
Luminescent hexagonal microtubes prepared through water-induced self-assembly of a polymorphic organoboron compound: formation mechanism and waveguide behaviour†
Pradip A.
Gaikwad
 a,
Prodipta
Samadder
b,
Shubham
Som
a,
Deepak
Chopra
*a,
Prakash P.
Neelakandan
*b and
Aasheesh
Srivastava
*a
a,
Prodipta
Samadder
b,
Shubham
Som
a,
Deepak
Chopra
*a,
Prakash P.
Neelakandan
*b and
Aasheesh
Srivastava
*a
aDepartment of Chemistry, Indian Institute of Science Education and Research (IISER) Bhopal, Bhopal Bypass Road, Bhauri, Bhopal 462 066, Madhya Pradesh, India. E-mail: dchopra@iiserb.ac.in; asrivastava@iiserb.ac.in
bInstitute of Nano Science and Technology, Sector – 81, Mohali 140306, Punjab, India. E-mail: ppn@inst.ac.in
First published on 8th August 2023
Abstract
Tetra-coordinated organoboron (TCOB) compounds are promising candidates for developing high-performance optical devices due to their excellent optoelectronic performance. Fabricating TCOB-based nanomaterials of controlled and defined morphology through rapid and easy-to-execute protocols can significantly accelerate their practical utility in the aforesaid applications. Herein, we report water-induced self-assembly (WISA) to convert a polymorphic TCOB complex (HNBI-B, derived from a 2-(2′-hydroxy-naphthyl)-benzimidazole precursor) into two unique nanomorphologies viz. nanodiscoids (NDs) and fluorescent microtubes with hexagonal cross-sections (HMTs). Detailed electron microscopic investigations revealed that oriented assembly and fusion of the initially formed NDs yield the blue emissive HMTs (SSQY = 26.7%) that exhibited highly promising photophysical behaviour. For example, the HMTs outperformed all the crystal polymorphs of HNBI-B obtained from CHCl3, EtOAc and MeOH in emissivity and also exhibited superior waveguide behaviour, with a much lower optical loss coefficient α′ = 1.692 dB mm−1 compared to the rod-shaped microcrystals of HNBI-B obtained from MeOH (α′ = 1.853 dB mm−1). Thus, this work reports rapid access to high performance optical nanomaterials through WISA, opening new avenues for creating useful nanomaterial morphologies with superior optical performance.
Introduction
Organic scaffolds with high-efficiency solid-state emission are being aggressively pursued for diverse optoelectronic devices such as organic light-emitting diodes (OLEDs),1,2 sensors,3,4 lasers,5 photonic circuits6,7 and microscale optical waveguides.8,9 The optical properties of these scaffolds can be tuned by altering their molecular structure and packing modes in the solid state.10–16 Organoboron compounds show excellent luminescence behaviour such as sharp emission and absorption bands, high fluorescence quantum yields, large Stokes shifts and high thermal and photochemical stability.17–21 They are also capable of efficient waveguiding.22,23 These useful properties of organoboron compounds make them highly attractive targets for a wide variety of photonic applications.24–26 The instability of B–F bonds in boron-dipyrromethene (BODIPys) towards irradiation and solvent polarity has led researchers to focus towards TCOB complexes.27 The B–O and B–N bonds of such TCOB molecules have a greater covalent character compared to the B–F bond found in the BODIPy class of fluorophores.28,29 Specifically, tetra-coordinated organoboron (TCOB) compounds with extended π-conjugation and structural rigidity exhibit notable Stokes shifts, improved fluorescence emission (both in solid and solution states) and offer a broad range of molecular diversity through chemical synthesis.30 Hydroxyquinolate,31,32 pyridylphenolate,33,34 benzimidazolephenolate,35 and similar bidentate heterocyclic ligands are often used to prepare TCOB complexes.The deployment of these TCOB complexes as emitters and electron transport materials for organic field effect transistors, OLEDs, sensory and bioimaging materials has drawn intense attention.36–38 Neelakandan and co-workers reported the waveguiding abilities of TCOB complexes derived from salicylideneimine and naphthalideneimine residues.39,40 Zhang et al. prepared several TCOB complexes of 2-hydroxyphenyl imidazole that exhibited excellent electroluminescent properties, high thermal stability and acted as active component for a deep blue emitting OLED material.41,42 Such benzimidazole based TCOBs with extended π-conjugation and structural rigidity are considered ideal for developing bright, blue-emissive materials, and as promising candidates for the development of sensors and solar cells.43,44
Despite the high potential of these TCOB complexes in optoelectronics, controlled synthesis of the nano/micro architectures derived from them remain unexplored.22 It can be envisaged that an enriching diversity of nano-morphologies can be accessed through careful modulation of their intermolecular interactions and by application of the self-assembly principles. Accessible protocols for preparing high-performance optoelectronic nanomaterials from TCOB complexes will allow rapid realization of their potential. Considering the above, we explored solvent-induced modulation of crystal structure packing and nanomaterial morphology in a representative benzimidazole containing TCOB complex, HNBI-B. We had previously demonstrated the utility of water-induced self-assembly (WISA) for creating diverse nanoarchitectures of amino acid derivatives.45 Herein, we show that the WISA protocol can be utilized to gain facile access to nano-discoid (ND) and hollow hexagonal microtube (HMT) morphologies of HNBI-B.46–48 The excellent photophysical properties of these nanomaterials and the utility of HMTs as optical waveguides is also presented.
Results and discussion
The TCOB complex viz. HNBI-B employed in this study was readily prepared from the precursor 2-hydroxynaphtalene benzimidazole (HNBI) using a reported protocol.49,50 The product HNBI-B was well characterized by standard characterization techniques (NMR, ESI-HRMS and SC-XRD, ESI). Since HNBI-B contains both H-bond donor (imidazole –NH) and acceptor (phenolic oxygen) residues and has rigid molecular structure, it could be easily crystallized from different solvents viz. CHCl3, EtOAc or MeOH upon exposure to hexane vapours at 296–298 K with 60% relative humidity by vapor diffusion method (Fig. 1a). These solvents were chosen based on their differential polarities and H-bonding abilities that increase from CHCl3 to MeOH. Interesting differences in the morphology and crystal packing was observed for these crystals.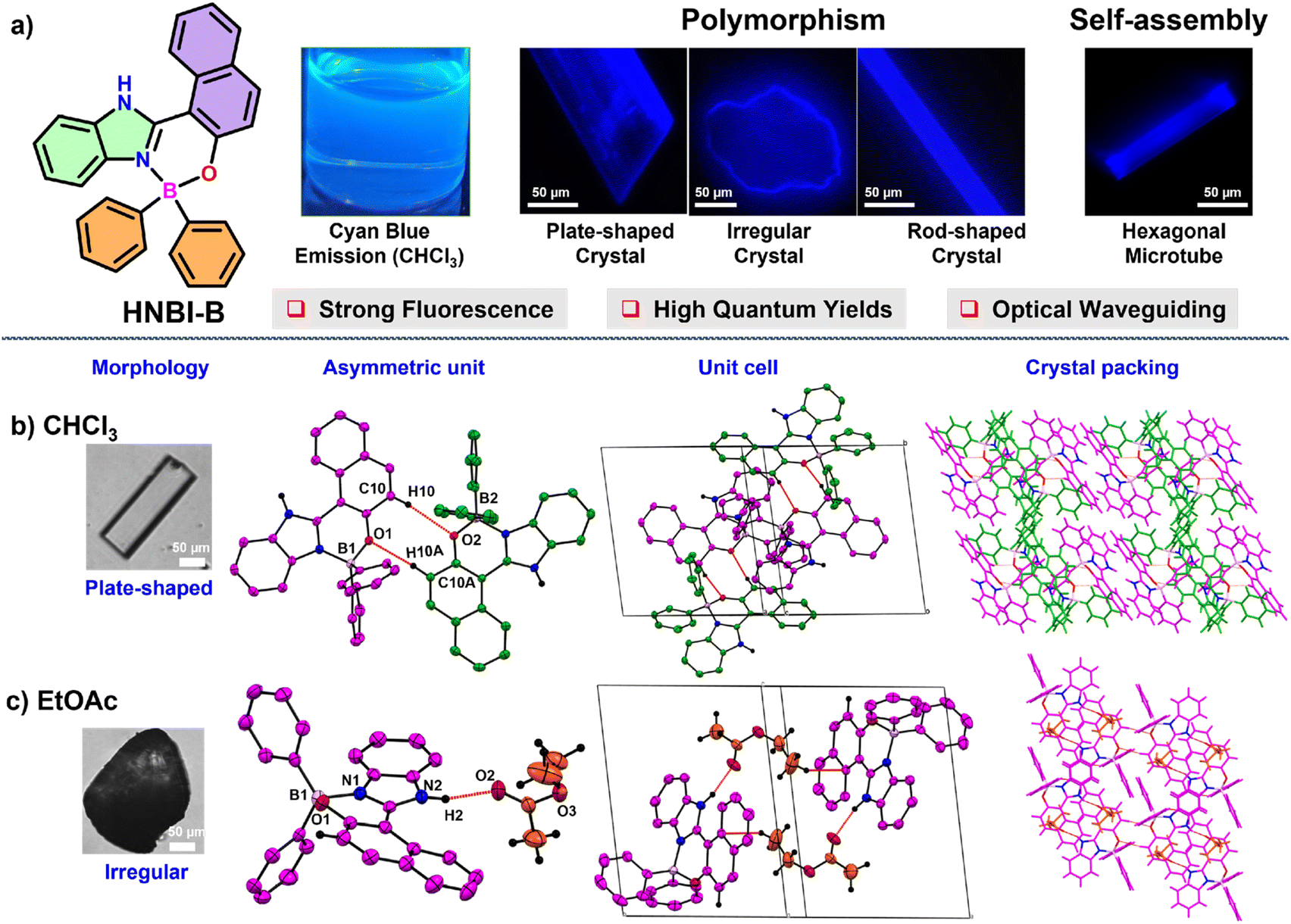 | ||
| Fig. 1 (a) HNBI-B displays a strong cyan-blue fluorescence in both the solution and the solid states. It exhibits solvent-dependent crystal polymorphism and self-assembles into hexagonal microtubes (HMTs). The rod-shaped crystals formed in MeOH and the HMTs display optical waveguiding behaviour. (b) and (c) Crystal morphology and structural details obtained from SCXRD studies for HNBI-B crystals obtained from (b) CHCl3 and (c) EtOAc solutions, when exposed to hexane vapours at 296–298 K. The ORTEP diagrams are displayed at 50% probability. The asymmetric unit, unit cell and molecular packing along the axes b and a, respectively in the crystals are shown in different panels. The C–H⋯π, C–H⋯O, and aromatic π–π interactions dominate in the crystal obtained from CHCl3 whereas H-bonding interactions between HNBI-B and EtOAc (red dotted lines) are evident in the latter. | ||
While flat plate-like crystals were obtained from CHCl3 (Fig. 1b, left panel), EtOAc produced irregular crystals (Fig. 1c, left panel), while MeOH yielded elongated rod-shaped crystals (see next paragraph). The plate-shaped crystals of HNBI-B obtained from CHCl3 belonged to a triclinic crystal system with a P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. The asymmetric unit of this crystal contained two HNBI-B arranged in head-to-tail manner without the presence of any solvent molecule. The HNBI-B molecules engaged in C–H⋯π, C–H⋯O and aromatic π–π interactions with each other through the C10–H10⋯O2 (2.61(2) Å) and C10A–H10A⋯O1 (2.46 (2) Å) hydrogen bonding interactions (Fig. 1b, middle-left panel). The unit cell includes four HNBI-B molecules, and the cell-density and cell-volume were 1.342 g cm3 and 2100.37(7) Å3, respectively (Fig. 1a, middle-right panel). Furthermore, HNBI-B displays layer-to-layer crystal packing along b-axis, where each layer involved only in weak –C–H⋯π interactions (Fig. 1b, right panel). Similarly, from EtOAc an irregular shaped crystal (Fig. 1c, left panel) belonging to the P triclinic space group was obtained. Strong hydrogen-bonding interactions between the carbonyl group of EtOAc and –NH group of HNBI-B (N2–H2⋯O2, 1.91(2) Å, Fig. 1c, middle-left panel) were evident in the SCXRD studies. The unit cell of this crystal also consisted of four molecules, two molecules each of HNBI-B and EtOAc with cell density and cell volume of this crystal being 1.254 g cm−3 and 1357.02(15) Å3, respectively (Fig. 1c, middle-right panel). The crystal packing of molecules along a-axis showed the directional arrangement of HNBI-B molecules with solvent (EtOAc) molecules bridging the adjacent HNBI-B molecules (Fig. 1c, right panel).
space group. The asymmetric unit of this crystal contained two HNBI-B arranged in head-to-tail manner without the presence of any solvent molecule. The HNBI-B molecules engaged in C–H⋯π, C–H⋯O and aromatic π–π interactions with each other through the C10–H10⋯O2 (2.61(2) Å) and C10A–H10A⋯O1 (2.46 (2) Å) hydrogen bonding interactions (Fig. 1b, middle-left panel). The unit cell includes four HNBI-B molecules, and the cell-density and cell-volume were 1.342 g cm3 and 2100.37(7) Å3, respectively (Fig. 1a, middle-right panel). Furthermore, HNBI-B displays layer-to-layer crystal packing along b-axis, where each layer involved only in weak –C–H⋯π interactions (Fig. 1b, right panel). Similarly, from EtOAc an irregular shaped crystal (Fig. 1c, left panel) belonging to the P triclinic space group was obtained. Strong hydrogen-bonding interactions between the carbonyl group of EtOAc and –NH group of HNBI-B (N2–H2⋯O2, 1.91(2) Å, Fig. 1c, middle-left panel) were evident in the SCXRD studies. The unit cell of this crystal also consisted of four molecules, two molecules each of HNBI-B and EtOAc with cell density and cell volume of this crystal being 1.254 g cm−3 and 1357.02(15) Å3, respectively (Fig. 1c, middle-right panel). The crystal packing of molecules along a-axis showed the directional arrangement of HNBI-B molecules with solvent (EtOAc) molecules bridging the adjacent HNBI-B molecules (Fig. 1c, right panel).
Contrastingly, with MeOH elongated rod-shaped crystals belonging to orthorhombic space group Pbca were obtained (Fig. 2a). The asymmetric unit of these crystals consisted of three HNBI-B molecules and one MeOH molecule (Fig. 2b). Here, two types of H-bonding interactions were evident: those between HNBI-B molecules themselves and those between MeOH and HNBI-B as shown in Fig. 2b and c. MeOH molecules present in the crystal acted as both H-bond acceptor as well as a donor – the methanolic oxygen accepts hydrogen of one HNBI-B unit (N6–H6⋯O4 (1.87(3) Å)) and the solvent also act as proton donor to phenolic oxygen present in another HNBI-B unit (O4–H4B⋯O3 (2.07(4) Å) along crystallographic a and c axes (Fig. 2c). Thus, the H-bonding interactions of MeOH linked two molecules of HNBI-B (showed in violet color). The neighbouring molecules of HNBI-B (purple and green colored) were mutually perpendicular to each other with π-stacked naphthalene residues seen in the packing diagram. These two molecules are majorly held together by strong H-bonding N4–H4⋯O1 (2.12(3) Å) and aromatic π⋯π interactions along the c direction. The crystal packing of HNBI-B indicates the longitudinal growth as a result of various C–H⋯π, N–H⋯O, O–H⋯O and π⋯π interactions present (Fig. 2d). The unit cell of this crystal contained a total of 32 molecules (24 HNBI-B and 8 MeOH) and had a substantial cell volume of 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 967.3(2) Å3 and a high cell density of 1.337 g cm−3. Data for single-crystal X-ray diffraction (SC-XRD) study on these crystals is provided in ESI (Tables S1–S3†) along with the significant intermolecular interactions observed (Tables S4–S6, ESI†).
967.3(2) Å3 and a high cell density of 1.337 g cm−3. Data for single-crystal X-ray diffraction (SC-XRD) study on these crystals is provided in ESI (Tables S1–S3†) along with the significant intermolecular interactions observed (Tables S4–S6, ESI†).
 | ||
| Fig. 2 The SC-XRD analysis of HNBI-B crystallized from MeOH. ORTEP's were displayed with 50% probability. (a) Image of rod-shaped crystal in MeOH taken under optical microscope. (b) The asymmetric unit contains three HNBI-B molecules and one solvent molecule (shown in different colors for easy visualization). The neighbouring HNBI-B molecules are held together by strong intermolecular hydrogen bonding interactions (N4–H4⋯O1, 2.12(3) Å). (c) MeOH interacting with two HNBI-B molecules through strong H-bonding interactions (shown by red dotted lines). Vertical blue dotted lines are visual aid to indicate the linear organization of the solvent molecules in the crystal. MeOH acts as hydrogen bond donor and a hydrogen bond acceptor as well. (d) The packing of HNBI-B along the bc plane indicates unidirectional growth of the molecular assemblies in the crystal. The unit cell contains 24 HNBI-B and 8 MeOH molecules (light grey box). | ||
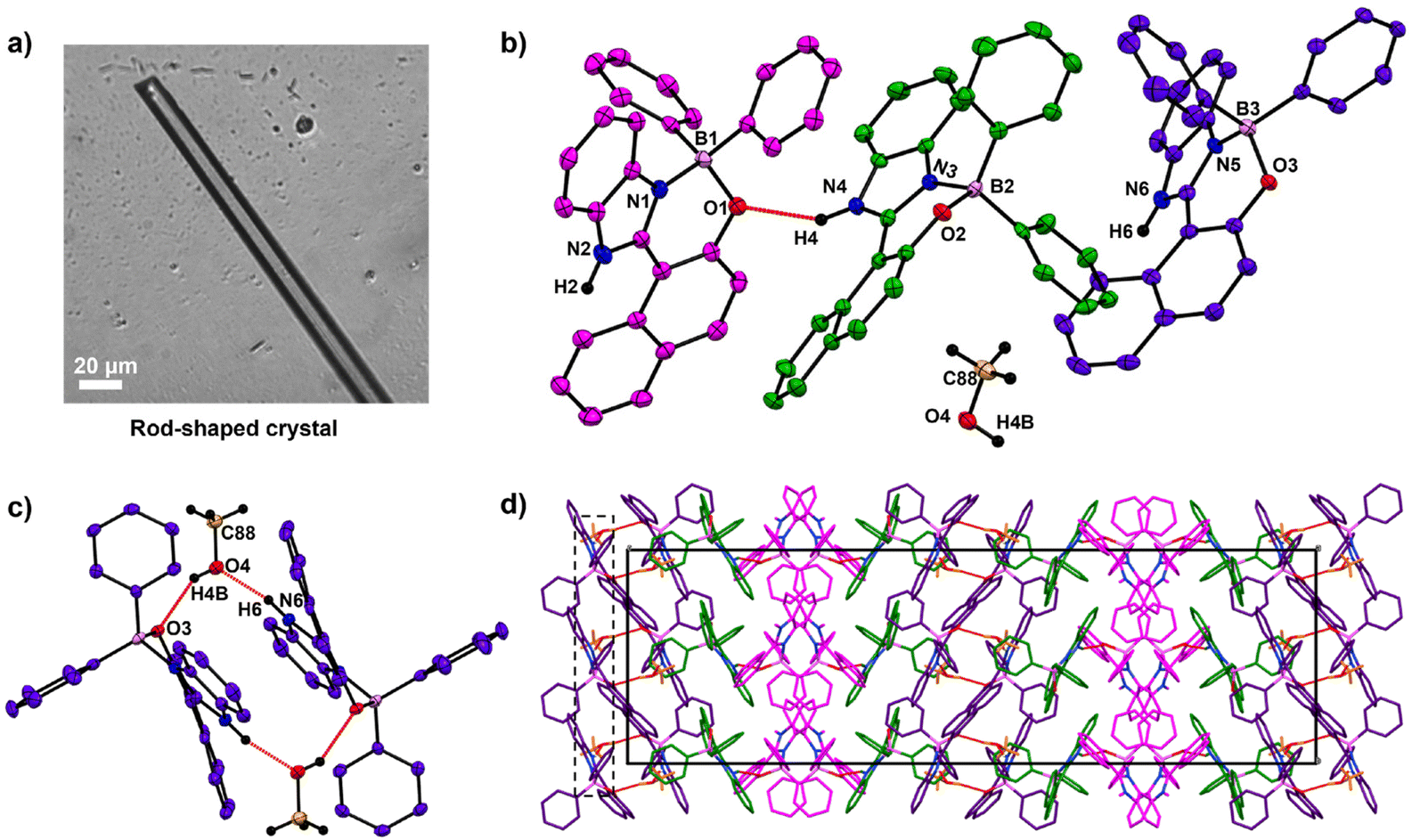
The crystal structure studies demonstrated that alteration of H-bonding interactions between the solute (HNBI-B) and the solvent generates different polymorphs of HNBI-B. Unidirectional assembly of HNBI-B was observed in the polar protic solvent, MeOH. Considering this, we next introduced water into the system, since H2O offers even greater opportunity for H-bonding interactions with HNBI-B. Additionally, water will also accentuate the hydrophobic interactions in HNBI-B, facilitating its self-assembly. Excitingly, when water was added to the methanolic solution of HNBI-B (50 μM), two distinct morphologies emerged rapidly through this water-induced self-assembly (WISA). At 20% v/v (H2O/MeOH), a turbid fluorescent solution containing HNBI-B aggregates with nanodiscoids (NDs) morphology was obtained immediately (Fig. 3, left panels). Electron microscopy (SEM and TEM) studies showed that the NDs have a diameter of 300–600 nm and had an outer shell of thickness 50–100 nm (Fig. 3b, panels marked (b–e)). Upon increasing the water content to 50% v/v, some fluorescent flocculant was observed floating over the solution (Fig. 3, right panels). Upon harvesting it by skimming and observing under microscope, we noticed the presence of hollow microtubules with hexagonal cross-sections (HMTs) (Fig. 3a). These HMTs were 10–50 μm long, and 2–3 μm in width, and had a thickness of 200–300 nm (Fig. 3, panels marked (f–i)). However, ageing of the 20%v/v H2O/MeOH solution of HNBI-B for 3 days also yielded HMTs as well. The HMTs obtained after 3 days from NDs did not have uniform dimensions. Thus, the NDs are likely the initially formed kinetic intermediates during the WISA of HNBI-B that gradually evolve to form HMTs (see below for detailed discussion). We explored the diffractability of the harvested HMTs towards both the X-ray and electrons to obtain detailed information of molecular interactions between HNBI-B molecules in HMTs form. However, PXRD of the HMTs was rather broad and featureless, indicating their amorphous nature (Fig. S4, ESI†). This was further supported by the selected area electron diffraction (SAED) pattern collected for several HMTs that exhibited no diffraction peaks, thereby, confirming the amorphous nature of these assemblies (Fig. S5, ESI†). The HMTs were thermally stable below 130 °C, with putative loss of the entrapped water molecules resulting in their structural collapse (Fig. S6, ESI†).
 | ||
| Fig. 3 (a) Preparation of nanodiscoids (NDs) and hexagonal microtubes (HMTs) through water-induced self-assembly (WISA) of HNBI-B (50 μM in MeOH). While the NDs were stably suspended in solution, the HMTs were seen floating over the solution. (b) SEM image, (c) TEM image, (d and e) Optical microscopy image in bright field and fluorescence micrograph of NDs (size 300–600 nm and thickness 50–100 nm). (f) SEM, (g) TEM, (h) bright field optical micrograph, and (i) Ffluorescence micrograph of the HMTs (length 10–50 μm, diameter 2–3 μm, and thickness 200–300 nm). Gradual conversion of NDs into HMTs upon ageing the solution was also observed. | ||
The formation of hollow HMTs through WISA was quite intriguing and offers easy access to such exquisite architectures. Therefore, we investigated the formation mechanism of HMTs in detail through time-dependent electron microscopy studies (Fig. 4a and b), which indicated that the formation of HMTs proceeds through the following sequential steps: (1) initially, rapid aggregation of HNBI-B molecules occurred to form spherical nano-discoids (NDs) which matured over the next one hour (panel marked t = 1 h); (2) subsequently, these NDs underwent oriented self-assembly to produce longitudinal morphologies (panel marked t = 2 h); (3) thereafter, these NDs fused to form tubular nanostructures (panel marked t = 3 h); (4) finally, reconstruction of these tubular precursors produced the well-faceted microtubes with hexagonal cross-sections (HMTs, panel marked t = 4 h). Fig. 4c is a schematic representation of the formation process of HMTs and NDs through WISA. In the first step of this self-assembly process, the non-solvent water induced rapid aggregation of HNBI-B into hollow nano-droplets due to an increase in glass transition temperature (Tg) while the molecules in the interior of the droplet remained plasticized by MeOH. These particles underwent oriented aggregation through the involvement of multiple intermolecular interactions such as π–π stackings, H-bonding interactions between HNBI-B molecules themselves and between HNBI-B and solvent molecules. The directional nature of these interactions likely guided the oriented self-assembly of the NDs to the HMTs. These NDs gradually burst open from both sides and HNBI-B assemblies reorganized to form the HMTs. It is worth mentioning that HMTs of the desired dimensions can be prepared by controlling the volume ratio of MeOH and H2O, concentration of HNBI-B and the duration of the growth of self-assemblies.
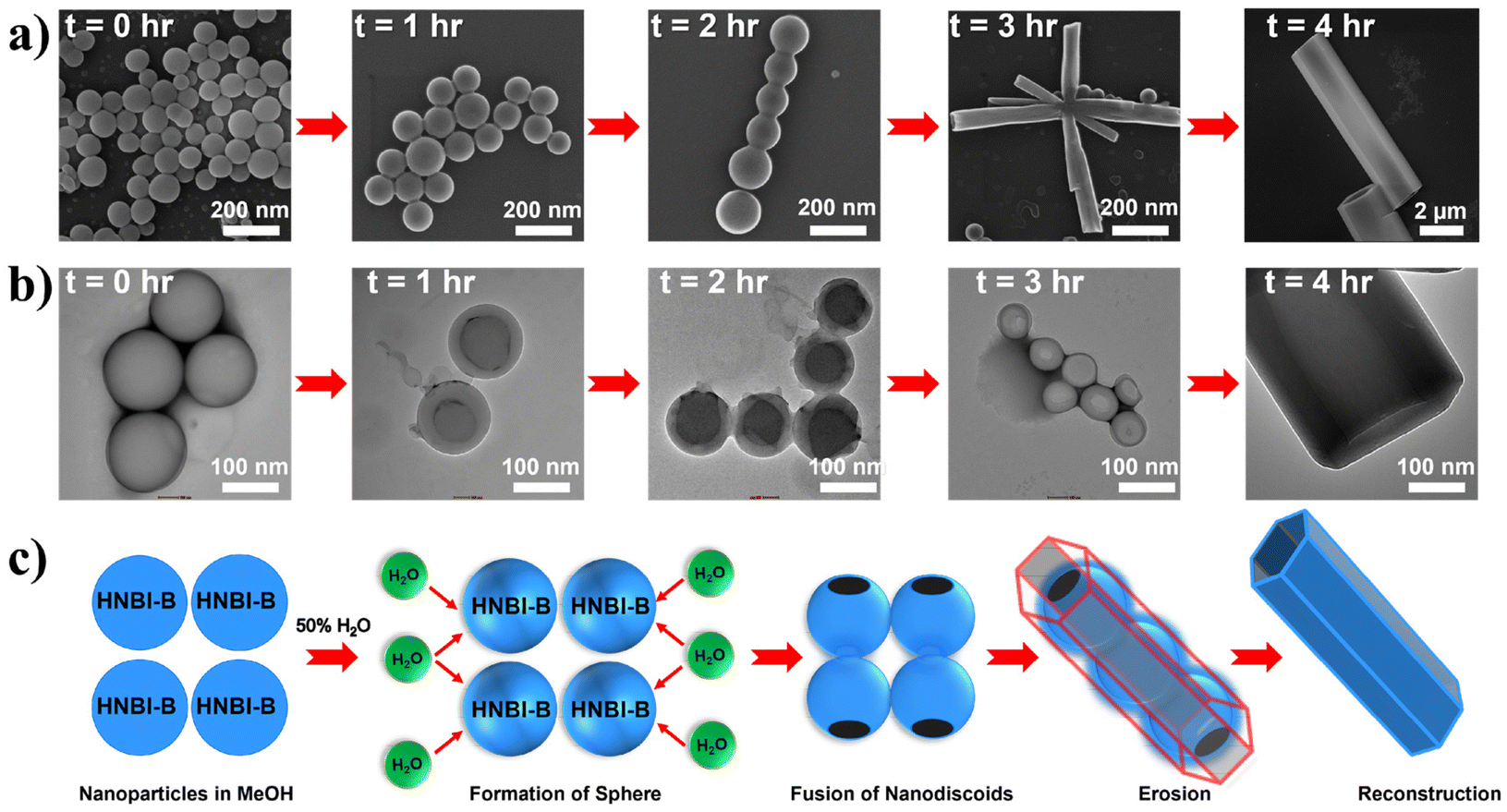 | ||
| Fig. 4 (a and b) Time-dependent SEM and TEM imaging shows the formation of HMTs from NDs. (c) Schematic of proposed sequence of molecular self-assembly to form HMTs with the increasing time after addition of water. Initially, HNBI-B particles are observed that starts to form spheres upon addition of water. In 50% v/v H2O, the spheres rupture from two sides and subsequently merge to form hexagonal microtubes. | ||
Photophysical investigations on HNBI-B in its dissolved as well as solid-state were undertaken thereafter. The molecule showed absorption λmax at ∼380 nm and emission in the range of 430–460 nm in solvents like CHCl3, EtOAc and MeOH (Fig. 5a and b) with relative quantum yields of 92%, 87% and 77%, respectively (Fig. S7, ESI†). Photophysical studies in solid-state were undertaken in the as-prepared powder form, crystals obtained from CHCl3, EtOAc, MeOH, and the HMTs form. The solid-state absorption and emission spectra of HNBI-B are depicted in Fig. 5c and d. The crystal obtained in different solvents and HMTs exhibits enhanced absorption compared to the as-prepared powder form (Fig. 5c). The solid-state absorption of any material is dependent on their physical properties and structural forms. In crystals, the highly ordered arrangement of constituent molecules with their well-defined orientation leads to more efficient absorption of the incident photons, which is seen as enhanced absorption and reduces scattering in the UV-vis spectrum. On the contrary, the random orientation of the molecular aggregates in powder form along with the irregular arrangement of the constituent molecules in these aggregates leads to predominance of scattering over absorption. The as-prepared powder form of HNBI-B as well as the crystals obtained from CHCl3 and EtOAc exhibited broad emission in the 400–700 nm range, while the emission spectra for the rod-shaped crystals obtained from MeOH and HMTs showed sharper emission profiles with full width at half maximum (FWHM) of ca. 50 nm. The solid-state quantum yield (SSQY) for as-prepared powder form, crystals obtained from CHCl3 and EtOAc is 1.5%, 5.9% and 13.2% respectively. The rod-shaped crystals from MeOH and the HMTs showed significantly higher SSQY of 17.1% and 26.7%, respectively (Fig. 5e). Thus, the oriented assemblies of HNBI-B molecules into rod-shaped crystals and HMTs produced superior photophysical properties over the other crystal morphologies of HNBI-B.
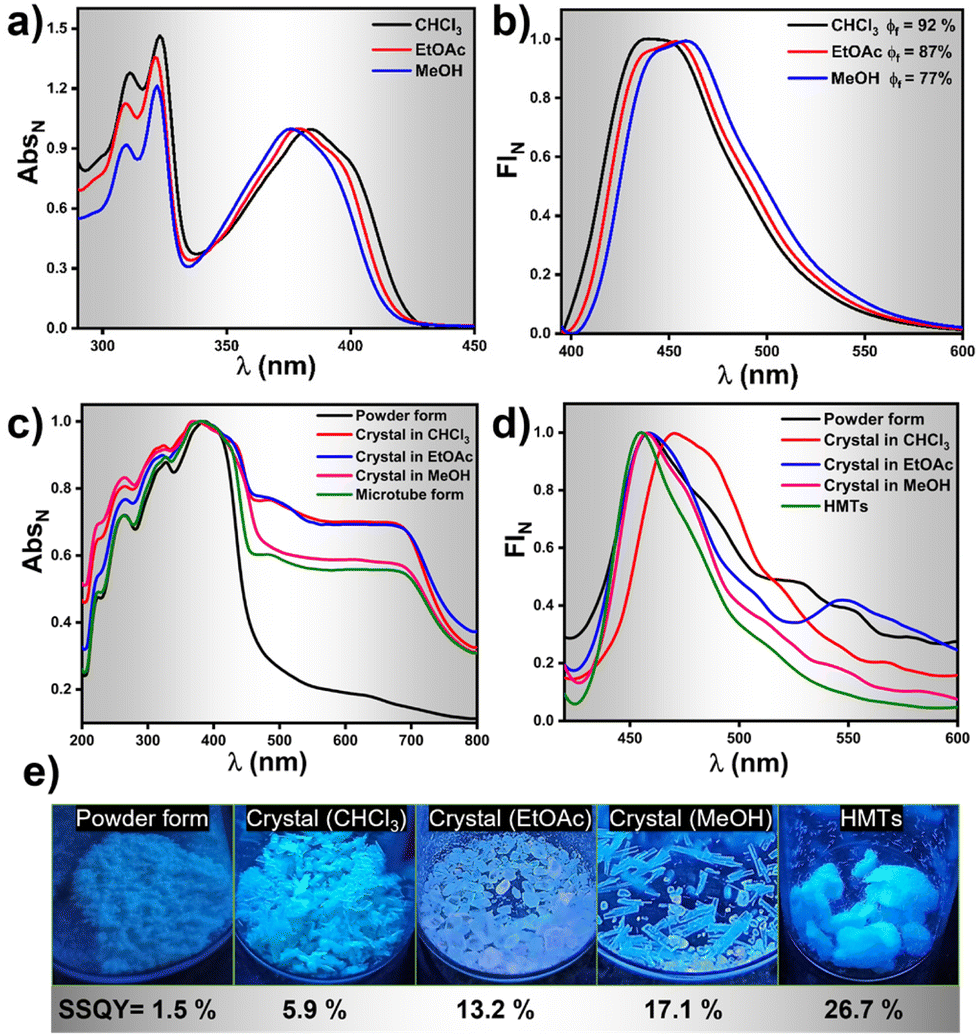 | ||
| Fig. 5 Photophysical properties of HNBI-B in solution and solid state. (a and b) Emission and absorption spectra of HNBI-B in CHCl3, EtOAc and MeOH (λex-390 nm, 5 μM conc.). (c and d) Solid state absorption and emission spectra of HNBI-B in different forms viz. As-prepared powder form, crystals obtained from CHCl3, EtOAc, MeOH and HMTs form. (e) Solid state quantum yield of HNBI-B in each form and photographs taken under UV light. | ||
While recording the fluorescence micrographs of the rod-shaped crystals and the HMTs, we noticed a strong blue emission at the tip of these assemblies. This pointed towards the possibility of these architectures acting as waveguides. We therefore investigated the optical waveguiding capabilities on these two samples using 405 nm light from a Ti:sapphire laser at different positions along the longitudinal length. When the light input was given at one end of the crystal or the HMT, blue fluorescence was clearly observed at the other end. The emission from the far end of the crystal and HMTs was collected upon excitation at different locations along the long axes (Fig. 6a and d). The corresponding excitation position-dependent emission spectra for the crystal and microtube at various excitation positions is shown in Fig. 6b and e. The fluorescence intensity at the tip of the microtube (Itip) steadily increased as we moved the excitation position closer to the collection point. The single-exponential fit of the spectra (Fig. 6c and f) was used to determine the optical waveguiding efficiency of the both the architectures, using the equation Itip/Ibody = exp(−α′D), where D is the distance between the tip and the excitation point and α′ is the optical loss coefficient.51 Accordingly, the calculated α′ value for the crystal was 1.853 dB mm−1 whereas for HMTs it was found to be 1.692 dB mm−1. Such low α′ value suggests promising waveguide behaviour of both the rod-like crystals and the HMTs which is attributed to their unique nanoarchitecture and the oriented molecular organization in them. Moreover, the HMTs showed lower optical loss compared to the microrod crystals. Earlier studies have also demonstrated that hollow microstructures display superior photophysical properties compared to their solid counterparts due to strong confinement of light inside high refractive index medium.52,53 Moreover, we believe that the hollow architectures also reduce the scattering losses significantly in comparison to the packed counterparts during the propagation of the optical wave through the sample. Thus, it is evident that the WISA allows facile access to nanostructures of fluorescent organic molecules with superior photophysical outcomes. We believe the straight-forward nature of this approach will find a wider adoption by researchers exploring rapid protocols for creating high-performance optical nanostructures.
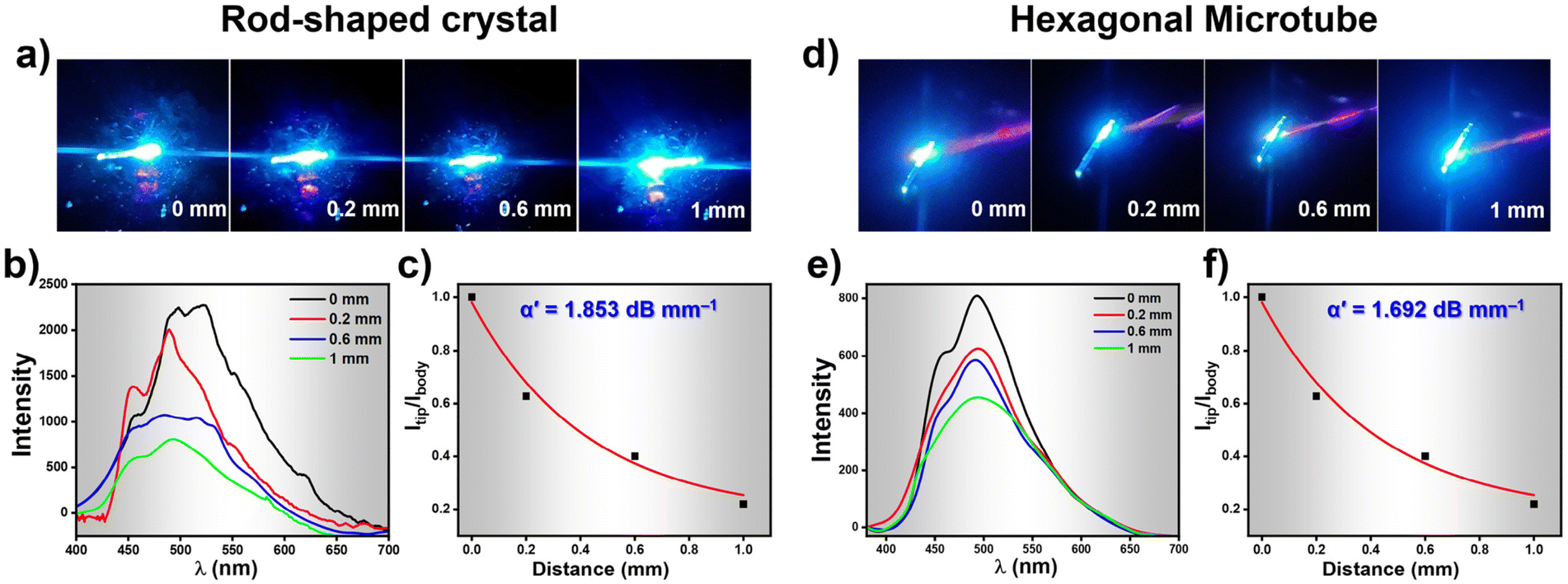 | ||
| Fig. 6 (a) Optical images of the rod-shaped crystal showing excitation at different positions indicating the waveguide nature. (b) Excitation position dependent FL spectra collected at various positions shown in (a). (c) Optical loss calculated from the plot of Itip/Ibodyvs. distance plot for the rod-shaped crystals. (d) Optical images of the HMTs showing excitation at different locations indicating the waveguide nature. (e) Excitation position dependent FL spectra collected at various position shown in (d). (f) Optical loss calculated from the plot of Itip/Ibodyvs. distance plot for the HMTs. | ||
Conclusions
Thus, in this paper we explored the polymorphic crystallization of a tetra-coordinated organoboron (TCOB) compound viz. HNBI-B by employing solvents of different polarity and H-bonding ability, establishing the involvement of solute-solvent H-bonding in the crystals of HNBI-B. This guided us to explore the water-induced self-assembly (WISA) of HNBI-B, which provided rapid and easy access to two different nano-morphologies viz. nanodiscoids (NDs) and hexagonal microtubes (HMTs). Consistent dimensions of these nanoarchitectures could be prepared by optimizing the concentration of HNBI-B, solvent composition, and growth duration. Detailed time-dependent microscopy investigations provided key insights into oriented self-assemblies of HNBI-B to form the HMTs. The rod-shaped crystals and HMTs exhibited higher solid-state quantum yields compared to the as prepared HNBI-B solid. Excellent optical waveguiding was achieved from the HMTs as well rod-shaped crystals. Notably, HMTs showed lower optical loss than crystals presumably due to reduced scattering losses and better confinement of light wave in such hollow nanostructures. We believe this study will propel researchers to explore the WISA protocol to create diverse nanomorphologies of other relevant molecules for optical and biomedical applications.Author contributions
The manuscript was written by A. S. and P. A. G. through the inputs of all the co-authors. Preparation of HNBI-B, its self-assembly and formation mechanism studies along with photophysical characterization were conducted by P. A. G. of the material. P. S. carried out the waveguide experiment of HMTs and rod-shaped crystal under the guidance of P. P. N. S. S. and D. C. helped in the SCXRD studies.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank SERB India for funding this work through project no. CRG/2021/007029. We thank IISER Bhopal for the intramural funds and facilities. We also thank the FIST-funded TEM facility available in the Department of Chemistry, IISER Bhopal. P. A. G. and S. S. thank UGC, India for fellowships. This article is dedicated to Prof. Santanu Bhattacharya on his 65th birthday.References
- S.-J. Zou, Y. Shen, F.-M. Xie, J.-D. Chen, Y.-Q. Li and J.-X. Tang, Mater. Chem. Front., 2020, 4, 788–820 RSC.
- D. Li, H. Zhang and Y. Wang, Chem. Soc. Rev., 2013, 42, 8416–8433 RSC.
- K. Dhanunjayarao, V. Mukundam and K. Venkatasubbaiah, Inorg. Chem., 2016, 55, 11153–11159 CrossRef CAS.
- X. Zhang, X. Liu, R. Lu, H. Zhang and P. Gong, J. Mater. Chem., 2011, 22, 1167–1172 RSC.
- J.-J. Wu, H. Gao, X.-D. Wang, Y. Wu, L. Jiang and L.-S. Liao, Adv. Opt. Mater., 2023, 11, 2200815 CrossRef CAS.
- C. Zhang, Y. S. Zhao and J. Yao, Phys. Chem. Chem. Phys., 2011, 13, 9060–9073 RSC.
- Y. Yan and Y. S. Zhao, Chem. Soc. Rev., 2014, 43, 4325–4340 RSC.
- K. Takazawa, Y. Kitahama, Y. Kimura and G. Kido, Nano Lett., 2005, 5, 1293–1296 CrossRef CAS.
- S. Kim and R. Yan, J. Mater. Chem. C, 2018, 6, 11795–11816 RSC.
- S. Ito, CrystEngComm, 2022, 24, 1112–1126 RSC.
- R. R. Cui, Y. C. Lv, Y. S. Zhao, N. Zhao and N. Li, Mater. Chem. Front., 2018, 2, 910–916 RSC.
- P. Srujana, P. Sudhakar and T. P. Radhakrishnan, J. Mater. Chem. C, 2018, 6, 9314–9329 RSC.
- I. Hussain, S. Sahoo, M. S. Sayed, M. Ahmad, M. S. Javed, C. Lamiel, Y. Li, J.-J. Shim, X. Ma and K. Zhang, Coord. Chem. Rev., 2022, 458, 214429 CrossRef CAS.
- B. Lu, X. Fang and D. Yan, ACS Appl. Mater. Interfaces, 2020, 12, 31940–31951 CrossRef CAS.
- C. Park, J. E. Park and H. C. Choi, Acc. Chem. Res., 2014, 47, 2353–2364 CrossRef CAS PubMed.
- J. Wang, S. Xu, A. Li, L. Chen, W. Xu and H. Zhang, Mater. Chem. Front., 2021, 5, 1477–1485 RSC.
- G. Meng, L. Liu, Z. He, D. Hall, X. Wang, T. Peng, X. Yin, P. Chen, D. Beljonne, Y. Olivier, E. Zysman-Colman, N. Wang and S. Wang, Chem. Sci., 2022, 13, 1665–1674 RSC.
- Y. Wu, H. Lu, S. Wang, Z. Li and Z. Shen, J. Mater. Chem. C, 2015, 3, 12281–12289 RSC.
- A. C. Shaikh, D. S. Ranade, S. Thorat, A. Maity, P. P. Kulkarni, R. G. Gonnade, P. Munshi and N. T. Patil, Chem. Commun., 2015, 51, 16115–16118 RSC.
- W. Fang, Y. Zhang, G. Zhang, L. Kong, L. Yang and J. Yang, CrystEngComm, 2017, 19, 1294–1303 RSC.
- X. Liu, J. Zhou, H. Zheng, J. Liu, Z. Liu, L. Ni, X. Kong, C. Zhang and H. Cao, Chin. J. Chem., 2023, 41, 924–930 CrossRef CAS.
- P.-Z. Chen, H. Zhang, L.-Y. Niu, Y. Zhang, Y.-Z. Chen, H.-B. Fu and Q.-Z. Yang, Adv. Funct. Mater., 2017, 27, 1700332 CrossRef.
- P. Galer, R. C. Korošec, M. Vidmar and B. Šket, J. Am. Chem. Soc., 2014, 136, 7383–7394 CrossRef CAS PubMed.
- D. Frath, J. Massue, G. Ulrich and R. Ziessel, Angew. Chem., Int. Ed., 2014, 53, 2290–2310 CrossRef CAS PubMed.
- C. B. Fialho, T. F. C. Cruz, A. I. Rodrigues, M. J. Calhorda, L. F. V. Ferreira, P. Pander, F. B. Dias, J. Morgado, A. L. Maçanita and P. T. Gomes, Dalton Trans., 2023, 52, 4933–4953 RSC.
- I. Debnath, T. Roy, J. Matern, S. A. H. Jansen, G. Fernández and K. Mahata, Org. Chem. Front., 2021, 8, 5432–5439 RSC.
- A. Nagai, K. Kokado, Y. Nagata, M. Arita and Y. Chujo, J. Org. Chem., 2008, 73, 8605–8607 CrossRef CAS PubMed.
- C. Goze, G. Ulrich, L. J. Mallon, B. D. Allen, A. Harriman and R. Ziessel, J. Am. Chem. Soc., 2006, 128, 10231–10239 CrossRef CAS.
- K. Paramasivam, C. B. Fialho, T. F. C. Cruz, A. I. Rodrigues, B. Ferreira, C. S. B. Gomes, D. Vila-Viçosa, A. Charas, J. M. S. S. Esperança, L. F. V. Ferreira, M. J. Calhorda, A. L. Maçanita, J. Morgado and P. T. Gomes, Inorg. Chem. Front., 2021, 8, 3960–3983 RSC.
- R. Deng, L. Li, M. Song, S. Zhao, L. Zhou and S. Yao, CrystEngComm, 2016, 18, 4382–4387 RSC.
- G. Y. Ruelas-Álvarez, A. J. Cárdenas-Valenzuela, L. L. Galaviz-Moreno, A. Cruz-Enríquez, J. J. Campos-Gaxiola, H. Höpfl, J. Baldenebro-López, E. C. Vargas-Olvera, V. Miranda-Soto, B. A. G. Grajeda and D. Glossman-Mitnik, Crystals, 2022, 12, 783 CrossRef.
- K. Durka, I. Głowacki, S. Luliński, B. Łuszczyńska, J. Smętek, P. Szczepanik, J. Serwatowski, U. E. Wawrzyniak, G. Wesela-Bauman, E. Witkowska, G. Wiosna-Sałyga and K. Woźniak, J. Mater. Chem. C, 2015, 3, 1354–1364 RSC.
- M. Urban, K. Durka, P. Górka, G. Wiosna-Sałyga, K. Nawara, P. Jankowski and S. Luliński, Dalton Trans., 2019, 48, 8642–8663 RSC.
- P. Li, H. Chan, S.-L. Lai, M. Ng, M.-Y. Chan and V. W.-W. Yam, Angew. Chem., 2019, 131, 9186–9192 CrossRef.
- P. A. A. M. Vaz, J. Rocha, A. M. S. Silva and S. Guieu, Materials, 2021, 14, 4298 CrossRef CAS PubMed.
- J. Shi, Z. Ran and F. Peng, Dyes Pigm., 2022, 204, 110383 CrossRef CAS.
- A. C. Murali, P. Nayak and K. Venkatasubbaiah, Dalton Trans., 2022, 51, 5751–5771 RSC.
- Y.-L. Rao and S. Wang, Inorg. Chem., 2011, 50, 12263–12274 CrossRef CAS PubMed.
- K. Naim, M. Singh, S. Sharma, R. V. Nair, P. Venugopalan, S. C. Sahoo and P. P. Neelakandan, Chem. – Eur. J., 2020, 26, 11979–11984 CrossRef CAS PubMed.
- K. Naim, S. C. Sahoo, P. Venugopalan and P. P. Neelakandan, Cryst. Growth Des., 2021, 21, 3798–3806 CrossRef CAS.
- Z. Zhang, H. Zhang, C. Jiao, K. Ye, H. Zhang, J. Zhang and Y. Wang, Inorg. Chem., 2015, 54, 2652–2659 CrossRef CAS PubMed.
- Z. Zhang, Z. Zhang, H. Zhang and Y. Wang, Dalton Trans., 2017, 47, 127–134 RSC.
- N. Borah, B. Nayak, A. Gogoi and G. Das, New J. Chem., 2019, 43, 16497–16505 RSC.
- M. Más-Montoya, L. Usea, A. E. Ferao, M. F. Montenegro, C. R. de Arellano, A. Tárraga, J. N. Rodríguez-López and D. Curiel, J. Org. Chem., 2016, 81, 3296–3302 CrossRef.
- S. D. Bhagat and A. Srivastava, CrystEngComm, 2016, 18, 4369–4373 RSC.
- X. Wang, J. Feng, Y. Bai, Q. Zhang and Y. Yin, Chem. Rev., 2016, 116, 10983–11060 CrossRef CAS PubMed.
- H. Xia, Y. Chen, G. Yang, G. Zou, Q. Zhang, D. Zhang, P. Wang and H. Ming, ACS Appl. Mater. Interfaces, 2014, 6, 15466–15471 CrossRef CAS PubMed.
- S. Shah, P. Marandi and P. P. Neelakandan, Front. Chem., 2021, 9, 708854 CrossRef CAS.
- B. Eren and Y. Bekdemir, Quím. Nova, 2014, 37(4), 643–647 CrossRef CAS.
- V. S. Sadu, H.-R. Bin, D.-M. Lee and K.-I. Lee, Sci. Rep., 2017, 7, 242 CrossRef PubMed.
- L. Catalano, D. P. Karothu, S. Schramm, E. Ahmed, R. Rezgui, T. J. Barber, A. Famulari and P. Naumov, Angew. Chem., Int. Ed., 2018, 57, 17254–17258 CrossRef CAS PubMed.
- Y. S. Zhao, J. Xu, A. Peng, H. Fu, Y. Ma, L. Jiang and J. Yao, Angew. Chem., Int. Ed., 2008, 47, 7301–7305 CrossRef CAS PubMed.
- M.-P. Zhuo, Y.-C. Tao, X.-D. Wang, S. Chen and L.-S. Liao, J. Mater. Chem. C, 2018, 6, 9594–9598 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2150129, 2207239 and 2208755. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3nr02903d |
| This journal is © The Royal Society of Chemistry 2023 |