
meso-Carbazole decorated BODIPYs – an electron donor–acceptor system with excellent fluorosolvato/vapochromic behavior, aggregation-induced emission, and antileishmanial activity†
Diana
Mathew
ac,
Santanu
Sasidharan
b,
Prakash
Saudagar
b,
Subramaniam
Sujatha
*a and
Pattiyil
Parameswaran
 *c
*c
aDepartment of Chemistry, Bioinorganic Materials Research Laboratory, National Institute of Technology Calicut, Kozhikode, 673 601, India. E-mail: ssinorgsuji@gmail.com
bDepartment of Biotechnology, National Institute of Technology Warangal, Warangal, 506004, India
cDepartment of Chemistry, Theoretical and Computational Chemistry Laboratory, National Institute of Technology Calicut, Kozhikode, 673 601, India. E-mail: param@nitc.ac.in
First published on 29th March 2023
Abstract
We report the synthesis and photophysical properties of meso-carbazole BODIPY C1 and its β-monobromo (C2) and β,β′-dibromo (C3) substituted derivatives. Since carbazole is electron-rich as compared to BODIPY, the compounds C1 and C2 act as electron donor–acceptor systems and show red-shifted fluorosolvato/vapochromism with a Stokes shift of 170–180 nm as the dielectric constant of the solvent increases. This fluorosolvato/vapochromism is attributed to twisted intramolecular charge transfer (TICT), as confirmed by viscosity-dependent studies. The fluorescence emission and solvent polarity-dependent fluorochromism are hardly observed in the dibromo derivative C3 due to the enhanced intersystem crossing (ISC), which is evident from DPBF titrations and the molecular orbital analysis. All three luminogens show excellent aggregation-induced emission (AIE) behavior and act as red-light-emitting aggregates. Thus, the low-intensity TICT-induced red emission can be enhanced by aggregate formation. Moreover, here we report for the first time the anti-leishmanial activity of carbazole BODIPYs against Leishmania donovani promastigotes, which can be taken forward as potential anti-leishmanial compounds.
Introduction
Organic luminescent materials have received multifaceted interest in scientific and technological fields due to their potential applications as light-harvesting devices,1 tunable organic laser dyes,2 fluorescent sensors,3 biological labeling and imaging dyes.4–6 Among them, the boron dipyrromethene (BODIPY) fluorescent dyes have been the focus of considerable research over the last three decades.7 Their photophysical properties can be modified by the nature of the substituents and their position on the BODIPY core. Moreover, the functionalized derivatives, such as halogenated BODIPYs, are key building blocks for synthesizing substituted BODIPYs. Indeed, they generate longer wavelength BODIPY dyes and are potential sensitizers for photodynamic therapy (PDT).8,9 Thus, the effect of halogen atoms such as bromine on the structural and spectral properties of BODIPYs is quite interesting.10–12Although the structural versatility of BODIPYs benefits tuning their photophysical and chemical properties,11 the major challenge in applying this family of dyes is the aggregation caused quenching (ACQ) in their solid-state and small Stokes shift of the α,β-unsubstituted derivatives.13 One approach to achieving a large Stokes shift is to incorporate electron-rich moieties at the meso-position of BODIPYs selectively. This will induce a charge transfer process and exhibit excellent solvatochromic and vapochromic luminescence effects.16–18 However, the solvent polarity-dependent red-shifted emission is usually weakened in solution due to intramolecular charge transfer (ICT). On the other hand, the red emission can be revived by the formation of aggregates. Restricting intramolecular motion in the aggregates enhances the luminance efficiency in the condensed state (aggregation-induced emission (AIE)).14,15 Hence, appending bulky groups at the BODIPY core is suitable for generating twisted structures, which hampers the π–π interactions between the adjacent molecules.
Hence, we have selected the carbazole unit, which has an electron-rich and bulky aromatic fragment, having good absorption and photoluminescence behavior for the construction of a donor–acceptor system.19–21 Carbazole is known for its unique properties such as excellent thermal and chemical stability, low redox potential, facile functionalization of the aromatic ring, hole transport properties, and medicinal activity.22–24,33 It is reported that the absorption and emission behavior of BODIPYs can be modified by the presence of an electron-rich carbazole moiety.25,26 The linkage and the position at which carbazole is attached to the BODIPY scaffold play a major role. The structural diversity of the carbazole-BODIPY conjugates includes the BODIPY derivatives with carbazole rings at the alpha, beta, and meso-positions of the BODIPY skeleton.27–32,38 Recently, Kesavan et al. reported that the carbazole at the meso-position altered the electronic properties of the BODIPYs.28 Misra et al. studied the donor–acceptor interactions between carbazole and BODIPY,34 while Giribabu and coworkers described the photoinduced energy transfer in carbazole-BODIPY dyads.25 Though the reports on carbazole-substituted BODIPYs have increased considerably, an unambiguous report of the AIE effect of carbazole-BODIPYs is not yet reported.
Considering the unique photophysical properties of carbazole, we combined BODIPY with carbazole to design an electron donor–acceptor system with dual photoresponse to solvent polarity and luminogen aggregation. The red-shifted and quenched emission in polar solvents caused by TICT can be revived by the AIE behavior of meso-carbazole BODIPYs in aqueous mixtures. We further discuss the role of bromine substitution in generating triplet photosensitizers.
The details of the antileishmanial activity of BODIPY dyes are very limited in the literature. Leishmaniasis is an intracellular protozoal parasitic infection that causes a spectrum of diseases in humans. Leishmaniasis remains in the cutaneous form to the fatal visceral form worldwide because of the unavailability of safe and cost-effective drugs for treatment. Inspired by the reports on the antileishmanial activity of carbazole-based compounds,35–37 we attempted antileishmanial studies of the synthesized carbazole-BODIPY derivatives towards Leishmania donovani promastigotes.
Results and discussion
Syntheses and characterization of meso-carbazole BODIPY and its brominated derivatives
The meso-carbazole substituted BODIPY (C1) was synthesized in a one-pot reaction, which involves the synthesis of dipyrromethane (3) followed by oxidation with 3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) and further complexation with borontrifluoroetherate (BF3OEt2) (Scheme 1). The meso-carbazole appended dipyrromethane (3) was obtained by trifluoroacetic acid (TFA)-assisted condensation between pyrrole (1) and N-ethyl carbazole-3-carboxaldehyde (2) as per the literature.11,21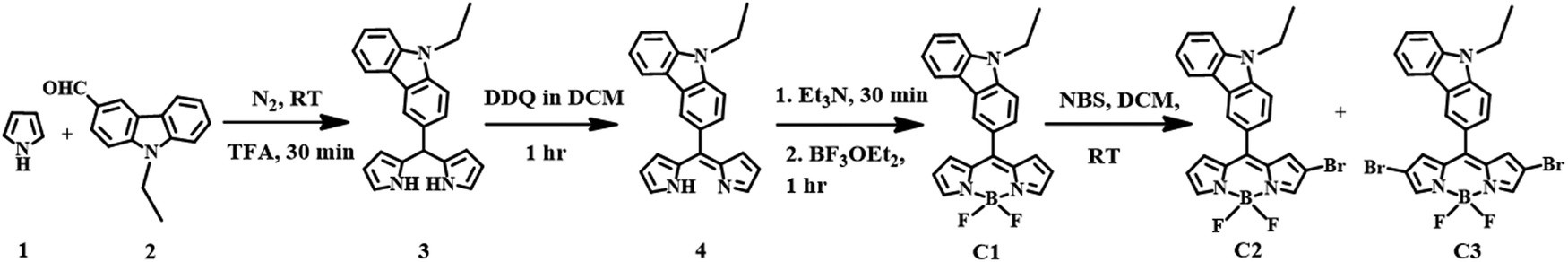 | ||
| Scheme 1 Synthesis of meso-carbazole substituted BODIPY (C1) and its mono- (C2) and dibromo derivatives (C3). | ||
The dipyrromethane formed was immediately subjected to oxidation with DDQ in DCM at room temperature for an hour after removing the excess pyrrole. The resulting dipyrrin (4) was complexed with BF3OEt2. The reaction was achieved within an hour, and the corresponding meso-carbazole appended BODIPY (C1) was isolated by silica gel column chromatography as a reddish-orange powder with a 35% yield.
The regioselective bromination of α,β-unsubstituted BODIPY C1 was done by the reaction with N-bromosuccinimide (NBS) in two different equivalences (Scheme 1).39,40 The treatment of C1 with 1.2 equivalents of NBS resulted in the formation of 75% C2 along with unreacted C1. However, the addition of 2.4 equivalents gave 25% C3 along with a 15% yield of C2.
The synthesized meso-carbazole BODIPY derivatives were quite soluble in common organic solvents and were characterized by FTIR, 1H, and 13C NMR spectroscopic methods, high-resolution mass spectrometry (HRMS), and XRD analysis, as well as by UV-visible and fluorescence spectroscopic techniques. The IR spectra of the synthesized compounds show the characteristic aromatic ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C−H vibrational peak at 2923 cm−1 and the vibrational peak at 1537 and 1735 cm−1 corresponding to B–F and CN stretching in the BODIPY core (Fig. S1, ESI†). The B–N stretching frequency is observed at 1068 cm−1. The strong vibrations around 1342–1266 cm−1 correspond to the C–N stretching of the carbazole moiety. The β-C–H stretching vibration of the pyrrole ring observed at 770 cm−1 in unbrominated BODIPY C1 is absent in the brominated derivative C3. The absorption in the range of 500–600 cm−1 indicates the presence of the C–Br stretching frequency of aryl bromides in brominated BODIPYs C2 and C3.
C−H vibrational peak at 2923 cm−1 and the vibrational peak at 1537 and 1735 cm−1 corresponding to B–F and CN stretching in the BODIPY core (Fig. S1, ESI†). The B–N stretching frequency is observed at 1068 cm−1. The strong vibrations around 1342–1266 cm−1 correspond to the C–N stretching of the carbazole moiety. The β-C–H stretching vibration of the pyrrole ring observed at 770 cm−1 in unbrominated BODIPY C1 is absent in the brominated derivative C3. The absorption in the range of 500–600 cm−1 indicates the presence of the C–Br stretching frequency of aryl bromides in brominated BODIPYs C2 and C3.
In the 1H NMR spectra of the synthesized BODIPYs, the proton intensities and chemical shifts agree with the expected structure. The 1H NMR spectra of BODIPY C1 show two peaks of two proton intensities at δ 6.57 ppm and δ 7.04 ppm, which corresponds to two sets of β-pyrrolic hydrogens and the α-pyrrolic protons appear as a singlet at δ 7.94 ppm (Fig. S2, ESI†). The carbazole aromatic protons appear as five sets of signals between δ 7.31 and δ 8.35 ppm. The N-ethyl aliphatic protons appear as a triplet of three hydrogen intensities at δ 1.52 ppm and a quartet of two hydrogen intensities at δ 4.46 ppm. The absence of proton signals corresponding to β-pyrrole hydrogens at the 2- and 2,6-positions indicates the successful bromination at the β-pyrrole carbon in C2 and C3, respectively (Fig. S2b and S2c, ESI†). The 13C NMR spectra of BODIPYs C1–C3 recorded in CDCl3 show peaks at δ 148.1, 146.1, 145.2, 141.1 ppm, corresponding to the aromatic pyrrolic carbon signals. The aromatic carbazole peaks appear between δ 133.3 and δ 103.7 ppm. The two aliphatic carbon atoms of the N-ethyl substituent at the carbazole unit appear around δ 14.1 and δ 39.5 ppm in the 13C NMR spectra of the BODIPYs (Fig. S3, ESI†). The mass spectrum of the compounds discloses the peak representing the molecular ion at m/z = 385.1573, 465.0664, and 542.9751, for C1, C2, and C3, which represents the protonated molecular ion (Fig. S4, ESI†).
Single crystal X-ray diffraction studies
The single crystal of the BODIPY C1 was obtained by the slow evaporation of hexane into chloroform for two weeks. The BODIPY C1 crystallizes in a monoclinic crystal system with a P21/c space group (Fig. 1a and Tables S1, S2, ESI†). The two pyrrole rings and the boron-containing central six-membered ring are coplanar (Φpyrrole = 178.5°), while fluorine atoms are in different planes. The boron atom in BODIPY C1 exhibits distorted tetrahedral geometry with an N1–B–N2 angle of 106.0° and F1–B–F2 angle of 108.5° (F1–B–N1 = 110.16°).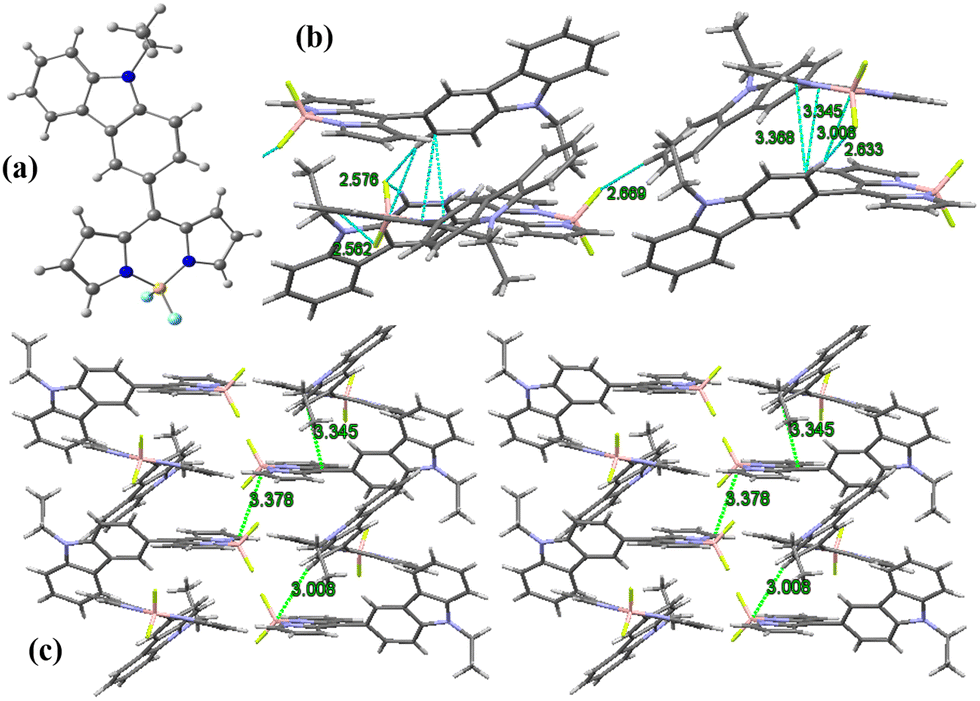 | ||
| Fig. 1 (a) X-Ray crystal structure of BODIPY C1; (b) unit cell and intermolecular interactions in C1; (c) molecular crystal packing of BODIPY C1. | ||
The dihedral angle between the meso-carbazole substituent and the BODIPY core is midway between planar and perpendicular conformation. This non-planar arrangement of meso-carbazole substituent with the BODIPY core limits the possible resonance between the BODIPY core and the carbazole substituent, which is confirmed by the presence of CBODIPY–CCarbazole single-bond distance (1.472 Å). The molecular crystal packing of C1 exhibits a parallel 3D arrangement of molecules stabilized by (BODIPY)B⋯H–C(carbazole) interaction (B1–H10 = 3.008 Å), (pyr)C⋯C(carbazole) π–π interactions (C13–C10 = 3.345 Å, C14–C10 = 3.368 Å) and (BODIPY)F⋯H–C(carbazole) intermolecular hydrogen bonding interactions (F2–H10 = 2.633 Å, F1–H3 = 2.669 Å, F1–H22 = 2.562 Å, F2–H9 = 2.576 Å) (Fig. 1b and c).
Photophysical properties
The photophysical properties of carbazole and the synthesized compounds were studied in solvents of different polarities and tabulated in Tables S3, and S4 (ESI†). The aromatic π → π* transitions of the carbazole chromophore are attributed to the peaks at 293, 333, and 342 nm (Fig. 2).25,26 The synthesized meso-carbazole substituted BODIPY C1–C3 shows a well-resolved and intense absorption band in the 490–535 nm region with a shoulder centered around 480 nm and a weak broad absorption band around ∼330–390 nm.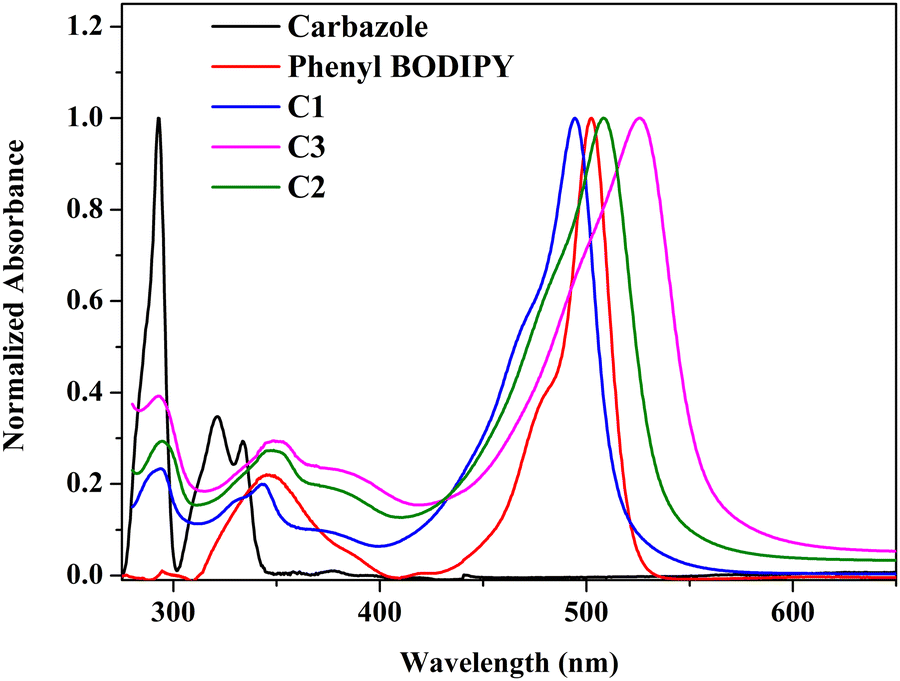 | ||
| Fig. 2 Normalized absorption spectra of carbazole, phenyl-BODIPY, and meso-carbazole BODIPYs–C1, C2 and C3 in hexane (10−5 M) at room temperature. | ||
Introducing a carbazole moiety at the meso-position of the BODIPY chromophore did not cause any drastic spectral change in the UV-vis spectra compared to meso-phenyl BODIPY except for a peak around 290 nm in the UV region corresponding to the carbazole unit. This is due to the non-planar arrangement of the meso-carbazole substituent with the aromatic BODIPY core, which prevents electron π-conjugation, which is evident in the crystal structure. On the other hand, the substitution of bromine at the pyrrole carbon produced a redshift of ∼10 nm in the λmax, which is consistent with the reported literature.11 The λmax is red-shifted from 499 nm in C1 to 516 nm in C2 in hexane, and is further shifted to 533 nm in dibromo derivative C3 (Fig. S5a, ESI†).
The fluorescence analysis of meso-carbazole substituted BODIPYs shows that C1 gives an emission at 507 nm in hexane (λexc = 480 nm) (Fig. S5b, ESI†). A significant red-shift of ∼20 nm was observed in the emission maximum through each attached bromine in brominated derivatives C2 (λmax = 527 nm) and C3 (λmax = 549 nm), along with a decrease in fluorescence quantum yield. The fluorescence quantum yield in hexane decreased from 0.51 (C1), to 0.4 (C2) to 0.11 (C3) in unbrominated and brominated derivatives, owing to the heavy halogen effect, which was more prominent in dibromo derivative C3.11 The synthesized meso-carbazole substituted BODIPYs C1 and C2 show remarkable fluorosolvatochromism (vide infra). Hence, the fluorescence efficiency of the BODIPY can be modulated by incorporating the carbazole unit at the meso-position.
The BODIPYs C1, C2, and C3 are strongly emissive in their solid state. The emission peaks are observed at 626, 597, and 620 nm for C1, C2, and C3 (Fig. S5c, ESI†). The placement of the bulky carbazole group on the BODIPY core structure hinders π–π stacking between the neighboring molecules and enhances the emission in the solid state. The solid-state spectrum of the compounds shows notable emission at a longer wavelength, as expected, considering the stronger intermolecular interactions.49
Theoretical calculations
We have performed quantum mechanical calculations to understand the structural and electronic properties of the synthesized meso-carbazole-substituted BODIPYs. The geometrical optimizations were carried out in the gas phase by choosing the initial coordinates from the X-ray crystallography data (Fig. 3).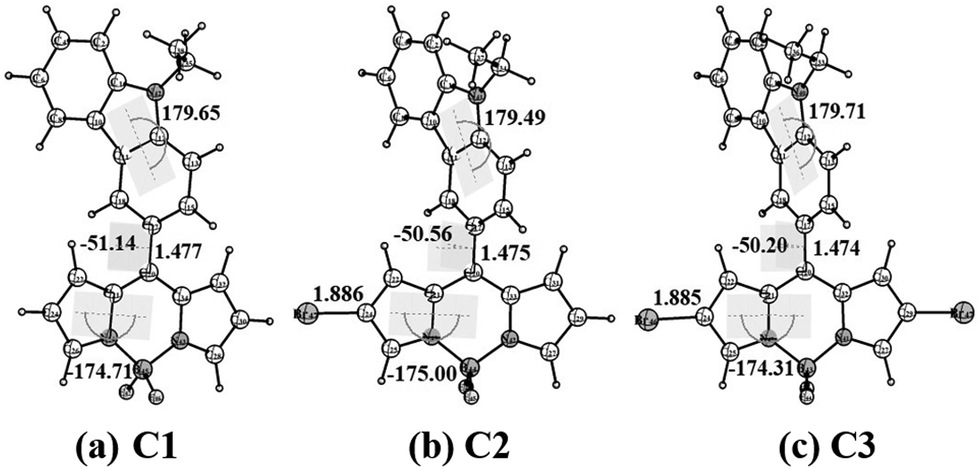 | ||
| Fig. 3 Optimized geometry of meso-carbazole substituted BODIPY C1, C2, and C3 at the BP86/def2-TZVPP level of theory at the ground state in the gas phase. All angles are given in degrees and bond lengths in Å. | ||
The geometrical parameters of C1 agree with the single-crystal data. The carbazole ring and the BODIPY core are non-planar to each other (dihedral angle between the carbazole and BODIPY plane Φcarb = 51°) and the bromine substitution at the β-pyrrolic carbon does not yield a significant geometrical change. The β-pyrrolic carbon atoms of BODIPY C1 are comparatively nucleophilic, which validates well the molecular electrostatic potential (MESP) values on the van der Waals surface of the carbon atoms (Fig. S6, ESI†). The MESP values on the β-pyrrolic carbon atom C24 (−10.04 kcal mol−1) have more negative potential as compared to the carbazole carbon atoms C2, C13, and C18 (−4.50, −0.89, and −3.66 kcal mol−1, respectively) (Table S5, ESI†). This explains the successful electrophilic bromination at β-pyrrolic carbon atoms other than at the carbazole ring.
The frontier molecular orbital analysis of C1–C3 in the singlet state in DMSO shows that the highest occupied molecular orbital (HOMO) is mainly localized on the carbazole unit, and the lowest unoccupied molecular orbital (LUMO) is on the BODIPY core. The HOMO−1 orbital is localized on the BODIPY core with the contribution from bromine in C2 and C3 (Fig. 4).
 | ||
| Fig. 4 Selected molecular orbitals and important electronic transitions of C1, C2, and C3 computed using TDDFT at the M06/def2-TZVPP//BP86/def2-TZVPP level of theory in DMSO using the PCM solvent model. Eigenvalues are given in eV. The surfaces are plotted at the iso-surface value of 0.03. The corresponding wavelength (λ) in nm and oscillator strength (f) are given. | ||
To study the electronic absorption behavior and the effect of solvents on the photophysical properties of the BODIPYs C1–C3, time-dependent density functional theory (TD-DFT) calculations were performed in DMSO using the PCM solvent model at the M06/Def2-TZVPP//BP86/Def2-TZVPP level of theory. The computed vertical transitions and their oscillator strengths directly correlate with the experimental results. The HOMO−1, HOMO, and LUMO molecular orbitals are mainly involved in the transitions. The most probable transition in DMSO is from HOMO−1 → LUMO in C1 (λmax calc. = 427 nm, f = 0.37), C2 (λmax calc. = 444 nm, f = 0.37), and C3 (λmax calc. = 461 nm, f = 0.41), which corresponds to π → π* transition in the BODIPY core (Table S6, ESI† and Fig. 4). There is one more transition at a higher wavelength having appreciable oscillator strength (C1: λmax calc. = 490 nm f = 0.25, C2: λmax calc. = 508 nm f = 0.26, and C3: λmax calc. = 526 nm f = 0.27), which corresponds to the π → π* transition between the carbazole and the BODIPY and denotes a charge-transfer transition. Note that the oscillator strength of this transition is reduced considerably when the TDDFT calculations are performed on a structure with Φcarb = 90° (Table S7 and Fig. S7, ESI†). This matches with the experimentally observed absorption spectra that the charge transfer is not observed in the ground state geometry. This is because the free rotation of carbazole with respect to the BODIPY core is possible between the dihedral angle 51° to 128.8° (51° and 128.8° are equivalent structures) through 90° as it can easily overcome the energy barrier of 1.46 kcal mol−1 at room temperature (Fig. S21 – rotational energy plot, ESI†) (vide infra).
Moreover, the bromine substitution at the pyrrolic carbon has imparted stabilization in the molecular orbitals in C2 and C3, and subsequently, the HOMO−1 → LUMO energy gap is reduced from 3.46 eV in C1 to 3.38 eV in C2 and 3.31 eV in C3 (Fig. S8, ESI†). This explains the bathochromic shift in λmax observed in the absorption spectra upon bromination. These results indicate that the electronic structure of these compounds can be influenced by regioselective bromination, which can tune their photophysical properties.
Intramolecular excitation energy transfer
Intramolecular excitation energy transfer is observed in the meso-carbazole BODIPYs. The parameters such as solvent used, concentration, and slit width were kept constant in this study to compare the results at different emission wavelengths.The excitation of meso-carbazole BODIPY at 290 nm in CHCl3 results in the HOMO → LUMO+1 transition, where both orbitals are in the carbazole unit. Interestingly, the emission occurs from the BODIPY unit at 500 nm in C1, and the carbazole emission was significantly diminished in the spectra. Similarly, C2 emits at 505 nm, and C3 gives a weak emission at 513 nm upon excitation at 290 nm (Fig. 5a). The emission at 500 nm occurs from the LUMO → HOMO−1 orbital localized on the BODIPY core (Fig. 5b and Fig. S9, ESI†). Hence, the photophysical mechanism can be explained as follows. The S0 → S2 absorption occurs by the absorption of radiation at 290 nm, which then undergoes internal conversion to the S1 state and the emission occurs from the S1 state. These findings are consistent with the energy transfer efficiency calculated in carbazole BODIPY by Gupta et al.21
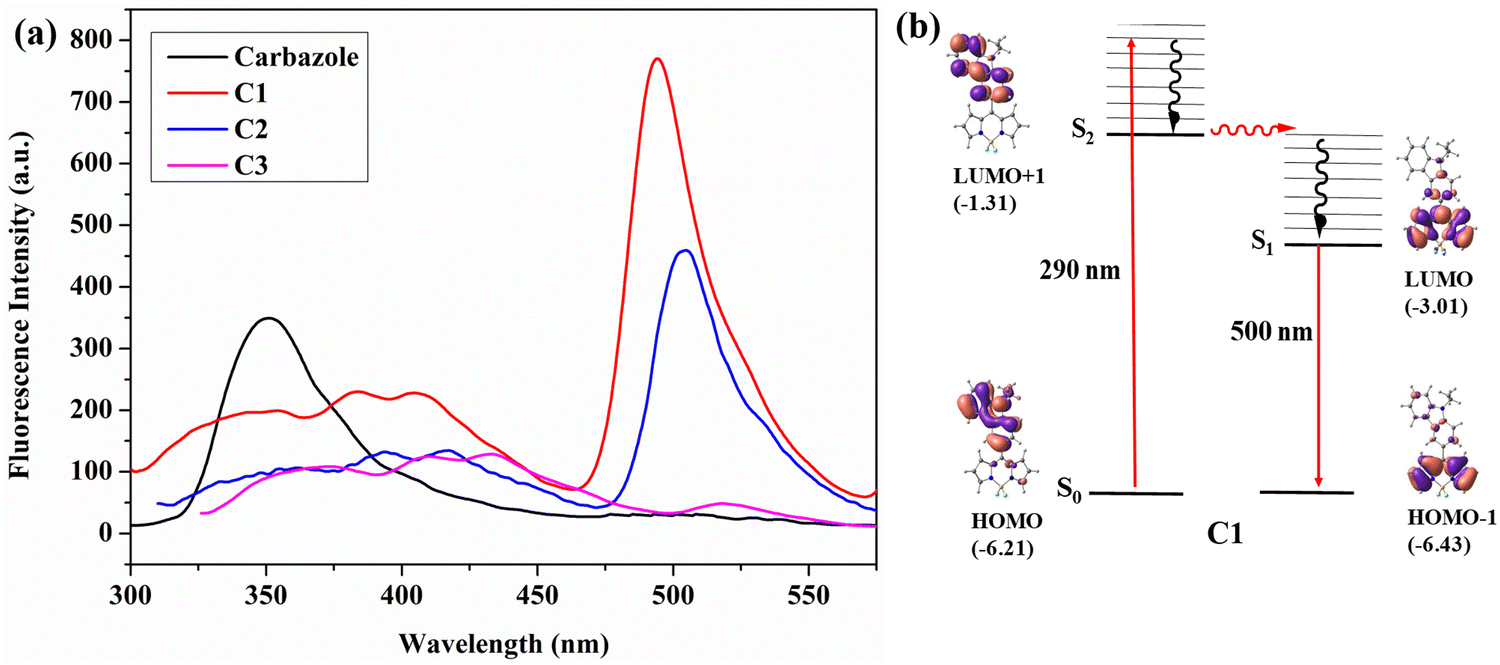 | ||
| Fig. 5 (a) Fluorescence spectrum of meso-carbazole substituted BODIPYs C1, C2, and C3 (10−7 M, λex = 290 nm) and carbazole (10−7 M) in chloroform at room temperature; (b) important molecular orbitals and electronic transitions occurring in C1 calculated using TDDFT at the M06/Def2-TZVPP//BP86/Def2-TZVPP level of theory. Eigenvalues are given in eV. | ||
Effect of solvents on photophysical properties of meso-carbazole BODIPYs
The absorption and emission properties of the compounds were studied in solvents of different polarities (Tables S3 and S4, ESI†). The compounds C1, C2, and C3 showed similar absorption profiles in solvents of different polarities (Fig. 6a). This indicates that the ground state dipole moment is not affected by solvent polarity. In contrast, the emission spectra of the compounds show strong solvent polarity dependence (Fig. 6b and Fig. S10, ESI†). In non-polar solvents like hexane, the emission band is structured and has a mirror-image relationship with their absorption spectra. This is because of the similar geometries of the absorbing and emitting species in the non-polar solvents,41 and the emission occurs from the S1 state (LUMO → HOMO−1 transition). As the solvent polarity increased, the fluorescence band became broadened and red-shifted. The compound C1 in hexane exhibited emission at 507 nm, with a fluorescence quantum yield (Φf) of 0.51 (Fig. 7a and Table S4, ESI†). In THF, a broad and red-shifted peak is formed at 607 nm with a fluorescence quantum yield of 0.21.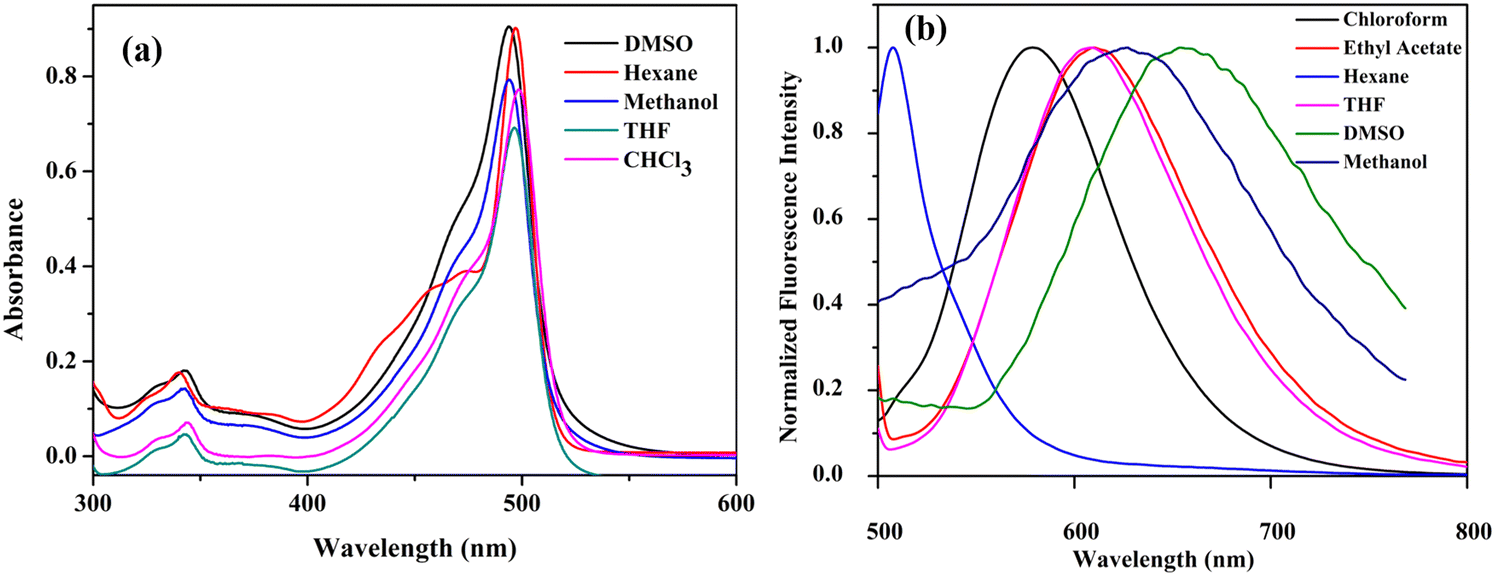 | ||
| Fig. 6 (a) UV-visible spectrum (10−5 M) and (b) fluorescence spectrum (10−7 M, λex = 480 nm) (normalized) of C1 in various solvents at room temperature. | ||
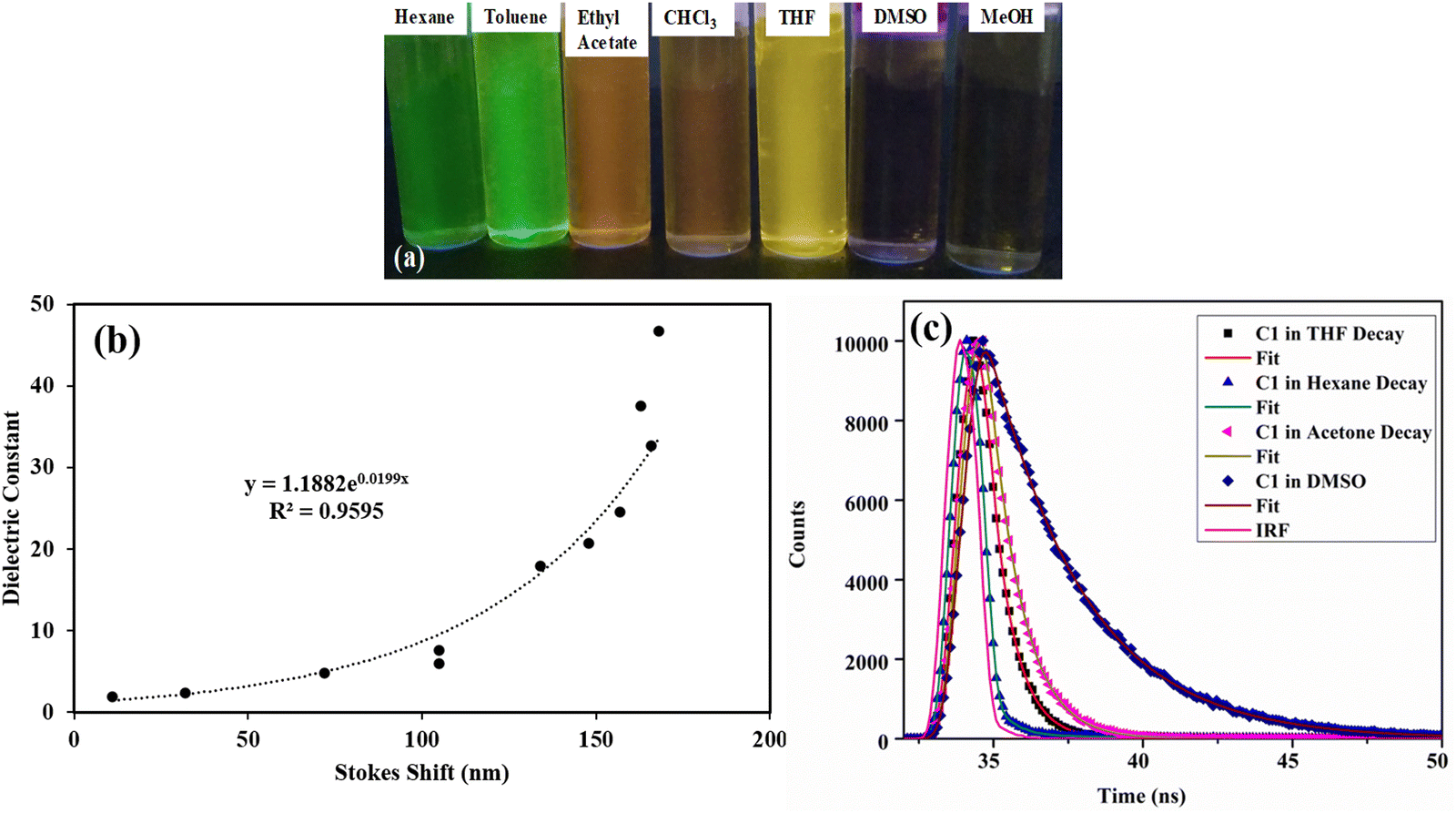 | ||
| Fig. 7 (a) Photograph of C1 in various solvents under UV irradiation-solvatochromism; (b) the plot of the dielectric constant vs. Stokes shift (nm) for C1; (c) the fluorescence decay profiles of C1 in different solvents. The instrument response function (IRF) is shown in the red line. | ||
Dual emission is observed in highly polar solvents like DMSO and methanol, which are weakened in intensity with Φf < 0.1. A new red-shifted emission band is observed at 675 nm for C1 in DMSO (LUMO → HOMO transition), attaining a Stokes shift of 182 nm. This is a characteristic of the twisted intramolecular charge transfer (TICT) effect42,43 in BODIPYs, where the emission happens from two different states. Similar behaviour is observed for mono-bromo derivative C2, which also produces a solvent-dependent emission (Fig. S10b and S11a, ESI†). The emission wavelength is red-shifted by nearly 168 nm, from 527 nm in hexane to 676 nm in DMSO, and the fluorescence quantum yield decreased from 0.4 in hexane to 0.01 in DMSO (Table S4, ESI†). The fluorescence quenching efficiency, Q, can be estimated from the fluorescence quantum yield of carbazole (Φ(reference)) and the fluorescence quantum yield of BODIPY (Φ(BODIPY)) (Tables S3 and S4, ESI†), which increases with solvent polarity.25
The relationship between the dielectric property of the solvent and the corresponding Stokes shift for C1, C2, and C3 was studied (Fig. 7b and Fig. S12, ESI†). For C1 and C2, the Stokes shift shows an exponential increase with the dielectric constant of the solvent. The dielectric constant of non-polar solvents is almost proportional to the Stokes shift. Furthermore, as the dielectric constant increases with increasing solvent polarity, there is a sharper increase in the Stokes shift. This is because the dielectric properties of the polar environment strongly affect the charge transfer processes. No such correlation is observed for dibromo derivative C3 (Fig. S12b, ESI†).
For a better understanding of the solvatochromic behavior, the fluorescence lifetimes (τ) of these dyes were measured in solvents of different polarities (Tables S4 and S8, ESI†). They were best fitted with a biexponential function with a lifetime in the nanosecond range. Fitted decay time (τi), pre-exponential coefficients (αi), and chi-squared (χ2) values and residuals are given in Table S8 and Fig. S13 (ESI†). The fluorescence lifetime gradually increases with increasing solvent polarity for C1 (0.2 ns in hexane, 3.14 ns in DMSO) and C2 (0.147 ns in hexane, 2 ns in DMSO), which confirms the excited state stabilization in polar solvents (Fig. 7c and Fig. S13a, ESI†). Furthermore, the radiative (kr) and nonradiative (knr) decay rates51 calculated from Φf and τ indicate that the radiative decay rate decreases sharply with increasing solvent polarity for both C1 and C2 (Table S4, ESI†). This strongly suggests that the decrease in fluorescence quantum yield is due to the nonradiative relaxation in polar solvents.
The meso-carbazole substituted BODIPY C1 and the mono-bromo derivative C2 exhibit superior solvatochromic fluorescence features. While the dibromo derivative C3 is non-emissive in all solvents with a small Stokes shift, no notable solvatochromism is observed (Fig. S10c, S11b, S12b and Table S4, ESI†). Moreover, the fluorescence lifetime is shorter for C3 and not dependent on solvent polarity (0.65 in hexane, 0.61 in DMSO) (Fig. S13b, ESI†).
Solvatochromic analysis for the estimation of the excited-state dipole moments
The sensitivity of the fluorophores to solvent polarity is studied in terms of the difference in the dipole moment between the ground and excited states. The solvent polarity-dependent increase in Stokes shift proposes an increase in the dipole moment upon excitation. This can be estimated from the Lippert–Mataga plot, which is the plot of Stokes shift (Δν) versus orientational polarizability (Δf), which is given as follows:47where c is the velocity of light, h is Planck's constant, μe − μg is the dipole moment difference between the excited and the ground states, respectively (Δμ), and a0 is the radius of the Onsager cavity around the BODIPYs, which is considered as the sphere. The dielectric constant (ε) and refraction index (η) of the solvent are grouped as Δf, which is known as orientation polarizability. The radius of the Onsager cavity is calculated at the M06/Def2-TZVPP level of theory (Table 1). The ground-state dipole moment for the compounds was calculated from the optimized ground-state geometry at the BP86/Def2-TZVPP level of theory in the gas phase. For different solvents, the Stokes shift is fitted to a Lippert–Mataga relationship and the difference between the ground and excited-state dipole moments (Δμ) was calculated from the slopes of Lippert–Mataga plots (Fig. 8). The linear correlation between the Stokes shift and the orientation polarizability of C1 and C2 suggests the higher polarization of the excited states than the ground states. There is a significant increase in the dipole moment in the excited state (Table 1), and the larger excited state dipole moment for both C1 and C2 (Fig. 8 and Fig. S14a, ESI†) reflects the charge transfer characteristics of these dyes in the excited state.
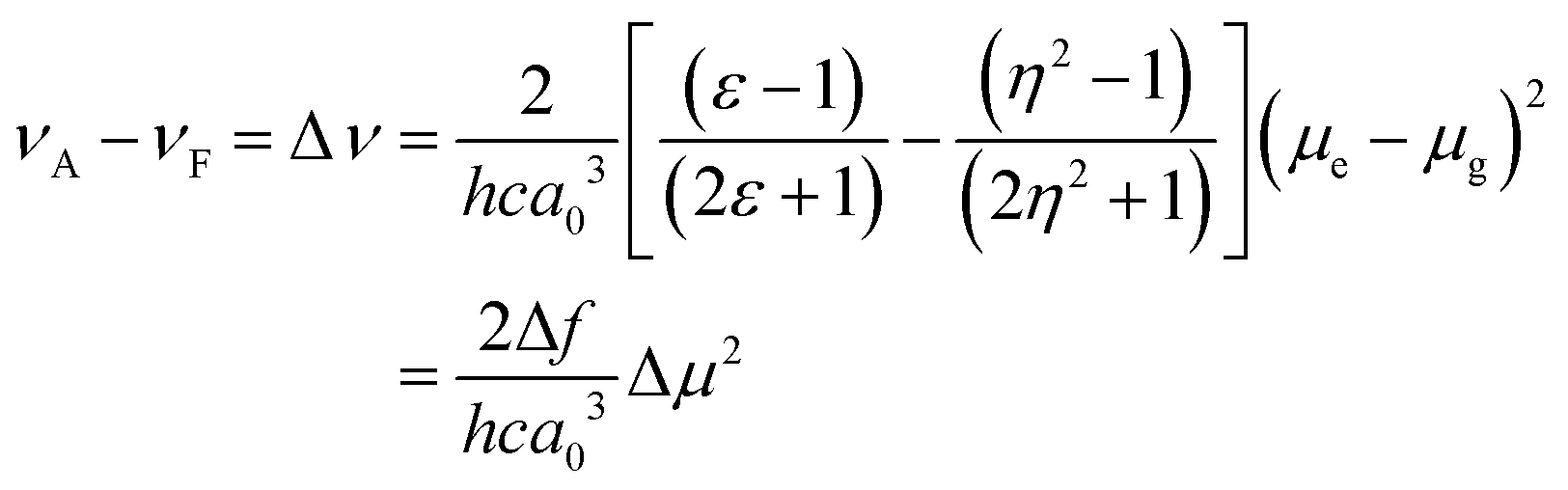
| BODIPY | Onsager radius, a0 (Å) calculated | μ g (Debye) calculated | Δμ (Debye) from Lippert–Mataga plot | μ e (Debye) = (Δμ + μg) |
|---|---|---|---|---|
| C1 | 4.82 | 8.56 | 14.7 | 23.91 |
| C2 | 5.47 | 9.21 | 12.8 | 21.36 |
| C3 | 6.17 | 9.28 | 5.3 | 14.58 |
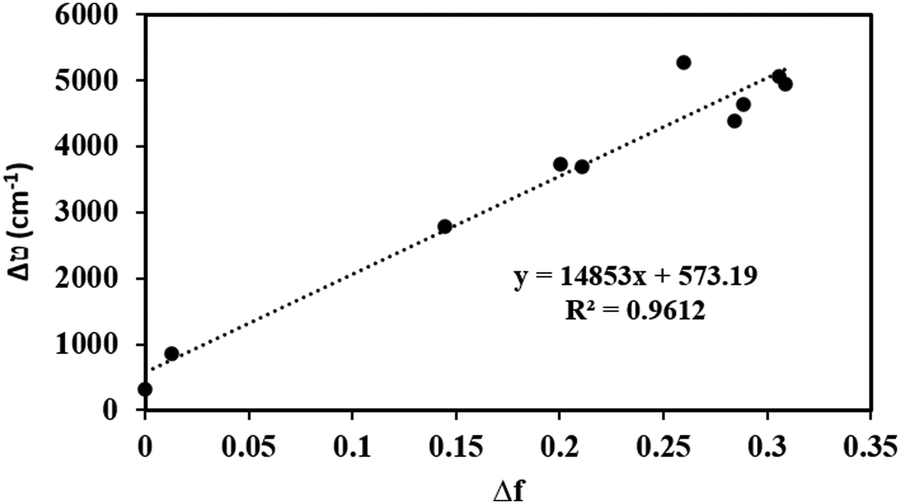 | ||
| Fig. 8 Lippert–Mataga plot: dependence of Stokes shifts on the solvent polarity function, Δf for C1. | ||
The direction of the dipole moment in the ground state is from the electron-rich carbazole to the electron-deficient BODIPY core (Fig. S15, ESI†). The decreased value of the excited-state dipole moment for C3 (Fig. S14b, ESI†) obtained from the Lippert–Mataga plot might be due to noticeable intersystem crossing due to the heavy atom effect.
Twisted intramolecular charge transfer process
To verify the TICT in meso-carbazole BODIPY derivatives, we studied the solvent polarity and viscosity effect on the emission spectra. The solvent polarity was systematically varied by mixing the polar THF (ε = 7.5) and non-polar hexane (ε = 1.8) in different ratios and recording the emission spectra of the BODIPY derivatives (Fig. S16a, ESI†). In 100% THF, BODIPY C1 has a red-shifted and broad emission at 627 nm. As the hexane fraction in the THF/hexane mixture is increased from 0 to 100%, the emission is blue-shifted and intensified. In 90% hexane, the peak in the longer wavelength region at 627 nm disappeared, and the sharp emission in the shorter wavelength region, ∼500 nm became prominent. The effect of viscosity was examined in an ethanol (η = 1.08 cP)/glycerol (η = 934 cP) mixture (Fig. S16b, ESI†). There is dual emission observed in pure ethanol. The intensity of the emission band around 500 nm increases as the fraction of glycerol increases and the longer wavelength emission band completely disappeared in 100% glycerol.The observed increase in emission at a higher wavelength with the decrease in viscosity and increase in polarity suggests that the charge transfer in the excited state is accompanied by a conformational change leading to the formation of a twisted intramolecular charge transfer (TICT) state. The non-polar or viscous environments prevent the rotation of the carbazole about the CBODIPY–CCarbazole bond connected to the BODIPY unit.
Thus, in the non-polar/viscous solvent, the emission occurs from the S1 state, giving a sharp emission band around 500 nm. In ethanol and THF, the intramolecular rotation brings the BODIPY to the TICT state, where the charge separation between the carbazole and BODIPY units occurs and obeys the minimum overlap rule.58 The twisted molecular conformation is stabilized by the solvating effect of the polar solvent. Thus, the TICT emission is red-shifted and weakened (Scheme 2).
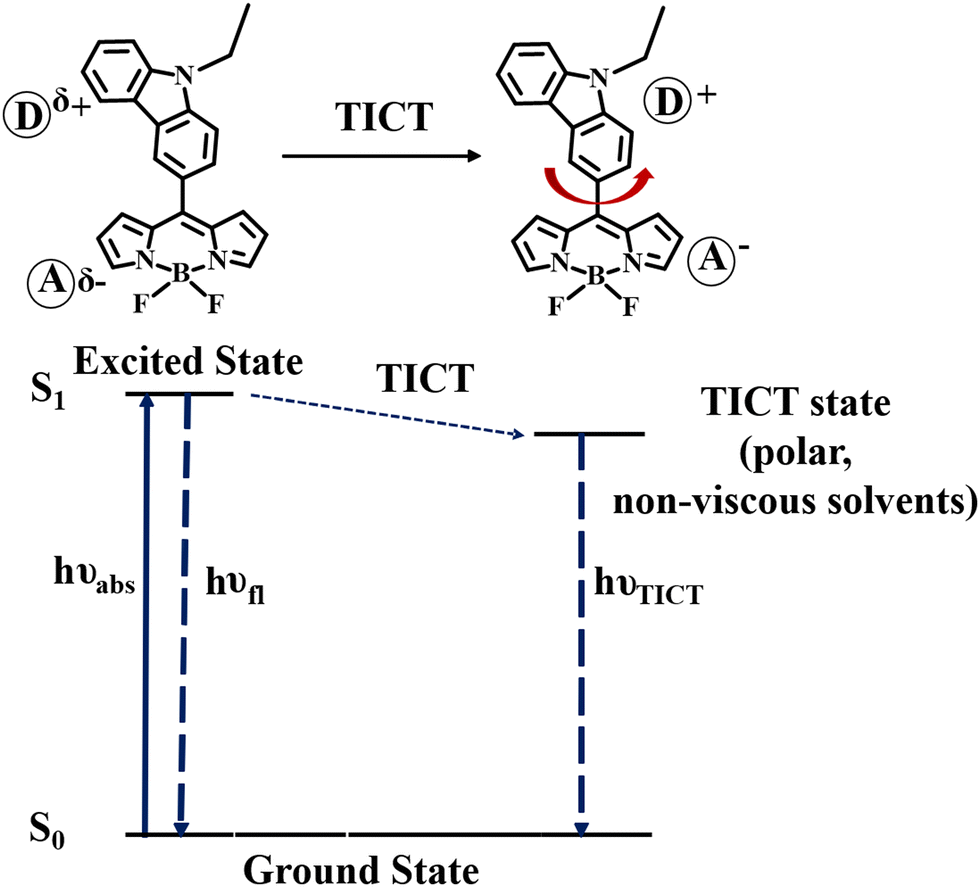 | ||
| Scheme 2 Schematic representation of TICT state formation in meso-carbazole BODIPY. | ||
Singlet oxygen generation: chemical quenching with DPBF
As discussed earlier, the non-emissive behavior of dibromo-BODIPY derivative C3 can be anticipated to be due to the intersystem crossing in C3 produced by heavy atom bromine. The enhanced intersystem crossing in dibromo derivative C3 is explained by calculating the singlet oxygen quantum yield by the chemical trapping method using diphenylisobenzofuran (DPBF) in DMSO.44 The DPBF shows a faster degradation in the presence of the dibromo derivative than that of mono-bromo as well as unbrominated BODIPY (Fig. 9b). The steady degradation of DPBF occurs in the presence of singlet oxygen, generated by the dibrominated derivative in the triplet state as per the widely studied mechanism given in Fig. 9a.45,46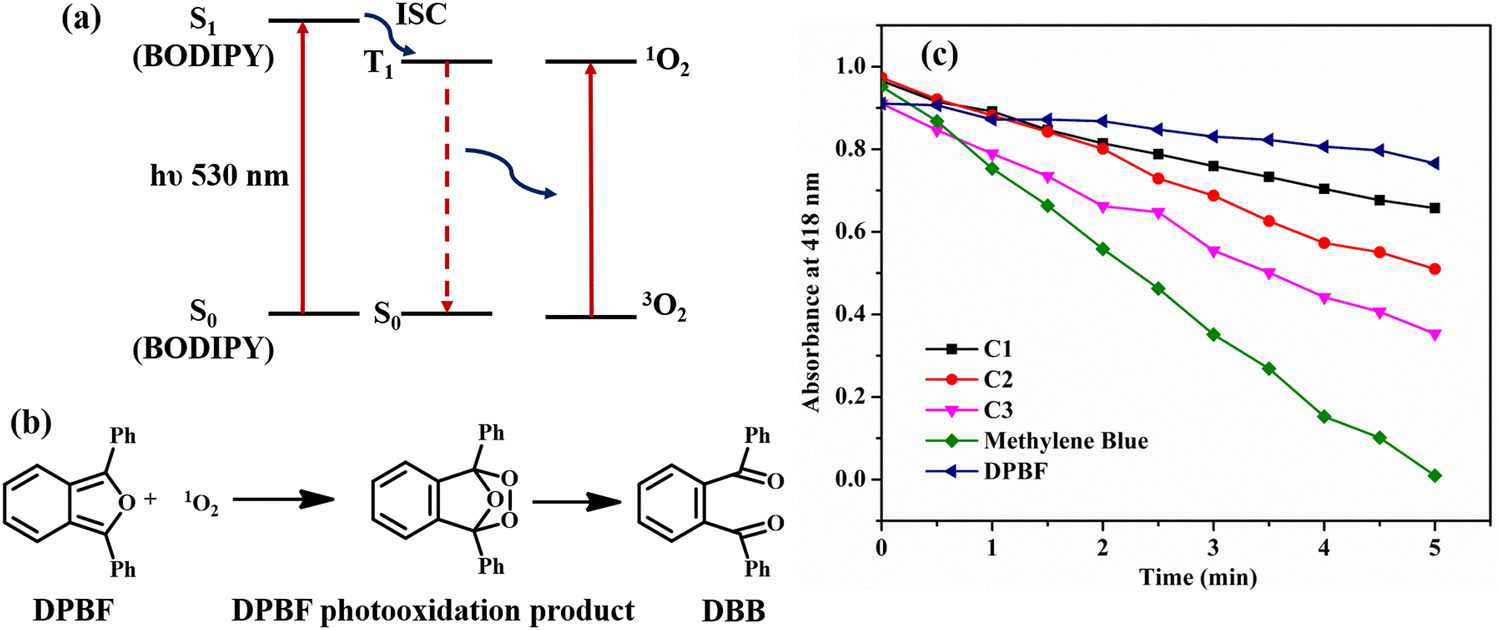 | ||
| Fig. 9 (a) The mechanism of generation of singlet oxygen by brominated BODIPYs and (b) the degradation of DPBF in the presence of 1O2; (c) the plot of DPBF absorbance at 418 nm vs. irradiation time in the presence of meso-carbazole substituted BODIPY C1, and the brominated derivatives C2 and C3. | ||
The singlet oxygen quantum yield is calculated using methylene blue as the reference and follows the order C3 (0.48) > C2 (0.23) > C1 (0.15). The amount of 1O2 produced is related to the population of BODIPY in the T1 state. There are reports that molecules with charge transfer characteristics exhibit higher singlet oxygen quantum yield than those molecules without a charge transfer character.61 As the solvent polarity is increased in DMSO, the charge transfer is enhanced in C1, and the efficiency of C1 for singlet oxygen generation is also increased significantly, resulting in a reasonable 1O2 quantum yield of 0.15. However, the singlet oxygen quantum yield of unbrominated derivative C1 is much less compared to dibromo derivative C3. Thus, incorporating heavy atom bromine at the 2,6-position of the BODIPY core could enhance the intersystem crossing (ISC), affectedly increase the singlet oxygen quantum yield, and decrease the fluorescence quantum yield in C3.
We calculated the energy difference between the singlet and triplet states of O2 and meso-carbazole substituted BODIPY at the M06/Def2-TZVPP//BP86/Def2-TZVPP level of theory (Table 2). A correlation in the singlet–triplet energy gap is observed between the dioxygen and the compounds, though the energy difference among the compounds is minimal. The energy gap of dioxygen (37.21 kcal mol−1) matches well with the energy difference between the triplet and singlet state of dibromo derivative C3 (37.57 kcal mol−1). The singly occupied molecular orbitals in the triplet state are SOMO and SOMO−1, which resulted from the excitation of an electron from the filled HOMO−1 orbital to the LUMO. The SOMO orbital is stabilized in C3 compared to C1 (SOMO: C1 (−4.74 eV), C2 (−4.84 eV), C3 (−4.93 eV)) (Fig S17, ESI†). This decreases the energy gap between the SOMO → SOMO−1 transition in C3 and matches with the triplet-singlet energy gap of O2. Thus, the enhanced S1 → T1 ISC increases the singlet oxygen quantum yield of C3, lowering the fluorescence quantum yield.
| Compound | ΔE (kcal mol−1) |
|---|---|
| E(1O2) − E(3O2) | 37.21 |
| E(T1) − E(S0) | |
| C1 | 38.52 |
| C2 | 38.27 |
| C3 | 37.57 |
Vapochromism
The vapour stimuli-responsive materials find applications in the detection of volatile organic compounds and the development of cost-effective and metal-free vapochromic organic materials is in great demand. We studied the vapochromic behavior of synthesized meso-carbazole BODIPY by exposing it to different volatile solvent vapors. The solvents of different polarities are taken in a screw-capped vial, and Whatman filter (10−5 M) paper coated with BODIPY is placed inside the cap. The vial is heated, and the color change is observed under a UV lamp (Fig. 10). The BODIPY C1 has a red-shifted emission as the solvent polarity is increased, and the emission color observed on the filter paper is at par with the solvatochromic behavior observed in the solution phase. | ||
| Fig. 10 The luminescence of BODIPY C1 on Whatman filter paper exposed to various solvent vapors taken under a UV lamp. | ||
The green emission in toluene changes to red emission in DCM, which is quenched in more polar solvent methanol (Fig. 10). This is because the intramolecular charge transfer in the molecule is activated in the presence of solvent vapor, which in turn results in the emission property. The original color is restored within seconds as the solvent is removed from the filter paper. Similar behavior was observed in mono-bromo derivative C2, while dibromo derivative C3 does not show any emission when stimulated by solvent vapor. The promising vaporesponsive property of meso-carbazole BODIPY C1 and C2 in the solid state can be utilized to develop sensors for volatile organic solvents.
Aggregation-induced emission studies on meso-carbazole BODIPY
Considering the high solid fluorescence of the meso-carbazole substituted BODIPYs (Fig. S5c, ESI†), we studied the AIE behavior of the dyes in the DMSO–water system, with different fractions of water (fw) as an antisolvent. The dyes C1, C2, and C3 are non-emissive in DMSO (Φf: C1 = 0.02, C2 = 0.01, C3 = 0.001) due to the TICT effect.As discussed above, in pure DMSO, C1 emits at 510 nm with TICT emission at 675 nm. As the amount of water in the DMSO increases, the TICT emission intensity at 675 nm decreases due to the further increase in the solvent polarity. When the water fraction increases to 70%, the formation of nano aggregates starts, and the aggregates exist in a less polar medium. This causes a blue shift in the TICT emission from 675 nm to 618 nm, along with an enhancement in emission efficiency, due to the restriction in intramolecular rotation of C1 in the aggregates (Fig. 11a).
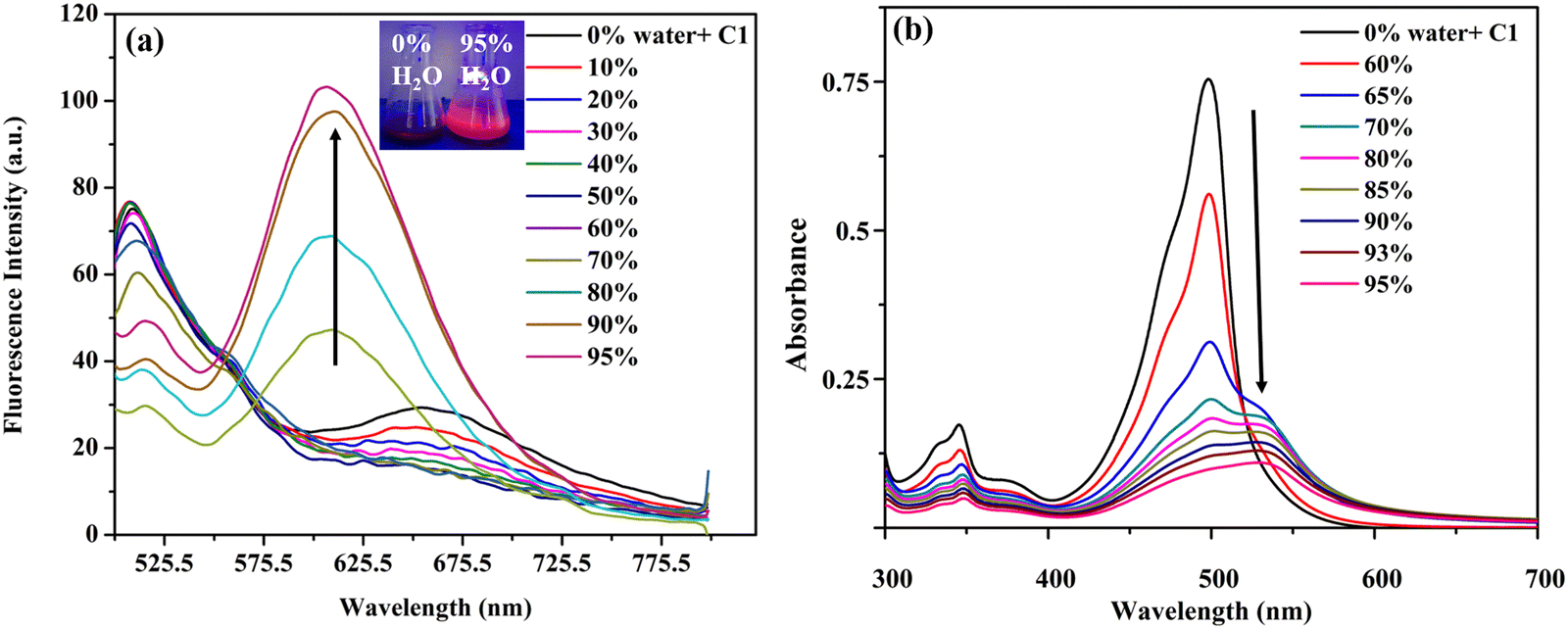 | ||
| Fig. 11 (a) Fluorescence spectra of C1 in DMSO–H2O mixtures with different water fractions (10−6 M, λexc = 490 nm) (inset: photograph of C1 in DMSO/H2O mixture (fw = 0% and 95%), taken under 365 nm excitation of a UV lamp; (b) absorption spectra of C1 in DMSO–H2O mixtures with different water fractions (10−5 M). | ||
When the fw in the DMSO solution reached 95%, the emission intensity of the dye at 618 nm was approximately 5-fold higher than that in pure DMSO, and a strong red fluorescence was observed under UV light (Fig. 11b).
The AIE characteristics of brominated BODIPY C2 in DMSO/H2O mixtures were similar to those of C1 (Fig. S18b, ESI†). The dibromo derivative C3, on the contrary, shows less enhancement in the fluorescence intensity at 630 nm, still, the solution is luminescent (Fig. S19b, ESI†). The quantitative enhancement of emission was estimated from the AIE factor (αAIE) (Table S9, ESI†). The AIE factor is calculated by comparing the quantum yields in solution and the aggregated state (Φf when fw = 95%: C1 = 0.52, C2 = 0.21, C3 = 0.015) and is found to be 26, 21, and 15, respectively, for C1, C2, and C3. This indicates that the meso-carbazole substituted BODIPYs, C1, C2, and C3, are AIE-active, being non-fluorescent in solution, and emitting intensely in their aggregated state.
The formation of nano-aggregates was confirmed by absorption spectral studies. It was observed that the absorption peak of C1 was slightly red-shifted and broadened from 499 nm to 527 nm as fw increased from 0% to 95% (Fig. 11b). There is a regular decrease in the absorbance along with the disappearance of the fine structure, due to the decrease in the concentration of DMSO solution of BODIPY and the formation of nano-aggregates. A levelling-off tail is observed in the longer wavelength region of the absorption spectra. This is because of the decrease in the light transmission of the mixture due to the light scattering of the nanoaggregate suspensions in the aqueous mixture (Mie effect).48 This is direct evidence of particle formation. Although similar spectral changes are observed for BODIPYs C2 and C3, the red shift in the absorption spectra is not very significant (Fig. S18a and S19a, ESI†).
The size, shape, and morphology of the aggregates formed with the increase in water fraction were studied using DLS and SEM analyses. In the aqueous mixture of C1 with 70% water, aggregates with an average diameter of 157 nm are formed (Fig. S20, ESI†).
The brominated derivatives C2 and C3 form aggregates with average diameters of 201 nm and 217 nm, respectively. The SEM images of the compounds obtained by drop-casting 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 water–DMSO mixtures gave a clear representation of the morphology of the aggregates at the onset of aggregation. The BODIPY C1 assembled into belt-like nanostructures, while C2 had a flower-shaped crystalline structure. Also, aggregates of C3 were structurally spherical at 70% water (Fig. 12).
:3 water–DMSO mixtures gave a clear representation of the morphology of the aggregates at the onset of aggregation. The BODIPY C1 assembled into belt-like nanostructures, while C2 had a flower-shaped crystalline structure. Also, aggregates of C3 were structurally spherical at 70% water (Fig. 12).
 | ||
| Fig. 12 SEM images of meso-carbazole substituted BODIPYs (10−5 M) in DMSO–water mixtures (30/70 v/v): (a) C1; (b) C2; (c) C3; magnification: 2.5k, scale bar: 20 μm. | ||
Single crystal structure analysis and theoretical calculations
The single-crystal XRD analysis explains the packing of the molecules during aggregation (Fig. 1). The molecules of BODIPY C1 have a twisted conformation, which effectively inhibits the emission quenching caused by the unfavorable π–π interactions between the dye molecules. As discussed earlier, the packing is stabilized by multiple B⋯H(carbazole), (Pyr)C⋯C(carbazole) interactions and C–H⋯F hydrogen bonding interactions between the adjacent molecules. These multiple non-covalent interactions rigidify the molecular conformation in the aggregate state and restrict the intramolecular rotation of the carbazole unit, allowing the BODIPYs to emit strongly.To better understand the optical behavior of the dye aggregates, we calculated the rotational energy. As discussed earlier, the free rotation of the carbazole concerning the BODIPY core around the single bond is possible in an isolated molecule as it easily overcomes the barrier of 1.46 kcal mol−1 at room temperature. This free rotation is restricted upon aggregation resulting in enhanced emission (Fig. S21, ESI†). The stable geometry has the dihedral angle −51.1°, 51.1°, and 128.8° between the planes of carbazole and BODIPY. We measured the HOMO–LUMO energy gap at different dihedral angles and found it to be less when the dihedral angle is −51.1° (which is a conformation similar to the crystal structure), compared to other dihedral angles at which the carbazole unit has a free rotation (Fig. 13). Thus, the HOMO–LUMO gap decreases and nearly overlaps in the aggregates, generating a radiative emission pathway and producing a bright red fluorescence.
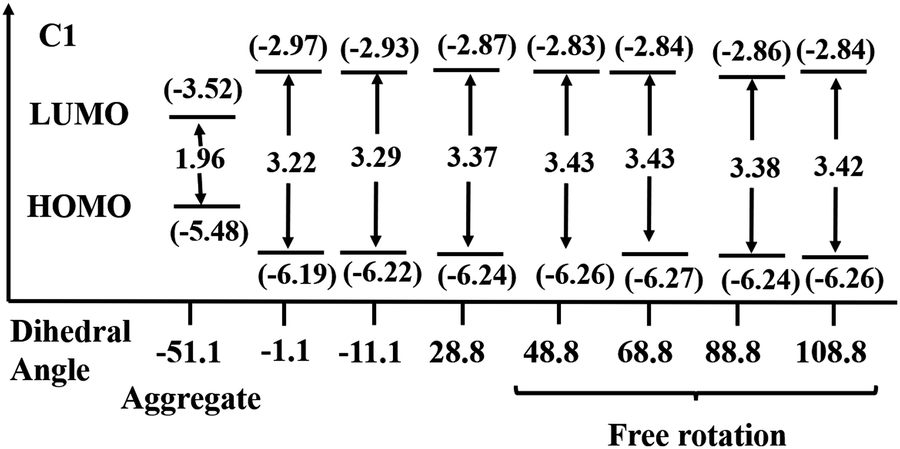 | ||
| Fig. 13 HOMO–LUMO energy gap in C1 at different dihedral angles at the BP86/Def2-TZVPP level of theory. Eigenvalues are given in eV. | ||
Antileishmanial studies
The inhibitory activity of the meso-carbazole substituted BODIPYs on the promastigotes of Leishmania donovani was carried out using the MTT assay. The promastigote form was chosen since it is the active form of the parasite observed in the human host. The IC50 was calculated by plotting the concentration of the compounds versus the percentage viability, as shown in Fig. 14. The detailed methodology for the anti-leishmanial study is given in the experimental section (vide infra).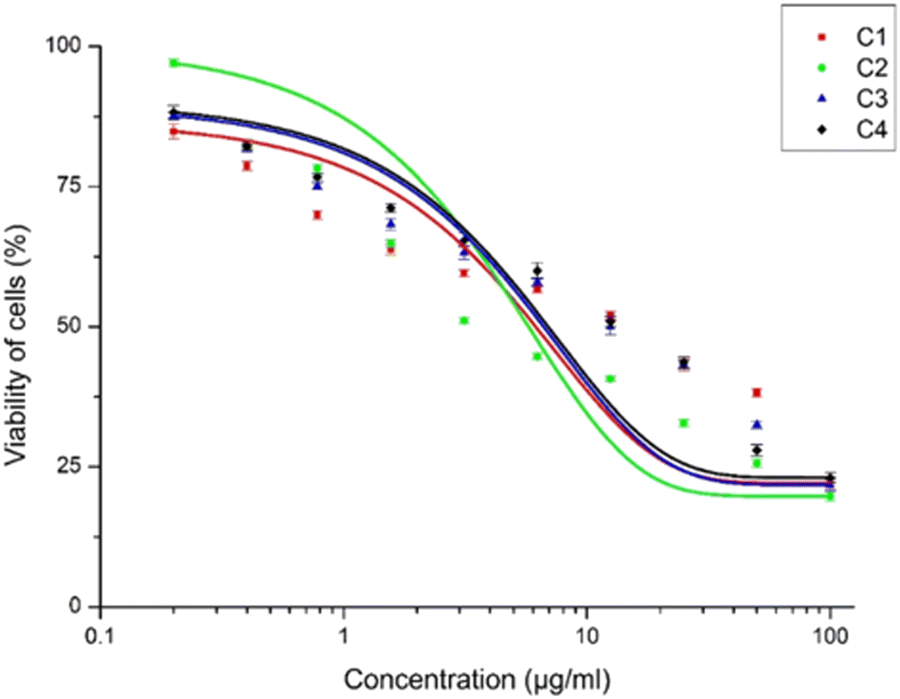 | ||
| Fig. 14 The percentage viability of Leishmania promastigotes for different concentrations of compounds C1, C2, C3, and C4 (0.2–100 μg mL−1). C4 is the electron-rich carbazole compound. The symbols represent the mean of three independent experiments and the error bars give the standard error of the mean. The higher activity of the C2 compound relative to C1 and C3 can be clearly observed in the drug–response curves. | ||
Interestingly, all the synthesized carbazole derivative BODIPYs exhibited anti-leishmanial activity against Leishmania promastigotes, and BODIPY C2 has the highest activity. The IC50 values of the compounds C1, C2, and C3 were found to be 9.309 ± 2.567 μg mL−1, 1.963 ± 0.272 μg mL−1, and 9.285 ± 1.728 μg mL−1 respectively and following the order C1–C3 > C2. The IC50 value of C2 is very low compared to the carbazole (C4), which has an IC50 value of 11.28 ± 1.791 μg mL−1. It is understood from the anti-leishmanial assay that the compounds C1–C3 exhibit increased activity, while the parent compound C4, from which the compounds C1–C3 were derived, had lower activity.
While C2 with a mono-bromine substitution at the meso-position has higher activity, the dibromo derivative did not show an additive effect and rather had activity similar to C1. Therefore, the C2 compound had the optimal functional groups at the desired positions that can bring about the necessary activity against the promastigotes of Leishmania. It has been reported earlier that a carbazole alkaloid, mahanine, induces ROS-mediated apoptosis in Leishmania donovani promastigotes.36 We hypothesize that a similar mechanism of action might be the cause of the viability reduction of C1–C3 compounds.
Contrary to our expectations, there was no change in the anti-leishmanial activity of the compounds in the presence of light. A possible reason for this might be the BODIPY moiety in the compounds that do not essentially contribute to the anti-leishmanial activity. It has been reported earlier that BODIPY compounds do not exhibit any anti-leishmanial activity but are able to maintain the activity of the anti-leishmanial miltefosine compound.60 Similarly, our studies indicate that the activity of the compounds C1–C3 is not derived exclusively from the BODIPY moiety alone but is a combinatorial effect of the carbazole, BODIPY and bromine substitutions. From the current study, these carbazole-derived compounds with mono and disubstituted Br groups at the meso-position have significant anti-leishmanial activity and can be taken forward as potential anti-leishmanial compounds.
Conclusions
In summary, meso-carbazole substituted BODIPY (C1), and its brominated derivatives (C2 and C3) were facilely synthesized. Carbazole substitution on the meso-position of the BODIPY core does not alter the energy levels of the π-conjugated system; however, the bromine substitution at the pyrrole carbon produces a red-shift in the absorption and emission spectra. The BODIPYs C1 and C2 display a prominent solvatofluorochromic effect. Their emission is tuned in the entire visible region with a large Stokes shift due to the TICT effect. The fluorescence emission and solvatochromism are not prominent in the dibromo BODIPY derivative C3 due to the heavy atom effect of bromine. Supportive evidence for enhanced ISC was obtained from the DPBF titration profiles and the molecular orbital analysis. Moreover, the synthesized compounds are strongly luminescent in aggregates. Increasing solvent polarity causes a large bathochromic shift in the emission. However, there is a dramatic decrease in the emission efficiency, and the red emission is greatly improved by aggregate formation. Thus, the AIE can enhance the TICT-induced red emission. Also, we propose the application of these compounds as a potential antileishmanial agent.Experimental section
Materials and methods
Reagent-grade chemicals were used for the synthesis of all compounds. Pyrrole and boron trifluoride etherate (BF3OEt2) were purchased from Spectrochem (India) and were purified by passing through neutral alumina before use. N-Ethyl carbazole-3-carboxaldehyde purchased from TCI Chemicals (India) Pvt. Ltd and 2,3-dichloro-5,6-dicyano-p-benzoquinone (DDQ) obtained from Sigma Aldrich were used as received. Trifluoroacetic acid (TFA) was purchased from Merck (India) and was distilled over P2O5. Triethylamine (Et3N) purchased from Qualigens was used as received. N-Bromosuccinimide (NBS) was purchased from Merck (India) and dried at 80 °C for 6 h after recrystallizing from hot water. Dimethylsulphoxide (DMSO), dimethylformamide (DMF), toluene, and acetonitrile were purchased from Fischer Scientific and were used as received. Chloroform, dichloromethane, hexane, and ethyl acetate were purchased from Avra Synthesis Pvt. Ltd. And were purified by distilling over K2CO3. Column chromatography was performed on silica gel (230–400 mesh) obtained from Avra.FTIR data were collected on a JASCO FT/IR-4700 with KBr pellets. 1H and 13C NMR spectra were recorded with a Bruker 400 MHz FT-NMR spectrometer and Bruker AVIII 500 MHz FT NMR Spectrometer in CDCl3 using TMS as the internal reference. UV-visible spectra were recorded on a JASCO V-750 instrument using quartz cuvettes at room temperature. Fluorescence spectra were recorded on a PerkinElmer LS-55/PerkinElmer LS-45 luminescence spectrophotometer with a slit width of 15. High-Resolution MS data were recorded on a Shimadzu model 8045 LC-Q-TOF mass spectrometer. X-Ray diffraction data for C1 were collected on a Bruker Kappa APEXII diffractometer. The fluorescence lifetime is measured using JOBIN-VYON M/S with Pulsed-Diode excitation sources using the TCSPC method (λexc = 460 nm). SEM images are obtained using an FEI-Quanta FEG high-resolution scanning electron microscope. DLS measurements were done on the HORIBA SZ-100 nanoparticle analyzer. The DPBF titrations were carried out using a light source of λ = 400–750 nm, 15 J cm−2 from Marutek.
Synthesis of meso-carbazole BODIPY (C1)
meso-Carbazole substituted BODIPY was synthesized in a one-pot reaction, as described in Scheme 1, starting from acid-catalyzed condensation of N-ethyl carbazole-3-carboxaldehyde and pyrrole. To the N-ethyl carbazole-3-carboxaldehyde (1 equiv.), freshly distilled pyrrole (20 equiv.) and a catalytic amount of trifluoroacetic acid (TFA) were added under a N2 atmosphere. Upon completion of the reaction in 30 minutes, the excess pyrrole is removed. The corresponding dipyrromethane (100 mg, 2 mmol) in DCM is immediately treated with an equimolar amount of 3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) (75 mg, 2 mmol) under nitrogen atmosphere for 1 h at room temperature. TLC indicated the formation of dipyrromethene, which was further treated with triethylamine (5 mL) and complexed with borontrifluoroetherate (5 mL) in DCM for 30–45 minutes. The crude product was then washed with water, extracted with CHCl3, filtered, and the solvent was removed under reduced pressure. This was further purified by silica gel column chromatography using chloroform/hexane (8:2) to afford an orange-red powder.
Synthesis of brominated BODIPYs (C2 and C3)
To the solution of BODIPY (200 mg, 0.0052 moles) dissolved in dry DCM, 2.4 equivalents of recrystallized N-bromosuccinimide (NBS) (184 mg, 0.00156 moles) in dry DCM were added dropwise and stirred at room temperature for about 15 hours for the completion of the reaction. The reaction mixture was washed with water, extracted with chloroform, and evaporated. The purification was performed by column chromatography on silica gel using CHCl3/hexane as eluent, which gave two products, mono-brominated (C2) and di-brominated (C3) derivatives, along with a minor amount of unreacted BODIPY C1.ε) = 498 nm (4.42); fluorescence (CHCl3): λem = 578 nm (λexc = 490 nm). 1H NMR (500 MHz, CDCl3, δ in ppm): 1.52 (t, 3H, –CH3), 4.46 (q, 2H, –CH2), 6.57 (d, 2H, Py-H), 7.04 (d, 2H, Py-H), 7.94 (s, 2H, Py-H), 8.35 (s, 1H, Ar-H), 8.14 (d, 1H, Ar-H), 7.73 (m, 2H, Ar-H), 7.53 (d, 2H, Ar-H), 7.71 (d, 1H, Ar-H). 13C NMR (125 MHz, CDCl3, δ in ppm): δ 146.1, 145.2, 141.1, 136.4, 133.3, 134.3, 132.2, 130.16, 130.4, 131.3, 129.6, 120.61, 110.2, 106.9, 39.5, 14.1. IR (KBr, cm−1): 2922, 2364, 1596, 1531, 1380, 1257, 1068, 983, 770. HRMS (ESI-TOF) m/z [M + H]+: 385.1573 (calc. = 384.1495).
ε) = 515 nm (4.24); fluorescence (CHCl3): λem = 587 nm (λexc = 490 nm). 1H NMR (500 MHz, CDCl3, δ in ppm): 1.52 (t, 3H, –CH3), 4.46 (q, 2H, –CH2), 6.63 (s, 1H, Py-H), 6.97 (s, 1H, Py-H), 7.07 (s, 1H, Py-H), 7.38 (m, 1H, Ar-H), 7.53 (m, 1H, Ar-H), 7.63 (d, 1H, Ar-H), 7.69 (d, 1H, Ar-H), 7.79 (s, 1H, Ar-H), 8.00 (s, 1H, Py-H), 8.19 (d, 1H, Ar-H), 8.26 (s, 1H, Py-H), 8.33 (s, 1H, Ar-H). 13C NMR (125 MHz, CDCl3, δ in ppm): δ 148.1, 147.2, 143.1, 136.4, 135.3, 134.3–134.2, 132.16, 131.4, 131.3, 129.6, 120.61, 106.9, 39.5, 14.1. IR (KBr, cm−1): 2913, 2350, 1745, 1551, 1465, 1366, 1224, 1087, 988, 818, 718, 576. HRMS (ESI-TOF) m/z [M]+: 465.0664 (calc. = 465.0623).
ε) = 534 nm (4.32); fluorescence (CHCl3): λem = 551 nm (λexc = 490 nm). 1H NMR (500 MHz, CHCl3, δ in ppm): 1.57 (3H, d, –CH3), 4.56 (2H, q, –CH2), 7.55 (2H, m, Ar-H), 7.61 (1H, m, Ar-H), 8.32 (1H, s, Ar-H), 8.19 (1H, d, Ar-H), 7.84 (2H, s, Py-H), 8.39 (2H, d, Py-H), 7.69 (1H, d, Ar-H). 13C NMR (125 MHz, CDCl3, δ in ppm): 145.5, 143.7, 141.7, 133.3, 132.1, 131.2, 128.4–128.3, 123.6, 117.3, 103.7, 38.2, 15.2. IR (KBr, cm−1): 2926, 2355, 1730, 1541, 1466, 1367, 1262, 1122, 983, 578. HRMS (ESI-TOF) m/z [M + H]+: 542.9751 (calc. = 541.9681).
Measurement of fluorescent quantum yield
The fluorescence quantum yield (Φf) is measured using the equation given below:where FS and FR are the integrated fluorescence intensities of the sample and reference, AS and AR are the absorbances of the sample and reference at the excitation wavelength, and ηS and ηR are the refractive indices of the solvents used for the sample and reference. Fluorescein in 0.1 M NaOH solution was used as the reference (Φreff = 0.90, λex = 480 nm).
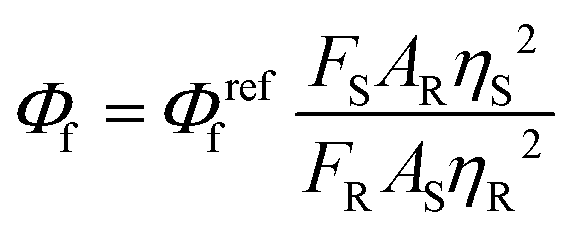
Measurement of singlet oxygen quantum yield (ΦΔ)
The singlet oxygen (1O2) acceptor, DPBF (1,3-diphenylisobenzofuran), (4.9 × 10−5 M) with and without BODIPYs (10−5 M) in DMSO was prepared in the dark and was irradiated using the light source of λ > 450 nm at room temperature under stirring. The singlet oxygen generated by BODIPYs degrades the DPBF to 1,2-dibenzoylbenzene (DBB). At regular irradiation intervals, the degradation of the DPBF molecules was monitored by measuring the decrease in absorbance at 418 nm. A plot of absorption maxima of DPBF at 418 nm vs irradiation time is drawn. The singlet oxygen quantum yield is calculated using the equation given below:where k and kref are the DPBF photo-bleaching rate constants in the presence of the samples and reference, respectively, which are obtained from the slope of the plot of irradiation time vs. absorption maxima of DPBF at 418 nm; I and Iref are the rates of light absorption at the irradiation wavelength by the samples and reference, respectively. Methylene blue was used as the reference (ΦΔ = 0.52).
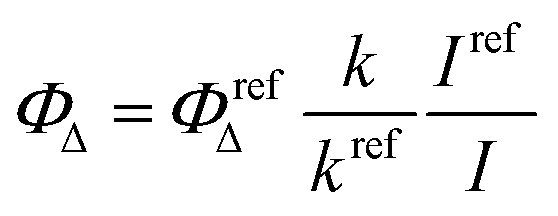
Preparation of aggregates
The stock solution of the compounds was prepared in DMSO (10−3 M). The portions of stock solution were pipetted out into a vial, along with an adequate amount of water (0–95%) under vigorous stirring to furnish 10−5 M solutions. The UV-vis and fluorescence measurements of the freshly prepared solutions were performed.Computational methodology
The quantum mechanical calculations were carried out with the Gaussian 0952 software package. The ground-state geometry optimizations were performed at the DFT50 level of theory using the exchange of Becke in conjunction with the correlation functional of Perdew (BP86).51 The basis set used is def2-TZVPP.54 The single-point calculations on the resulting optimized geometries have also been carried out using meta-GGA exchange–correlation functional M0653 with the Def2-TZVPP basis set. The electronic energy from the single point calculation at the M06/Def2-TZVPP level of theory and the zero-point correction at the BP86/Def2-TZVPP level of theory are summed to obtain the total electronic energy of the molecule. The molecular electrostatic potential (MESP) maps were calculated on the van der Waals surface at the same level of theory and analyzed using the Multiwfn software package.57 The time-dependent density functional theory (TDDFT)55 calculations were performed at the M06/Def2-TZVPP level of the Gaussian 09 program using the PCM model56 in DMSO.Antileishmanial studies: methodology
The antileishmanial activity of the compounds C1, C2, C3, and C4 (carbazole) was investigated by an MTT cell proliferation assay.59 The assay is based on the colorimetric deduction of the reduced product of MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] dye. The reduction is caused by various enzymes in the mitochondria wherein the tetrazolium dye is converted into purple insoluble formazan crystals in viable cells. Initially, the compounds were serially diluted in a concentration range from 0.2–100 μg mL−1 to a total volume of 100 μL in a 96 well culture plate. To this, exponential growth phase promastigote cells (1 × 106 cell mL−1) grown in RPMI-1640 media supplemented with 15% FBS (Foetal Bovine Serum) were added to the 96 well culture plate and allowed to grow for 24 h at 25 °C. After the incubation, MTT was added to the 96-well culture plate and incubated in the dark for 4 h at 25 °C until the formation of insoluble formazan crystals. The insoluble formazan crystals were solubilized by the addition of 10% SDS and then incubated for 4 h. The absorbance was then measured at 570 nm using a 96-well plate reader. The calculated IC50 is the concentration of the compounds required for a 50% reduction in cell viability.Conflicts of interest
There are no conflicts to declare.Acknowledgements
DM thanks MoE and the NIT Calicut for the financial support. The Centre for Material Characterization and Centre for Computational Modelling and Simulations, NIT Calicut, is acknowledged for their research assistance. We thank SAIF, IIT Madras for NMR, HRMS, lifetime, single crystal, and SEM analysis. Thanks go to MG University for the DLS analysis.References
- K. V. Rao, A. Jain and S. J. George, J. Mater. Chem. C, 2014, 2, 3055–3064 RSC.
- A. Maity, A. Sarkar, S. Bhaktha and S. K. Patra, New J. Chem., 2020, 44, 14650–14661 RSC.
- K. P. Carter, A. M. Young and A. E. Palmer, Chem. Rev., 2014, 114, 4564–4601 CrossRef CAS PubMed.
- T. T. Divya, D. Raghav, K. Rathinasamy and L. Chakkumkumarath, New J. Chem., 2019, 43, 16349–16358 RSC.
- Y. Ni and J. Wu, Org. Biomol. Chem., 2014, 12, 3774–3791 RSC.
- A. Brzeczek, K. Piwowar, W. Domagala, M. M. Mikołajczyk, K. Walczak and P. Wagner, RSC Adv., 2016, 6, 36500–36509 RSC.
- P. Rybczynski and A. Kaczmarek-Kedziera, Struct. Chem., 2021, 32, 953–965 CrossRef CAS.
- L. Jiao, W. Pang, J. Zhou, Y. Wei, X. Mu, G. Bai and E. Hao, J. Org. Chem., 2011, 76, 9988–9996 CrossRef CAS PubMed.
- M. Obłoza, Ł. Łapok, T. Pędziński and M. Nowakowska, Asian J. Org. Chem., 2019, 8, 1879–1892 CrossRef.
- V. Lakshmi and M. Ravikanth, Dalton Trans., 2012, 41, 5903–5911 RSC.
- D. Prasannan, D. Raghav, S. Sujatha, H. Hareendrakrishna Kumar, K. Rathinasamy and C. Arunkumar, RSC Adv., 2016, 6, 80808–80824 RSC.
- R. Prieto-Montero, A. Prieto-Castaneda, R. Sola-Llano, A. R. Agarrabeitia, D. Garcıa-Fresnadillo, I. Lopez-Arbeloa, A. Villanueva, M. J. Ortiz, S. Moya and V. Martınez-Martınez, Photochem. Photobiol., 2020, 96, 458–477 CrossRef CAS PubMed.
- Y. Gawale and N. Sekar, J. Photochem. Photobiol. B: Biol., 2018, 178, 472–480 CrossRef CAS PubMed.
- H. Wang, E. Zhao, J. W. Y. Lam and B. Z. Tang, Mater. Today, 2015, 18, 365–377 CrossRef CAS.
- J. Mei, N. L. Leung, R. T. Kwok, J. W. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- E. Li, K. Jie, M. Liu, X. Sheng, W. Zhu and F. Huang, Chem. Soc. Rev., 2020, 49, 1517–1544 RSC.
- O. S. Wenger, Chem. Rev., 2013, 113, 3686–3733 CrossRef CAS PubMed.
- S. Yokoyama, H. Asahara and N. Nishiwaki, Cryst. Growth Des., 2020, 20, 1383–1387 CrossRef CAS.
- Y. Li, J. Ding, M. Day, Y. Tao, J. Lu and M. D'iorio, Chem. Mater., 2004, 16, 2165–2173 CrossRef CAS.
- M. S. Mudadu, A. N. Singh and R. P. Thummel, J. Org. Chem., 2008, 73, 6513–6520 CrossRef CAS PubMed.
- P. E. Kesavan and I. Gupta, Dalton Trans., 2014, 43, 12405–12413 RSC.
- V. Promarak, M. Ichikawa, T. Sudyoadsuk, S. Saengsuwan, S. Jungsuttiwong and T. Keawin, Thin Solid Films, 2008, 516, 2881–2888 CrossRef CAS.
- Z.-S. Wang, N. Koumura, Y. Cui, M. Takahashi, H. Sekiguchi, A. Mori, T. Kubo, A. Furube and K. Hara, Chem. Mater., 2008, 20, 3993–4003 CrossRef CAS.
- A. Głuszyńska, Eur. J. Med. Chem., 2015, 94, 405–426 CrossRef PubMed.
- G. Reddy, N. Duvva, S. Seetharaman, F. D’Souza and L. Giribabu, Phys. Chem. Chem. Phys., 2018, 20, 27418–27428 RSC.
- S. Wanwong, P. Khomein and S. Thayumanavan, Chem. Cent. J., 2018, 12, 60–69 CrossRef CAS PubMed.
- D. Zhang, Y. Wang, Y. Xiao, S. Qian and X. Qian, Tetrahedron, 2009, 65, 8099–8103 CrossRef CAS.
- I. Gupta and P. E. Kesavan, Front. Chem., 2019, 7, 841–872 CrossRef CAS PubMed.
- H. Y. Lin, W. C. Huang, Y. C. Chen, H. H. Chou, C. Y. Hsu, J. T. Lin and H.-W. Lin, Chem. Commun., 2012, 48, 8913–8915 RSC.
- P. Yang, W. Wu, J. Zhao, D. Huang and X. Yi, J. Mater. Chem., 2012, 22, 20273–20283 RSC.
- Y. Ooyama, M. Kanda, T. Enoki, Y. Adachi and J. Ohshita, RSC Adv., 2017, 7, 13072–13081 RSC.
- J. Strahan, B. C. Popere, P. Khomein, C. A. Pointer, S. M. Martin, A. N. Oldacre, S. Thayumanavan and E. R. Young, Dalton Trans., 2019, 48, 8488–8501 RSC.
- X. Xia and Y. Qian, Analyst, 2018, 143, 5218–5224 RSC.
- R. Misra, T. Jadhav, B. Dhokale, P. Gautam, R. Sharma, R. Maragani and S. M. Mobin, Dalton Trans., 2014, 43, 13076–13086 RSC.
- J. J. Brendle, A. Outlaw, A. Kumar, D. W. Boykin, D. A. Patrick, R. R. Tidwell and K. A. Werbovetz, Antimicrob. Agents Chemother., 2002, 46, 797–807 CrossRef CAS PubMed.
- S. Roy, D. Dutta, E. M. Satyavarapu, P. K. Yadav, C. Mandal, S. Kar and C. Mandal, Sci. Rep., 2017, 7, 4141–4148 CrossRef PubMed.
- S. M. Thomas, A. Purmal, M. Pollastri and K. Mensa-Wilmot, Sci. Rep., 2016, 6, 32083–32093 CrossRef CAS PubMed.
- I. F. Sengul, E. Okutan, H. Kandemir, E. Astar and B. Çosut, Dyes Pigm., 2015, 123, 32–38 CrossRef CAS.
- Y. Hayashi, S. Yamaguchi, W. Y. Cha, D. Kim and H. Shinokubo, Org. Lett., 2011, 13, 2992–2995 CrossRef CAS PubMed.
- V. Lakshmi, M. R. Rao and M. Ravikanth, Org. Biomol. Chem., 2015, 13, 2501–2517 RSC.
- Y. J. Chen, Y. Ling, L. Ding, C. L. Xiang and G. Zhou, J. Mater. Chem. C, 2016, 4, 8496–8505 RSC.
- S. K. Saha, P. Purkayastha and A. B. Das, J. Photochem. Photobiol., A, 2008, 195, 368–377 CrossRef CAS.
- (a) R. Hu, E. Lager, A. Aguilar-Aguilar, J. Liu, J. W. Y. Lam, H. H. Y. Sung, I. D. Williams, Y. Zhong, K. S. Wong, E. Pena-Cabrera and B. Z. Tang, J. Phys. Chem. C, 2009, 113, 15845–15853 CrossRef CAS; (b) A. Kundu, S. Karthikeyan, Y. Sagara, D. Moon and S. P. Anthony, ACS Omega, 2019, 4, 5147–5154 CrossRef CAS PubMed.
- X.-F. Zhang and X. Yang, J. Phys. Chem. B, 2013, 117, 5533–5539 CrossRef CAS PubMed.
- C. Wang and Y. Qian, Org. Biomol. Chem., 2019, 17, 8001–8007 RSC.
- R. T. Kuznetsova, I. V. Aksenova, D. E. Bashkirtsev, A. S. Shulev, E. V. Antina, M. B. Berezin and N. A. Bumagina, High Energy Chem., 2017, 51, 175–181 CrossRef CAS.
- (a) V. E. Z. Lippert, Elektrochem, 1957, 61, 962–975 Search PubMed; (b) N. Mataga, Y. Kaifu and M. Koizumi, Bull. Chem. Soc. Jpn., 1956, 29, 465–470 CrossRef CAS.
- C. Y. K. Chan, J. W. Y. Lam, Z. Zhao, S. Chen, P. Lu, H. H. Y. Sung, H. S. Kwok, Y. Ma, I. D. Williams and B. Z. Tang, J. Mater. Chem. C, 2014, 2, 4320–4327 RSC.
- P. Das, A. Kumar, A. Chowdhury and P. S. Mukherjee, ACS Omega, 2018, 3, 13757–13771 CrossRef CAS PubMed.
- (a) W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133–A1138 CrossRef; (b) P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- (a) A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS PubMed; (b) J. P. Perdew, Phys. Rev. B, 1986, 33, 8822–8824 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- F. Weigend, M. Häser, H. Patzelt and R. Ahlrichs, Chem. Phys. Lett., 1998, 294, 143–152 CrossRef CAS.
- A. Dreuw and M. Head-Gordon, Chem. Rev., 2005, 105, 4009–4037 CrossRef CAS PubMed.
- M. R. P. Long and C. M. Isborn, J. Phys. Chem. B, 2017, 121, 10105–10117 CrossRef PubMed.
- (a) T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed; (b) T. Lu and F. Chen, J. Mol. Graphics Modell., 2012, 38, 314–323 CrossRef CAS PubMed.
- (a) Z. R. Grabowski and J. Dobkowski, Pure & Appl. Chem., 1983, 55, 245–252 Search PubMed; (b) H. Sinha, S. Muralidharaan and K. Yates, Can. J. Chem., 1992, 70, 1932–1938 CrossRef CAS; (c) C. Zhong, Phys. Chem. Chem. Phys., 2015, 17, 9248–9257 RSC.
- S. Sasidharan and P. Saudagar, Acta Trop., 2022, 231, 106448 CrossRef CAS PubMed.
- V. Hornillos, E. Carrillo, L. Rivas, F. Amat-Guerri and A. U. Acuña, Bioorg. Med. Chem., 2008, 18, 6336–6339 CrossRef CAS PubMed.
- W. Hu, Y. Lin, X.-F. Zhang, M. Feng, S. Zhao and J. Zhang, Dyes Pigm., 2019, 164, 139–147 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2235136. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3nj00747b |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2023 |