
Influence of surfactant on glass transition temperature of poly(lactic-co-glycolic acid) nanoparticles†
Guangliang
Liu
a,
Roberto
Martinez
a,
Anika
Bhatnagar
b and
Kathleen
McEnnis
 *a
*a
aOtto H. York Department of Chemical and Materials Engineering, New Jersey Institute of Technology, Newark, NJ 07102, USA. E-mail: mcennis@njit.edu
bMiddlesex Academy for Allied Health and Biomedical Sciences, Woodbridge, NJ 07095, USA. E-mail: bhatnagara@mcmsnj.net
First published on 3rd July 2023
Abstract
Poly(D,L-lactic-co-glycolic acid) (PLGA) is one of the most commonly used drug carriers in nanomedicines because of its biodegradability, biocompatibility and low toxicity. However, the physico-chemical characterization and study of drug release are often lacking the investigation of the glass transition temperature (Tg), which is an excellent indicator of drug release behavior. In addition, the residual surfactant used during the synthesis of nanoparticles will change the glass transition temperature. We thus prepared PLGA nanoparticles with polymeric (poly(vinyl alcohol) (PVA)) and ionic (didodecyldimethylammonium bromide (DMAB)) surfactant to investigate their influence on the glass transition temperature. Determination of Tg in dry and wet conditions were carried out. The use of concentrated surfactant during synthesis resulted in a larger amount of residual surfactant in the resulting particles. Increasing residual PVA content resulted in an increase in particle Tg for all but the most concentrated PVA concentrations, while increasing residual DMAB content resulted in no significant change in particle Tg. With the presence of residual surfactant, the Tg of particle and bulk samples measured in wet conditions is much lower than that in dry conditions, except for bulk PLGA containing the ionic surfactant, which may be related to the plasticizing effect of the DMAB molecules. Notably, the Tg of both particles in wet conditions is approaching physiological temperatures where subtle changes in Tg could have dramatic effects on drug release properties. In conclusion, the selection of surfactant and the remaining amount of surfactant are crucial parameters to utilize in designing the physico-chemical properties of PLGA particles.
1. Introduction
Polymeric particles are adaptable vehicles for drug delivery since the drug release kinetics can be purposely designed by modifying chemical properties, such as monomer composition and molecular weight, as well as physical properties, such as particle size, morphology, and porosity of the formulation. Among polymeric particles, poly(lactic-co-glycolic acid) (PLGA) has proven to be a very successful drug delivery system in the past few decades.1–4 PLGA is a random copolymer consisting of a defined ratio of poly(lactic acid) and poly(glycolic acid) that has been applied extensively in biomedical products due to its biocompatibility, biosafety, and degradability. Many PLGA formulations, from millimeter to nanometer size, are available by different manufacturing processes,5–9 and PLGA particles can encapsulate a wide range of drug molecules.PLGA particles are usually characterized by particle size, size distribution, zeta potential, morphology, and drug loading efficiency, as these are crucial factors that contribute to the drug release profiles.10 Glass transition temperature (Tg), however, is an underutilized property in PLGA particle characterization.11 When the surrounding temperature of PLGA is higher than the glass transition temperature, more drug is rapidly released shortly after incubation as a burst release of drug. This increase in burst release is because the PLGA transforms from a glassy solid into a soft rubbery material while above the Tg, allowing for more drug loaded inside the PLGA to be suddenly released due to the enhanced polymer chain mobility. The long-term drug release is based on PLGA's biodegradation. The kinetics of drug release associated with biodegradation is affected by many factors, such as the molecular weight of the copolymer,12 monomer ratio, hydrophobicity of drug, manufacturing process, and release medium.5,6,13 It is also indicated that the closer the Tg is to body temperature, the worse long-term drug release will be.14
The analysis of Tg in PLGA particles is complicated by the presence of a surfactant. PLGA particle preparation techniques, such as nanoemulsion, require the use of a surfactant to stabilize the system and prevent the aggregation of the particles. Polyvinyl alcohol (PVA) is the most commonly used surfactant in PLGA particle formulation. However, previous studies have shown that a portion of PVA stays with PLGA nanoparticles even after washing.15,16 Notably, residual PVA can change the particle Tg by introducing a second polymeric component to the system. As an alternative surfactant, didodecyldimethylammonium bromide (DMAB) is also widely implemented to generate PLGA particles.17–20 DMAB is a cationic surfactant that will impart a positive charge on the particle surface, which has been proposed to enhance interactions between cells and nanoparticles. For instance, Jin et al. made drug loaded 100 nm PLGA nanoparticles with DMAB and achieved a significant increase in cellular uptake due to the positively charged surface.19 However, residual surfactant on the surface of a NP can alter the interfacial interactions and mobility of the polymer chain, thus affecting Tg and therefore drug release properties.
Another complication in analyzing polymeric particles’ Tg is the addition of small molecules which can act as plasticizers.21,22 The mechanical properties of polymers can be changed through the addition of small compounds that can embed themselves into the polymer matrix and distribute throughout it. Plasticizers can lower the intermolecular interactions of polymers, resulting in an increase in mobility of polymer chains, and as a result, a decrease in Tg.23,24 When PLGA nanoparticles are administered as drug delivery vehicles in biological fluids, the presence of water can have a significant impact on the Tg of PLGA, as water can act as a plasticizer and increase the mobility of the polymer chains. Therefore, it is important to measure Tg of PLGA nanoparticles in wet conditions to gain a better understanding of their behavior during drug delivery and to optimize their design for biomedical applications.
In the present study, we prepared PLGA nanoparticles with PVA (PVA-PLGA NPs) and DMAB (DMAB-PLGA NPs) and determined the amount of residual surfactant present. Additionally, the Tg of PVA-PLGA NPs and DMAB-PLGA NPs in both dry and wet conditions were determined. Furthermore, the Tg of the bulk was compared to the Tg of the nanoparticles to illustrate the deviations due to 3D confinement and the importance of measuring Tg for designing drug delivery nanoparticles.
2. Methodology
2.1 Materials
The PLGA (85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15, molecular weight: 50000–75000 Da), PVA (molecular weight: 32000–50000, 87–89% hydrolyzed), and chloroform (HPLC grade, amylene stabilized) were purchased from Sigma-Aldrich. DMAB (99% purity), DMSO-d6 (methyl sulfoxide, 99.9% atom D), and chloroform-d (99.8% atom D, 0.03 v/v% TMS) were purchased form Thermo Scientific. Ultrapure water (Thermo Scientific Smart2Pure 3 UV/UF) was applied in particle preparation and purification.
:15, molecular weight: 50000–75000 Da), PVA (molecular weight: 32000–50000, 87–89% hydrolyzed), and chloroform (HPLC grade, amylene stabilized) were purchased from Sigma-Aldrich. DMAB (99% purity), DMSO-d6 (methyl sulfoxide, 99.9% atom D), and chloroform-d (99.8% atom D, 0.03 v/v% TMS) were purchased form Thermo Scientific. Ultrapure water (Thermo Scientific Smart2Pure 3 UV/UF) was applied in particle preparation and purification.
2.2 Method
DMAB-PLGA NPs was synthesized by modifying the method presented by Kwon. et al.17 In detail, 20 mg of PLGA was dissolved in 1 ml chloroform. Then, the organic phase was added into 4 ml of aqueous solution with varying amounts of DMAB (0.25–2 w/v%). The mixture was emulsified by tip sonication under 100 amplitude for 20 minutes (1 second on and 4 second off, 100 minutes of total run time). The obtained emulsion was poured into 8 ml of ultrapure water and stirred overnight. The particles were washed by five centrifugation cycles using an Eppendorf Centrifuge 5430 R at 22136 RCF for one hour. After each centrifugation cycle, the supernatant was removed and the particles were resuspended in pure water.
For measuring the Tg of non-PVA PLGA nanoparticles, nanoprecipitation was used to produce surfactant free nanoparticles. Briefly, 20 mg of PLGA was measured and dissolved in 2 ml of acetone to form the organic phase. Subsequently, the organic phase was introduced into 100 ml of ultrapure water with stirring. The resulting mixture was stirred overnight to allow complete evaporation of the organic solvent. Finally, the samples were filtered using a 40 μm filter to eliminate any large debris.
Scanning electron microscopy (SEM, JSM-7900F) was used to observe the morphology of PVA-PLGA and DMAB-PLGA NPs. 10 μl of particle suspension was deposited on a small piece of aluminium foil and dried in the fume hood. Then, the sample was coated with carbon to generate a conductive surface and imaged by SEM.
3. Results and discussion
3.1 Characterization of PLGA nanoparticle diameter
PLGA nanoparticles were prepared with different polymeric (PVA) and cationic (DMAB) surfactant concentration. The mean size as a function of the surfactant concentration used during synthesis is displayed in Fig. 1A. Representative SEM images of particles prepared by PVA (Fig. 1B) and DMAB (Fig. 1C) confirmed that particles were successfully synthesized. For PVA-PLGA NPs, PVA concentrations (CPVA) of 0.5%, 1%, 1.5%, 2% and 2.5% in the aqueous phase were used during nanoemulsion preparation of the particles. PVA plays an important role in nanoparticle preparation by preventing aggregation of the PLGA.27,28 The particle diameter shows a steady decrease as higher PVA concentration was used during particle synthesis, with mean diameters of 146.4 ± 5.1 nm, 136.0 ± 4.7 nm, 125.1 ± 3.3 nm, and 120.7 ± 6.0 nm, with increasing CPVA from 0.5 v/w% to 2.0 v/w%. The particle diameter with 2.5 w/v% initial PVA of 118.4 ± 4.0 nm is statistically similar to that of 2.0 w/v% initial PVA (Fig. S2, ESI†). Miladi et al. has reported a decrease in the mean diameter of nanoparticles with increasing PVA concentrations, explained by increasing viscosity of the aqueous phase with increasing PVA concentration, preventing coalescence of emulsion droplets formed by emulsification.29 Therefore, a rise in the concentration of PVA ensures a greater stability of the system, and as a result, a reduction in the coalescence of the emulsion.28 It has also been suggested that when increasing the surfactant concentration, a relatively small amount of stabilizer is adsorbed at the interface, while the surplus remains within the aqueous phase without exerting any notable influence on the emulsification process,17,30 which explains the similar particle diameters found with 2.0 w/v% and 2.5 w/v% initial PVA concentration. Moreover, Sahoo et al. discovered a positive correlation between initial PVA concentration used during nanoemulsion particle synthesis and residual surfactant present on the resulting nanoparticles; even after purification, a certain amount of residual PVA was associated with the nanoparticles. As a result, increased residual PVA will stabilize the emulsion droplets, leading to smaller nanoparticles.15 For DMAB-PLGA nanoparticles, the diameter fluctuates as the initial DMAB concentration increases (Fig. 1A). The error bars from each data point suggest that the particle diameter of DMAB-PLGA particles is not as stable as PVA-PLGA particles. It has been reported that the utilization of 0.5% DMAB in ethyl acetate during the nanoemulsion process resulted in the production of particles of approximately 50 nm. Subsequently, an increase in DMAB concentration to 1% led to a corresponding increase in particle size of 102 nm, suggesting that the cationic nature of DMAB favors its retention in the aqueous phase rather than participating in the emulsification process, resulting in DMAB playing a less significant role in emulsification.31 In Cooper et al.'s study, DMAB-stabilized PLGA nanoparticles were synthesized with concentrations ranging from 0.1% to 1%. The investigation demonstrated that the nanoparticle size was maximized when utilizing DMAB concentrations between 0.25% and 0.75% w/v, with particle aggregation observed at DMAB concentrations exceeding 1%.32 However, Kwon et al. reported that DMAB-PLGA particle diameter decreases substantially from 1 w/v% to 2 w/v% initial DMAB concentration.17 The results presented here also support a significant decrease in diameter occurring at higher initial DMAB concentration of 1.5 w/v%. However, the trend does not continue at 2 w/v% initial DMAB concentration. | ||
| Fig. 1 Size of PLGA nanoparticles made using PVA (blue) and DMAB (green) (mean ± SD, n = 3). Size decreases with increasing PVA concentration while particles prepared with DMAB have no clear trend and large error bars, indicating that the size of DMAB-PLGA particles is more variable. SEM image of PVA-PLGA NPs (B) and SEM image of DMAB-PLGA NPs (C). | ||
3.2 Evaluation of residual surfactant
A significant amount of residual surfactant is known to remain on the PLGA particle surface or within the particle,15 despite washing, and this has been shown to alter particle attributes such intracellular uptake and hydrophobicity. Typical washing procedures of PLGA NPs usually consists of 3 washing cycles.33 However, our Tg results of 3-times washed particles showed a second Tg around 68 °C (Fig. 2), corresponding to the Tg of PVA, which indicated that there was still a significant amount of non-blended PVA in PLGA NPs. Thus, increasing the washing cycles was needed to remove free PVA from PLGA nanoparticles so only one Tg was observed. | ||
| Fig. 2 mDSC thermograph of PVA-PLGA NPs washed 3 times (A) and 5 times (B). A second transition was observed from 3-times washed PLGA particles (A). Unblended PVA was removed after 5 washes (B). | ||
1H NMR was used to determine the amount of residual surfactant. The 1H NMR spectrum of 2% PVA-PLGA is shown in Fig. 3, where peak A at 5.2 ppm is related to the CH of the lactide unit while peak B around 4.8 ppm corresponds to the CH2 of the glycolide unit.34,35 The peak (C) around 3.8 ppm is assigned to the CH from PVA. The peaks at 3.3 ppm and 2.5 ppm are assigned to H2O and DMSO, respectively. Additionally, 1H NMR was also performed on DMAB-PLGA nanoparticles. The spectrum of 2% DMAB-PLGA is presented in Fig. 3, where the peak (D) at 3.4 ppm is assigned to the two methyl groups in the DMAB molecule. The sharp peak at 7.3 ppm is related to chloroform. The following equations were performed to calculate residual surfactant:
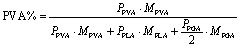 | (1) |
 | (2) |
 | ||
| Fig. 3 NMR spectrums of 2% PVA-PLGA NPs in DMSO-d6 (red) and 2% DMAB-PLGA NPs in CDCl3 (blue). | ||
 | ||
| Fig. 4 Residual surfactant evaluation by mDSC and NMR (A). Residual PVA was calculated by the linear equation and Fox equation (B) which determined the lower and upper limits of weight percentage. NMR was also used to determine residual DMAB (C) and residual PVA (D) (mean ± SD, n = 3). | ||
Despite the presence of one Tg in the DSC scan, residual PVA still remains but has blended with the PLGA, increasing the Tg. Thus, mDSC can also be used to estimate the quantity of residual PVA that has blended with the PLGA in the particles (Fig. 4A). Using mDSC to determine the Tg of PVA bulk, PVA-PLGA nanoemulsion particles, and non-surfactant PLGA particles, the amount of residual surfactant can be estimated. For this analysis, only the third heating curves of the mDSC scans were analyzed, which was confirmed to be only from PLGA bulk (Fig. 5), thus the influences from the confinement as a particle, interfacial effects, and the thermal history on the Tg were able to be eliminated. The bulk Tg of PVA-PLGA particles is greater than the bulk Tg for surfactant-free PLGA particles, indicating that the PVA (Tg: 68.8 °C) had blended with the PLGA. Accordingly, the following formulas were used to calculate the predicted weight % of residual PVA:
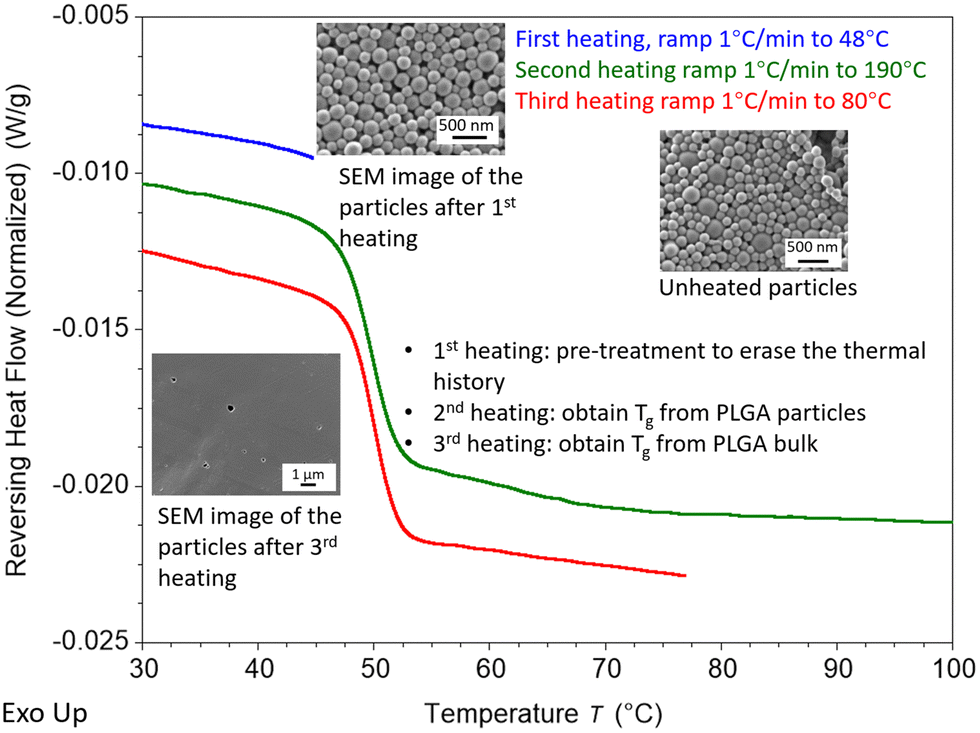 | ||
| Fig. 5 DSC procedure and SEM images of PLGA nanoparticles. The blue curve represents the pre-treatment to erase thermal history while keeping the morphology of particles. Particle Tg was obtained from the second heating cycle (green curve). Bulk Tg was obtained from the third heating cycle (red curve). The SEM image of the sample after the third heating confirmed no particles existed in the last heating cycle. Curves shifted vertically for easier viewing. | ||
Linear equation:
| Tg,blend = w1·T1 + w2·T2 | (3) |
Fox equation:36
 | (4) |
3.3 Determination of mDSC annealing temperature
Modulated DSC (mDSC) was used to determine the nanoparticle Tg and the bulk Tg of the sample. The ΔTg is calculated as the nanoparticle Tg - bulk Tg. A pre-treatment step was applied for Tg measurement of PLGA nanoparticles and bulk before the heating ramp to erase the thermal history. The pre-treatment step is similar to that which Zhang et al. and Christie et al. implemented in order to determine the Tg of PS particles; the samples are subjected to a pre-treatment step at a temperature that is relatively close to the Tg in order to erase the thermal history of the samples, but the samples will continue to exist in the form of particles.38,39 In this study, a pre-treatment temperature of 48 °C is used for erasing the thermal history of the particles in dry conditions and 38 °C for particles in wet conditions. The morphology of particles after pre-treatment, shown in the SEM inserts in Fig. 5, confirms that particles still remain after the pre-treatment step. The pre-treatment process is included in the mDSC run as the first heating ramp. The nanoparticle Tg with thermal history erased is determined by using the second heating ramp. During the second heating, the sample is heated to 190 °C in dry conditions to complete the transformation of the sample into bulk PLGA. Even though crystallization is not anticipated in the PLGA used in this study, the second heating cycle brings the sample above the Tm of PLGA in order to guarantee that the bulk state is attained. To determine the bulk PLGA Tg, the third heating ramp is brought up to 80 °C for a clear analysis of the Tg. Similarly, for wet measurements the Tg of the nanoparticles is obtained from the second heating cycle of the mDSC measurements. To obtain the Tg of wet bulk, samples were freeze-dried in the pan to remove water and then heated to 190 °C to completely transform the particles to the bulk stage. Lastly, the post-treated sample was measured in wet conditions to obtain the corresponding Tg of the bulk sample. From intrinsic viscosity measurements it was determined that the DSC procedure did not result in significant degradation (Fig. S1, ESI†).3.4 T g of PVA-PLGA and DMAB-PLGA nanoparticles
Fig. 6 shows the Tg of PVA-PLGA NPs and DMAB-PLGA NPs in dry conditions with varying weight fraction of residual surfactant (as determined by NMR) and the Tg with the corresponding particle diameters. In Fig. 6A, Tg increases with increasing residual PVA, except the Tg from particles synthesized by 2.5 w/v% PVA in the final data point with a weight fraction of 0.087 residual PVA. PVA-PLGA NPs prepared with concentrated surfactant have more residual surfactant, which results in an increased overall Tg of the binary system since the Tg of PVA is higher than PLGA's (Fig. S6, ESI†). However, phase separation can occur if the second polymeric component reaches its solubility limit, which suggests that not all the PVA is blended with the PLGA for the last data point. Fig. 6B indicates an opposite trend in Tgvs. weight fraction of residual surfactant because the mean diameter of the NPs decreases with increasing PVA concentration. The statistical analysis revealed a significant difference between the initial and final data points. The error bars, which represent the standard deviation from a sample size of n = 3, indicate consistent Tg results, despite the small magnitude of difference of approximately one degree between groups. The mDSC technique has been shown to yield more accurate results than standard DSC due to the removal of thermal relaxation which would result in Tg shift.26Fig. 6C shows no clear trend between the Tg of DMAB-PLGA NPs in dry conditions and residual DMAB, as one-way ANOVA and Tukey's post-test show no statistically significant difference among this data (Fig. S5, ESI†). Similarly, no significant difference is observed in Fig. 6D with particle diameter ranging from 110 nm to 158 nm. | ||
| Fig. 6 T g of dry particles vs. residual surfactant (A and C) and Tg of dry particles vs. particle diameter (B and D) (mean ± SD, n = 3). | ||
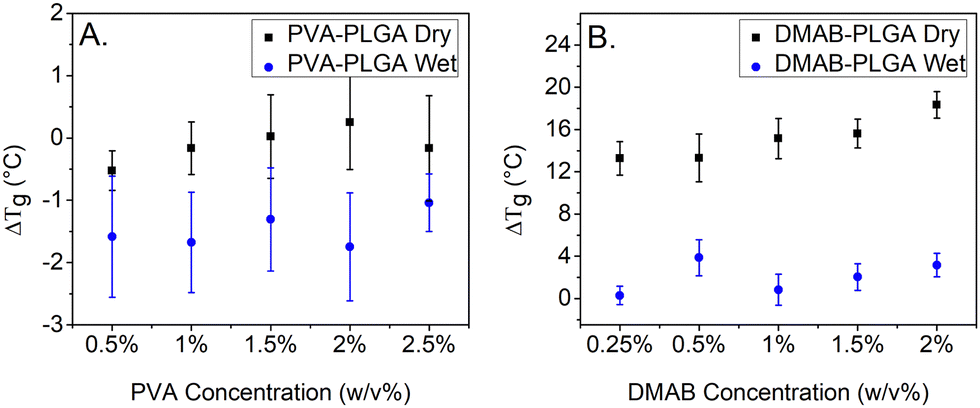
As shown in Fig. 7A, the Tg of PVA-PLGA bulk in dry conditions experiences a subtle increase from 0.5% to 2.0% initial PVA concentration and a decrease from 2.0% to 2.5% initial PVA concentration compared to the Tg of NPs, but a trend still exists. The ΔTg of PVA-PLGA NPs in dry conditions gives a similar trend. However, the Tg of DMAB-PLGA bulk experiences a significant reduction (Fig. 7C) after the second heating cycle, which could be explained by the heating process promoting the penetration of DMAB into the PLGA, plasticizing the system. The decreasing trend is caused by the larger amount of residual DMAB in the particles. The positive correlation of ΔTg and DMAB concentration results from the opposite trends present in the Tg of DMAB-PLGA dry bulk and DMAB-PLGA dry nanoparticles. The ΔTg of PVA-PLGA samples (Fig. 7B) would suggest no clear confinement effect on Tg, which in contrast to the Tg results for polystyrene nanoparticles.38–40 However, the size range of PLGA nanoparticles in this study is relatively narrow, within 50 nm, which limits the ability to observe a significant size effect. Additionally, the presence of residual surfactant affecting the Tg may be masking any confinement effects. Specifically, the ΔTg between PVA-PLGA and DMAB-PLGA is markedly different in the presence of surfactant.
 | ||
| Fig. 7 Particle Tg and bulk Tg measured in dry condition for both PVA-PLGA (A) and DMAB-PLGA (C). ΔTg of PVA-PLGA (B) and DMAB-PLGA (D) in dry conditions (mean ± SD, n = 3). | ||
Water is well-known to plasticize many polymers, including PLGA, and will therefore have a significant impact on Tg when present.12,41 Similarly, the results presented here with PLGA particles show that the Tg of nanoparticles synthesized with PVA (Fig. 8A) and DMAB (Fig. 8C) is much lower when evaluated in water compared to dry conditions, suggesting that these materials will undergo plasticization when exposed to fluids during use as a drug delivery vehicle. Zhang et al. reported that polystyrene nanoparticles exhibit a Tg reduction in wet measurement conditions, which was caused by a mobile layer on the particle surface (∼30 nm thickness).38 It is likely that a similar phenomenon may be occurring here where the presence of water penetrates the surface of PLGA nanoparticles freeing up the polymer chains and increasing the chain mobility. They also suggested that significant Tg reduction was observed when the particle diameter was less than 600 nm. Given that wet conditions are crucial for drug delivery particles, the Tg's proximity to body temperature makes it more likely that the particle will experience an undesired burst release of its drug payload. A Tg of PLGA nanoparticles that is around 37 °C is important to consider for drug delivery because it is close to the physiological temperature of the human body. This means that when PLGA-based drug delivery systems are administered in the body, the polymer will transition from a glassy, solid state to a rubbery, flexible state, which allows for rapid release of the drug. Notably, the Tg of DMAB-PLGA bulk is around 37 °C (Fig. 8D) even measured in dry conditions, which is significantly lower than that of PVA's (Fig. 8B). One possible explanation is that the PLGA became more liquid-like during the second heating, allowing the surface DMAB molecules to penetrate the particle matrix. Unlike PVA, which can blend with PLGA in the nanoparticles to increase the Tg, DMAB molecule with positively charged quaternary ammonium groups can be adsorbed by PLGA with the negatively charged carboxylate groups,42 increasing the free volume between polymer chains and increasing polymer mobility. This plasticizing effect can make the PLGA polymer more flexible. Thus, DMAB is a small molecule that can act as a plasticizer to lower the Tg. Representative curves of the mDSC scans are plotted in Fig. 9A, and a clear Tg shift is observed from the DMAB bulk (green dotted line). Fig. 9B shows the ΔTg of DMAB-PLGA is significantly higher than that of PVA-PLGA.
 | ||
| Fig. 8 T g of particles in dry and wet conditions vs. surfactant concentrations (A and C). Tg of bulk in dry and wet condition vs. surfactant concentrations (B and D) (mean ± SD, n = 3). | ||
 | ||
| Fig. 9 Examples of mDSC plots (A) of PLGA nanoparticles (blue line) and bulk (blue dotted line) for 2% PVA-PLGA NPs (blue) and 2% DMAB-PLGA NPs (green), and (B) ΔTg of dry PLGA nanoparticles compared to the bulk Tg for nanoparticles made with varying amounts of PVA (blue) and DMAB (green). ΔTg of DMAB-PLGA nanoparticles is significantly higher than the ΔTg of PVA-PLGA nanoparticles (mean ± SD, n = 3). Curves shifted vertically for easier viewing. | ||
Analysis of the Tg of PLGA nanoparticles in this study (Fig. 10A and B) also show that the ΔTg is smaller in water compared to dry for both sets of particles, indicating that the shift in nanoparticle Tg in water is attributable to interfacial effects in addition to plasticization, suggesting that further investigation of surfactant free PLGA nanoparticles is needed. Notably, ΔTg of dry DMAB samples is significantly lower compared to other ΔTg's, in part because the Tg bulk of dry DMAB samples was particularly low. The possible reason is described above which is due to the plasticizing effect of DMAB.
 | ||
| Fig. 10 ΔTg of PLGA nanoparticles made with varying amounts of PVA (A) and DMAB (B) measured by mDSC either dry or in water. ΔTg represents the difference between nanoparticle Tg and bulk Tg (mean ± SD, n = 3). | ||
4. Conclusion
The influence of surfactant on the Tg of PLGA nanoparticles was investigated using mDSC. Residual surfactant exhibited a positive correlation with initial surfactant concentration, which was also confirmed by H NMR. In Tg measurements in dry conditions, DMAB-PLGA NPs showed a significantly large ΔTg, notably from the decrease in Tg of the bulk, likely due to the penetration of DMAB molecules into the polymer matrix, plasticizing the sample. Additionally, a Tg reduction in wet measurement was observed, confirming that water acts as a plasticizer in PLGA. The decrease in Tg in wet conditions for both surfactants is noteworthy as the Tg is approaching physiological temperature, resulting in a greater burst release of drug for drug loaded particles.Interestingly, the ΔTg in water was less than the ΔTg in dry conditions, suggesting that there are also interfacial effects from the different external environments. However, the presence of surfactant in PLGA nanoparticles complicates investigation of the interfacial effect. The difference in behavior exhibited by PLGA's ΔTg in comparison to other commonly studied polymers, such as PS, raise interesting questions regarding the polymer physics of PLGA in confined environments. Beyond the investigation of Tg for drug delivery applications, further study of surfactant free PLGA particles are warranted to comprehensively explain this phenomenon.
Author contributions
K. M. conceived and directed the study. G. L. and R. M. prepared PLGA nanoparticles and measured Tg. G. L. acquire the NMR spectrums and SEM images and analyzed the resulting data. R. M. performed the intrinsic viscosity measurements. A. B. assisted with data analysis. All authors contributed to giving valuable suggestions and discussion. K. M. oversaw the project and led the research activity planning and execution.Conflicts of interest
There are no conflicts to declare.References
- C. Zhang, L. Yang, F. Wan, H. Bera, D. Cun, J. Rantanen and M. Yang, Int. J. Pharm., 2020, 585, 119441 CrossRef CAS PubMed.
- K. Xu, N. An, H. Zhang, Q. Zhang, K. Zhang, X. Hu, Y. Wu, F. Wu, J. Xiao, H. Zhang, R. Peng, H. Li and C. Jia, J. Drug Delivery Sci. Technol., 2020, 55, 101405 CrossRef CAS.
- S. Shakeri, M. Ashrafizadeh, A. Zarrabi, R. Roghanian, E. G. Afshar, A. Pardakhty, R. Mohammadinejad, A. Kumar and V. K. Thakur, Biomedicines, 2020, 8, 13 CrossRef CAS.
- C.-Y. Cheng, Q.-H. Pho, X.-Y. Wu, T.-Y. Chin, C.-M. Chen, P.-H. Fang, Y.-C. Lin and M.-F. Hsieh, Polymers, 2018, 10, 519 CrossRef PubMed.
- H. K. Makadia and S. J. Siegel, Polymers, 2011, 3, 1377–1397 CrossRef CAS PubMed.
- L. I. Cabezas, I. Gracia, A. De Lucas and J. F. Rodríguez, Ind. Eng. Chem. Res., 2014, 53, 15374–15382 CrossRef CAS.
- L. Y. Lee, S. H. Ranganath, Y. Fu, J. L. Zheng, H. S. Lee, C.-H. Wang and K. A. Smith, Chem. Eng. Sci., 2009, 64, 4341–4349 CrossRef CAS.
- M. Alonso-Sande, A. Des Rieux, V. Fievez, B. Sarmento, A. Delgado, C. Evora, C. Remuñán-López, V. Préat and M. J. Alonso, Biomacromolecules, 2013, 14, 4046–4052 CrossRef CAS PubMed.
- S. Fredenberg, M. Wahlgren, M. Reslow and A. Axelsson, Int. J. Pharm., 2011, 415, 34–52 CrossRef CAS PubMed.
- R. Karnik, F. Gu, P. Basto, C. Cannizzaro, L. Dean, W. Kyei-Manu, R. Langer and O. C. Farokhzad, Nano Lett., 2008, 8, 2906–2912 CrossRef CAS PubMed.
- G. Liu and K. Mcennis, Polymers, 2022, 14, 993 CrossRef CAS PubMed.
- H. Levine and L. Slade, Water Sci. Rev., 1988, 3, 79–185 Search PubMed.
- Sonam, H. Chaudhary, V. Arora, K. Kholi and V. Kumar, Polym. Rev., 2013, 53, 546–567 CrossRef CAS.
- P. In Pyo Park and S. Jonnalagadda, J. Appl. Polym. Sci., 2006, 100, 1983–1987 CrossRef.
- S. K. Sahoo, J. Panyam, S. Prabha and V. Labhasetwar, J. Controlled Release, 2002, 82, 105–114 CrossRef CAS.
- S. Spek, M. Haeuser, M. M. Schaefer and K. Langer, Appl. Surf. Sci., 2015, 347, 378–385 CrossRef CAS.
- H.-Y. Kwon, J.-Y. Lee, S.-W. Choi, Y. Jang and J.-H. Kim, Colloids Surf., A, 2001, 182, 123–130 CrossRef CAS.
- V. Bhardwaj, D. D. Ankola, S. C. Gupta, M. Schneider, C.-M. Lehr and M. N. V. R. Kumar, Pharm. Res., 2009, 26, 2495–2503 CrossRef CAS PubMed.
- Y. Jin, A. Xu, M. Yao, B. Li, Y. Jin and J. Ying, Int. J. Nanomed., 2012, 3547, DOI:10.2147/ijn.s32188.
- R. Gossmann, K. Langer and D. Mulac, PLoS One, 2015, 10, e0127532 CrossRef PubMed.
- C. Bouissou, J. J. Rouse, R. Price and C. F. Van Der Walle, Pharm. Res., 2006, 23, 1295–1305 CrossRef CAS.
- J. U. Menon, S. Kona, A. S. Wadajkar, F. Desai, A. Vadla and K. T. Nguyen, J. Biomed. Mater. Res., Part A, 2012, 100A, 1998–2005 CrossRef CAS PubMed.
- P. P. Simon and H. J. Ploehn, J. Rheol., 2000, 44, 169–183 CrossRef CAS.
- E. A. Dimarzio and J. H. Gibbs, J. Polym. Sci., Part A: Gen. Pap., 1963, 1, 1417–1428 CrossRef CAS.
- P. G. Royall, D. Q. M. Craig and C. Doherty, Pharm. Res., 1998, 15, 1117–1121 CrossRef CAS PubMed.
- C. Leyva-Porras, P. Cruz-Alcantar, V. Espinosa-Solís, E. Martínez-Guerra, C. I. Piñón-Balderrama, I. Compean Martínez and M. Z. Saavedra-Leos, Polymers, 2019, 12, 5 CrossRef PubMed.
- H. Rachmawati, Sci. Pharm., 2016, 84, 191–202 CrossRef CAS PubMed.
- J. Vysloužil, P. Doležel, M. Kejdušová, E. Mašková, J. Mašek, R. Lukáč, V. Košťál, D. Vetchý and K. Dvořáčková, Acta Pharm., 2014, 64, 403–417 CrossRef PubMed.
- K. Miladi, S. Sfar, H. Fessi and A. Elaissari, Ind. Crops Prod., 2015, 72, 24–33 CrossRef CAS.
- R. Jalil and J. Nixon, J. Microencapsulation, 1990, 7, 25–39 CrossRef CAS PubMed.
- M. Khemani, M. Sharon and M. Sharon, Int. Scholarly Res. Not., 2012, 2012, 187354 Search PubMed.
- D. L. Cooper and S. Harirforoosh, PLoS One, 2014, 9, e87326 CrossRef PubMed.
- K. Y. Hernández-Giottonini, R. J. Rodríguez-Córdova, C. A. Gutiérrez-Valenzuela, O. Peñuñuri-Miranda, P. Zavala-Rivera, P. Guerrero-Germán and A. Lucero-Acuña, RSC Adv., 2020, 10, 4218–4231 RSC.
- J. Sun, J. Walker, M. Beck-Broichsitter and S. P. Schwendeman, Drug Delivery Transl. Res., 2022, 12, 720–729 CrossRef CAS PubMed.
- S. Khaledi, S. Jafari, S. Hamidi, O. Molavi and S. Davaran, J. Biomater. Sci., Polym. Ed., 2020, 31, 1107–1126 CrossRef CAS PubMed.
- P. Bonardelli, G. Moggi and A. Turturro, Polymer, 1986, 27, 905–909 CrossRef CAS.
- L. Robeson, Polymers, 2014, 6, 1251–1265 CrossRef.
- C. Zhang, Y. Guo and R. D. Priestley, Macromolecules, 2011, 44, 4001–4006 CrossRef CAS.
- D. Christie, C. Zhang, J. Fu, B. Koel and R. D. Priestley, J. Polym. Sci., Part B: Polym. Phys., 2016, 54, 1776–1783 CrossRef CAS.
- S. Feng, Z. Li, R. Liu, B. Mai, Q. Wu, G. Liang, H. Gao and F. Zhu, Soft Matter, 2013, 9, 4614 RSC.
- N. Passerini and D. Q. M. Craig, J. Controlled Release, 2001, 73, 111–115 CrossRef CAS PubMed.
- T. Miyazawa, M. Itaya, G. C. Burdeos, K. Nakagawa and T. Miyazawa, Int. J. Nanomed., 2021, 16, 3937–3999 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sm00082f |
| This journal is © The Royal Society of Chemistry 2023 |