
Nanopore actuation of a DNA-tracked nanovehicle†
Wei
Si
 *a,
Xiaojing
Lin
a,
Liwei
Wang
a,
Gensheng
Wu
b,
Yin
Zhang
a,
Yunfei
Chen
a and
Jingjie
Sha
*a
*a,
Xiaojing
Lin
a,
Liwei
Wang
a,
Gensheng
Wu
b,
Yin
Zhang
a,
Yunfei
Chen
a and
Jingjie
Sha
*a
aJiangsu Key Laboratory for Design and Manufacture of Micro-Nano Biomedical Instruments, School of Mechanical Engineering, Southeast University, Nanjing 211100, China. E-mail: wei.si@seu.edu.cn; major212@seu.edu.cn
bSchool of Mechanical and Electronic Engineering, Nanjing Forestry University, Nanjing 210037, China
First published on 14th August 2023
Abstract
As a kind of nanomachine that has great potential for applications in nanoscale sensing and manipulation, nanovehicles with unique shapes and functions have received extensive attention in recent years. Different from the existing common method of using synthetic chemistry to design and manufacture a nanovehicle, here we theoretically report a molecularly assembled DNA-tracked nanovehicle that can move on a solid-state surface using molecular dynamics simulations. A graphene membrane with four nanopores acts as the chassis of the nanoscale vehicle, and two circular ssDNAs across the nanopores serve as the wheels. The electroosmotic flows induced by independently charged nanopores with different surface charge densities under external electric fields were found to be the main power to actuate the controlled rotary motion of circular ssDNAs across every two nanopores. By tuning the rotary speed of each circular ssDNA, the linear and turning movements of the designed nanovehicle were realized. The designed nanovehicle makes it possible to have access to almost everywhere in the human body, which would lead to significant breakthroughs in the fields of nanoscale surgery, drug delivery and so on. The research not only enriches the family of nanorobots, but also opens another way for designing nanovehicles.
Introduction
Molecular motion widely exists in living organisms and undertakes important cellular functions. Molecular transports frequently occur within or between cells through various molecular motors like myosin, dynein, and kinesin that can move along actin filaments or act on microtubules.1 These molecular machines are the products of the amazing nature, and researchers have never stopped being curious about the mechanisms of their biological microscopic functions, and much effort has been devoted to exploring whether similar micro-functions can be artificially realized. With the continuous development of physical and biological science and technology, the microscopic world has been deeply explored. It is now possible to control material transport, energy conduction and information transmission at the molecular level.2 In the realization of these capabilities, many nanomolecular devices, for example motors,3,4 rotors,5 scissors,6 barrows,7 elevators,8 shuttles,9 hinges,10 switches11 and other nanoscale devices, have been gradually invented and designed to mimic or replace the functions of some biomolecules in living organisms. At the same time, as a crucial biological function, molecular transport has aroused widespread interest, resulting in various nanoscale mechanical designs such as nanowalkers,12,13 nanocars,14 and nanotrucks15 to realize the delivery of various cargos. Nanocars mentioned here are an application of molecular motors to convert mechanical energy for nanoscale transport.16 The structure of a nanocar is similar to a real car; however, it is mainly composed of wheels and a chassis.2 In 2005, the first generation nanocar,17 which consisted of spherical fullerene wheels, alkyne-based free-rotating shafts, and a molecular chassis, was successfully designed and synthesized. Tour et al. successfully achieved its movement by rolling it on a gold surface using a spherical fullerene-C60, and the direction in which the nanomolecule walked was also controlled by precise design. The reported results then validated the feasibility of controlling the directionality of motion in molecular-sized nanostructures through precise molecular synthesis.However, it was found that fullerenes had comparably low solubility during the process of synthesizing nanocars, the yield of the substitution reaction was low, and it was also difficult to carry out research on light as a driving energy source.18 In order to overcome these problems encountered in the synthesis of fullerene-wheeled nanocars, second generation nanocars, namely carborane-wheeled nanocars,19 were designed and synthesized. Compared with fullerenes, the synthesis of nanocars bearing p-carborane required fewer synthesis steps. With the continuous and deep research studies and experiments carried out by researchers, nanocars with wheels composed of different molecules have also been designed, such as adamantane-wheeled nanocars20 and organometallic-wheeled nanocars.21
Unfortunately, most of the synthesized nanocars mentioned above cannot move independently, and their motions are mainly manipulated on a solid-state surface by scanning tunneling microscopy (STM) with high atomic resolution. Usually, there is a significant electric field (E) near the STM probe so that the nanocars can get rid of the strong surface confinement22 and successful driving of the nanovehicle could happen; however charge transfer and dynamic redistribution will unavoidably occur on the nanovehicle wheels under the electric force. Besides, the molecular details during the manipulation of the nanovehicle are difficult to obtain from STM images23 as obtaining high-quality STM images are very challenging, especially for moving nanovehicles.20 What's more, using such a huge and heavy instrument of STM to actuate nanovehicles significantly limits the practical applications of nanotransportation in vivo. Recent studies have found that light24,25 can also stimulate the motion of nanocars, while the smallest size of the focusing spot was about hundreds of nanometers according to the wavelength of selected different color light, which can not guarantee the high accuracy of nanoscale manipulations. Comparably, electric fields may be the easiest and most convenient way to control the motion of nanocars on a surface.22,26
For designing new functional nanovehicles,27 all-atom molecular dynamics (MD) simulation is used by researchers as it provides a simple, convenient, and structurally accurate way to explore the mechanisms of actuating a nanovehicle. MD simulations also make it possible to visualize the movement of a nanovehicle at single atom resolution, which is very significant in constructing and actuating a nanovehicle with a new driving method.28 Akimov et al.29 created a molecular dynamics model of a nanocar and actuated it on a surface by heat, and the theoretical results successfully reproduced the kinetic characteristics of the designed nanocars and unveiled the mechanisms of experimentally observed thermal activation. Similarly, Nemati et al.30 used all-atom MD simulations to study the characteristics of nanocars at different temperatures and analyzed the influence of the wheel rolling mechanism on the motion of nanocars.
As a simple and readily available biocompatible material, in recent years DNA has been widely used in the field of nanorobots that have great potential in applications such as drug delivery for healthcare. Based on DNA self-assembly techniques, DNA molecules have been successfully applied in the design of nanowalkers,31 assembly lines,32 movable robotic arms,33 and so on. When DNA molecules were tethered to a nanoparticle, multiple-leg DNA–nanoparticle assembled nanorobots34,35 were designed by our group, and multi-mode manipulations including capturing, releasing, jumping, and crawling of the nanorobots were realized. For our previously designed nanorobots, ssDNAs acted as the legs of the nanorobots as they were not annular. If circular DNAs were used, then it might be possible to mimic the tracked wheels of a nanovehicle. As validated by the research performed by our group36,37 and other groups,38,39 the hydrophobic parts of biomolecules like DNA and protein would have strong interactions with hydrophobic surfaces like graphene and other two-dimensional membranes. Also, similar to the studies performed previously by our group,34,35 the controlled electroosmotic flows (EOFs) could be the main power to actuate the rolling of circular DNA across the nanopores.40 Thus, it actually allows the DNA-tracked wheel stably rolling on a solid-state surface just like already reported nanocars that used rotation of spherical nano-molecules to achieve controllable surface movement. According to this concept, in this paper, we theoretically designed a DNA-tracked nanovehicle with a quad-nanopore graphene membrane acting as the chassis and two circular DNAs acting as the wheels across every two nanopores. Systematic MD simulations and theoretical analysis were performed in this work to validate the feasibility of using controlled electroosmotic force as well as electrophoretic force for actuating and manipulating the nanovehicle.
Results and discussion
The designed nanovehicle consists of a chassis (the quad-nanopore graphene membrane) and two wheels (the circular ssDNAs). Each circular ssDNA was across two nanopores whose surface charge density (σ) could be tuned by the mimicked electrode independently, see Fig. 1a. The negative and positive charges were assigned to green atoms and red atoms respectively to control the rolling of the circular ssDNAs. The diameters of the nanopores are all 1.4 nm, this dimension not only allows the successful moving of DNA but also produces well controlled electroosmotic flow through the nanopores, which was already validated in our recent works.34,35,41 Once the nanopore was charged, obvious electroosmotic flow would be generated. As shown in Fig. 1b, when the nanopores were negatively (or positively) charged and an electric field along the positive z direction was applied, EOF would move upward (or downward). If a circular ssDNA1 was placed across the negatively charged pore1 and positively charged pore2, the rolling of ssDNA1 could be realized due to the cooperation of the electroosmotic flows through these two nanopores. Similarly, ssDNA2 could also roll according to the torque generated by the opposite EOF through pore3 and pore4. Thus by setting a suitable external E and σ of each nanopore, the synchronous and asynchronous rolling of the two circular DNAs across the charged nanopores may induce the straight and steering movement of the DNA-tracked nanovehicle, which will be discussed in detail below. Here, as already reported by our previous work that ssDNA would translocate in the form of base-constriction-base meshing and ratcheting across the nanopore constriction42 which actually facilitated stepwise translocation43,44 of ssDNA through ultra-thin nanopores like graphene, see Fig. 1c, and this indeed makes the synchronous and asynchronous motions of the two ssDNA-tracked wheels more controllable.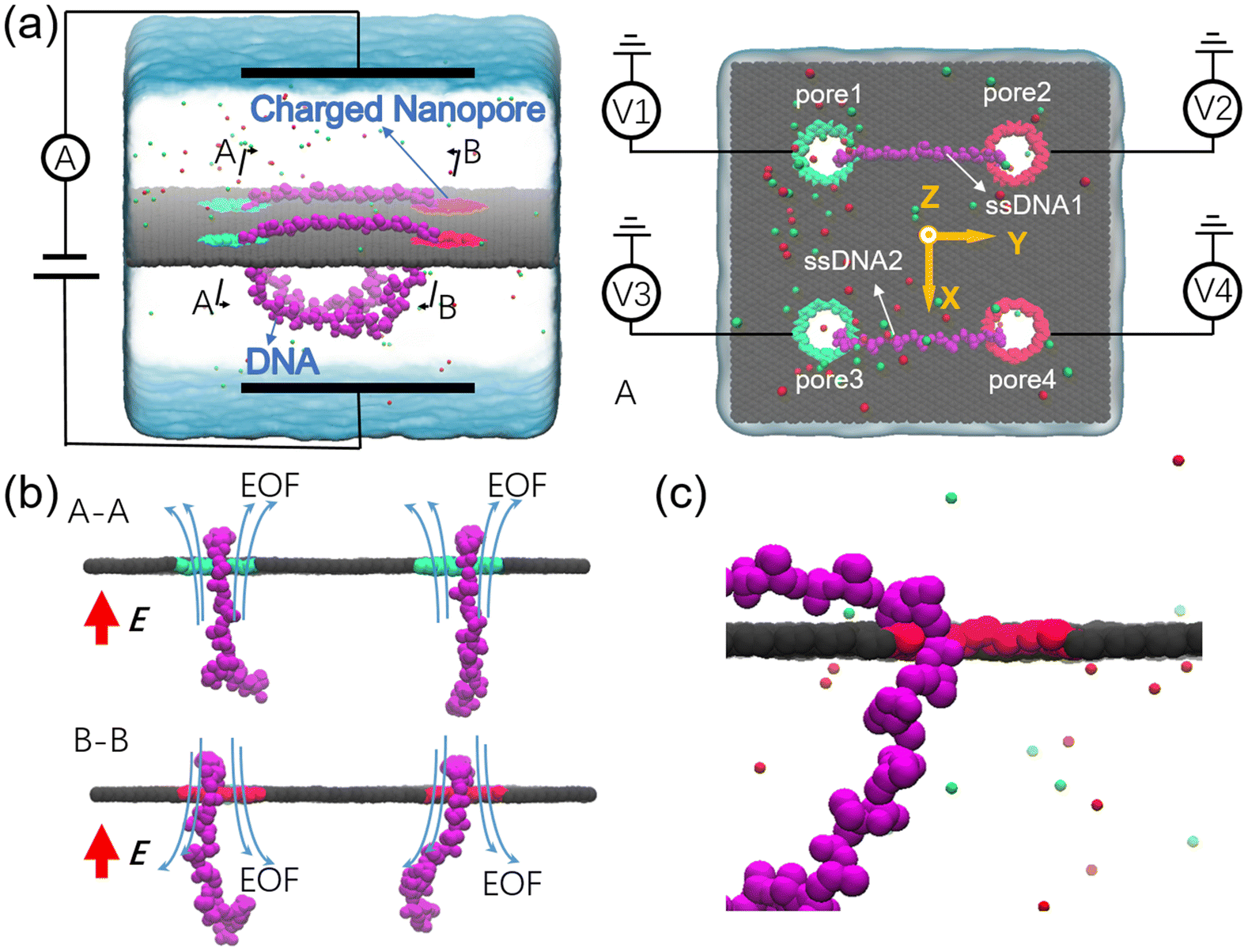 | ||
| Fig. 1 Schematic illustration of the simulation setup for actuating a DNA-tracked nanovehicle. (a) A typical simulation system setup. The DNA-tracked nanovehicle is composed of two ssDNAs acting as the wheels and a quad-nanopore graphene membrane acting as the chassis. Atoms of the graphene membrane are shown as gray spheres. The ssDNAs are shown in purple using VDW representations. Potassium and chloride ions are shown as magenta and green spheres, respectively. The ionic solution is shown as an ice blue transparent surface. The right panel also shows the top view of the system. The red atoms and green atoms at the nanopore boundaries in the membrane are connected to mimicked electrodes to independently tune the nanopore surface charge densities. The red and green colors of the nanopores indicate that the nanopores are positively and negatively charged, respectively. (b) Section views of A–A and B–B cutting planes shown in panel (a). The bluish arrows indicate the electroosmotic flows through each nanopores. (c) One of typical conformations of the ssDNA permeating through a positively charged nanopore. | ||
To investigate the feasibility of rolling the circular ssDNA across two differently charged nanopores, the system shown in Fig. 2a was first set up, the simulation details are described in ESI Method 1.† When the nanopores were not charged or of the same charge and an external electric field was applied along the z axis, no rolling movement of the circular ssDNA was observed, see Fig. S1 of the ESI.† As already reported by both our group45 and other groups,46 there is an access resistance of solid-state nanopores, and the potential mainly drops across the nanopore. If both nanopores were not charged, since the two nanopores have the same dimensions and are symmetrically arranged in the membrane, the potential drops across the nanopores would be the same, which would induce consistent electric forces exerted on the nucleotides residing inside the nanopores. Thus there was almost no net force for driving the rolling movement of the circular ssDNA. It could be found that once the nanopores were differently charged, the electroosmotic flows through the nanopores would break the balances of the electric forces exerted on the nucleotides inside the nanopores. Fig. 2a also shows two snapshots of the circular ssDNA across the negatively charged pore1 and positively charged pore2. The obvious shifting of the marked light blue nucleotide indicates the successful rolling manipulation of the circular DNA due to the imbalance of the net forces through the two differently charged nanopores. A typical example of the circular ssDNA rolling trajectory is also shown in ESI Movie 1.† For example, when pore1 and pore2 have a surface charge density of −0.105 e Å−2 and 0.105 e Å−2, respectively, and an electric field of 0.15 V Å−1 along the z direction is applied to the system, the circular ssDNA starts rolling across the two nanopores, see the y component of the center of mass (CoM) of the marked nucleotide versus simulation time shown in the top panel of Fig. 2b. Here, due to the imbalance between the electroosmotic force and electrophoretic force, an obvious conformation change was found along the z direction, Fig. 2a, and the nucleotides above the membrane was pushed tightly on the surface while the other nucleotides below the membrane were pulled away from the membrane. The mean total ionic currents (I) and the mean ionic currents induced by potassium (K+) and chloride (Cl−) ions separately under different electric fields through each nanopore are shown in ESI Fig. S2,† and the corresponding ionic current traces are also shown in ESI Fig. S3.† It is clear that ionic currents associated with coions were much smaller than that induced by counterions, which was due to the strong electroosmotic flow formed in the charged nanopores. In particular, for the negatively charged pore1, chloride ions were found to move in the same direction as potassium ions because of the high selectivity of potassium ions. Slight current variations are also observed in ESI Fig. S3,† which were caused by the conformation as well as orientation changes of the nucleotides inside the nanopores, which have already been discussed in detail in our previous works.42,47 The top panel of Fig. 2b shows three cycles of rolling movement when continuing the simulation for up to 48 ns, and the circular ssDNA took ∼12 ns to finish a complete cycle of rolling movement. Here, halting of the circular ssDNA during the rolling process was also found, see the plateau of the traces of displacement in the y direction shown in Fig. 2b. Some haltings would last even longer than 6 ns, and this was actually due to the strong binding of the nucleotides to the membrane surface38,39 during the stepwise translocation of ssDNA through the nanopore. When the electric field was increased to 0.18 V Å−1, faster rolling of the circular ssDNA was observed, see the bottom panel of Fig. 2b, and the halting time was also significantly decreased. Fig. 2c shows the relationship between the external E and the mean sliding velocity (vy) of the circular ssDNA along the y direction on the membrane surface, it was found that vy increased with E, and this was indeed attributed to the strong imbalance of the net forces exerted on the nucleotides inside the nanopores, which was influenced by the corresponding electroosmotic flows through the two differently charged nanopores. Here, it is worth recalling that the rolling velocity of the circular ssDNA was not a constant, see the plots of the y component of the instantaneous velocity of the marked nucleotide versus time under different E values shown in ESI Fig. S4.† This was due to the common Brownian dynamics48,49 and the stepwise rolling of the circular ssDNA that was induced by the strong binding of DNA to the graphene membrane.38,39 It was also found that, for the amplitude of surface charge density of 0.105 e Å−2, E should be larger than 0.14 V Å−1, and then it was possible to generate EOF with sufficient strength so as to actuate the rolling of the circular ssDNA (ESI, S4 and S5†). Besides the electric field, the effect of the absolute surface charge density of nanopores on the rolling manipulation of circular ssDNA was also investigated, Fig. 2d and e. Here, it should be noted that the surface charge density was calculated using the total charge Q (obtained by 35 atoms that are homogeneously charged at the nanopore boundary) divided by the nanopore surface area S. The estimated voltage of a battery needed to charge the pore atoms was calculated using the two formulas: V = Q/C = σS/C and C = 2Lπεε0/ln(R/Rpore), where C, L, ε and ε0 are the capacitance, length, and relative and absolute dielectric constants, respectively; R and Rpore are the outer and inner diameters of the effective charged cylinder.50 Here, under an applied electric field of ∼0.16 V Å−1, increasing the amplitude of σ from 0.035 e Å−2 (corresponding to a bias of ∼0.4 V applied to the electrode using a previously reported theory50) to 0.105 e Å−2 would make the rolling of circular ssDNA faster across the two nanopores, Fig. 2d and ESI Movie 2.† This was due to the stronger electroosmotic flows moving oppositely through the two differently charged nanopores, which formed a higher torque exerted on the circular ssDNA. However, further increasing the amplitude of surface charge density would not accelerate the rolling movement of circular ssDNA, Fig. 2e, and interestingly vy of the circular ssDNA was found to decrease with σ when the surface charge density was larger than 0.105 e Å−2, and this was reasonable and understandable as the high positive charge of pore2 would attract the negatively charged DNA tightly, and the strong binding would play the major role affecting the rolling of circular ssDNA and deaccelerate the movement. Details of the current traces and water fluxes are shown in ESI Fig. S6 and S7.† More displacements under different simulation conditions are shown in ESI Fig. S8–S10.†
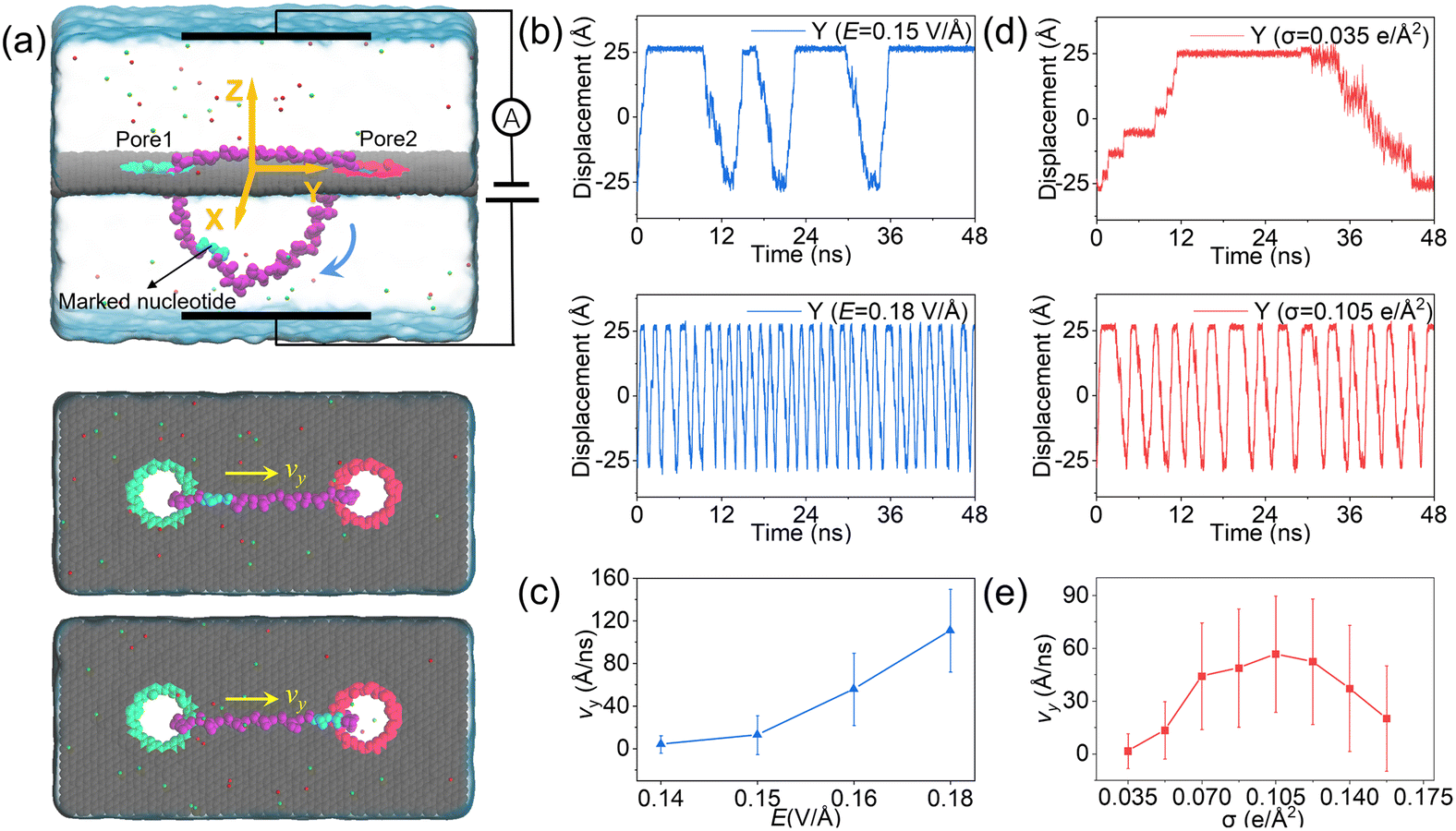 | ||
| Fig. 2 MD simulations of a circular ssDNA rolling across two independently charged nanopores. (a) Schematic illustration of the simulation system setup. The red color and green color of the nanopores indicate that the nanopore is positively and negatively charged, respectively. The blue arrow indicates the rolling direction of the circular ssDNA. Two typical conformations of the circular DNA were also shown below to indicate the two positions where the marked light blue nucleotide is located. (b) The y component of CoM of the marked nucleotide versus simulation time. The displacement traces were shown for the electric fields of 0.15 V Å−1 and 0.18 V Å−1 as two examples. The two nanopores have the same amplitude of the surface charge density (0.105 e Å−2) but different signs. (c) The effect of external E on the mean sliding velocity of the circular ssDNA along the membrane surface. The velocities (along y direction) of nucleotides within 8 Å of the center of the graphene membrane were calculated and averaged using the entire trajectories. (d) The same as panel (b) but for absolute surface charge densities of 0.035 e Å−2 and 0.105 e Å−2, respectively. The electric field was 0.16 V Å−1. (e) The same as panel (c) but for the effect of σ on the mean sliding velocity of the circular ssDNA along the membrane surface. | ||
In the above simulations of a circular ssDNA rolling across two independently charged nanopores, the obvious current difference induced by different ion species and water fluxes through the charged nanopores was observed, and the asymmetric ion and water fluxes through the two differently charged nanopores caused actuation of the circular ssDNA by electroosmotic flow driving. To further investigate the rolling mechanism of the circular ssDNA, the simulation system shown in Fig. 3a was set up, the transport dynamics of different species including K+ and Cl− and water molecules through the differently charged nanopores were studied, and the simulation details are described in the ESI Method 2.† Due to the positive charges of pore2, the potential around the nanopore was higher than that around the negatively charged pore2. When an external bias was applied along the z axis, though the potential redistributed it was still found to mainly drop across the two nanopores regardless of their charged state, Fig. 3b, which was consistent with the results from previous studies conducted by our group51 and other groups.43 The higher and lower potentials around the positively and negatively charged nanopores led to selective ion transport through them, which was proved by the obvious distinct ionic currents induced by K+ and Cl− separately through the two nanopores, Fig. 3c. The total currents through the two nanopores are shown in ESI Fig. S11.† For both nanopores, the charged nanopore would attract counterions and repel coions; thus the ionic currents induced by the counterions were much larger than that induced by coions. For example, more potassium ions translocated through the negatively charged pore1, and very less chloride ions could pass through it. Opposite results were found for the positively charged pore2. Due to the high selectivity of ion transport through the charged nanopore, uncharged water molecules were also found to permeate through the nanopore which formed the strong electroosmotic flow, and the moving direction of water molecules was opposite in the two differently charged nanopores, Fig. 3c. The electroosmotic flow was also found when lowering E to 0.02 V Å−1, ESI Fig. S12,† indicating the intrinsic property of the ion selectivity of a charged nanopore. Fig. 3d clearly shows the steady-state local densities of ionic current and water fluxes on the y–z cross-section of the double-nanopore system, and obvious ion and water fluxes were all found around the nanopores due to the local ion concentration redistributions shown in ESI Fig. S13.† In particular, the opposite water flux through the two differently charged nanopores would form a torque exerted on the circular ssDNA when it was across the two nanopores due to the two coupled opposite electroosmotic forces. Thus the mechanism of actuating the rolling movement of the circular ssDNA across the two differently charged nanopores was unveiled here.
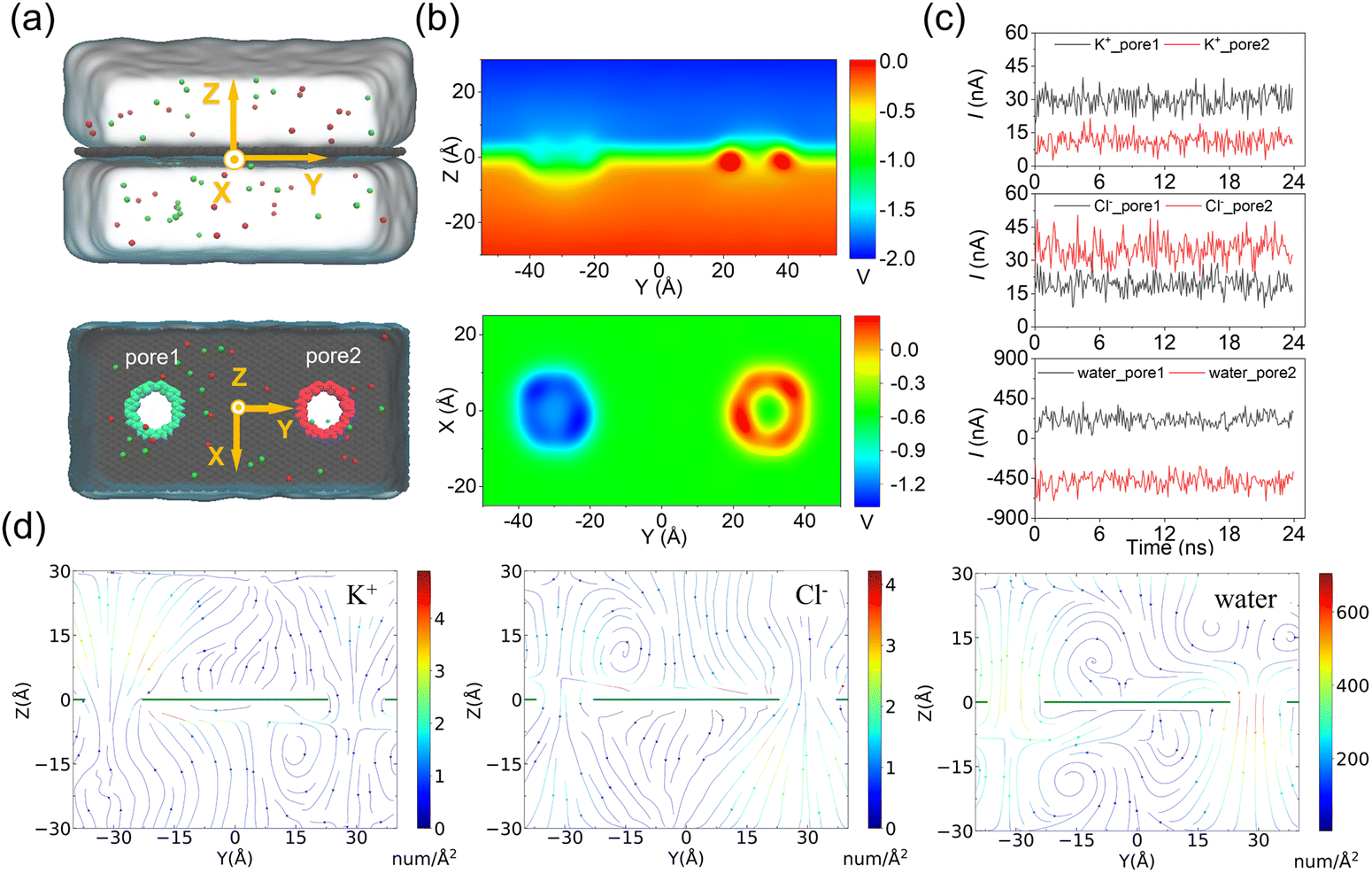 | ||
| Fig. 3 MD simulations of selective ion transport through the two differently charged nanopores. (a) The simulation system setup viewed from the side (up) and top (down). The colored nanopores indicate that they are positively (red) or negatively (green) charged, and the amplitude of the nanopore surface charge density is set to 0.105 e Å−2. (b) The two-dimensional potential distributions on the y–z plane (up) and x–y plane (down) under an E value of 0.02 V Å−1. (c) The traces versus time for ionic currents that are induced by different ions (K+ – up panel, Cl− – middle panel) and the trace versus time for the number of water molecules (down) through each charged nanopore. The negative value of the number of water molecules indicate that they move in the negative z direction. All the data were block averaged in 0.096 ns blocks. (d) Steady-state local densities of ionic current and water fluxes (streamlines) on the y–z cross section of the double-nanopore system. Flux maps of the ionic currents for K+ and Cl− are shown in the left and middle panels, respectively. The water flux map is shown in the right panel. The arrow indicates the direction of the local flux, and the color indicates the flux's magnitude. The maps were computed from a 24 ns-long MD trajectory sampled with a frequency of 4.8 ps. | ||
In the above simulations, circular ssDNA was successfully manipulated to roll across the two charged nanopores due to the well controlled electroosmotic flows through the nanopores. Based on this interesting finding, if circular ssDNAs serve as tracked wheels and the graphene membrane acts as the chassis, a proof of concept design of DNA-tracked nanovehicles was proposed, which is already shown in Fig. 1, and the simulation details are described in ESI Method 3.† Here, two DNA-tracked wheels were selected, it is because this setting not only made the designed nanovehicle closer to the appearance of a real vehicle but also supported the chassis stably to avoid rollover of it compared to one DNA-tracked wheel. The DNA-tracked nanovehicle would move straightly when the two DNA-tracked wheels roll synchronously; in contrast, the asynchronous rolling manipulations of the two circular ssDNAs will induce turning movement of the nanovehicle. As shown in Fig. 4a, similar to the results found in Fig. 3b, obvious potential redistributions were found around the four independently charged nanopores, which would induce the selective ion transport through those nanopores and affect the rolling velocities of the circular ssDNAs. According to the setup of the designed nanovehicle shown in Fig. 1a, after assigning the same amplitude (0.105 e Å−2) of σ to the four nanopores, making pore1/pore3 and pore2/pore4 negatively and positively charged, respectively, the two circular ssDNAs were observed to roll synchronously due to the same conditions applied to each nanopore pair (pore1–pore2, pore3–pore4), see the two marked light blue nucleotides shown in Fig. 4b, a typical example of the synchronous rolling trajectory for the two circular ssDNAs is shown in ESI Movie 3.† The actuation of the circular ssDNAs was further validated to be induced by the ion selectivity of the independently charged nanopores. Fig. 4c shows the mean total ionic currents and ionic currents caused by Cl− and K+ separately through each charged nanopore shown in Fig. 4b. The traces versus time for ionic currents that are induced by different ions and the trace versus time for the number of water molecules through each charged nanopore are shown in ESI Fig. S14.† By decreasing the amplitude of σ of pore1 and pore2 from 0.105 e Å−2 to 0.07 e Å−2, the asynchronous rolling movements of the two ssDNAs were found, Fig. 4d and ESI Movie 4.† Slower rolling of ssDNA1 was found compared to ssDNA2 as the pore1–pore2 nanopore pair has a low σ compared to the pore3–pore4 nanopore pair. Similar to Fig. 4c, Fig. 4e also shows the mean total ionic currents and ionic currents caused by Cl− and K+ separately through each charged nanopore shown in Fig. 4d. The y components of displacements of the marked nucleotides in ssDNA1 and ssDNA2 versus simulation time for the synchronous rolling manipulations of circular ssDNAs are shown in Fig. 4f. The same number of rolling cycles of each circular ssDNA was found for the two nanopore pairs due to the similar rolling velocity of each circular ssDNA shown in Fig. 4g. While for the asynchronous rolling manipulations of circular ssDNAs, the y components of displacements of the marked nucleotides in ssDNA1 and ssDNA2 versus simulation time are shown in Fig. 4h. Quite a different number of rolling cycles of each circular ssDNA was found for the two nanopore pairs due to the different rolling velocities of each circular ssDNA as shown in Fig. 4i. To further test the generality of the interesting results shown in Fig. 4, additional independent MD simulations with different E and σ values were performed, ESI Fig. S15–S18†. These results again validated the feasibility of synchronous and asynchronous rolling manipulations of the two circular ssDNAs. It will induce the straight and turning movements of DNA-tracked nanovehicles travelling on a solid-state membrane surface, see the detailed discussion below.
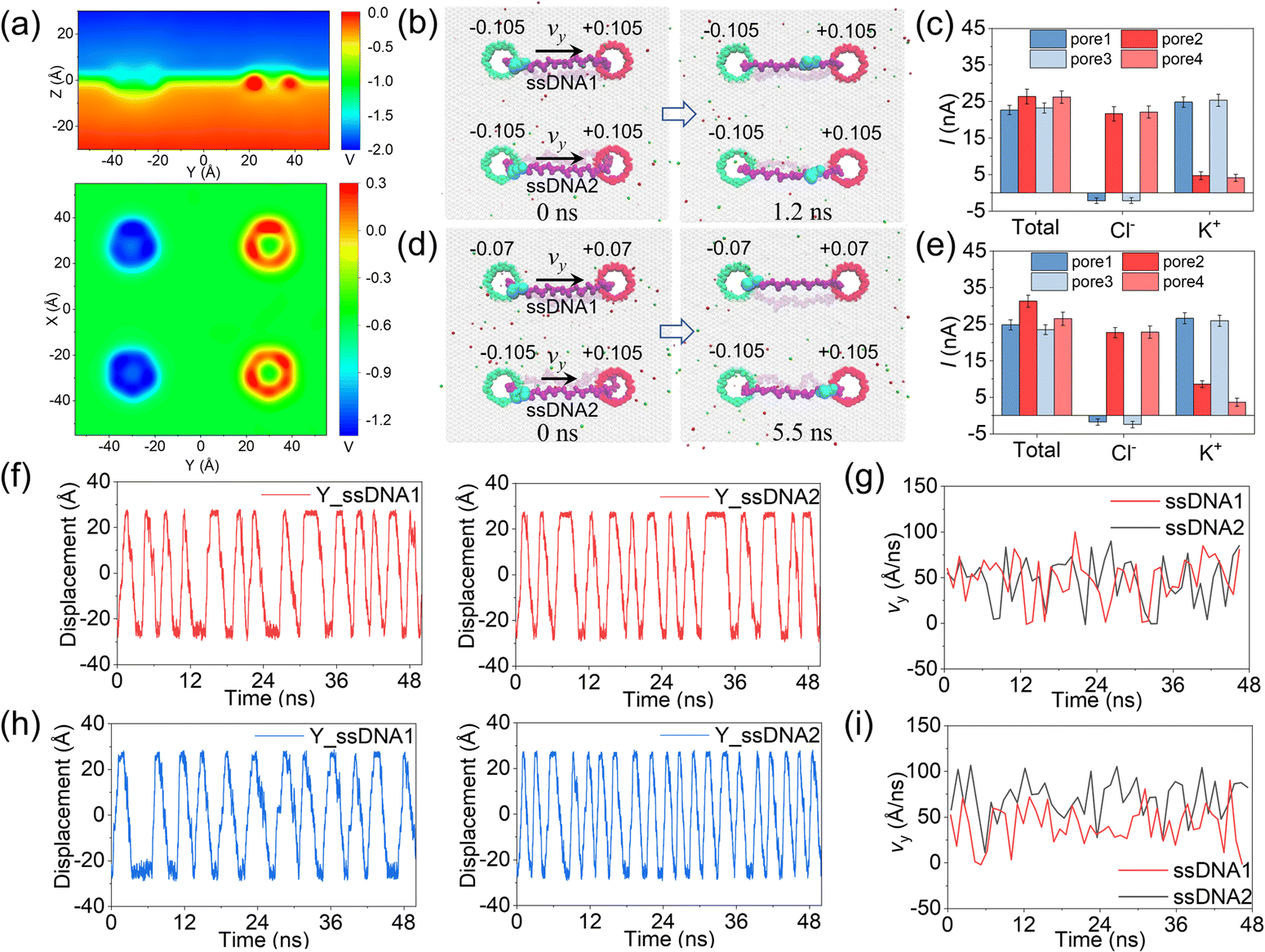 | ||
| Fig. 4 MD simulations of synchronous and asynchronous rolling of the two circular DNAs across the charged nanopores. (a) The two-dimensional potential distributions on the y–z plane (up) and x–y plane (down) under an E value of 0.02 V Å−1. (b) Two typical conformations of the DNA-tracked nanovehicle for synchronous rolling of the two circular DNAs across the charged nanopores. Green and red colors indicate the negative and positive surface charge densities, respectively. The σ values are shown above each nanopore. E is 0.14 V Å−1. The graphene membrane is set transparent to clearly show the whole DNA-tracked wheels. The two marked light blue nucleotides move synchronously with time. (c) The mean total ionic currents and ionic currents caused by Cl− and K+ separately through each charged nanopore shown in panel (b). The error bars represent the standard errors. (d) The same as panel (b) but for asynchronous rolling of the two circular DNAs across the charged nanopores. (e) The same as panel (c) but for the nanopores shown in panel (d). (f) The y components of displacements of the marked nucleotides in ssDNA1 and ssDNA2 versus simulation time for the synchronous rolling manipulations of circular ssDNAs shown in panel (b). (g) The real-time sliding velocities of the circular ssDNAs along the y direction on the membrane surface for the synchronous rolling manipulations of circular ssDNAs shown in panel (b). (h) The same as panel (f) but for the asynchronous rolling manipulations of circular ssDNAs shown in panel (d). (i) The same as panel (g) but for the asynchronous rolling manipulations of circular ssDNAs shown in panel (d). | ||
As discussed clearly above, by tuning well the surface charge density of each nanopore, synchronous and asynchronous rolling manipulations of the two circular ssDNAs were realized. To further confirm that the synchronous and asynchronous rolling manipulations of the two circular ssDNAs will induce straight and turning movements of the DNA-tracked nanovehicle, a graphene substrate was put under the designed nanovehicle to mimic the road, Fig. 5, and the simulation details are provided in ESI Method 4.† The graphene road was chosen because of the strong binding38,43 between it and the DNA molecule; thus the friction between them can facilitate the travelling of the nanovehicle on the graphene membrane. Here, torques of T1 and T2 were independently exerted on the ssDNA1 and ssDNA2, respectively, and Fa (perpendicular to the graphene road) is also applied to the nucleotides that are in touch with the membrane surface to avoid detaching of the wheels from the road, see Fig. 5a and the methods described in the Experimental section. Just as expected, when T1 was the same as T2, the DNA-tracked nanovehicle moved forward, and when the directions of the torques were reversed, the nanovehicle was observed to move backward, see the projections of CoM of the nanovehicle on the x–y plane shown in Fig. 5b. Fig. 5c shows the y components of displacements of the DNA-tracked nanovehicle versus simulation time during its forward and backward movements. The uneven speed of the nanovehicle was also observed, which was actually due to the stepwise translocations of the ssDNAs and the strong binding of nucleotides to the membrane which have already been discussed in detail above. ESI Fig. S19† shows a sequence of microscopic configurations of the DNA-tracked nanovehicle moving forward and backward. When torque T1 was smaller than T2, circular ssDNA2 rolled faster than ssDNA1, and the nanovehicle was observed to turn left during its travel. To describe the turning degree of the nanovehicle, angle θ was defined, it reflected the rotation angle measured counterclockwise from the positive x-axis in the x–y plane, Fig. 5d. The projections of CoM of the nanovehicle on the x–y plane in Fig. 5e clearly show the entire arcuate travelling traces, indicating successful direction turning of the designed nanovehicle, which was also proved by the rotation angle θ versus time shown in Fig. 5f. ESI Fig. S19† also shows a sequence of microscopic configurations of the DNA-tracked nanovehicle turning left and right. Here, it should be noted that the trajectories of the travelling nanovehicle were not that smooth, Fig. 5b, c and e, f, and sometimes the nanovehicle would pause for a while, and when it restarted moving the wheel skidding was also observed. This was actually due to the binding as well as friction between the DNA strands and the graphene membrane surface which have been discussed in detail above. Anyway, according to the analysis here, the rectilinear and steering movements of the DNA-tracked nanovehicle were proved to be feasible by the synchronous and asynchronous rolling manipulations of the two DNA-tracked wheels during its travel.
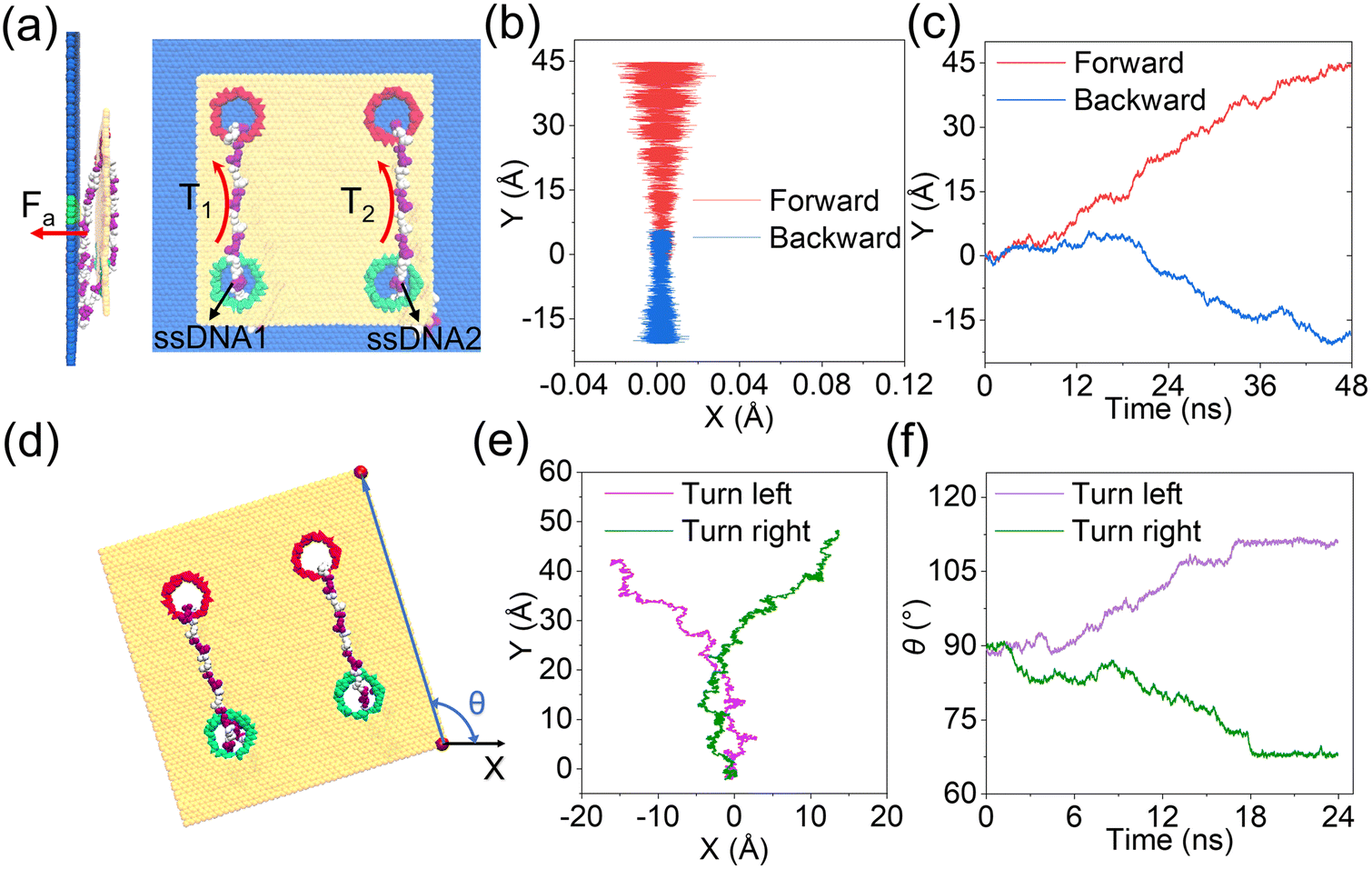 | ||
| Fig. 5 MD simulations of the DNA-tracked nanovehicle traveling on the surface of the graphene membrane. (a) The system setup viewed from the side (left) and the top (right). Atoms of the graphene chassis are shown as yellow spheres. The red-and-white circular ssDNAs are put across every two nanopores. Fa is used to keep the ssDNA in touch with the membrane surface, while T1 and T2 are torques exerted on the two ssDNAs independently to actuate their rolling movements, see methods described in the Experimental section. (b) Projection of CoM of the DNA-tracked nanovehicle during its forward and backward movements. (c) The y components of displacements of the DNA-tracked nanovehicle versus simulation time during its forward and backward movements. (d) Schematic illustration of the DNA-tracked nanovehicle with a turning angle of θ. θ is defined as the rotation angle measured counterclockwise from the positive x-axis in the x–y plane. (e) The same as panel (b) but for the processes of the DNA-tracked nanovehicle turning left and right. (f) Turning angle of the DNA-tracked nanovehicle versus simulation time during the processes of the DNA-tracked nanovehicle turning left and right. | ||
Conclusions
In this paper, we theoretically designed a DNA-tracked nanovehicle that is composed of a quad-nanopore graphene chassis and two circular ssDNA wheels. By strategically tuning the surface charge densities of the nanopores and the applied electric fields, controllable electroosmotic flows were generated independently through each charged nanopore due to the rearrangements of the potential as well as ion concentration around the nanopores that induced selective ion transports through them. The competition and/or combination of the electroosmotic force and electrophoretic force would cause force coupling exerted on the circular ssDNA wheel, and the torque formed by the coupled forces then induced the rolling movement of the circular ssDNA across a nanopore pair. By independently tuning the surface charge densities of the two nanopore pairs in the quad-nanopore graphene chassis, synchronous and asynchronous rolling manipulations of the two DNA-tracked wheels were realized, which led to the rectilinear and steering movements of the DNA-tracked nanovehicle during its travel. By simply tuning the magnitudes of the electric fields as well as surface charge densities of the nanopores, the rolling speeds of the DNA-tracked wheels could be independently controlled, and therefore the designed nanovehicle could be driven in desired directions and speeds as expected, which has advantages in wide applications like long-distance transportation. In a typical task, the DNA-tracked nanovehicle can be quickly delivered to target sites for accurate drug delivery. It should be noted that the transport of ssDNA through a graphene nanopore is still challenging and so far most experimental groups failed to do so.52 Thus, in real experiments, silicon based materials were suggested to use to validate the feasibility of the designed device and get some very preliminary results. With the fast development of nanofabrication technology, the nanopore arrays could be fabricated,53 and nanoelectrodes could also be embedded in the solid-state membrane.54 Also, the synthesis of a circular DNA is also feasible based on the developed biotechnology.55 However, the assembly of the quad-nanopore membrane chassis, independent nanoelectrodes and circular DNAs was quite challenging, which is the future experimental work we will focus on. It should also be noted that the fabrication and assembly of the designed nanovehicle is really challenging based on existing science and technology, let alone its application in recent years. However, if the global researchers could work together and solve the technical issues in the manufacturing and manipulation of nanovehicles, we believe the proof-of-concept design of the DNA-tracked nanovehicle would lead to significant breakthroughs in the fields of nanoscale surgery, drug delivery and so on.Experimental section
In this paper, all simulations with a time step of 2 fs were performed using the program NAMD2;56 periodic boundary conditions along the x, y and z axes were applied. The DNA-tracked nanovehicle is composed of a graphene membrane with four charged nanopores and two single-stranded DNA (dA22) molecules. The ssDNA, graphene membrane, TIP3P water, and ions were described by the CHARMM36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 57 force field with CUFIX58 corrections. The ion concentrations for the simulation systems were all 2 M, and some potassium ions or chloride ions were added or removed so as to neutralize the system. VMD59 and Tcl scripts were used to view and analyze the trajectories of all the simulated systems. For details of the MD simulations see ESI 1–4.†
57 force field with CUFIX58 corrections. The ion concentrations for the simulation systems were all 2 M, and some potassium ions or chloride ions were added or removed so as to neutralize the system. VMD59 and Tcl scripts were used to view and analyze the trajectories of all the simulated systems. For details of the MD simulations see ESI 1–4.†
Author contributions
Wei Si: conceptualization, resources, formal analysis, supervision, writing – original draft, and writing – review & editing; Xiaojing Lin: investigation, methodology, writing – original draft, and writing – review & editing; Liwei Wang: writing – review & editing; Gensheng Wu: writing – review & editing; Yin Zhang: writing – review & editing; Yunfei Chen: writing – review & editing; Jingjie Sha: conceptualization and writing – review & editing.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors are thankful for the financial support from the National Natural Science Foundation of China (Grant No. 52275558 and 51905097). Dr Wei Si acknowledges financial support from the Zhishan Young Scholar Award Program of Southeast University and the Fundamental Research Funds for the Central Universities (Grant No. 2242021R41168). The authors also acknowledge the supercomputing time at the Beijing Computing Center.References
- I. Rayment, Structure, 1996, 4, 501–504 CrossRef CAS PubMed.
- Y. Shirai, A. J. Osgood, Y. Zhao, Y. Yao, L. Saudan, H. Yang, H. Y. Chiu, L. B. Alemany, T. Sasaki, J. F. Morin, J. M. Guerrero, K. F. Kelly and J. M. Tour, J. Am. Chem. Soc., 2006, 128, 4854–4864 CrossRef CAS PubMed.
- A. A. Kulago, E. M. Mes, M. Klok, A. Meetsma, A. M. Brouwer and B. L. Feringa, J. Org. Chem., 2010, 75, 666–679 CrossRef CAS PubMed.
- M. Klok, N. Boyle, M. T. Pryce, A. Meetsma, W. R. Browne and B. L. Feringa, J. Am. Chem. Soc., 2008, 130, 10484–10485 CrossRef CAS PubMed.
- N. Koumura, R. W. Zijlstra, R. A. van Delden, N. Harada and B. L. Feringa, Nature, 1999, 401, 152–155 CrossRef CAS PubMed.
- T. Muraoka, K. Kinbara, Y. Kobayashi and T. Aida, J. Am. Chem. Soc., 2003, 125, 5612–5613 CrossRef CAS PubMed.
- G. Rapenne and G. Jimenez-Bueno, Tetrahedron, 2007, 63, 7018–7026 CrossRef CAS.
- J. D. Badjic, V. Balzani, A. Credi, S. Silvi and J. F. Stoddart, Science, 2004, 303, 1845–1849 CrossRef CAS PubMed.
- K. Wozniak, B. Korybut-Daszkiewicz, R. Bilewicz, A. Wieckowska and S. Domagala, Acta Crystallogr., Sect. A: Found. Crystallogr., 2004, 60, S32 Search PubMed.
- Y. Norikane and N. Tamaoki, Org. Lett., 2004, 6, 2595–2598 CrossRef CAS PubMed.
- Y. Zhang, Z. Chang, H. Zhao, S. Crespi, B. Feringa and D. P. Zhao, Chem, 2020, 6, 2420–2429 CAS.
- M. Lv, J. Liu, R. Yu and J. Jiang, Chem. Sci., 2020, 11, 10361–10366 RSC.
- X. Chen, Y. Rong, H. Wang, H. Zong and W. Li, Anal. Chim. Acta, 2021, 1182, 338937 CrossRef CAS PubMed.
- M. Li, S. Li, K. Zhang, X. Chi, H. Zhou, H. Xu, Y. Zhang, Q. Li, D. Wang and M. Zeng, Nanoscale, 2021, 13, 16748–16754 RSC.
- T. Sasaki, J. F. Morin, M. Lu and J. M. Tour, Tetrahedron Lett., 2007, 48, 5817–5820 CrossRef CAS.
- S. Kassem, T. van Leeuwen, A. S. Lubbe, M. R. Wilson, B. L. Feringa and D. A. Leigh, Chem. Soc. Rev., 2017, 46, 2592–2621 RSC.
- Y. Shirai, A. J. Osgood, Y. Zhao, K. F. Kelly and J. M. Tour, Nano Lett., 2005, 5, 2330–2334 CrossRef CAS PubMed.
- J. F. Morin, Y. Shirai and J. M. Tour, Org. Lett., 2006, 8, 1713–1716 CrossRef CAS PubMed.
- J. F. Morin, T. Sasaki, Y. Shirai, J. M. Guerrero and J. M. Tour, J. Org. Chem., 2007, 72, 9481–9490 CrossRef CAS PubMed.
- P. L. E. Chu, L. Wang, S. Khatua, A. B. Kolomeisky, S. Link and J. M. Tour, ACS Nano, 2013, 7, 35–41 CrossRef CAS PubMed.
- G. Vives and J. M. Tour, Tetrahedron Lett., 2009, 50, 1427–1430 CrossRef CAS.
- A. V. Akimov and A. B. Kolomeisky, J. Phys. Chem. C, 2012, 116, 22595–22601 CrossRef CAS.
- A. Nemati, N. H. Pishkenari, A. Meghdari and S. Sohrabpour, Phys. Chem. Chem. Phys., 2017, 20, 332–344 RSC.
- A. Saywell, A. Bakker, J. Mielke, T. Kumagai, M. Wolf, V. García-López, P.-T. Chiang, J. M. Tour and L. Grill, ACS Nano, 2016, 10, 10945–10952 CrossRef CAS PubMed.
- P. T. Chiang, J. Mielke, J. Godoy, J. M. Guerrero, L. B. Alemany, C. J. Villagómez, A. Saywell, L. Grill and J. M. Tour, ACS Nano, 2012, 6, 592–597 CrossRef CAS PubMed.
- A. van Venrooy, V. GarcíaLópez, J. T. Li, J. M. Tour and A. V. Dubrovskiy, J. Org. Chem., 2020, 85, 13644–13654 CrossRef CAS PubMed.
- Y. Shirai, K. Minami, W. Nakanishi, Y. Yonamine, C. Joachim and K. Ariga, Jpn. J. Appl. Phys., 2016, 55, 1102A1102 CrossRef.
- A. Nemati, H. N. Pishkenari, A. Meghdari and S. S. Ge, J. Phys. Chem. C, 2019, 123, 26018–26030 CrossRef CAS.
- A. V. Akimov, A. V. Nemukhin, A. A. Moskovsky, A. B. Kolomeisky and J. M. Tour, J. Chem. Theory Comput., 2008, 4, 652–656 CrossRef CAS PubMed.
- M. Vaezi, N. H. Pishkenari and A. Nemati, Comput. Mater. Sci., 2022, 207, 111317 CrossRef CAS.
- A. J. Thubagere, W. Li, R. F. Johnson, Z. Chen, S. Doroudi, Y. L. Lee, G. Izatt, S. Wittman, N. Srinivas, D. Woods, E. Winfree and L. Qian, Science, 2017, 357, eaan6558 CrossRef PubMed.
- X. Zhang, X. Ding, J. Zou and H. Gu, Methods Mol. Biol., 2017, 1500, 257–268 CrossRef CAS PubMed.
- E. Kopperger, J. List, S. Madhira, F. Rothfischer, D. C. Lamb and F. C. Simmel, Science, 2018, 359, 296–301 CrossRef CAS PubMed.
- W. Si, Z. Zhu, G. Wu, Y. Zhang, Y. Chen and J. Sha, Small Methods, 2022, 6, e2200318 CrossRef PubMed.
- W. Si, M. Yu, G. Wu, C. Chen, J. Sha, Y. Zhang and Y. Chen, ACS Nano, 2020, 14, 15349–15360 CrossRef CAS PubMed.
- W. Si, R. Yuan, G. Wu, Y. Kan, J. Sha, Y. Chen, Y. Zhang and Y. Shen, J. Phys. Chem. Lett., 2022, 13, 3863–3872 CrossRef CAS PubMed.
- W. Si, Q. Y. Sun, C. Chen, M. Yu, J. J. Sha, Y. Zhang, Y. J. Kan and Y. F. Chen, Small Methods, 2020, 4, 1900822 CrossRef CAS.
- M. Shankla and A. Aksimentiev, Nat. Nanotechnol., 2019, 14, 858–865 CrossRef CAS PubMed.
- B. Luan and R. Zhou, Nat. Commun., 2019, 10, 4610 CrossRef PubMed.
- B. Luan and A. Aksimentiev, Phys. Rev. E, 2008, 78, 021912 CrossRef PubMed.
- W. Si, Y. Zhang, J. Sha and Y. Chen, Nanoscale, 2018, 10, 19450–19458 RSC.
- M. Yu, W. Si, T. Zeng, C. Chen, X. Lin, Z. Ji, F. Guo, Y. Li, J. Sha and Y. Dong, J. Phys. Chem. Lett., 2021, 12, 9132–9141 CrossRef CAS PubMed.
- M. Shankla and A. Aksimentiev, Nat. Commun., 2014, 5, 5171 CrossRef CAS PubMed.
- D. B. Wells, M. Belkin, J. Comer and A. Aksimentiev, Nano Lett., 2012, 12, 4117–4123 CrossRef CAS PubMed.
- W. Si, H. J. Yang, K. Li, G. S. Wu, Y. Zhang, Y. J. Kan, X. Xie, J. J. Sha, L. Liu and Y. F. Chen, Sci. China: Technol. Sci., 2017, 60, 552–560 CrossRef CAS.
- C. Hyun, R. Rollings and J. L. Li, Small, 2012, 8, 385–392 CrossRef CAS PubMed.
- W. Si and A. Aksimentiev, ACS Nano, 2017, 11, 7091–7100 CrossRef CAS PubMed.
- J. Comer and A. Aksimentiev, J. Phys. Chem. C, 2012, 116, 3376–3393 CrossRef CAS PubMed.
- C. J. F. Solano, K. R. Pothula, J. D. Prajapati, P. M. De Biase, S. Y. Noskov and U. Kleinekathöfer, J. Chem. Theory Comput., 2016, 12, 2401–2417 CrossRef CAS PubMed.
- S.-W. Nam, M. J. Rooks, K.-B. Kim and S. M. Rossnagel, Nano Lett., 2009, 9, 2044–2048 CrossRef CAS PubMed.
- W. Si, H. Yang, G. Wu, Y. Zhang and J. Sha, Nanoscale, 2021, 13, 15352–15361 RSC.
- A. Smolyanitsky and B. Luan, Phys. Rev. Lett., 2021, 127, 138103 CrossRef CAS PubMed.
- S. Pud, S. H. Chao, M. Belkin, D. Verschueren, T. Huijben, C. van Engelenburg, C. Dekker and A. Aksimentiev, Nano Lett., 2016, 16, 8021–8028 CrossRef CAS PubMed.
- K.-H. Paik, Y. Liu, V. Tabard-Cossa, M. J. Waugh, D. E. Huber, J. Provine, R. T. Howe, R. W. Dutton and R. W. Davis, ACS Nano, 2012, 6, 6767–6775 CrossRef CAS PubMed.
- E. O. Prakash, D. Eric and D. Pascal, J. Org. Chem., 2003, 68, 8708–8710 CrossRef PubMed.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kalé and K. Schulten, J. Comput. Chem., 2005, 26, 1781–1802 CrossRef CAS PubMed.
- K. Vanommeslaeghe, E. Hatcher, C. Acharya, S. Kundu, S. Zhong, J. Shim, E. Darian, O. Guvench, P. Lopes, I. Vorobyov and A. D. Mackerell, Jr., J. Comput. Chem., 2010, 31, 671–690 CAS.
- J. Yoo and A. Aksimentiev, J. Phys. Chem. Lett., 2012, 3, 45–50 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nr02633g |
| This journal is © The Royal Society of Chemistry 2023 |