-Douri and A. F. A. El-Rehim, J. Phys. Chem. Solids, 2022, 167, 110795 CrossRef CAS.
- M. A. Ghebouli, T. Chihi, B. Ghebouli and M. Fatmi, Chin. J. Phys., 2018, 56, 323–330 CrossRef CAS.
- 6.4 The Solar Spectrum | METEO 300: Fundamentals of Atmospheric Science, https://www.e-education.psu.edu/meteo300/node/683, (accessed 23 August 2023).
- Y. Dai and Y. Xiong, Nano Res. Energy, 2022, 1, e9120006 CrossRef.
- Z. Wu, H. Tüysüz, F. Besenbacher, Y. Dai and Y. Xiong, Nanoscale, 2023, 15, 5598–5622 RSC.
- C. Acar, I. Dincer and G. F. Naterer, Int. J. Energy Res., 2016, 40, 1449–1473 CrossRef CAS.
- I. Dincer and C. Acar, Int. J. Hydrogen Energy, 2015, 40, 11094–11111 CrossRef CAS.
- F. Qi, Y. Pu, D. Wu, X. Tang and Q. Huang, Chemical Record, 2023, e202300078 CrossRef CAS.
- G. H. Han, J. Bang, G. Park, S. Choe, Y. J. Jang, H. W. Jang, S. Y. Kim and S. H. Ahn, Small, 2023, 19, 2205765 CrossRef CAS.
- E. Gong, S. Ali, C. B. Hiragond, H. S. Kim, N. S. Powar, D. Kim, H. Kim and S. Il In, Energy Environ. Sci., 2022, 15, 880–937 RSC.
- M. Shi, G. Li, W. Tian, S. Jin, X. Tao, Y. Jiang, E. A. Pidko, R. Li, C. Li, M. Shi, X. Tao, Y. Jiang, R. Li, C. Li, G. Li, E. A. Pidko, W. Tian and S. Jin, Adv. Mater., 2020, 32, 2002137 CrossRef CAS.
- G. Chen, P. Wang, Y. Wu, Q. Zhang, Q. Wu, Z. Wang, Z. Zheng, Y. Liu, Y. Dai, B. G. Huang Chen, P. Wang, Y. Wu, Q. Zhang, Z. Wang, Z. Zheng, Y. Liu, B. Huang, Q. Wu and Y. Dai, Adv. Mater., 2020, 32, 2001344 CrossRef CAS.
- A. H. Slavney, L. Leppert, D. Bartesaghi, A. Gold-Parker, M. F. Toney, T. J. Savenije, J. B. Neaton and H. I. Karunadasa, J. Am. Chem. Soc., 2017, 139, 5015–5018 CrossRef CAS.
- J. Shang, W. Hao, X. Lv, T. Wang, X. Wang, Y. Du, S. Dou, T. Xie, D. Wang and J. Wang, ACS Catal., 2014, 4, 954–961 CrossRef CAS.
- Z. He, Q. Tang, X. Liu, X. Yan, K. Li and D. Yue, Energy Fuels, 2021, 35, 15005–15009 CrossRef CAS.
- T. Wang, D. Yue, X. Li and Y. Zhao, Appl. Catal., B, 2020, 268, 118399 CrossRef CAS.
- Y. Jiang, K. Li, X. Wu, M. Zhu, H. Zhang, K. Zhang, Y. Wang, K. P. Loh, Y. Shi and Q. H. Xu, ACS Appl. Mater. Interfaces, 2021, 13, 10037–10046 CrossRef CAS PubMed.
- X. Li, X. Lv, N. Li, J. Wu, Y. Z. Zheng and X. Tao, Appl. Catal., B, 2019, 243, 76–85 CrossRef CAS.
- Y. Zhang, Z. Sun, Z. Wang, Y. Zang and X. Tao, Int. J. Hydrogen Energy, 2022, 47, 8829–8840 CrossRef CAS.
- Q. Huang, Y. Guo, J. Chen, Y. Lou and Y. Zhao, New J. Chem., 2022, 46, 7395–7402 RSC.
- Q. Xu, L. Zhang, B. Cheng, J. Fan and J. Yu, Chem, 2020, 6 CAS.
- L. Zhou, Y. Li, S. Yang, M. Zhang, Z. Wu, R. Jin and Y. Xing, Chem. Eng. J., 2021, 420, 130361 CrossRef CAS.
- F. Xu, K. Meng, B. Cheng, S. Wang, J. Xu and J. Yu, Nat. Commun., 2020, 11, 4613 CrossRef CAS PubMed.
- H. Lv, H. Yin, N. Jiao, C. Yuan, S. Weng, K. Zhou, Y. Dang, X. Wang, Z. Lu and Y. Zhang, Small Methods, 2023, 7, 2201365 CrossRef CAS PubMed.
- L. Zhou, Y. F. Xu, B. X. Chen, D. Bin Kuang and C. Y. Su, Small, 2018, 14, 1703762 CrossRef PubMed.
- Z. Liu, H. Yang, J. Wang, Y. Yuan, K. Hills-Kimball, T. Cai, P. Wang, A. Tang and O. Chen, Nano Lett., 2021, 21, 1620–1627 CrossRef CAS PubMed.
- J. Ran, M. Jaroniec and S. Z. Qiao, Adv. Mater., 2018, 30 Search PubMed.
- J. Pi, X. Jia, Z. Long, S. Yang, H. Wu, D. Zhou, Q. Wang, H. Zheng, Y. Yang, J. Zhang and J. Qiu, Adv. Energy Mater., 2022, 12, 2202074 CrossRef CAS.
- T. Chen, M. Zhou, W. Chen, Y. Zhang, S. Ou and Y. Liu, Sustain Energy Fuels, 2021, 5, 3598–3605 RSC.
- S. Kumar, I. Hassan, M. Regue, S. Gonzalez-Carrero, E. Rattner, M. A. Isaacs and S. Eslava, J. Mater. Chem. A, 2021, 9, 12179–12187 RSC.
- F. Yang, A. O. Elnabawy, R. Schimmenti, P. Song, J. Wang, Z. Peng, S. Yao, R. Deng, S. Song, Y. Lin, M. Mavrikakis and W. Xu, Nat. Commun., 2020, 11, 1088 CrossRef CAS PubMed.
- J. Cui, M. Daboczi, M. Regue, Y. C. Chin, K. Pagano, J. Zhang, M. A. Isaacs, G. Kerherve, A. Mornto, J. West, S. Gimenez, J. S. Kim and S. Eslava, Adv. Funct. Mater., 2022, 32, 2207136 CrossRef CAS.
- M. S. Sena, J. Cui, Y. Baghdadi, E. Rattner, M. Daboczi, A. L. Lopes-Moriyama, A. G. dos Santos and S. Eslava, ACS Appl. Energy Mater., 2023, 6(20), 10193–10204 CrossRef CAS.
- B. Anasori, M. R. Lukatskaya and Y. Gogotsi, Nat. Rev. Mater., 2017, 2 Search PubMed.
- G. Eda and S. A. Maier, ACS Nano, 2013, 7 Search PubMed.
- Z. Zhang, B. Wang, H. B. Zhao, J. F. Liao, Z. C. Zhou, T. Liu, B. He, Q. Wei, S. Chen, H. Y. Chen, D. Bin Kuang, Y. Li and G. Xing, Appl. Catal., B, 2022, 312, 121358 CrossRef CAS.
- M. Ebihara, T. Ikeda, S. Okunaka, H. Tokudome, K. Domen and K. Katayama, Nat. Commun., 2021, 12, 3716 CrossRef CAS.
- P. Zhou, J. Yu and M. Jaroniec, Adv. Mater., 2014, 26 CAS.
- A. Mahmoud Idris, S. Zheng, L. Wu, S. Zhou, H. Lin, Z. Chen, L. Xu, J. Wang and Z. Li, Chem. Eng. J., 2022, 446, 137197 CrossRef CAS.
- L. Wu, S. Zheng, H. Lin, S. Zhou, A. Mahmoud Idris, J. Wang, S. Li and Z. Li, J. Colloid Interface Sci., 2023, 629, 233–242 CrossRef CAS PubMed.
- Y. Ren, D. Zeng and W. J. Ong, Cuihua Xuebao, 2019, 40, 289–319 CAS.
- Y. Wang, H. Huang, Z. Zhang, C. Wang, Y. Yang, Q. Li and D. Xu, Appl. Catal., B, 2021, 282, 119570 CrossRef CAS.
- P. Li, Y. Lin, M. Ma, M. Zhang, J. Li, Z. Wang, V. Maheskumar, Z. Y. Jiang and R. Zhang, ACS Appl. Energy Mater., 2022, 5, 1942–1952 CrossRef CAS.
- X. Wang, B. Hu, Y. Li and G. Zhang, Solar RRL, 2023, 7(18), 2300410 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |