
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Connecting metal–organic cages (MOCs) for CO2 remediation
Javier
Martí-Rujas

Dipartimento di Chimica Materiali e Ingegneria Chimica. ‘‘Giulio Natta’’, Politecnico di Milano, Via L. Mancinelli 7, 20131 Milan, Italy. E-mail: javier.marti@polimi.it
First published on 5th September 2023
Abstract
In this Perspective article, recent developments in the self-assembly of supramolecular porous materials made of zero-dimensional (0D) porous metal organic cages (MOCs), connected by different approaches, and their application in CO2 remediation are reviewed. The connection of MOCs carried out by coordination-bond driven linking, covalent bond linking and mechanical bond formation, leads to the formation of novel smart materials ranging from solid to gel states of matter, and in some cases, porous materials with hierarchical porosity stemming from the intrinsic MOC porosity and the voids generated among the connected MOCs. Both porosities can be tuned depending on the MOC sizes and on the way the cages are connected (i.e., coordination-driven and covalent bond linking). In general, the supramolecular, often networked, materials arising from the connection of MOCs tend to show diffuse scattering, denoting only short-range order, making their structural elucidation very challenging or simply not possible. Thus, the bulk structure of materials formed by connected MOCs is often deduced from information obtained through other techniques like powder XRD, pair distribution function (PDF) analysis, solid-state NMR, dynamic light scattering (DLS), and positron annihilation lifetime spectroscopy (PALS). Materials obtained upon the connection of MOCs are usually in the form of amorphous solids, gels, xerogels and porous liquids (PLs) but have shown to outperform the CO2 capacity and improved mechanical properties when compared to the single MOCs. The preparation of materials at the interface between solids and liquids is important to create functional materials displaying unique properties such as porosity in liquids and gels arising from the MOCs and their networked assembly, hence allowing CO2 gas diffusion into the cage's voids where CO2 can be trapped or catalytically transformed into other molecules (i.e., formic acid (HCOOH)). Thus, materials containing connected MOCs can be exploited where MOFs cannot be used due to their brittle nature, typical of solid crystalline materials, for instance in applications where the porous material has to fit to different shapes for example those required to fit in tubes or shapes that usually crystalline solids cannot adapt easily, thus expanding the applications of connected MOC materials in areas involving fluids.
1. Introduction
Combustion of fossil fuels, such as natural gas, petroleum, and coal in order to meet the energy demands of our past and recent societies, has contributed to an atmospheric rise in carbon dioxide (CO2), and hence the Earth's temperature increases due to the greenhouse effect.1 The rise of atmospheric CO2 is threatening the Earth equilibrium including the natural environment and the human society. Therefore, technologies on carbon capture and conversion approaches are being proposed to decrease the CO2 atmospheric emission.2,3Metal organic frameworks (MOFs) as post zeolitic materials are highly crystalline materials obtained by self-assembling organic ligands and metal ions or clusters of metals (called secondary building units or SBUs) “stuck” together by coordination bonds.4–8 MOFs are also regarded as coordination polymers or coordination networks. The possibility of synthesizing porous MOFs and fine-tuning their porosity has given the chance to exploit such materials for gas adsorption, and in particular MOFs and MOF-based materials have emerged as good candidates to capture CO29 and are also suitable for CO2 transformation into other molecules that are not harmful to the environment.2
MOF's structural properties including porosity, designability, crystallinity, flexibility and well-defined structures obtained from single crystal X-ray diffraction data (SC-XRD)10 and powder X-ray diffraction data (powder XRD)11 have contributed to the rapid advancement of MOFs as functional materials for CO2 remediation.9 The atomic resolution 3D crystal structure allows not only the direct visualization of the interior of the pores and their surfaces, but also the in situ observation of chemical intermediates in reactions that in traditional solution reactions cannot be isolated.12–14 Hence, concerning gas adsorption, the availability of an X-ray crystal structure gives the opportunity to better understand where the CO2 molecules can be adsorbed. Crucially, 3D X-ray crystallographic analysis is fundamental to perform a combined theoretical–experimental structural analysis on CO2 adsorption of a given porous material allowing rationalization of the adsorption process.9
Another class of hybrid metal organic materials are the so-called metal organic cages (MOCs)15–22 (also known as metal organic polyhedra (MOPs)), that are porous structures used in many applications such as in molecular sensing,23 stabilization of unstable species,20,21 and catalysis18 but also for CO2 capture and transformation.24MOCs are discrete (0D) structures with well-defined internal voids that can be exploited as host–guest systems. In the solid-state, MOCs must maintain their internal voids to efficiently use their host–guest properties, for instance in heterogeneous gas–solid reactions where the cages are the solid phases used to trap CO2.
The focus of this Perspective article is not on isolated MOCs, but on materials that are formed by “connected” MOCs25–27 as this linking of MOCs yields functional novel materials like amorphous polymeric porous MOCs, supramolecular gels, aerogels and porous liquids (PLs) for CO2 applications (vide ante). Unusual physical phenomena ranging from controlled microporosity in soft matter yielding soft supramolecular materials which are flexible and permanently porous, at the interface between solids and liquids, have been recently reported. The strategy of linking MOCs is interesting because it exploits the MOC's properties while also combining features of new materials.25–27 Usually, the new materials formed after linking the single MOCs display properties that belong to soft matter rather than pure solids which have led to the design of self-healing porous polymers and materials targeted for drug delivery.27 Reviews about MOCs linked in different ways such as chemical bonds including coordination and covalent bonds using organic molecules, or mechanical bonds forming polymeric structures of MOCs are very few25–27 and reviews of such materials for CO2 adsorption and fixation are nonexistent to the best of our knowledge. Therefore, this Perspective article will be of interest to many researchers working on the host–guest chemistry of polymerized MOCs covering a wide range of areas going from fundamental to the applied sides of materials sciences.
2. Metal organic cages (MOCs) as host structures for inclusion properties in supramolecular materials formed of connected MOCs
MOCs are hybrid metal–organic materials self-assembled with metal ions and organic ligands which do not form extended structures but discrete zero-dimensional (0-D) complexes with 3D internal voids.15–19,28 Crucially, the size and shape of the MOCs can be tuned by choosing ligands with various sizes and shapes which in combination with transition metals can give rise to different metal organic polyhedric structures. This was developed by several research groups15,16,19,21,22 and a seminal contribution has been the work by Fujita,29 which used pyridinic ligands and encapsulated Pd(II) and Pt(II) salts to block the polymerization towards MOFs formation (i.e., Cd(NO3)5,29vs. (en)Pd(NO3)2),30 thus constraining the self-assembling products towards discrete structures (convergent way) including squares,30 cages31 and bowls.32 In general, the polyhedric structures are highly symmetric and are classified as Archimedean or Platonic solids.26 Recently new trends have been developed to synthesize less symmetric MOCs to mimic the asymmetric binding pockets where enzymes carry on biocatalytic reactions in proteins.33The increased interest in MOCs research is because they offer a wide variety of host–guest chemistry applications taking advantage of the internal space of the nanocages which can be used as a nanocontainer in relevant industrial applications.15,18,20,21 This is especially important because molecules in a confined space can show a different behavior from that observed in the bulk solution and unusual reactions can be carried out within the cages.18,34 Also, the internal space of the cages can be chemically functionalized to create specific binding sites that can be used for molecular recognition in such a way that can be applied for trapping gas molecules such as H2, N2 or CO2 as small gases, but also volatile organic compounds (VOCs) such as large aromatic compounds, drug molecules or macromolecules like proteins35 just to mention a few.
Much of the work involving MOCs is being done in solution because the self-assembled cages are stable in the liquid media, and in particular in water,36 in which many host–guest processes can be monitored by solution NMR spectroscopy. On the other hand, working with MOCs in the solid-state, for instance in the molecular recognition of aromatic guests, the trapping of small gases like CO2, or in the study of catalytic reactions, the integrity (i.e., porosity) of the metal–organic cages should be maintained. The stability of the MOC's architecture is fundamental if the cages must be used in separation applications such as CO2 adsorption because they need to be activated. That is to create the empty space, by removing the guest component (usually a solvent used for the crystallization), which has the role of templating agent.31 Importantly, in the activation process also water and/or solvent molecules coordinated to metal centers are removed, which can lead to uncoordinated metal atoms (nodes) that are used as catalytic reaction sites. However, upon thermal treatment, MOCs might collapse and therefore lose the intrinsic porosity and hence their potential function as molecular adsorbents. This structural collapse must be avoided if the guest molecules should be included in the MOCs.
2.1. Different strategies to link MOCs for CO2 applications
MOCs can be defined as “macromolecules” because of their discrete (0D) nature and, in many cases are defined as porous due to their internal voids and gas adsorption abilities. In the crystalline state, electrostatic interactions hold MOCs together, giving them a periodic nature. Thus, it can be envisaged that their inherent porosity might be maintained if monomeric MOCs are linked together through organic ligands in a supramolecular structure. However, the probability to obtain the linked MOCs in a crystalline and ordered state is difficult. On the other hand, it is also important to design strategies leading to porous materials that behave more like soft matter rather than crystalline (i.e., rigid structure), because soft mater materials can be used in applications in the liquid or gel states. Therefore, not only to increase the MOCs’ stability and flexibility, but to explore the formation of new and large supramolecular entities, strategies such as linking MOCsvia coordination of bridging ligands to unsaturated metal sites; connecting the 0D cages upon covalent bond formation; or by interlocking MOCs using mechanical bonds, have been recently developed (Fig. 1).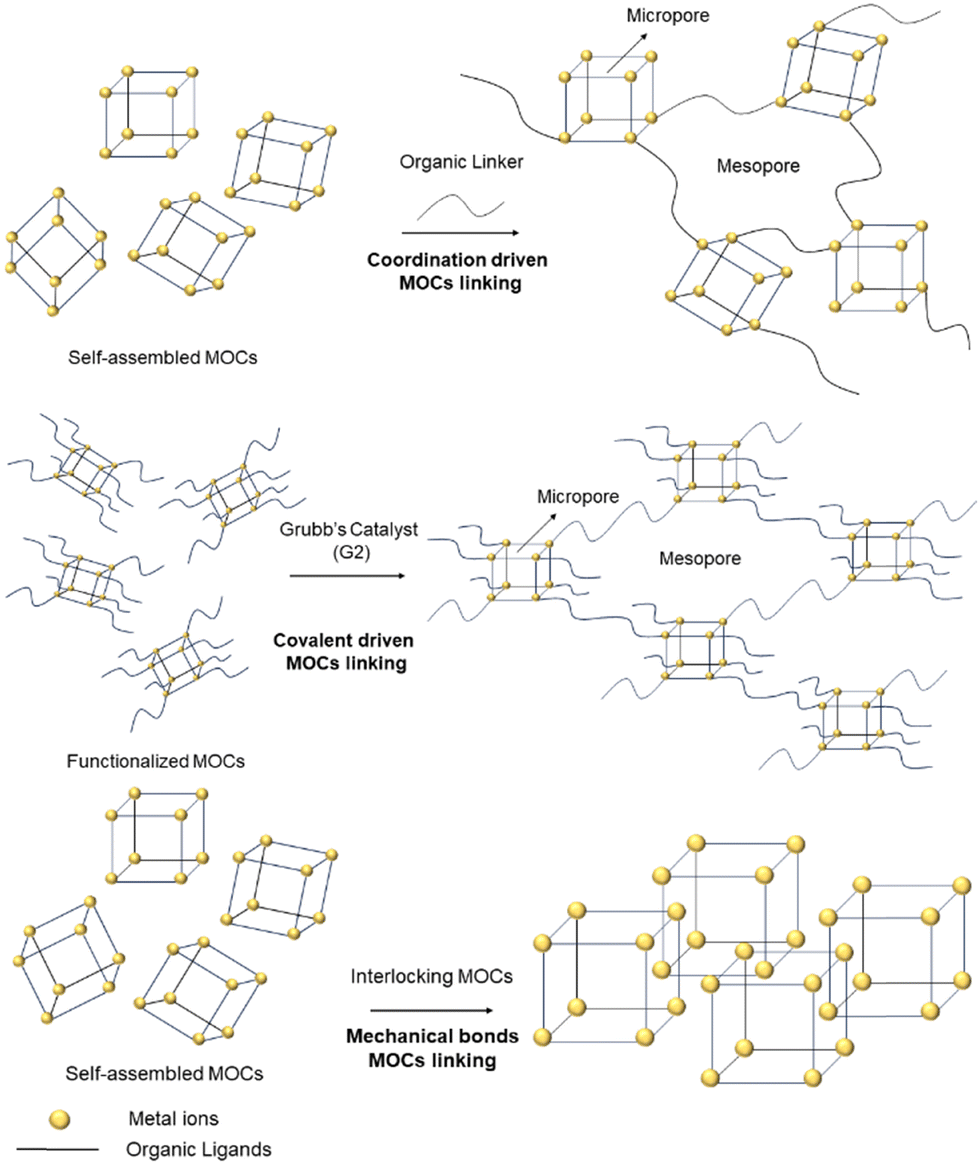 | ||
| Fig. 1 Cartoon showing the stepwise synthesis of connected MOCsvia ligand coordination (top), covalent bond formation from functionalized MOCs (middle) and interlocking of MOCsvia mechanical bonds (bottom). | ||
Two MOCs can be connected in different manners. In the next examples the synthetic approaches shown in Fig. 1 have been applied to crosslink MOCs. In this way, polymeric structures made of discrete MOCs are produced. This strategy uses MOCs for obtaining supramolecular polymers with different types of porosities (i.e., hierarchical porosity) generated from intrinsic microporosity inside the cage cavity and mesoporosity between the cages generated after the linkage of the MOCs (Fig. 1).
Crucially, the mesoporosity will depend also on the length, the flexibility and shape of the linker molecule connecting the MOCs. The bulk porous nature of the material can be used for trapping CO2 or for transforming CO2 into non-harmful substances. The new supramolecular materials formed by connected MOCs showed that CO2 can be adsorbed outperforming the CO2 adsorption capacity and catalytic activity of non-connected MOCs. Moreover, the overall stability and flexibility of the linked MOCs is also enhanced with respect to the original molecular MOCs.
2.2. The low crystallinity of solid materials formed of connected MOCs and their structural characterization
The preparation of large supramolecular entities, such as those arising from the connection of MOCs, often in between the solid and liquid states (i.e., soft matter), yields solids that are poorly crystalline, with only short-range order, or just amorphous, in which the absence of Bragg diffraction does not allow full structure determination using single crystal X-ray diffraction (SC-XRD)10 or powder XRD structure solution.11 The entrapment of crystallization solvent inside the MOCs or in the spaces generated between the connected cages, tend to form amorphous materials. It is important to mention that in some cases, depending on the way the self-assembly takes place such as when fast crystallization processes are required to control the crystallization product (i.e., crystallization under kinetic control), often result as microcrystals or amorphous phases which complicates the structure elucidation by SC-XRD (vide ante).The impossibility in the understanding of the 3D atomic arrangement in the self-assembled material does not allow us to make efficient progress in the design and fine tuning for improving the properties of materials made of connected MOCs. Thus, understanding their functional properties without detailed structural characterization is often very challenging. However, techniques such as X-ray diffraction (SAXS), pair distribution function (PDF) analysis, combined with solution and solid-state NMR, dynamic light scattering (DLS), and positron annihilation lifetime spectroscopy (PALS) can be used to gain information on the supramolecular superstructure formed upon linking MOCs.
Despite the difficulties in the structural characterization of some of the supramolecular materials obtained by connecting monomeric MOCs, a myriad of new properties were discovered, such as softness and processability of porous materials, making the effort of studying new avenues of assembling individual MOCs into polymeric materials worth exploring. As most porous materials are used in the solid-state, they cannot be used in common flow processes. Supramolecular MOC polymers displaying characteristic features of liquid or liquid-like materials such as gels27 are clearly interesting with huge potential in industrial applications.
3. Polymeric structures generated by linking MOCs with ligands via coordination bonds
3.1. Formation of a supramolecular solid polymer connecting MOCsvia coordination bonds with hierarchical porosity
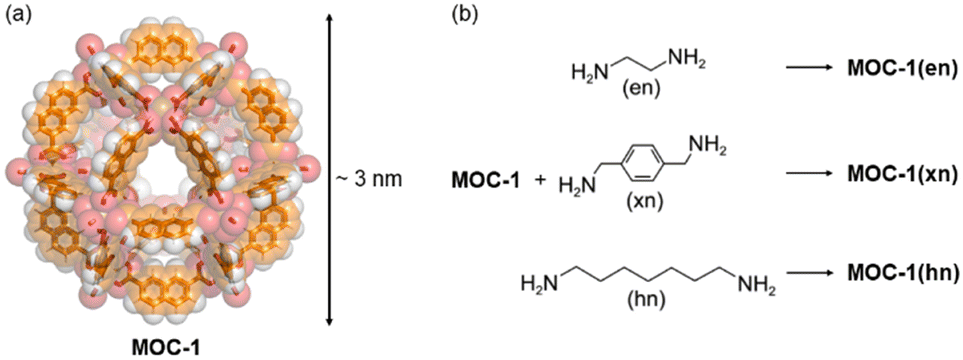
An example showing how MOCs can be connected through covalent bonds using bidentate ligands was reported in 2011 by Chun and Moon in which 2,7-napthalenedicarboxylate and Cu2+ were self-assembled into a large MOC with 12 Cu–Cu paddlewheel units, [Cu24(2,7-ndc)24(DMF)10(H2O)14 (MOC-1).37 The SC-XRD structure was determined by synchrotron radiation and the self-assembled material is a 3.3 nm cuboctahedron (Archimedean solid) as shown in Fig. 2. In the crystalline state, the cuboctahedrons are in close contact with adjacent ones via the triangular windows packing in a body-centered cubic structure (Im![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group). The MOC is very large with its 68% of space occupied by solvent molecules, which is 22
m space group). The MOC is very large with its 68% of space occupied by solvent molecules, which is 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 147 Å3 of its total unit cell volume. The cuboctahedron can be considered nearly spherical with an internal diameter of an imaginary sphere of 1.8 nm (Fig. 2a) falling in the microporous regime. The as synthesized MOC-1 is crystalline but becomes amorphous in the activation process, upon evacuation of guests, because they act as templating agents. However, the amorphous phase of MOC-1 shows an amorphous-to-crystalline (AC) transformation once it traps guest molecules. MOC-1 can trap CO2 which shows reversible type I isotherms without significant hysteresis because only the internal voids of amorphous MOC-1 (microporous regime) are available for gas inclusion.37 One approach to see if there is an increase in CO2 adsorption, with respect to isolated MOC-1, can be carried out by synthesizing a polymeric structure of connected MOC-1, but the intrinsic porosity of individual MOCs must be maintained.
147 Å3 of its total unit cell volume. The cuboctahedron can be considered nearly spherical with an internal diameter of an imaginary sphere of 1.8 nm (Fig. 2a) falling in the microporous regime. The as synthesized MOC-1 is crystalline but becomes amorphous in the activation process, upon evacuation of guests, because they act as templating agents. However, the amorphous phase of MOC-1 shows an amorphous-to-crystalline (AC) transformation once it traps guest molecules. MOC-1 can trap CO2 which shows reversible type I isotherms without significant hysteresis because only the internal voids of amorphous MOC-1 (microporous regime) are available for gas inclusion.37 One approach to see if there is an increase in CO2 adsorption, with respect to isolated MOC-1, can be carried out by synthesizing a polymeric structure of connected MOC-1, but the intrinsic porosity of individual MOCs must be maintained.
 | ||
| Fig. 2 (a) Single crystal XRD structure of the cuboctahedral MOC-1.37 (b) Chemical reaction of MOC-1 with the diamine spacers used to form the supramolecular material made of connected MOC-1.37 | ||
The linkers used to connect the MOC-1 were diamine spacers with different lengths and flexibility such as ethylenediamine (en), xylenediamine (xn) and diaminoheptane (hn) (Fig. 2b). Crosslinking such ligands with MOC-1, results in a porous material that has two pore regimes: the intrinsic MOC belonging to the microporous regime and the one created among the linked cages, the mesoporous regime. The supramolecular material with ethylenediamine linker shows some crystallinity while the solid materials including xylenediamine and diaminoheptane are amorphous phases. This is probably due to the higher flexibility of xn and hn.
Interestingly, CO2 sorption experiments at 195 K and 1 bar are systematically lowered in going from purely microporous MOC-1 to mesoporous materials MOC-1(en), MOC-1(xn) and MOC-1(hn). This behavior is explained as the adsorption of small gases is favoured in micropores showing strong fluid wall interactions. Although the mesoporous materials did not perform better than MOC-1 for CO2 adsorption, this example demonstrates the potential to prepare bimodal porous materials (hierarchical porosity) whose porosities can be tuned by selecting different cages and linkers. In this way, the preparation of supramolecular hybrid metal organic materials that are more in the regime of soft-matter (i.e., supramolecular polymers with intrinsic (micro)porosity) than conventional solid MOFs or MOCs can be synthesized.
3.2. Synthesis of a supramolecular porous aerogel (SAG) for CO2 adsorption connecting MOCs through coordination bonds
When thinking about materials used as adsorbents, what comes to mind are solid crystalline materials which are brittle, such as MOFs. Porous materials displaying properties like processability and softness are important as they can be used in applications where crystalline, rigid, and porous MOFs cannot be exploited. Furukawa and co-workers using derivatives of the highly stable cuboctahedral MOC [Rh2(bdc)2]12 (MOC-2)38 (where H2bdc – benzene-1,3-dicarboxylic acid) self-assembled using a dirhodium paddlewheel motif and a dicarboxylic acid (Fig. 3a) could synthesize amorphous soft porous materials.39 Because of the different lability among equatorial (poorly labile) and apical (highly labile) dirhodium paddlewheel atoms in the MOC-2, the cages can be linked via coordination bonds using appropriate ligands (vide ante). The icosahedral MOC-2 has high structural stability upon desolvation and guest removal due to the Rh–Rh single bond and strong Rh–carboxylate coordination bond.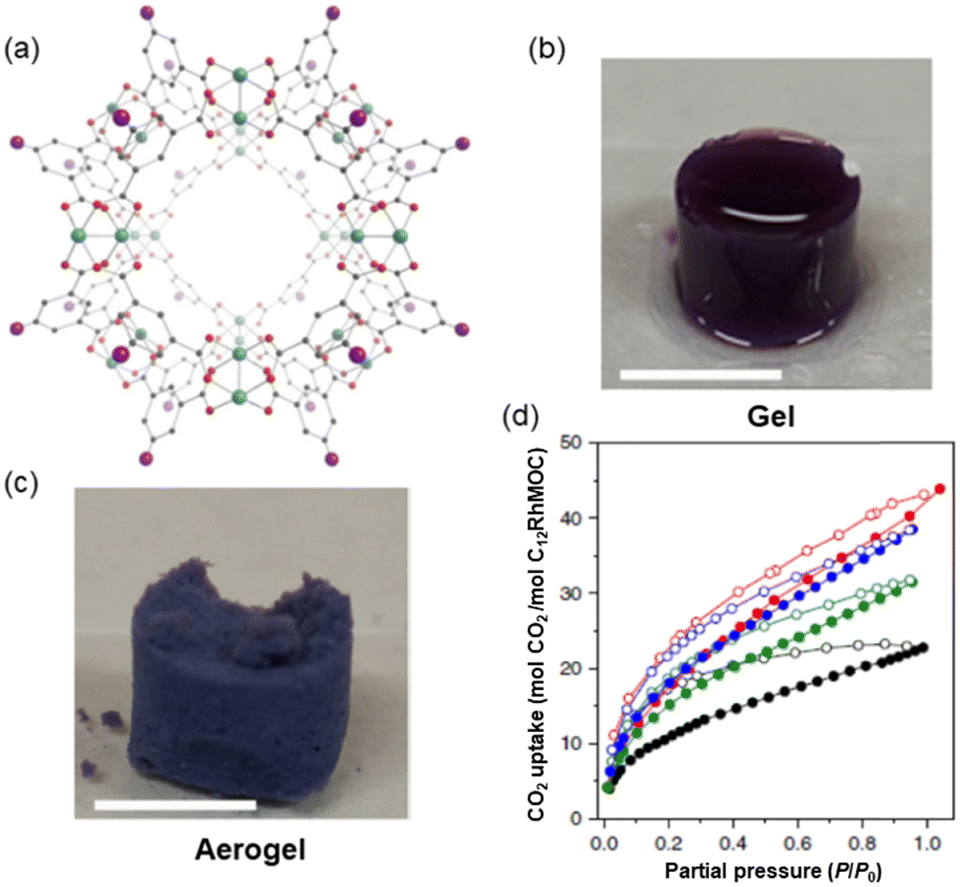 | ||
| Fig. 3 (a) Structure of the C12RhMOF porous monomer (MOC-3) obtained from the related [Rh2(bdc)2]12 crystal structure formed by rhodium ions (green) coordinated to the oxygen atoms (red) of H2bdc-C12 ligand in gray, with the appended aliphatic chain simplified as a purple sphere. (b) Gel and (c) aerogel obtained from the connection of monomeric MOC-3. (d) CO2 adsorption isotherm at 195 K of SAG-1 (red), supramolecular MOC polymer (blue), supramolecular MOC polymer of bigger dimensions (green) and C12RhMOP (black). Adsorption and desorption experiments are represented with filled and empty symbols respectively. Reproduced from ref. 40. | ||
The authors demonstrated how a supramolecular MOC polymer obtained by linking a derivative dirhodium MOC-3, in which H2bdc ligand is substituted by 5-dodecoxybenzene-1,3-dicarboxylic acid = H2bdc-C12 to increase its solubility, using linkers with two imidazole functionalities (Fig. 4).40 To polymerize the C12RhMOP monomers (MOC-3), the imidazole linker 1,4-bis(imidazole-1-ylmethyl)benzene (bix) was used. The addition (stepwise) of bix into a DMF solution of MOC-3 was monitored by dynamic light scattering (DLS) giving a maximum of particle sizes of 78 nm that were revealed to be spherical as shown by field emission scanning electron microscopy (FESEM). Larger particles can be obtained adding less molecular equivalents of bix. Powder XRD experiments show that the spherical particles are amorphous as no Bragg diffraction is observed.
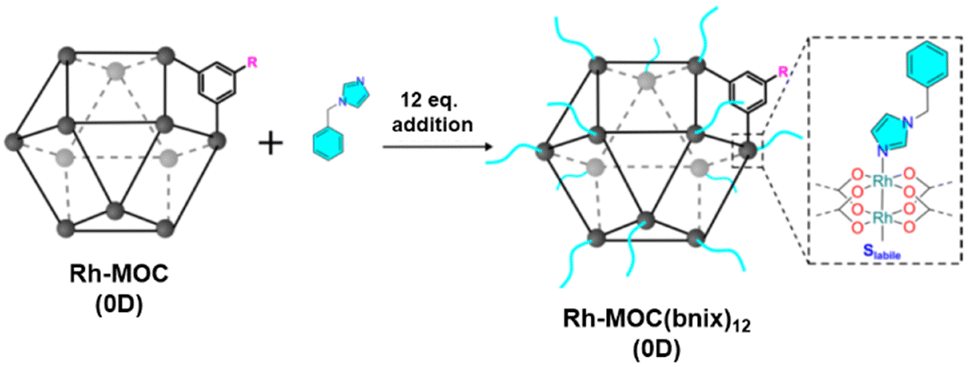 | ||
| Fig. 4 Cartoon showing the functionalization of Rh-MOC (0D) with bnix ligand to mimic the external surface of supramolecular colloid C12Rh-CPP and gel C12Rh-SAG. Reproduced from ref. 47 with permission from ACS, copyright 2022. | ||
Kinetic control in the synthesis of porous extended MOF structures41–43 and MOCs44,45 has been proved to be a good strategy to selectively obtain the desired products. In this vein, applying kinetic control by excess of bix molecules to a MOC-3 solution and cooling from 80 °C to room temperature, it was possible to isolate a MOC monomer with the (C12RhMOC)(bix)12 composition of 5.2 ± 1.2 nm size. After 1d, no aggregation was observed by keeping the suspension at room temperature. Because all the coordination sites are occupied by bix ligand, the only way to link the cages is by heating the solution containing the isolated MOCs (fully coordinated with bix) in order to release bix ligands and thus trigger the polymerization of (C12RhMOC)(bix)12 into larger particles of 22 ± 7 nm. The supramolecular colloidal gel (SCG) was obtained upon incubation (Fig. 3b). The macromolecular polymeric structure of linked MOCs did not form sufficient electrostatic interactions to pack the building components in an ordered manner, resulting in an amorphous phase as shown by powder XRD.
The porosity of the supramolecular polymer was evidenced by CO2 gas adsorption by transforming first the SCG to the aerogel (treated with supercritical CO2 and then heating in the vacuum) SAG-1 (where SAG = supramolecular aerogel). SAG-1 was composed of a hierarchical microporous structure of fused particles of 38 ± 11 nm in size (Fig. 3c).
The supramolecular material contains a robust RhMOC backbone with permanent microporosity in an amorphous state. The intrinsic porosity of MOC-3 is preserved after polymerization as shown by the type I isotherm at 195 K (Fig. 3d). Crucially, the uptake at P/P0 = 0.95 of SAG-1 is superior to that observed for the porous monomer with 68.64 cm3 g−1 and 40.23 mol CO2/mol C12RhMOC vs. 46.01 cm3 g−1 and 22.20 mol CO2/mol C12RhMOC respectively. Thus, using kinetic control, it is possible to prepare a supramolecular material with two different macroscopic morphologies including spherical MOCs particles and 3D interconnected colloidal gels.
3.3. Synthesis of a supramolecular porous aerogel (SAG) for photocatalytic reduction of CO2 into formic acid
As mentioned, one efficient method to reduce the amount of CO2 gas is by transforming it into molecules that can be used in other applications.46 The versatility of SAG-1 has also been exploited as a heterogeneous catalyst in the photocatalytic reduction of carbon dioxide into formic acid (HCOOH).47 Crucially, the catalytic activity was better than the single MOCs paddlewheel complexes and other heterogeneous photocatalysts.For the catalytic study, the authors exploited both MOCs, MOC-2 and MOC-3, using isophthalic acid and 5-dodecoxybenzene-1,3-dicarboxylic acid respectively, and by using bix the MOCs were self-assembled into supramolecular polymers in the form of colloids or gels depending on the quantity of bix used. Unfortunately, the supramolecular polymers are amorphous showing only short-range order.
Interestingly, both the colloidal and the gel evidenced catalytic activity for the transformation of CO2 into formic acid as the only product. Both C12Rh-CPP (colloid) and C12Rh-SAG show the same catalytic activity (TOF = 59 h−1) despite having different morphology. Importantly, the catalytic activity is a bit higher than the molecular building block C12RhMOC (TOF = 52 h−1). To understand the catalytic behavior, pristine C12RhMOC was functionalized with monodentate 1-benzylimidazole (bnix) to yield [Rh2(C12-bdc)2]12(bnix)12 (C12RhMOCbnix) to mimic the external surface of the MOCs in C12Rh-CPP and C12Rh-SAG (Fig. 4). Interestingly, the catalytic activity is increased with values very similar to those observed in the supramolecular structures with linked MOCs. From those results, first, the authors concluded that the catalytic activity is not limited to surface effects in the heterogeneous polymers and, second, that the internal part of the cages is accessible to the reactants. The presence of the coordinated bix and bnix ligands slightly increases the electron density around the Rh centers, resulting in an increase in the catalytic activity.
The self-assembled supramolecular catalysts have demonstrated high TOF up to 60 h−1 outperforming by 30% the RhMOC and other state-of-the-art heterogeneous photocatalyst systems including Rh-MOFs by at least a factor of 12. The optimal catalytic activity has been rationalized because of the high number of Rh atoms per gram of the catalyst but also favoured by the accessibility inside the supramolecular cage. Structural aspects focussing on the inter-atomic distances between atoms were determined using PDF analysis, regarding the stability of the MOCs linked by bix ligands. Both samples, pristine and spent samples show that the G(r) curves demonstrate that at the molecular level, there is no distortion or decomposition of the paddlewheel Rh sites at the Rh–Rh nodes in the MOC and no presence of Rh0 species was detected.
3.4. Synthesis of a supramolecular porous aerogel IL by connecting MOCsvia coordination bonds and its CO2 adsorption capacity
The integration of porous liquids (PLs) in the fabrication of gels can yield new functional materials displaying new properties arising from the combination of porosity and structural tunability. Functional gels combine properties of solids and liquids which so far have been synthesized using microporous MOCs,48 metal–organic frameworks (MOFs),49 and covalent-organic frameworks (COFs).50 They become porous after solvent removal forming porous aerogels but they lose the initial gel properties. To avoid the loss of gel–liquid properties (soft materials) whilst maintaining the porous properties, PL-like gels or porous gels are supposed to be a promising alternative to PLs for some useful applications.More recently, the exploitation of MOC-2 gel has been reported, through guest exchange, during which the original DMF guests are replaced by the ionic liquid (IL) 1-butyl-3-methylimidazolium tetrafluoroborate ([BMIM]+[BF4]−).51 The IL is too large to enter in the cage cavities once the gel is activated but it enters into the hierarchichal porosity (i.e., the larger pores). Upon heating, the solvents can be removed giving the obtained ionic liquid gel accessible microporosity with good CO2 performances. The liquid exchange process is possible due to the robust nature of the MOCs that maintain the gel structural properties (i.e., it does not deform or collapse). The new material is claimed to be in between porous solids and porous liquids. The new pore-networked gels have an increased gas sorption capacity as a result of the accessible porosity in the MOC network.
The rhodium-based cuboctahedral MOC-2, [Rh2(bdc)]12 (bdc = benzene-1,3-dicarboxylate) was selected for the porous building block for the formation of gel networks as it has 12 available coordination sites at each axial position of the dirhodium paddlewheel moiety for further coordination-driven crosslinking, and its thermal stability yields structural integrity under harsh activation conditions. To link the MOCs by coordination bonds and the gel formation, the bidentate ligand 1,4-bis(imidazole-1-ylmethyl)benzene (btx) was used.
The Gel_IL microporosity was measured using CO2 adsorption. The microporous of the MOCs were filled with DMF or acetone used in the self-assembling process, which are removed upon heating at 120 °C in a vacuum overnight. After the thermal treatment, the material did not collapse, and the porosity of the supramolecular structure was tested by CO2 adsorption at 303 K. The isotherms were compared with the IL [BMIM]+[BF4]−. As shown in Fig. 5a, neat IL has a lower adsorption capacity compared to all the Gel_IL tested (i.e., with different concentration of MOC networks).
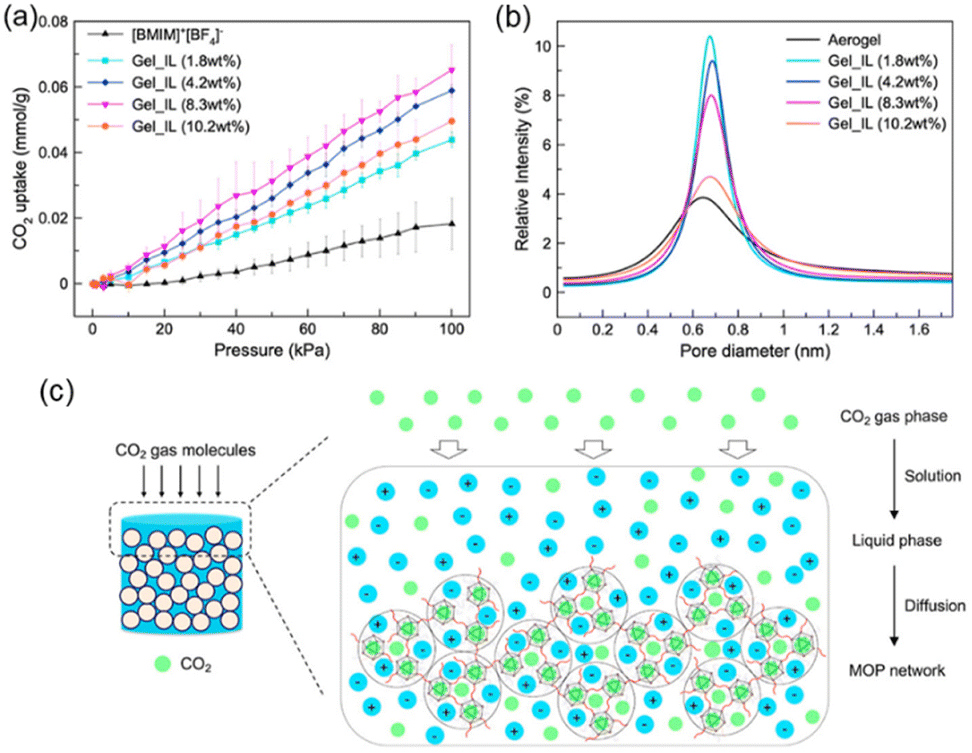 | ||
| Fig. 5 (a) CO2 uptake isotherms of Gel_IL at different concentrations of MOC-2 networks at 303 K under 100 kPa. (b) Particle size distribution of Gel_IL with different concentrations of MOC-2 networks derived from PALS experiments. (c) Cartoon representing the CO2 gas diffusion pathway within the wet sample of connected MOCs. Reproduced from ref. 51 with permission from ACS, copyright 2023. | ||
The CO2 sorption at ca. 100 kPa in the gel with 1.8 wt% adsorbed 0.044 mmol CO2 per gram sample which is almost two times larger than that of pure [BMIM]+[BF4]−. If the concentration of MOC-2 in the gel is increased then the CO2 adsorption increases too, showing that the MOC network is accessible in the wet gel state. To investigate more deeply the porosity of Gel_IL, positron annihilation lifetime spectroscopy (PALS) experiments were performed as it allows the detection of voids in liquid samples. Exposing the ionic liquid sample to a positron source like 22Na generates the ortho-positron (i.e., a parallel spin complex between e+ and e−). Measuring the lifetime of o-positron it is possible to estimate the pore distribution size (PDS) of the liquid sample. That is, larger pores correspond to slower decay rates and hence longer lifetimes. The pore size distribution in the wet Gel_IL by measuring the lifetime of the o-positron was ca. 2.7 ns corresponding to a pore size of ca. 0.68 nm (Fig. 5b), which is the size in between the MOC network (solid aerogel) and that of the ionic liquid [BMIM]+[BF4]−. Increasing the concentration of the MOC network does not increase the PDS but decreases the intensity of the peak due to lower mobility of the gel in the concentrated gel. The CO2 uptake in the gel material follows the solution-diffusion model (Fig. 5c), where the guest CO2 molecules reach the interior of the connected MOCs. Thus, even in the wet state the supramolecular material is porous.
4. From MOCs to polymers: covalent cross-linking of MOCs to form supramolecular flexible MOC materials
4.1. Connecting MOCs by covalent bond formation for CO2 adsorption
A different strategy to link MOCs, to those observed so far, was reported by Shimizu and co-workers by using the modified Cu24 MOC cuboctahedral Cu24 isophthalate by attaching in the 5-position of the isophthalate linkers in the external part of the cages with long alkyl chains having terminal alkenes (MOC-4) (Fig. 6a).52 Using olefin metathesis reactions, the functionalized MOC-4 were then connected. In the new synthesized system, the MOCs are bridged by –OC18H34O– units among the isophthalate ligands (Fig. 6b). Synchrotron single crystal X-ray crystallography revealed the structure which shows that the Cu24 metals linked by the organic ligand yield a cuboctahedral of ca. 2 nm packing in the Pnnm space group.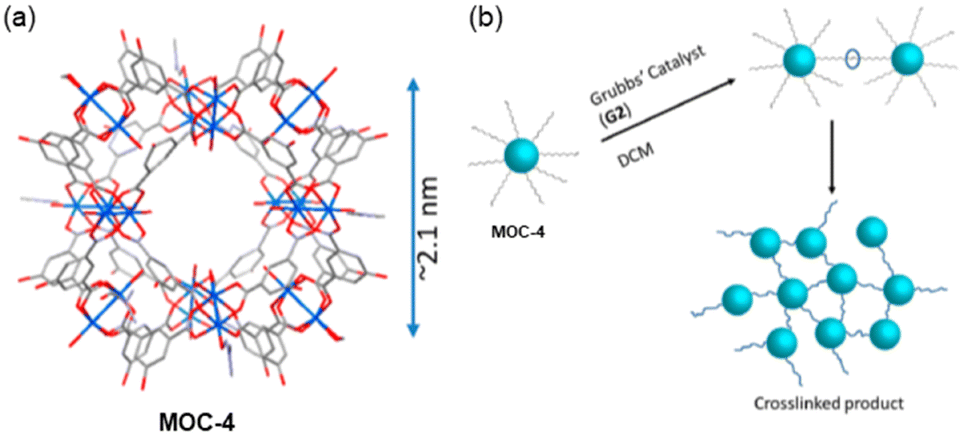 | ||
| Fig. 6 (a) Synchrotron single crystal X-ray structure of MOC-4 displaying the core MOC as the disordered alkyl chains could not be resolved crystallographically. (b) Cartoon depicting the cross-linking process via G2 Grubb's catalyst in DCM. Reproduced from ref. 52 with permission from ACS, copyright 2019. | ||
The MOC structure remains stable when dissolved in organic solvents but not in water. The functionalized MOC were linked via olefin metathesis using Grubb's second-generation catalyst (G2) by solubilizing the cage in DCM under atmosphere conditions. In this work, three MOC-4 cross-linked versions with ≈20%, ≈40% and ≈80% degrees of cross-linking were studied. The three products referred to as MOC-4 × 20, MOC-4 × 40 and MOC-4 × 80 were indicated by solution 1H NMR. Upon crosslinking, the solid (MOC-4 × 40) and (MOC-4 × 80) appeared from the solutions in common organic solvents (i.e., DCM, chloroform, ethyl acetate, THF and DMF) as microcrystalline samples (Fig. 7a). The partial solubility of MOC-4 × 20 suggested that the cross-linking reaction did not proceed sufficiently due to inhomogeneous local cross-linking.
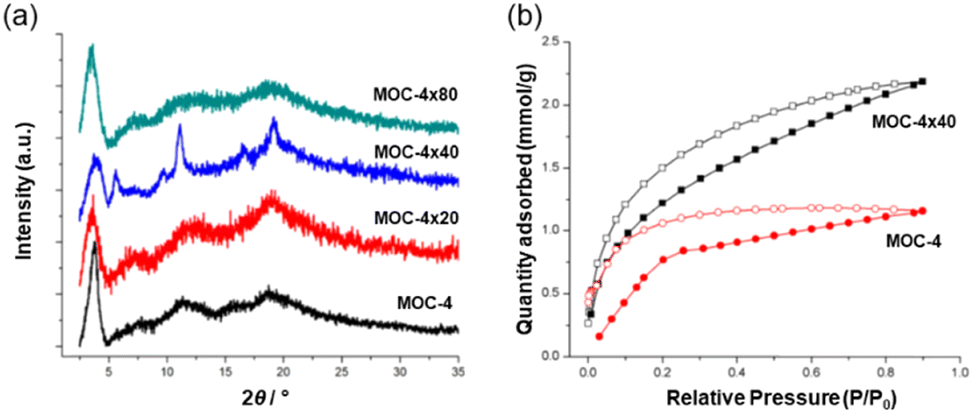 | ||
| Fig. 7 (a) Powder XRD patterns of the MOC-4 and the supramolecular cross-linked products. (b) CO2 adsorption experiments carried out at 195 K and 1.2 bar. Reproduced from ref. 52 with permission from ACS, copyright 2019. | ||
The most homogeneously cross-linked MOC-4 solid (MOC-4 × 40) was used for CO2 gas adsorption experiments at 195 K/1.2 bar, and adsorbed 2.18 mmol g−1 of CO2 while monomeric MOC-4 adsorbed 1.16 mmol g−1. Thus, the presence of additional voids is a result of the cross-linking process, and this allows for a higher CO2 adsorption in the solid with “connected” MOCs than the single MOC-4. Interestingly, CO2 adsorption at 298 K shows the same trend as seen for the 195 K experiments only with a slight decrease in the uptake of CO2 of 2.3 mmol g−1 for MOC × 40 and 1.3 mmol g−1 for MOC-4 (Fig. 7b).
5. From MOCs to polycatenanes or [n]-catenane networks
5.1. Linking MOCsvia mechanical bonds to form polycatenanes made of cages
The last approach to connecting MOCs in a polymeric manner outlined in this Perspective is by linking them not by coordination bonds, or by covalent bonds as recently seen, but by mechanical bonds.53–56 Although studies of CO2 adsorption in poly-catenanes formed of interlocked MOCs are very few,57 clearly the intriguing structural properties of cage-catenanes make such materials potential candidates for exploitation in CO2 remediation. Polycatenanes made of rings (i.e., not by MOCs) can be defined as mechanically linked polymers derived from catenane subunits. The mechanical properties of catenanes are important because the individual ring components in the catenane can have rotations, twisting and elongation movements giving the mechanical molecule important functional properties.56 In order to separate two rings in a catenane it is necessary to break chemical bonds even though they are not connected via chemical bonds. If instead of using rings for the formation of the catenane chains, MOCs are used, then cage-catenanes or cage-polycatenanes can be generated.58,59 However, the synthesis of the cage-catenanes is challenging because several aspects must be considered for appropriate cage interlocking.First, the size and number of windows and the size of the cavity should be large enough to allow the formation of the mechanical bond.59 Second, suitable metal atoms with a certain lability are needed for the error checking in the reversible coordination bond formation involving intermediate structures. Third, the presence of guest molecules that act as templating agents are needed to obtain crystalline structures.59 Fourth, depending if the catenation is forming 1D-chains, 2D layers or 3D cage catenation, the electrostatic forces keeping the chains or layers will influence their crystallinity, their dynamic behavior upon external stimuli, and hence the gas adsorption properties. Thus, catenanes formed of interlocked MOCs is another degree of structural complexity.
5.1. Mechanically interlocked tetrahedral M6L4MOCs yielding 3D cage polycatenanes
The example of Lu and co-workers reported a polycatenane self-assembly of tetrahedral MOCs prepared using six [NiL]2+ cations (where L = 1,4,8,11-tetraazaundecane) and four tri(4-carboxy-benzyl)amine ligand (H3tcba) (Fig. 8a and b).57 The crystal structure was determined using an initial model obtained from SC-XRD but refined using powder XRD data by Rietveld refinement as suitable crystals for SC-XRD were not available. Four tcba3− anions link six [NiL]2+ cations via its three carboxy-benzyl groups leading to tetrahedral cages with the formula [NiL]6(tbca)4 (MOC-5). In MOC-5, each Ni(II) is six-coordinated to four N atoms from L and two carboxylate O atoms from two individual tcba3− anions, and each tcba3− connects three [NiL]2+. The MOC is formed of four tcba3− anions located at the vertexes of the cage and six Ni(II) metals situated at the edges of the cage resulting in a M6L4 nanocage (Fig. 8c and 8d). The size of the cage considering the distances among edges is 16.64 Å. In the center of the cage there is a ClO4− anion. The solvent accessible volume is ca. 10% of the total unit cell volume. The large openings of the M6L4MOC-5 nanocages allows a quadruple catenation in which one cage is mechanically interlocked with another four through all its four vertices (Fig. 8d). The overall structure is a 3D extended polycatenane remaining one of the few of such structures reported.60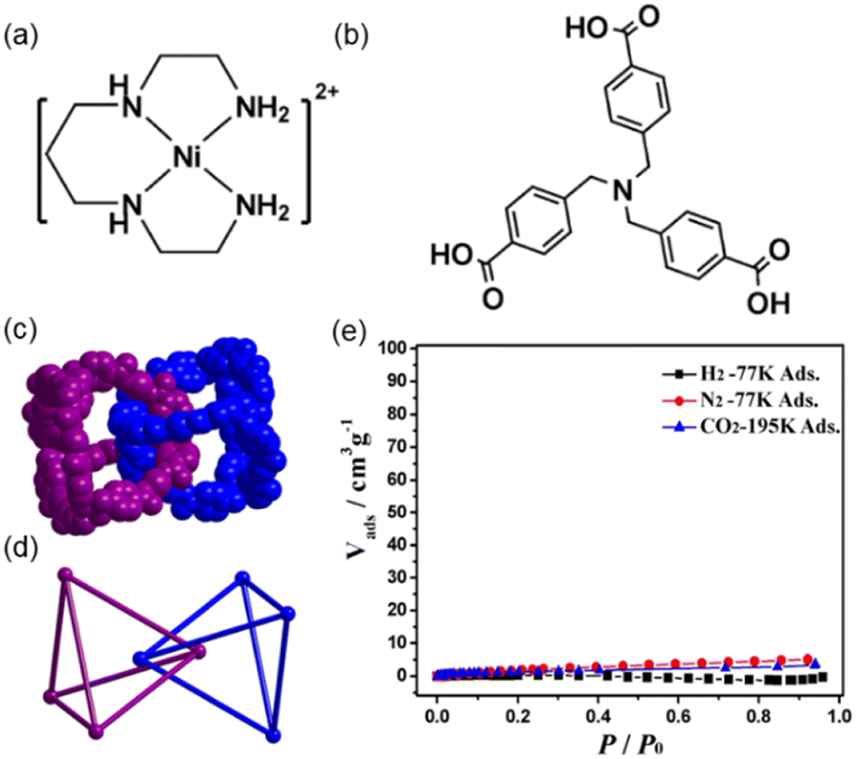 | ||
| Fig. 8 (a) Structure of cation [NiL]2+ and (b) ligand H3tcba used for self-assembling MOC-5. (c) Space-filling and (d) ball-and-stick representations of two interlocked MOC-5 nanocages, showing the interlocked corners in face-to-face stacking fashion with a distance of 6.219 Å between two nitrogen atoms of two tcba3−. A single cage is interlocked by other four cages (not shown). (e) Sorption isotherms of N2, H2 and CO2 for dehydrated mechanically interlocked MOC-5. Reproduced from ref. 57. | ||
Gas adsorption experiments show that the polycatenane formed of interlocked tetrahedral cages is nonporous for CO2 at 195 K (Fig. 8e), because it becomes amorphous upon guest water release (activation process). However, it shows that for small alcohols such as methanol, ethanol i-propanol and i-butanol it is porous. The sorption isotherms show that the smaller the alcohol the better the adsorption capacity per cage. Thus, after interlocking of the tetrahedral MOCs there is enough space for adsorbing gas molecules (i.e., alcohols).
5.2. Mechanically interlocked icosahedral M12L8MOCs yielding 1D cage polycatenanes
Another type of poly-[n]-catenane structure that can be a potential candidate for CO2 adsorption is the 1D chain structure of interlocked icosahedral M12L8 nanocages which have huge internal voids of ca. 2700 Å3. So far, the work done on such materials has been using the tridentate and rigid ligands 2,4,6-tris-(4-pyridyl)pyridine (TPP),61,62 (MOC-6) and 2,4,6-tris-(4-pyridyl)benzene (TPB),44,45,63–65 (MOC-7) in which one single icosahedral M12L8 nanocage is interlocked by two adjacent cages forming 1D chains along the c-crystallographic direction (Fig. 9a). Single crystals of the M12L8 poly-[n]-catenanes are stable at room temperature and have shown to behave dynamically upon external stimuli. In particular, the heterogeneous gas–solid adsorption of aromatic molecules via molecular recognition, has been demonstrated by multiple single-crystal-to-single-crystal reactions.66 However, CO2 adsorption has not yet been tested for such materials involving MOC-7. The mechanically interlocked material shown in Fig. 9b might show CO2 uptake.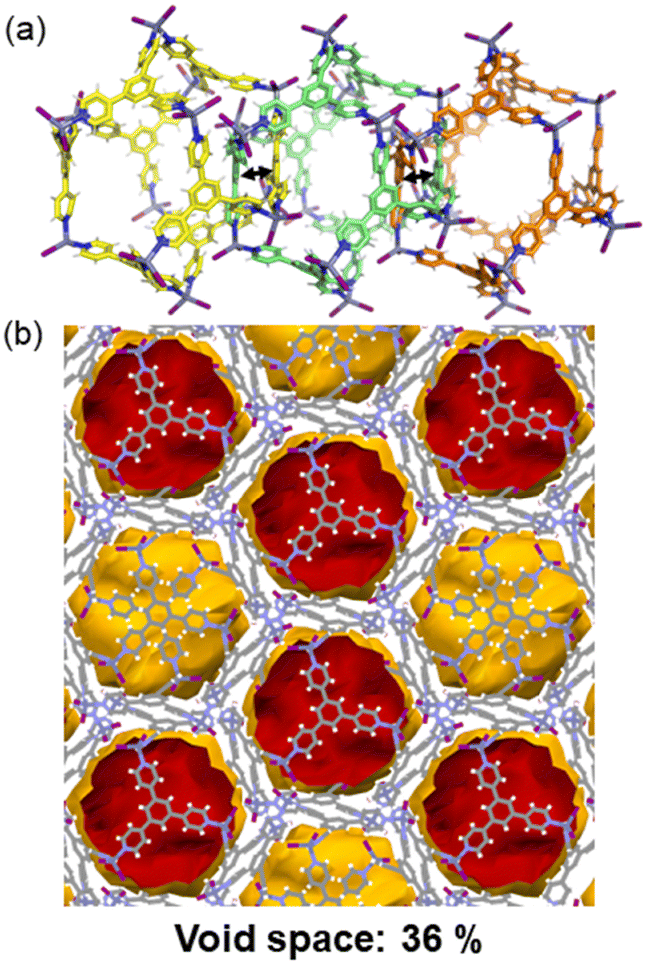 | ||
| Fig. 9 (a) Interlocking of three M12L8 nanocages expanding along the c-axis. To differentiate the individual M12L8 cages the carbon atoms are green, yellow and orange. (b) View of the isolated voids in the M12L8MOCs after manually removing the solvent molecules. Reproduced from ref. 45. | ||
So far, such MOC-7 within the polymeric structures can be suitable for gas adsorption because they are interlocked in the absence of solvent molecules. It has been demonstrated that the amorphous phase of the M12L8 poly-[n]-catenane can adsorb, from the gas phase, aromatic molecules as evidenced by powder XRD analysis, thus indicating a significant dynamic behavior of the non-ordered phase containing the interlocked M12L8 nanocages.63 It has also been demonstrated that in a heterogeneous solid–liquid phase process, methanol can be trapped in the amorphous nanocages producing a swelling effect on the material.63 Thus, CO2 could also be entrapped in the interlocked MOC-7 cages.
6. Benefits of linking MOCs for CO2 remediation
The beneficial aspects of synthesizing new materials by connecting MOCs for CO2 remediation are numerous and important ranging from structural to functional aspects. The most salient features described in this article are highlighted below.First, the crosslinking of MOCs gives the possibility of synthesizing porous materials with hierarchical porosity including microporosity and mesoporosity with applications in CO2 remediation that can outperform those of isolated MOCs.
Second, the formation of supramolecular metal–organic architectures upon connecting MOCs yields materials that are soft-mater-like materials such as supramolecular porous gels and aerogels which find applications in areas involving fluids.
Third, as shown, the CO2 uptake performance of MOC-3 is preserved after cross-linking (i.e., the original porous properties of MOC-3 are maintained) and in SAG-1 the CO2 uptake at P/P0 = 0.95 is higher compared to that of the isolated MOC-3 (40.23 mol CO2/mol C12RhMOC (SAG-1) vs. 22.20 mol CO2/mol C12RhMOC (MOC-3)) showing the benefit of connecting isolated MOC-3.
Fourth, improved photocatalytic reduction of CO2 into formic acid using the supramolecular aerogel SAG-1 is significantly improved, outperforming by 30% that of single MOCs paddlewheel complexes and other heterogeneous photocatalysts.
Fifth, novel materials that are between porous solids and porous liquids can be synthesized (by connecting MOC-2 with the introduction of IL in the mesopores), showing about double CO2 sorption capacity at ca. 100 KPa when compared to the IL. This is explained due to the accessible porosity in the soft material of connected MOCs.
Sixth, using a different cross-linking mechanism by covalent bond formation, and not by means of coordination bonds as in the previous cases, linked MOC-4 (at 192 K/1.2 bar) shows improved gas uptake of 2.18 mmol g−1 of CO2, while monomeric MOC-4 adsorbed 1.16 mmol g−1.
Mechanically interlocked MOCs are far less explored for CO2 adsorption, but the dynamic behavior and the internal voids left after the MOC's interlocking show potential in CO2 remediation applications.
7. Conclusions
Crystal engineering and soft matter engineering strategies aimed to synthesize new materials embedding MOCs linked through coordination driven and covalent cross-linking, have shown promising structural and functional properties. Materials formed of connected MOCs have been exploited for the specific application of CO2 adsorption and its catalytic transformation into organic compounds. In this Perspective, it has been shown that for the few examples of MOCs linked by coordination bonds or covalent bonds, in most of the cases, the CO2 adsorbed is superior when the cages are connected compared to the monomer MOCs not forming part of the supramolecular polymer. Photocatalytic CO2 reduction to formic acid has been shown to work well in a supramolecular aerogel of connected MOCs. Gel-like materials prepared by connecting MOCs can be exploited in applications where MOFs cannot be used due to their brittle nature, typical of solid crystalline materials. Semi-solid materials like gels containing strongly connected MOCs are processable, soft, and can be shaped and used for instance in application shapes that usually crystalline solids cannot adapt easily, thus expanding the applications of connected MOCs materials in areas involving fluids. Mechanically interlocked tetrahedral MOCs have been tested also for CO2 adsorption. Despite not showing CO2 uptake they can take up other small molecules like alcohols. Larger interlocked cages such as M12L8, due to their dynamic behaviour upon external stimuli, are potential candidates which are worth trying in applications of CO2 remediation. The potential of linking different types of MOCs using coordination driven, covalent linking or mechanical bonds, into supramolecular porous materials, really shows how versatile this research area is. Supramolecular materials including linked MOCs, like the ones reviewed in this Perspective, will soon produce novel materials with unexplored functional properties not only in CO2 remediation but in many other applications from regenerative medicine to soft robotics.Author contributions
J. M.-R. conceived and wrote the article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. M.-R. thanks Politecnico di Milano for funding (Fondo Chiamata Diretta Internazionalizazzione. Prg. Id. 61566).Notes and references
- E. S. Sanz-Pérez, C. R. Murdock, S. A. Didas and C. W. Jones, Chem. Rev., 2016, 116, 11840–11876 CrossRef PubMed.
- L. Zhang, Z.-J. Zhao and J. Gong, Angew. Chem., Int. Ed., 2017, 56, 11326–11353 CrossRef CAS PubMed.
- M. Ding, R. W. Flaig, H.-L. Jiang and O. M. Yaghi, Chem. Soc. Rev., 2019, 48, 2783–2828 RSC.
- B. F. Hoskins and R. Robson, J. Am. Chem. Soc., 1990, 112, 1546–1554 CrossRef CAS.
- M. Fujita, Y. J. Kwon, S. Wahsizu and K. Ogura, J. Am. Chem. Soc., 1994, 115, 1151 CrossRef.
- O. M. Yaghi and H. Li, J. Am. Chem. Soc., 1995, 117, 10401 CrossRef CAS.
- M. Kondo, T. Yoshitomi, K. Seki, H. Matsuzaka and S. Kitagawa, Angew. Chem., Int. Ed. Engl., 1997, 36, 1725 CrossRef CAS.
- L. R. MacGillivray, Metal Organic Frameworks: Design and Application, Wiley, 2010 Search PubMed.
- B. Lin, T. T. T. Nguyen, R. Vaidhyanathan, J. Burner, J. M. Taylor, H. Durekova, F. Akhtar, R. K. Mah, O. G. Nik, S. Marx, N. Fylstra, S. S. Iremonger, K. W. Dawson, P. Sarkar, P. Hovington, A. Rajendran, T. K. Woo and G. K. H. Shimizu, Science, 2021, 374, 1464–1469 CrossRef PubMed.
- M. Kawano and M. Fujita, Coord. Chem. Rev., 2007, 251, 2592–2605 CrossRef CAS.
- J. Martí-Rujas, Dalton Trans., 2020, 49, 13897–13916 RSC.
- T. Haneda, M. Kawano, T. Kawamichi and M. Fujita, J. Am. Chem. Soc., 2008, 130, 1578–1579 CrossRef CAS PubMed.
- T. Kawamichi, T. Haneda, M. Kawano and M. Fujita, Nature, 2009, 462, 633–635 CrossRef PubMed.
- R. Kubota, S. Tashiro, M. Shiro and M. Shionoya, Nat. Chem., 2014, 6, 913–918 CrossRef CAS PubMed.
- M. Fujita, D. Oguro, M. Miyazawa, H. Oka, K. Yamaguchi and K. Ogura, Nature, 1995, 378, 469 CrossRef CAS.
- B. Olenyuk, J. A. Whiteford, A. F. Etter and P. J. Stang, Science, 1999, 398, 796–799 CAS.
- M. Fujita, N. Fujita, K. Ogura and K. Yamaguchi, Nature, 1999, 400, 52–55 CrossRef CAS.
- M. Yoshizawa, M. Tamura and M. Fujita, Science, 2006, 312, 251–254 CrossRef CAS PubMed.
- D. Caulder and K. N. Raymond, Acc. Chem. Res., 1999, 32, 975–982 CrossRef CAS.
- D. Fiedler, R. G. Bergman and K. N. Raymond, Angew. Chem., Int. Ed., 2006, 45, 745–748 CrossRef CAS PubMed.
- P. Mal, B. Brenier, K. Rissanen and J. R. Nitschke, Science, 2009, 324, 1697–1699 CrossRef CAS PubMed.
- S. Pullen and G. H. Clever, Acc. Chem. Res., 2018, 51, 3052–3064 CrossRef CAS PubMed.
- D. Zhang, T. K. Ronson, Y.-Q. Zou and J. R. Nitschke, Nat. Rev. Chem., 2021, 5, 168–182 CrossRef CAS PubMed.
- A. C. Sudik, A. R. Millward, N. W. Ockwig, A. P. Côté, J. Kim and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 7110–7118 CrossRef CAS PubMed.
- M. Hosono and S. Kitagawa, Acc. Chem. Res., 2018, 51, 2437–2446 CrossRef PubMed.
- E. M. El-Sayed and D. Yuan, Chem. Lett., 2020, 49, 28–53 CrossRef CAS.
- I. Jahovic, Y.-Q. Zou, S. Adorinni, J. R. Nitschke and S. Marchesan, Matter, 2021, 4, 2123–2140 CrossRef CAS.
- J. E. M. Lewis, Chem. Commun., 2022, 58, 13873–13886 RSC.
- O. Ohmori and M. Fujita, Chem. Commun., 2004, 1586 RSC.
- M. Fujita, J. Yazaki and K. Ogura, J. Am. Chem. Soc., 1990, 112, 5645 CrossRef CAS.
- T. Kusukawa and M. Fujita, J. Am. Chem. Soc., 2002, 125, 13576 CrossRef PubMed.
- S.-Y. Yu, T. Kusukawa, K. Biradha and M. Fujita, J. Am. Chem. Soc., 2000, 122, 2665–2666 CrossRef CAS.
- C. F. Espinosa, T. N. Ronson and J. N. Nitschke, J. Am. Chem. Soc., 2023, 145, 9965–9969 CrossRef CAS PubMed.
- Y. Nishioka, T. Yamaguchi, M. Yoshizawa and M. Fujita, J. Am. Chem. Soc., 2007, 129, 7000–7001 CrossRef CAS PubMed.
- T. Nakama, A. Rossen, R. Ebihara, M. Yagi-Utsumi, D. Fujita, K. Kato, S. Sato and M. Fujita, Chem. Sci., 2023, 14, 2910–2914 RSC.
- E. G. Percástegui, T. N. Ronson and J. R. Nitschke, Chem. Rev., 2020, 120, 13480–13544 CrossRef PubMed.
- H.-J. Jung, D.-H. Moon and H.-P. Chun, Bull. Korean Chem. Soc., 2011, 32, 2489 CrossRef CAS.
- S. Furukawa, N. Horike, M. Kondo, Y. Hijikata, A. Carne-Sanchez, P. Larpent, N. Louvain, S. Diring, H. Sato, R. Matsuda, R. Kawano and S. Kitagawa, Inorg. Chem., 2016, 55, 10843–10846 CrossRef CAS PubMed.
- A. Carné-Sánchez, G. A. Craig, P. Larpent, V. Guillerm, K. Urayama, D. Maspoch and S. Furukawa, Angew. Chem., Int. Ed., 2019, 58, 6347–6350 CrossRef PubMed.
- A. Carne-Sanchez, G. A. Crag, P. Larpent, T. Hirose, M. Higuchi, S. Kitagawa, K. Matsuda, K. Urayama and S. Furukawa, Nat. Commun., 2018, 9, 2506 CrossRef PubMed.
- M. Kawano, T. Haneda, D. Hashizume, F. Izumi and M. Fujita, Angew. Chem., Int. Ed., 2008, 47, 1269–1271 CrossRef CAS PubMed.
- J. Martí-Rujas, N. Islam, D. Hashizume, F. Izumi, M. Fujita, H. J. Song, H. C. Choi and M. Kawano, Angew. Chem., Int. Ed., 2011, 50, 6105–6108 CrossRef PubMed.
- J. Martí-Rujas and M. Kawano, Acc. Chem. Res., 2013, 46, 493–505 CrossRef PubMed.
- S. Torresi, S. Famulari and J. Martí-Rujas, J. Am. Chem. Soc., 2020, 142, 9537 CrossRef CAS PubMed.
- J. Martí-Rujas, S. Elli and A. Famulari, Sci. Rep., 2023, 13, 5605 CrossRef PubMed.
- X. B. Lu and J. Darensbourg, Chem. Soc. Rev., 2012, 41, 1462–1484 RSC.
- A. C. Ghosh, A. Legrand, R. Rajapaksha, G. A. Craig, C. Sassoye, G. Balázs, D. Farrusseng, S. Furukawa, J. Canivet and F. M. Wisser, J. Am. Chem. Soc., 2022, 144, 3626–3636 CrossRef CAS PubMed.
- C. Lu, M. Zhang, D. Tang, X. Yan, Z. Zhang, Z. Zhou, B. Song, H. Wang, X. Li, S. Yin, H. Sepehrpour and P. J. Stang, J. Am. Chem. Soc., 2018, 140, 7674–7680 CrossRef CAS PubMed.
- B. Bueken, N. Van Velthoven, T. Willhammar, T. Stassin, I. Stassen, D. A. Keen, G. V. Baron, J. F. M. Denayer, R. Ameloot, S. Bals, D. De Vos and T. D. Bennett, Chem. Sci., 2017, 8, 3939–3948 RSC.
- D. Zhu, Y. Zhu, Q. Yan, M. Barnes, F. Liu, P. Yu, C.-P. Tseng, N. Tjahjono, P.-C. Huang, M. M. Rahman, E. Egap, P. M. Ajayan and R. Verduzco, Chem. Mater., 2021, 33, 4216–4224 CrossRef CAS.
- Z. Wang, A. Ozcan, G. A. Craig, F. Haase, T. Aoyama, D. Poloneeva, K. Horio, K. Horio, M. Higuchi, M.-S. Yao, C. M. Doherty, G. Maurin, K. Urayama, A. Bavykina, S. Horike, J. Gascon, R. Semino and S. Furukawa, J. Am. Chem. Soc., 2023, 145, 14456–14465 CrossRef CAS PubMed.
- G. Lal, M. Derakhshandeh, F. Akhtar, D. M. Spasyuk, J.-B. Lin, M. Trifkovic and G. K. H. Shimizu, J. Am. Chem. Soc., 2019, 141, 1045–1053 CrossRef CAS PubMed.
- J. F. Stoddart, Chem. Soc. Rev., 2009, 38, 1802–1820 RSC.
- G. Gil-Ramírez, D. A. Leigh and A. J. Stephens, Angew. Chem., Int. Ed., 2015, 54, 6110–6150 CrossRef PubMed.
- C. J. Bruns and J. F. Stoddart, The Nature of the Mechanical Bond: From Molecules to Machines, Wiley, Hoboken, NJ, 2016 Search PubMed.
- Q. Wu, P. M. Rauscher, X. Lang, R. L. Wojtecki, J. J. de Pablo, M. J. A. Hore and S. J. Rowan, Science, 2017, 358, 1434–1439 CrossRef CAS PubMed.
- L. Jiang, P. Ju, X.-R. Meng, X.-J. Kuang and T.-B. Lu, Sci. Rep., 2012, 2, 668–672 CrossRef PubMed.
- Y. Yamauchi, M. Yoshizawa and M. Fujita, J. Am. Chem. Soc., 2008, 130, 5832–5833 CrossRef CAS PubMed.
- L. Chen, Q. Chen, M. Wu, F. Jiang and M. Hong, Acc. Chem. Res., 2015, 48, 201–210 CrossRef CAS PubMed.
- X. Kuang, X. Wu, R. Yu, J. P. Donahue, J. Huang and C.-Z. Lu, Nat. Chem., 2010, 2, 461 CrossRef CAS PubMed.
- J. Heine, J. Schmedt auf der Gunne and S. Dehnen, J. Am. Chem. Soc., 2011, 133, 10018 CrossRef CAS PubMed.
- E. C. Constable, G. Zhang, C. E. Housecroft and J. Zampese, CrystEngComm, 2011, 13, 6864 RSC.
- J. Martí-Rujas, S. Elli, A. Sacchetti and F. Castiglione, Dalton Trans., 2022, 51, 53–58 RSC.
- J. Martí-Rujas and A. Famulari, Cryst. Growth Des., 2022, 22, 4494–4502 CrossRef.
- J. Martí-Rujas, S. Ma and A. Famulari, Inorg. Chem., 2022, 61, 10863–10871 CrossRef PubMed.
- J. Martí-Rujas, S. Elli, A. Zanotti, A. Famulari and F. Castiglione, Chem. – Eur. J. DOI:10.1002/chem.202302025.
| This journal is © The Royal Society of Chemistry 2023 |