
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal-doped nickel-based chalcogenides and phosphochalcogenides for electrochemical water splitting
Dmitrii A.
Rakov
 ab
ab
aInstitute for Frontier Materials, Deakin University, Geelong, Victoria 3217, Australia. E-mail: dmitrii.rakov@research.deakin.edu.au
bARC Centre of Excellence for Electromaterials Science (ACES), Deakin University, Burwood, Victoria 3125, Australia
First published on 20th December 2022
Abstract
The rational design of an affordable, stable, and active electrocatalyst is essential for a sustainable hydrogen economy. Recently, great progress has been achieved with nickel-based chalcogenides and phosphochalcogenides for both hydrogen and oxygen evolution reactions, and the metal doping strategy has been demonstrated to have a substantial impact on the performance of electrocatalysts. Common approaches to simultaneously improve the intrinsic activity of nickel-based (phospho)chalcogenides by metal doping and increase the active surface area of the electrocatalyst bring some challenges to deciding accurately whether metal doping will improve the electrocatalytic activity or not. This review highlights the influence of 3d block metal doping (Cu, Co, Fe and Mn) on the electrocatalytic performance of these phases with reference to the electronic structure of the materials. Such aspects as the electrical conductivity, hydrogen and oxygen intermediate adsorption mechanism, surface bifunctionality, role of chalcogenide/phosphochalcogenide surface functional groups, electrocatalytic stability and related changes upon metal doping are discussed in this review article. This work discusses current gaps that need to be filled to develop a systematic strategy in the modulation of the electronic/structural properties of metal-doped nickel-based (phospho)chalcogenides for their effective use in electrochemical water splitting.
 Dmitrii A. Rakov | Dmitrii Rakov was awarded his PhD degree from Deakin University (Australia) in Prof. Maria Forsyth's group. He has worked in NASA Ames research centre with Dr John W. Lawson within Universities Space Research Association program. He is currently a postdoctoral research fellow at the University of Queensland (Australia) in Prof. Chengzhong Yu's group. His research focuses on computational/experimental electromaterials science, in particular, on the optimization of the charge transfer mechanism in energy storage and conversion devices. |
Introduction
In recent years, great attention has been devoted to renewable energy sources (e.g. solar, wind, tidal, geothermal, etc.) due to rising concerns about the energy crisis, global warming and the related use of fossil fuels.1–3 Among many, hydrogen, as a carbon-free and high energy density (146 kJ g−1) source, has become the main candidate for future alternative fuels.3 Nowadays, hydrogen is mainly produced in three ways, namely methane steam reforming, coal gasification, and water electrolysis. Water electrolysis is one of the most environmentally friendly means of hydrogen generation.4 The current development of hydrogen production through water splitting at the industrial scale requires a non-expensive, robust, and efficient catalyst. A typical cell for electrochemical water splitting consists of electrodes performing two half-reactions, the oxygen evolution reaction (OER) at the cathode and the hydrogen evolution reaction (HER) at the anode.| 4H+(aq) + 4e− → 2H2(g) E = (−0.059pH) V vs. RHE (reduction) | (1) |
| H2O → O2(g) + 4H+(aq) + 4e− E = (1.23 V − 0.059pH) V vs. RHE (oxidation) | (2) |
| 2H2O → O2(g) + H2(g) ΔE = 1.23 V | (3) |
Recently, enormous progress has been made towards the development of a noble metal-free catalyst for electrochemical water splitting, though the underlying strategy for rational design and optimization of an affordable, stable and efficient electrocatalyst is still under development.4,7–10 Multiple strategies have been suggested to improve the activity and stability of electrocatalysts, such as increasing the number of active sites, improving the intrinsic activity, and modifying the electrolyte composition.11–17 The increase in the intrinsic activity of surface sites is a more effective means to optimize the overall activity of the electrocatalyst compared to the nanofabrication strategy.11 The elemental doping has a significant impact on the overall electronic properties of the bulk phase as well as on the surface electrocatalytic activity.17–24 Nickel-based transition metal chalcogenides (MaXb, where X = S, Se, Te) have been pointed out as promising noble-metal-free electrocatalysts for water splitting due to their high efficiency, tunable electrocatalytic behaviour, and reasonable cost of raw materials.25–37 Another family of electrocatalysts such as transition metal phosphochalcogenides, e.g., nickel phosphochalcogenides (NiPX3, where X = S, Se), also so-called lamellar phases, consist of metal cation-supporting [P2S6]4− or [P2Se6]4− units weakly bonded to each other via van der Waals forces. These compounds are structurally similar to transition metal chalcogenides, and upon reduced dimensionality, they show a range of unique properties suitable for application in the electronic industry,38,39 energy storage,40–42 and catalysis;31,43–45 more comprehensive reviews about the structure and application of MPX3 materials can be found in the literature.29,46,47
Here, we summarize recent advances in transition metal (TM) doping of nickel chalcogenides and phosphochalcogenides for electrochemical water splitting. This review emphasizes the major aspects of elemental doping that should be considered for the rational optimization of the electrocatalytic activity and stability of catalysts for both the HER and OER. Fig. 1 shows unit cells of metal-doped nickel phosphochalcogenides and some typical nickel chalcogenides discussed in this review. To minimize the scope of this work, the review will consider only several 3d transition metals such as Cu, Co, Fe, and Mn.
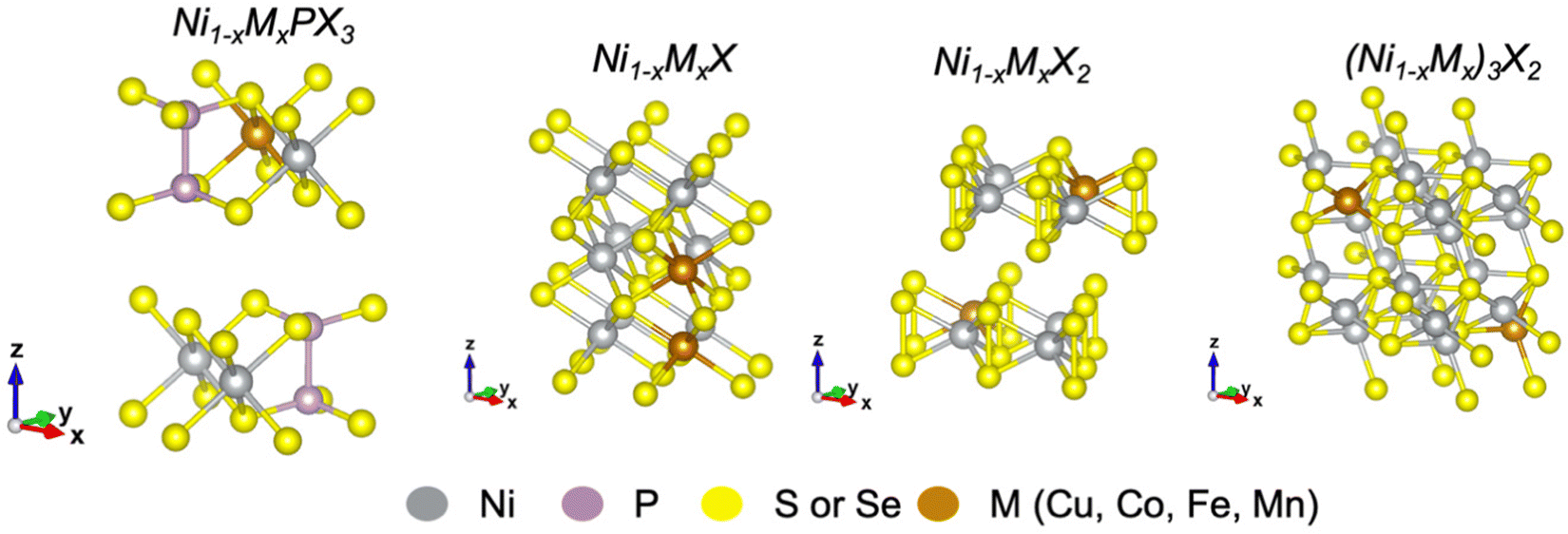 | ||
| Fig. 1 Visual representation of unit cells for metal-doped (left) phosphochalcogenides (monoclinic, symmetry space group P1), nickel monochalcogenides (hexagonal, symmetry space group P1), nickel dichalcogenides (hexagonal, symmetry space group P1), and (right) tri nickel dichalcogenides (trigonal, symmetry space group P1). Axes XYZ correspond to periodic cell dimensions (ABC). | ||
Mechanism of electrochemical water splitting and related factors
Regardless of the acidity of the electrolyte, the thermodynamic voltage for water splitting is 1.23 V at 25 °C and 1 atm. However, an extra potential, often referred to as an overpotential (η), is required to overcome activation energy barriers resulting from the additional resistance of the electrode, electrolyte and impurities (ions, dissolved gases), solid/gas interface and contacts resistance as the overall mechanism of the charge transfer at the electrocatalyst/electrolyte interface.4,6,12,14,48–50 The reaction rate for both the HER and OER processes is greatly affected by the acidity of the electrolyte and the electrocatalyst nature.51,52 Therefore, chemically and morphologically stable, and efficient noble-metal-free catalysts that can perform well towards both half-reactions in the same media are still highly required for electrochemical water splitting. Usually, the HER rate decreases in solutions with a high pH value due to the absence of free protons, while the electrocatalyst chemical stability, e.g., against corrosion and elemental etching, and kinetics of the counter OER half-reaction are more efficient at higher pH due to partial surface oxidation and facile formation of oxygen intermediate species;11,53–56 however, the water splitting mechanism in neutral and alkaline media is even more complex due to an extra step involving water dissociation. Recently, it was shown that not only the medium pH but also the electrode/electrolyte interfacial structure of solvated ions is important for the HER as it contributes to interfacial transport and the overall energy barrier for a conversion reaction, and therefore new strategies to optimize the arrangement of interfacial water molecules and their short-range order near the charged surface by tuning the cation chemistry should be considered.57The mechanism of electrochemical water splitting in different media is summarized in Tables 1 and 2.
| Step | HER in acid | OER in acid |
|---|---|---|
| Overall | 2H+(aq) + 2e− = H2(g) | 2H2O = 4H+(aq) + O2(g) + 4e− |
| (1) | H+(aq) + e− = H*(ad) | H2O = OH(ad) + H+(aq) + e− |
| (2) | H+(aq) + e− + H*(ad) = H2(g) | OH(ad) = O(ad) + H+(aq) + e− |
| (3) | H*(ad) + H*(ad) = H2(g) | O(ad) + H2O = OOH(ad) + H+(aq) + e− |
| (4) | OOH(ad) = H+(aq) + O2(g) + e− |
| Step | HER in neutral/alkaline | OER in neutral | OER in alkaline |
|---|---|---|---|
| Overall | 2H2O + 2e− = H2(g) + 2OH−(aq) | 4OH−(aq) = 2H2O + O2(g) + 4e− | 4OH−(aq) = 2H2O + O2(g) + 4e− |
| (1) | H2O + e− = H*(ad) + OH−(aq) | 2H2O = OH(ad) + H2O + H+(aq) + e− | OH−(aq) = OH(ad) + e− |
| (2) | H2O + e− + H*(ad) = H2(g) + OH−(aq) | OH(ad) + H2O = O(ad) + H2O + H+(aq) + e− | OH−(aq) + OH(ad) = O(ad) + H2O + e− |
| (3) | H*(ad) + H*(ad) = H2(g) | O(ad) + H2O = OOH(ad) + H+(aq) + e− | O(ad) + OH−(aq) = OOH(ad) + e− |
| (4) | OOH(ad) = H+(aq) + O2(g) + e− | OOH(ad) + OH−(aq) = O2(g) + H2O + e− |
The electrochemical catalyst minimizes the energy barrier to proceed through the chemical process compared to an identical non-catalytic reaction that does not affect the chemical equilibrium. The concept of “active sites” is often used to describe the surface atomic spot contributing the most towards the adsorption of intermediates during the electrochemical water splitting process. However, the mechanism of both the HER and OER depends on the acidity of the electrolyte, which might consist of multiple steps also involving a water dissociation. In this case, the active site will be a combination of several surface centers, not only one, with different adsorption properties to facilitate the optimum rate of all involved steps, including water surface dissociation. Therefore, to further address the activity of electrocatalysts in different media, this review will focus on a combination of several factors affecting the HER and OER processes for metal chalcogenides and phosphochalcogenides rather than looking for a true active site/sites of these materials. Namely, the role of electric conductivity (metallicity), adsorption kinetics of both hydrogen and oxygen intermediates, bifunctionally of the surface and surface functional groups (P, S or Se-based groups) on the electrochemical water splitting will be further discussed in this review.
Electrical conductivity and metallicity
The electric conductivity of the catalyst has a strong influence on its overall electrocatalytic activity; however, the electric conductivity may be relevant for the dry materials as some changes in the surface chemistry are expected after the contact with the electrolyte. The changes in oxidation state and presence of surface functional groups can significantly affect the charge transfer process; therefore, the effective conductivity as the HER and OER descriptor should be measured only for activated catalysts directly from the solution.58 The electrical conductivity might have an impact on the surface wettability of the electrocatalyst, which has to be investigated. In Fig. 2a, Mattia et al. showed that the contact angle between the carbon substrate and polar water or non-polar solvents depends on the annealing temperature of the pre-treated carbon material; however, in these conditions, it is difficult to separate the contribution of surface functional groups and electrical conductivity of the material.59 Usually, the material with higher bulk electrical conductivity is more electrocatalytically active compared to the low electric conductive system (Fig. 2b); however, the surface reactivity towards adsorption/desorption of intermediates should be considered as well.25,31,60–64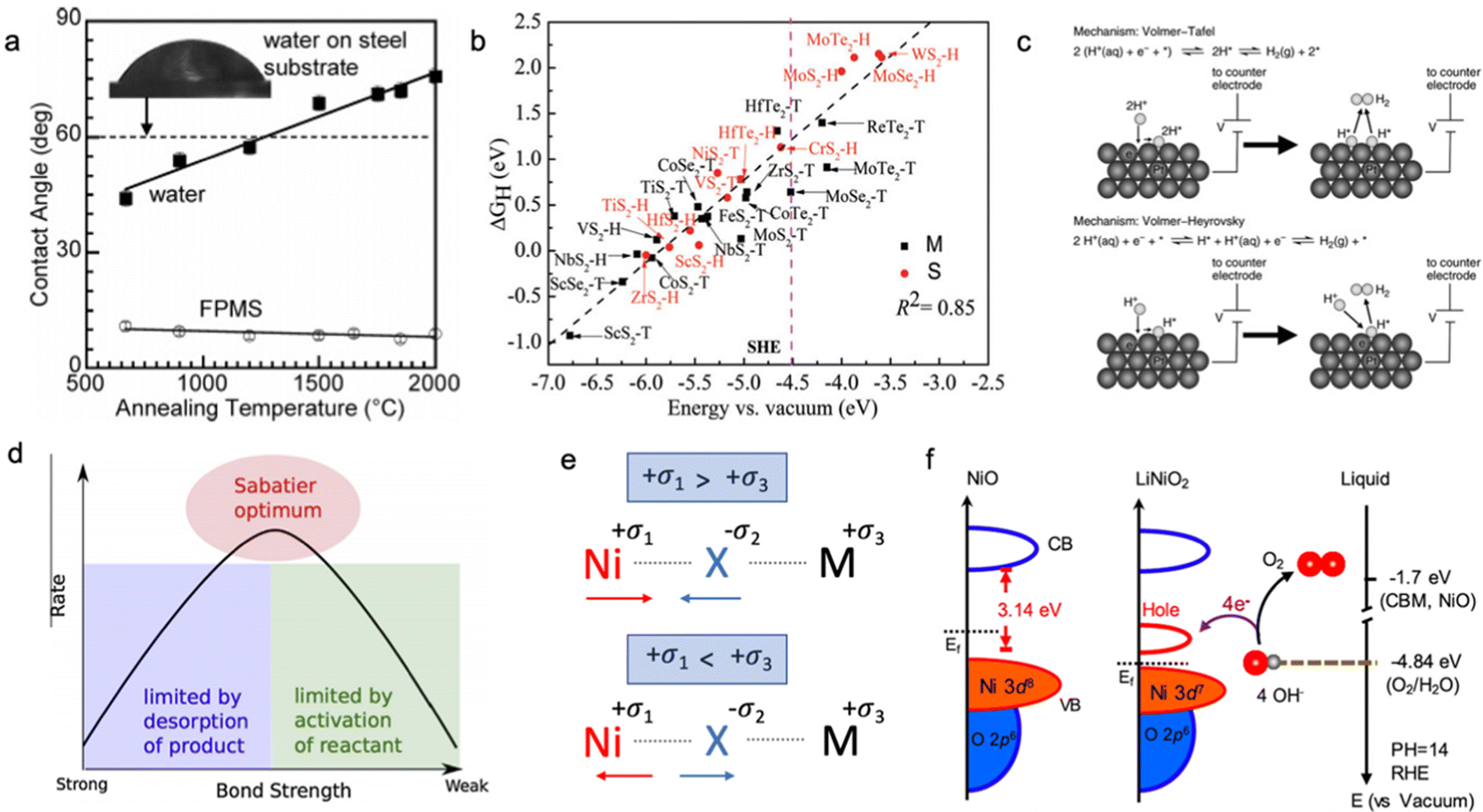 | ||
Fig. 2 (a) Contact angle on CVD-carbon film deposited on glassy carbon:![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) contact angle for water and nonpolar fluorosilicone (FPMS) as a function of annealing temperature and electrical conductivity of the carbon substrate. Reproduced with permission from ref. 59. Copyright © 2006, American Chemical Society. (b) The hydrogen adsorption free energy ΔGH as a function of the conduction band minimum Ecv for a 2 × 2 supercell of transition metal dichalcogenides. The pink dashed lines indicate the SHE, and S and M indicate semiconductor and metallic, respectively. Reproduced with permission from ref. 60. Copyright © 2017, the Royal Society of Chemistry. (c) Volmer–Tafel and Volmer–Heyrovsky hydrogen evolution reaction mechanism in acid and alkaline media. Reproduced with permission from ref. 79. Copyright © 2012, American Chemical Society. (d) Sabatier principal volcano plot for the binding energy of adsorbed intermediates. Reproduced with permission from ref. 80. Copyright 2015 Elsevier Ltd. (e) Schematic illustration of chemical bond hybridization between chalcogen (X) and nickel (Ni) atoms upon doping of metal (M). (f) Schematic diagram for electronic structures of NiO and LiNiO2 (left) and energy diagram at the oxide–liquid interface on the vacuum level scale at pH = 14 (right); the redox level for O2/H2O (red dashed line) is −4.84 eV, the conduction band minimum (CBM) level for NiO is −1.7 eV according to the formula: ECBM = Ebandgap − Eionization, (Ebandgap = 3.7 eV, Eionization = 5.4 eV). Reproduced with permission from ref. 78. Copyright © 2019, American Chemical Society. contact angle for water and nonpolar fluorosilicone (FPMS) as a function of annealing temperature and electrical conductivity of the carbon substrate. Reproduced with permission from ref. 59. Copyright © 2006, American Chemical Society. (b) The hydrogen adsorption free energy ΔGH as a function of the conduction band minimum Ecv for a 2 × 2 supercell of transition metal dichalcogenides. The pink dashed lines indicate the SHE, and S and M indicate semiconductor and metallic, respectively. Reproduced with permission from ref. 60. Copyright © 2017, the Royal Society of Chemistry. (c) Volmer–Tafel and Volmer–Heyrovsky hydrogen evolution reaction mechanism in acid and alkaline media. Reproduced with permission from ref. 79. Copyright © 2012, American Chemical Society. (d) Sabatier principal volcano plot for the binding energy of adsorbed intermediates. Reproduced with permission from ref. 80. Copyright 2015 Elsevier Ltd. (e) Schematic illustration of chemical bond hybridization between chalcogen (X) and nickel (Ni) atoms upon doping of metal (M). (f) Schematic diagram for electronic structures of NiO and LiNiO2 (left) and energy diagram at the oxide–liquid interface on the vacuum level scale at pH = 14 (right); the redox level for O2/H2O (red dashed line) is −4.84 eV, the conduction band minimum (CBM) level for NiO is −1.7 eV according to the formula: ECBM = Ebandgap − Eionization, (Ebandgap = 3.7 eV, Eionization = 5.4 eV). Reproduced with permission from ref. 78. Copyright © 2019, American Chemical Society. | ||
Hydrogen adsorption
The binding strength of adsorbed hydrogen on the active site of the catalyst is an important descriptor of the electrocatalytic HER activity of the material in acid media (Fig. 2). The catalyst with a strong binding strength of adsorbed hydrogen intermediates limits the desorption step (Tafel/Heyrovsky step), while the weak binding leads to poor adsorption-activation behaviour (Volmer step) according to the Sabatier principle (Fig. 2d). As shown in Fig. 2b, materials with a more metallic character often form stronger bonding with hydrogen compared to semiconductors upon adsorption of a proton. However, this binding strength of hydrogen intermediates (H*) does not solemnly depend on the electrical conductivity of the electrocatalyst, but is also influenced by its position in the conduction band and the Fermi level at the solid–liquid interface. The localized density of state measurement is an important way to understand the excess and/or deficiency of electrons at the specific surface centres,65,66 which can be an indicator of the adsorption strength of the intermediates. Apart from that, the nature of the surface termination of the catalyst should be also considered due to the difference in electrocatalytic activity of exposed adsorption sites.67,68 Specifically, the HER occurring predominantly through the chalcogenide site makes NiS2 more active compared to NiSe2, whereas the opposite trend can be observed for the HER occurring on the metal site.69,70 It was shown that the position of the conduction band of the active site located above the standard hydrogen electrode (SHE) leads to weak bonding with adsorbed hydrogen, while a conduction band lower than SHE shows the opposite trend with strong adsorbed H* intermediates.60 This is also related to the occupancy of the conduction band and type of semiconductor; the very high occupancy of the conduction band is unfavourable for the acceptance of one extra electron from the hydrogen, and subsequently, this leads to weak bonding (anti-bonding).Therefore, the electrocatalyst should have a partially occupied and low-energy conduction band for stronger hydrogen bonding; and modification of the material between n-type and p-type semiconductors might be an effective strategy.71–74
Oxygen intermediate adsorption
Speaking of the oxygen evolution reaction, this process is controlled by the binding strength of oxygen-based intermediates (O*, OH*, OOH*), and the ideally performing water oxidation electrocatalyst binds these species neither too strongly nor too weakly.61,63 Previously, it was mentioned that the space charge layer at the surface of the electrode affects the resulting potential drop of the Helmholtz layer along with the energy barrier for the charge transfer kinetics, and, therefore, it directly influences the concentration of hydroxyl anions and adsorption strength of oxygen-containing intermediates.63 In this regard, it is believed that p-type semiconductors are more suitable catalysts for the OER compared to n-type ones, as p-type semiconductive materials possess more positive near-surface potential with large availability of holes on the electrode surface for stabilization of adsorbed oxygen-based intermediates.75,76 As shown in Fig. 2e, metal doping has a profound impact on the hybridization state of the metal–chalcogenide bond; namely, for doping metals in an oxidation state less positive than that of nickel, nickel shares more electrons with the chalcogenide making the metal–chalcogenide bond shorter and the chalcogenide more negative. Meanwhile, doping Ni–X with highly positive M leads to an opposite effect, namely, the metal–chalcogenide bond gets longer with higher electron occupancy on the Ni site. According to the theory of charge transfer,77 electron transfer can take place only from any occupied state to an unoccupied state if their energies are matched. For reduction, the electrons transfer from the occupied state of the electrode to the unoccupied state of the electroreactant, while for oxidation the receiving state will be on the electrode. The strong overlap of the occupied and unoccupied states near the Fermi level on the electrocatalytic electrode will minimize the overpotential needed for charge transfer. Suntivich et al. have shown this for perovskite oxides and another work has been done for the LixNi1−xO catalyst, which shows higher electrocatalytic activity towards the OER upon Li doping due to hybridization of the Ni–O bond and the creation of more vacancies for electron acceptance upon adsorption of oxygen-based intermediates, as shown in Fig. 2f, where a new unoccupied state emerges near the Fermi level.61,78Bifunctionality of the surface
As mentioned above, the acidity of the electrolyte has a strong influence on the electrocatalyst rate performance, and hydrogen evolution in neutral/alkaline media requires an overpotential associated with a decrease in a number of electroactive species, namely H+, according to the Nernst equation.81 So, an extra step such as “water dissociation” takes place to generate electroactive species for both cathodic and anodic reactions. In this regard, the proper combination of surface neighbours, i.e., active sites, for facile water deprotonation, recombination of intermediates, and gas desorption can improve the efficiency of the HER rate even at high pH values and/or improve the OER in acidic media.22,51,82–84 This phenomenon is called “bifunctionality”, namely, the catalyst should possess both strong and weak bonding active sites responsible for the adsorption of O*-based intermediates and recombination/desorption of H* intermediates (Fig. 3). This can be achieved by TM doping with different oxophilicity; for example, the Ni–Pt(111) surface shows lower HER overpotential in alkaline media than that of non-doped Pt(111).85 In this case, the hydrophilicity of Pt(111) improves with Ni doping and more protons can be electrogenerated upon adsorption of water molecules. A similar observation was done for TM doping in the NiPS3 phase, where a proper combination of TM with different oxophilicity and doping ratio leads to more active HER and OER behaviour.32,86 Also, highly oxophilic vanadium was successfully used in the Co1−xVxPS electrocatalyst to accelerate the water dissociation kinetics.87 Thus, both the H* and O* intermediate bonding strength should be taken into account to comprehensively evaluate the impact of the electrocatalyst nature on the water splitting mechanism.82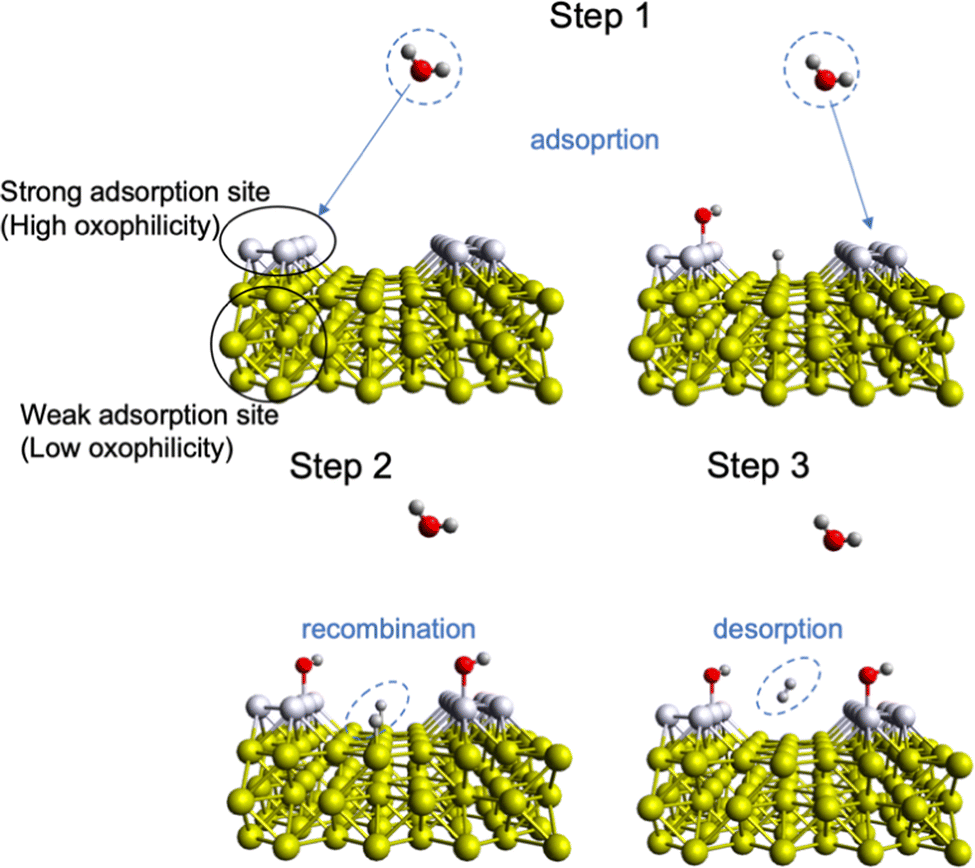 | ||
| Fig. 3 Mechanism of surface bifunctionality for the Volmer–Tafel HER reaction in neutral/basic media where water dissociation takes place. | ||
Surface functional groups (P, S or Se-based groups)
Anion nature and its chemical response to changes in the acidity of the electrolyte can significantly affect the reactivity of the electrocatalyst. It was shown that the MoP(001) surface shows high activity towards the HER in neutral media, where Mo sites help to adsorb water and MoP sites recombine H* into H2.88 Meanwhile, an excess of H+ or OH− alternates the surface and block sites for the dissociation of water, which leads to poor electrocatalytic behaviour. Fang et al. show that P-doping improves the H* desorption/recombination process in bimetallic NiCo alloy, while Se and O-doping show the opposite trend.89 This is due to the difference in electronegativity of the anion, which follows the trend O (3.44) > Se (2.55) > P (2.19), and both O and Se demonstrate very strong hydrogen adsorption enthalpies. Li et al. showed that incorporating Se-vacancies into Ni2P (Ni2P/Ni3Se4) significantly improves the HER activity due to an increase in adsorption energy for the H* intermediate on the Se site.90 In the case of bimetallic nickel–iron oxide, the introduction of sulphur to oxygen vacancies also showed surface charge delocalization to iron sites, i.e., more electrons on iron sites after sulphur doping, which is again due to the difference in the electronegativity of the anions (S (2.58) vs. O (3.44)).65 Such changes in surface charge between neighbours contribute a significant role to the bifunctionality of the electrocatalyst as well as its electronic conductivity (band gap value).Regarding the effect of anion chemistry on the OER behaviour, Wu et al. show that S2−-vacancies in NiFe are responsible for high water oxidation activity, which changes to SOX form after long-term cycling. This process was attributed to surface poisoning and deterioration of the OER activity of the electrocatalyst. However, Hu et al. showed that the chalcogenide anion of CuO@CuSx leaches into the electrolyte during cycling and SO42− is formed.91 The presence of SO42− in the solution and the oxidized CuO@CuOOH surface leads to an interfacial abundance of OH− which improves the OER activity. The leaching of chalcogenide anions during the OER was seen in many literature reports.92–95 However, some reports focus unjustifiably too much on the structure of the pre-catalyst to describe the OER mechanism rather than comprehensively investigating the activated surface of the electrocatalyst.96 As for phosphide and phosphate anions, the electrocatalysts with these compounds also demonstrated an improved behaviour for water oxidation;97,98 however, again more studies are needed to understand the difference between pre-catalyst and real active sites.
Metal-doped nickel phosphochalcogenides (NiPX3)
MPS3 (M = Cu, Ni, Fe, Co, Mn) are semiconductors with a band gap in the range of 1.3–3.5 eV, whereas MPSe3 (M = Cu, Ni, Fe, Co, Mn) show more metallic behaviour29,64,99 and the conduction band is mainly formed by [P2S6]4− or [P2Se6]4− frameworks where the metal contributes to the occupancy of the states.100 In general, MPSe3 phases outperform MPS3 phases in electrocatalytic activity due to higher electrical conductivity leading to better charge transfer kinetics,32,36,64,101,102 where the MxPy phase is found to be more electrocatalytically active than the MxSy/MxSey phase for both HER and OER processes.103–105 As shown in Fig. 4a and b, electrocatalytic hydrogen evolution was studied for MPS3 bulk phases, and the lowest overpotential was found for NiPS3; meanwhile, CoPS3 shows superior OER performance to other MPS3 materials.31 As said above, the HER activity greatly depends on the adsorption strength of H* intermediates to the catalyst surface, and a rational combination of both weak and strong adsorption centres can be an effective strategy to optimize the electrocatalyst performance.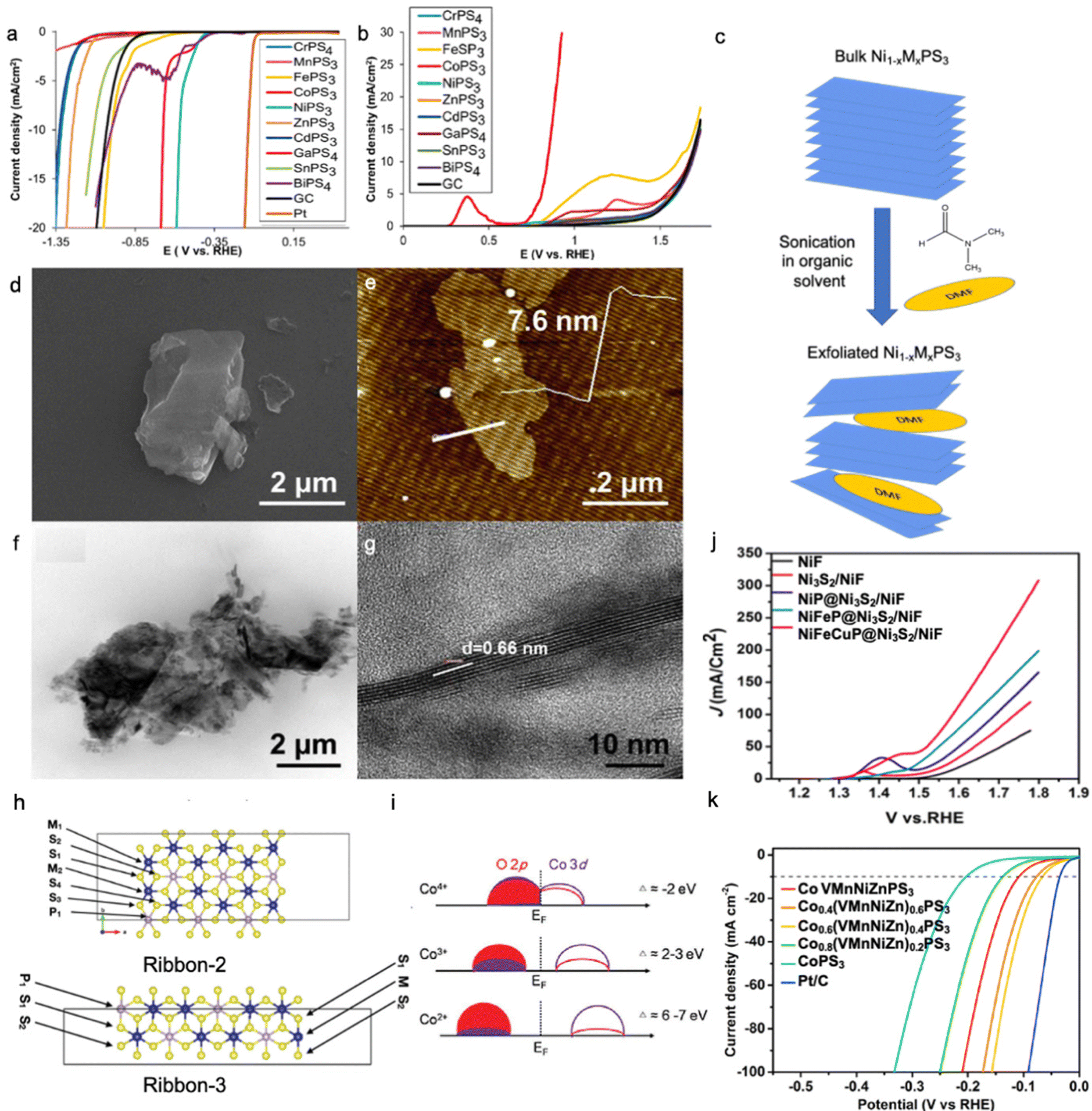 | ||
| Fig. 4 LSV curves for (a) HER and (b) OER activity of bulk MPS3 phases. Reproduced with permission from ref. 31. Copyright © 2017, American Chemical Society. (c) Schematic illustration of Ni1−xMxPS3 (where M is a doping metal) nanosheet preparation by exfoliation of bulk crystals in organic solvent (N,N-dimethylformamide (DMF)). Images of Mn-doped NiPS3 nanosheets prepared by the synthetic route in Fig. 3c were obtained by (d) SEM, (e) AFM, and (f and g) TEM. Reproduced with permission from ref. 22. Copyright 2018 Elsevier Ltd. (h) Possible edges of the MPS3 material with adsorption sites. Reproduced with permission from ref. 122. Copyright © 2021, the Royal Society of Chemistry. (i) Charge transfer model of Co2+, Co3+, Co4+ in a covalent system. Reproduced with permission from ref. 123. Copyright 2021, Wiley-VCH. (j) LSV curves for the OER activity of trimetallic phosphides grown on Ni3S2/Ni foam. Reproduced with permission from ref. 140. Copyright © 2020, American Chemical Society. (k) LSV curves for the HER activity of high entropy CoPS3-based alloy. Reproduced with permission from ref. 121. Copyright © 2022, American Chemical Society. All reported polarization curves were recorded in 1.0 M KOH. | ||
Song et al. showed that NiPS3 possesses both weak and strong H* adsorption centres, whereas FePS3 has only weak H* centres with ΔGH > 0 eV.32 The electronic structure analysis shows that the lowest unoccupied molecular orbital (LUMO) at the conduction band, which is responsible for the electron acceptance, is located at a different position for MPS3 (M = Ni, Fe, Mn).106 Specifically, the LUMO of NiPS3 represented by the eg orbital is at relatively higher energy than t2g of FePS3 and MnPS3, therefore showing that NiPS3 will be in favour of the Tafel/Heyrovsky desorption step compared to other MPS3 phases due to weaker electron affinity.31 The excellent water oxidative activity of CoPS3 is the most likely due to higher CoxPy activity compared to the NixPy phase.107–109 The band gap of NiPS3 was found to be 1.4–1.8 eV experimentally,110,111 while density functional theory gives a wider range of values 1.0–2.19 eV resulting from the choice of the basis set, temperature effect and zero-point motion correction term.43,110,112 The valence band of NiPS3 consists of up-spin 3d eg of nickel, 3s of phosphorous, and 3p of sulphur, whereas the conduction band is dominated by down-spin 3d eg of nickel.113 The position of the metal d-band near the Fermi level means that the nickel centre will likely accept an electron during the cathodic reaction.
Metal-doped NiPS3 phase can be synthesized via different methods such as solid-state synthesis,22 solvothermal,114 and chemical vapour deposition;43,115 however, precise control of stoichiometry can be better realized only through solid-state synthesis in an inert atmosphere. In this method, elemental powders are mixed in their stoichiometric ratio and annealed at high temperatures in a degassed quartz tube. To further expose the active surface area, the as-synthesized bulk phase can be sonicated in an organic solvent to allow exfoliation across a van der Waals gap (Fig. 4c), where the degree of exfoliation can be controlled by the nature of the organic solvent and sonication conditions.116,117Fig. 4d–g show the morphology of Mn-doped NiPS3 nanosheets prepared by solid-state synthesis and exfoliation with N,N-dimethylformamide (DMF). Table 3 shows the summary of some synthesis methods for metal phosphochalcogenides.
| Material | Metal precursors | Source of P/S | Method of synthesis | Ref. |
|---|---|---|---|---|
| Bulk MPS3 (M = Ni, Co, Fe, Mn) | Metal powders | Phosphorous, sulfur | Sealed under high vacuum and annealed in a quartz glass ampule at 650/750 °C for 120/288 h | 31 |
| Bulk MPSe3 (M = Ni, Co, Fe, Mn) | Metal powders | Phosphorous, selenium | Sealed under high vacuum and annealed in a quartz glass ampule at 650 °C for 240 h | 45 |
| Exfoliated MnPX3 (X = S, Se) | Metal powders | Phosphorous, sulfur | Similar to ref. 31, then sonicated in sodium cholate at 750 W for 1 h and collected after centrifugation | 36 |
| Exfoliated FePSe3 | Metal powders | Phosphorous, sulfur | Similar to ref. 31, then sonicated in acetone–water mixture for 4 h and collected after centrifugation | 36 |
| Exfoliated NiPS3 | Metal powders | Phosphorous, sulfur | Similar to ref. 31, then sonicated in CTAB mixture for 10 h and collected after centrifugation | 118 |
| NiPS3/C or CoNiPS3/C | Ni–Co Prussian blue analogue | Phosphorous, sulfur | Prepared Ni–Co PBA placed in a tube furnace for annealing. Synthesised samples were washed with CS2 and HCCl3 | 43 |
| Bulk Cu2P2S6 | Cu2S | P2S5 | Stoichiometric mixture was vacuum sealed in a quartz tube and annealed at 623 K for 24 h | 119 |
| Cu3PS4 | Cu powder | Phosphorous, sulfur | Stoichiometric mixture was vacuum sealed in a quartz tube and annealed at 800 K for one week | 120 |
| Ni0.7Fe0.3PS3/Mxene | Ni(NO3)2·6H2O (≥98.5%), Fe(NO3)3·9H2O (≥98%) | Phosphorous, sulfur | Transitional metal layered hydroxide aqueous solution was mixed with Ti3C2Tx solution, and the mixture was ultra-sonicated for 1 h. The as-obtained nanocomposites were preheated in an Ar atmosphere at 300 °C for 1 h, then mixed with red phosphorus and sulphur powder in a molar ratio of 1:3 and vacuum sealed in a quartz tube for annealing |
44 |
| Metal doped NiPS3 | Metal powders | Phosphorous, sulfur | Sealed under high vacuum and annealed in a quartz glass ampule at 650/750 °C for 120/288 h | 22, 32, 86 and 121 |
Alam et al. investigate the nature of the active sites in CoPS3 and NiPS3 monolayer sheets, which demonstrated the highest activity on the edges compared to that on the plane, similar to the MoS2 phase.122 Detailed analysis of the hydrogen adsorption free energy on each atom of the investigated edge ribbons (Fig. 4h) shows that during both the first and second hydrogen adsorption steps, CoPS3 shows stronger adsorption on both metallic and sulphur/phosphorous sites compared to that of NiPS3. Metallic adsorption sites of NiPS3 show a positive free energy value unlike that for CoPS3, whereas sulphur and phosphorus always interact strongly with adsorbed hydrogen for these two phases. These differences in adsorption sites indicate why the HER for CoPS3 occurs via the Volmer–Tafel step with high coverage of adsorbed hydrogen, while that in NiPS3 happens through Volmer–Heyrovsky with low coverage of adsorbed hydrogen.
Considering the difference in electrocatalytic activity of NiPS3 and CoPS3 and their difference in the adsorption strength of hydrogen, it is important to ask why NiPS3 outperforms CoPS3 in the HER but underperforms that in the OER. This is related to the formation of high valence metallic states such as Co3+/Co4+ during the water oxidation, which strongly absorbs H2O and OH− as well as a higher degree of covalency between the metal and oxygen, which decreases the value of the charge transfer due to the hybridization of 3d and 2p orbitals (Fig. 4i).123
The Xu group studied the Co-doped NiPS3 phase towards hydrogen evolution activity, and Ni0.95Co0.05PS3 nanosheets appeared to be the catalyst with an optimal composition having an overpotential of 71 mV vs. RHE at −10 mA cm−2 and Tafel slope of 77 mV dec−1 at 1.0 M KOH,86 and the Co-doped sample was found to be more electrically conductive than the NiPS3 phase by three orders of magnitude. This agrees with a decreased value of band gap that was previously seen for Co-doped nickel sulphides from ∼1.4/1.2 eV to 0.17/0.08 eV.124 The nickel phosphides show a metallic nature with a metal d-band located near the Fermi level125,126 with phosphorous 3s and 3p states located mostly in the conduction band much lower than the Fermi level. The Co-doping to nickel phosphide phase shifts the nickel 3d band to the Fermi level, which creates a hole state, optimizing the value of free energy for hydrogen adsorption compared to other undoped Co/Ni.127,128 This also corresponds to the lower band gap for the Co-doped NiPS3 phase, e.g., 1.47 eV for CoNiPS3vs. 1.50 eV for NiPS3 eV, which demonstrates excellent HER/OER activity on the N-doped carbon substrate compared to non-doped pure NiPS3 phase.43
Fe-doped NiPS3 phase has been widely studied for electrochemical water splitting, and the Ni0.9Fe0.1PS3 phase demonstrated excellent HER (72 mV vs. RHE at −10 mA cm−2 and Tafel slope of 73 mV dec−1) and OER (329 mV vs. RHE at 20 mA cm−2 and Tafel slope of 69 mV dec−1) in 1 M KOH.32 The same composition demonstrated good HER activity of 196 mV@10 mA cm−2 on MXene nanosheets, while the OER activity was optimized with a relatively larger Fe content of Ni0.7Fe0.3PS3@MXene (282 mV@10 mA cm−2).44 For the HER, it was shown that Fe-doping narrows the band gap of the material, which leads to better charge transfer as well as optimizes the H* binding energy (ΔGH ∼ 0.00 eV).32,129 Meanwhile, the enhancement in the OER activity upon Fe-doping is most likely due to the formation of highly active Ni(Fe)OOH species.32,44,130 The catalyst surface undergoes severe oxidation within an anodic scan, and the metal oxide phase will contribute a major part to the OER performance of the catalyst due to reduced charge trasnfer resistance.118
R. Subbaraman et al. showed that the OH–M2+δ bond is a fundamental descriptor of the HER/OER activity of metal oxide-based electrocatalysts, and this bond strength increases in the following trend Mn > Fe > Co > Ni, which corresponds to a decrease in catalyst reactivity, namely, the reactivity falls in accordance with Ni > Co > Fe > Mn.131 Despite successful NiPS3 doping by Co and Fe, the HER activity was not improved upon Mn doping,22 which might be related to the large band gap of MnPS3 and relatively strong H2O adsorption due to the low position of the LUMO within the conduction band.29,106,132 Apart from that, the MnP phase does not show any active sites with ideal ΔGH (within ±0.2 eV) compared to those of NiP, CoP and FeP analogues.133 The corresponding decrease in OER activity of NiPS3 upon Mn doping is in good agreement with the formation of MnO2 which strengthens the OH–M2+δ bond and hinders the Tafel/Heyrovsky recombination–desorption step. Besides, the Mn doping also increases the level of metal–oxygen covalency due to 3d and 2p hybridization (Ni–O is 2.09 Å vs. Mn–O is 1.92 Å), namely, the 2p hole state near the Fermi level disappears with an increase of Mn content, which leads to stronger interactions with adsorbed intermediates.134 The stronger adsorption character and lower electrical conductivity correlate with increased electron affinity for the Mn4+ state affecting the charge transport.135 This is also evidenced by the increasing dominance of the Volmer step for both HER and OER processes with Mn-doped NiPS3.22 In this sense, the NiPS3 doping with low oxophilic metals, such as Cu, should be considered to further expand the role of oxophilicity on the performance of the NiPS3 electrocatalyst,119,136 especially knowing that a successful synergetic effect can be achieved in their mixed phosphides.136 Also, Cu doping in the NiS@Ni2P phase demonstrated a positive effect on the electrocatalytic HER activity of the nickel-foam-based catalyst.137 The 10 weight % Cu doping in NiPS3 showed a stable photocathodic HER behaviour in 0.5 M H2SO4,138 and the electronic structure of this doped phosphochalcogenide is a p-type semiconductor, which might be beneficial for the electrochemical OER as mentioned above. Cu alloying with sulphur and phosphorus sources can give Cu3PS4 and Cu2P2S6 phases,119 and the DFT calculation for the CuPS3 monolayer revealed its small band gap of 1.63 eV.139 Although, CuPS3 has never been reported for electrochemical water splitting, it will likely be less active than pristine NiPS3; however, the presence of Ni in the CuPS3 phase can give a very low value for hydrogen adsorption energy, e.g., 0.09 eV similar to that of Pt surfaces of −0.09 eV.139 Therefore, it will be interesting to see some work on the synthesis and application of the Cu-doped NiPS3 phase towards the HER and OER.
Last but not least, it might be also useful to consider three or more metal-based MPX3 phases for electrochemical water splitting (Fig. 4j and k) to properly adjust the surface bifunctionality and electronic properties.121,140,141 The electrocatalytic activities of selected metal phosphochalcogenides towards the HER and OER are summarised in Tables 4 and 5.
| Materials | Loading mass, μg cm−2 | j@η, mA cm−2@mV | Electrolyte | Ref. |
|---|---|---|---|---|
| NSs NiPS3 (thickness = 4 nm) | 510 ± 10 | 10@193 | 1.0 M KOH | 32 |
| NSs CoPS3 (thickness = 2.8 ± 0.7 nm) | 350.0a | 10@202.8 | 1.0 M KOH | 121 |
| NSs FePS3 (thickness = 0.7 to 1.7 nm) | 150 | 10@337 | 0.5 M KOH | 142 |
| NSs FePSe3 (thickness = 0.7 to 1.7 nm) | 150 | 10@309 ± 2 | 0.5 M KOH | 101 |
| NSs FePSe3 (thickness = 0.7 to 1.7 nm) | 150 | 10@195 ± 3 | 0.5 M H2SO4 | 101 |
| NSs MnPS3 (thickness = 38.1 ± 11.2 nm) | 141.5 | 10@1090 ± 71 | 1.0 M KOH | 36 |
| NSs MnPS3 (thickness = 38.1 ± 11.2 nm) | 141.5 | 10@835 ± 68 | 0.5 M H2SO4 | 36 |
| NSs MnPSe3 (thickness = 16.7 ± 9.3 nm) | 141.5 | 10@992 ± 56 | 1.0 M KOH | 36 |
| NSs MnPSe3 (thickness = 16.7 ± 9.3 nm) | 141.5 | 10@640 ± 87 | 0.5 M H2SO4 | 36 |
| NSs MnPS3 (thickness = 7.6 nm) | 1250 | 10@1140 | 1.0 M KOH | 22 |
| NSs Co-doped NiPS3 (0% Co doping) (thickness = 6.7 nm) | 510 ± 10 | 10@∼220 | 1.0 M KOH | 86 |
| NSs Co-doped NiPS3 (5% Co doping) (thickness = 6.7 nm) | 510 ± 10 | 10@71 | 1.0 M KOH | 86 |
| NSs Co-doped NiPS3 (9% Co doping) (thickness = 6.7 nm) | 510 ± 10 | 10@145 | 1.0 M KOH | 86 |
| NSs Fe-doped NiPS3 (0% Fe doping) (thickness = 4 nm) | 510 ± 10 | 10@193 | 1.0 M KOH | 32 |
| NSs Fe-doped NiPS3 (5% Fe doping) (thickness = 4 nm) | 510 ± 10 | 10@130 | 1.0 M KOH | 32 |
| NSs Fe-doped NiPS3 (10% Fe doping) (thickness = 4 nm) | 510 ± 10 | 10@72 | 1.0 M KOH | 32 |
| NSs Fe-doped NiPS3 (15% Fe doping) (thickness = 4 nm) | 510 ± 10 | 10@152 | 1.0 M KOH | 32 |
| NSs Mn-doped NiPS3 (0% Mn doping) (thickness = 7.6 nm) | 1250 | 10@320 | 1.0 M KOH | 22 |
| NSs Mn-doped NiPS3 (5% Mn doping) (thickness = 7.6 nm) | 1250 | 10@380 | 1.0 M KOH | 22 |
| NSs Co0.6(VMnNiZn)0.4PS3 (thickness 2.8 nm) | 350.0a | 10@65.9 | 1.0 M KOH | 121 |
| NiPS3/C nanoshape | 300 | 10@∼330 | 1.0 M KOH | 43 |
| CoNiPS3/C nanoshape | 300 | 10@∼140 | 1.0 M KOH | 43 |
| Materials | Loading mass, μg cm−2 | j@η, mA cm−2@mV | Electrolyte | Ref. |
|---|---|---|---|---|
| NSs NiPS3 (thickness = 0.64 nm) | 382 | 10@350 | 0.1 M KOH | 118 |
| NSs CoPS3 | 10@378 | 1.0 M KOH | 143 | |
| NiPS3/C nanoshape | 300 | 30@∼330 | 1.0 M KOH | 43 |
| CoNiPS3/C nanoshape | 30@∼285–250 | 1.0 M KOH | 43 | |
| NSs rGO-FePSe3 | 150 | 10@430 | 0.5 M KOH | 101 |
| Ni0.7Fe0.3PS3@MXene | 250 | 10@282 | 1.0 M KOH | 44 |
| NSs Fe-doped NiPS3 (0% Fe doping) (thickness = 4 nm) | 510 ± 10 | 20@437 | 1.0 M KOH | 32 |
| NSs Fe-doped NiPS3 (5% Fe doping) (thickness = 4 nm) | 510 ± 10 | 20@359 | 1.0 M KOH | |
| NSs Fe-doped NiPS3 (10% Fe doping) (thickness = 4 nm) | 510 ± 10 | 20@329 | 1.0 M KOH | |
| NSs Fe-doped NiPS3 (15% Fe doping) (thickness = 4 nm) | 510 ± 10 | 20@356 | 1.0 M KOH | |
| NSs Mn-doped NiPS3 (0% Mn doping) (thickness = 7.6 nm) | 1250 | 10@295 | 1.0 M KOH | 22 |
| NSs Mn-doped NiPS3 (5% Mn doping) (thickness = 7.6 nm) | 1250 | 10@350 | 1.0 M KOH |
Metal doped nickel chalcogenides (NiaXb, X = S, Se)
Nickel chalcogenides, such as sulphides and selenides, are found in different stoichiometric and non-stoichiometric compositions, namely, NiX, NiX2, Ni3X2, and Ni9X8. Both sulphides and selenides are insoluble in water and stable within neutral and near-neutral pH; also, their surfaces are covered with oxide/hydroxide films under harsh alkaline conditions, making them highly suitable for HER electrocatalysts and OER precatalysts.26 However, these compounds are highly unstable within acidic conditions due to the dissolution of chalcogenides.26 It was found that the HER electrocatalytic activity of nickel selenides changes in the following order Ni3S2 > NiS2 > NiS, which was attributed to the higher electrical conductivity of Ni3S2.144 Meanwhile, the opposite result showed that NiS outperforms the Ni3S2 catalyst,145 which is probably related to different phases of NiS and Ni3S2 having different exposed adsorption sites. As for selenides, NiSe2 was found to be superior to other nickel selenide phases for both the HER and OER in 1.0 KOH.146,147 A more comprehensive review of nickel chalcogenides and their synthesis, including tellurides, as catalysts for electrochemical water splitting, can be found in the literature.26,148 Synthesis methods of some metal chalcogenides are shown in Table 6.| Material | Metal precursors | Source of S/Se | Method | Ref. |
|---|---|---|---|---|
| MX2 (M = Ni, Fe, Co, and X = S, Se) | A metal film by e-beam evaporation | Selenium/sulphur power | Selenium/sulphur power and metal precursor heated with Ar flow in a tube furnace | 149 and 150 |
| Fe–NiS2/CF | Ni(NO3)2·6H2O (0.3 mmol), Fe(NO3)3·9H2O (0.1 mmol) | Sulphur powder | Layered metal hydroxides are grown on a carbon fibre. Then, sulphur power and metal precursor are heated with Ar flow in a tube furnace | 151 |
| FexNi1−xSe2 nanoparticles | Nickel acetylacetonate and iron acetylacetonate | Selenium powder | A mixture of 0.1 mmol of Ni(acac)2, 0.1 mmol of Fe(acac)2 and 0.4 mmol of selenium powder was dissolved in the mixed solution of organic solvents. Then, solvothermal synthesis was performed in the autoclave. Particles were collected by centrifugation | 152 |
| MS (M = Ni, Co, Fe, Mn) | Metal hexacyanoferrate (MHCF) (M = Ni, Mn, Fe, Co) | Na2S | Electrodeposition of metal hexacyanoferrate on the surface of the GC electrode by applying potential from 0 V to 1 V for 20 cycles. Then, the same electrodeposition with 10 mM of Na2S solution | 153 |
| Metal doped NiX/NF (X = S, Se) | Metal salts, nickel foam | Thiourea, selenium powered | Solvothermal synthesis in an autoclave with/without the addition of a reducing agent | 92 and 154 |
Wang et al. estimated the hydrogen evolution activity for different metal dichalcogenides, and it was found that CoX2 has the lowest absolute value of H* adsorption.60 As shown in Fig. 5a and b, Kong et al. demonstrated that the HER activity in 0.5 M H2SO4 decreases in the order of CoX2 > NiX2 > FeX2,149 which is most likely due to a decrease in electrical conductivity in CoX2 > NiX2 > FeX2 and partial occupation of the conduction band in CoX2 and NiX2.155,156 A similar trend for the dichalcogenide phase was observed towards the OER process in 1.0 M KOH on different substrates.151,157,158 Regarding monochalcogenides, NiX and CoX also exhibited superior HER and OER activity in an alkaline medium;153,159,160 however, FeS has also demonstrated superior activity towards the OER process, which might be due to different surface area and roughness factors between the examined metal monosulfides.150,161–163 However, it is important to accurately estimate the active surface area of the catalyst to provide a meaningful comparison of the effect of the chemical nature of metal chalcogenides on their electrocatalytic activity.164,165 The HER activity of both MnS2 and MnS is the lowest compared to other chalcogenides due to weak hydrogen bonding to the surface resulting from the smallest DOS occupation near the Fermi level,153,166,167 and the same conclusion was done towards the OER activity of manganese sulphide, which is most likely due to the strong OH–M2+δ bond (Fig. 5c).131
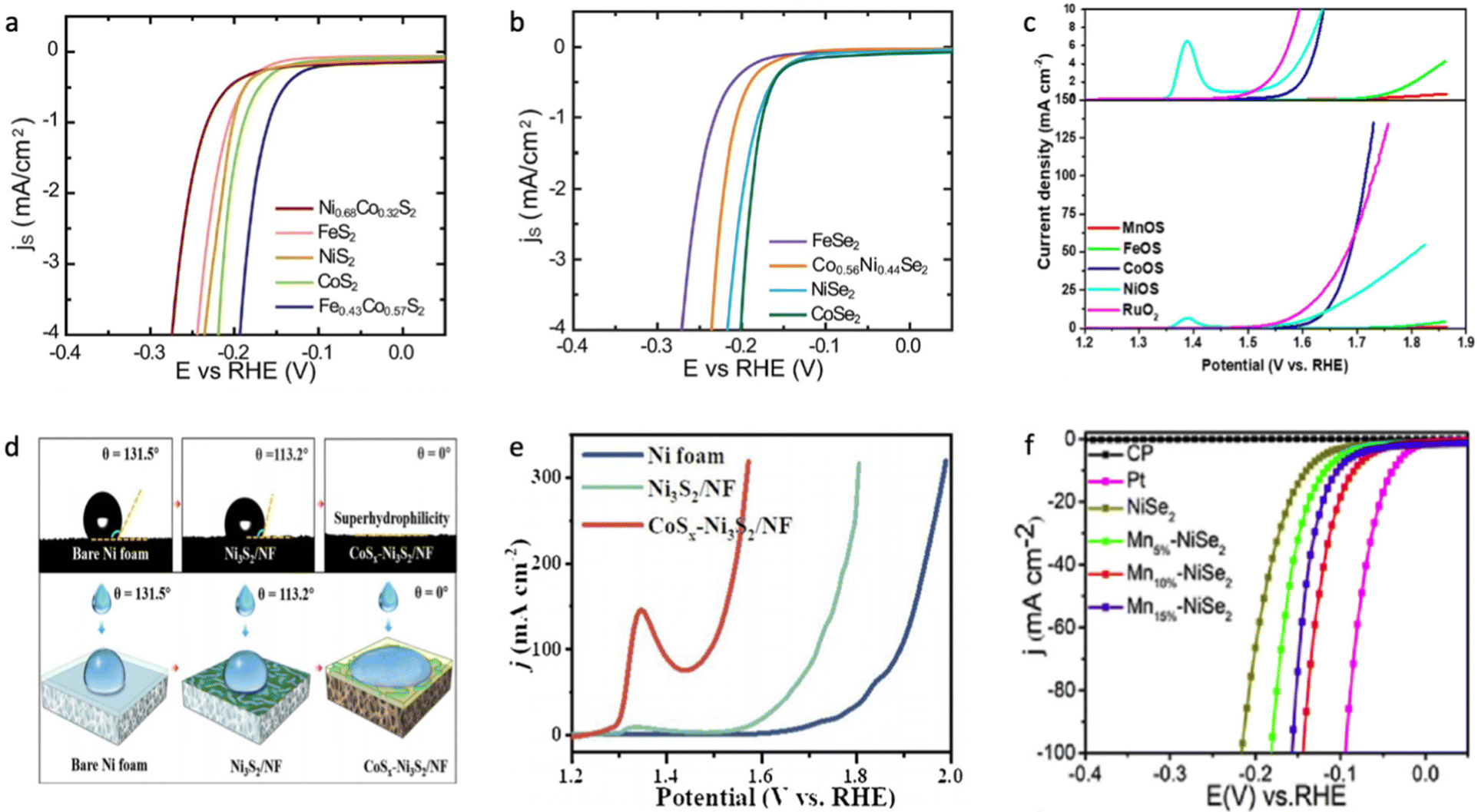 | ||
| Fig. 5 (a) LVS curves of transition metal disulphides in which surface-area-normalized current density is plotted against the potential in 0.5 M H2SO4. (b) LVS curves of transition metal diselenides in which surface-area-normalized current density is plotted against the potential in 0.5 M H2SO4. Reproduced with permission from ref. 149. Copyright © 2013, the Royal Society of Chemistry. (c) LSV for sulphur-containing transition metal (hydro)oxides compared with commercial RuO2 at 1.0 M KOH. Reproduced with permission from ref. 199. Copyright © 2020 Wiley Publications. (d) Contact angles of bare nickel foam (NF), Ni2S3/NF and CoSx–Ni2S3/NF, where CoSx–Ni2S3/NF shows superhydrophilic behaviour. (e) LVS for bare nickel foam (NF), Ni2S3/NF and CoSx–Ni2S3/NF during the OER process in 1.0 M KOH. Reproduced with permission from ref. 172. Copyright 2020 Elsevier Ltd. (f) LSV curves of NiSe, Mn–NiSe, Pt/C and bare carbon fibre paper in 0.5 M H2SO4. Reproduced with permission from ref. 186. Copyright 2020 Elsevier Ltd. | ||
It was shown that Co-doping in NiaXb improves both the HER and OER performance, which is in good agreement with the optimized H* adsorption energy and higher electrical conductivity of CoX2.149,155,168–171 As shown in Fig. 5d and e, surface wettability plays a crucial role in attenuating the HER/OER overpotential, which mitigates near-surface insulating bubble adhesion due to the formation of a very low contact angle at the electrolyte/surface interface.172,173 This phenomenon is significantly influenced by surface oxophilicity, e.g., the ability to connect with oxygen-based species either through adsorption or formation of hydrogen/ionic bonds, which helps to improve the kinetics of the adsorption step.174 However, when comparing any electrocatalyst grown on a 3D substrate it is important to study the electrochemical activity normalized by active surface area, not just by geometric surface area, to avoid misleading observations.164,165 The improved HER performance, resulting from the increase in oxophilic high valence states decreasing the activation energy barrier, was shown for various Fe-doped NiaXb as nanoparticles deposited on the electrode or a composite architecture grown on a conductive substrate.175–179 The excellent OER performance of Fe-doped NiaXb was attributed to the formation of the Ni(Fe)OOH phase, which possesses high water oxidative activity due to the bifunctionality of the surface in the alkaline medium.130,151,158,180–182 It was also shown that even the presence of 1.0 mM Fe3+ impurities in alkaline electrolyte significantly increases the OER activity of the NiOxHy phase.183 This outstanding OER activity was attributed to the formation of a high oxidation Ni and Fe state, where Fe is an actual active site with an unoccupied state at the minimum of the conduction band.183,184 In contrast to the poor HER/OER activity of Mn-doped NiPS3, Mn-doped NiaXb materials show improved electrocatalytic behaviour compared to the non-doped sample (Fig. 5f),185,186 and this is apparently due to the absence of electrocatalytically poor NiMnP and MnP phases.187–189 It was shown that Mn doping in nickel sulphide leads to stronger H2O interaction energy and optimized H* adsorption energy on the catalyst surface, subsequently enhancing the HER performance in neutral and alkaline solution.190 Although, the Mn doping in NiS leads to a large band gap from 2.05 eV for NiS to 2.31 eV for Mn0.5Ni0.5S, respectively.124,191 It was also shown that Mn-doping in nickel-based oxides or chalcogenides shows a worse effect on the electrochemical water splitting performance than Co or Fe doping, which correlates with the poor conductivity and very strong oxophilicity of Mn-doped nickel-based catalysts.192,193 It is likely that the successful Mn doping to nickel-based chalcogenides was just observed due to the overestimation of the active surface area of the electrocatalyst, especially for those samples prepared not through solid-state synthesis, although the positive effect of the oxophilic Mn states in nickel oxides or chalcogenides for the water dissociation step might take place. More investigation in this direction is needed to properly answer this question.
Yin et al. demonstrated that Cu, as well as Co, can improve the HER activity of NiS in 1.0 M KOH, which is due to changes in Ni electronic structure enabling the lower charge transfer resistance,169 namely the doping makes the eg–t2g gap (exited–transition state) for the 3d orbital small enough for fast charge transfer and increases the Ni3+ content for a better adsorption mechanism. The CuNiS composite also demonstrated an outstanding OER activity in alkaline media with stronger H2O adsorption energy for Cu–NiOOH compared to the NiOOH phase.194
Wang et al. calculated ΔGH for various metal-doped NiSe2, and, in general, it was found that on both nickels, selenium, and the dopant site of (001), (110), and (111) plane Fe, Co, and Cu doping improves H* adsorption much better compared to other metals (ΔGH ∼ 0.0 eV).179 Besides, considering a trimetallic composition to optimize electrocatalyst performance again might be an effective strategy.195–198 The electrocatalytic activities of selected metal chalcogenides towards the HER and OER are summarised in Tables 7 and 8.
| Materials | Loading mass, μg cm−2 | j@η, mA cm−2@mV | Electrolyte | Ref. |
|---|---|---|---|---|
| NiS | — | 10@169 | 1.0 M NaOH | 153 |
| CoS | — | 10@251 | 1.0 M NaOH | |
| FeS | — | 10@329 | 1.0 M NaOH | |
| MnS | — | 10@589 | 1.0 M NaOH | |
| NSs NiS2 | — | 10@172 | 1.0 M KOH | 169 |
| NSs Co–NiS2 | — | 10@80 | 1.0 M KOH | |
| NSs Fe–NiS2 | — | 10@192 | 1.0 M KOH | |
| NSs Cu–NiS2 | — | 10@143 | 1.0 M KOH | |
| NSs NiS2 | 200 | 10@249 | 0.5 M H2SO4 | 175 |
| NSs Fe–NiS2 | 200 | 10@121 | 0.5 M H2SO4 | |
| NSs NiSe2/CF | 2000 | 10@207 | 1.0 M KOH | 186 |
| NSs Mn–NiSe2/CF (10% Mn doping) | 2000 | 10@128 | 1.0 M KOH |
| Materials | Loading mass, μg cm−2 | j@η, mA cm−2@mV | Electrolyte | Ref. |
|---|---|---|---|---|
| NiOS/C | 240 | 10@420 | 1.0 M KOH | 199 |
| CoOS/C | 240 | 10@400 | 1.0 M KOH | |
| FeOS/C | 240 | 10@490 | 1.0 M KOH | |
| MnOS/C | 240 | 10@600 | 1.0 M KOH | |
| Fe–NiS2/CF | 1410 | 20@243 | 1.0 M KOH | 151 |
| NiSe2 | 450 | 10@305 | 1.0 M KOH | 152 |
| FeSe2 | 450 | 10@332 | 1.0 M KOH | |
| Ni0.25Fe0.75Se2 | 450 | 10@271 | 1.0 M KOH | |
| Ni0.5Fe0.5Se2 | 450 | 10@235 | 1.0 M KOH | |
| Ni0.75Fe0.25Se2 | 450 | 10@250 | 1.0 M KOH | |
| Ni2S3 | — | 100@450 | 1.0 M KOH | 200 |
| Co–Ni2S3 | — | 100@380 | 1.0 M KOH | |
| Fe–Ni2S3 | — | 100@270 | 1.0 M KOH | |
| Cu–Ni2S3 | — | 100@430 | 1.0 M KOH | |
| NiSe/NF | — | 50@420 | 1.0 M KOH | 201 |
| Co–NiSe/NF | — | 330@420 | 1.0 M KOH | |
| Cu–NiSe/NF | — | 380@420 | 1.0 M KOH |
Electrocatalytic stability
Several processes take place upon electrochemical water splitting, which directly affects the durability of the catalyst; for example, oxidation, elemental leaching, structural distortion and so on. Therefore, the rational design of electrocatalysts should consider the link between the electrocatalytic activity and the chemical stability of materials against degradation. Fig. 6 shows the relationships between electrocatalytic activity and chemical stability of transition metal dichalcogenides (TMDs) with different electronic structures.25 The basal plane of TMDs with metallic character possesses higher electrocatalytic activity (ΔGH ∼ 0 eV) than that of semiconducting materials (ΔGH > 0 eV), and the transition metal nature of metallic TMDs does not influence their activity much compared to that of semiconductors. Accordingly, metal doping for metallic TMDs might not be an effective strategy to optimize the performance of electrocatalysts, as it strongly influences the chemical stability of the material. As shown in Fig. 6b, most of the TMD edge sites are stable against the dissolution of chalcogens into the electrolyte (ΔGHX < 0 eV); therefore, the increase in the number of exposed edge sites of the catalyst is highly desirable to optimize the electrocatalytic activity. The chemical stability of electrocatalysts can be described according to their density of state in Fig. 6a, which shows that semiconductive materials with a higher occupancy in the valence band are more likely to transit electrons to the conduction band, which leads to higher activity and lower chemical stability. For the metallic TMDs, the occupancy near the Fermi level as well as the vacancy of unoccupied orbitals plays the determining role in the catalyst stability. Additionally, the covalency of the metal–anion bond has to be taken into account to predict possible changes in oxidation states and phase transition during electrochemical water splitting; however, a compromise between the structural stability and high electrocatalytic activity of the material should be found.134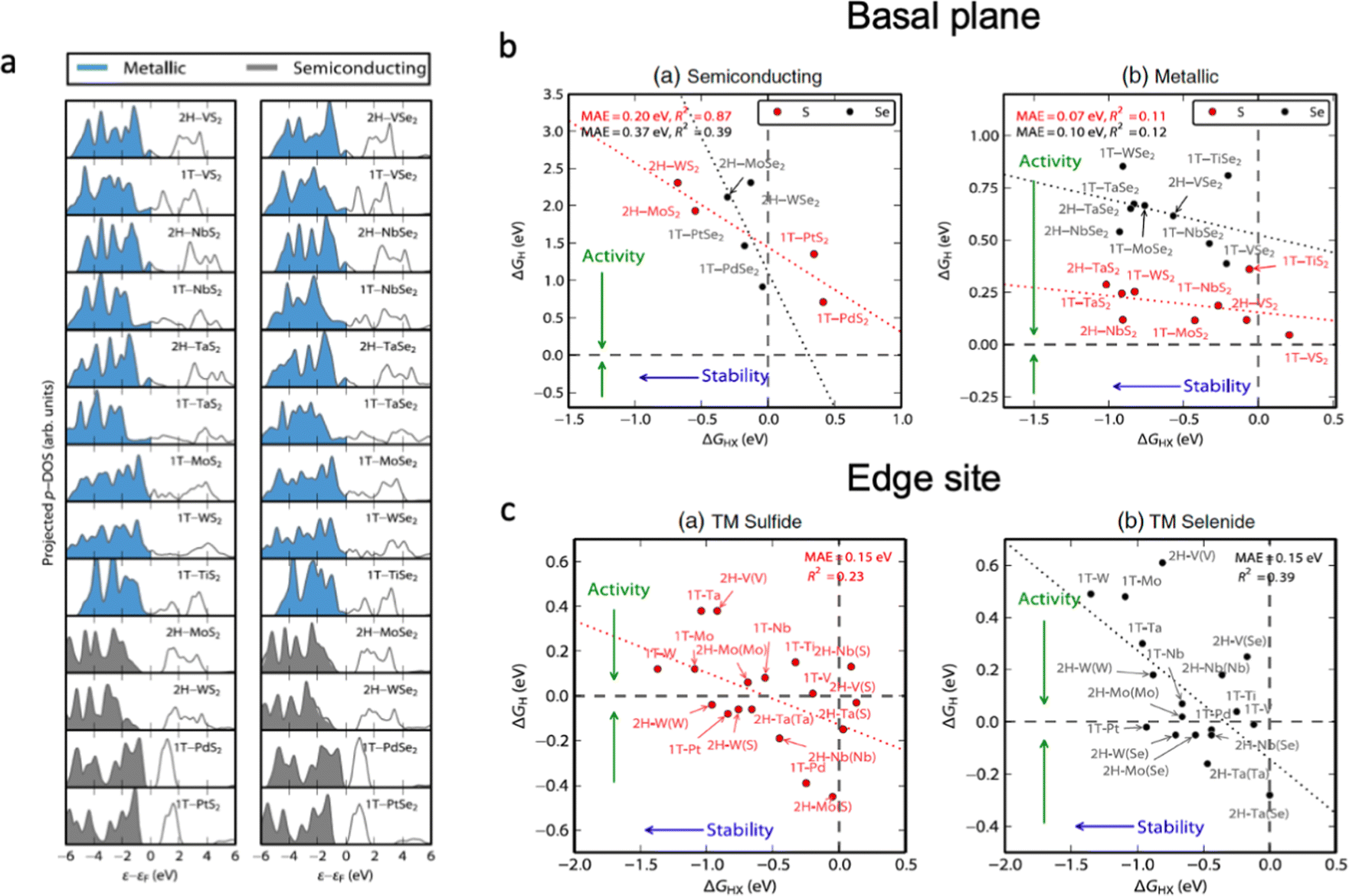 | ||
| Fig. 6 (a) Projected density of states on the chalcogen atom (S or Se) for TMDs relative to the Fermi level, Ef. The blue densities of states indicate that the basal plane is metallic, whereas the grey ones are semiconducting. (b) Semiconducting single-layered TMD basal planes. The plot of the hydrogen adsorption free energy, ΔGH, as a function of the HX adsorption free energy, ΔGHX (X = S or Se). (c) The differential hydrogen adsorption free energy ΔGH as a function of the HX adsorption free energy at a corrosion resistance of 10−6 bar H2X (X = S or Se). The dashed lines indicate ΔGH = 0 eV and ΔGHX = 0 eV. The data span a narrow range of ΔGH and the mean absolute errors are small, but the R2 values are low due to the significant scatter. Reproduced with permission from ref. 25. Copyright 2015 Elsevier Ltd. | ||
The mechanical damage caused by involving gas bubbles on the surface of the electrocatalyst is another problem, as the gas departure induces an opposite force towards the surface, which is proportional to the size of the bubble.50,202,203 This might significantly influence the morphology of the electrocatalyst and its electrocatalytic stability; therefore, a super hydrophilic surface design with mechanically stable morphology, e.g., preferably flat, should take place to facilitate gas detachment upon wetting of the nanopores. Other methods which can be utilized to enhance bubble detachment, such as the application of an external magnetic, ultrasonic fields, etc., are thoughtfully discussed in the literature.50
Summary and perspectives
Electrochemical water splitting has attracted enormous attention in recent years due to the rise of the fuel cell-based industry and global concern about climate change. Transition metal van der Waals materials, such as chalcogenides and phosphochalcogenides, have demonstrated a substantial electrocatalytic performance, which might be comparable with noble metal-based electrocatalysts upon surface defect engineering, high surface area morphology, and structure modification. Transition metal doping seems to be one of the best strategies to further enhance the performance of the host phase through overall electrical conductivity changes, bifunctionality of the surface, aerophobicity (hydrophilicity), etc.; however, this approach stills lacks a fundamental theory of “why and how” that can guide rational electrocatalyst design.This review attempts to summarize recent advances in metal doping (Co, Fe, Mn, Cu) of nickel-based chalcogenides (NiaXb, X = S and Se) and phosphochalcogenides (NiPX3, X = S and Se) as a catalyst for electrochemical water splitting. This literature briefly introduced the main concepts of electrochemical water splitting as well as transition metal chalcogenides and phosphochalcogenides as a family of electrocatalysts. We explained the effect of anion nature on the electrocatalytic activity as well as its stability against leaching or oxidation. It is found that the relationship between electronic structure and electrocatalytic activity/stability for metal-doped nickel-based chalcogenides (NiaXb, X = S and Se) and phosphochalcogenides (NiPX3, X = S and Se) is not well conceptualized. The current practice to improve the catalyst activity/stability possesses an arbitrary character that relies on empirical experience to optimize surface wettability, bifunctionality, electric conductivity, etc. Here, we show that doping of considered metals (Mn, Fe, Co, Cu) can improve the electrocatalytic behaviour of NiaXb either due to bifunctionality, e.g., facile water dissociation step, and/or improving electrical conductivity except for Mn-doping, whereas doping in (NiPX3, X = S and Se) shows more complex behaviour. Namely, the electrocatalytic activity of NiPS3 is proportional to the activity of two different phases, e.g., NixSy and NixPy. The metal phosphide phase is more electrocatalytically active than the chalcogenide phase, and MnP is the least active phase amongst its counterparts, which results in Mn doping having a deteriorating impact on the HER/OER activity of (NiPX3, X = S and Se) materials. Other considered doping metals (Co, Fe, Cu) except Mn unanimously demonstrated either success or promise for effective electrochemical water splitting with both chalcogenides and phosphochalcogenides. However, the doping ratio should be considered carefully to adjust the surface bifunctionality as well as the conductivity of the material. Mn doping for electrochemical water splitting should be considered very carefully over such doping as Cu, Co, and Fe in nickel (phospho)chalcogenides/oxides phases; this is due to a very high difference in the electron affinity and oxophilicity between Mn and Ni sites, which might lead to poor charge transport and dominant adsorption behavior during electrochemical water splitting.
This review explains the relationships between computational data for the electronic structure and experimental electrocatalytic changes in nickel-based chalcogenide and phosphochalcogenide electrocatalysts upon transition metal doping. Apart from the general suggestion to consider a larger variety of doping metals as well as to use more than one metal for doping to properly adjust the bifunctionality and electrical conductivity of the material, some gaps in the systematization of the current results should be considered for accurate comparison.
On the theoretical side, more systematic studies are needed to fulfil the gap between the correlation of electronic structure and electrocatalytic performance of transition metal-doped NiaXb and NiPX3. When describing the free energy of the electrocatalyst during HER/OER processes, it is important to report the energy of the CB/VB regarding the vacuum reference to accurately compare it with SHE potential at the same scale. Electronic structure studies should consider a higher level of density functional theory than a local-density and generalized gradient approximation, such as Hubbard correction approaches, to accurately represent the density of states profile for semiconductive phases. More attention should be dedicated to studies of the thermodynamic stability of metal-doped NiaXb and NiPX3 by accurate comparison of their band gap and referenced position of the Fermi level, as well as the role of electrolyte species on the thermodynamics of the conversion reaction. Picosecond scale ab initio molecular dynamic approaches will be highly useful for this purpose.
On the experimental side, a broader variety of metals should be considered for doping of NiaXb and NiPX3 within the same conditions for comparison, including the accurate correction of their active surface area. A commonly accepted strategy, to increase the surface area of electrocatalysts through morphology changes, makes it more complicated to understand the true nature of the electrocatalytic activity/stability of the material, especially for foam-based electrocatalysts, as their electrochemical data corrected onto a geometric surface area might overestimate the real activity. The active surface area and active surface area normalized voltammetry data should be reported to accurately describe the trend in intrinsic activity and stability of different metal-doped nickel-based catalysts. It is compulsory to use a fixed metal doping ratio whenever accurate data for different metal-doped NiaXb and NiPX3 is reported. Apart from that, it is preferably to compare as-synthesized crystals with the same dominant facet structure in order to avoid contribution of polycrystallinity in the electrocatalysts. In this regard, it makes sense to conduct more fundamental experimental studies on metal doping in nickel-based (phospho)chalcogenide phases for bulk materials with the same symmetry of crystals or preferably in single crystal form. It will also be important to conduct additional studies for Cu, Ni, Co, Fe and Mn-based (phospho)chalcogenides/oxides, to understand the maximum difference in the oxophilicity of transition metals, which can enable efficient charge transport and bifunctionality in these systems. New precise experimental methods, as well as real-time testing to detect the true electrocatalytic activity and stability of the active site of the material, should be considered more often. A phase transformation upon metal doping and/or applied current might take place; therefore, the precise structure with its physical–chemical properties of newly formed phases (oxides, etc.) need to be examined carefully, at least using chelating agents, along with electrochemical testing which selectively reacts with metals in a particular oxidation state or more direct synchrotron-based techniques.
Conflicts of interest
The author declares no competing financial interest.References
- T. Ahmad and D. Zhang, Energy Rep., 2020, 6, 1973–1991 CrossRef.
- N. Shen, R. Deng, H. Liao and O. Shevchuk, Util. Policy, 2020, 64, 101055 CrossRef.
- A. Ugurlu and S. Oztuna, Int. J. Hydrogen Energy, 2015, 40, 11178–11188 CrossRef CAS.
- X. Zou and Y. Zhang, Chem. Soc. Rev., 2015, 44, 5148–5180 RSC.
- J. Zhu, L. Hu, P. Zhao, L. Y. S. Lee and K.-Y. Wong, Chem. Rev., 2020, 120, 851–918 CrossRef CAS PubMed.
- M. Tahir, L. Pan, F. Idrees, X. Zhang, L. Wang, J.-J. Zou and Z. L. Wang, Nano Energy, 2017, 37, 136–157 CrossRef CAS.
- B. Xiong, L. Chen and J. Shi, ACS Catal., 2018, 8, 3688–3707 CrossRef CAS.
- Y. Wang, B. Kong, D. Zhao, H. Wang and C. Selomulya, Nano Today, 2017, 15, 26–55 CrossRef CAS.
- M. Gong, D.-Y. Wang, C.-C. Chen, B.-J. Hwang and H. Dai, Nano Res., 2016, 9, 28–46 CrossRef CAS.
- M. Gong and H. Dai, Nano Res., 2015, 8, 23–39 CrossRef CAS.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- S. Klaus, Y. Cai, M. W. Louie, L. Trotochaud and A. T. Bell, J. Phys. Chem. C, 2015, 119, 7243–7254 CrossRef CAS.
- C. Spöri, J. T. H. Kwan, A. Bonakdarpour, D. P. Wilkinson and P. Strasser, Angew. Chem., Int. Ed., 2017, 56, 5994–6021 CrossRef PubMed.
- P. Wang, M. Yan, J. Meng, G. Jiang, L. Qu, X. Pan, J. Z. Liu and L. Mai, Nat. Commun., 2017, 8, 645 CrossRef PubMed.
- J. Zaffran, M. B. Stevens, C. D. M. Trang, M. Nagli, M. Shehadeh, S. W. Boettcher and M. Caspary Toroker, Chem. Mater., 2017, 29, 4761–4767 CrossRef CAS.
- J. A. Arminio-Ravelo, A. W. Jensen, K. D. Jensen, J. Quinson and M. Escudero-Escribano, ChemPhysChem, 2019, 20, 2956–2963 CrossRef CAS PubMed.
- F. Lu, M. Zhou, Y. Zhou and X. Zeng, Small, 2017, 13, 1701931 CrossRef PubMed.
- A. Ciechan, M. J. Winiarski and M. Samsel-Czekała, Intermetallics, 2013, 41, 44–50 CrossRef CAS.
- J. R. Swierk, S. Klaus, L. Trotochaud, A. T. Bell and T. D. Tilley, J. Phys. Chem. C, 2015, 119, 19022–19029 CrossRef CAS.
- J. Jiang, F. Sun, S. Zhou, W. Hu, H. Zhang, J. Dong, Z. Jiang, J. Zhao, J. Li, W. Yan and M. Wang, Nat. Commun., 2018, 9, 2885 CrossRef PubMed.
- W. Gao, M. Yan, H.-Y. Cheung, Z. Xia, X. Zhou, Y. Qin, C.-Y. Wong, J. C. Ho, C.-R. Chang and Y. Qu, Nano Energy, 2017, 38, 290–296 CrossRef CAS.
- D. Rakov, Y. Li, S. Niu and P. Xu, J. Alloys Compd., 2018, 769, 532–538 CrossRef CAS.
- D. Zhou, Z. Cai, Y. Jia, X. Xiong, Q. Xie, S. Wang, Y. Zhang, W. Liu, H. Duan and X. Sun, Nanoscale Horiz., 2018, 3, 532–537 RSC.
- Y. Wang, R. Zhang, J. Li, L. Li and S. Lin, Nanoscale Res. Lett., 2014, 9, 46 CrossRef PubMed.
- C. Tsai, K. Chan, J. K. Nørskov and F. Abild-Pedersen, Surf. Sci., 2015, 640, 133–140 CrossRef CAS.
- S. Anantharaj, S. Kundu and S. Noda, J. Mater. Chem. A, 2020, 8, 4174–4192 RSC.
- J. Luxa, Š. Cintl, L. Spejchalová, J.-Y. Lin and Z. Sofer, ACS Appl. Energy Mater., 2020, 3, 11992–11999 CrossRef CAS.
- J. Wang, X. Li, B. Wei, R. Sun, W. Yu, H. Y. Hoh, H. Xu, J. Li, X. Ge, Z. Chen, C. Su and Z. Wang, Adv. Funct. Mater., 2020, 30, 1908708 CrossRef CAS.
- F. Wang, T. A. Shifa, P. Yu, P. He, Y. Liu, F. Wang, Z. Wang, X. Zhan, X. Lou, F. Xia and J. He, Adv. Funct. Mater., 2018, 28, 1802151 CrossRef.
- A. P. Tiwari, D. Kim, Y. Kim, O. Prakash and H. Lee, Nano Energy, 2016, 28, 366–372 CrossRef CAS.
- C. C. Mayorga-Martinez, Z. Sofer, D. Sedmidubský, Š. Huber, A. Y. S. Eng and M. Pumera, ACS Appl. Mater. Interfaces, 2017, 9, 12563–12573 CrossRef CAS PubMed.
- B. Song, K. Li, Y. Yin, T. Wu, L. Dang, M. Cabán-Acevedo, J. Han, T. Gao, X. Wang, Z. Zhang, J. R. Schmidt, P. Xu and S. Jin, ACS Catal., 2017, 7, 8549–8557 CrossRef CAS.
- Z. Liang, Z. Yang, J. Dang, J. Qi, H. Yuan, J. Gao, W. Zhang, H. Zheng and R. Cao, Chem. – Eur. J., 2019, 25, 621–626 CAS.
- S. Wang, B. Xiao, S. Shen, K. Song, Z. Lin, Z. Wang, Y. Chen and W. Zhong, Nanoscale, 2020, 12, 14459–14464 RSC.
- J. Chang, G. Wang, A. Belharsa, J. Ge, W. Xing and Y. Yang, Small Methods, 2020, 4, 1900632 CrossRef CAS.
- R. Gusmão, Z. Sofer and M. Pumera, Adv. Funct. Mater., 2019, 29, 1805975 CrossRef.
- J. Yu, W.-J. Li, H. Zhang, F. Zhou, R. Li, C.-Y. Xu, L. Zhou, H. Zhong and J. Wang, Nano Energy, 2019, 57, 222–229 CrossRef CAS.
- R. N. Jenjeti, R. Kumar and S. Sampath, J. Mater. Chem. A, 2019, 7, 14545–14551 RSC.
- A. Hashemi, H.-P. Komsa, M. Puska and A. V. Krasheninnikov, J. Phys. Chem. C, 2017, 121, 27207–27217 CrossRef CAS.
- Y. V. Kuzminskii, B. M. Voronin and N. N. Redin, J. Power Sources, 1995, 55, 133–141 CrossRef CAS.
- Y. Sang, L. Wang, X. Cao, G. Ding, Y. Ding, Y. Hao, N. Xu, H. Yu, L. Li and S. Peng, J. Alloys Compd., 2020, 831, 154775 CrossRef CAS.
- Q. Liang, Y. Zheng, C. Du, Y. Luo, J. Zhang, B. Li, Y. Zong and Q. Yan, Small Methods, 2017, 1, 1700304 CrossRef.
- Q. Liang, L. Zhong, C. Du, Y. Zheng, Y. Luo, J. Xu, S. Li and Q. Yan, Adv. Funct. Mater., 2018, 28, 1805075 CrossRef.
- C.-F. Du, K. N. Dinh, Q. Liang, Y. Zheng, Y. Luo, J. Zhang and Q. Yan, Adv. Energy Mater., 2018, 8, 1801127 CrossRef.
- R. Gusmão, Z. Sofer, D. Sedmidubský, Š. Huber and M. Pumera, ACS Catal., 2017, 7, 8159–8170 CrossRef.
- R. Samal, G. Sanyal, B. Chakraborty and C. S. Rout, J. Mater. Chem. A, 2021, 9, 2560–2591 RSC.
- M. Zhu, H. Kou, K. Wang, H. Wu, D. Ding, G. Zhou and S. Ding, Mater. Horiz., 2020, 7, 3131–3160 RSC.
- A. C. Garcia, T. Touzalin, C. Nieuwland, N. Perini and M. T. M. Koper, Angew. Chem., Int. Ed., 2019, 58, 12999–13003 CrossRef CAS PubMed.
- N. Mahmood, Y. Yao, J.-W. Zhang, L. Pan, X. Zhang and J.-J. Zou, Adv. Sci., 2018, 5, 1700464 CrossRef PubMed.
- G. B. Darband, M. Aliofkhazraei and S. Shanmugam, Renewable Sustainable Energy Rev., 2019, 114, 109300 CrossRef CAS.
- D. Strmcnik, M. Uchimura, C. Wang, R. Subbaraman, N. Danilovic, D. van der Vliet, A. P. Paulikas, V. R. Stamenkovic and N. M. Markovic, Nat. Chem., 2013, 5, 300–306 CrossRef CAS PubMed.
- M. Görlin, M. Gliech, J. F. de Araújo, S. Dresp, A. Bergmann and P. Strasser, Catal. Today, 2016, 262, 65–73 CrossRef.
- T. Reier, H. N. Nong, D. Teschner, R. Schlögl and P. Strasser, Adv. Energy Mater., 2017, 7, 1601275 CrossRef.
- C. C. L. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef CAS PubMed.
- B. Zhang, X. Zheng, O. Voznyy, R. Comin, M. Bajdich, M. García-Melchor, L. Han, J. Xu, M. Liu, L. Zheng, F. P. García de Arquer, C. T. Dinh, F. Fan, M. Yuan, E. Yassitepe, N. Chen, T. Regier, P. Liu, Y. Li, P. De Luna, A. Janmohamed, H. L. Xin, H. Yang, A. Vojvodic and E. H. Sargent, Science, 2016, 352, 333–337 CrossRef CAS PubMed.
- L. D. Wickramasinghe, R. Zhou, R. Zong, P. Vo, K. J. Gagnon and R. P. Thummel, J. Am. Chem. Soc., 2015, 137, 13260–13263 CrossRef CAS PubMed.
- Y. H. Wang, S. Zheng, W. M. Yang, R. Y. Zhou, Q. F. He, P. Radjenovic, J. C. Dong, S. Li, J. Zheng, Z. L. Yang, G. Attard, F. Pan, Z. Q. Tian and J. F. Li, Nature, 2021, 600, 81–85 CrossRef CAS PubMed.
- M. S. Burke, L. J. Enman, A. S. Batchellor, S. Zou and S. W. Boettcher, Chem. Mater., 2015, 27, 7549–7558 CrossRef CAS.
- D. Mattia, M. P. Rossi, B. M. Kim, G. Korneva, H. H. Bau and Y. Gogotsi, J. Phys. Chem. B, 2006, 110, 9850–9855 CrossRef CAS PubMed.
- J. Wang, J. Liu, B. Zhang, X. Ji, K. Xu, C. Chen, L. Miao and J. Jiang, Phys. Chem. Chem. Phys., 2017, 19, 10125–10132 RSC.
- W. T. Hong, M. Risch, K. A. Stoerzinger, A. Grimaud, J. Suntivich and Y. Shao-Horn, Energy Environ. Sci., 2015, 8, 1404–1427 RSC.
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- Y. Matsumoto and E. Sato, Mater. Chem. Phys., 1986, 14, 397–426 CrossRef CAS.
- P. Sen and R. K. Chouhan, Electron. Struct., 2020, 2, 25003 CrossRef CAS.
- J. Jin, J. Yin, H. Liu, B. Huang, Y. Hu, H. Zhang, M. Sun, Y. Peng, P. Xi and C. Yan, Angew. Chem., 2021, 133, 14236–14242 CrossRef.
- J. Yin, J. Jin, M. Lu, B. Huang, H. Zhang, Y. Peng, P. Xi and C.-H. Yan, J. Am. Chem. Soc., 2020, 142, 18378–18386 CrossRef CAS PubMed.
- D. Jia, L. Han, Y. Li, W. He, C. Liu, J. Zhang, C. Chen, H. Liu and H. L. Xin, J. Mater. Chem. A, 2020, 8, 18207–18214 RSC.
- J. Wang, A. Yang, J. Li, K. Su, Y. Tang and X. Qiu, Appl. Catal., B, 2022, 300, 120727 CrossRef CAS.
- Y. Ge, S.-P. Gao, P. Dong, R. Baines, P. M. Ajayan, M. Ye and J. Shen, Nanoscale, 2017, 9, 5538–5544 RSC.
- A. Long, W. Li, M. Zhou, W. Gao, B. Liu, J. Wei, X. Zhang, H. Liu, Y. Liu and X. Zeng, J. Mater. Chem. A, 2019, 7, 21514–21522 RSC.
- J. You, C. Si, J. Zhou and Z. Sun, J. Phys. Chem. C, 2019, 123, 3719–3726 CrossRef CAS.
- S. R. Kadam, A. N. Enyashin, L. Houben, R. Bar-Ziv and M. Bar-Sadan, J. Mater. Chem. A, 2020, 8, 1403–1416 RSC.
- X. Chia, N. A. A. Sutrisnoh, Z. Sofer, J. Luxa and M. Pumera, Chem. – Eur. J., 2018, 24, 3199–3208 CrossRef CAS PubMed.
- Y. Song, X. Wang and W. Mi, Phys. Rev. Mater., 2017, 1, 74408 CrossRef.
- H. B. Tao, L. Fang, J. Chen, H. Bin Yang, J. Gao, J. Miao, S. Chen and B. Liu, J. Am. Chem. Soc., 2016, 138, 9978–9985 CrossRef CAS PubMed.
- E. Fabbri, A. Habereder, K. Waltar, R. Kötz and T. J. Schmidt, Catal. Sci. Technol., 2014, 4, 3800–3821 RSC.
- A. J. Bard and L. R. Faulkner, Fundamentals and Applications, Springer, New York, Wiley, 2002 Search PubMed.
- G. Fu, X. Wen, S. Xi, Z. Chen, W. Li, J.-Y. Zhang, A. Tadich, R. Wu, D.-C. Qi, Y. Du, J. Cheng and K. H. L. Zhang, Chem. Mater., 2019, 31, 419–428 CrossRef CAS.
- A. B. Laursen, A. S. Varela, F. Dionigi, H. Fanchiu, C. Miller, O. L. Trinhammer, J. Rossmeisl and S. Dahl, J. Chem. Educ., 2012, 89, 1595–1599 CrossRef CAS.
- A. J. Medford, A. Vojvodic, J. S. Hummelshøj, J. Voss, F. Abild-Pedersen, F. Studt, T. Bligaard, A. Nilsson and J. K. Nørskov, J. Catal., 2015, 328, 36–42 CrossRef CAS.
- H. L. Lord, W. Zhan and J. Pawliszyn, Fundamentals and applications of needle trap devices, 2012, vol. 2 Search PubMed.
- W. Luc, Z. Jiang, J. G. Chen and F. Jiao, ACS Catal., 2018, 8, 9327–9333 CrossRef CAS.
- Q. Sun, Y. Dong, Z. Wang, S. Yin and C. Zhao, Small, 2018, 14, 1704137 CrossRef PubMed.
- M. K. Kundu, R. Mishra, T. Bhowmik and S. Barman, J. Mater. Chem. A, 2018, 6, 23531–23541 RSC.
- D. Strmcnik, P. P. Lopes, B. Genorio, V. R. Stamenkovic and N. M. Markovic, Nano Energy, 2016, 29, 29–36 CrossRef CAS.
- K. Li, D. Rakov, W. Zhang and P. Xu, Chem. Commun., 2017, 53, 8199–8202 RSC.
- N. Q. Tran, V. Q. Bui, H. M. Le, Y. Kawazoe and H. Lee, Adv. Energy Mater., 2018, 8, 1702139 CrossRef.
- B. Kim, T. Kim, K. Lee and J. Li, ChemElectroChem, 2020, 7, 3578–3589 CrossRef CAS.
- Z. Fang, L. Peng, Y. Qian, X. Zhang, Y. Xie, J. J. Cha and G. Yu, J. Am. Chem. Soc., 2018, 140, 5241–5247 CrossRef CAS PubMed.
- Y. Li, Y. Gao, S. Yang, C. Wu and Y. Tan, J. Mater. Chem. A, 2020, 8, 12013–12027 RSC.
- J. Hu, A. Al-Salihy, J. Wang, X. Li, Y. Fu, Z. Li, X. Han, B. Song and P. Xu, Adv. Sci., 2021, 8, 2103314 CrossRef CAS PubMed.
- D. Rakov, C. Sun, Z. Lu, S. Li and P. Xu, Adv. Energy Sustainable Res., 2021, 2, 2100071 CrossRef.
- X. Xu, F. Song and X. Hu, Nat. Commun., 2016, 7, 1–7 Search PubMed.
- T. X. Nguyen, Y. Su, C. Lin and J. Ting, Adv. Funct. Mater., 2021, 31, 2106229 CrossRef CAS.
- O. Mabayoje, A. Shoola, B. R. Wygant and C. B. Mullins, ACS Energy Lett., 2016, 1, 195–201 CrossRef CAS.
- Q. Chen, L. Huang, Q. Kong, X. An, X. Wu, W. Yao and C. Sun, J. Electroanal. Chem., 2022, 907, 116047 CrossRef CAS.
- C.-Z. Yuan, Y.-F. Jiang, Z. Wang, X. Xie, Z.-K. Yang, A. Bin Yousaf and A.-W. Xu, J. Mater. Chem. A, 2016, 4, 8155–8160 RSC.
- Z. Liu, G. Zhang, K. Zhang, H. Liu and J. Qu, ACS Sustainable Chem. Eng., 2018, 6, 7206–7211 CrossRef CAS.
- V. Grasso, S. Santangelo and M. Piacentini, Solid State Ionics, 1986, 20, 9–15 CrossRef CAS.
- Y. Ohno and K. Hirama, J. Solid State Chem., 1986, 63, 258–266 CrossRef CAS.
- D. Mukherjee, P. M. Austeria and S. Sampath, ACS Appl. Energy Mater., 2018, 1, 220–231 CrossRef CAS.
- J. Yang, Y. Zhou, Q. Guo, Y. Dedkov and E. Voloshina, RSC Adv., 2020, 10, 851–864 RSC.
- Z. Liu, Y. Wang, R. Chen, C. Chen, H. Yang, J. Ma, Y. Li and S. Wang, J. Power Sources, 2018, 403, 90–96 CrossRef CAS.
- J. Zhuo, M. Cabán-Acevedo, H. Liang, L. Samad, Q. Ding, Y. Fu, M. Li and S. Jin, ACS Catal., 2015, 5, 6355–6361 CrossRef CAS.
- G.-F. Chen, T. Y. Ma, Z.-Q. Liu, N. Li, Y.-Z. Su, K. Davey and S.-Z. Qiao, Adv. Funct. Mater., 2016, 26, 3314–3323 CrossRef CAS.
- V. Zhukov, S. Alvarez and D. Novikov, J. Phys. Chem. Solids, 1996, 57, 647–652 CrossRef CAS.
- B. Qiu, L. Cai, Y. Wang, Z. Lin, Y. Zuo, M. Wang and Y. Chai, Adv. Funct. Mater., 2018, 28, 1706008 CrossRef.
- S. Fu, C. Zhu, J. Song, M. H. Engelhard, X. Li, D. Du and Y. Lin, ACS Energy Lett., 2016, 1, 792–796 CrossRef CAS.
- S. Shanmugam, A. Sivanantham, M. Matsunaga, U. Simon and T. Osaka, Electrochim. Acta, 2019, 297, 749–754 CrossRef CAS.
- S. Y. Kim, T. Y. Kim, L. J. Sandilands, S. Sinn, M.-C. Lee, J. Son, S. Lee, K.-Y. Choi, W. Kim and B.-G. Park, Phys. Rev. Lett., 2018, 120, 136402 CrossRef CAS PubMed.
- C.-H. Ho, T.-Y. Hsu and L. C. Muhimmah, npj 2D Mater. Appl., 2021, 5, 1–10 CrossRef.
- M. Yan, Y. Jin, Z. Wu, A. Tsaturyan, A. Makarova, D. Smirnov, E. Voloshina and Y. Dedkov, J. Phys. Chem. Lett., 2021, 12, 2400–2405 CrossRef CAS PubMed.
- N. Kurita and K. Nakao, J. Phys. Soc. Jpn., 1989, 58, 232–243 CrossRef CAS.
- Y.-Z. Xu, C.-Z. Yuan, Z.-W. Liu and X.-P. Chen, Catal. Sci. Technol., 2018, 8, 128–133 RSC.
- C.-F. Du, N. D. Khang, Q. Liang, Y. Zheng, Y. Luo, J. Zhang and Q. Yan, Adv. Energy Mater., 2018, 8, 1801127, DOI:10.1002/aenm.201801127.
- A. Jawaid, D. Nepal, K. Park, M. Jespersen, A. Qualley, P. Mirau, L. F. Drummy and R. A. Vaia, Chem. Mater., 2016, 28, 337–348 CrossRef CAS.
- E. D. Grayfer, M. N. Kozlova and V. E. Fedorov, Adv. Colloid Interface Sci., 2017, 245, 40–61 CrossRef CAS PubMed.
- B. Konkena, J. Masa, A. J. R. Botz, I. Sinev, W. Xia, J. Koßmann, R. Drautz, M. Muhler and W. Schuhmann, ACS Catal., 2017, 7, 229–237 CrossRef CAS.
- A. Kuhn, L. M. Schoop, R. Eger and B. V. Lotsch, Z. Anorg. Allg. Chem., 2016, 642, 356–360 CrossRef CAS.
- D. Tripathy and S. Sampath, J. Power Sources, 2020, 478, 229066 CrossRef CAS.
- R. Wang, J. Huang, X. Zhang, J. Han, Z. Zhang, T. Gao, L. Xu, S. Liu, P. Xu and B. Song, ACS Nano, 2022, 16, 3593–3603 CrossRef CAS PubMed.
- K. Alam, T. Das, S. Chakraborty and P. Sen, Phys. Chem. Chem. Phys., 2021, 23, 23967–23977 RSC.
- H. Sun, X. Xu, Y. Song, W. Zhou and Z. Shao, Adv. Funct. Mater., 2021, 31, 2009779 CrossRef CAS.
- Z. Xiao, L. Yan, Q. Hu, B. Xiang, Y. Wang, J. Hao, X. Zou, W. Li and S. Wei, Dalton Trans., 2022, 51, 8318–8326 RSC.
- R. E. N. Jun, J.-G. Wang, J.-F. Li and Y.-W. Li, J. Fuel Chem. Technol., 2007, 35, 458–464 CrossRef.
- D. Zhang, X. Zhou, J. Liu, L. Dong, J. Zhao, Y. Xie and W. Sun, Mater. Sci. Eng., B, 2021, 273, 115389 CrossRef CAS.
- H. Liang, A. N. Gandi, D. H. Anjum, X. Wang, U. Schwingenschlögl and H. N. Alshareef, Nano Lett., 2016, 16, 7718–7725 CrossRef CAS PubMed.
- D. Liu, H. Ai, M. Chen, P. Zhou, B. Li, D. Liu, X. Du, K. H. Lo, K. Ng and S. Wang, Small, 2021, 17, 2007557 CrossRef CAS PubMed.
- Q. Liang, L. Zhong, C. Du, Y. Luo, Y. Zheng, S. Li and Q. Yan, Nano Energy, 2018, 47, 257–265 CrossRef CAS.
- F. Dionigi and P. Strasser, Adv. Energy Mater., 2016, 6, 1600621 CrossRef.
- R. Subbaraman, D. Tripkovic, K.-C. Chang, D. Strmcnik, A. P. Paulikas, P. Hirunsit, M. Chan, J. Greeley, V. Stamenkovic and N. M. Markovic, Nat. Mater., 2012, 11, 550–557 CrossRef CAS PubMed.
- S.-S. Sun, J. Mater. Sci.: Mater. Electron., 2007, 18, 1143–1146 CrossRef CAS.
- T.-W. Wang, T.-L. Wang, W.-J. Chou, L.-F. Wu and S.-H. Lin, Phys. Chem. Chem. Phys., 2021, 23, 2305–2312 RSC.
- H. Liu, J. Zhou, L. Zhang, Z. Hu, C. Kuo, J. Li, Y. Wang, L. H. Tjeng, T.-W. Pi, A. Tanaka, L. Song, J.-Q. Wang and S. Zhang, J. Phys. Chem. C, 2017, 121, 16079–16087 CrossRef CAS.
- M. N. Grisolia, J. Varignon, G. Sanchez-Santolino, A. Arora, S. Valencia, M. Varela, R. Abrudan, E. Weschke, E. Schierle, J. E. Rault, J. P. Rueff, A. Barthélémy, J. Santamaria and M. Bibes, Nat. Phys., 2016, 12, 484–492 Search PubMed.
- L. Yu, J. Zhang, Y. Dang, J. He, Z. Tobin, P. Kerns, Y. Dou, Y. Jiang, Y. He and S. L. Suib, ACS Catal., 2019, 9, 6919–6928 Search PubMed.
- H. Li, G. Gao, H. Zhao, W. Wang, Y. Yang, Y. Du, S. Li, Y. Liu and L. Wang, Int. J. Hydrogen Energy, 2021, 46, 33078–33086 CrossRef CAS.
- A. Aruchamy, H. Berger and F. Lévy, J. Electrochem. Soc., 1989, 136, 2261 CrossRef CAS.
- Y. Sun, A. Huang, Z. Li, Y.-Q. Fu and Z. Wang, Electrocatalysis, 2022, 13, 494–501 CrossRef CAS.
- M. Khodabakhshi, S. Chen, T. Ye, H. Wu, L. Yang, W. Zhang and H. Chang, ACS Appl. Mater. Interfaces, 2020, 12, 36268–36276 CrossRef CAS PubMed.
- J. Xu, J. Li, D. Xiong, B. Zhang, Y. Liu, K.-H. Wu, I. Amorim, W. Li and L. Liu, Chem. Sci., 2018, 9, 3470–3476 RSC.
- D. Mukherjee, P. M. Austeria and S. Sampath, ACS Energy Lett., 2016, 1, 367–372 CrossRef CAS.
- P. Liu and Y. Pu, Int. J. Hydrogen Energy, 2022, 47, 197–202 CrossRef CAS.
- N. Jiang, Q. Tang, M. Sheng, B. You, D. Jiang and Y. Sun, Catal. Sci. Technol., 2016, 6, 1077–1084 RSC.
- D. Y. Chung, J. W. Han, D.-H. Lim, J.-H. Jo, S. J. Yoo, H. Lee and Y.-E. Sung, Nanoscale, 2015, 7, 5157–5163 RSC.
- J. Zhu and Y. Ni, CrystEngComm, 2018, 20, 3344–3352 RSC.
- J. Masud, M. Nath, U. De Silva and D. Allada, ECS Meet. Abstr, 2018,(29), 1674, DOI:10.1149/ma2018-01/29/1674.
- K. S. Bhat and H. S. Nagaraja, Mater. Res. Innovations, 2021, 25, 29–52 CrossRef CAS.
- D. Kong, J. J. Cha, H. Wang, H. R. Lee and Y. Cui, Energy Environ. Sci., 2013, 6, 3553–3558 RSC.
- M. S. Faber, M. A. Lukowski, Q. Ding, N. S. Kaiser and S. Jin, J. Phys. Chem. C, 2014, 118, 21347–21356 CrossRef CAS PubMed.
- C. Yu, H. Huang, S. Zhou, X. Han, C. Zhao, J. Yang, S. Li, W. Guo, B. An, J. Zhao and J. Qiu, Nano Res., 2018, 11, 3411–3418 CrossRef CAS.
- Y. Du, G. Cheng and W. Luo, Catal. Sci. Technol., 2017, 7, 4604–4608 RSC.
- G. Kalaiyarasan, K. Aswathi and J. Joseph, Int. J. Hydrogen Energy, 2017, 42, 22866–22876 CrossRef CAS.
- Y. Wang, N. Chen, X. Du, X. Han and X. Zhang, J. Alloys Compd., 2022, 893, 162269 CrossRef CAS.
- T. A. Bither, R. J. Bouchard, W. H. Cloud, P. C. Donohue and W. J. Siemons, Inorg. Chem., 1968, 7, 2208–2220 CrossRef CAS.
- S. Suga, K. Inoue, M. Taniguchi, S. Shin, M. Seki, K. Sato and T. Teranishi, J. Phys. Soc. Jpn., 1983, 52, 1848–1856 CrossRef CAS.
- J.-Q. Chi, K.-L. Yan, Z. Xiao, B. Dong, X. Shang, W.-K. Gao, X. Li, Y.-M. Chai and C.-G. Liu, Int. J. Hydrogen Energy, 2017, 42, 20599–20607 CrossRef CAS.
- Y. Du, G. Cheng and W. Luo, Catal. Sci. Technol., 2017, 7, 4604–4608 RSC.
- X. Wang, Y. Zhou, M. Liu, C. Chen and J. Zhang, Electrochim. Acta, 2019, 297, 197–205 CrossRef CAS.
- X. Cao, Y. Hong, N. Zhang, Q. Chen, J. Masud, M. A. Zaeem and M. Nath, ACS Catal., 2018, 8, 8273–8289 CrossRef CAS.
- A. Shankar, R. Elakkiya and G. Maduraiveeran, New J. Chem., 2020, 44, 5071–5078 RSC.
- X. He, X. Zhao, F. Yin, B. Chen, G. Li and H. Yin, Int. J. Energy Res., 2020, 44, 7057–7067 CrossRef CAS.
- X. Luan, H. Du, Y. Kong, F. Qu and L. Lu, Chem. Commun., 2019, 55, 7335–7338 RSC.
- W. Zheng, M. Liu and L. Y. S. Lee, ACS Energy Lett., 2020, 5, 3260–3264 CrossRef CAS.
- E. Cossar, M. S. E. Houache, Z. Zhang and E. A. Baranova, J. Electroanal. Chem., 2020, 870, 114246 CrossRef CAS.
- M.-H. Wu, W.-J. Chou, J.-S. Huang, D. B. Putungan and S.-H. Lin, Phys. Chem. Chem. Phys., 2019, 21, 21561–21567 RSC.
- R. Gómez-Balderas, R. Oviedo-Roa, J. M. Martínez-Magadán, C. Amador and D. A. Dixon, Surf. Sci., 2002, 518, 163–173 CrossRef.
- D. Liang, J. Mao, P. Liu, J. Li, J. Yan and W. Song, Int. J. Hydrogen Energy, 2020, 45, 27047–27055 CrossRef CAS.
- J. Yin, J. Jin, H. Zhang, M. Lu, Y. Peng, B. Huang, P. Xi and C.-H. Yan, Angew. Chem., Int. Ed., 2019, 58, 18676–18682 CrossRef CAS PubMed.
- Y.-Z. Xu, C.-Z. Yuan and X.-P. Chen, RSC Adv., 2016, 6, 106832–106836 RSC.
- Q. Wang, P. Liu, Y. Liu, X. Peng, Y.-L. Men, Y.-B. Li and Y.-X. Pan, Ind. Eng. Chem. Res., 2019, 58, 22977–22983 CrossRef CAS.
- L. Jiang, N. Yang, C. Yang, X. Zhu, Y. Jiang, X. Shen, C. Li and Q. Sun, Appl. Catal., B, 2020, 269, 118780 CrossRef CAS.
- H. Li, S. Chen, Y. Zhang, Q. Zhang, X. Jia, Q. Zhang, L. Gu, X. Sun, L. Song and X. Wang, Nat. Commun., 2018, 9, 2452 CrossRef PubMed.
- P. Farinazzo Bergamo Dias Martins, P. Papa Lopes, E. A. Ticianelli, V. R. Stamenkovic, N. M. Markovic and D. Strmcnik, Electrochem. Commun., 2019, 100, 30–33 CrossRef.
- J. Yan, H. Wu, P. li, H. Chen, R. Jiang and S. (Frank) Liu, J. Mater. Chem. A, 2017, 5, 10173–10181 RSC.
- B. Fei, Z. Chen, J. Liu, H. Xu, X. Yan, H. Qing, M. Chen and R. Wu, Adv. Energy Mater., 2020, 10, 2001963 CrossRef CAS.
- S. Dutta, A. Indra, Y. Feng, T. Song and U. Paik, ACS Appl. Mater. Interfaces, 2017, 9, 33766–33774 CrossRef CAS PubMed.
- Z. Zou, X. Wang, J. Huang, Z. Wu and F. Gao, J. Mater. Chem. A, 2019, 7, 2233–2241 RSC.
- T. Wang, D. Gao, W. Xiao, P. Xi, D. Xue and J. Wang, Nano Res., 2018, 11, 6051–6061 CrossRef CAS.
- W. Dai, Y. Pan, K. Ren, Y. Zhu and T. Lu, Electrochim. Acta, 2020, 355, 136821 CrossRef CAS.
- G. Zhang, Y.-S. Feng, W.-T. Lu, D. He, C.-Y. Wang, Y.-K. Li, X.-Y. Wang and F.-F. Cao, ACS Catal., 2018, 8, 5431–5441 CrossRef CAS.
- L. Lv, Z. Li, K.-H. Xue, Y. Ruan, X. Ao, H. Wan, X. Miao, B. Zhang, J. Jiang, C. Wang and K. (Ken) Ostrikov, Nano Energy, 2018, 47, 275–284 CrossRef CAS.
- M. B. Stevens, C. D. M. Trang, L. J. Enman, J. Deng and S. W. Boettcher, J. Am. Chem. Soc., 2017, 139, 11361–11364 CrossRef CAS PubMed.
- B. M. Hunter, N. B. Thompson, A. M. Müller, G. R. Rossman, M. G. Hill, J. R. Winkler and H. B. Gray, Joule, 2018, 2, 747–763 CrossRef CAS.
- Y. Gong, Y. Zhi, Y. Lin, T. Zhou, J. Li, F. Jiao and W. Wang, Dalton Trans., 2019, 48, 6718–6729 RSC.
- X. Wang, H. Tian, M. Pi, D. Zhang and S. Chen, Int. J. Hydrogen Energy, 2020, 45, 12237–12243 CrossRef CAS.
- H.-W. Man, C.-S. Tsang, M. M.-J. Li, J. Mo, B. Huang, L. Y. S. Lee, Y. Leung, K.-Y. Wong and S. C. E. Tsang, Chem. Commun., 2018, 54, 8630–8633 RSC.
- H.-W. Man, C.-S. Tsang, M. M.-J. Li, J. Mo, B. Huang, L. Y. S. Lee, Y. Leung, K.-Y. Wong and S. C. E. Tsang, Appl. Catal., B, 2019, 242, 186–193 CrossRef CAS.
- M. Wang, W. Fu, L. Du, Y. Wei, P. Rao, L. Wei, X. Zhao, Y. Wang and S. Sun, Appl. Surf. Sci., 2020, 515, 146059 CrossRef CAS.
- L. Zeng, Z. Liu, K. Sun, Y. Chen, J. Zhao, Y. Chen, Y. Pan, Y. Lu, Y. Liu and C. Liu, J. Mater. Chem. A, 2019, 7, 25628–25640 RSC.
- A. Manikandan, E. Hema, M. Durka, M. Amutha Selvi, T. Alagesan and S. Arul Antony, J. Inorg. Organomet. Polym. Mater., 2015, 25, 804–815 CrossRef CAS.
- L. Zeng, K. Zhou, L. Yang, G. Du, L. Liu and W. Zhou, ACS Appl. Energy Mater., 2018, 1, 6279–6287 CrossRef.
- C. Qin, A. Fan, D. Ren, C. Luan, J. Yang, Y. Liu, X. Zhang, X. Dai and M. Wang, Electrochim. Acta, 2019, 323, 134756 CrossRef CAS.
- N. Chen, Y. Wang, X. Du and X. Zhang, Dalton Trans., 2021, 50, 2964–2972 RSC.
- J.-J. Duan, Z. Han, R.-L. Zhang, J.-J. Feng, L. Zhang, Q.-L. Zhang and A.-J. Wang, J. Colloid Interface Sci., 2021, 588, 248–256 CrossRef CAS PubMed.
- J.-F. Qin, M. Yang, S. Hou, B. Dong, T.-S. Chen, X. Ma, J.-Y. Xie, Y.-N. Zhou, J. Nan and Y.-M. Chai, Appl. Surf. Sci., 2020, 502, 144172 CrossRef CAS.
- M. A. Ashraf, C. Li, B. T. Pham and D. Zhang, Int. J. Hydrogen Energy, 2020, 45, 24670–24683 CrossRef CAS.
- M. Cui, C. Yang, B. Li, Q. Dong, M. Wu, S. Hwang, H. Xie, X. Wang, G. Wang and L. Hu, Adv. Energy Mater., 2021, 11, 2002887 CrossRef CAS.
- F. Zeng, C. Broicher, J. P. Hofmann, S. Palkovits and R. Palkovits, ChemCatChem, 2020, 12, 710–716 CrossRef CAS.
- W. Wen, X. Du and X. Zhang, J. Alloys Compd., 2022, 918, 165739 CrossRef CAS.
- X. Du, G. Ma and X. Zhang, ACS Sustainable Chem. Eng., 2019, 7, 19257–19267 CrossRef CAS.
- W. Xu, Z. Lu, X. Sun, L. Jiang and X. Duan, Acc. Chem. Res., 2018, 51, 1590–1598 CrossRef CAS PubMed.
- X. Shan, J. Liu, H. Mu, Y. Xiao, B. Mei, W. Liu, G. Lin, Z. Jiang, L. Wen and L. Jiang, Angew. Chem., Int. Ed., 2020, 59, 1659–1665 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |