
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal–organic framework-derived single atom catalysts for electrocatalytic reduction of carbon dioxide to C1 products
Xu
Han†
a,
Ting
Zhang†
a and
Jordi
Arbiol
 *ab
*ab
aCatalan Institute of Nanoscience and Nanotechnology (ICN2), CSIC and BIST, Campus UAB, Bellaterra, Barcelona, 08193, Catalonia, Spain. E-mail: arbiol@icrea.cat
bICREA, Pg. Lluís Companys 23, Barcelona, 08010, Catalonia, Spain
First published on 30th November 2022
Abstract
Electrochemical carbon dioxide reduction reaction (eCO2 RR) is an efficient strategy to relieve global environmental and energy issues by converting excess CO2 from the atmosphere to value-added products. Single-atom catalysts (SACs) derived from metal–organic frameworks (MOF), which feature unique active sites and adjustable structures, are emerging as extraordinary materials for eCO2 RR. By modulating the MOF precursors and their fabrication strategy, MOF-derived SACs with specific-site coordination configuration have been recently designed for the conversion of CO2 to targeted products. In the first part of this review, MOF synthesis routes to afford well-dispersed SACs along with the respective synthesis strategy have been systematically reviewed, and typical examples for each strategy have been discussed. Compared with traditional M-N4 active sites, SACs with regulated coordination structures have been rapidly developed for eCO2 RR. Secondly, the relationship between regulation of the coordination environment of the central metal atoms, including asymmetrical M-Nx sites, hetero-atom doped M-Nx sites, and dual-metal active sites (M–M sites), and their respective catalytic performance has been systematically discussed. Finally, the challenges and future research directions for the application of SACs derived from MOFs for eCO2 RR have been proposed.
1. Introduction
A high concentration of carbon dioxide (CO2) in the atmosphere caused by burning fossil fuels since the industrial revolution has gradually triggered increasing environmental issues, such as global warming, extreme weather, species extinction and rising of the sea levels.1–3 To reduce the CO2 concentration in the atmosphere, significant efforts have been undertaken to maintain the carbon cycle, which largely relies on accelerating the development of a large number of technologies. Two main approaches have been recently proposed to address the above issues: (1) the physical capture and storage of CO2 and (2) the chemical conversion of CO2 into useful chemicals.4,5 Compared to the former, converting CO2 to renewable chemicals/fuels is an attractive and effective method, because fossil fuels are still the main source for providing energy, and therefore conversion of CO2 can not only mitigate CO2 emission but also alleviate the energy shortage (Fig. 1).4,6,7 Over the past few decades, CO2 conversion technologies have included photochemical,8 electrochemical9 and biological10 related processes. Among them, the electrochemical reduction of CO2 (eCO2 RR) driven by renewable power (like wind and solar) to produce higher-value chemical products is especially desirable in terms of energy efficiency and cost because of (1) controllable electrode potentials and ambient reaction temperatures as well as pressures; (2) recycled electrolytes, minimizing overall chemical consumption; (3) reduced generation of new CO2 sources due to electricity as the driver; and (4) easy scale-up applications.6,11,12 Therefore, eCO2 RR provides a promising way for generating chemical energy, which can be regarded as a form of renewable energy storage.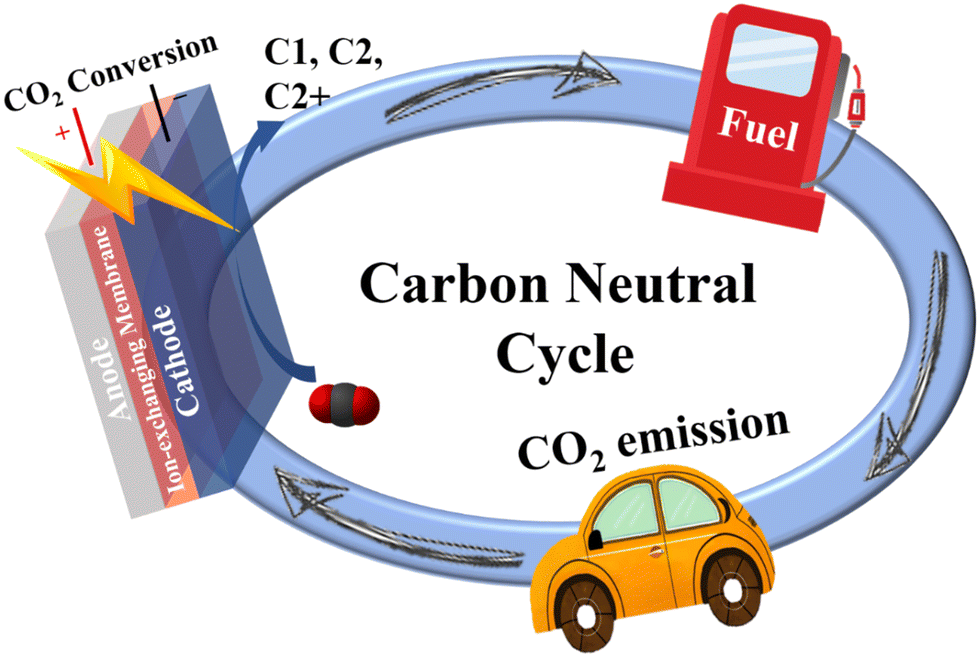 | ||
| Fig. 1 The schematic illustration of a carbon neutral cycle based on electrochemical CO2 reduction reaction. | ||
The eCO2 RR process is divided into three main steps, including the adsorption of CO2 on active sites for CO2* formation, electron/proton transfer for cleaving the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond and producing different intermediates, and desorption and diffusion of products from active sites.13–18 Until now, eCO2 RR has faced many problems due to the thermodynamic stability and chemical inertness of CO2 as a reactant (the dissociation energy of a CO bond is about 806 kJ mol−1).19 The first activation step of CO2 to form CO2*−, occurring at −1.9 V versus standard hydrogen electrode (SHE) (Table 1), leads to a high CO2 activation barrier and insufficient efficiencies.20,21 In addition, the thermodynamical potentials for producing different products are very close accompanied with the hydrogen evolution reaction (HER, Table 1), resulting in an inevitable effect on the desirable product selectivity.22,23 Based on the number of carbon atoms, the eCO2 RR products are classified into the following categories: C1 products, including carbon monoxide (CO), formate/formic acid (HCOOH), methanol (CH3OH) and methane (CH4); C2 products, including ethylene (C2H4), ethanol (C2H5OH) and acetate (CH3COOH); C3 products such as propylene (C3H6) and n-propanol (C3H7OH); and long-chain products.17 Although significant progress has been made in the eCO2 RR process, it is still limited by the lack of high-efficient electrocatalysts, showing a low energy efficiency and activity as well as poor selectivity because of the complex product distributions. Designing eCO2 RR electrocatalysts with high activity and selectivity for a particular product is thus highly desirable to speed up the sluggish eCO2 RR and solve the above problems.
O bond and producing different intermediates, and desorption and diffusion of products from active sites.13–18 Until now, eCO2 RR has faced many problems due to the thermodynamic stability and chemical inertness of CO2 as a reactant (the dissociation energy of a CO bond is about 806 kJ mol−1).19 The first activation step of CO2 to form CO2*−, occurring at −1.9 V versus standard hydrogen electrode (SHE) (Table 1), leads to a high CO2 activation barrier and insufficient efficiencies.20,21 In addition, the thermodynamical potentials for producing different products are very close accompanied with the hydrogen evolution reaction (HER, Table 1), resulting in an inevitable effect on the desirable product selectivity.22,23 Based on the number of carbon atoms, the eCO2 RR products are classified into the following categories: C1 products, including carbon monoxide (CO), formate/formic acid (HCOOH), methanol (CH3OH) and methane (CH4); C2 products, including ethylene (C2H4), ethanol (C2H5OH) and acetate (CH3COOH); C3 products such as propylene (C3H6) and n-propanol (C3H7OH); and long-chain products.17 Although significant progress has been made in the eCO2 RR process, it is still limited by the lack of high-efficient electrocatalysts, showing a low energy efficiency and activity as well as poor selectivity because of the complex product distributions. Designing eCO2 RR electrocatalysts with high activity and selectivity for a particular product is thus highly desirable to speed up the sluggish eCO2 RR and solve the above problems.
| Half-electrochemical reactions | Standard potential (V vs. SHE) |
|---|---|
| 2H+ + 2e− → H2 | −0.42 |
| CO2 + 2H+ + 2e− → CO + H2O | −0.52 |
| CO2 + 2H+ + 2e− → HCOOH | −0.61 |
| CO2 + 4H+ + 4e− → HCHO + H2O | −0.51 |
| CO2 + 6H+ + 6e− → CH3OH + H2O | −0.38 |
| CO2 + 8H+ + 8e− → CH4 + 2H2O | −0.24 |
| 2CO2 + 12H+ + 12e− → C2H4 + 4H2O | −0.34 |
| 2CO2 + 12H+ + 12e− → C2H5OH + 3H2O | −0.33 |
| CO2 + e− → CO2*− | −1.90 |
Single-atom catalysts (SACs) with atomically dispersed catalytic sites, which are anchored on a specific support, were first reported by Zhang and co-workers in 2011.24–26 Due to their unique electronic and geometric structures, SACs inherit the advantages of both heterogeneous and homogeneous catalysts, and have recently emerged as an up-to-date frontier in materials science. They have shown great potential in energy-related catalysis owing to the (1) maximization of the percentage of metal atoms as active sites; (2) increase of the effective active site density; (3) regulation of the central metal atoms and coordination environments; and (4) adjustment of the electronic structure.27–34 Benefiting from the simple and precise single metal-based active sites, their catalytic performance can be easily regulated, which helps to overcome the high activation barriers and the sluggish kinetics in eCO2 RR, and therefore SACs have recently been developed in eCO2 RR with excellent selectivity and activity, opening a way to improve the CO2 conversion. In addition, their simplicity has allowed one to deeply and comprehensively understand the relationship between the structures and their catalytic performance.35–39Fig. 2 summarizes different reported metal centers in SACs for eCO2 RR, which include three types based on the nature of the metal centers: (1) transition metals (Co,40,41 Fe,42,43 Ni,44,45 Cu,46,47 Zn48,49 and Mo50,51), (2) noble metals (Pd,52,53 Au54 and Ag55,56) and (3) p-block elements (Sn57–59 and Bi60).
 | ||
| Fig. 2 The reported products on SACs with different metal centers for eCO2 RR. | ||
Although there have been some significant efforts in the design of SACs for eCO2 RR, due to their high surface energies when dispersing catalysts at the atomic level, the fabrication of SACs still suffers from low porosity structures and low metal loading (<1.0 wt%), as well as the tendency for metal aggregation.61–64 As a result, exploring a controlled strategy to construct SACs with suitable supports to bear porous structures, uniform atomic sites, high metal content and strong coordination environments is of critical importance to avoid agglomeration during preparation and subsequent reaction efficiency improvement.
Several synthetic methodologies have been developed and explored in order to obtain SACs. The main ones have been coprecipitation,65 impregnation,66 atomic layer deposition67 and the use of metal–organic frameworks (MOFs) as templates.60,68–74 Among them, the use of MOFs as templates, with regular frame structures and uniform pores constructed by coordinating bridging organic ligands and metal centers, has been considered as one of the most promising approaches for preparing high quality SACs.61–64,72,75 Owing to the spatial separation effect of MOFs, single metal sites with uniform dispersion can be accommodated into their metal nodes, organic ligands or pores at different locations.61,63,64,76 For example, ZIF-8 as the “star” MOF, with a pore diameter of 3.4 Å, which can be used to prepare nanoporous carbon materials with an unexpectedly high surface area by direct pyrolysis,77 has been widely used as a template to prepare a large number of SACs, by efficiently encapsulating different metal atom species.37,78,79 After pyrolysis, the single atom (SA) embedded into the carbon support derived from ZIF-8 is generated via coordination bonds with surrounding chelating atoms, such as N, which in this case come from 2-methylimidazole. Functional UiO-66-NH2 can also coordinate and confine the metal ions in the skeletons through uncoordinated amino groups, further inhibiting the migration of metal species.80 Followed by a thermal treatment, different SACs have been also obtained by using UiO-66-NH2 as a MOF template. In most cases, compared to other SAC preparation approaches, the SACs derived from MOFs have many merits: (1) due to pyrolysis at a high temperature, such SACs can be used in harsher conditions with long-term stability compared to those obtained by other conventional synthetic methods; (2) such SACs have a large surface area and high porosity inherited from parent MOFs, which could facilitate electron and mass transport as well as increase the accessibility to active SA sites, leading to an unexpected eCO2 RR performance.28,61,75,81,82 Therefore, tremendous efforts have been devoted for optimizing MOF-derived SACs. In Fig. 3, a brief history of the development and the latest progress of MOF derived SACs for eCO2 RR is presented. In addition, MOF-derived SACs have been divided into two categories according to their synthetic approaches.
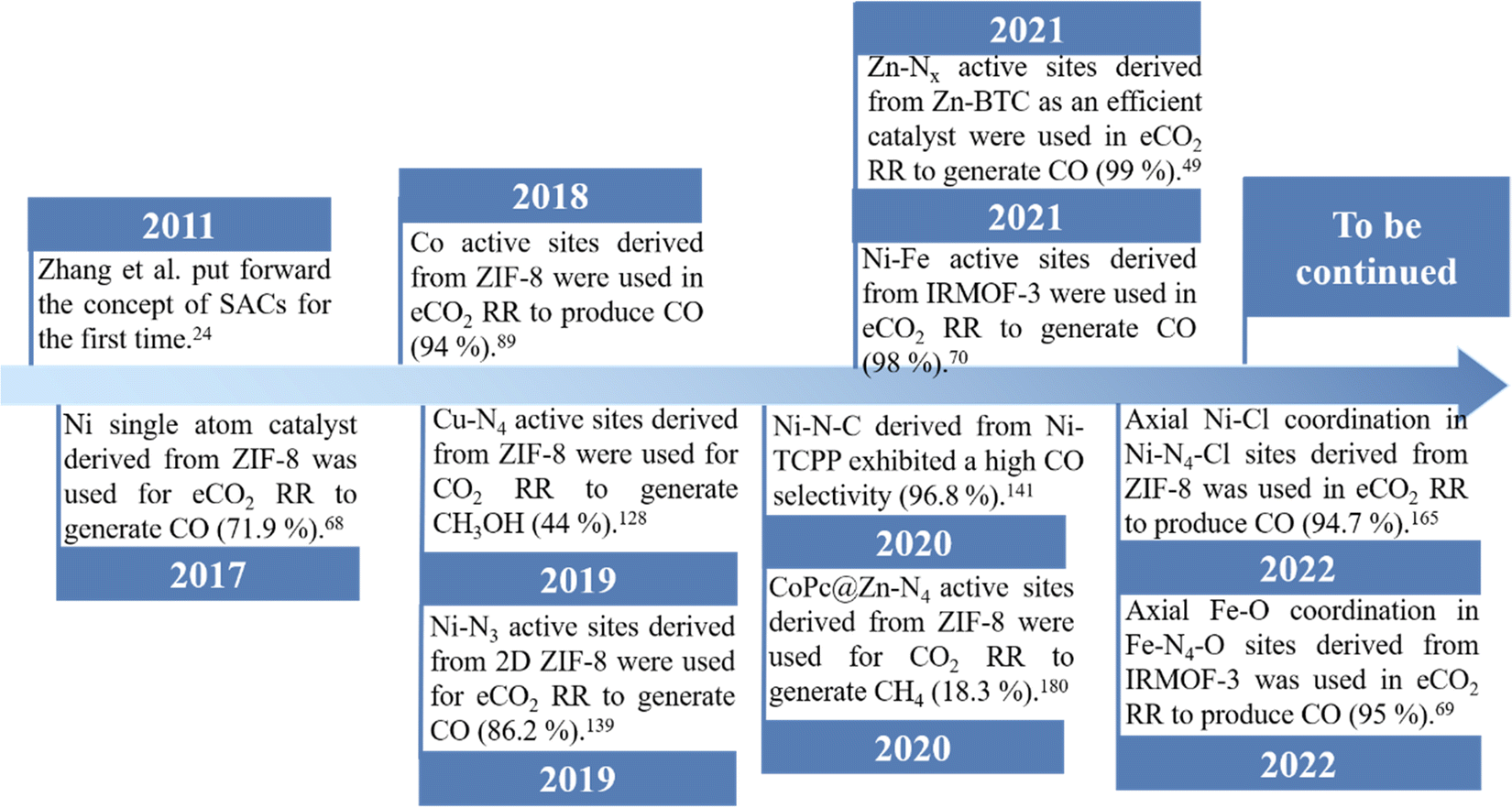 | ||
| Fig. 3 A brief history of the development of SACs derived from MOFs. | ||
1.1 Formation of SACs by direct pyrolysis of MOFs
MOFs are composed of various metal nodes and organic ligands; thus, the atomically dispersed metal species can be derived from the metal nodes in the frameworks after pyrolysis. In this strategy, suitable MOFs have been chosen to form the targeted SACs after thermal, acid-etching or activation treatments. For instance, Jiang et al. prepared a Zn SAC derived from ZIF-8 with an ultrahigh Zn loading mass (11.3 wt%).83 However, during the direct preparation, metal nanoparticles are also generated when using this method, accompanied by the formation of SACs. In this way, a post-acid treatment is usually used to effectively remove the metal particles, leaving the atomically dispersed metal sites anchored on the carbon-based substrates.84–86 For instance, a highly dispersed manganese (Mn) SAC was reported by Yang et al. by directly pyrolyzing Mn-based MOFs.85 The former procedure allowed the removal of the residual MnO particles created by applying an acid-etching process: in parallel, the same process generated a porous structure. Despite substantial achievements made in this strategy, the universality of this approach is still limited by the tedious acid treatment or activation process, which not only increases the synthetic complexity, but also leads to a low metal loading.1.2 Formation of SACs by pyrolysis of MOFs with the second metal addition
In this strategy, additional targeted metals are introduced into the metal nodes or pores of MOF precursors for forming SACs after pyrolysis. Up to now, among many MOF precursors, Zn-based MOFs with nitrogen-rich organic linkers are the ones preferred. On the one hand, it is feasible to form the metal–nitrogen coordination interaction after pyrolysis, which can stabilize the SACs. On the other hand, because of the relatively low boiling point of Zn (906 °C), most of the Zn metal atoms are evaporated as a self-sacrificial metal during pyrolysis, which can avoid the formation of excess metal hybrids, leaving the targeted metals atomically dispersed on the porous carbon.68,87–90 For example, second metal ions (like Co2+,87,91,92 Ni2+,68,93 Fe3+,69,94etc.) have been successfully implanted into Zn-based MOFs with homogeneous dispersion in metal nodes or pores, resulting in a much longer spatial distance between each other. By this means, single-atom Co, Ni, and Fe sites anchored on a N-doped carbon support have been successfully synthesized by pyrolysis of predesigned bimetallic MOFs.The utilization of SACs in eCO2 RR has made progress in recent years, owing to the use of MOFs as templates to synthesize these SACs, opening a new chapter for eCO2 RR. Although many reviews on SACs for eCO2 RR have been reported recently, the study of eCO2 RR catalyzed by MOF-derived SACs influenced by the coordinated environment is still in its infancy. Therefore, this review will not only focus on the development of SACs derived from MOFs, but also give a discussion of the relationship between the coordination environments of MOF-derived SACs and their catalytic performance. Due to the simple separated active sites in SACs, the carbon–carbon coupling is difficult. Therefore, C1 products are the preferred ones over most SACs derived from MOFs, instead of longer hydrocarbon chains. In this way, we will mainly focus on C1 products, including CO, HCOOH, CH3OH and CH4. In addition, some challenges in such field are also provided at the end of the present work. This review is intended to provide an overview for the future design of MOF-derived SACs for eCO2 RR via the accurate elucidation of their structure–activity relationship.
2. Fundamentals of the eCO2 RR
ECO2 RR occurs on the cathode surface and involves a multi-electron/proton transfer process. The main C1 products, CO, HCOOH, CH3OH and CH4, can be obtained by transferring 2, 4, 6, and 8 electrons, respectively, with the consequent reduction of CO2 based on the applied potential. The possible pathways for the main C1 products are given below. CO2 reduction to CO and HCOOH can start with a one proton/electron transfer to a CO2 molecule followed by the stabilization of COOH* and OCHO* intermediates.36,95,96 Subsequently, for CO generation, COOH* is transformed to CO* through a dehydrogenation reaction, and then, the CO* desorbs from the catalysts’ surfaces, forming the final CO product.19,97 In addition, *CO can be further reduced to generate multi-electron products if it binds to the catalyst surface strongly. With a succession of proton-coupled electron transfers, CH3OH and CH4 can be generated.2,98 It is worth mentioning that the energy barriers to produce CH3OH are high, making CH3OH generation rather difficult.96 In the case of HCOOH generation, the OCHO* intermediates desorb from the metal surface to form HCOOH.2.1 CO2-to-CO pathway
Among all the eCO2 RR products, from a commercial point of view, CO is considered as the most attractive targeted product because it can be produced with a high faradaic efficiency (FE) exceeding the hitherto reported value of 90% and only requires a simple two-electron/proton transfer pathway with low energy barriers.19,99–103 Meanwhile, CO and H2, as syngas, can be utilized to produce various commodity chemicals in industry via the Fischer–Tropsch process.17,104 So far, the majority of reported SACs have shown high catalytic activity towards CO2-to-CO reaction, which is comparable to that of noble metal catalysts, such as Au and Ag. The proposed mechanisms for CO2-to-CO reaction on SACs are often divided into two categories: (1) the pathway of CO2 RR to CO via a carboxyl (*COOH) intermediate and (2) a proton decoupled electron transfer related mechanism (Fig. 4).48,105,106 These proposed reaction mechanisms merely represent the reaction pathway for a specific catalyst and condition.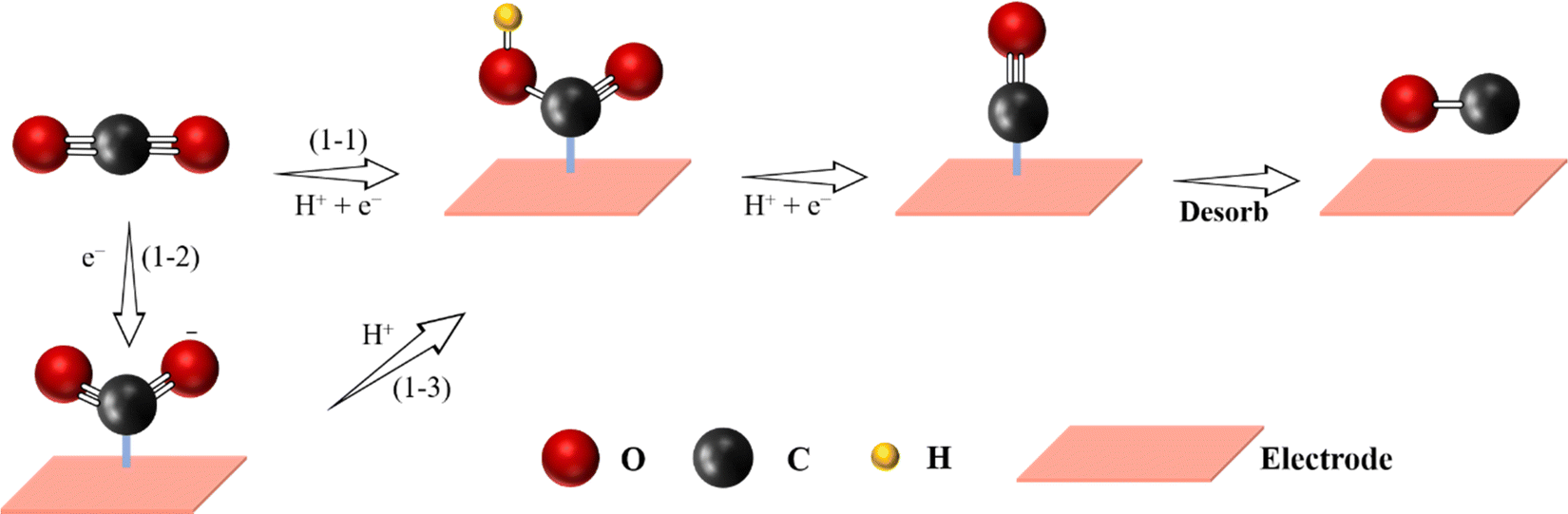 | ||
| Fig. 4 Proposed reaction pathway for electrochemical CO2-to-CO reaction (C atoms: gray; O atoms: red; H atoms: yellow). | ||
The first conversion mechanism consisting of the pathway of CO2 RR to CO via a carboxyl (*COOH) intermediate is proposed to occur in three steps:
(i) CO2(g) + e− + H+ → *COOH;
(ii) *COOH + e− + H+ → *CO + H2O;
(iii) *CO → CO + *;
where * indicates that the molecule is adsorbed on the surface of the catalysts.107,108 *COOH forms via a concerted protonation and electron transfer. Then, the *COOH intermediate converts to *CO via further proton–electron transfer. Finally, *CO dissociates from the active sites to generate CO. For instance, the formation of the Fe-N4-COOH intermediate from CO2via a concerted protonation and electron transfer process has been proposed by Chen et al. on FeN4 sites.109
The second mechanism, which consists of proton decoupled electron transfer, includes the following steps:
(i) formation of an adsorbed CO2˙−via one-electron transfer;
(ii) a protonation process of CO2˙− to form *COOH;
(iii) *COOH + e− +H+ → *CO +H2O;
(iv) *CO → CO + *;
where * indicates that the molecule is adsorbed on the surface of the catalysts.110,111 Such a mechanism was proposed on a Fe-based SAC, consisting of a homogeneous molecular catalyst with metal–N macrocyclic complexes. Despite the above proposed mechanisms, the development and improvement of advanced characterization methods, especially operando spectroscopic techniques, to investigate in more detail the mechanisms of eCO2 RR, are still needed.
2.2 CO2-to-HCOOH pathway
As an essential energy-dense carrier for many industrial processes,112,113 formic acid/formate (HCOOH/HCOO−) (HCOO− exists at pH > 4.1, while HCOOH exists at pH < 4.1) can be sustainably produced from eCO2 RR,114–116 which is also a two-electron process consisting of 3 steps37,100 (Fig. 5):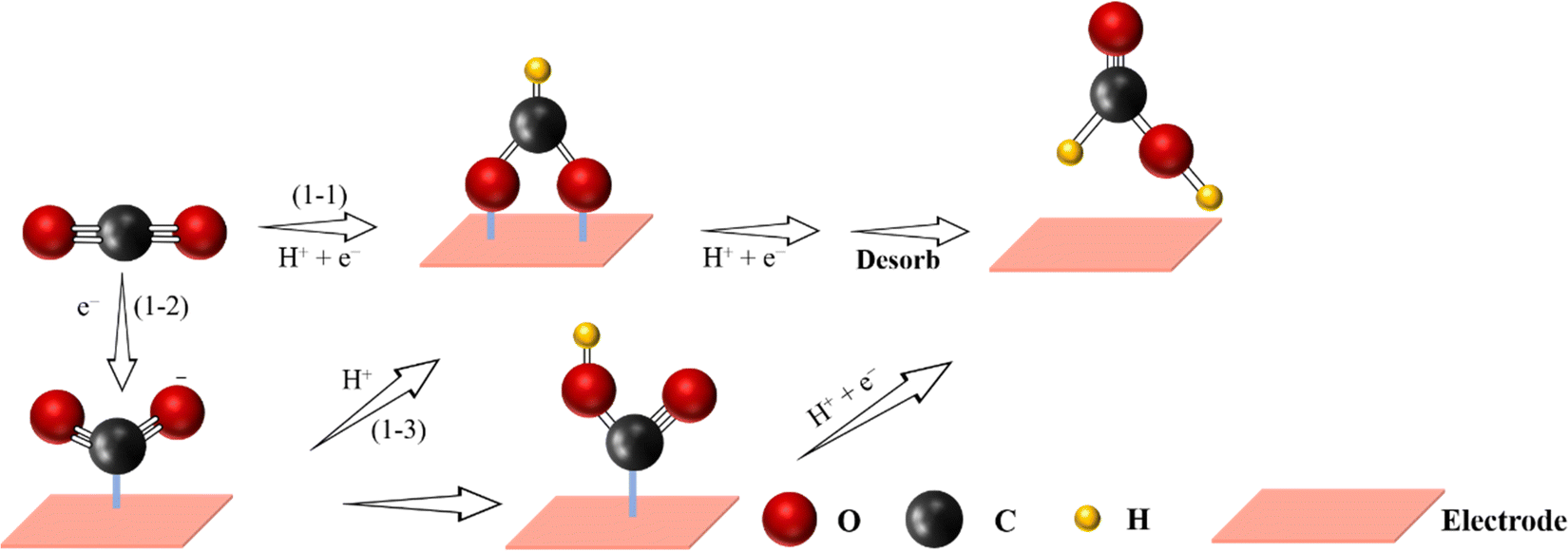 | ||
| Fig. 5 Proposed reaction pathway for the electrochemical CO2-to-HCOOH reaction (C atoms: gray; O atoms: red; H atoms: yellow). | ||
(i) CO2 + H+ + e− → *OCHO;
(ii) *OCHO + H+ + e− → *HCOOH;
(iii) *HCOOH → * + HCOOH.
Similarly, the first conversion mechanism step for the formation of HCOOH in eCO2 RR consists of the generation of *CO2˙−via one-electron transfer. Secondly, the formation of the *OCHO intermediate involves a protonation process. The same intermediate (*OCHO) through a proton-coupled electron transfer (PCET) step can be formed after direct gaseous CO2 conversion. An alternative process is the generation of *OCHO through the attack and insertion of the CO2 molecule on *H species. Finally, through another PCET step, the *OCHO intermediate is converted to HCOOH or HCOO−.117 Generally, Sn,118 Pb,117 Bi,119 In,120 Pd,100 and other metal-free catalysts121 possess high selectivity towards HCOOH or HCOO− formation. In a similar way to the bulk catalysts, in some cases, SACs with the above metal centers also showed HCOOH selectivity in eCO2 RR. For instance, single In active sites were supported on a MOF-derived N-doped carbon matrix and used to trigger the CO2-to-HCOOH reaction.122 In comparison to the N–C sample, In-based SACs exhibited a higher FE (80%) towards HCOOH under a low applied potential of −0.80 V vs. RHE. In addition, by replacing the In metal centers by Sn,123 these Sn-based SACs also exhibited good selectivity (62%) towards HCOOH. In this way, the strong electronic interaction between Sn single-atoms and the N-doped carbon materials could increase the bonding strength of the CO2˙− intermediate, thereby promoting the conversion of CO2 to HCOOH.
2.3 CO2-to-CH3OH pathway
As a basic organic chemical raw material, methanol has been widely used in many fields, including the production of fine chemicals, plastics or medicines.124 In addition, due to its high energy density (4.8 kW h L−1), methanol is considered as a promising fuel.95 Nowadays, the most common method for CH3OH formation is mixing CO and H2 under high temperature (250–300 °C) and pressure (3.5–10 MPa).37,95 Unfortunately, such a method usually requires high-security equipment. The eCO2-to-CH3OH process provides an alternative way to produce CH3OH under moderate conditions. Compared to CO-to-HCOOH reaction, the eCO2-to-CH3OH reaction is a more complex process involving 6-electron transfer: CO2 + 6H+ + 6e− → CH3OH + 2H2O, making the kinetics of the reduction reaction slow, because it needs a high energy barrier to produce *CH2OH or *CH3O intermediates in the reaction, and therefore there is still a great challenge for large-scale production of CH3OH at industry levels (Fig. 6).2,125 Nevertheless, some interesting examples have been studied in order to materialize this reaction. For example, a CoN4/graphene catalyst was simulated, revealing the best way to produce CH3OH by following the next steps: CO2 (g) → COOH* → CO* → CHO* → CH2O* → *OCH3 → CH3OH, where the energy barrier for reducing CHO* to CH2O* was 0.53 eV, being the speed determining step.126 Following this idea, Wu et al. immobilized a cobalt-based molecular catalyst with CoN4 active sites on carbon nanotubes showing a CH3OH selectivity of 40%.127 Through the above-mentioned mechanism, it is well established that *CO is an important intermediate for the formation of CH3OH. If the adsorption energy of *CO on the catalyst's surface is high enough, the reaction further proceeds by following multiple PCET steps to generate CH3OH. Although many efforts have been devoted to fabricate SACs for the generation of CH3OH, the obtained activity and selectivity are still insufficient. Yang et al. could improve the selectivity (FEmethanol = 44%) by designing Cu-based SACs coated on carbon nanofibers, which is a great advancement but still not sufficient for large scale industrial production.128 Therefore, understanding the structural characteristics for designing efficient SACs for CH3OH production is still at the early stage, mainly relying on Cu-based SACs, because generation of CH3OH (*CH2OH and *CH3O) presents very high energy barriers and thus its production remains a big challenge.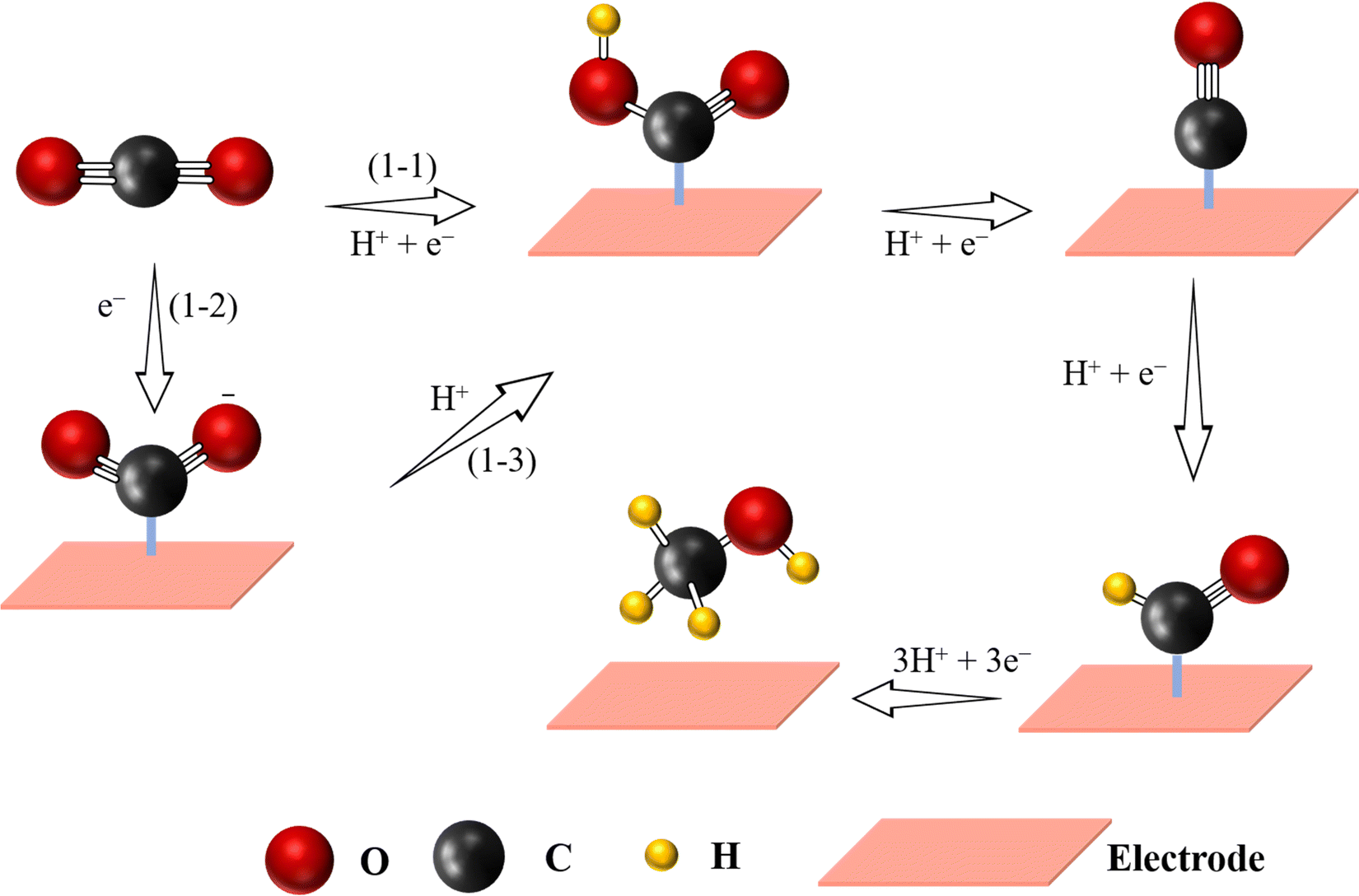 | ||
| Fig. 6 Proposed reaction pathway for the electrochemical CO2-to-CH3OH reaction (C atoms: gray; O atoms: red; H atoms: yellow). | ||
2.4 CO2-to-CH4 pathway
Methane, widely found in natural gas, is considered as a high-quality gas fuel due to the high gravimetric energy density (13.9 kW h kg−1), and it can be further utilized as an energy carrier to replace traditional fossil fuels.37,95,124 However, the electrochemical CO2-to-CH4 process is more difficult as it requires the transfer of 8 electrons and 8 protons: CO2 + 8H+ + 8e− → CH4 + 2H2O (Fig. 7). Therefore, the activity and selectivity for the formation of CH4 are low.2 Moreover, the presence of the *CO intermediate on the active sites is important in order to generate CH4. Furthermore, the generation of *CHO intermediates via protonation of *CO needs to overcome a large energy barrier, thus leading to a higher negative potential. As illustrated above, following the formation of *CHO, an adsorbed carbon species (*C) is formed. After four continuous PCET steps, CH4 can be finally generated. By changing the pyrolysis temperature and coordination numbers of N, a series of Cu-based SACs with promising efficiency towards CH4 have been prepared. Among them, isolated Cu–N2 active sites have been shown to be favorable for CH4 generation with the highest FECH4 of 38.6% at −1.6 V vs. RHE.129 Moreover, Han et al. designed ZnN4 active sites anchored in microporous nitrogen-doped carbon, which reached a FECH4 of 85%, inhibiting the reduction of CO2 to CO.130 Notice that on the ZnN4 site, the rate-limiting step for the reduction of CO2 to CH4 was the transformation of *OCHOH to *CHO.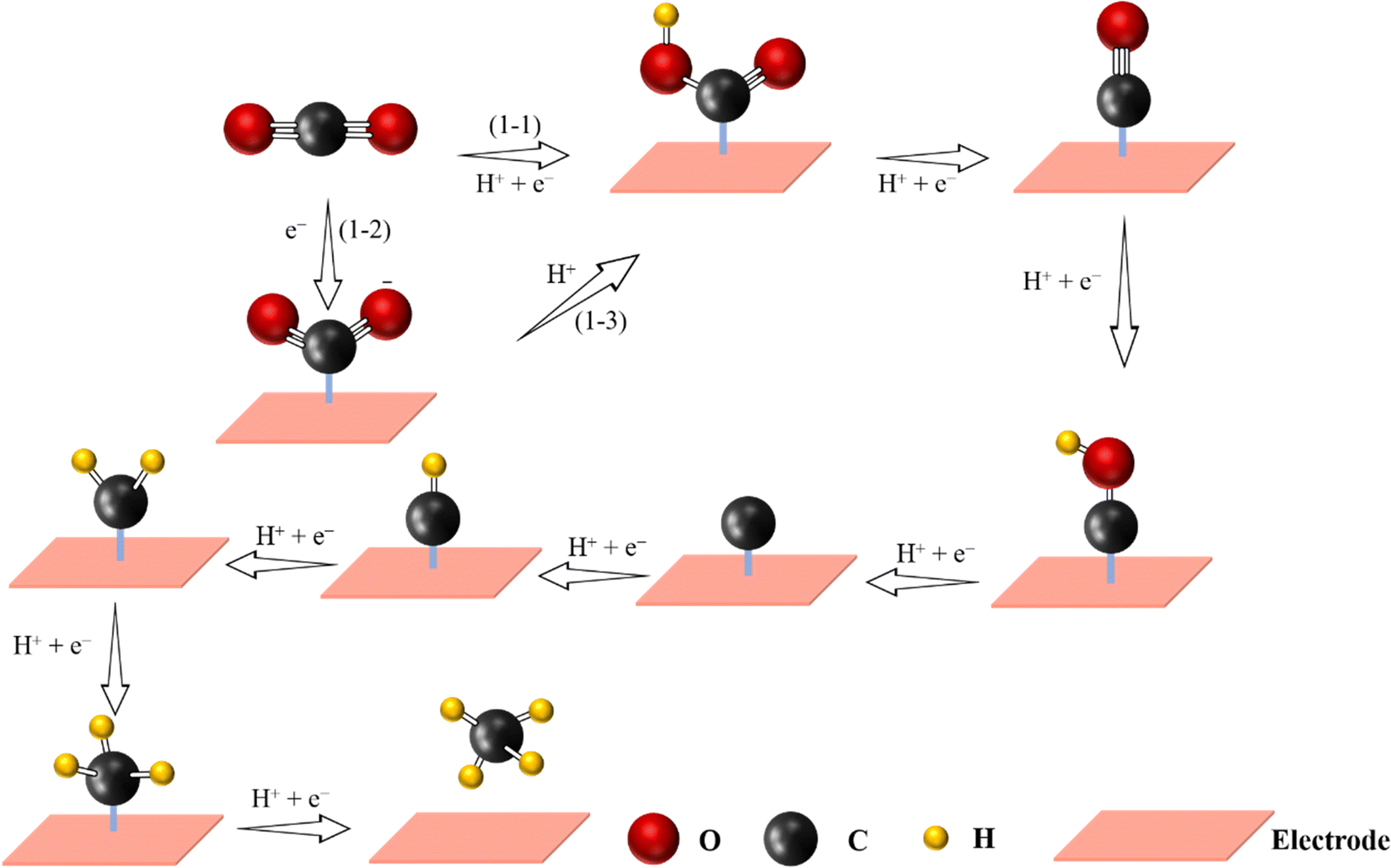 | ||
| Fig. 7 Proposed reaction pathway for electrochemical CO2-to-CH4 reaction (C atoms: gray; O atoms: red; H atoms: yellow). | ||
3. C1 products on MOF-derived SACs
SACs have been reported as effective catalysts due to their four-coordinated M-N4 active sites with a symmetric coordination environment.131–134 Due to their adjustable geometry and electronic properties, and the high exposure of active sites, M-N4 moieties are considered one of the main active sites for eCO2 RR catalytic performance. Therefore, it can be concluded that different activity and selectivity of the products are mainly influenced by the nature of the central metal atoms. Previous theoretical and experimental results proved that the metal-complex (M-N4) site in SACs is the main active center of the catalytic reaction. Different central metals have different binding energies with intermediates such as *CO and *COOH, which may lead to different product selectivity. For example, Hu et al. supported that the selectivity and activity of M-N-C catalysts towards CO were generally ordered as Ni > Fe ≫ Co and Ni, Fe ≫ Co, respectively.105 Different catalysts with M-N4 active sites in a symmetric coordination environment have been reported for electrochemical CO2-to-CO reaction, as shown in Table 2.| Configuration | Method | Main product | FEmax@potential (vs. RHE) | Ref. |
|---|---|---|---|---|
| Ni-N4 | MOF-derived | CO | 71.9%@−0.89 V | 68 |
| Ni-N4 | MOF-derived | CO | 98%@−1.0 V | 138 |
| Ni-N4 | MOF-derived | CO | 97%@−0.9 V | 139 |
| Ni-N4 | MOF-derived | CO | 96.2%@−1.0 V | 140 |
| Ni-N4 | MOF-derived | CO | 96.8%@−0.8 V | 141 |
| Ni-N4 | MOF-derived | CO | 97%@−0.79 V | 142 |
| Ni-N4 | MOF-derived | CO | 92–98%@−1.03 V | 143 |
| Fe3+-N4 | MOF-derived | CO | >80%@−0.47 V | 42 |
| Fe-N4 | MOF-derived | CO | 94%@−0.46 V | 144 |
| Fe-N4 | MOF-derived | CO | >90%@−0.8 V | 145 |
| Co-N4 | MOF-derived | CO | 82%@−1.0 V | 146 |
| Co-N4 | MOF-derived | CO | 98.7%@−0.6 V | 147 |
| Cu-N4 | MOF-derived | CH3OH | 44%@−0.9 V | 128 |
| Cu-N4/N2 | MOF-derived | C2H4 | 24%@−1.4 V | 129 |
| Cu-N4 | MOF-derived | C2H5OH | 55%@−1.2 V | 148 |
| Cu-N4 | MOF-derived | CO | 98%@−0.9 V | 149 |
| Co-N4 | MOF-derived | CO | 82% @−0.8 V | 146 |
| Zn-N3/N4 | MOF-derived | CO | 100%@−0.57 V | 49 |
| In-N4 | MOF-derived | CO | 97.2%@−0.7 V | 150 |
| In-N4 | MOF-derived | HCOOH | 96%@−0.65 V | 151 |
| Bi-N4 | MOF-derived | CO | >97% | 60 |
| Ni-N4/Cu-N4 | MOF-derived | CO | 99%@−0.97 V | 152 |
However, theoretical and experimental results revealed that the high structure/electron symmetry of the M-N4 moiety, resulting from its symmetrical planar structure, makes it chemically inert to a certain extent, thus reducing the kinetics of the catalytic reaction and hindering it.39,132,135,136 Therefore, in order to tune the performance of M-N4 active sites for eCO2 RR, breaking the symmetrical electronic structure of M-N4via a reasonable adjustment of the coordination structure to optimize the electronic structure of the M-N4, and subsequently affecting the binding energies for eCO2 RR intermediates have caught researchers’ attention and have been developed in full swing. By tuning the coordination numbers and introducing second atoms (metal or non-metal), the d orbital of the metal site can be significantly tuned, affecting the catalytic activity for eCO2 RR.137 In the last few years, researchers have prepared a variety of novel SACs derived from MOFs, with most of them displaying more ideal eCO2 RR performance. Therefore, in this section, we will discuss the influence of the alteration of coordination components, including M-Nx SACs, M-Nx-heteroatom SACs and M–M SACs, on the formation of different C1 products (Fig. 8).
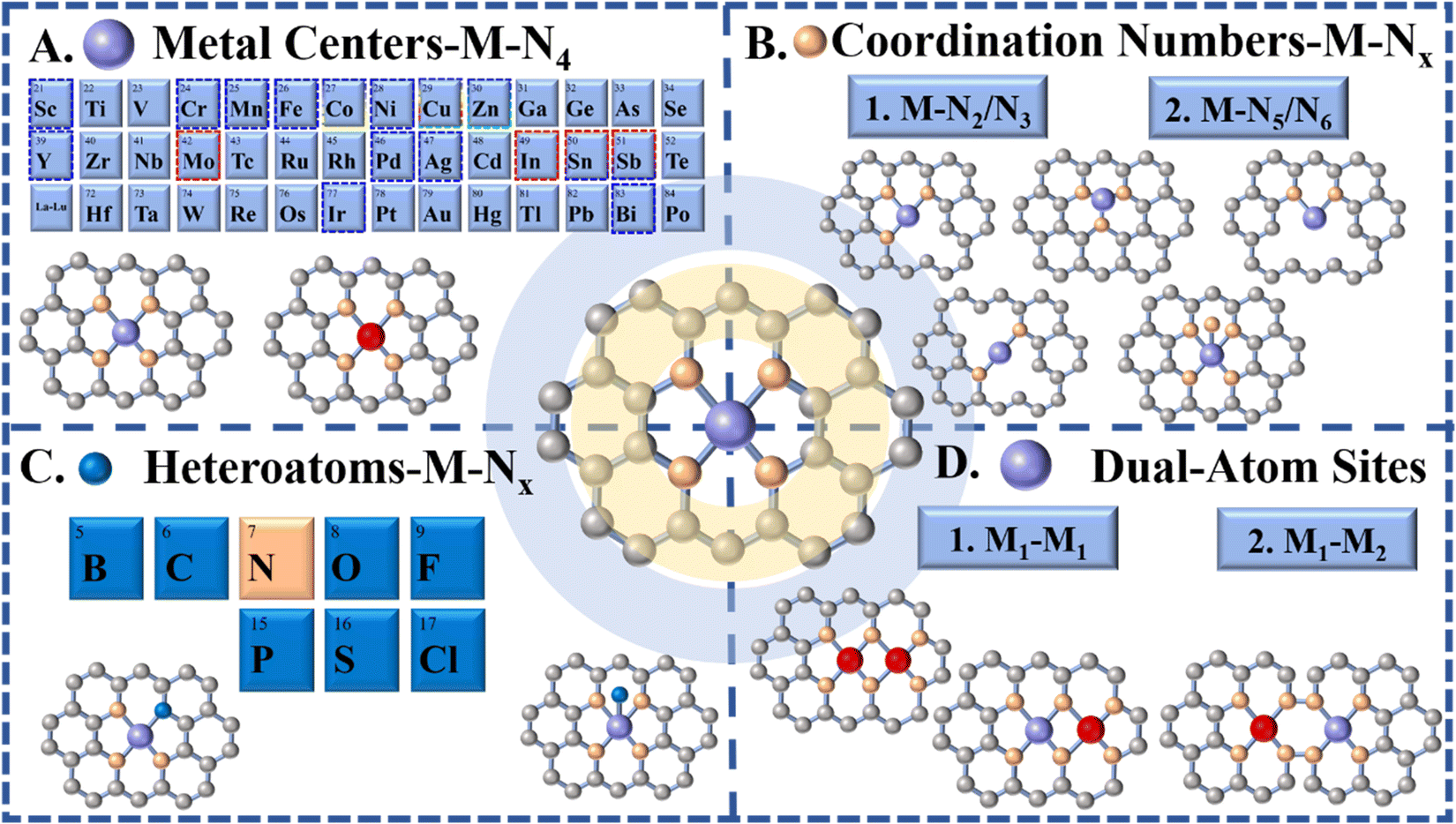 | ||
| Fig. 8 Schematic illustration showing regulation of central metal atoms and the local atomic environment in M-N-C catalysts (C atoms: gray; N atoms: orange; metal atoms: purple/red; heteroatoms: blue). | ||
3.1 M-Nx(x<4)
In most cases, SACs derived from MOFs form a stable M-N4 coordination after pyrolysis at a high temperature due to their N-rich organic ligands. However, compared to traditional M-N4 active sites, current works have revealed that the coordination number of the central metal atoms is also a key parameter, which affects the activation or intermediate dissociation of CO2, thus giving us the possibility to tailor it in order to improve the catalytic activity and selectivity of SACs.21,37,39,75,124 According to previous results, via adjusting the synthesis conditions, the number of nitrogen atoms coordinated with metal atoms could be regulated to realize M-Nx(1≤x≤5)-C-like active sites, such as M-N1/N2/N3/N4/N5-C.153,154 Such regulated active sites can lead to a stronger or weaker interaction with the eCO2 RR intermediate (*COOH or *CO), and thus exhibit higher or lower eCO2 RR activity and selectivity. Especially, SACs with low coordination numbers exhibited higher catalytic performance towards CO generation. Generally, the pyrolysis temperature is one of the main conditions that we can modify in order to affect the coordination numbers of SACs (Fig. 9A). The nitrogen coordination number tends to gradually decrease with the increase of the pyrolysis temperature.125 Furthermore, the formed M-Nx with defects can significantly improve the catalytic performance for eCO2 RR. For example, Jiang et al. prepared MOF-74-derived Ni SA-N2-C with lower N coordination numbers by controlling the pyrolysis temperature and achieved a high CO faradaic efficiency (98%) at −0.80 V vs. RHE and turnover frequency (1622 h−1), which is better than the efficiency shown by similar metal catalysts with higher N coordination numbers (e.g. Ni SA-N3-C and Ni SA-N4-C) (Fig. 9B).155 Theoretical calculations indicated that a lower coordination number could change the electronic structure of the active site, which is favorable to the formation of the COOH* intermediate, thereby enhancing the electroreduction activity of CO2. Similarly, it has been shown that ZIF-8 derived FeN3 active sites possess balanced adsorption energies for *COOH and *CO intermediates, facilitating CO formation, and thus, exhibiting superior eCO2 RR performance with CO faradaic efficiency up to 96% at −0.5 V vs. RHE, a turnover frequency of 2225 h−1, and an outstanding stability, which further support the superiority of M-Nx sites with low coordination numbers.156 More importantly, the FeN3 catalyst possessed excellent stability during the 48 h test. In addition, atomically dispersed Co active sites with two-coordinated nitrogen atoms, fabricated via pyrolysis of bimetallic Co/Zn ZIFs, showed high selectivity and superior activity with 94% FE(CO) at an overpotential of 520 mV as well as excellent stability (60 h). Such unsaturated Co-N2 sites could promote the activation of CO2 to the CO2˙− intermediate and hence enhance the CO2 electroreduction activity.89 Effective conversion of CO2 to CO on some Cu-N4 active sites is more likely to occur.149 Compared to Cu-N4 active sites, the CO desorption on Cu-N3 sites is difficult due to their high energy barrier, leading to higher selectivity towards CH4.157,158 These results further confirm the promising use of MOFs for preparing low coordination number M-Nx sites, which facilitate the activation of CO2-to-CO. For other C1 products, SACs with low coordination number M-Nx sites still show insufficient selectivity.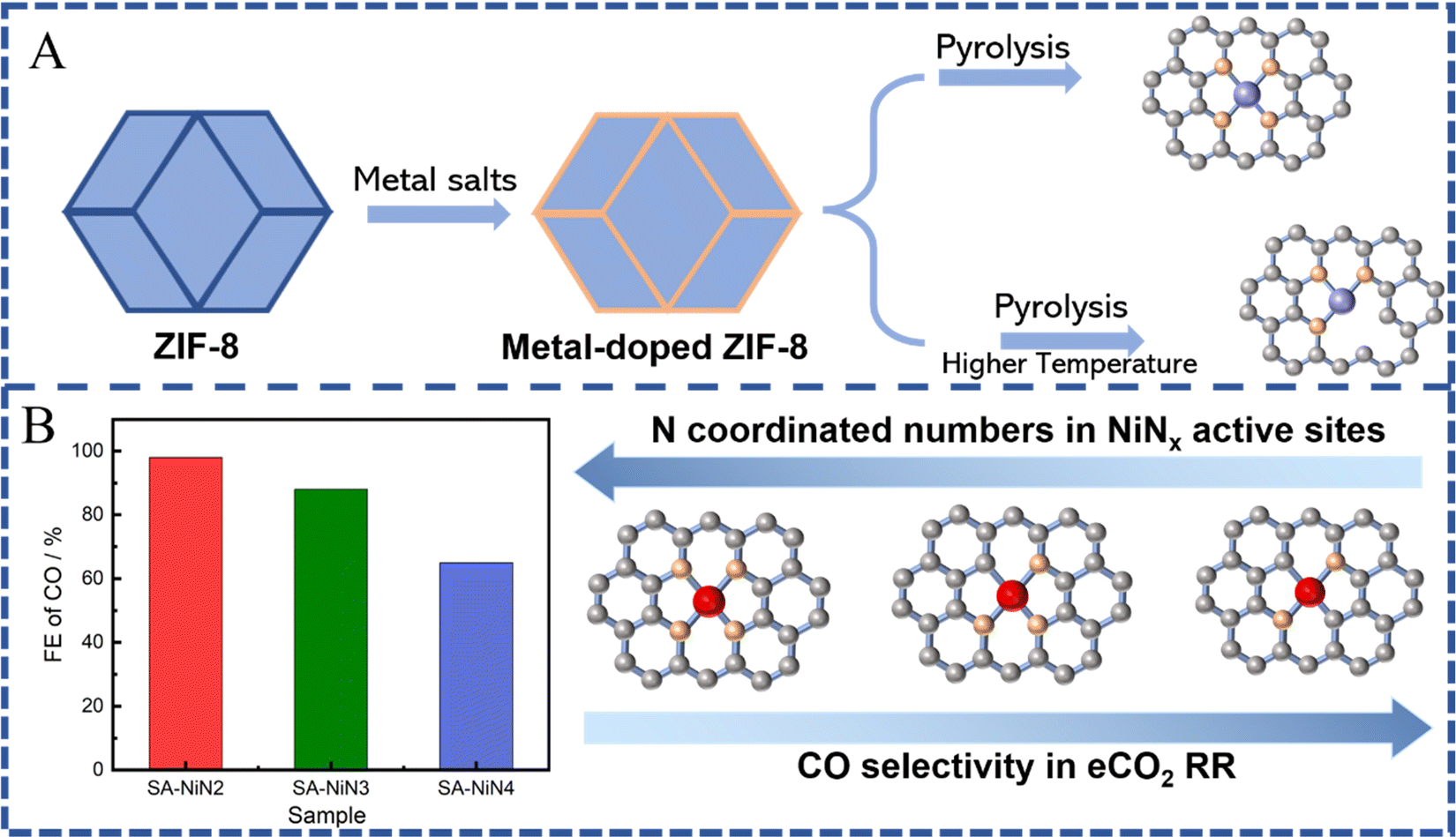 | ||
| Fig. 9 (A) Schematic illustration of ZIF-8 derived SACs with different N coordination numbers (C atoms: gray; N atoms: orange; metal atoms: purple/red). (B) FE of CO (at −0.80 V vs. RHE) obtained on Ni-SACs with different N coordination numbers. | ||
3.2 M-Nx-heteroatoms
Apart from the alteration of coordination numbers in SACs, the coordination environment of the metal centers is another important factor, which can also regulate the electronic and geometric structures of the SACs to influence the catalytic performance towards eCO2 RR.39,136,159,160 Due to the strong electronegative coordination of different heteroatoms, the strength of electronic metal–support interaction (EMSI) can be directly influenced, thus changing the valence electron distribution of SA sites, which can decrease the binding energy of the intermediates during the adsorption or desorption processes.39,159 Based on this idea, in addition to the N atoms, the site modification by using some other hybrid coordinated atoms (O,69,161,162 S,163 or Cl,164,165 among others) has also been investigated by researchers. The relative low electronegativity of the above mentioned hybrid coordinated atoms results in an electron transfer inside the active site of SACs with a heteroatom coordination, which consequently influences the binding energy for the different intermediates during their adsorption or desorption processes.39 For example, Chen et al. introduced an axial Cl atom into the FeN4 sites to modulate the electronic structure by following two steps: (1) pyrolyzing two-dimensional ZIF nanosheets with loaded Fe and (2) performing a low-temperature incubation in hydrochloric acid solution. The obtained FeN4Cl active sites facilitated the desorption of CO* and inhibited the adsorption of H*, resulting in an improved FE(CO) of 90.5% and a low overpotential of 490 mV in eCO2 RR.164 Sn-based SACs consisting of atomically dispersed SnN3O1 active sites supported on a N-rich carbon matrix (Sn-NOC) were also prepared for efficient eCO2 RR. Compared to the classic Sn-N4 configuration, which produced HCOOH and H2 as predominant products, SnN3O1 generated CO as the main product with a FECO of 94% at −0.7 V vs. RHE. DFT calculations showed that the atomic arrangement of SnN3O1 reduces the activation energy for *COO and *COOH formation, while increasing the energy barrier for HCOO* formation significantly, thereby facilitating CO2-to-CO conversion and suppressing HCOOH production.59 The axial halogen atoms with distinct electronegativity can break the symmetric charge distribution of MN4 active sites and facilitate the *COOH formation, leading to high selectivity towards CO.165 Therefore, a series of Ni-SACs with different halogen doping atoms were prepared by the pyrolysis of ZIF-8, in which due to the most electronegative Cl atoms, NiNC-Cl exhibited the highest FE of CO (94.7%), outperforming NiNC-Br and NiNC-I samples. DFT calculations revealed that the axial coordinated halogen atoms could facilitate *COOH formation, thereby boosting the CO2 reduction reaction (Fig. 10). In addition to the two-step introduction, the coordination of heteroatoms can be easily achieved after pyrolysis via the rational design of organic linkers with functional groups, providing a feasible strategy to dope the hybrid atoms into the SACs. Following the above idea, we have recently reported a facile route via using oxygen- and nitrogen-rich MOFs (IRMOF-3) as precursors to obtain the Fe–O and Fe–N chelation simultaneously, resulting in atomically dispersed axial O-coordinated FeN4 active sites.69 Compared to the FeN4 active sites without O coordination, the formed FeN4-O sites exhibit much better catalytic performance toward CO, reaching a maximum FECO of 95% at −0.50 V versus RHE and 30 h stability. Density functional theory calculations indicated that the axial O-coordination regulates the binding energy of intermediates in the reaction pathways, resulting in a smoother desorption of CO and increased energy for the competitive hydrogen production.69 Similarly, the introduction of oxygen ligands also brought remarkably high FE (78%) for the electrochemical conversion of CO2 to CH4 on Cu-SACs, surpassing most of the reported SACs, which stop at two-electron transfer related reduction processes. Theoretical calculations revealed that the high selectivity on formed CuN2O2 active sites with an oxygen ligand is due to the properly elevated CH4 and H2 energy barriers and the fine-tuned electronic structure of the Cu active sites.47 In summary, precisely controlling the coordination environment of SACs by heteroatom dopants has a significant effect on the catalytic performance towards eCO2 RR.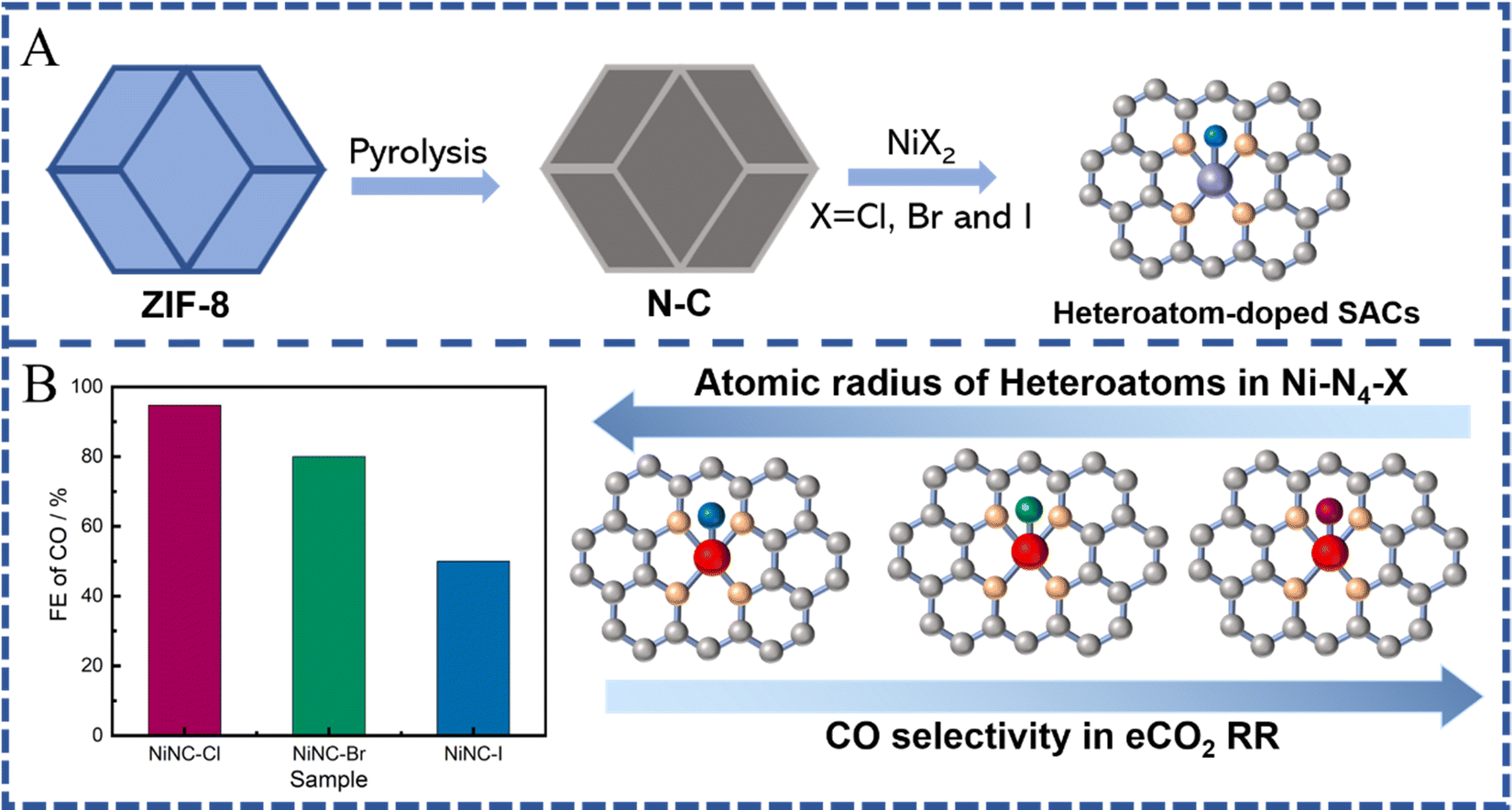 | ||
| Fig. 10 (A) Schematic illustration of ZIF-8 derived SACs with different heteroatom dopants (C atoms: gray; N atoms: orange; metal atoms: purple/red; heteroatoms: blue). (B) FE of CO (at −0.7 V vs. RHE) obtained on Ni-SACs with different heteroatom dopants (C atoms: gray; N atoms: orange; Ni atoms: red; Cl atom: dark red; Br atom: green; I atom: dark blue). | ||
3.3 M-M SACs
Although several reported monometallic active sites have already exhibited high selectivity for CO, they still suffer from sluggish kinetics for different steps of the eCO2 RR. For instance, Fe-N4 and Co-N4 sites have shown low onset potential for CO2 RR because of favorable kinetics for *COOH formation, but a relatively low FE for CO due to the strong binding energy of CO to the single Fe- or Co-atom sites.70,166–169 In addition, traditional SACs are sporadically dispersed on substrates without interaction between each other, leading to problems for C–C coupling reactions during eCO2 RR. To address the above issues, the design and synthesis of a multi-metal active center, possessing flexible active centers and synergy between adjacent metal sites, can bring unexpected catalytic performance to SACs.170–172 With potential abundant metal sites and nanopores, MOFs are certified as ideal templates for stabilizing and preparing dual-atom metal sites. In this section, MOF-derived dual-active sites will be divided into two types including binuclear homologous dual active sites (M1–M1) and binuclear heterologous dual active sites (M1–M2).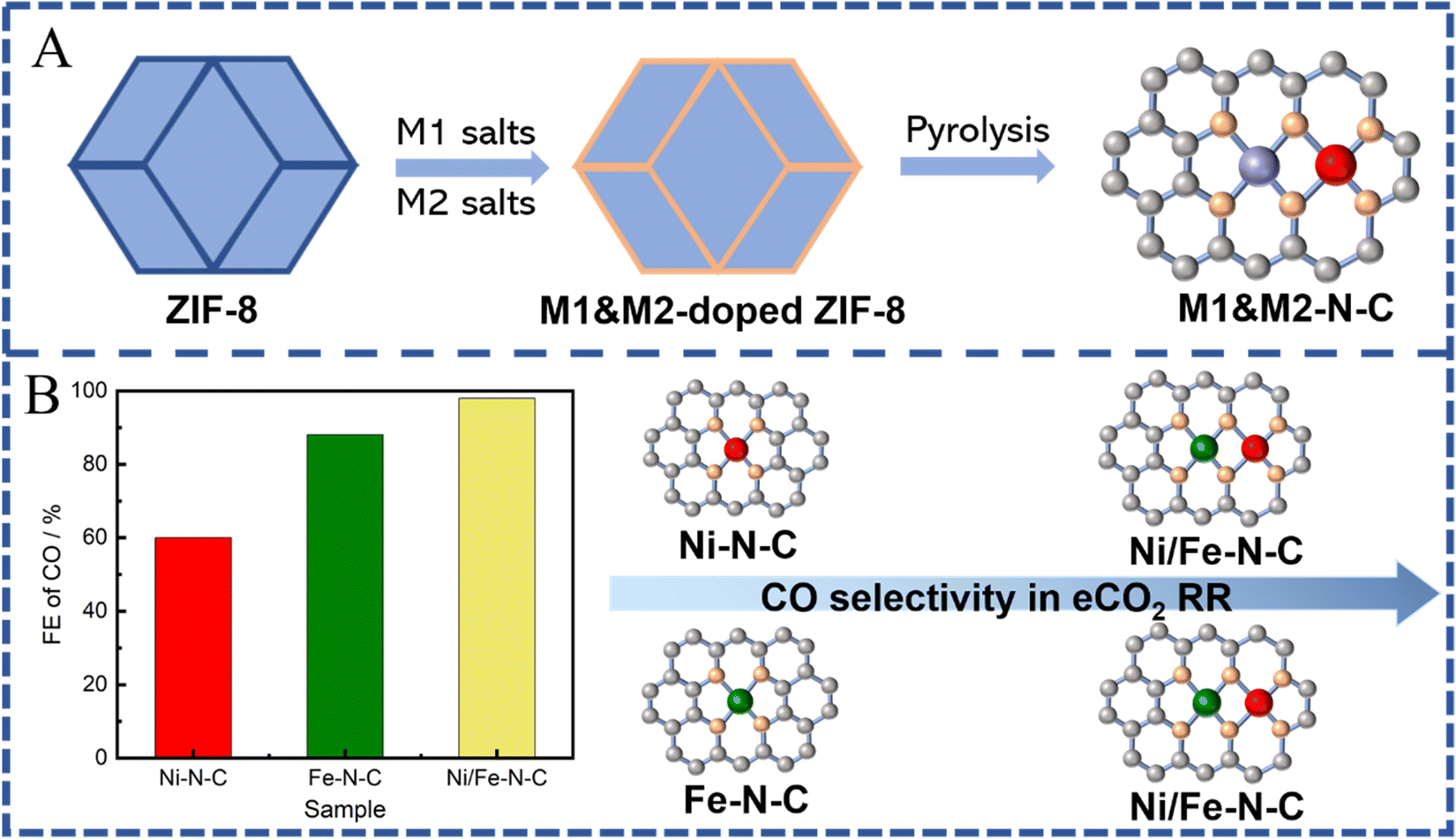 | ||
| Fig. 11 (A) Schematic illustration of ZIF-8 derived SACs with two metal sites (C atoms: gray; N atoms: orange; metal atoms: purple/red; heteroatoms: blue). (B) FE of CO (at −0.50 V vs. RHE) obtained on Ni-N-C, Fe-N-C and Ni/Fe-N-C samples (C atoms: gray; N atoms: orange; Ni atoms: red; Fe atom: green). | ||
4. Summary and outlook
Benefiting from their high-efficiency catalytic ability, SACs derived from MOFs are one of the most promising electrocatalyst types. In fact, they are expected to replace traditional metal catalysts for eCO2 RR. By using strategies that can regulate the electronic structure, the binding energy of adsorption/desorption for intermediates can be effectively adjusted and the energy barrier can be lowered, thus tuning the selectivity to achieve the targeted product. Although much progress has been made in the field of SAC preparation, many challenges remain. Firstly, from the practical application, the scaling up of synthesis strategies with commercial reactants and under lower temperature conditions needs to be further developed. The inferior accessibility due to the low density of single-atom active sites significantly hampers the catalytic performance in SACs. Increasing the metal load while avoiding agglomeration will be a bottleneck that needs to be surpassed in the development of new synthesis methods. In this exploration, we believe that the utilization of MOFs is, in this way, an attractive strategy. In addition, the enhancement of the intrinsic activity of the single-atom sites could also promote a better catalytic performance. The choice of the metal center and the regulation of its geometric and electronic environment will still be a key point to optimize the adsorption/desorption energy of the intermediates during eCO2 RR. In this way, the varied porous architectures and environment of MOFs provide more possibilities for enhancing the intrinsic nature of the single-atom sites.For the SACs derived from MOFs, some points still need to be given more attention during the design: (1) more suitable MOFs with a precise structure and a variety of functional groups are highly needed for creating SACs. (2) Heteroatom coordinated SACs should also be developed through heteroatom-rich MOF precursors to overcome the limitation from N-coordinated SAC materials. (3) Due to their complicated synthesis, the high price of ligands, and the limited stability of MOFs, we should also focus on the improved development of MOF precursors.
In addition, developing accurate characterization methodologies is urgently needed in order to better understand the local structure of SACs. In this way, for most of the available characterization methods, precise characterization down to the atomic scale still remains a challenge. The average structure rather than the local structure brings more uncertainties in the optimization of the preparation process. Moreover, without a clear knowledge of the nature of the active sites, research about catalytic mechanisms and theoretical computation is in a quandary. To understand the nature of single-atom sites and explore their reaction mechanisms, we will need to combine different advanced characterization methodologies, such as electron related microscopy and spectroscopy at the nano- and atomic scale with synchrotron X-ray experiments, and even take advantage of theoretical simulations (e.g. DFT) and artificial intelligence (AI) automated data analysis.181–183
Furthermore, we should take into account that during reaction processes, the environment around the active sites has dynamic variations, which have impacts on the properties of these active sites. In situ and operando techniques, such as in situ electron microscopy, in situ X-ray absorption spectroscopy and in situ X-ray photoelectron spectroscopy, may help to unearth SACs’ properties, which change during the catalytic reactions, to gain a comprehensive understanding of the catalysts and reaction mechanisms.
Likewise, as most of the current research on SACs has been limited to C1 products, more complex and higher value products should be explored. For more complex C2 products, which include more intermediates, protonation and electron transfers, the separated SACs serving as an optimized active site for a single process could not implement the multiple-intermediate conversion processes. We have shown that dual- and multiple-atom sites can show catalytic synergistic effects. Therefore, these synergistic properties should be used to achieve the more complex C2 product transformation. Additionally, the substrates interacting with SACs could regulate their geometric and electronic structure via the functionalization of the substrate precursor.
Finally, most of the SACs have shown their eCO2 RR performance in H-cells. The low solubility of CO2 in aqueous-fed systems limits the current density of CO2 conversion to less than 35 mA cm−2 which could not satisfy the requirements for industrial application. Additionally, the optimization of the catalysts only at low current density may improve the catalyst only for the wrong environment. Using flow cells with a gas-diffusion layer could increase the current densities under a more realistic operating condition.
From our point of view, all the above progress will contribute in the future to the final optimization of the eCO2 RR to meet large-scale industrial production.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
ICN2 acknowledges funding from Generalitat de Catalunya 2017 SGR 327. This study was supported by MCIN with funding from European Union NextGenerationEU (PRTR-C17.I1) and Generalitat de Catalunya. The authors acknowledge support from the project NANOGEN (PID2020-116093RB-C43), funded by MCIN/AEI/10.13039/501100011033/and by “ERDF A way of making Europe”, by the “European Union”. ICN2 is supported by the Severo Ochoa program from Spanish MCIN/AEI (Grant No. CEX2021-001214-S) and is funded by the CERCA Programme/Generalitat de Catalunya.References
- D. Voiry, H. S. Shin, K. P. Loh and M. Chhowalla, Nat. Rev. Chem., 2018, 2, 0105 CrossRef.
- G. Wang, J. Chen, Y. Ding, P. Cai, L. Yi, Y. Li, C. Tu, Y. Hou, Z. Wen and L. Dai, Chem. Soc. Rev., 2021, 50, 4993–5061 RSC.
- S. Navarro-Jaén, M. Virginie, J. Bonin, M. Robert, R. Wojcieszak and A. Y. Khodakov, Nat. Rev. Chem., 2021, 5, 564–579 CrossRef.
- M. Ding, R. W. Flaig, H.-L. Jiang and O. M. Yaghi, Chem. Soc. Rev., 2019, 48, 2783–2828 RSC.
- J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 RSC.
- P. Zhang, J. Tong and K. Huang, Materials and Processes for CO2 Capture, Conversion, and Sequestration, 2018, pp. 213–266 DOI:10.1002/9781119231059.ch5.
- R. Snoeckx and A. Bogaerts, Chem. Soc. Rev., 2017, 46, 5805–5863 RSC.
- S. C. Shit, I. Shown, R. Paul, K.-H. Chen, J. Mondal and L.-C. Chen, Nanoscale, 2020, 12, 23301–23332 RSC.
- J.-P. Jones, G. K. S. Prakash and G. A. Olah, Isr. J. Chem., 2014, 54, 1451–1466 CrossRef CAS.
- Z. Yuan, M. R. Eden and R. Gani, Ind. Eng. Chem. Res., 2016, 55, 3383–3419 CrossRef CAS.
- J. Liu, C. Guo, A. Vasileff and S. Qiao, Small Methods, 2017, 1, 1600006 CrossRef.
- Y. Wu, S. Cao, J. Hou, Z. Li, B. Zhang, P. Zhai, Y. Zhang and L. Sun, Adv. Energy Mater., 2020, 10, 2000588 CrossRef.
- Y. Wang, P. Han, X. Lv, L. Zhang and G. Zheng, Joule, 2018, 2, 2551–2582 CrossRef.
- D. D. Zhu, J. L. Liu and S. Z. Qiao, Adv. Mater., 2016, 28, 3423–3452 CrossRef.
- L. Zhang, Z.-J. Zhao and J. Gong, Angew. Chem., Int. Ed., 2017, 56, 11326–11353 CrossRef.
- A. D. Handoko, F. Wei, Jenndy, B. S. Yeo and Z. W. Seh, Nat. Catal., 2018, 1, 922–934 CrossRef.
- Y. Zheng, A. Vasileff, X. Zhou, Y. Jiao, M. Jaroniec and S.-Z. Qiao, J. Am. Chem. Soc., 2019, 141, 7646–7659 CrossRef CAS.
- X. Han, T. Zhang, M. Biset-Peiró, X. Zhang, J. Li, W. Tang, P. Tang, J. R. Morante and J. Arbiol, ACS Appl. Mater. Interfaces, 2022, 14, 32157–32165 CrossRef CAS PubMed.
- S. Jin, Z. Hao, K. Zhang, Z. Yan and J. Chen, Angew. Chem., Int. Ed., 2021, 60, 20627–20648 CrossRef CAS PubMed.
- E. E. Benson, C. P. Kubiak, A. J. Sathrum and J. M. Smieja, Chem. Soc. Rev., 2009, 38, 89–99 RSC.
- Q. Sun, C. Jia, Y. Zhao and C. Zhao, Chin. J. Catal., 2022, 43, 1547–1597 CrossRef CAS.
- C. S. Diercks, Y. Liu, K. E. Cordova and O. M. Yaghi, Nat. Mater., 2018, 17, 301–307 CrossRef CAS PubMed.
- Y.-J. Zhang, V. Sethuraman, R. Michalsky and A. A. Peterson, ACS Catal., 2014, 4, 3742–3748 CrossRef.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef.
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef PubMed.
- L. Liu and A. Corma, Chem. Rev., 2018, 118, 4981–5079 CrossRef.
- X. Wang, Y. Zhang, J. Wu, Z. Zhang, Q. Liao, Z. Kang and Y. Zhang, Chem. Rev., 2022, 122, 1273–1348 CrossRef PubMed.
- M. B. Gawande, P. Fornasiero and R. Zbořil, ACS Catal., 2020, 10, 2231–2259 CrossRef.
- D. Zhao, Z. Zhuang, X. Cao, C. Zhang, Q. Peng, C. Chen and Y. Li, Chem. Soc. Rev., 2020, 49, 2215–2264 RSC.
- B. Chang, L. Zhang, S. Wu, Z. Sun and Z. Cheng, Chem. Soc. Rev., 2022, 51, 3688–3734 RSC.
- J. Xia, B. Wang, J. Di, Y. Li, S.-Z. Yang, H. Li and S. Guo, Mater. Today, 2022, 53, 217–237 CrossRef.
- Y. Yang, Y. Yang, Z. Pei, K.-H. Wu, C. Tan, H. Wang, L. Wei, A. Mahmood, C. Yan, J. Dong, S. Zhao and Y. Chen, Matter, 2020, 3, 1442–1476 CrossRef.
- Y. Wang, H. Su, Y. He, L. Li, S. Zhu, H. Shen, P. Xie, X. Fu, G. Zhou, C. Feng, D. Zhao, F. Xiao, X. Zhu, Y. Zeng, M. Shao, S. Chen, G. Wu, J. Zeng and C. Wang, Chem. Rev., 2020, 120, 12217–12314 CrossRef PubMed.
- X. Li, L. Liu, X. Ren, J. Gao, Y. Huang and B. Liu, Sci. Adv., 2020, 6, eabb6833 CrossRef PubMed.
- T. N. Nguyen, M. Salehi, Q. V. Le, A. Seifitokaldani and C. T. Dinh, ACS Catal., 2020, 10, 10068–10095 CrossRef.
- L. Wang, W. Chen, D. Zhang, Y. Du, R. Amal, S. Qiao, J. Wu and Z. Yin, Chem. Soc. Rev., 2019, 48, 5310–5349 RSC.
- D. Zhou, X. Li, H. Shang, F. Qin and W. Chen, J. Mater. Chem. A, 2021, 9, 23382–23418 RSC.
- F. Doherty, H. Wang, M. Yang and B. R. Goldsmith, Catal. Sci. Technol., 2020, 10, 5772–5791 RSC.
- Q. Qu, S. Ji, Y. Chen, D. Wang and Y. Li, Chem. Sci., 2021, 12, 4201–4215 RSC.
- Y. Pan, R. Lin, Y. Chen, S. Liu, W. Zhu, X. Cao, W. Chen, K. Wu, W.-C. Cheong, Y. Wang, L. Zheng, J. Luo, Y. Lin, Y. Liu, C. Liu, J. Li, Q. Lu, X. Chen, D. Wang, Q. Peng, C. Chen and Y. Li, J. Am. Chem. Soc., 2018, 140, 4218–4221 CrossRef PubMed.
- H.-n Zhang, H.-q Wang, S.-p Jia, Q. Chang, N. Li, Y. Li, X.-l Shi, Z.-y Li and S.-l Hu, New Carbon Mater., 2022, 37, 734–742 CrossRef.
- J. Gu, C.-S. Hsu, L. Bai, H. M. Chen and X. Hu, Science, 2019, 364, 1091–1094 CrossRef PubMed.
- X. Qin, S. Zhu, F. Xiao, L. Zhang and M. Shao, ACS Energy Lett., 2019, 4, 1778–1783 CrossRef.
- H. B. Yang, S.-F. Hung, S. Liu, K. Yuan, S. Miao, L. Zhang, X. Huang, H.-Y. Wang, W. Cai, R. Chen, J. Gao, X. Yang, W. Chen, Y. Huang, H. M. Chen, C. M. Li, T. Zhang and B. Liu, Nat. Energy, 2018, 3, 140–147 CrossRef.
- X. Rong, H.-J. Wang, X.-L. Lu, R. Si and T.-B. Lu, Angew. Chem., Int. Ed., 2020, 59, 1961–1965 CrossRef.
- J. Jiao, R. Lin, S. Liu, W.-C. Cheong, C. Zhang, Z. Chen, Y. Pan, J. Tang, K. Wu, S.-F. Hung, H. M. Chen, L. Zheng, Q. Lu, X. Yang, B. Xu, H. Xiao, J. Li, D. Wang, Q. Peng, C. Chen and Y. Li, Nat. Chem., 2019, 11, 222–228 CrossRef PubMed.
- Y. Cai, J. Fu, Y. Zhou, Y.-C. Chang, Q. Min, J.-J. Zhu, Y. Lin and W. Zhu, Nat. Commun., 2021, 12, 586 CrossRef PubMed.
- F. Yang, P. Song, X. Liu, B. Mei, W. Xing, Z. Jiang, L. Gu and W. Xu, Angew. Chem., Int. Ed., 2018, 57, 12303–12307 CrossRef.
- S. Li, S. Zhao, X. Lu, M. Ceccato, X.-M. Hu, A. Roldan, J. Catalano, M. Liu, T. Skrydstrup and K. Daasbjerg, Angew. Chem., Int. Ed., 2021, 60, 22826–22832 CrossRef.
- S. Baskaran and J. Jung, Appl. Surf. Sci., 2022, 592, 153339 CrossRef.
- P. Huang, M. Cheng, H. Zhang, M. Zuo, C. Xiao and Y. Xie, Nano Energy, 2019, 61, 428–434 CrossRef.
- Q. He, J. H. Lee, D. Liu, Y. Liu, Z. Lin, Z. Xie, S. Hwang, S. Kattel, L. Song and J. G. Chen, Adv. Funct. Mater., 2020, 30, 2000407 CrossRef.
- N. Zhang, X. Zhang, Y. Kang, C. Ye, R. Jin, H. Yan, R. Lin, J. Yang, Q. Xu, Y. Wang, Q. Zhang, L. Gu, L. Liu, W. Song, J. Liu, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 13388–13393 CrossRef PubMed.
- J. Bok, S. Y. Lee, B.-H. Lee, C. Kim, D. L. T. Nguyen, J. W. Kim, E. Jung, C. W. Lee, Y. Jung, H. S. Lee, J. Kim, K. Lee, W. Ko, Y. S. Kim, S.-P. Cho, J. S. Yoo, T. Hyeon and Y. J. Hwang, J. Am. Chem. Soc., 2021, 143, 5386–5395 CrossRef PubMed.
- N. Zhang, X. Zhang, L. Tao, P. Jiang, C. Ye, R. Lin, Z. Huang, A. Li, D. Pang, H. Yan, Y. Wang, P. Xu, S. An, Q. Zhang, L. Liu, S. Du, X. Han, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 6170–6176 CrossRef CAS.
- Y. Li, C. Chen, R. Cao, Z. Pan, H. He and K. Zhou, Appl. Catal. B: Environ., 2020, 268, 118747 CrossRef CAS.
- W. Ni, Y. Gao, Y. Lin, C. Ma, X. Guo, S. Wang and S. Zhang, ACS Catal., 2021, 11, 5212–5221 CrossRef CAS.
- X. Zu, X. Li, W. Liu, Y. Sun, J. Xu, T. Yao, W. Yan, S. Gao, C. Wang, S. Wei and Y. Xie, Adv. Mater., 2019, 31, 1808135 CrossRef.
- J. Guo, W. Zhang, L.-H. Zhang, D. Chen, J. Zhan, X. Wang, N. R. Shiju and F. Yu, Adv. Sci., 2021, 8, 2102884 CrossRef CAS PubMed.
- E. Zhang, T. Wang, K. Yu, J. Liu, W. Chen, A. Li, H. Rong, R. Lin, S. Ji, X. Zheng, Y. Wang, L. Zheng, C. Chen, D. Wang, J. Zhang and Y. Li, J. Am. Chem. Soc., 2019, 141, 16569–16573 CrossRef CAS.
- C.-C. Hou, H.-F. Wang, C. Li and Q. Xu, Energy Environ. Sci., 2020, 13, 1658–1693 RSC.
- Y. Chen, S. Ji, C. Chen, Q. Peng, D. Wang and Y. Li, Joule, 2018, 2, 1242–1264 CrossRef CAS.
- L. Jiao and H.-L. Jiang, Chem, 2019, 5, 786–804 CAS.
- X. Liang, S. Ji, Y. Chen and D. Wang, iScience, 2022, 25, 104177 CrossRef CAS.
- Q. Fu, H. Saltsburg and M. Flytzani-Stephanopoulos, Science, 2003, 301, 935–938 CrossRef CAS.
- S. F. J. Hackett, R. M. Brydson, M. H. Gass, I. Harvey, A. D. Newman, K. Wilson and A. F. Lee, Angew. Chem., Int. Ed., 2007, 46, 8593–8596 CrossRef CAS.
- S. Sun, G. Zhang, N. Gauquelin, N. Chen, J. Zhou, S. Yang, W. Chen, X. Meng, D. Geng, M. N. Banis, R. Li, S. Ye, S. Knights, G. A. Botton, T.-K. Sham and X. Sun, Sci. Rep., 2013, 3, 1775 CrossRef.
- C. Zhao, X. Dai, T. Yao, W. Chen, X. Wang, J. Wang, J. Yang, S. Wei, Y. Wu and Y. Li, J. Am. Chem. Soc., 2017, 139, 8078–8081 CrossRef CAS.
- T. Zhang, X. Han, H. Liu, M. Biset-Peiró, J. Li, X. Zhang, P. Tang, B. Yang, L. Zheng, J. R. Morante and J. Arbiol, Adv. Funct. Mater., 2022, 32, 2111446 CrossRef CAS.
- T. Zhang, X. Han, H. Liu, M. Biset-Peiró, X. Zhang, P. Tan, P. Tang, B. Yang, L. Zheng, J. R. Morante and J. Arbiol, Energy Environ. Sci., 2021, 14, 4847–4857 RSC.
- J. Gao, Y. Hu, Y. Wang, X. Lin, K. Hu, X. Lin, G. Xie, X. Liu, K. M. Reddy, Q. Yuan and H.-J. Qiu, Small, 2021, 17, 2104684 CrossRef CAS.
- J. Ye, J. Yan, Y. Peng, F. Li and J. Sun, Catal. Today, 2023, 410, 68–84 CrossRef CAS.
- S. S. A. Shah, T. Najam, M. Wen, S.-Q. Zang, A. Waseem and H.-L. Jiang, Small Struct., 2022, 3, 2100090 CrossRef CAS.
- Y.-Z. Chen, R. Zhang, L. Jiao and H.-L. Jiang, Coord. Chem. Rev., 2018, 362, 1–23 CrossRef CAS.
- Z. Song, L. Zhang, K. Doyle-Davis, X. Fu, J.-L. Luo and X. Sun, Adv. Energy Mater., 2020, 10, 2001561 CrossRef CAS.
- H. Huang, K. Shen, F. Chen and Y. Li, ACS Catal., 2020, 10, 6579–6586 CrossRef CAS.
- H.-L. Jiang, B. Liu, Y.-Q. Lan, K. Kuratani, T. Akita, H. Shioyama, F. Zong and Q. Xu, J. Am. Chem. Soc., 2011, 133, 11854–11857 CrossRef CAS PubMed.
- L. Jiao, G. Wan, R. Zhang, H. Zhou, S.-H. Yu and H.-L. Jiang, Angew. Chem., Int. Ed., 2018, 57, 8525–8529 CrossRef CAS.
- L. Jiao, R. Zhang, G. Wan, W. Yang, X. Wan, H. Zhou, J. Shui, S.-H. Yu and H.-L. Jiang, Nat. Commun., 2020, 11, 2831 CrossRef CAS PubMed.
- W. Chen, J. Pei, C.-T. He, J. Wan, H. Ren, Y. Wang, J. Dong, K. Wu, W.-C. Cheong, J. Mao, X. Zheng, W. Yan, Z. Zhuang, C. Chen, Q. Peng, D. Wang and Y. Li, Adv. Mater., 2018, 30, 1800396 CrossRef.
- A. Han, B. Wang, A. Kumar, Y. Qin, J. Jin, X. Wang, C. Yang, B. Dong, Y. Jia, J. Liu and X. Sun, Small Methods, 2019, 3, 1800471 CrossRef.
- J. Hwang, Korean J. Chem. Eng., 2021, 38, 1104–1116 CrossRef CAS.
- Q. Yang, C.-C. Yang, C.-H. Lin and H.-L. Jiang, Angew. Chem., Int. Ed., 2019, 58, 3511–3515 CrossRef CAS PubMed.
- A. Zitolo, V. Goellner, V. Armel, M.-T. Sougrati, T. Mineva, L. Stievano, E. Fonda and F. Jaouen, Nat. Mater., 2015, 14, 937–942 CrossRef CAS PubMed.
- Y. Yang, K. Mao, S. Gao, H. Huang, G. Xia, Z. Lin, P. Jiang, C. Wang, H. Wang and Q. Chen, Adv. Mater., 2018, 30, 1801732 CrossRef.
- L. Fan, P. F. Liu, X. Yan, L. Gu, Z. Z. Yang, H. G. Yang, S. Qiu and X. Yao, Nat. Commun., 2016, 7, 10667 CrossRef CAS PubMed.
- Y.-Z. Chen, C. Wang, Z.-Y. Wu, Y. Xiong, Q. Xu, S.-H. Yu and H.-L. Jiang, Adv. Mater., 2015, 27, 5010–5016 CrossRef CAS.
- P. Yin, T. Yao, Y. Wu, L. Zheng, Y. Lin, W. Liu, H. Ju, J. Zhu, X. Hong, Z. Deng, G. Zhou, S. Wei and Y. Li, Angew. Chem., Int. Ed., 2016, 55, 10800–10805 CrossRef CAS PubMed.
- X. Wang, Z. Chen, X. Zhao, T. Yao, W. Chen, R. You, C. Zhao, G. Wu, J. Wang, W. Huang, J. Yang, X. Hong, S. Wei, Y. Wu and Y. Li, Angew. Chem., Int. Ed., 2018, 57, 1944–1948 CrossRef CAS PubMed.
- X. Dai, Z. Chen, T. Yao, L. Zheng, Y. Lin, W. Liu, H. Ju, J. Zhu, X. Hong, S. Wei, Y. Wu and Y. Li, Chem. Commun., 2017, 53, 11568–11571 RSC.
- Z. Li, Y. Chen, X. Lu, H. Li, L. Leng, T. Zhang and J. H. Horton, Nano Res., 2022, 15, 4023–4031 CrossRef CAS.
- S. Yuan, Z. Pu, H. Zhou, J. Yu, I. S. Amiinu, J. Zhu, Q. Liang, J. Yang, D. He, Z. Hu, G. Van Tendeloo and S. Mu, Nano Energy, 2019, 59, 472–480 CrossRef CAS.
- L. Jiao, J. Zhu, Y. Zhang, W. Yang, S. Zhou, A. Li, C. Xie, X. Zheng, W. Zhou, S.-H. Yu and H.-L. Jiang, J. Am. Chem. Soc., 2021, 143, 19417–19424 CrossRef CAS.
- M. Xiao, J. Zhu, L. Ma, Z. Jin, J. Ge, X. Deng, Y. Hou, Q. He, J. Li, Q. Jia, S. Mukerjee, R. Yang, Z. Jiang, D. S. Su, C. Liu and W. Xing, ACS Catal., 2018, 8, 2824–2832 CrossRef CAS.
- G. Li, Y. Liu, Q. Zhang, Q. Hu, W. Guo, X. Cao, Y. Dou, L. Cheng, Y. Song, J. Su, L. Huang and R. Ye, J. Mater. Chem. A, 2022, 10, 19254–19277 RSC.
- J. Zhang, W. Cai, F. X. Hu, H. Yang and B. Liu, Chem. Sci., 2021, 12, 6800–6819 RSC.
- Y. J. Sa, C. W. Lee, S. Y. Lee, J. Na, U. Lee and Y. J. Hwang, Chem. Soc. Rev., 2020, 49, 6632–6665 RSC.
- T. K. Todorova, M. W. Schreiber and M. Fontecave, ACS Catal., 2020, 10, 1754–1768 CrossRef CAS.
- M. Li, H. Wang, W. Luo, P. C. Sherrell, J. Chen and J. Yang, Adv. Mater., 2020, 32, 2001848 CrossRef CAS.
- D. Gao, H. Zhou, F. Cai, J. Wang, G. Wang and X. Bao, ACS Catal., 2018, 8, 1510–1519 CrossRef CAS.
- C. Yang, Y. Wang, L. Qian, A. M. Al-Enizi, L. Zhang and G. Zheng, ACS Appl. Energy Mater., 2021, 4, 1034–1044 CrossRef CAS.
- W. Wang, S. Wang, X. Ma and J. Gong, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC.
- K. Fan, Y. Jia, Y. Ji, P. Kuang, B. Zhu, X. Liu and J. Yu, ACS Catal., 2020, 10, 358–364 CrossRef CAS.
- C. Ding, A. Li, S.-M. Lu, H. Zhang and C. Li, ACS Catal., 2016, 6, 6438–6443 CrossRef CAS.
- X.-M. Hu, H. H. Hval, E. T. Bjerglund, K. J. Dalgaard, M. R. Madsen, M.-M. Pohl, E. Welter, P. Lamagni, K. B. Buhl, M. Bremholm, M. Beller, S. U. Pedersen, T. Skrydstrup and K. Daasbjerg, ACS Catal., 2018, 8, 6255–6264 CrossRef CAS.
- P. Hou, W. Song, X. Wang, Z. Hu and P. Kang, Small, 2020, 16, 2001896 CrossRef CAS.
- T. Liu, G. Wang and X. Bao, J. Phys. Chem. C, 2021, 125, 26013–26020 CrossRef CAS.
- M. D. Hossain, Y. Huang, T. H. Yu, W. A. Goddard Iii and Z. Luo, Nat. Commun., 2020, 11, 2256 CrossRef CAS PubMed.
- S. Chen, X. Li, C.-W. Kao, T. Luo, K. Chen, J. Fu, C. Ma, H. Li, M. Li, T.-S. Chan and M. Liu, Angew. Chem., Int. Ed., 2022, 61, e202206233 CAS.
- Z. Zhang and D. Wang, J. Mater. Chem. A, 2022, 10, 5863–5877 RSC.
- S. Liu, H. B. Yang, S.-F. Hung, J. Ding, W. Cai, L. Liu, J. Gao, X. Li, X. Ren, Z. Kuang, Y. Huang, T. Zhang and B. Liu, Angew. Chem., Int. Ed., 2020, 59, 798–803 CrossRef CAS PubMed.
- Y. Li, Y. Yan, Y. He and S. Du, ACS Appl. Mater. Interfaces, 2022, 14, 11457–11464 CrossRef CAS PubMed.
- Z. Ma, U. Legrand, E. Pahija, J. R. Tavares and D. C. Boffito, Ind. Eng. Chem. Res., 2021, 60, 803–815 CrossRef CAS.
- P. Bumroongsakulsawat and G. H. Kelsall, Electrochim. Acta, 2014, 141, 216–225 CrossRef CAS.
- X. Yu and P. G. Pickup, J. Power Sources, 2008, 182, 124–132 CrossRef CAS.
- M. Grasemann and G. Laurenczy, Energy Environ. Sci., 2012, 5, 8171–8181 RSC.
- S. Back, J.-H. Kim, Y.-T. Kim and Y. Jung, Phys. Chem. Chem. Phys., 2016, 18, 9652–9657 RSC.
- S. Zhao, S. Li, T. Guo, S. Zhang, J. Wang, Y. Wu and Y. Chen, Nano-Micro Lett., 2019, 11, 62 CrossRef.
- Y.-X. Duan, Y.-T. Zhou, Z. Yu, D.-X. Liu, Z. Wen, J.-M. Yan and Q. Jiang, Angew. Chem., Int. Ed., 2021, 60, 8798–8802 CrossRef CAS PubMed.
- H. Wu, Z. Li, Y. Liu, X. Zou, L. Yin and S. Lin, Sustain. Energy Fuels, 2021, 5, 5798–5803 RSC.
- W.-B. Li, C. Yu, X.-Y. Tan, S. Cui, Y.-F. Zhang and J.-S. Qiu, New Carbon Mater., 2022, 37, 277–287 CrossRef.
- P. Lu, X. Tan, H. Zhao, Q. Xiang, K. Liu, X. Zhao, X. Yin, X. Li, X. Hai, S. Xi, A. T. S. Wee, S. J. Pennycook, X. Yu, M. Yuan, J. Wu, G. Zhang, S. C. Smith and Z. Yin, ACS Nano, 2021, 15, 5671–5678 CrossRef CAS.
- Y. Zhao, J. Liang, C. Wang, J. Ma and G. G. Wallace, Adv. Energy Mater., 2018, 8, 1702524 CrossRef.
- T. Wang, J. Zhang, F. Li, B. Liu and S. Kawi, Mater. Rep.: Energy, 2022, 2, 100140 Search PubMed.
- T. Tang, Z. Wang and J. Guan, Adv. Funct. Mater., 2022, 32, 2111504 CrossRef CAS.
- Z. Wang, J. Zhao and Q. Cai, Phys. Chem. Chem. Phys., 2017, 19, 23113–23121 RSC.
- Y. Wu, Z. Jiang, X. Lu, Y. Liang and H. Wang, Nature, 2019, 575, 639–642 CrossRef CAS PubMed.
- H. Yang, Y. Wu, G. Li, Q. Lin, Q. Hu, Q. Zhang, J. Liu and C. He, J. Am. Chem. Soc., 2019, 141, 12717–12723 CrossRef CAS.
- A. Guan, Z. Chen, Y. Quan, C. Peng, Z. Wang, T.-K. Sham, C. Yang, Y. Ji, L. Qian, X. Xu and G. Zheng, ACS Energy Lett., 2020, 5, 1044–1053 CrossRef CAS.
- L. Han, S. Song, M. Liu, S. Yao, Z. Liang, H. Cheng, Z. Ren, W. Liu, R. Lin, G. Qi, X. Liu, Q. Wu, J. Luo and H. L. Xin, J. Am. Chem. Soc., 2020, 142, 12563–12567 CrossRef CAS PubMed.
- R. Liu and C. Streb, Adv. Energy Mater., 2021, 11, 2101120 CrossRef CAS.
- W. Xu, H. Tang, H. Gu, H. Xi, P. Wu, B. Liang, Q. Liu and W. Chen, J. Mater. Chem. A, 2022, 10, 14732–14746 RSC.
- F. Pan, H. Zhang, K. Liu, D. Cullen, K. More, M. Wang, Z. Feng, G. Wang, G. Wu and Y. Li, ACS Catal., 2018, 8, 3116–3122 CrossRef CAS.
- X. Yang, J. Cheng, X. Xuan, N. Liu and J. Liu, ACS Sustainable Chem. Eng., 2020, 8, 10536–10543 CrossRef CAS.
- X. Li, H. Rong, J. Zhang, D. Wang and Y. Li, Nano Res., 2020, 13, 1842–1855 CrossRef CAS.
- X. Liu, Y. Liu, W. Yang, X. Feng and B. Wang, Chem. – Eur. J., 2022, 28, e202201471 CAS.
- W. I. Choi, B. C. Wood, E. Schwegler and T. Ogitsu, Adv. Energy Mater., 2015, 5, 1501423 CrossRef.
- Y. Hou, Y.-L. Liang, P.-C. Shi, Y.-B. Huang and R. Cao, Appl. Catal. B: Environ., 2020, 271, 118929 CrossRef CAS.
- P. Lu, Y. Yang, J. Yao, M. Wang, S. Dipazir, M. Yuan, J. Zhang, X. Wang, Z. Xie and G. Zhang, Appl. Catal. B: Environ., 2019, 241, 113–119 CrossRef CAS.
- Z. Chen, X. Zhang, W. Liu, M. Jiao, K. Mou, X. Zhang and L. Liu, Energy Environ. Sci., 2021, 14, 2349–2356 RSC.
- L. Jiao, W. Yang, G. Wan, R. Zhang, X. Zheng, H. Zhou, S.-H. Yu and H.-L. Jiang, Angew. Chem., Int. Ed., 2020, 59, 22408–22413 CrossRef.
- H. Wang, G. Liu, C. Chen, W. Tu, Y. Lu, S. Wu, D. O’Hare and R. Xu, ACS Sustainable Chem. Eng., 2021, 9, 3792–3801 CrossRef CAS.
- F. Dvořák, M. Farnesi Camellone, A. Tovt, N.-D. Tran, F. R. Negreiros, M. Vorokhta, T. Skála, I. Matolínová, J. Mysliveček, V. Matolín and S. Fabris, Nat. Commun., 2016, 7, 10801 CrossRef PubMed.
- Y. Wang, M. Wang, Z. Zhang, Q. Wang, Z. Jiang, M. Lucero, X. Zhang, X. Li, M. Gu, Z. Feng and Y. Liang, ACS Catal., 2019, 9, 6252–6261 CrossRef CAS.
- N. Mohd Adli, W. Shan, S. Hwang, W. Samarakoon, S. Karakalos, Y. Li, D. A. Cullen, D. Su, Z. Feng, G. Wang and G. Wu, Angew. Chem., Int. Ed., 2021, 60, 1022–1032 CrossRef CAS PubMed.
- Z. Geng, Y. Cao, W. Chen, X. Kong, Y. Liu, T. Yao and Y. Lin, Appl. Catal. B: Environ., 2019, 240, 234–240 CrossRef CAS.
- Y. Zhou, L. Zheng, D. Yang, H. Yang, Q. Lu, Q. Zhang, L. Gu and X. Wang, Small Methods, 2021, 5, 2000991 CrossRef CAS.
- D. Karapinar, N. T. Huan, N. Ranjbar Sahraie, J. Li, D. Wakerley, N. Touati, S. Zanna, D. Taverna, L. H. Galvão Tizei, A. Zitolo, F. Jaouen, V. Mougel and M. Fontecave, Angew. Chem., Int. Ed., 2019, 58, 15098–15103 CrossRef CAS PubMed.
- H. Cheng, X. Wu, X. Li, X. Nie, S. Fan, M. Feng, Z. Fan, M. Tan, Y. Chen and G. He, Chem. Eng. J., 2021, 407, 126842 CrossRef CAS.
- W. Guo, X. Tan, J. Bi, L. Xu, D. Yang, C. Chen, Q. Zhu, J. Ma, A. Tayal, J. Ma, Y. Huang, X. Sun, S. Liu and B. Han, J. Am. Chem. Soc., 2021, 143, 6877–6885 CrossRef CAS.
- H. Shang, T. Wang, J. Pei, Z. Jiang, D. Zhou, Y. Wang, H. Li, J. Dong, Z. Zhuang, W. Chen, D. Wang, J. Zhang and Y. Li, Angew. Chem., Int. Ed., 2020, 59, 22465–22469 CrossRef CAS.
- H. Cheng, X. Wu, M. Feng, X. Li, G. Lei, Z. Fan, D. Pan, F. Cui and G. He, ACS Catal., 2021, 11, 12673–12681 CrossRef CAS.
- Y. Zhang, L. Jiao, W. Yang, C. Xie and H.-L. Jiang, Angew. Chem., Int. Ed., 2021, 60, 7607–7611 CrossRef CAS.
- X. Song, H. Zhang, Y. Yang, B. Zhang, M. Zuo, X. Cao, J. Sun, C. Lin, X. Li and Z. Jiang, Adv. Sci., 2018, 5, 1800177 CrossRef PubMed.
- Y.-N. Gong, L. Jiao, Y. Qian, C.-Y. Pan, L. Zheng, X. Cai, B. Liu, S.-H. Yu and H.-L. Jiang, Angew. Chem., Int. Ed., 2020, 59, 2705–2709 CrossRef CAS.
- T. Wang, X. Sang, W. Zheng, B. Yang, S. Yao, C. Lei, Z. Li, Q. He, J. Lu, L. Lei, L. Dai and Y. Hou, Adv. Mater., 2020, 32, 2002430 CrossRef CAS PubMed.
- S. Chen, Y. Li, Z. Bu, F. Yang, J. Luo, Q. An, Z. Zeng, J. Wang and S. Deng, J. Mater. Chem. A, 2021, 9, 1705–1712 RSC.
- X. Xuan, J. Cheng, X. Yang and J. Zhou, ACS Sustainable Chem. Eng., 2020, 8, 1679–1686 CrossRef CAS.
- Y. Wang, Y. Liu, W. Liu, J. Wu, Q. Li, Q. Feng, Z. Chen, X. Xiong, D. Wang and Y. Lei, Energy Environ. Sci., 2020, 13, 4609–4624 RSC.
- M. Ma and Q. Tang, J. Mater. Chem. C, 2022, 10, 15948–15956 RSC.
- Z. Chen, A. Huang, K. Yu, T. Cui, Z. Zhuang, S. Liu, J. Li, R. Tu, K. Sun, X. Tan, J. Zhang, D. Liu, Y. Zhang, P. Jiang, Y. Pan, C. Chen, Q. Peng and Y. Li, Energy Environ. Sci., 2021, 14, 3430–3437 RSC.
- X. Hu, S. Yao, L. Chen, X. Zhang, M. Jiao, Z. Lu and Z. Zhou, J. Mater. Chem. A, 2021, 9, 23515–23521 RSC.
- Y. Wu, C. Chen, X. Yan, X. Sun, Q. Zhu, P. Li, Y. Li, S. Liu, J. Ma, Y. Huang and B. Han, Angew. Chem., Int. Ed., 2021, 60, 20803–20810 CrossRef CAS PubMed.
- Z. Li, R. Wu, S. Xiao, Y. Yang, L. Lai, J. S. Chen and Y. Chen, Chem. Eng. J., 2022, 430, 132882 CrossRef CAS.
- J.-X. Peng, W. Yang, Z. Jia, L. Jiao and H.-L. Jiang, Nano Res., 2022, 15, 10063–10069 CrossRef CAS.
- L. Meng, E. Zhang, H. Peng, Y. Wang, D. Wang, H. Rong and J. Zhang, ChemCatChem, 2022, 14, e202101801 CrossRef CAS.
- R. Li and D. Wang, Adv. Energy Mater., 2022, 12, 2103564 CrossRef CAS.
- Y. Li, W. Shan, M. J. Zachman, M. Wang, S. Hwang, H. Tabassum, J. Yang, X. Yang, S. Karakalos, Z. Feng, G. Wang and G. Wu, Angew. Chem., Int. Ed., 2022, 61, e202205632 CAS.
- Y. Hua, B. Zhang, W. Hao and Z. Gao, Cell Rep. Phys. Sci., 2022, 3, 100703 CrossRef CAS.
- T. He, A. R. P. Santiago, Y. Kong, M. A. Ahsan, R. Luque, A. Du and H. Pan, Small, 2022, 18, 2106091 CrossRef CAS PubMed.
- Z. Liang, L. Song, M. Sun, B. Huang and Y. Du, Sci. Adv., 2021, 7, eabl4915 CrossRef CAS PubMed.
- D. Wu, B. He, Y. Wang, P. Lv, D. Ma and Y. Jia, J. Phys. D: Appl. Phys., 2022, 55, 203001 CrossRef.
- N. Zhang, X. Zhang, Y. Kang, C. Ye, R. Jin, H. Yan, R. Lin, J. Yang, Q. Xu, Y. Wang, Q. Zhang, L. Gu, L. Liu, W. Song, J. Liu, D. Wang and Y. Li, Angew. Chem., Int. Ed., 2021, 60, 13388–13393 CrossRef CAS.
- J. Zhao, J. Zhao, F. Li and Z. Chen, J. Phys. Chem. C, 2018, 122, 19712–19721 CrossRef CAS.
- X. Zhao, K. Zhao, Y. Liu, Y. Su, S. Chen, H. Yu and X. Quan, ACS Catal., 2022, 12, 11412–11420 CrossRef CAS.
- P. He, H. Feng, S. Wang, H. Ding, Y. Liang, M. Ling and X. Zhang, Mater. Adv., 2022, 3, 4566–4577 RSC.
- S. Li, A. Guan, C. Yang, C. Peng, X. Lv, Y. Ji, Y. Quan, Q. Wang, L. Zhang and G. Zheng, ACS Mater. Lett., 2021, 3, 1729–1737 CrossRef CAS.
- L. Lin, H. Li, C. Yan, H. Li, R. Si, M. Li, J. Xiao, G. Wang and X. Bao, Adv. Mater., 2019, 31, 1903470 CrossRef CAS.
- W. Xie, H. Li, G. Cui, J. Li, Y. Song, S. Li, X. Zhang, J. Y. Lee, M. Shao and M. Wei, Angew. Chem., Int. Ed., 2021, 60, 7382–7388 CrossRef CAS PubMed.
- L. Lin, T. Liu, J. Xiao, H. Li, P. Wei, D. Gao, B. Nan, R. Si, G. Wang and X. Bao, Angew. Chem., Int. Ed., 2020, 59, 22408–22413 CrossRef CAS.
- M. Botifoll, I. Pinto-Huguet and J. Arbiol, Nanoscale Horiz., 2022, 7, 1427–1477 RSC.
- Z. Liang, J. Wang, P. Tang, W. Tang, L. Liu, M. Shakouri, X. Wang, J. Llorca, S. Zhao, M. Heggen, R. E. Dunin-Borkowski, A. Cabot, H. B. Wu and J. Arbiol, Appl. Catal. B: Environ., 2022, 314, 121451 CrossRef CAS.
- Z. Liang, T. Zhang, P. Cao, T. Yoshida, W. Tang, X. Wang, Y. Zuo, P. Tang, M. Heggen, R. E. Dunin-Borkowski, J. R. Morante, A. Cabot, M. Yamashita and J. Arbiol, Chem. Eng. J., 2022, 442, 136129 CrossRef CAS.
Footnote |
| † Equal contribution. |
| This journal is © The Royal Society of Chemistry 2023 |