
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnveiling polyamorphism and polyamorphic interconversions in pharmaceuticals: the peculiar case of hydrochlorothiazide†
Inês C. B.
Martins
 *a,
Anders S.
Larsen
a,
Anders Ø.
Madsen
a,
Olivia Aalling
Frederiksen
b,
Alexandra
Correia
c,
Kirsten M. Ø.
Jensen
b,
Henrik S.
Jeppesen
d and
Thomas
Rades
*a
*a,
Anders S.
Larsen
a,
Anders Ø.
Madsen
a,
Olivia Aalling
Frederiksen
b,
Alexandra
Correia
c,
Kirsten M. Ø.
Jensen
b,
Henrik S.
Jeppesen
d and
Thomas
Rades
*a
aDepartment of Pharmacy, University of Copenhagen, Universitetsparken 2, 2100 Copenhagen, Denmark. E-mail: ines.martins@sund.ku.dk; thomas.rades@sund.ku.dk
bDepartment of Chemistry, University of Copenhagen, Universitetsparken 5, 2100 Copenhagen, Denmark
cDivision of Pharmaceutical Chemistry and Technology, University of Helsinki, Viikinkaari 5, 00790 Helsinki, Finland
dDeutsches Elektronen-Synchrotron (DESY), Notkestrasse 85, D-22607 Hamburg, Germany
First published on 28th September 2023
Abstract
Polyamorphism has been a controversial and highly debated solid-state phenomenon in both material and pharmaceutical communities. Although some evidence of this fascinating phenomenon has been reported for several inorganic systems, and more recently also for a few organic compounds, the occurrence of polyamorphism is poorly understood and the molecular-level organization of polyamorphic forms is still unknown. Here we have investigated the occurrence of polyamorphism and polyamorphic interconversions in hydrochlorothiazide (HCT), using both experimental and computational methods. Three distinct HCT polyamorphs, presenting distinct physical and thermal stabilities as well as distinct relaxation properties, were systematically prepared using spray-drying (SD), quench-cooling (QC) and ball milling (BM) methods. HCT polyamorph II (obtained by QC) was found to be more physically stable than polyamorphs I and III (obtained by SD and BM, respectively). Furthermore, polyamorphs I and III could be converted into polyamorph II after QC, while polyamorph II did not convert to any other polyamorph after SD or BM. Molecular dynamics simulations show that HCT dihedral angle distributions are significantly different for polyamorphs I and II, which is postulated as a possible explanation for their different physicochemical properties.
Introduction
Most low molecular weight active pharmaceutical ingredients (APIs), especially in the development of oral dosage forms, such as tablets, show a too low aqueous solubility for the expected dose.1,2 Thus one of the greatest challenges in pharmaceutical development is to find strategies to improve the solubility and bioavailability of APIs, without compromising their safety and effectiveness.3 Converting crystalline APIs into their amorphous forms offers one of the best solutions for this solubility problem.4–6 Amorphous forms are high energy solids, which often coincides with an increase in solubility and thus potentially oral bioavailability.7 They are characterized by the existence of a glass transition temperature (Tg) at which a transition from a glass to a viscous or a rubbery form (known as the super-cooled melt) occurs.8Amorphous forms can be prepared via the thermodynamic path, where spray-drying (SD) and quench-cooling (QC) preparation methods are mostly used, or via the kinetic path, with ball-milling (BM) being the mostly used method.8–10 Published studies demonstrate that the preparation method used can significantly influence the physicochemical properties of amorphous forms. For example, amorphous indomethacin prepared by QC and SD presents significant differences in its physical stability and dissolution behavior.11–13 Another example with significant differences in their pharmaceutical performance are amorphous beta-lactam antibiotics prepared by freeze-drying and QC.14 More recently, amorphous forms of several APIs, including sulfathiazole, glibenclamide and hydrochlorothiazide, prepared by SD and QC, were found to present different physical stabilities and distinct Tg values.15
These reports led us to raise a fundamental question regarding the amorphous state: How do two amorphous forms of the same API, presenting different physicochemical properties, differ from each other?
This question can be easily answered for crystalline materials, as different crystal structures of the same API give rise to different polymorphic forms with unique structures and properties as well as detectable conversion processes.16 In disordered amorphous materials, no changes in long-range order can lead to a phenomenon like this, yet differently prepared amorphous forms do (sometimes) exhibit different physicochemical properties. It seems clear that differences in the physicochemical behavior between distinct amorphous forms may be fundamentally related with the intricacies of their molecular arrangements.
To address this unsolved question, we have investigated a phenomenon termed polyamorphism.17,18 First introduced in 1970 by Angell et al., polyamorphism is defined as the formation of different amorphous forms of a single compound, separated by a phase transition.19,20 The observation of this phenomenon has been documented for inorganic systems, such as SiO2, GeO2 and K2Sb8Se13, and also for water, where differences in their densities and glass structures, under certain temperature and pressure conditions, were first reported.21–25
Few claims about polyamorphism in organic systems, such as D-mannitol, triphenyl phosphite and paracetamol, have been described.26–29 However, its occurrence is poorly understood and the structural elucidation (i.e., molecular-level organization) of polyamorphic forms is unknown. Similar to polymorphism in crystalline materials,30 proving the true existence of polyamorphism will require knowing the molecular arrangement (structures) of the distinct amorphous forms as well as the conditions (e.g. temperature and humidity) at which these forms can possibly interconvert.
Herein we will present, for the first time, a thorough and comprehensive study, using a combination of thermal analyses modulated differential scanning calorimetry (mDSC), thermogravimetric analyses (TGA) and isothermal calorimetry, and scattering methods (X-ray diffraction and total scattering by means of pair distribution function (PDF) analysis), with molecular dynamics (MD) simulations to elucidate the formation mechanism of polyamorphism in the API hydrochlorothiazide (HCT). We show that, not only distinct amorphous HCT forms can be systematically obtained using different preparation methods (SD, QC and BM) but also polyamorphic interconversions can occur when a specific amorphous HCT form is submitted to temperature or milling treatments (Fig. 1). The use of PDF combined with MD simulations provided information about the HCT molecular dihedral angle distribution, which was found to be different for two of the polyamorphs.
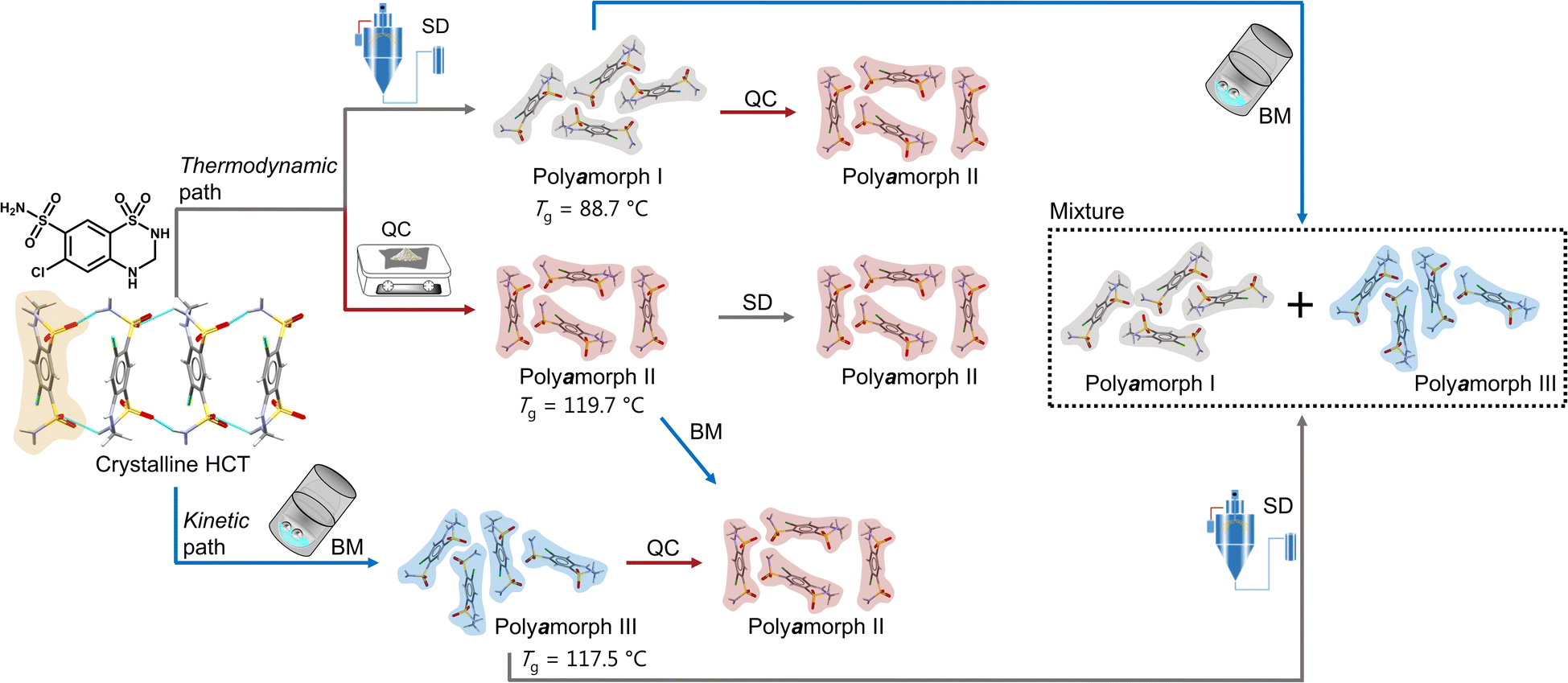 | ||
| Fig. 1 Schematic representation of the formation of HCT polyamorphs and their respective polyamorphic interconversions via both thermodynamic and kinetic pathways. | ||
Results and discussion
Isolation and stability of polyamorphs
HCT polyamorphs were obtained by SD, QC and BM of the stable crystalline form of the API (polymorph I of HCT in P21 space group). For the SD method, ethanol was found to be the best solvent to allow for a full amorphization of HCT. This amorphous phase, termed as polyamorph I had a Tg of 88.7 °C (after implementing a second heating cycle to allow full evaporation of any residual solvent or moisture) and a recrystallization temperature of ca. 135.2 °C (Fig. 2a and b). Before performing a mDSC analysis, the samples were dried over 10 days in a desiccator containing phosphorous pentoxide and activated silica. TGA analysis (Fig. S1†) showed that a residual moisture/solvent content of 3.05% was still present in the sample. The Tg obtained in a first heating cycle in the DSC was therefore affected by the presence of moisture/solvent and was found to be at ca. 72 °C. This value is in agreement with the one reported by Edueng et. al., where a Tg of 76 °C was measured after drying the sample in a desiccator for 2 days.15 Therefore, to obtain the real Tg value (measured to be 88.7 °C), we have implemented a second heating cycle to remove the remaining solvent moisture content (Fig. 2b).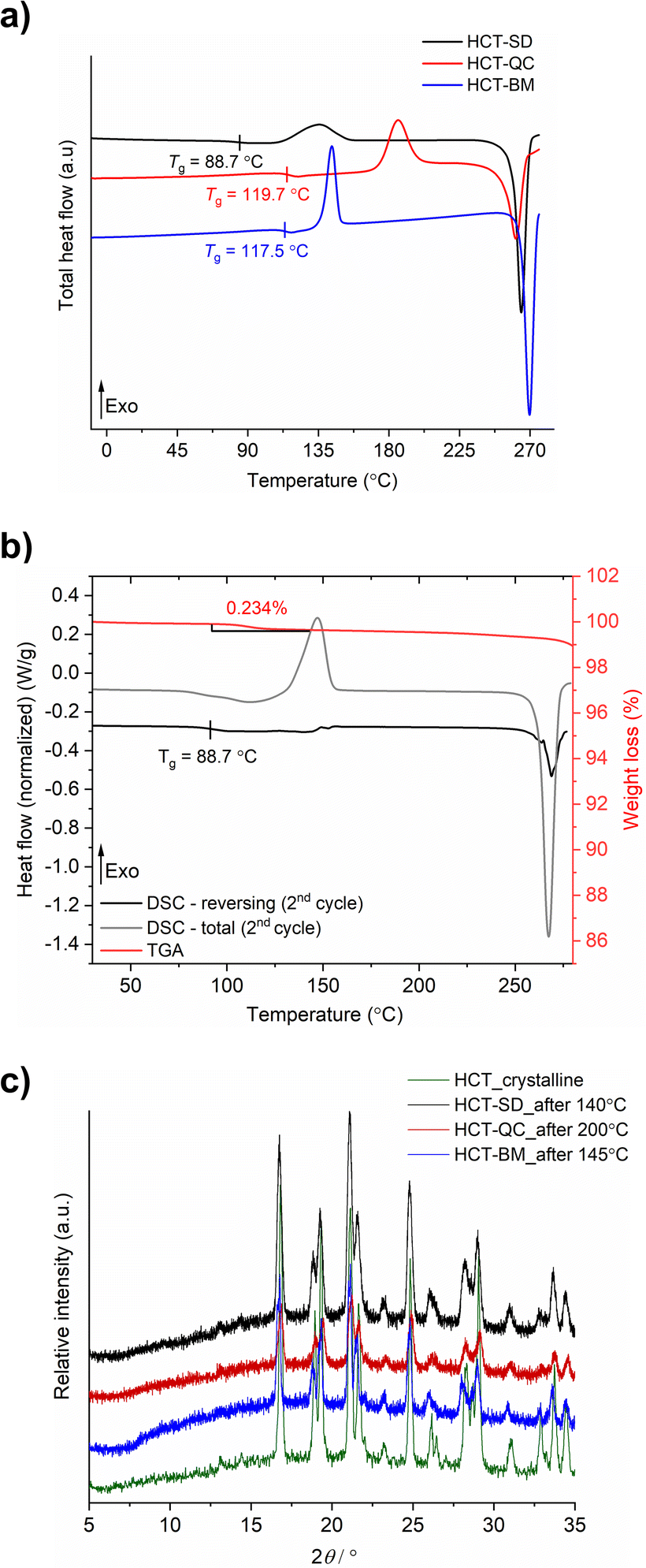 | ||
| Fig. 2 (a) mDSC thermograms of HCT polyamorphs obtained by SD, QC and BM after a second heating cycle (total heat flow); (b) mDSC (total and reversing heat flow) and TGA of polyamorph I (obtained by SD) after the second heating cycle; (c) PXRD data of all HCT polyamorphs obtained after the recrystallization temperature. | ||
Polyamorph II was obtained by heating HCT until its melting point (275 °C), without reaching its decomposition temperature (ca. 283 °C), followed by fast cooling until room temperature at different rates. The determined Tg value is substantially higher than the one found in polyamorph I (119.7 °C, in agreement with the value reported by Edueng et. al.), presenting also a higher recrystallization temperature (TR = 186.1 °C). For this polyamorph, no significant moisture was detected in the sample (Fig. S2†), and the measured Tg was taken from the second heating cycle as well. Using different cooling rates did not significantly affected the Tg, having values between 118.4 °C and 119.2 °C (Fig. S3†).
For the amorphous HCT prepared by BM (polyamorph III), different milling times were used at the same milling frequency (30 Hz) in order to study the amorphization rate. Complete amorphization was found after 60 min for 100 mg of HCT (Fig. S4a†). The Tg was detected at 117.5 °C with a recrystallization temperature of 143.6 °C (Fig. 2a). Although the Tg value is closer to the one found for polyamorph II, its recrystallization temperature is very different, suggesting that the amorphous form could also be different. Furthermore, when using longer milling times (75, 90, 105 and 120 min), an interesting increase in the Tg value from 115.7 °C to 118.9 °C and 119.4 °C (matching the Tg of polyamorph II) was observed (Fig. S4b†). However, the recrystallization temperatures were similar for all milled samples and also different from the one found on polyamorph II. This suggests that polyamorphs II and III are indeed different.
After submitting each polyamorph to an increased temperature until their recrystallization points, the obtained crystalline HCT phase was found to be the same (Fig. 2c), corresponding to the stable commercially available polymorph I (P21 space group).
The shelf-life stability against crystallization was evaluated by PXRD, after submitting the samples to two different conditions: (i) room temperature and humidity; and (ii) 0% humidity (using phosphorous pentoxide with activated silica). HCT polyamorphs follow the shelf-life stability order of I < III < II, as expected from their Tg values. Polyamorph I recrystallized after 3 days under condition (i) and after 70 days under condition (ii), while the most stable polyamorph II recrystallized after 90 days under condition (i) and after 300 days under condition (ii) (Fig. S5–S7†).
The purity of all freshly prepared HCT polyamorphs was measured using HPLC (Fig. S8–S24†). The results show no contamination after heating (QC) or mechanical (BM) treatment, assuring a high degree of purity in the samples. Karl Fischer titration experiments were performed to all samples after drying them in a desiccator. The results show a residual percentage of water present in all samples and ranging from 0.1% to 0.6% (Table S1†). Furthermore, three iterative QC experiments at 275 °C as well as additional experiments to purposely degrade the samples by QC at different temperatures (275 °C, 280 °C and 290 °C) during different time periods (10 min, 30 min and 2 h), were conducted. HPLC and FTIR-ATR results, presented in ESI† (Fig. S25–S35) show that the degradation only occurs at 290 °C.
Polyamorphic interconversions
Proving the existence of phase conversions between distinct amorphous phases, is a step further to understand the true existence of polyamorphism. Similar to polymorphism in crystalline materials, where under certain temperature, humidity and/or pressure conditions a polymorphic conversion occurs,30 in polyamorphism such conversions may also occur.We have explored possible polyamorphic conversions by submitting each isolated polyamorph to different stress conditions (Fig. 1): (a) polyamorph I obtained by SD was submitted to QC (in situ in the DSC) and BM; (b) polyamorph II obtained by QC was further submitted to SD and BM; and (c) polyamorph III obtained by BM was submitted to SD and QC (in situ in the DSC).
mDSC data shows successful conversions of polyamorphs I and III to polyamorph II after QC (Fig. S36 and S37†), while no conversion from polyamorph II into polyamorph I is observed after SD (Fig. 3a). These results indicate that temperature plays a critical role in the phase conversion, supporting that polyamorph II is the most stable amorphous form. Furthermore, the presence of ethanol during SD does not appear to be a critical parameter for inducing a phase conversion in polyamorph II. In this way, ethanol seems to be a ‘spectator’ or a ‘stabilizer’ of the initial amorphous form used. Surprisingly, when SD polyamorph III, only a partial amount of the material was converted to polyamorph I, while the other portion remained as polyamorph III. The same result was also obtained when BM polyamorph I. From the mDSC data (Fig. 3b and S38†) it is possible to observe the presence of two Tg values, where the one corresponding to polyamorph I is shifted by 10 °C to a higher temperature (98.2 °C) and the other one, corresponding to polyamorph III, is shifted ca. 3 °C to lower temperature (114.7 °C). These are very interesting and unique results as such type of effects (presence of two Tg values with shifts with respect to the pure compounds) have been only observed on amorphous solid dispersions.31 In these multicomponent amorphous systems, composed of a polymer and a drug, when both components are interacting (i.e., intermolecular interactions), it is common to obtain a shift in both polymer and drug Tg values (one increases its temperature while the other one decreases).31 In our molecular homogenous systems, there is a clear phase separation between both HCT polyamorphs, suggesting that both phases are distinct (‘heterogeneous’), i.e., they have different molecular-level organizations. We hypothesize that at the interface of these two polyamorphs, intermolecular interactions may occur, being responsible for the observed Tg shifts. However, a thorough and dedicated investigation is necessary to understand this phenomenon and it is being conducted in a separated project.
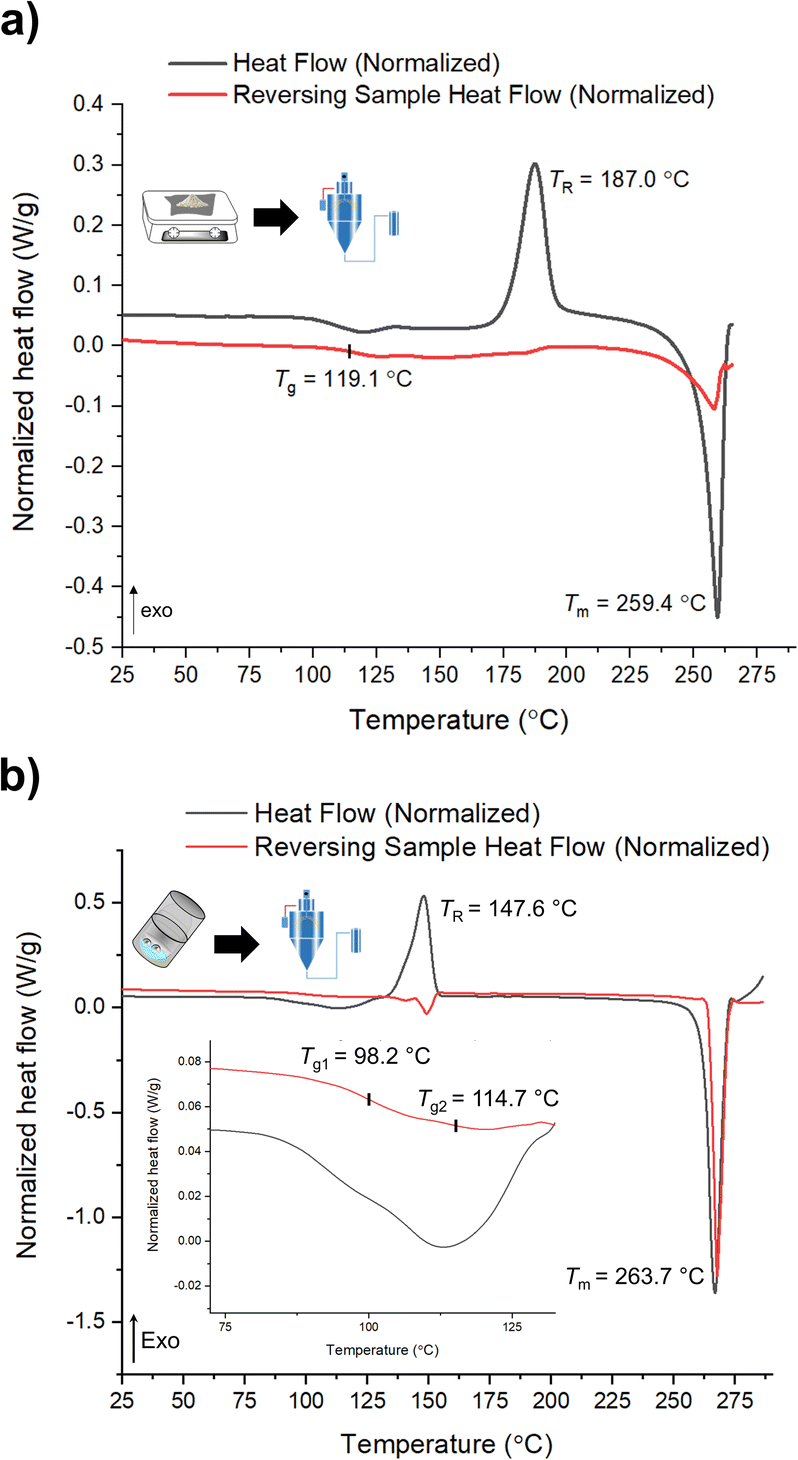 | ||
| Fig. 3 mDSC data of (a) SD of polyamorph II obtained by QC; and (b) SD of polyamorph III obtained by BM. | ||
Considering the current investigation and results, polyamorph II appears to be the most favourable form to occur rather than polyamorphs I and III.
Molecular-level elucidation
From the above data it becomes clear that distinct types of disorder must be present in the amorphous phases prepared by different methods, which also seem to be responsible for affecting their physicochemical properties. What is not clear is what those differences are and how they can affect the molecular-level organization in the amorphous state.PDF analysis has been used to directly compare changes in intramolecular bond distances and intermolecular packing and to track processing and aging induced changes of amorphous pharmaceutical systems.32–41
X-ray total scattering data was collected for all HCT amorphous phases produced by SD, QC and BM at the P02.1 beamline, at the DESY synchrotron (Hamburg, Germany). The short wavelength used (0.207 Å), combined with an appropriate data collection strategy, enabled data to be recorded over an acceptable Q-range (in this case, Qmax = 16 Å−1) to provide the necessary resolution in real-space for structural analysis. The reduced total scattering structure function, F(Q), and the reduced PDF, G(r), were obtained and are presented in Fig. 4a and b, respectively. Surprisingly, the PDFs were similar for each HCT polyamorph (Fig. S39† for the difference curve between all Experimental data), suggesting similar intramolecular and intermolecular distributions. The three PDFs show sharp structural peaks until 8 Å, which agrees with the HCT molecular size. Broad features are observed above 8 Å most likely arising from intermolecular correlations. A zoom-in on the high r-region, 7–30 Å (Fig. S40†) highlights the absence of significant differences in the on-average intermolecular packing of each HCT polyamorph (i.e., all three PDFs show similar features at 10, 14 and 18 Å). A comparison between the simulated PDF of the single HCT molecule and the PDF of polyamorph II (Fig. S41†) shows a match between data and single structure, suggesting that the PDF peaks represent only intramolecular correlations. This also indicates that all HCT samples are completely disordered (fully amorphous) with lack of mid to long-range periodicity.
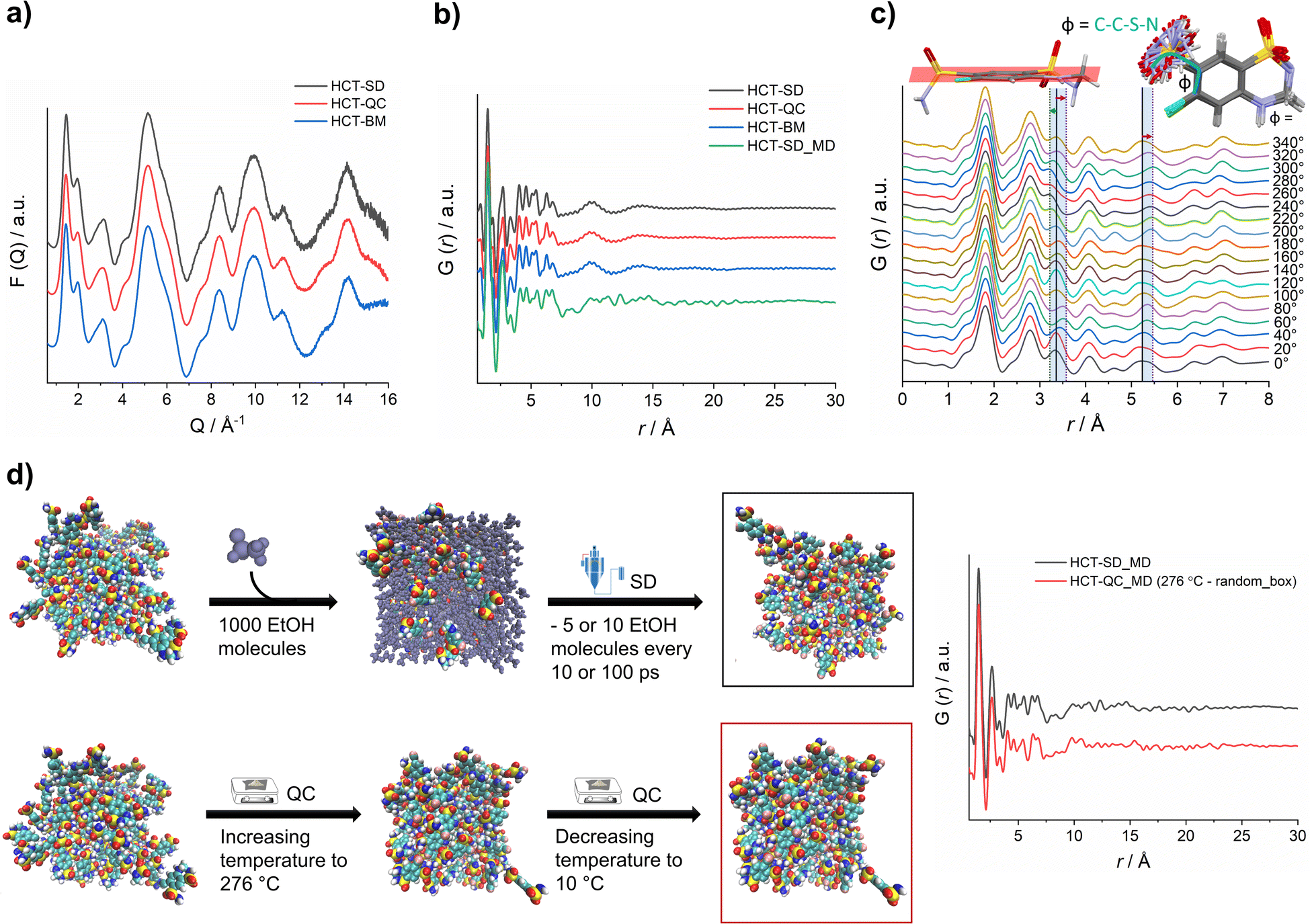 | ||
| Fig. 4 (a) F(Q) and (b) PDF of experimental HCT polyamorphs and simulated QC method (green curve) with an offset in the y-axis for better clarity; (c) simulated PDF of HCT single molecule when varying the dihedral angle Φ; and (d) schematic representation of the MD simulations of both SD and QC methods with the corresponding simulated PDF curves in black and red, respectively (offset in the y-axis for better clarity). EtOH stands for ethanol. | ||
It has been previously demonstrated that in materials where the intramolecular component is predominant (typically in the range of 1.5 Å to 4 Å), it is possible to identify the presence of distinct molecular conformations.35,42,43 In HCT, conformational modifications can occur in two groups of the molecule: (1) the sulfonamide group can freely rotate, changing the Φ angle (C–C–S–N) (Fig. 4c); and (2) the second sulfonamide group, integrated in the six-member ring, can change its configuration, standing up or below the molecular plane (Fig. 4c). A closer examination on the PDF generated from distinct rotational Φ angles shows a small shift (to the left or right) of the signals located at ca. 3.4 Å and 5.3 Å. When looking back to the PDFs of all polyamorphs (Fig. 4B), it becomes obvious that no particular configuration is obtained; instead an average of all possible configurations is present. However, it is not clear how many molecules exist in each configuration and how this can influence the stability of each polyamorph. To better understand this aspect, we have in silico prepared the amorphous materials using MD simulations (Fig. 4d).
We have simulated the experimental conditions to produce polyamorphs I (SD) and II (QC) and obtained reliable molecular models. For this, two main strategies were implemented: (i) SD – starting with 100 randomly inserted HCT molecules in the MD box, 1000 ethanol molecules were also inserted to create a solution. At every 10 or 100 ps of simulation, 5 or 10 ethanol molecules were removed at a temperature of 80 °C (the experimental inlet temperature used for SD); and (ii) QC – starting from randomly inserted 100 HCT molecules in the MD box, a cycle consisting of increasing the temperature until 276 °C followed by a decrease until room temperature was implemented; and starting from a supercell crystal structure, different ‘defects’ (number of molecules removed from the crystal lattice) were created to allow its melting when increasing the temperature until 276 °C or higher (maximum 426 °C). A higher number of defects corresponds to a lower melting temperature and vice versa. In Fig. 4d a schematic representation of the SD and one of the QC approaches, considering the random box approach, is illustrated. Details about the MD simulations can be found in the Experimental section of ESI.† For polyamorph III, no robust and reliable method could be implemented to simulate milling conditions and therefore this amorphous form could not further be considered in the computational experiments.
PDFs were simulated for each MD model obtained from SD and QC (random box approach) methods (Fig. 4d). For the two MD models, we observe a similar structural ordering up to 8 Å, suggesting similar molecular arrangements. Furthermore, a direct comparison of the simulated PDFs from MD with the experimental PDFs (Fig. 4b) shows a good agreement in the low r-range up to 8 Å (Fig. S42† shows the difference curve between simulated and experimental PDFs). These results do not only agree with the experimental PDF trend observed for each polyamorph, i.e., similar PDFs for all polyamorphs (Fig. 4b), but also confirm that the obtained models are reliable on representing the molecular environment of each polyamorph. The PDF generated from the QC simulations at 326 °C and 476 °C and presented in Fig. S43a and b† are also similar to the experimental PDF data. However, for the system obtained after QC at 476 °C, small changes start to appear at ca. 5 Å probably as a result of the overestimated melting temperature. For both QC approaches at 276 °C and SD (5 ethanol molecules removed at every 100 ps of simulation), two replicas were performed and the simulated PDFs are presented in Fig. 4d and S44.†
Additional calculation of radial distribution function curves for key NH⋯O hydrogen bonds interactions present in the HCT crystalline structure (Fig. S45†), shows that the probability of finding those specific interactions is similar for both polyamorphs obtained by QC and SD, also supporting the experimental and simulated PDF results.
To answer our key question “How do two amorphous forms of the same API, presenting different physicochemical properties, differ from each other?” we have calculated the dihedral angle distribution for all molecules used in the MD simulations of both SD and QC. For that, we considered both functional groups where changes in the dihedral angles C–C–S–N and N–C–N–C can occur, i.e., the sulfonamide group alone and integrated in the six-member ring. The 2D plots containing the distribution of both angles are presented in Fig. 5. According to the HCT crystalline structure (stable P21 polymorph I), either both N–C–N–C and C–C–S–N dihedral angles are negative (Fig. 5, blue triangle) or both are positive. On the other hand, for the metastable HCT polymorph (P21/c polymorph II), both negative and positive values are present for both N–C–N–C and C–C–S–N dihedral angles (Fig. 5, pink triangle) as a result of the implementation of the mirror plane c crystallographic operation. Although no symmetry operations occur between the molecules in the amorphous state, there are also internal molecular symmetry elements to consider. If we take the aromatic ring plane of HCT as a reference, it is easily seen that when both dihedral angles are negative and both are positive, the conformational result is the same for interatomic distance probabilities (PDF). Therefore, for the sake of simplicity, in our discussion we will mainly focus on two sets of dihedral angles: (i) negative N–C–N–C and negative C–C–S–N; and (ii) positive N–C–N–C and negative C–C–S–N.
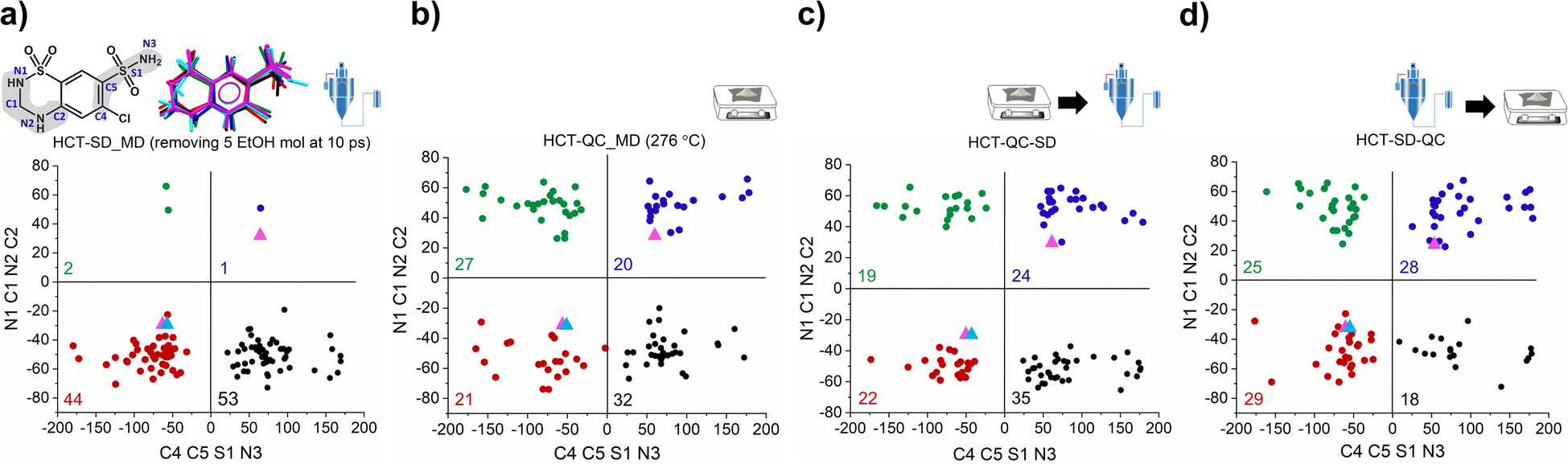 | ||
| Fig. 5 2D plot of N–C–N–C and C–C–S–N dihedral angle distributions of: (a) polyamorph I obtained by SD; (b) polyamorph II obtained by QC; (c) SD of polyamorph II; and (d) QC of polyamorph I. HCT structures representative of each dihedral angle combinations is also presented. The blue triangle corresponds to the structure in P21 space group while the pink triangle corresponds to the other polymorph in P21/c space group. EtOH stands for ethanol. | ||
After performing the SD simulation, negative values for the dihedral angle N–C–N–C were favorably retained, with 44% of the molecules presenting also a negative value for the C–C–S–N dihedral angle, and 53% presenting a positive one (Fig. 5a). This result indicates two important aspects: (1) the sulfonamide directly connected to the aromatic ring freely rotates, inducing a broad distribution of C–C–S–N dihedral angles composed by both negative and positive values; and (2) the ethanol molecules used during the simulation help in stabilizing the initial HCT configuration used (P21 polymorph I), i.e., a negative N–C–N–C dihedral angle. Furthermore, changing the evaporation rate, by either increasing the number of ethanol molecules to be removed or increasing the time of removing, did not affect the dihedral angle distribution that remains practically the same (Fig. S46 and S47†).
On the other hand, when performing QC simulations, and regardless of the chosen starting point (supercell or random box), the N–C–N–C dihedral angle is evenly distributed, being 27% positive and 21% negative (Fig. 5b and S48†). This result indicates that the high temperature used to melt HCT is sufficient to induce a change in the chain configuration, bringing the initial negative N–C–N–C dihedral angle to a positive value. For the QC simulations, performed at higher temperatures (326 °C and 426 °C), similar dihedral angle distributions were also obtained (Fig. S49†).
As experimentally demonstrated, polyamorphic interconversions occur under specific conditions. Polyamorph I obtained by SD converts to polyamorph II after QC, whereas polyamorph II does not convert to polyamorph I after SD (Fig. 1, 3a, and S36†). We have further investigated these polyamorphic transformations by MD simulations to check the dihedral angle distribution after QC the SD polyamorph I and after SD the QC polyamorph II. Details about the MD simulations can be found in the Experimental section of ESI.†
Interestingly, when SD polyamorph II obtained via QC, the dihedral angle distribution did not change, compared to the initial distribution of polyamorph II (Fig. 5b and c). This supports the claim that ethanol molecules behave either as ‘spectators’ or as contributors to the stabilization of the initial HCT configurations (in this case the configuration obtained after QC). When QC polyamorph I (Fig. 5d), the dihedral angle distribution changes and it is similar to the QC performed on the crystalline HCT. Regardless of which starting configurations are considered, after heating the system to the melting temperature of 276 °C and cooling it to room temperature, the N–C–N–C configuration changes to a point where half of the population occupies positive values and the other half negative values. The obtained MD results thus strongly support the experimental results previously discussed.
Relaxation studies
Amorphous forms may crystallize over time but even if crystallization does not take place at experimental time scales, their structures will ‘relax’ towards an equilibrium supercooled melt state.44–46 As an amorphous solid moves towards the equilibrium state, energy decreases, free volume decreases, and structural order increases in a phenomenon termed relaxation.46,47 Several studies indicate that measuring the relaxation process in freshly prepared amorphous forms can be used as a good surrogate to characterize their thermal history, thermodynamic properties and determine their physical stability.44,48 The structural relaxation time (τDβ, from eqn (3) in Experimental section of ESI†) was calculated for all HCT polyamorphs to determine their rate of structural relaxation and degree of molecular mobility. A small value of τDβ suggests a fast structural relaxation and a high degree of molecular mobility. As can be observed in Fig. 6, polyamorph I (obtained from SD) has the highest degree of molecular mobility and the fastest structural relaxation, followed by polyamorph III (obtained from BM) and polyamorph II (obtained from QC). The trend of the calculated τDβ values also follows the trend of the measured Tg (Fig. 2a) confirming the stability of each HCT polyamorph, which follows the order: polyamorph I < polyamorph III < polyamorph II.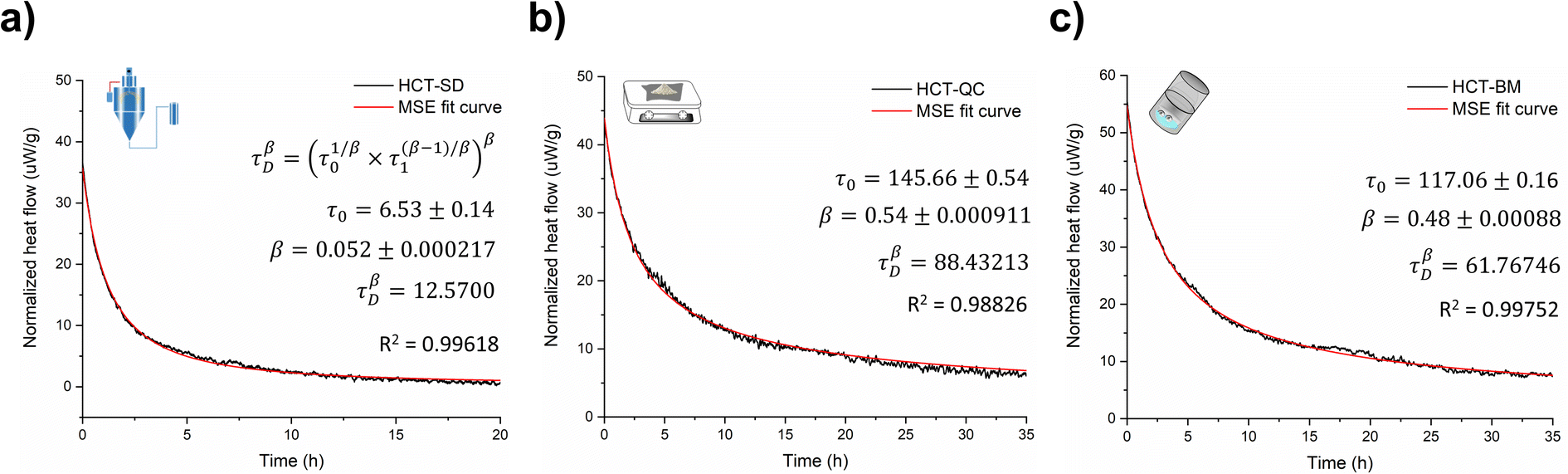 | ||
| Fig. 6 Relaxation curves and respective τDβ values calculated for (a) polyamorph I obtained by SD; (b) polyamorph II obtained by QC and (c) polyamorph III obtained by BM. | ||
The previously discussed polyamorphic interconversions, i.e., polyamorph I → polyamorph II and polyamorph III → polyamorph II, were confirmed by calculating the τDβ values (Fig. S50a†). It was found that the amorphous form obtained after QC of both polyamorph I and polyamorph III corresponds to polyamorph II as the relaxation time was observed to be very similar to the one obtained when QC crystalline HCT (Fig. 6b). Furthermore, the relaxation time obtained for polyamorph II after SD (Fig. S50b†) is also similar to polyamorph II after QC of the crystalline phase, confirming our previous results and claim that the ethanol used during SD acts as a ‘spectator’ or ‘stabilizer’ of the initial HCT configuration.
Conclusions
We have demonstrated, for the first time, the occurrence of polyamorphism and direct-phase polyamorphic interconversions for an organic pharmaceutical system. Hydrochlorothiazide (HCT) was successfully amorphized by SD, QC and BM methods, producing polyamorphs with distinct physical and thermal stabilities as well as relaxation behaviors. Polyamorph II, obtained by QC, was found to be more physically stable followed by polyamorphs III and I (obtained by BM and SD, respectively). Furthermore, conversion of polyamorphs I and III to polyamorph II were found to occur after QC treatment, while polyamorph II did not convert into polyamorphs I and III after SD and BM, proving it to be the ‘most stable’ amorphous form.Although similar experimental and simulated PDFs were obtained for all polyamorphs, distinct dihedral angle distributions of HCT (N–C–N–C) were found after MD simulations of QC and SD methods. In particular, for polyamorph I (generated after SD simulation), a negative N–C–N–C angular distribution was observed, while for polyamorph II (generated after QC simulation) an equal split of positive and negative N–C–N–C angular distributions was obtained. Furthermore, when investigating the polyamorphic interconversion, the simulated SD of polyamorph II (obtained by QC) and the QC of polyamorph I (obtained by SD) gave a similar dihedral angle distribution as observed for QC of crystalline HCT. A trend was then observed between the physical stability, Tg, relaxation time (τDβ) and HCT dihedral angle distribution: polyamorph I with the lowest physical stability, Tg and τDβ values, has a negative N–C–N–C dihedral angle distribution, similar to the molecular conformation found in the crystalline state (for both HCT polymorphs), while polyamorph II with the highest physical stability, Tg and τDβ values, has both a positive and negative (50–50%) N–C–N–C dihedral angle distribution.
This work demonstrates the complexity of the polyamorphism phenomenon and its molecular-level investigation. For HCT, the structural differences between distinct polyamorphs were only possible to be detected using the support of MD simulations. This approach has provided us with consistent results where distinct dihedral angle distributions were obtained for the most stable and less stable polyamorphs, being the key indicator of structural differences.
The occurrence of polyamorphism in organic pharmaceutical molecules is a possibility that deserves to be better explored in other systems. We can anticipate future discoveries of new polyamorphs that will possibly contribute to shaping the future of drug design when searching and manufacturing new amorphous drug formulations.
Data availability
The ESI† contains detailed description for all experimental techniques and computational methods used. Supplementary TGA, DSC, PXRD, HPLC, PDF, radial distribution function and dihedral angle distributions data are also provided.Author contributions
The manuscript was written by I. C. B. Martins and revised by all authors. The project was conceived by I. C. B. Martins and T. Rades. All experiments were performed by I. C. B. Martins. HPLC and FTIR-ATR experiments were performed by A. C. MD simulations were performed by I. C. B. Martins with support of A. S. Larsen and A. Ø. Madsen. PDF data collection was performed by I. C. B. Martins, A. Ø. Madsen and H. S. Jeppesen. PDF data treatment was performed by I. C. B. Martins, O. A. Frederiksen, K. M. Ø. Jensen and H. S. Jeppesen. All data analysis was performed by I. C. B. Martins. All images, including the TOC graphic, were created by I. C. B. Martins.Conflicts of interest
The authors declare no conflict of interests.Acknowledgements
The authors acknowledge the Independent Research Fund Denmark for funding the project 0135-00103A “Organic Polyamorphism: Existence – Formation – Relevance”, NordForsk (Nordic POP) for financial support travelling to conferences and workshops, and DESY (Hamburg, Germany), a member of the Helmholtz Association HGF, for the provision of experimental facilities. PDF of this research were carried out at PETRA III beamline P02.1. Beamtime was allocated for proposal(s) I-20210421-EC.Notes and references
- P. J. Eddershaw, A. P. Beresford and M. K. Bayliss, Drug Discovery Today, 2000, 5, 409–414 CrossRef CAS PubMed.
- D. J. Hauss, Adv. Drug Delivery Rev., 2007, 59, 667–676 CrossRef CAS PubMed.
- S. Domingos, V. André, S. Quaresma, I. C. B. Martins, M. F. Minas da Piedade and M. T. Duarte, J. Pharm. Pharmacol., 2015, 67, 830–846 CrossRef CAS PubMed.
- J. Liu, H. Grohganz, K. Löbmann, T. Rades and N.-J. Hempel, Pharmaceutics, 2021, 13, 389 CrossRef CAS PubMed.
- G. Kasten, L. Lobo, S. Dengale, H. Grohganz, T. Rades and K. Löbmann, Eur. J. Pharm. Biopharm., 2018, 132, 192–199 CrossRef CAS PubMed.
- B. C. Hancock and M. Parks, Pharm. Res., 2000, 17, 397–404 CrossRef CAS PubMed.
- B. C. Hancock and G. Zografi, J. Pharm. Sci., 1997, 86, 1–12 CrossRef CAS PubMed.
- L. I. Blaabjerg, E. Lindenberg, T. Rades, H. Grohganz and K. Löbmann, Int. J. Pharm., 2017, 521, 232–238 CrossRef CAS PubMed.
- S. Bates, G. Zografi, D. Engers, K. Morris, K. Crowley and A. Newman, Pharm. Res., 2006, 23, 2333–2349 CrossRef CAS PubMed.
- N. S. Trasi and S. R. Byrn, AAPS PharmSciTech, 2012, 13, 772–784 CrossRef CAS PubMed.
- P. Karmwar, K. Graeser, K. C. Gordon, C. J. Strachan and T. Rades, Eur. J. Pharm. Biopharm., 2012, 80, 459–464 CrossRef CAS PubMed.
- P. Karmwar, J. P. Boetker, K. A. Graeser, C. J. Strachan, J. Rantanen and T. Rades, Eur. J. Pharm. Sci., 2011, 44, 341–350 CrossRef CAS PubMed.
- M. Yoshioka, B. C. Hancock and G. Zografi, J. Pharm. Sci., 1994, 83, 1700–1705 CrossRef CAS PubMed.
- M. J. Pikal, A. L. Lukes, J. E. Lang and K. Gaines, J. Pharm. Sci., 1978, 67, 767–773 CrossRef CAS PubMed.
- K. Edueng, C. A. S. Bergström, J. Gråsjö and D. Mahlin, Pharmaceutics, 2019, 11(9), 425 CrossRef CAS PubMed.
- N. Inamdar, D. Dhamecha, A. Rathi and M. Saifee, Int. J. Health Res., 2009, 2, 291–306 Search PubMed.
- P. H. Poole, T. Grande, C. A. Angell and P. F. McMillan, Science, 1997, 275, 322–323 CrossRef CAS.
- B. C. Hancock, E. Y. Shalaev and S. L. Shamblin, J. Pharm. Pharmacol., 2010, 54, 1151–1152 CrossRef PubMed.
- C. A. Angell and E. J. Sare, J. Chem. Phys., 1970, 52, 1058–1068 CrossRef CAS.
- R. Kurita and H. Tanaka, Science, 2004, 306, 845–848 CrossRef CAS PubMed.
- P. H. Poole, T. Grande, F. Sciortino, H. E. Stanley and C. A. Angell, Comput. Mater. Sci., 1995, 4, 373–382 CrossRef CAS.
- M. H. Grimsditch, Phys. Rev. Lett., 1984, 52, 2379–2381 CrossRef CAS.
- O. Mishima, L. D. Calvert and E. Whalley, Nature, 1984, 310, 393–395 CrossRef CAS.
- S. Aasland and P. F. McMillan, Nature, 1994, 369, 633–636 CrossRef CAS.
- S. M. Islam, L. Peng, L. Zeng, C. D. Malliakas, D. Y. Chung, D. B. Buchholz, T. Chasapis, R. Li, K. Chrissafis, J. E. Medvedeva, G. G. Trimarchi, M. Grayson, T. J. Marks, M. J. Bedzyk, R. P. H. Chang, V. P. Dravid and M. G. Kanatzidis, J. Am. Chem. Soc., 2018, 140, 9261–9268 CrossRef CAS PubMed.
- M. Zhu and L. Yu, J. Chem. Phys., 2017, 146, 244503 CrossRef PubMed.
- J. Wiedersich, A. Kudlik, J. Gottwald, G. Benini, I. Roggatz and E. Rössler, J. Phys. Chem. B, 1997, 101, 5800–5803 CrossRef CAS.
- Y. Nguyen Thi, K. Rademann and F. Emmerling, CrystEngComm, 2015, 17, 9029–9036 RSC.
- F. Walton, J. Bolling, A. Farrell, J. MacEwen, C. D. Syme, M. G. Jiménez, H. M. Senn, C. Wilson, G. Cinque and K. Wynne, J. Am. Chem. Soc., 2020, 142, 7591–7597 CrossRef CAS PubMed.
- A. J. Cruz-Cabeza, S. M. Reutzel-Edens and J. Bernstein, Chem. Soc. Rev., 2015, 44, 8619–8635 RSC.
- J. Sharma, B. Singh, A. K. Agrawal and A. K. Bansal, J. Pharm. Sci., 2021, 110, 1470–1479 CrossRef CAS PubMed.
- F. Atassi, C. Mao, A. S. Masadeh and S. R. Byrn, J. Pharm. Sci., 2010, 99, 3684–3697 CrossRef CAS PubMed.
- S. J. L. Billinge, T. Dykhne, P. Juhás, E. Božin, R. Taylor, A. J. Florence and K. Shankland, CrystEngComm, 2010, 12, 1366–1368 RSC.
- C. J. Benmore, J. K. R. Weber, A. N. Tailor, B. R. Cherry, J. L. Yarger, Q. Mou, W. Weber, J. Neuefeind and S. R. Byrn, J. Pharm. Sci., 2013, 102, 1290–1300 CrossRef CAS PubMed.
- M. W. Terban and S. J. L. Billinge, Chem. Rev., 2022, 122, 1208–1272 CrossRef CAS PubMed.
- A. R. Sheth, S. Bates, F. X. Muller and D. J. W. Grant, Cryst. Growth Des., 2005, 5, 571–578 CrossRef CAS.
- J. P. Bøtker, P. Karmwar, C. J. Strachan, C. Cornett, F. Tian, Z. Zujovic, J. Rantanen and T. Rades, Int. J. Pharm., 2011, 417, 112–119 CrossRef PubMed.
- J. P. Boetker, V. Koradia, T. Rades, J. Rantanen and M. Savolainen, Pharmaceutics, 2012, 4, 93–103 CrossRef CAS PubMed.
- T. Dykhne, R. Taylor, A. Florence and S. J. L. Billinge, Pharm. Res., 2011, 28, 1041–1048 CrossRef CAS PubMed.
- C. A. Reiss, A. Kharchenko and M. Gateshki, Z. Kristallogr., 2012, 227, 257–261 CrossRef CAS.
- M. W. Terban, L. Russo, T. N. Pham, D. H. Barich, Y. T. Sun, M. D. Burke, J. Brum and S. J. L. Billinge, Mol. Pharm., 2020, 17, 2370–2389 CrossRef CAS PubMed.
- Q. Mou, C. J. Benmore and J. L. Yarger, J. Appl. Crystallogr., 2015, 48, 950–952 CrossRef CAS.
- R. Berardi, F. Spinozzi and C. Zannoni, Mol. Cryst. Liq. Cryst. Sci. Technol., Sect. A, 1996, 290, 245–253 CrossRef CAS.
- S. Groel, T. Menzen and G. Winter, Pharmaceutics, 2021, 13, 1735 CrossRef CAS PubMed.
- S. L. Shamblin, B. C. Hancock and M. J. Pikal, Pharm. Res., 2006, 23, 2254–2268 CrossRef CAS PubMed.
- C. Bhugra, R. Shmeis, S. L. Krill and M. J. Pikal, Pharm. Res., 2006, 23, 2277–2290 CrossRef CAS PubMed.
- K. Kawakami and Y. Ida, Pharm. Res., 2003, 20, 1430–1436 CrossRef CAS PubMed.
- A. M. Abdul-Fattah, K. M. Dellerman, R. H. Bogner and M. J. Pikal, J. Pharm. Sci., 2007, 96, 1237–1250 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc02802j |
| This journal is © The Royal Society of Chemistry 2023 |