
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrocatalytic formate and alcohol oxidation by hydride transfer at first-row transition metal complexes
Navar M.
White
 and
Kate M.
Waldie
*
and
Kate M.
Waldie
*
Department of Chemistry and Chemical Biology, Rutgers, The State University of New Jersey, 123 Bevier Road, Piscataway, New Jersey 08854, USA. E-mail: kate.waldie@rutgers.edu
First published on 13th June 2024
Abstract
The electrocatalytic oxidation of carbon-based liquid fuels, such as formic acid and alcohols, has important applications for our renewable energy transition. Molecular electrocatalysts based on transition metal complexes provide the opportunity to explore the interplay between precise catalyst design and electrocatalytic activity. Recent advances have seen the development of first-row transition metal electrocatalysts for these transformations that operate via hydride transfer between the substrate and catalyst. In this Frontier article, we present the key contributions to this field and discuss the proposed mechanisms for each case. These studies also reveal the remaining challenges for formate and alcohol oxidation with first-row transition metal systems, for which we provide perspectives on future directions for next-generation electrocatalyst design.
 Navar M. White | Navar Mercer White received his B.A. in Chemistry with a Minor in Geometrical Mathematics from Vassar College in 2015. Mercer is currently pursuing his Ph.D. in Chemistry at Rutgers, The State University of New Jersey (New Brunswick) under the guidance of Prof. Kate M. Waldie. His current research focuses on the development of organometallic catalysts for electrocatalytic transformations and their application for sustainable chemical synthesis and renewable energy conversion. |
 Kate M. Waldie | Kate M. Waldie received her Ph.D. in Chemistry from Stanford University in 2016 under the guidance of Prof. Robert M. Waymouth. She completed her postdoctoral research with Professor Clifford P. Kubiak at the University of California San Diego. She is currently Assistant Professor in the Department of Chemistry and Chemical Biology at Rutgers, The State University of New Jersey (New Brunswick), where her group focuses on the design and study of molecular transition metal complexes for renewable energy storage & conversion and organic synthesis. |
Introduction
To reduce our reliance on fossil fuels while meeting our ever-increasing global energy demand, the use of electrochemical technologies to facilitate the interconversion of renewably-derived electricity, chemicals, and usable energy is a central strategy.1 In this regard, electrochemical oxidation of formate and alcohols is of great interest: (1) Direct formic acid or alcohol fuel cells for electricity generation.2–6 While significant attention has been placed on H2 fuel, liquid carbon fuels derived from CO2 or biomass resources offer several advantages, including higher volumetric energy densities, easier storage and transportation, and compatibility with established infrastructure.7,8 The direct use of formic acid or alcohol fuels is complementary to their indirect use as H2 carriers for onboard H2 delivery,9,10 although fugitive H2 emissions are mitigated by the direct approach.11 (2) Alternative anode reactions for cathodic fuel production.12–15 The electrochemical production of chemical fuels is traditionally paired with the anodic oxygen evolution reaction (OER), which requires a large overpotential. Instead, OER may be replaced by formate or alcohol oxidation that can be accessed at less positive potentials, improving the efficacy of fuel production cells. This prospect is particularly attractive for the co-production of fuels with value-added alcohol oxidation products.16 (3) Alcohol oxidation for fine chemical synthesis, where traditional stoichiometric chemical oxidants or hydrogen acceptors are replaced by electrochemical oxidation at the anode.17–19The direct oxidation of formate or alcohols at the electrode is typically associated with high overpotentials to promote single-electron transfer (SET).20 Thus, electrocatalysis is key to enabling substrate oxidation at milder potentials. With homogeneous transition metal complexes, investigations into electrocatalytic alcohol oxidation initially focused on metal–oxo complexes, which are strong oxidants and promote C–H oxidation via proton coupled electron transfer (PCET)21–31 but require very positive potentials for regeneration of the metal–oxo. In the past two decades, transition metal electrocatalysts for alcohol and formate oxidation have been developed that operate via a metal-hydride intermediate, offering non-oxo-based strategies for electro-oxidation and opening the door to a wider possibility of electrocatalyst structures.7,32–34 The metal-hydride intermediate may be formed via hydride transfer from the substrate, after which the starting complex must be regenerated via oxidation at the electrode to establish an electrocatalytic cycle (Scheme 1). Herein, we highlight the recent developments in formate and alcohol electro-oxidation via hydride transfer using homogeneous first-row transition metal complexes. Through these reports, we discuss the key challenges that remain in this emerging field and provide our perspective on possible strategies for future electrocatalyst development.
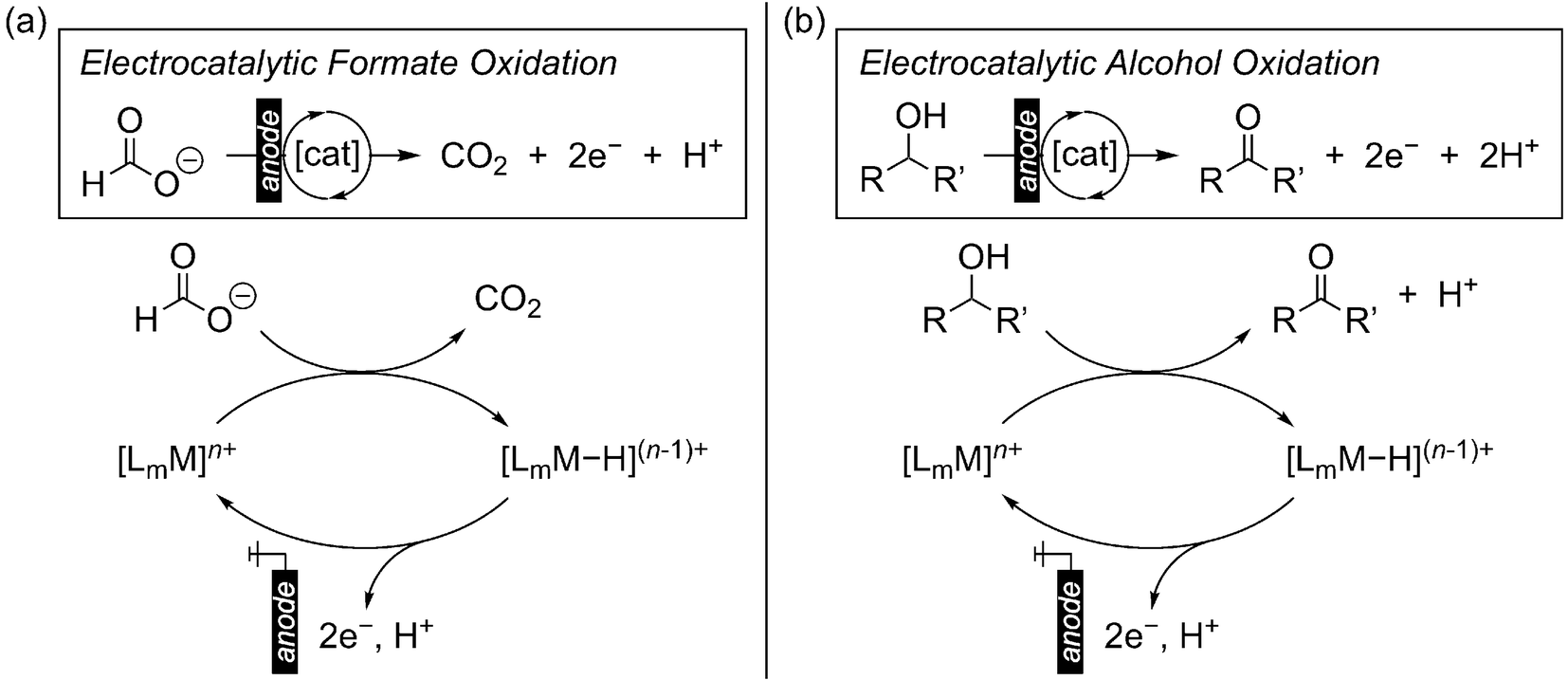 | ||
| Scheme 1 General scheme for electro-oxidation of (a) formate, or (b) alcohols via a metal-hydride intermediate. | ||
Thermodynamic hydricity
For catalysts that operate via hydride transfer (or net hydride transfer), the thermodynamic hydricity ΔGH− of the substrate and metal-hydride is a valuable consideration (Scheme 2). The hydricity of formate has been reported in water and acetonitrile: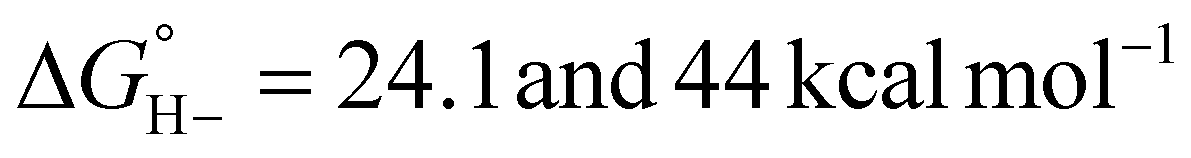 , respectively.35 While some aqueous hydricity measurements for metal-hydride complexes have been disclosed, a larger dataset is available in acetonitrile,35,36 which has the additional advantage of being a common electrochemical solvent.37 Thus, to favor formate oxidation via hydride transfer to a metal center,
, respectively.35 While some aqueous hydricity measurements for metal-hydride complexes have been disclosed, a larger dataset is available in acetonitrile,35,36 which has the additional advantage of being a common electrochemical solvent.37 Thus, to favor formate oxidation via hydride transfer to a metal center, 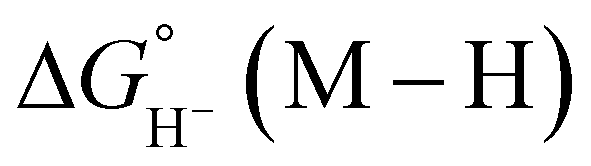 of the resulting metal-hydride must be greater than 44 kcal mol−1. We note that the favorability of metal–formate adduct formation and hydride transfer from this species may be relevant in certain cases.
of the resulting metal-hydride must be greater than 44 kcal mol−1. We note that the favorability of metal–formate adduct formation and hydride transfer from this species may be relevant in certain cases.
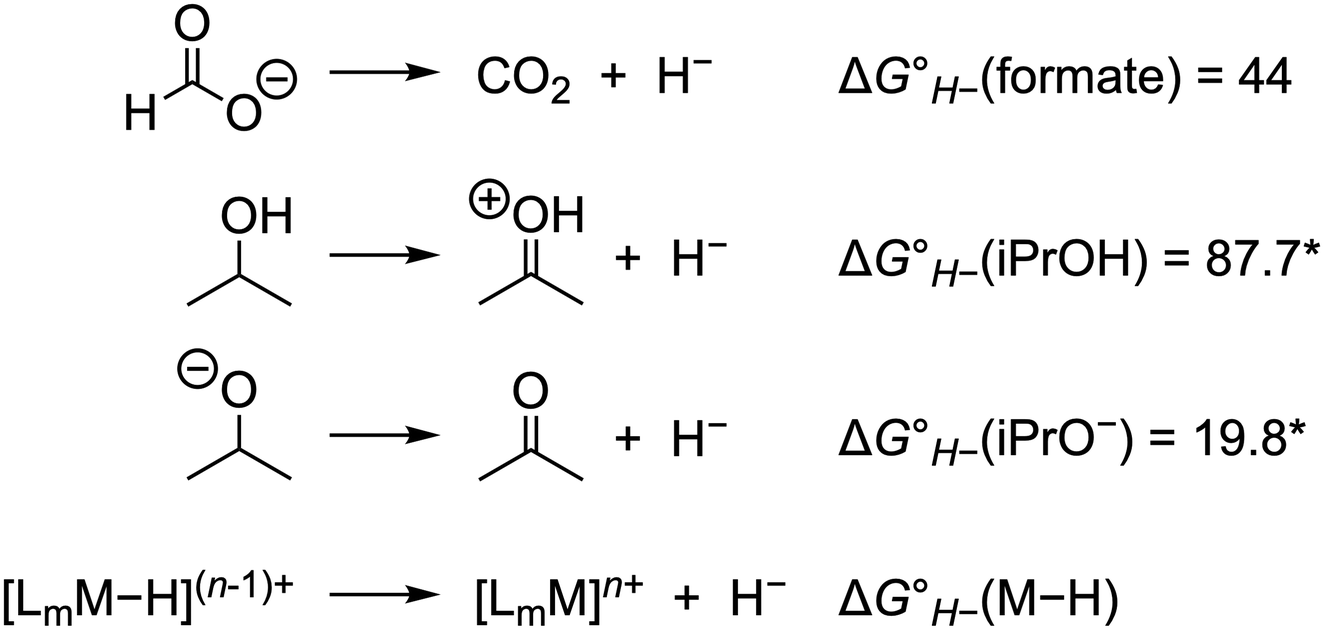 | ||
| Scheme 2 Thermodynamic hydricity values (kcal mol−1) in acetonitrile.35,38 *Estimated using computational methods.38 | ||
The thermodynamic hydricity of alcohols in acetonitrile is not well established. Estimated values for 2-propanol and 2-propoxide have been obtained from computational methods (Scheme 2): protonated acetone is the product of hydride loss from 2-propanol, and acetone is analogously obtained from 2-propoxide.38 In combination with the estimated pKa of protonated acetone (ca. 0.6),39 the former can be used to obtain an estimate for the thermodynamic potential  in acetonitrile (0.19 V vs. Fc+/0, the ferrocene/ferrocenium couple), which is in reasonable agreement with the value obtained using the gas-phase free energy for acetone hydrogenation (0.08 V vs. Fc+/0).40,41 As noted above, substrate coordination (alcohol or alkoxide) may be considered as well, or alcohol oxidation may proceed by a bifunctional outer-sphere mechanism involving concerted hydride and proton transfers.42–44
in acetonitrile (0.19 V vs. Fc+/0, the ferrocene/ferrocenium couple), which is in reasonable agreement with the value obtained using the gas-phase free energy for acetone hydrogenation (0.08 V vs. Fc+/0).40,41 As noted above, substrate coordination (alcohol or alkoxide) may be considered as well, or alcohol oxidation may proceed by a bifunctional outer-sphere mechanism involving concerted hydride and proton transfers.42–44
Electrocatalytic formate oxidation
Despite its relevance to renewable energy conversion, exploration of molecular transition metal complexes for electrocatalytic formate oxidation has been limited.45–49 The first report came in 2011 based on the first-row transition metal nickel, as opposed to a noble metal center.45 Given the hydricity of formate (vide supra), the authors reasoned that Ni(II) complexes with two P2N2 ligands were likely to serve as hydride acceptors from formate based on the bidirectional reactivity of these systems with H2.50,51 Indeed, the hydricity of the Ni(II)-hydride analogues for 1a–1h (Fig. 1) was measured to be greater than that of formate, ranging from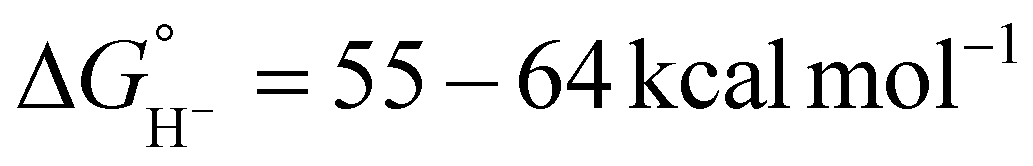 .35,45,52
.35,45,52
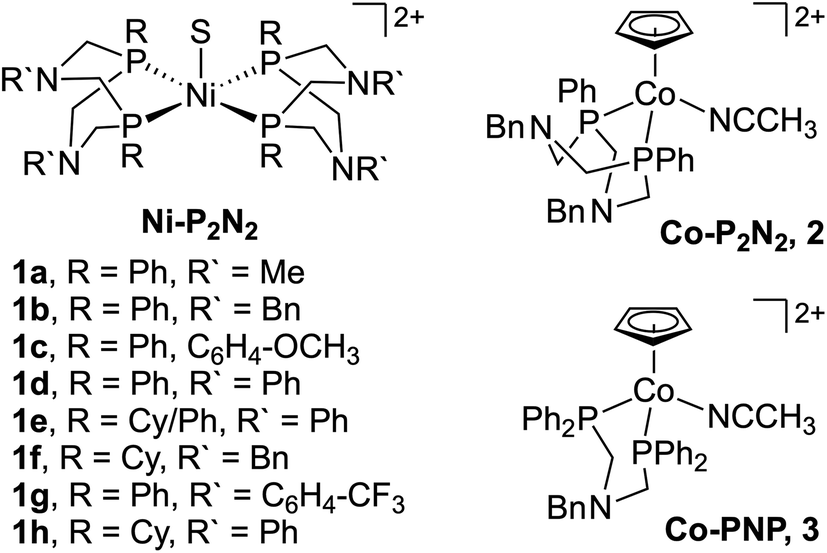 | ||
| Fig. 1 Ni and Co complexes investigated for electrocatalytic formate oxidation. S = acetonitrile. | ||
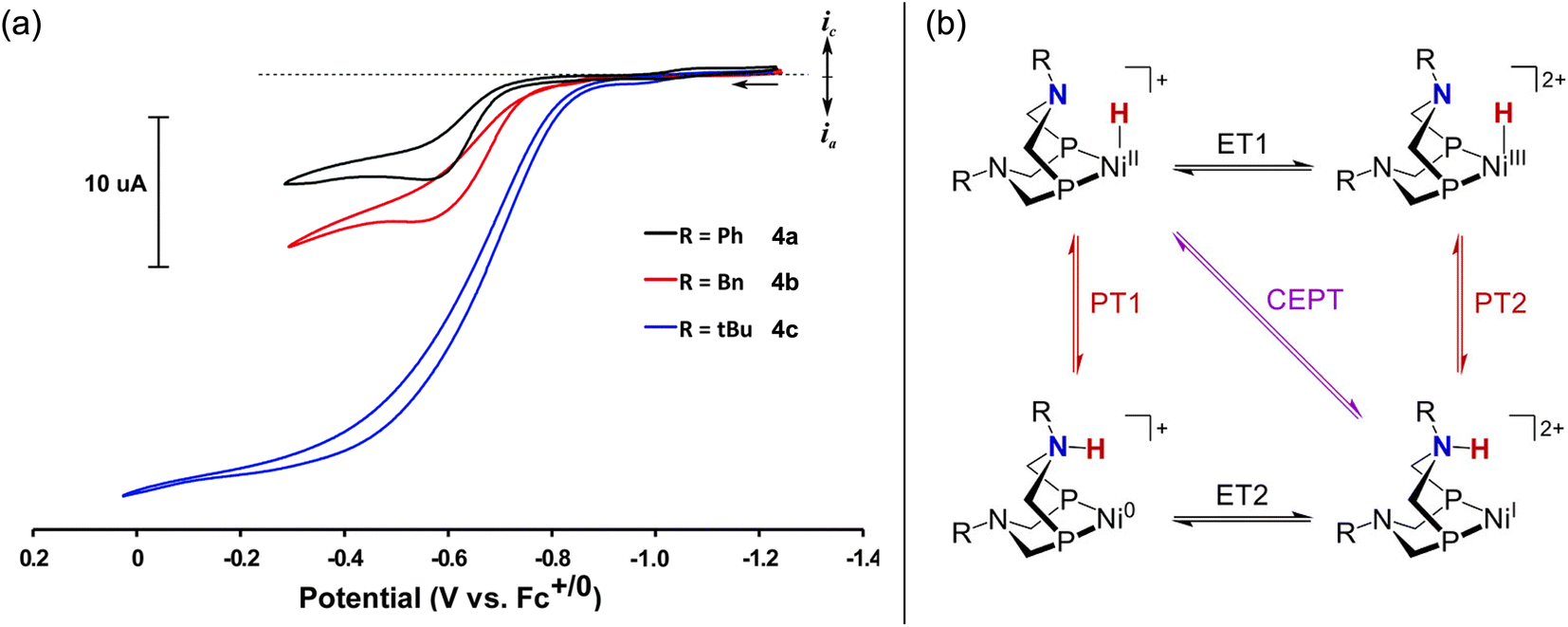
Treatment of the Ni–P2N2 complexes with 1 eq. formate led to Ni(II)-hydride formation by NMR, implying that hydride transfer is accessible. Cyclic voltammetry (CV) studies revealed electrocatalytic current near the Ni(I/II) couple (Fig. 2a), and CO2 production was confirmed in near-quantitative faradaic efficiency.45,46 The electrocatalytic TOF was found to depend on the P2N2 substituents, with the fastest electrocatalyst 1a having the least electron-rich phosphine (R = Ph) and the most basic amine (R′ = Me). Notably, the TOF was greater at smaller overpotentials η (Fig. 2b), counter to typical scaling relationships.53 In addition, because the potential of the Ni(I/II) couple and the hydricity of the corresponding Ni(II)-hydrides are correlated, faster catalysis was achieved using the most hydridic complex.45,52 However, other trends can similarly be constructed correlating TOF to the pendent amine basicity, making it difficult to draw conclusions about the mechanism from this data alone.
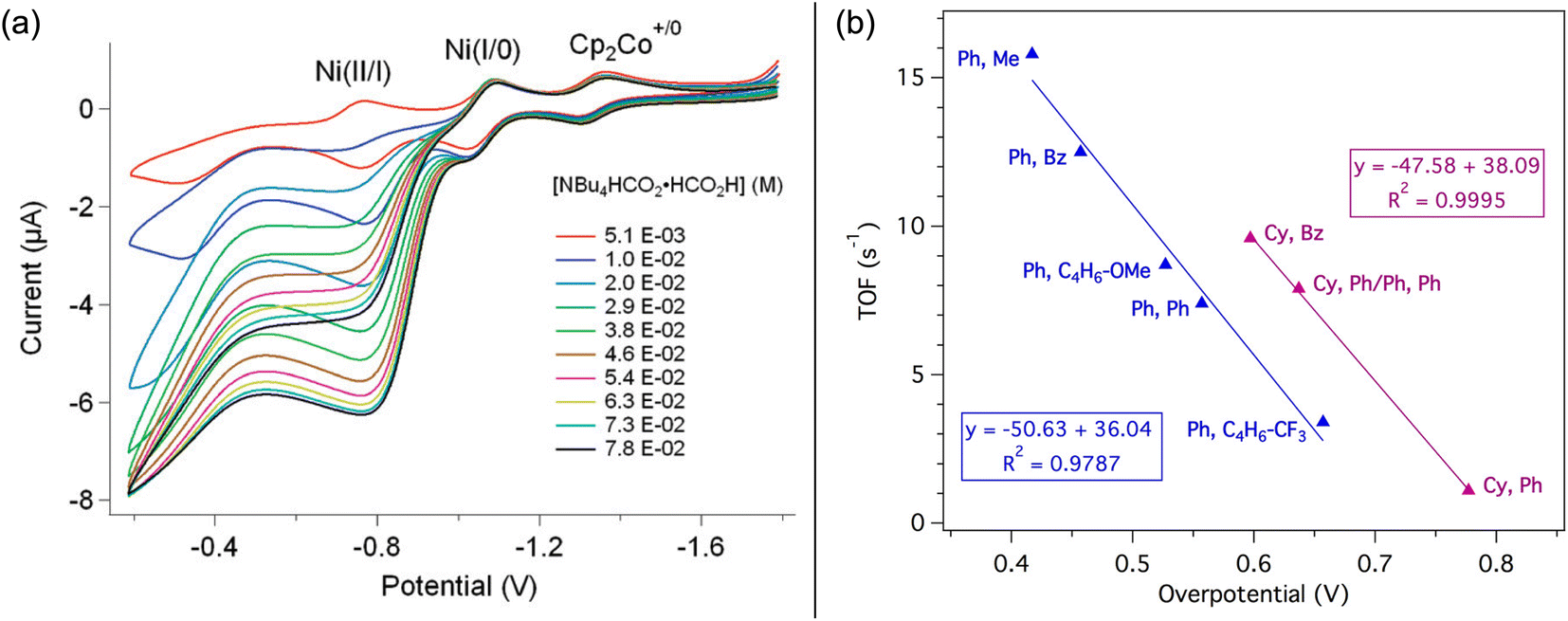 | ||
| Fig. 2 (a) CVs of 1e in benzonitrile showing electrocatalytic formate oxidation. Adapted with permission from ref. 45. Copyright 2011 American Chemical Society. (b) Correlation between TOF and overpotential for electrocatalytic formate oxidation with 1a–1h. Data labels indicate the PR2NR′2 R and R′ substituents, respectively. Adapted with permission from ref. 52. Copyright 2018 American Chemical Society. | ||
The correlation between TOF and amine basicity led the authors to propose formate oxidation by rate-limiting β-deprotonation of a Ni-formate by the pendent amine (Scheme 3, red).45,46 A subsequent study by Xue and Ahlquist used DFT-computational methods to probe accessible pathways for this transformation.54 Their results found that the barrier to β-deprotonation is significant and would lead to CO production, not CO2. Instead, the calculated lowest-energy pathway is direct hydride transfer from formate and intramolecular deprotonation of the resulting Ni(II)-hydride (Scheme 3, blue). The computed barriers for these steps are similar and depend on the ligand structure and are also likely influenced by the reaction conditions. While further work is needed to better elucidate the mechanism, the pendent amines are clearly crucial. Addition of formate to [Ni(depe)2] (depe = 1,2-bis(diethylphosphino)ethane) yielded the Ni(II)-hydride by CV,45 demonstrating stoichiometric formate oxidation 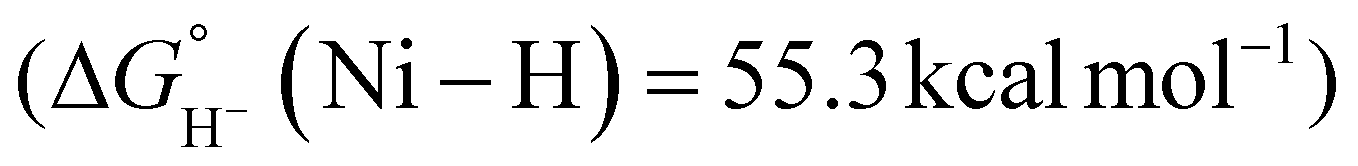 35,55,56 but electrocatalytic turnover was not observed. Despite the remaining questions, this story is an excellent demonstration of using thermodynamic analysis to guide electrocatalyst design, while also underscoring the importance of ligand functionality for achieving rapid reactivity.
35,55,56 but electrocatalytic turnover was not observed. Despite the remaining questions, this story is an excellent demonstration of using thermodynamic analysis to guide electrocatalyst design, while also underscoring the importance of ligand functionality for achieving rapid reactivity.
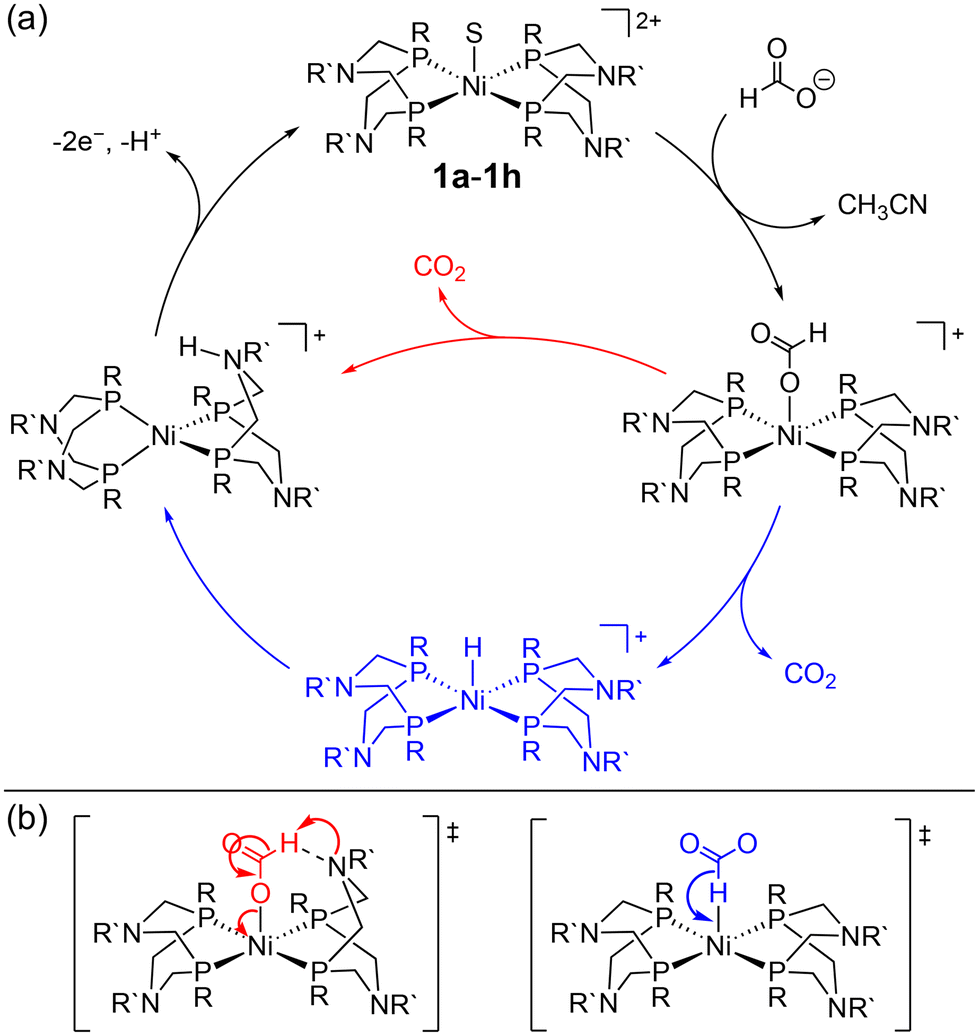 | ||
| Scheme 3 (a) Proposed mechanisms for formate oxidation by 1a–1h. (b) Proposed transition states via β-deprotonation (red, left) or direct hydride transfer (blue, right).45,46,54 | ||
Beyond the Ni–P2N2 systems, two noble metal catalysts (Ir and Pt) have been reported for electrocatalytic formate oxidation.47,48 In addition, we recently disclosed the electrocatalytic activity of Co–P2N22 and Co–PNP 3 for formate oxidation (Fig. 1).57 The hydricity of the corresponding Co(III)-hydrides was determined to be 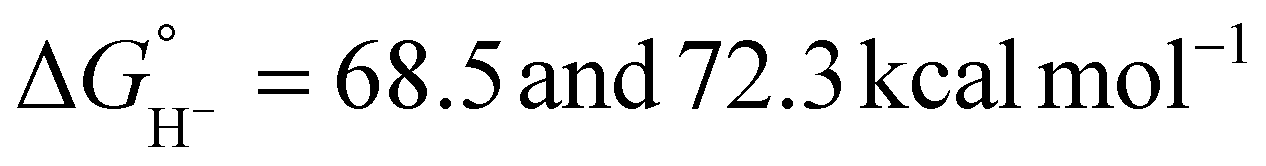 , respectively, indicating that 2 and 3 are thermodynamically primed for hydride transfer from formate. Indeed, by chronoamperometry, electrocatalytic current was observed for 3 near the Co(II/III) potential in the presence of formate. Controlled-potential electrolysis (CPE) confirmed quantitative faradaic efficiency for CO2 production.
, respectively, indicating that 2 and 3 are thermodynamically primed for hydride transfer from formate. Indeed, by chronoamperometry, electrocatalytic current was observed for 3 near the Co(II/III) potential in the presence of formate. Controlled-potential electrolysis (CPE) confirmed quantitative faradaic efficiency for CO2 production.
Formate oxidation is initiated by formation of a Co(III)-formate intermediate, observable by 1H NMR. Rate-limiting hydride transfer to cobalt then occurs, followed by deprotonation of the Co(III)-hydride and re-oxidation at the electrode. Interestingly, 3 exhibited a faster TOF than 2, which may be related to different hydrogen bonding interactions between the pendent amines and biformate buffer. Further studies to probe these effects are ongoing in our group. While 2 and 3 are less active than the Ni–P2N2 systems, the overpotentials are comparable (ca. 0.5 V) and all complexes operate using formate as the base. Complexes 2 and 3 represent the second class of first-row transition metal-hydride systems for formate oxidation, encouraging more studies to explore this reactivity at first-row metals including cobalt.
Electrocatalytic alcohol oxidation
The first example of electrocatalytic alcohol oxidation by a first-row transition metal complex was reported by Appel and co-workers.58,59 Inspired by the ability of pendent amine ligands to facilitate intramolecular proton shuttling,60 the authors selected the Ni–P2N2 framework (Fig. 3) – very similar to the above structures for formate oxidation.45,46 In their initial study, a series of Ni–P2N2 complexes were evaluated for chemical alcohol oxidation using triethylamine and decamethylferrocenium.58 Complex 4c showed the highest initial TOF for diphenylmethanol oxidation (TOF = 114 ± 5.2 h−1), while the absence of pendent amine in 5a or the presence of two P2N2 ligands in 1b and 1f led to poor activity. The latter may be due to greater steric hindrance at the metal, disfavoring alcohol binding.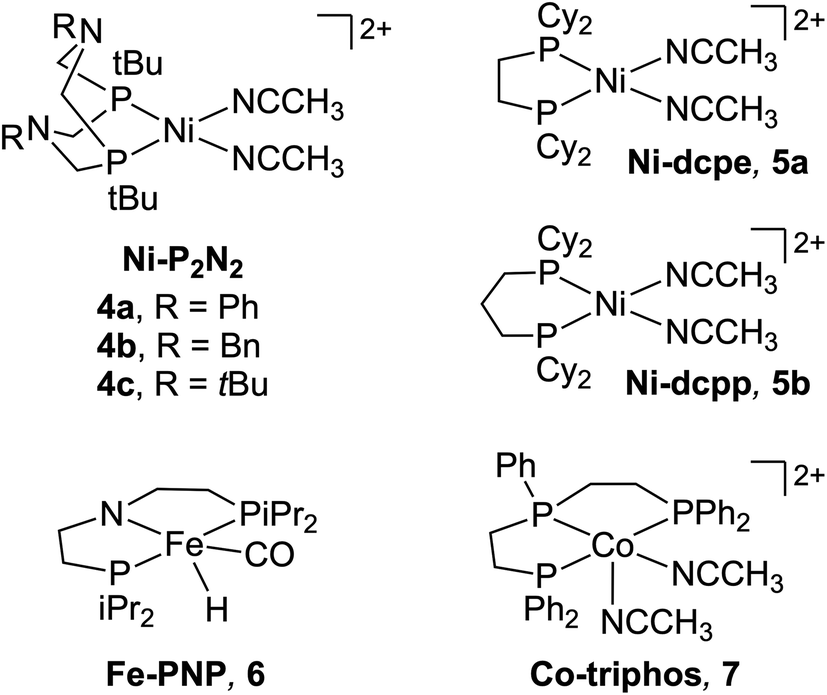 | ||
| Fig. 3 Ni, Fe, and Co complexes investigated for electrocatalytic alcohol oxidation. | ||
Appel and co-workers later demonstrated that 4c is a competent electrocatalyst for alcohol oxidation.59 As shown in Fig. 4a, electrocatalytic current was observed at Ecat/2 = −0.75 V vs. Fc+/0 in the presence of benzyl alcohol and triethylamine (30 eq.; TOF = 2.5 s−1). Selective formation of benzaldehyde was observed (≥90% faradaic efficiency). This landmark study was a significant step for the field of homogeneous electrocatalysis and showed the viability of first-row transition metal-hydrides for electrocatalytic alcohol oxidation. In addition, this study further expanded the electrocatalytic reactivity of Ni–P2N2 complexes, which had already been shown for H2 evolution, H2 oxidation, O2 reduction, and of course formate oxidation.60
 | ||
| Fig. 4 (a) CVs of 4a–4c in acetonitrile showing electrocatalytic oxidation of benzyl alcohol in the presence of triethylamine. Adapted from ref. 59 with permission from the Royal Society of Chemistry. (b) Possible PCET pathways for Ni(P2N2)-hydride oxidation. Adapted with permission from ref. 61. Copyright 2022 American Chemical Society. | ||
The pendent amines in the P2N2 ligand are crucial for electrocatalytic turnover with 4c – this was probed in greater detail by Hall, Wiedner, and co-workers, focusing on benzyl alcohol.61 Here, a new control complex 5b was evaluated for which the Ni(I/II) redox potential was better matched with 4c (ΔE1/2 = 30 mV) compared to 5a (ΔE1/2 = 200 mV). CV studies of 5b with benzyl alcohol and base did not show catalytic current; however, by NMR, 4c and 5b showed comparable rates of stoichiometric alcohol oxidation – the pendent amine is not needed for this process! Instead, the P2N2 ligand is thought to lower the potential for Ni-hydride oxidation via PCET (Fig. 4b), enabling electrocatalytic turnover at mild potentials with 4c (η = 0.39 V).40 This conclusion illuminates how ligand-promoted PCET may serve as a general strategy for improving the efficiency of metal-hydride catalysis.62–64 Similar effects are likely operative for formate oxidation with the Ni-bis(phosphine) analogues 1a–1h (vide supra).
Computational studies led to a more detailed picture of alcohol oxidation at 4c.61 Formation of the Ni-alkoxide is rate-limiting, which undergoes β-hydride elimination (Scheme 4a). The authors suggested that the P2N2 ligand may direct alcohol coordination via hydrogen bonding, but this interaction does not lower the overall barrier to Ni-alkoxide formation and deprotonation of the Ni–alcohol by the pendent amine is not accessible. These conclusions further corroborated their experimental results showing that the P2N2 is not necessary for rapid, stoichiometric alcohol oxidation (vide supra). The authors also identified an off-cycle Ni(II)–alcohol species 8, from which deprotonation is uphill and inhibits catalytic turnover (Scheme 4b). Ligand dissociation from 8 is required to access the Ni(II)-alkoxide. Clearly, speciation of this catalyst is complicated by the accessibility of multiple coordination sites and geometries. This poses a challenge for alcohol oxidation in the presence of other coordinating species, but, as the authors point out, knowledge of this off-cycle pathway lays the groundwork for future design strategies.
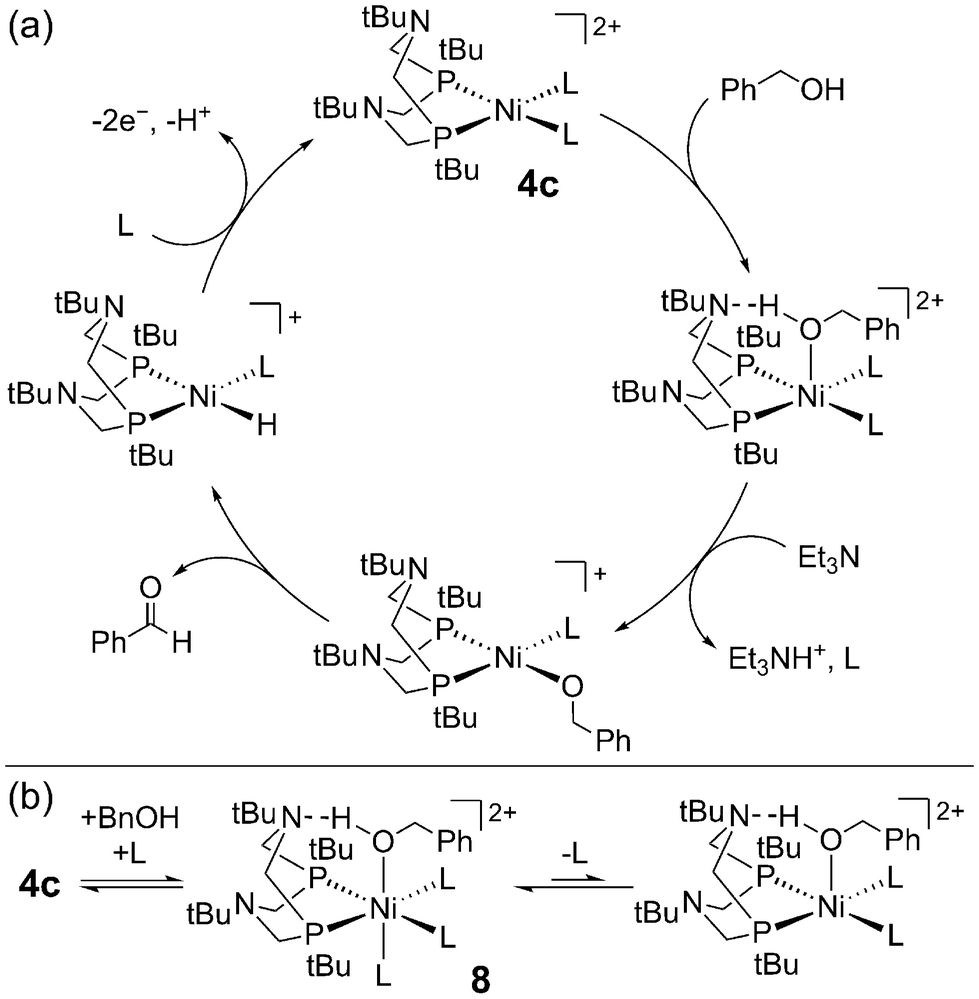 | ||
| Scheme 4 (a) Proposed mechanism for benzyl alcohol oxidation by 4c. (b) Formation of an off-cycle high-spin intermediate 8. L = acetonitrile, triethylamine.61 | ||
In 2020, Waymouth and co-workers reported the first Fe-hydride system for alcohol oxidation.65 Complex 6 and related compounds were previously shown to catalyze various hydrogen transfer reactions, including alcohol acceptorless dehydrogenation with impressive activity (TON ≥ 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) at elevated temperatures.66–72 The ligand amide group was proposed to be directly involved in alcohol oxidation via a bifunctional mechanism, where hydride and proton transfer from the substrate yields 9 (Fig. 5a).73,74 The authors65 underscored the anticipated similarities between acceptorless dehydrogenation and alcohol electro-dehydrogenation where the same bifunctional process could be followed by either H2 release or oxidation of the metal-hydride, respectively, reminiscent of their earlier work on the electrocatalytic activity of transfer hydrogenation catalysts.75,76
000) at elevated temperatures.66–72 The ligand amide group was proposed to be directly involved in alcohol oxidation via a bifunctional mechanism, where hydride and proton transfer from the substrate yields 9 (Fig. 5a).73,74 The authors65 underscored the anticipated similarities between acceptorless dehydrogenation and alcohol electro-dehydrogenation where the same bifunctional process could be followed by either H2 release or oxidation of the metal-hydride, respectively, reminiscent of their earlier work on the electrocatalytic activity of transfer hydrogenation catalysts.75,76
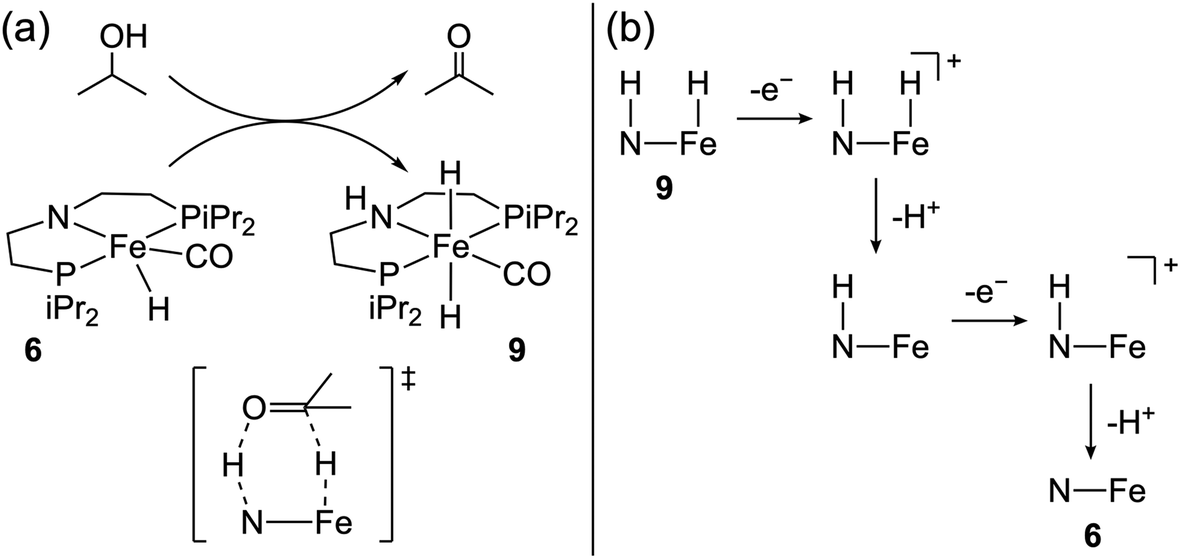 | ||
| Fig. 5 (a) Proposed bifunctional pathway for 2-propanol oxidation with 6. (b) ECEC oxidative deprotonation of 9.65 | ||
Complex 6 exhibited an electrocatalytic current by CV at Ecat/2 = −0.8 V vs. Fc+/0 in tetrahydrofuran with 2-propanol and phosphazene base (η = 1.1 V).40 The electrocatalytic TOF was estimated to be 1.7 s−1, and CPE studies showed quantitative faradaic efficiency for acetone. The authors showed that treatment of 9 with potassium t-butoxide and decamethylferrocenium led to the formation of 6, implying an overall 2e−/2H+ process for the oxidation of 9 under basic conditions. DFT calculations suggested an ECEC pathway for this oxidation, as shown in Fig. 5b. Experimentally, very strong bases are required for electrocatalytic turnover, although the relevant computed pKa values suggest weaker bases should promote the oxidative deprotonation of 9. Under electrocatalytic conditions, the lifetime of this catalyst is poor (TON = 1.9), perhaps due to formation of inert Fe(0) species.69 Nonetheless, this work established that acceptorless dehydrogenation catalysts can operate in an electrocatalytic regime, opening the door to the electrochemical evaluation of many other chemical catalysts.77–79
More recently, Hammes-Schiffer, Appel, and co-workers reported the electro-oxidation of benzyl alcohol using cobalt complex 7, which features a linear triphos ligand that lacks any proton-active sites.80 The authors posited that Co(II)-hydrides with phosphine ligands may be compatible with an alcohol oxidation scheme due to their mild potentials for oxidation and facile deprotonation of the resulting Co(III)-hydride.81,82 Electrocatalytic current with 7 in the presence of N,N-diisopropylethylamine was not observed on the CV timescale (TOF < 0.1 s−1); however, CPE conducted near the Co(I/II) couple revealed electrocatalytic production of benzaldehyde (97% faradaic efficiency, η = ca. 0.2 V).40 While the TON = 19.9 for 7 was greater than for 4c or 6, catalyst decomposition was still significant, with 90% decomposition during electrolysis.
The proposed mechanism is initiated by substrate coordination to 7 followed by intermolecular alcohol deprotonation, β-hydride elimination to the Co(II)-hydride, and oxidative deprotonation by an ECE pathway to regenerate 7 (Scheme 5a). A notable aspect of this system is the absence of pendent acid/base sites on the triphos ligand. Recalling the proposed mechanism for 4c, the pendent amines were not essential for stoichiometric alcohol oxidation and in fact did not increase the rate of this process.61 However, the electrocatalytic activity of 4c was dependent on oxidation of the Ni(II)-hydride via PCET enabled by the P2N2 ligand. For complex 7, 1e− oxidation of the Co(II)-hydride is facile and occurs at a more negative potential than Co(I/II) oxidation, indicating that pendent basic sites are not required for electrocatalysis. Nonetheless, it would be interesting to examine whether introduction of pendent bases into this architecture could enable Co(III)-hydride oxidation by a PCET pathway (Scheme 5b), analogous to the Ni–P2N2 systems. Such PCET reactivity would circumvent the original Co(I) intermediate and may shift the potential for oxidation of the Co(III)-hydride to a more negative value.
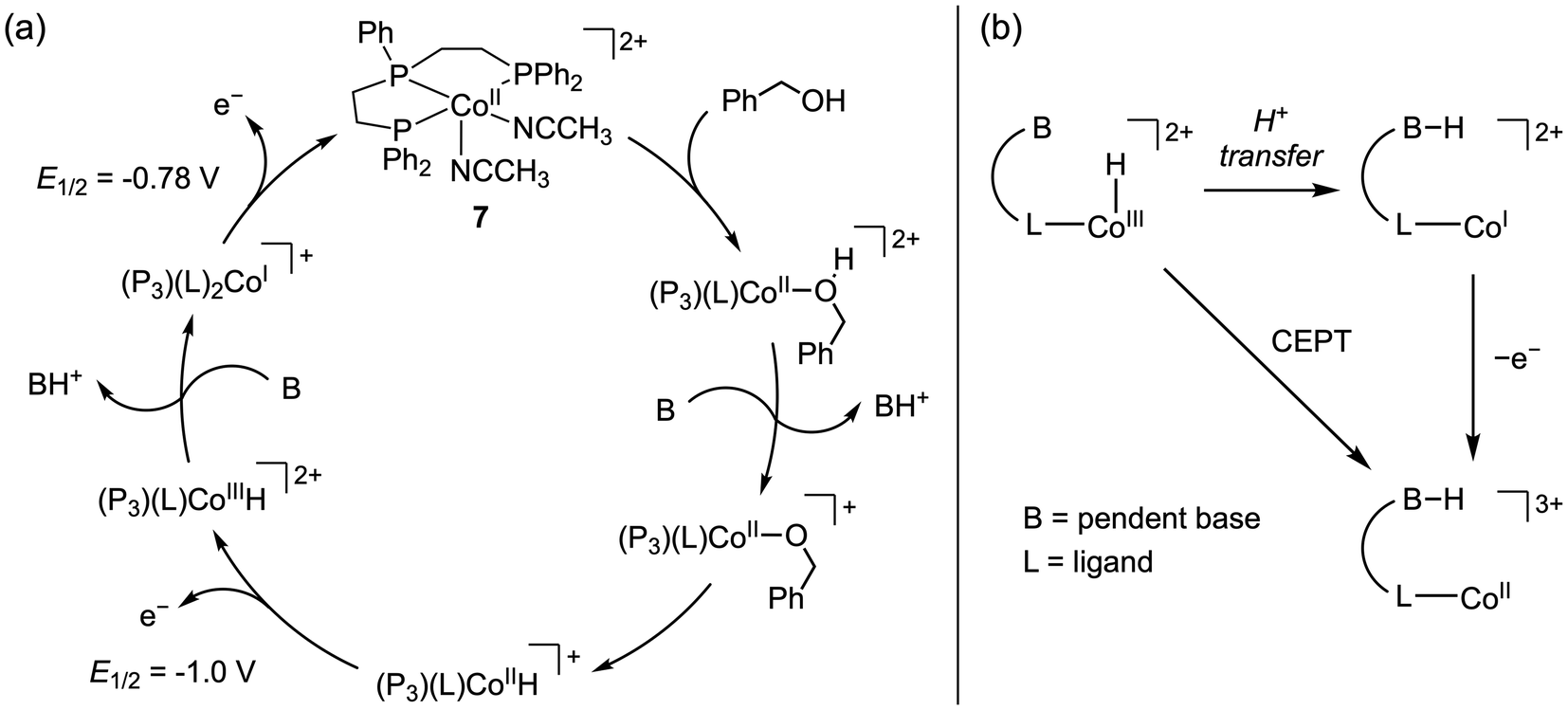 | ||
| Scheme 5 (a) Proposed mechanism for benzyl alcohol oxidation by 7 with iPr2EtN.80 (b) Possible ligand-promoted concerted electron–proton transfer (CEPT) pathway for Co(III)-hydride oxidation. | ||
Key challenges and future outlook
Reports of first-row transition metal-hydride complexes for formate and alcohol oxidation of are limited; nonetheless, these examples offer valuable insights into some of the remaining challenges to guide future research. Key performance metrics for these systems are summarized in Table 1, with comparison to select noble metal-hydride systems for both reactions.| Catalyst | Substrate | TOF (s−1) | TON | η (V) | FE (%) |
|---|---|---|---|---|---|
a For complex 1c.
b Estimated using 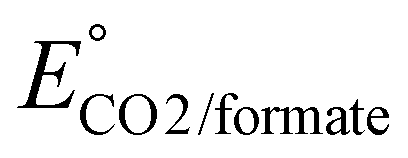 from ref. 36.
c For the 4e− oxidation to acetate and ethyl acetate. from ref. 36.
c For the 4e− oxidation to acetate and ethyl acetate.
|
|||||
| 1a 45 | Formate | 15.8 | >8 | 0.4252 | 93 ± 5a |
| 2 57 | Formate | <0.001 | >3.8 | 0.45 | 99 ± 3 |
| 3 57 | Formate | 0.04 | >10.7 | 0.57 | 96 ± 3 |
| [Ir(PONOP)(H)3]47 | Formate | 4.8 | n.d. | 1.25b | 87 ± 5 |
| [Pt(depe)]2+ (ref. 48) | Formate | <0.5 | >2.4 | 0.010 | 90 |
| 4c 59 | Benzyl alcohol | 2.5 | 3.1 | 0.3940 | ≥90 |
| 6 65 | 2-Propanol | 1.7 | 1.9 | 1.140 | 100 ± 15 |
| 7 80 | Benzyl alcohol | <0.1 | 19.9 | ∼0.240 | 97 |
| [Ru(CNN)(dppb)H]76 | 2-Propanol | 4.8 | >4.6 | 1.640 | 94 ± 5 |
| [Ru(NNP)(CO)ClH]33 | Ethanol | n.d. | 21c | n.d. | 62c |
| [Ir(PNP)(H)2]83 | 2-Propanol | n.d. | >4.0 | 1.540 | 78 |
A general mechanism for formate and alcohol oxidation involves metal-hydride formation via hydride transfer and electrochemical catalyst regeneration (Scheme 1). The overall substrate oxidation process is rate-limiting for the first-row systems in Table 1. Here, the hydride accepting ability of the metal is a valuable metric. The thermodynamic hydricity of the formate oxidation catalysts has been reported;45,52,57 however, values for the alcohol oxidation catalysts are lacking. Moving forward, as more hydricity measurements are reported, structure–activity relationships can be constructed to correlate hydride transfer energetics with electrocatalyst activity. The rate of hydride transfer, quantified as kinetic hydricity, is also an important yet understudied metric. Evaluating kinetic hydricity for the stoichiometric reaction of formate and alcohols with a series of metal complexes should reveal new design principles for promoting rapid hydride transfer, including the exploration of solvent effects that can significantly impact hydride transfer rates.84–87 In addition, the initial interaction of the substrate (hydride donor) with the metal (hydride acceptor) is proposed for these systems. Thus, increasing the metal Lewis acidity while maintaining an open coordination site is likely crucial, especially for alcohol oxidation given that substrate coordination to 4c and 7 is unfavorable.59,61,80 Greater Lewis acidity at the metal will also increase the acidity of the metal–alcohol intermediate. A simplistic approach may introduce more electron-withdrawing ligands, but such electronic changes will also affect other performance metrics (vide infra). An interesting prospect would be to explore redox-active ligands, which have been shown to enhance Lewis acidity at metal centers by serving as reversible electron reservoirs.79,88
The first-row examples in Table 1 largely feature pendent base ligands. Hydrogen-bonding interactions between the pendent amines and biformate may be relevant to the activity of 1–3, but the P2N2 ligand in 4c does not assist the alcohol oxidation steps. Looking to chemical dehydrogenation catalysts, many first-row systems utilize metal–ligand cooperativity (MLC) to enable rapid substrate oxidation,77,79 as with complex 6 where proton transfer to the ligand occurs with hydride addition at the metal. Future efforts should delve deeper into MLC pathways for first-row electrocatalysis, leveraging established ligand designs. Many of these chemical catalysts not only exhibit impressive activity but also catalyst lifetimes that far exceed the electrocatalytic examples. Rigid MLC pincer ligands may be beneficial to this end, though it will be important to evaluate stability under electrochemical conditions. A key challenge will be balancing productive substrate oxidation with the pKa requirements for electrocatalysis. As observed with 6, ligand deprotonation requires strong bases, translating to large overpotentials. Limited pKa measurements of such ligands have been reported,89–91 underscoring the need for further studies to elucidate the interplay between metal and ligand thermodynamic and kinetic properties for electrocatalysis.
The introduction of pendent hydrogen-bond donors, instead of pendent bases, offers another approach to improving the kinetics of substrate oxidation by stabilizing the hydride transfer transition state.77,79 Furthermore, pendent charged groups may deliver transition state stabilization through electrostatic interactions.92,93 The addition of exogenous Lewis acidic cations has been shown to increase the rate of formic acid dehydrogenation with iron catalysts.94,95 Intramolecular electrostatic effects via catalyst modification may provide even greater benefit.93,96–99
To establish an electrocatalytic cycle following substrate oxidation, 2e−/1H+ oxidative deprotonation of the metal-hydride is needed (Scheme 1). Shifting the requisite potential for catalyst regeneration to more negative values is desirable, but inductive ligand modifications will decrease the Lewis acidity of the metal center, resulting in weaker substrate interactions and lower catalytic rates. Therein lies a key challenge in molecular electrocatalysis: overcoming molecular scaling relationships to achieve high kinetic activity at small overpotentials. For formate and alcohol oxidation, notable advances have been made with the Ni–P2N2 systems. With 1a–1h and 4c, the pendent amines significantly lower the potential for metal-hydride oxidation by accessing PCET pathways. The pendent amine properties can be tuned largely independent of the metal center,60 allowing the amine basicity to be adjusted without compromising substrate activation via hydride transfer at the metal. This contributes to the impressive TOFs for these systems at modest overpotentials, even in comparison to noble-metal examples. However, extending this strategy to more robust complexes is still needed. Redox-active ligands and pendent charged groups, noted above as possible strategies for improving rates of substrate reactivity, may also engender different TOF-η trends by altering the correlation between the metal Lewis acidity to promote metal-hydride formation and the redox properties of the catalyst. Similar strategies have been applied for overcoming normal scaling relationships with other electrocatalytic reactions,100 and warrant exploration in the context of formate and alcohol electro-oxidation.
The Ni–P2N2 systems access fast rates at modest overpotentials thanks to intramolecular PCET pathways that enable oxidation of the metal-hydride at mild potentials. Future studies may explore related intermolecular approaches using hydrogen atom transfer (HAT) mediators to promote homolytic cleavage of the metal-hydride, bypassing higher-energy intermediates from stepwise electron and proton transfer (Scheme 6).101,102 Key to this approach is selecting HAT mediators for which hydrogen atom abstraction from the metal-hydride is favorable, and that can be electrochemically regenerated at a more negative potential than oxidation of the metal-hydride. This approach was recently demonstrated with noble-metal catalysts for alcohol oxidation.83,103 Through careful selection of HAT mediators, the electrocatalytic overpotentials were lowered by ca. 0.5 V while maintaining rapid TOFs, counter to normal scaling relationships.40 As additional thermodynamic metrics for first-row metal-hydride complexes become available, the rational selection of viable HAT mediators will become feasible – an exciting prospect for future investigations into oxidative electrocatalysis.
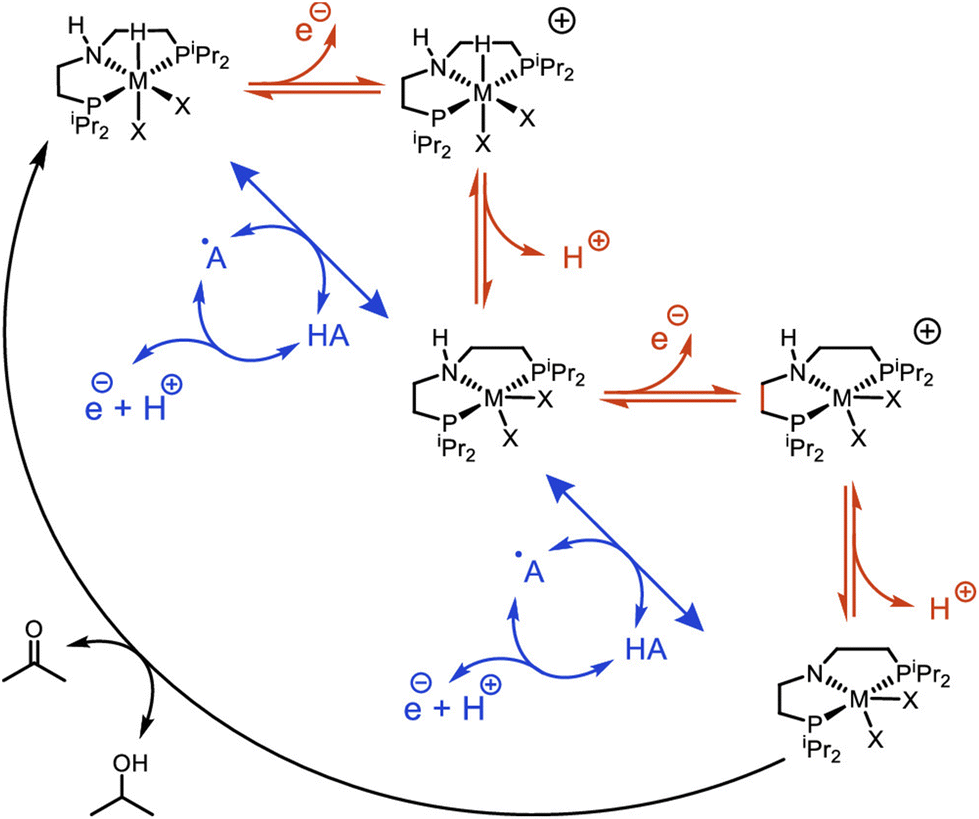 | ||
| Scheme 6 Metal-hydride oxidative deprotonation by stepwise (red) or mediated (blue) pathways. Reprinted with permission from ref. 83. Copyright 2020 American Chemical Society. | ||
While the studies highlighted here focused on catalysts involving discrete metal-hydride intermediates, alternative pathways that operate via net hydride transfer offer new opportunities for electrocatalyst design. Yang and co-workers posited that orthogonal hydride transfer, where the electron and proton transfer sites are not co-located as a metal-hydride, may have distinct advantages for the electrocatalytic interconversion of CO2 and formate, inspired by formate dehydrogenase.104 Very recently, a new iron electrocatalyst for formate oxidation was reported, for which the proposed mechanism invokes separate sites for proton and electron transfers to achieve net hydride transfer.49 The TOF is impressive (ca. 103 s−1), nearly two orders of magnitude greater than for 1a. However, the electrocatalytic potential is very high – more than 1.1 V positive of 1–3. It will be interesting to see whether these potentials can be shifted more negative while still accessing this orthogonal mechanism. It also remains to be seen whether alcohol oxidation may be accessible by an analogous net hydride transfer mechanism. We note the similarities to the reactivity of early metal–oxo electrocatalysts21–31 and the copper-nitroxyl radical co-catalytic system developed by Badalyan and Stahl,105 where net hydride abstraction is achieved in an orthogonal fashion.
An ideal electrocatalyst will operate near the standard thermodynamic potential for formate or alcohol oxidation, which depends on the solvent and solution basicity. For formate oxidation, the simplest system would utilize a formate buffer to provide the requisite basic conditions. Thus, catalyst design efforts should target electrocatalytic turnover with biformate near the defined thermodynamic potential in a particular solvent.106 Alcohol electro-oxidation also requires basic conditions, and thus the standard thermodynamic potential varies with base selection.40 In this regard, the base and solvent can be viewed as additional experimental handles for minimizing the electrocatalytic overpotential. However, the solution basicity must be matched with the pKa requirements of the catalytic mechanism, and the solvent must be compatible with the catalyst and base. Other parameters such as thermodynamic and kinetic hydricity are also solvent-dependent.35,84–87 Thus, a holistic approach where these factors are considered in parallel is recommended toward the design of optimized electrocatalysts and reaction conditions.
Author contributions
Conceptualization, K.M.W.; writing – original draft, N.M.W. and K.M.W.; writing – review and editing, N.M.W. and K.M.W.; supervision, K.M.W.Data availability
No primary research results, software, or code have been included and no new data were generated or analyzed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Rutgers, The State University of New Jersey.References
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- M. Grasemann and G. Laurenczy, Energy Environ. Sci., 2012, 5, 8171–8181 RSC.
- L. An and R. Chen, J. Power Sources, 2016, 320, 127–139 CrossRef CAS.
- D. M. Fadzillah, S. K. Kamarudin, M. A. Zainoodin and M. S. Masdar, Int. J. Hydrogen Energy, 2019, 44, 3031–3054 CrossRef CAS.
- S. S. Siwal, S. Thakur, Q. B. Zhang and V. K. Thakur, Mater. Today Adv., 2019, 14, 100182 CAS.
- A. Q. K. Nguyen, H. Q. Pham, S. T. M. Huynh and T. T. Huynh, Adv. Sustainable Syst., 2023, 7, 2300205 CrossRef CAS.
- A. W. Cook and K. M. Waldie, ACS Appl. Energy Mater., 2020, 3, 38–46 CrossRef CAS.
- S. Esquivel-Elizondo, A. H. Mejia, T. Sun, E. Shrestha, S. P. Hamburg and I. B. Ocko, Front. Energy Res., 2024, 11, 1207208 CrossRef.
- V. Yadav, G. Sivakumar, V. Gupta and E. Balaraman, ACS Catal., 2021, 11, 14712–14726 CrossRef CAS.
- J. Eppinger and K.-W. Huang, ACS Energy Lett., 2017, 2, 188–195 CrossRef CAS.
- I. Dutta, R. K. Parsapur, S. Chatterjee, A. M. Hengne, D. Tan, K. Peramaiah, T. I. Solling, O. J. Nielson and K.-W. Huang, ACS Energy Lett., 2023, 8, 3251–3257 CrossRef CAS.
- D. Wang, P. Wang, S. Wang, Y.-H. Chen, H. Zhang and A. Lei, Nat. Commun., 2019, 10, 2796 CrossRef PubMed.
- M. T. Bender, X. Yuan and K.-S. Choi, Nat. Commun., 2020, 11, 4594 CrossRef CAS PubMed.
- S. Verma, S. Lu and P. J. A. Kenis, Nat. Energy, 2019, 4, 466–474 CrossRef CAS.
- J. Na, B. Seo, K. J. C. W. Lee, H. Lee, Y. J. Hwang, B. K. Min, D. K. Lee, H.-S. Oh and U. Lee, Nat. Commun., 2019, 10, 5193 CrossRef PubMed.
- Y. Pan, H. Zhang, B. Zhang, F. Gong, J. Feng, H. Huang, S. Vanka, R. Fan, Q. Cao, M. Shen, Z. Li, Z. Zou, R. Xiao and S. Chu, Nat. Commun., 2023, 14, 1013 CrossRef CAS PubMed.
- B. A. Frontana-Uribe, R. D. Little, J. G. Ibanez, A. Palma and R. Vasquez-Medrano, Green Chem., 2010, 12, 2099–2119 RSC.
- E. J. Horn, B. R. Rosen and P. S. Baran, ACS Cent. Sci., 2016, 2, 302–308 CrossRef CAS PubMed.
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed.
- F. Wang and S. S. Stahl, Acc. Chem. Res., 2020, 53, 561–574 CrossRef CAS PubMed.
- M. S. Thompson and T. J. Meyer, J. Am. Chem. Soc., 1982, 104, 4106–4115 CrossRef CAS.
- L. Roecker and T. J. Meyer, J. Am. Chem. Soc., 1987, 109, 746–754 CrossRef CAS.
- W. K. Seok and T. J. Meyer, J. Am. Chem. Soc., 1988, 110, 7358–7367 CrossRef CAS.
- C.-M. Che, C. Ho and T.-C. Lau, J. Chem. Soc., Dalton Trans., 1991, 1901–1907 RSC.
- C.-K. Li, W.-T. Tang, C.-M. Che, K.-Y. Wong, R.-J. Wang and T. C. W. Mak, J. Chem. Soc., Dalton Trans., 1991, 1909–1914 RSC.
- Y.-K. Lai and K.-Y. Wong, Electrochim. Acta, 1993, 38, 1015–1021 CrossRef CAS.
- A. Gerli, J. Reedijk, M. T. Lakin and A. L. Speks, Inorg. Chem., 1995, 34, 1836–1843 CrossRef CAS.
- V. J. Catalano, R. A. Heck, C. E. Immoos, A. Öhman and M. G. Hill, Inorg. Chem., 1998, 37, 2150–2157 CrossRef CAS PubMed.
- M. Rodriguez, I. Romero, A. Llobet, A. Deronzier, M. Biner, T. Parella and H. Stoeckli-Evans, Inorg. Chem., 2001, 40, 4150–4156 CrossRef CAS PubMed.
- A. Paul, J. F. Hull, M. R. Norris, Z. Chen, D. H. Ess, J. J. Concepcion and T. J. Meyer, Inorg. Chem., 2011, 50, 1167–1169 CrossRef CAS PubMed.
- A. K. Vannucci, J. F. Hull, Z. Chen, R. A. Binstead, J. J. Concepcion and T. J. Meyer, J. Am. Chem. Soc., 2012, 134, 3972–3975 CrossRef CAS PubMed.
- M. Trincado, J. Bösken and H. Grützmacher, Coord. Chem. Rev., 2021, 443, 213967 CrossRef CAS.
- N. von Wolff, O. Rivalda-Wheelaghan and D. Tocqueville, ChemElectroChem, 2021, 8, 4019–4027 CrossRef CAS.
- I. Fokin, K.-T. Kuessner and I. Siewert, Synthesis, 2022, 54, 295–314 CrossRef CAS.
- E. S. Wiedner, M. B. Chambers, C. L. Pitman, R. M. Bullock, A. J. M. Miller and A. M. Appel, Chem. Rev., 2016, 116, 8655–8692 CrossRef CAS PubMed.
- K. M. Waldie, A. L. Ostericher, M. H. Reineke, A. F. Sasayama and C. P. Kubiak, ACS Catal., 2018, 8, 1313–1324 CrossRef CAS.
- S. Katipamula, N. M. White and K. M. Waldie, Chem. Catal., 2023, 3, 100561 CrossRef CAS.
- J. T. Muckerman, P. Achord, C. Creutz, D. E. Polyansky and E. Fujita, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15657–15662 CrossRef CAS PubMed.
- I. Fokin and I. Siewert, Chem. – Eur. J., 2020, 26, 14137–14143 CrossRef CAS PubMed.
- A. L. Speelman, J. B. Gerken, S. P. Heins, E. S. Wiedner, S. S. Stahl and A. M. Appel, Energy Environ. Sci., 2022, 15, 4015–4024 RSC.
- CRC Handbook of Chemistry and Physics, 81st Edition, ed. D. R. Lide, CRC Press, Boca Raton, FL, 2000 Search PubMed.
- R. Noyori, M. Yamakawa and S. Hashiguchi, J. Org. Chem., 2001, 66, 7931–7944 CrossRef CAS PubMed.
- P. A. Dub and J. C. Gordon, ACS Catal., 2017, 7, 6635–6655 CrossRef CAS.
- P. Chakraborty, S. Pradhan and B. Sundararaji, in Topics in Organometallic Chemistry, Springer, Berlin, Heidelberg, 2023 Search PubMed.
- B. R. Galan, J. Schöffel, J. C. Linehan, C. Seu, A. M. Appel, J. A. S. Roberts, M. L. Helm, U. J. Kilgore, J. Y. Yang, D. L. DuBois and C. P. Kubiak, J. Am. Chem. Soc., 2011, 133, 12767–12779 CrossRef CAS PubMed.
- C. Seu, A. M. Appel, M. D. Doud, D. L. DuBois and C. P. Kubiak, Energy Environ. Sci., 2012, 5, 6480–6490 RSC.
- J. Bi, P. Hou and P. Kang, ChemCatChem, 2019, 11, 2069–2072 CrossRef CAS.
- D. W. Cunningham, J. M. Barlow, R. S. Velazquez and J. Y. Yang, Angew. Chem., Int. Ed., 2020, 59, 4443–4447 CrossRef CAS PubMed.
- Y. Li, J.-Y. Chen, X. Zhang, Z. Peng, Q. Miao, W. Chen, F. Xie, R.-Z. Liao, S. Ye, C.-H. Tung and W. Wang, J. Am. Chem. Soc., 2023, 145, 26915–26924 CrossRef CAS PubMed.
- A. D. Wilson, R. H. Newell, M. J. McNevin, J. T. Muckerman, M. R. DuBois and D. L. DuBois, J. Am. Chem. Soc., 2005, 128, 358–366 CrossRef PubMed.
- M. Rakowski DuBois and D. L. DuBois, Chem. Soc. Rev., 2009, 38, 62–72 RSC.
- K. M. Waldie, F. M. Brunner and C. P. Kubiak, ACS Sustainable Chem. Eng., 2018, 6, 6841–6848 CrossRef CAS.
- M. L. Pegis, C. F. Wise, B. Koronkiewicz and J. M. Mayer, J. Am. Chem. Soc., 2017, 139, 11000–11003 CrossRef CAS PubMed.
- L. Xue and M. S. G. Ahlquist, Inorg. Chem., 2014, 53, 3281–3289 CrossRef CAS PubMed.
- D. E. Berning, B. C. Noll and D. L. DuBois, J. Am. Chem. Soc., 1999, 121, 11432–11447 CrossRef CAS.
- J. C. Calvin, M. Alex, W. W. Ellis and L. D. Daniel, J. Am. Chem. Soc., 2002, 124, 1918–1925 CrossRef PubMed.
- S. Katipamula, A. W. Cook, I. Niedzwiecki, T. J. Emge and K. M. Waldie, ChemRxiv, 2023 DOI:10.26434/chemrxiv-2023-tfn6t.
- C. J. Weiss, P. Das, D. L. Miller, M. L. Helm and A. M. Appel, ACS Catal., 2014, 4, 2951–2958 CrossRef CAS.
- C. J. Weiss, E. S. Wiedner, J. A. S. Roberts and A. M. Appel, Chem. Commun., 2015, 51, 6172–6174 RSC.
- E. S. Wiedner, A. M. Appel, S. Raugei, W. J. Shaw and R. M. Bullock, Chem. Rev., 2022, 122, 12427–12474 CrossRef CAS PubMed.
- T. Gunasekara, Y. Tong, A. L. Speelman, J. D. Erickson, A. M. Appel, M. B. Hall and E. S. Wiedner, ACS Catal., 2022, 12, 2729–2740 CrossRef CAS.
- C. J. Curtis, A. Miedaner, R. Ciancanelli, W. W. Ellis, B. C. Noll, M. Rakowski DuBois and D. L. DuBois, Inorg. Chem., 2003, 42, 216–227 CrossRef CAS PubMed.
- L. E. Fernandez, S. Horvath and S. Hammes-Schiffer, J. Phys. Chem. C, 2012, 116, 3171–3180 CrossRef CAS.
- S. Horvath, L. E. Fernandez, A. V. Soudackov and S. Hammes-Schiffer, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15663–15668 CrossRef CAS PubMed.
- E. A. McLoughlin, B. D. Matson, R. Sarangi and R. Waymouth, Inorg. Chem., 2020, 59, 1453–1460 CrossRef CAS PubMed.
- P. J. Bonitatibus, Jr., S. Chakraborty, M. D. Doherty, O. Siclovan, W. D. Jones and G. L. Soloveichik, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 1687–1692 CrossRef PubMed.
- E. Alberico, P. Sponholz, C. Cordes, M. Nielsen, H.-J. Drexler, W. Baumann, H. Junge and M. Beller, Angew. Chem., Int. Ed., 2013, 52, 14162–14166 CrossRef CAS PubMed.
- S. Werkmeister, K. Junge, B. Wendt, E. Alberico, H. Jiao, W. Baumann, H. Junge, F. Gallou and M. Beller, Angew. Chem., Int. Ed., 2014, 53, 8722–8726 CrossRef CAS PubMed.
- S. Chakraborty, P. O. Lagaditis, M. Förster, E. A. Bielinski, N. Hazari, M. C. Holthausen, W. D. Jones and S. Schneider, ACS Catal., 2014, 4, 3994–4003 CrossRef CAS.
- S. Chakraborty, W. W. Brennessel and W. D. Jones, J. Am. Chem. Soc., 2014, 136, 8564–8567 CrossRef CAS PubMed.
- S. Chakraborty, H. Dai, P. Bhattacharya, N. T. Fairweather, M. S. Gibson, J. A. Krause and G. Hairong, J. Am. Chem. Soc., 2014, 136, 7869–7872 CrossRef CAS PubMed.
- F. Schneck, M. Assmann, M. Balmer, K. Harms and R. Langer, Organometallics, 2016, 35, 1931–1943 CrossRef CAS.
- X. Yang, ACS Catal., 2013, 3, 2684–2688 CrossRef CAS.
- H. Jiao, K. Junge, E. Alberico and M. Beller, J. Comput. Chem., 2016, 37, 168–176 CrossRef CAS PubMed.
- K. R. Brownell, C. C. L. McCrory, C. E. D. Chidsey, R. H. Perry, R. N. Zare and R. M. Waymouth, J. Am. Chem. Soc., 2013, 135, 14299–14305 CrossRef CAS PubMed.
- K. M. Waldie, K. R. Flajslik, E. A. McLoughlin, C. E. D. Chidsey and R. M. Waymouth, J. Am. Chem. Soc., 2017, 139, 738–748 CrossRef CAS PubMed.
- G. A. Filonenko, R. van Putten, E. J. M. Hensen and E. A. Pidko, Chem. Soc. Rev., 2018, 47, 1459–1483 RSC.
- S. Budweg, K. Junge and M. Beller, Catal. Sci. Technol., 2020, 10, 3825–3842 RSC.
- L. Alig, M. Fritz and S. Schneider, Chem. Rev., 2019, 119, 2681–2751 CrossRef CAS PubMed.
- S. P. Heins, P. E. Schneider, A. L. Speelman, S. Hammes-Schiffer and A. M. Appel, ACS Catal., 2021, 11, 6384–6389 CrossRef CAS.
- R. Ciancanelli, B. C. Noll, D. L. DuBois and M. Rakowski DuBois, J. Am. Chem. Soc., 2002, 124, 2984–2992 CrossRef CAS PubMed.
- S. C. Marinescu, J. R. Winkler and H. B. Gray, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15127–15131 CrossRef CAS PubMed.
- C. M. Galvin and R. M. Waymouth, J. Am. Chem. Soc., 2020, 142, 19368–19378 CrossRef CAS PubMed.
- K. R. Brereton, N. E. Smith, N. Hazari and A. J. M. Miller, Chem. Soc. Rev., 2020, 49, 7929–7948 RSC.
- S. Ilic, A. Alherz, C. B. Musgrave and K. D. Glusac, Chem. Soc. Rev., 2018, 47, 2809–2836 RSC.
- M. R. Espinosa, M. Z. Ertem, M. Barakat, Q. J. Bruch, A. P. Deziel, M. R. Elsby, F. Hasanayn, N. Hazari, A. J. M. Miller, M. V. Pecoraro, A. M. Smith and N. E. Smith, J. Am. Chem. Soc., 2022, 144, 17939–17954 CrossRef CAS PubMed.
- M. R. Elsby, M. R. Espinosa, M. Z. Ertem, A. P. Deziel, N. Hazari, A. J. M. Miller, A. H. Paulus and M. V. Pecoraro, Organometallics, 2023, 42, 3005–3012 CrossRef CAS.
- V. Lyaskovskyy and B. de Bruin, ACS Catal., 2012, 2, 270–279 CrossRef CAS.
- D. Tocqueville, F. Crisanti, J. Guerrero, E. Nubret, M. Robert, D. Milstein and N. von Wolff, Chem. Sci., 2022, 13, 13220–13224 RSC.
- C. L. Mathis, J. Geary, Y. Ardon, M. S. Reese, M. A. Philliber, R. T. VanderLinden and C. T. Saouma, J. Am. Chem. Soc., 2019, 141, 14317–14328 CrossRef CAS PubMed.
- B. Askevold, A. Friedrich, M. R. Buchner, B. Lewall, A. C. Filippou, E. Herdtweck and S. Schneider, J. Organomet. Chem., 2013, 744, 35–40 CrossRef CAS.
- C. Costentin, S. Drouet, M. Robert and J.-M. Savéant, Science, 2012, 338, 90–94 CrossRef CAS PubMed.
- I. Azcarate, C. Costentin, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2016, 138, 16639–16644 CrossRef CAS PubMed.
- E. A. Bielinski, P. O. Lagaditis, Y. Zhang, B. Q. Mercado, C. Würtele, W. H. Bernskoetter, N. Hazari and S. Schneider, J. Am. Chem. Soc., 2014, 136, 10234–10237 CrossRef CAS PubMed.
- E. A. Bielinski, M. Förster, Y. Zhang, W. H. Bernskoetter, N. Hazari and M. C. Holthausen, ACS Catal., 2015, 5, 2404–2415 CrossRef CAS.
- D. J. Martin, B. Q. Mercado and J. M. Mayer, Sci. Adv., 2020, 6, eaaz3318 CrossRef CAS PubMed.
- R. Zhang and J. J. Warren, J. Am. Chem. Soc., 2020, 142, 13426–13434 CrossRef CAS PubMed.
- D. J. Martin and J. M. Mayer, J. Am. Chem. Soc., 2021, 143, 11423–11434 CrossRef CAS PubMed.
- N. D. Loewen, S. Pattanayak, R. Herber, J. C. Fettinger and L. A. Berben, J. Phys. Chem. Lett., 2021, 12, 3066–3073 CrossRef CAS PubMed.
- W. Nie and C. C. L. McCrory, Dalton Trans., 2022, 51, 6993–7010 RSC.
- M. Bourrez, R. Steinmetz, S. Ott, F. Gloaguen and L. Hammarström, Nat. Chem., 2015, 7, 140–145 CrossRef CAS PubMed.
- T. Huang, E. S. Rountree, A. P. Traywick, M. Bayoumi and J. L. Dempsey, J. Am. Chem. Soc., 2017, 140, 14655–14669 CrossRef PubMed.
- E. A. McLoughlin, K. C. Armstrong and R. M. Waymouth, ACS Catal., 2020, 10, 11654–11662 CrossRef CAS.
- J. Y. Yang, T. A. Kerr, X. S. Wang and J. M. Barlow, J. Am. Chem. Soc., 2020, 142, 19438–19445 CrossRef CAS PubMed.
- A. Badalyan and S. S. Stahl, Nature, 2016, 535, 406–410 CrossRef CAS PubMed.
- B. M. Stratakes, J. L. Dempsey and A. J. M. Miller, ChemElectroChem, 2021, 8, 4161–4180 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |