DOI:
10.1039/D3GC01025B
(Paper)
Green Chem., 2023,
25, 5712-5720
A novel tandem reactor design based on nano-Cu electrocatalysts and microbial biocatalysts for converting CO2 into ethylene and acetate†
Received
30th March 2023
, Accepted 20th June 2023
First published on 20th June 2023
Abstract
CO2 electrochemical conversion on copper electrocatalysts demonstrates selectivity and activity towards multicarbon compounds such as ethylene. However, the complex product distribution, as well as the wasted carbon and electrons in non-target products such as HCOOH, CO, and H2, can significantly increase the energy input and separation costs. As a result, we designed an electro-bio tandem reactor using copper electrocatalysts and microorganisms to transform CO2 into specific multicarbon compounds in both the gas and liquid phases. The unavoidable reducing products from electrocatalysis, including HCOOH in the liquid phase, and CO and H2 in the gas phase, can be consumed and recycled as electron donors or better carbon feedstocks for selective acetate synthesis in Moorella thermoacetica via the Wood–Ljungdahl pathway. In neutral electrolytes, the faradaic selectivity of acetate in liquid products is the highest (79.6%) for Cu-based CO2 electroreduction, and the electron conversion rate to ethylene and acetate is the highest for microbial electrosynthesis (8113.8 mmol h−1 m−2), demonstrating a green design for CO2 upcycling targeting multicarbon products via the parallel integration of electrocatalysis and fermentation.
1. Introduction
The massive consumption of fossil fuels and the resulting large carbon dioxide (CO2) emissions are the main reasons for the energy crisis and climate change.1 To solve this problem, it is preferred to recycle CO2 back into fuels and chemicals via environmentally friendly technologies.2–4 Electrocatalytic CO2 reduction (ECR) and biocatalytic CO2 reduction (BCR) are considered two feasible options.5,6 In electrocatalysis, controlling the selectivity of CO2 conversion products has been difficult.7,8 The most promising electrocatalyst candidate for C–C chemical bond formation is copper.9 Extensive research has been conducted on the design of copper catalysts,10 covering topics including morphology,11,12 sizes,13 lattice planes,14 and doping adjustments.15,16 Currently, ethylene is the most reported multicarbon product that can reach high faradaic efficiency (FE) in strong alkaline electrolytes.17 However, the inevitable by-products HCOOH, CO, and H2 are difficult to further convert or separate, which means energy loss or product separation costs in the electrolysis process. Furthermore, it is still difficult to target a specific hydrocarbon product in the electrolyte with an ECR scheme, such as acetic acid, which is an important chemical feedstock.18
BCR such as microbial electrosynthesis (MES) was developed, in which acetogen produces acetic acid autotrophically by direct electron transfer (DET).19 Microorganisms attach directly to the cathode and electrons are transferred via a short-distance mediator (or physically). The energy efficiency was high, reaching up to 90% for the conversion of electricity to the product.20 However, only a few microbes were capable of utilizing direct electrode electrons efficiently. And the close attachment of microbes to the cathode made it difficult to maximize the potential of the two systems operating independently when trying to optimize the conditions in terms of temperature, pH, electrolyte composition, and other factors.21
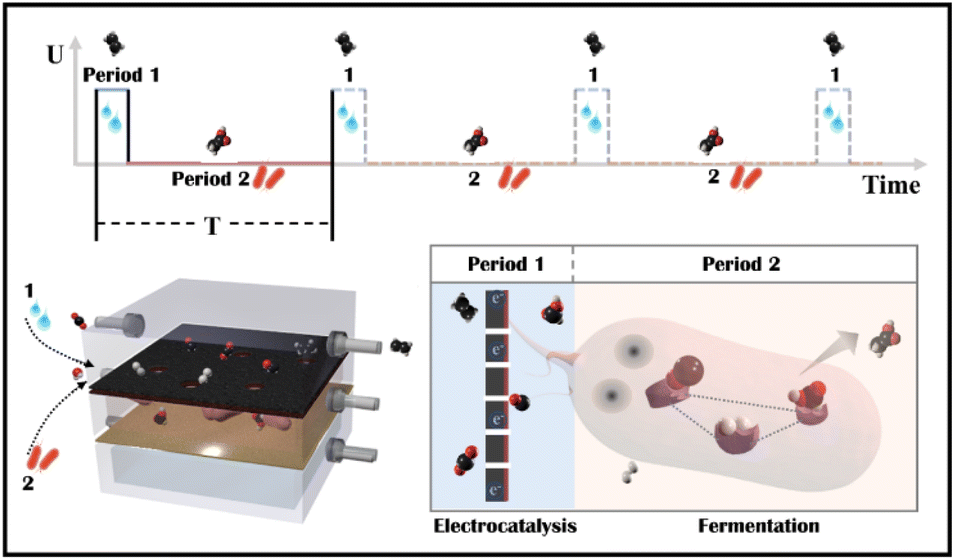
Tandem CO2 reduction strategies have been used in ECR and MES. Tandem ECR employs either a two-component catalyst or a two-step electrochemical tandem reaction to produce multicarbon products. For the former, the electrocatalyst consists of CO generation (Au/Ag/SACs) and C–C coupling components (Cu).22,23 The increased CO coverage on the Cu component promotes the generation of hydrocarbon and oxygenated products. However, similarly constructed bimetallic nanoparticles have been shown to favor different products.24,25 In a two-step tandem ECR, oxygenated products are favored by positioning a CO-producing catalyst (Au or Ag) near a Cu catalyst.26,27 The state-of-the-art two-step tandem catalysts, such as Cu/Fe–N–C, exhibit an enhanced FE to C2+ and an improved C2+ partial current density. However, CO and H2 in the gas phase remain unavoidable, particularly at high currents. The liquid products in a high concentration alkaline electrolyte (1 M KOH) are complicated, including HCOOH, CH3COOH, C2H5OH, and C3H7OH.28 Tandem MES is more selective, where electrodes produce H2, CO, and HCOOH as externally generated electron donors for microbes to use.29–31 Electron donors can be generated at current densities of hundreds of mA cm−2, whereas current densities demonstrated for cathodic microbial electrosynthesis are typically in the range of 1–20 mA cm−2.32 As a result, the overall throughput of tandem MES has been increased from the microbe side. For example, a biocompatible perfluorocarbon nanoemulsion was used to improve the accessibility of gas donors, H2.29,33 But, liquid donors may accumulate in the medium and potentially inactivate microorganisms (the starting value of HCOOH toxicity is 100 mM for acetogen).34 Thus, electrocatalysis is restricted to a low current density (4.2 mA cm−2).31 Alternatively, a CO2 electrolyzer was coupled to an amplified fermentation module (1 L), where CO2 was converted to acetate and ethanol with high carbon selectivity.30 However, continuous high-speed electrocatalytic reactions may result in low faradaic efficiency and waste electrocatalytic products (FEacetate was around 0.03% based on the electrocatalytic current, and the outgoing gas contained 61.9% H2 and 50.8% CO). In general, the electrocatalyst side in tandem MES has not been improved, although multicarbon products could be produced at a high rate on copper electrocatalysts.15,35 Besides, Moorella thermoacetica has been mentioned as an acetogen capable of metabolizing various electron donors (HCOOH,36,37 CO,38,39 H2,40,41 CH3OH, C2H5OH, C3H7OH) to acetate by the Wood–Ljungdahl metabolic pathway (Fig. S1†).42,43 CO and HCOOH have been considered better carboxyl or methyl precursors than CO2, respectively.44,45 However, the removal of ECR byproducts through multiple metabolic processes has not been studied. Herein, an electro-bio tandem reactor was investigated as a novel model that integrates ECR and MES in parallel mode (Scheme 1). A flow cell was used to conduct CO2 electroreduction on a typical nano-Cu electrocatalyst, producing ethylene at a high rate with inevitable by-products such as HCOOH, CO, and H2 (Period 1). HCOOH, CO, and H2 can then be consumed and recycled for selective acetate biosynthesis (Period 2). In comparison to ECR and tandem ECR (liquid products are always complex, and gas and liquid by-products remain unsolved) or MES (the rate is limited by slower fermentation) in a neutral electrolyte, this multi-line tandem reactor is a novel design to preserve the C–C coupling of fast electrocatalysis to obtain target products (ethylene) in the gas phase while satisfying the demand for microbial fermentation through non-target products and accumulating acetate in the liquid phase.
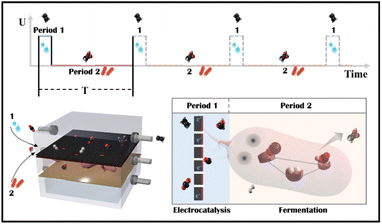 |
| | Scheme 1 Schematic illustration of an electro-bio tandem reactor assembled with copper nanoparticles and microbes. In one cycle time (T), copper was used as the model electrocatalyst to reduce CO2 to multicarbon products (ethylene) in a flow cell (Period 1). Then, Moorella thermoacetica metabolized the concomitant products such as HCOOH, CO, and H2 to acetate through the Wood–Ljungdahl pathway (Period 2). | |
2. Experimental
2.1 Synthesis of copper catalyst
Cu(OH)2 nanorods were prepared using the method previously reported by our group.46 1 g of Cu(NO3)2 was dissolved in 100 mL of distilled water, and 30 mL of NH3·H2O (0.15 M) solution was added to Cu(NO3)2 solution under constant stirring at room temperature. To adjust pH to 9–10, 10 mL of NaOH (1 M) solution was added dropwise (≈2 mL min−1) to the above solution, producing a blue Cu(OH)2 precipitate. After 30 minutes, the blue Cu(OH)2 precipitate was washed several times and harvested by centrifugation to obtain a solid product. It was then dried in a vacuum oven overnight. Nanoporous CuO was prepared by annealing the Cu(OH)2 nanorods under static air atmosphere at 500 °C for 2 h with a heating rate of 32 °C min−1. Finally, nanoporous copper was obtained by the electrochemical reduction of nanoporous CuO on GDL at a constant current density of 10 mA cm−2 in the flow cell as CO2 reduction.
2.2 Synthesis of medium
The defined, undefined, and heterotrophic media were prepared in a chemical fume hood (Table S1†). Deionized water was boiled with an indicator under a constantly purged N2 atmosphere until the color changed from blue to reddish pink. After cooling the solution to room temperature, the atmosphere was changed to CO2 and the medium was allowed to purge and equilibrate at pH 7 after adding the salt mixture, buffer, and ready-made vitamin and mineral solution. The pink color disappeared when the reducing solution was added during continuous gassing and boiling. The medium was dispensed in anaerobic bottles flushed with N2 gas, and butyl rubber and aluminum crimp seals were used to close the bottles. The media were then autoclaved for 15 minutes at 121 °C. Stock solutions of 240 g L−1 glucose, 150 g L−1 yeast extract, 0.5 M potassium formate, and 5.0 wt% reductant (a solution of Cys·HCl or Na2S·9H2O with pH adjusted to 7) were prepared by boiling deionized water for 5 minutes under N2, cooling to room temperature, dissolving the respective solutes, and sealing under N2 in 50 mL serum bottles with butyl stoppers and aluminum crimp seals. They were then autoclaved at 121 °C for 15 minutes. All concentrations of glucose, yeast extract, and reductant are given nominally, calculated from the initial volume of the media.
2.3 Bacterial strains
M. thermoacetica ATCC 39073 was cultured in a heterotrophic medium as an initial inoculum. Late log cultures were cryopreserved at −80 °C with a cryoprotectant of 20% sterilized glycerol until use. M. thermoacetica was cultured in 40 mL of the medium horizontally (without shaking) at 55 °C for three days. The bacterial cells were harvested anaerobically by centrifugation (6000 rpm, 5 min) under a N2 atmosphere and suspended in the defined medium for further use.
2.4 Metabolic experiments for liquid analysis
For CO or H2 group, culture tubes (12.5 mL) were pre-pressurized at room temperature with either 99.95% CO or 99.99% H2 to a final total pressure of atmospheric pressure (∼101 kPa). Culture tubes were pressurized to atmospheric pressure with 100% CO2 at room temperature for the HCOOH group. All tubes were autoclaved for 15 minutes at 121 °C. The undefined medium (4 mL per culture tube) was then injected and 0.5 mL of inoculum was added to specific tubes. Additionally, 0.2 mL of potassium formate solution was added to the undefined media at final concentrations of about 25 mM for the HCOOH group. The samples were cultured horizontally (without shaking) at 55 °C for 10 days. An automatic microplate reader was used to measure the growth in a 96-well plate at 600 nm (200 μL samples). The reference was an undefined medium without bacteria. A Bruker DRX 400 Advance MHz spectrometer was used to measure liquid products at various times. Typically, 0.5 mL of the medium was drawn into a centrifuge tube and centrifuged for 5 minutes at 6000 rpm. Then 200 μL of the supernatant was mixed with 100 μL of water, and 200 μL of sodium 3-(trimethylsilyl)-2,2′,3,3′-tetradeuteropropionate (TMSP-d4, Qingdao Tenglong Microwave Technology Co., Ltd) in D2O (Acros Organics, 99.9 atom % D) was added as the internal standard. The 1D 1H (400 MHz) spectrum was measured with water suppression by a presaturation method.
2.5 Metabolic experiments for gas analysis
Three parallel samples were set up for the HCOOH, CO, or H2 groups (using HCOOH, CO, or H2 as donors) as described above for gas analysis. The headspace gas was monitored by GC before (B) and after (A) culture. As a control, one additional sample (O) was cultured without any bacteria, and gas chromatography (GC) was used to identify the headspace gas after culture. The gas analysis method has been illustrated with simple diagrams (Fig. S7†). The needle was inserted into the anaerobic stopper, and a peristaltic pump was used to push the gas into the quantitative loop of the GC. The volume of headspace gas was approximately 8 mL. Due to the limited sample volume, the gas was allowed to return from the outlet. The peristaltic pump was operated for 2 minutes at a speed of 10 sccm to achieve an approximate concentration equilibrium balance in the pipeline. Then the gas was analyzed by GC. Given the errors introduced by the test method, we normalized the concentration obtained by the initial test filled into a tube as 100% and then calculated the proportion of the initial concentration consumed by the bacteria.
2.6 Characterization of the copper catalyst
The morphology and microstructure of the copper catalyst were characterized by SEM (JEOL, JSM-7800F) and transmission electron microscopy (TEM) (JEM 2100 200 kV), while high-resolution TEM was performed using a JEM 2800 TEM. The SEM images of the cathode were taken by directly sticking the gas diffusion layer (GDL) (Fuel Cell Store) on the sample stage. The X-ray diffraction (XRD) spectra were collected using XRD-6000 X-ray diffractometer (Shimadzu, Japan). X-ray photoelectron spectroscopy (XPS) data were obtained using a PHI 5000 Versa Probe detector for X-ray photoelectron spectroscopy. The solution pH was measured with a Thermo Scientific Orion VERSA STAR pH benchtop meter. Raman spectra were obtained using a confocal Raman spectrometer (Renishaw inVia-Reflex) under a laser wavelength of 633 nm. The surface area of the catalysts was tested using a nitrogen adsorption analyzer (ASAP 2020, Micromeritics) under the multipoint Brunauer–Emmett–Teller (BET) method, and the pore size analysis was performed by the Barrett–Joyner–Halenda method.
2.7 Characterization of M. thermoacetica
M. thermoacetica samples were fixed overnight in a solution of 5 vol% glutaraldehyde in 0.01 M phosphate-buffered saline (PBS) to prepare them for electron microscopy. Increasing ethanol concentrations in deionized water (0%, 25%, 50%, 75%, 90%, 100%, ∼10 mL with 10 minutes each) were used to wash the fixed samples. Before mounting on the scanning electron microscope (SEM) and field emission SEM imaging at 5 kV, approximately 5 nm of gold was sputtered onto the samples (JEOL, JSM-7800F).
2.8 Assembling the electro-bio tandem reactor
A flow cell was connected to a bacterial culture bottle, as shown in the diagram (Fig. S15†). In brief, typical electrolysis was performed as the anolyte and catholyte flowed in and out. GC was carried out to analyze the gas phase. After GC analysis confirmed that the electrocatalyst's performance was stable, the circuit was adjusted with a two- or three-way valve to allow fresh catholyte to circulate through the flow cell (the liquid volume distributed in the line was 10 mL). Electrolysis was carried out for 10 minutes. The gas line was then shut down, leaving gaseous products in the gas phase and liquid products in the catholyte. (The reason for 10 minutes is that the gas flow rate was set to 10 sccm, and 10 minutes is enough time to fill the flow pool's gas space, including the connected pipes. And the formate accumulated product was sufficient to support bacterial metabolism.) After the electrolysis was completed, the circuit was adjusted with a two- or three-way valve to introduce the defined medium with bacteria via a peristaltic pump. The peristaltic rate was kept constant at 1 mL min−1 until the gas or liquid was analyzed again by GC or nuclear magnetic resonance (NMR).
3. Results and discussion
3.1 Mechanism studies
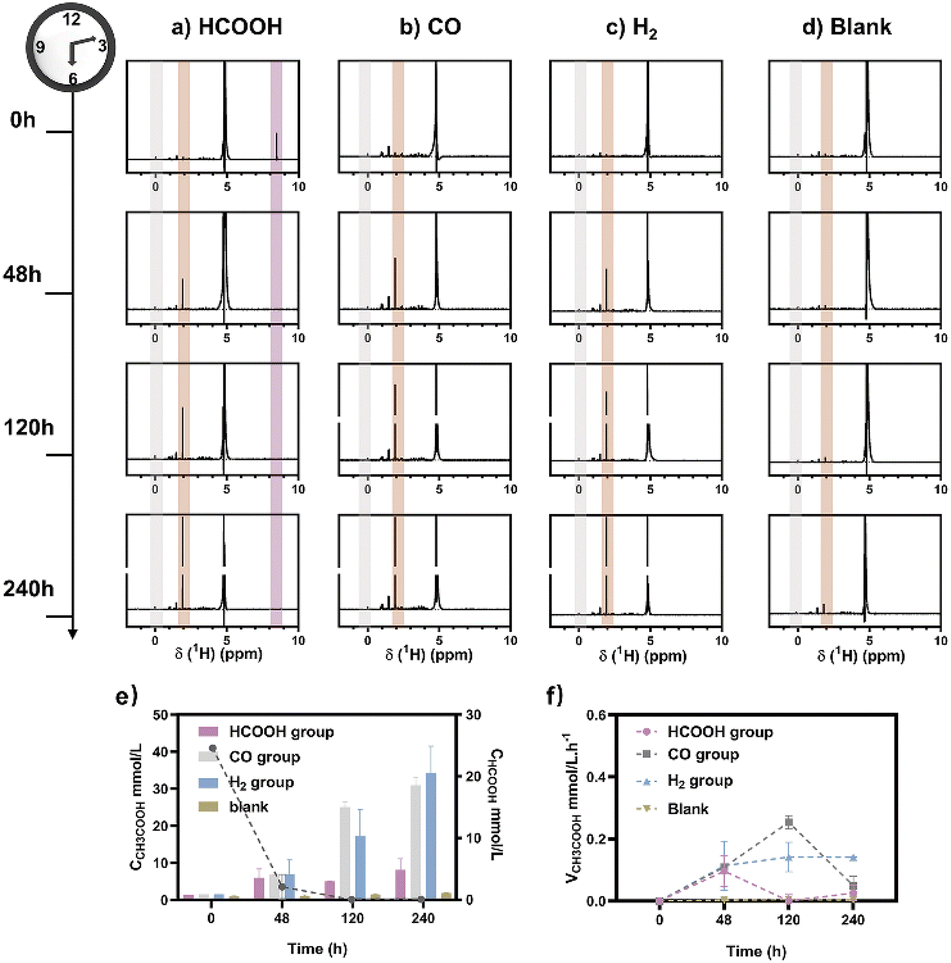
M. thermoacetica was cultured in our laboratory with a rod-like morphology and a length of 2.8 μm (Fig. S2†). The 16sRNA sequencing results agreed well with the literature and suggested a pure culture.47 The culture curve was acquired in a heterotrophic medium (Table S1†) with glucose as the carbon source (Fig. S3†). After 48 hours of proliferation, the OD600 increased to about 0.4. To investigate the metabolism of HCOOH, CO, and H2, we performed a series of control experiments in which HCOOH, CO, and H2 were provided separately in anaerobic tubes (Fig. 1). The undefined medium (Table S1†) contained carbonate as the auxiliary carbon source and one of the buffer pairs. HCOOH was added in the form of potassium formate to the HCOOH group. Here we do not distinguish between formic acid and formate, and they are collectively referred to as HCOOH. The concentration of HCOOH was easily adjustable; the chosen concentration of 25 mM was higher than the concentration produced by electrocatalysis but less than the toxic concentration. The maximum concentration of CO/H2 that electrolysis can produce is pure gas, but CO and H2 are insoluble and have approximate Henry's constants. Thus, their concentrations in the medium are comparable but significantly lower than that of HCOOH. In this case, CO or H2 was supplied as pure gas to be filled into the CO or H2 group's tube. In the experiments, we set the initial OD600 at about 0.5 to allow for enough bacterial quantity for fermentation. Despite inorganic carbon and electron donors providing insufficient support for cell growth,40 the final OD600 levels remained constant at 0.4 after 10 days (Fig. S4†). The majority of the energy was assumed to be used for metabolism rather than biomass synthesis. After two rounds of anaerobic washing of microorganisms, the initial concentration of acetate was considered to be low. The appearance of a small residual NMR peak was attributed to the yeast extract in the undefined medium (Fig. S5†). As measured by quantitative proton nuclear magnetic resonance (1H-qNMR) spectroscopy, acetate concentrations were all increased in the presence of HCOOH, CO, or H2 (Fig. 1a, b, and c). The final average acetate concentrations in the first 48 hours were 6.0, 6.8, and 7.0 mmol L−1 for the HCOOH, CO, and H2 groups, respectively (Fig. 1e). After 10 days, the acetate concentrations reached 6.8, 29.4, and 32.6 mmol L−1 for the HCOOH, CO, and H2 groups, respectively. But when HCOOH, CO, or H2 was removed, the concentration of acetate remained almost constant (Fig. 1d and e). The increasing concentration was only 0.1 mmol L−1 after 48 hours and 0.9 mmol L−1 after 10 days, indicating a low background contribution even with yeast extract as a potential carbon source. The acetate production rate for the three groups was not constant throughout the metabolic process (Fig. 1f). They all displayed a volcanic trend, from the initial adaptation period to the peak period and the delay period. The accumulation of acetate may cause rate reduction in the latter period.
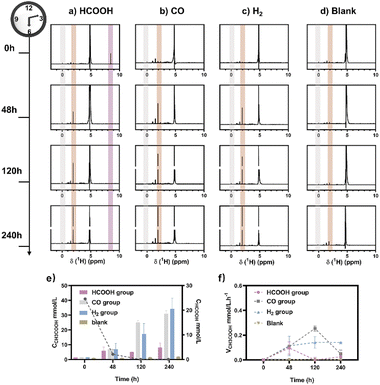 |
| | Fig. 1 Metabolic experiments were monitored using1H NMR spectra. CO2/CO32− was provided as a carbon source, and (a) HCOOH, (b) CO, and (c) H2 were provided as electron donors during fermentation. (d) No electron carriers were provided. The internal standard was sodium 3-(trimethylsilyl)-2,2′,3,3′-tetradeuteropropionate (TMSP, gray region). With the decrease of HCOOH (purple region), an increase in acetic acid was observed (brown region). (e) The calculated content of HCOOH (line chart, right axis) and acetate (histogram, left axis) was calculated for the HCOOH/CO/H2 group. (f) The production rate of acetate changed over time. | |
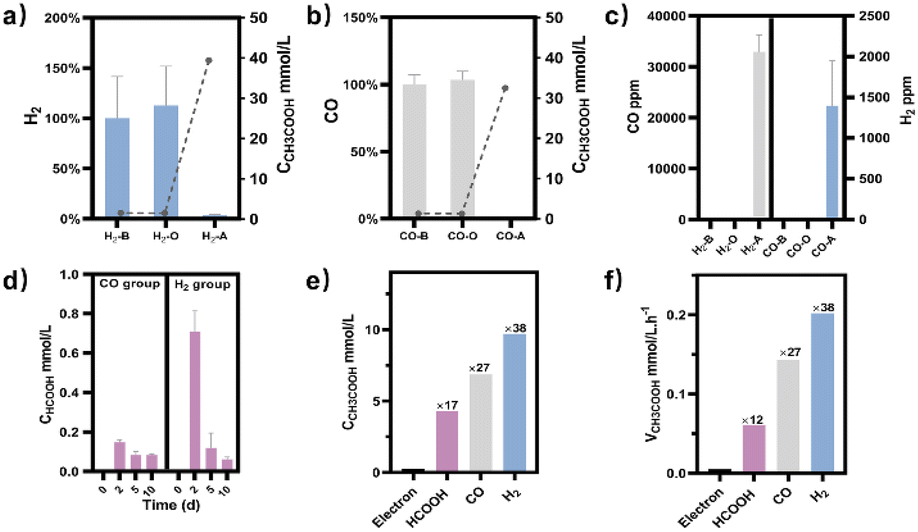
Meanwhile, NMR or GC analysis was conducted to track the variation in electron donor concentration (HCOOH, CO, and H2). After 10 days, all groups showed a decrease in HCOOH, CO, and H2 concentrations (Fig. 1e, 2a and b). HCOOH almost disappeared in the NMR results, with a final concentration of 0.07 mmol L−1. The initial CO and H2 concentrations in the tube were analyzed by GC and normalized to 100%, respectively. Ten days later, the content of CO and H2 was reduced to 0.03% and 3.6% on average. The results implied that 99.7%, 100.0%, and 96.4% of HCOOH, CO, or H2 had been fully metabolized after 10 days. In addition, for the HCOOH group, the conservation of electrons was verified. The electrons supplied by HCOOH were calculated and found to be 0.17 mmol, while the concentration of electrons required for the produced acetate was 0.14 mmol. The electron recovery efficiency for the HCOOH group was 80.2%, which was close to the reported value in the DET process.48 The extra electrons should be used to make biocarbon, which sustains the bacteria. Moreover, considering the possible contribution of yeast extract mentioned above, electron conservation was further confirmed without yeast extract (Fig. S6†). Product stoichiometries for CO and H2 have been reported.40 The pressure of gas in the anaerobic tube was not monitored in this work, and the concentration would be slightly diluted by the carrier gas at each turn of the six-way valve, limited by the testing mode (Fig. S7†). Therefore, electron conservation was not calculated for the CO or H2 group. Besides, it was worth noting that H2 (∼32![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 956 ppm) or CO (∼1397 ppm) was produced in the CO or H2 group, respectively (Fig. 2c). A trace amount of HCOOH was also found in the liquid, with a tendency to disappear over time (Fig. 2d). Peak contents were observed in the first 48 hours, 0.2 mmol L−1 for the CO group and 0.7 mmol L−1 for the H2 group. It was suspected that when one electron carrier was provided, a small number of other donors were generated to satisfy the biosynthesis requirements, relevant to the reversibility of the metabolic process.42,49 Then, the ability of M. thermoacetica to produce acetic acid autotrophically was tested by applying a negative cathode voltage (−0.4 V vs. RHE) for about 48 hours (Fig. S8†). The acetate production rate (0.0052 mM h−1) was higher than the reported value (0.001 mM h−1), but the calculated FE of 63.4% was slightly lower.48 Notably, the use of HCOOH, CO, and H2 as donors increased the final acetate concentration (Fig. 2e) and production rate (Fig. 2f), demonstrating its superiority under the same conditions (ambient temperature and pressure).
956 ppm) or CO (∼1397 ppm) was produced in the CO or H2 group, respectively (Fig. 2c). A trace amount of HCOOH was also found in the liquid, with a tendency to disappear over time (Fig. 2d). Peak contents were observed in the first 48 hours, 0.2 mmol L−1 for the CO group and 0.7 mmol L−1 for the H2 group. It was suspected that when one electron carrier was provided, a small number of other donors were generated to satisfy the biosynthesis requirements, relevant to the reversibility of the metabolic process.42,49 Then, the ability of M. thermoacetica to produce acetic acid autotrophically was tested by applying a negative cathode voltage (−0.4 V vs. RHE) for about 48 hours (Fig. S8†). The acetate production rate (0.0052 mM h−1) was higher than the reported value (0.001 mM h−1), but the calculated FE of 63.4% was slightly lower.48 Notably, the use of HCOOH, CO, and H2 as donors increased the final acetate concentration (Fig. 2e) and production rate (Fig. 2f), demonstrating its superiority under the same conditions (ambient temperature and pressure).
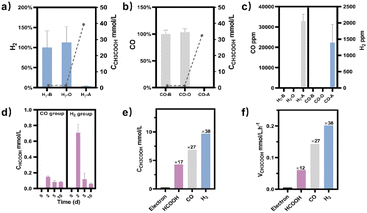 |
| | Fig. 2 (a) For the H2 group, the gas phase composition in the aerobic tube was analyzed before (H2-B), after the metabolic process (H2-A), or under the experiment setup without bacteria (H2-O). Acetate (line chart, right axis) was produced with the consumption of H2 (histogram, left axis). (b) For the CO group, the gas phase composition in the aerobic tube was analyzed before (CO-B), after the 10-day metabolic process (CO-A), or under the experiment setup without bacteria (CO-O). Acetate (line chart, right axis) was produced with the consumption of CO (histogram, left axis). (c) CO (left axis) or H2 (right axis) was produced during the metabolic process for the H2 or CO group, respectively. (d) HCOOH was produced for the CO and H2 groups during the metabolic process. (e) Comparison of the produced acetate concentration by applying a negative voltage (electron group) or by providing HCOOH, CO, and H2, respectively (ambient temperature and pressure). The numbers represent the multiple compared to the electron group. (f) Comparison of the acetate production rate by applying a negative voltage (electron group) or by providing HCOOH, CO, and H2, respectively (ambient temperature and pressure). The numbers represent the multiple compared to the electron group. | |
3.2 Electro-bio tandem reactor
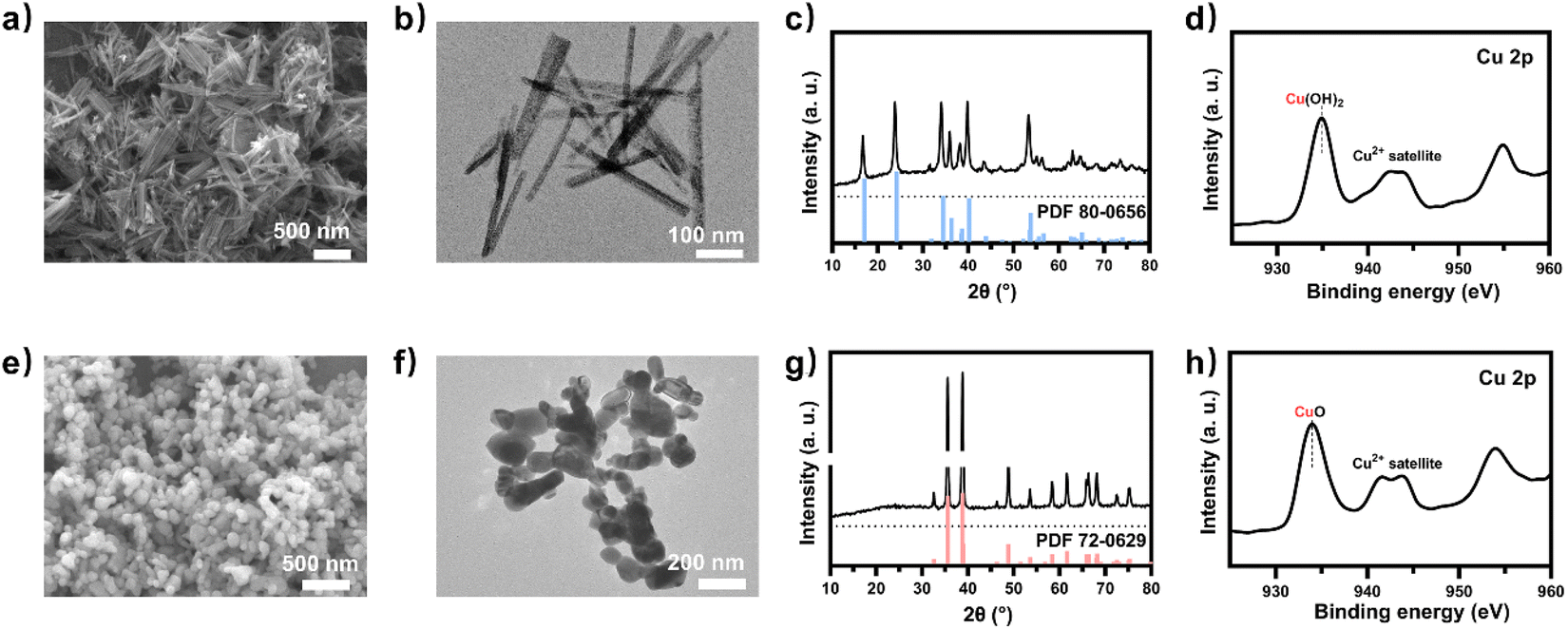
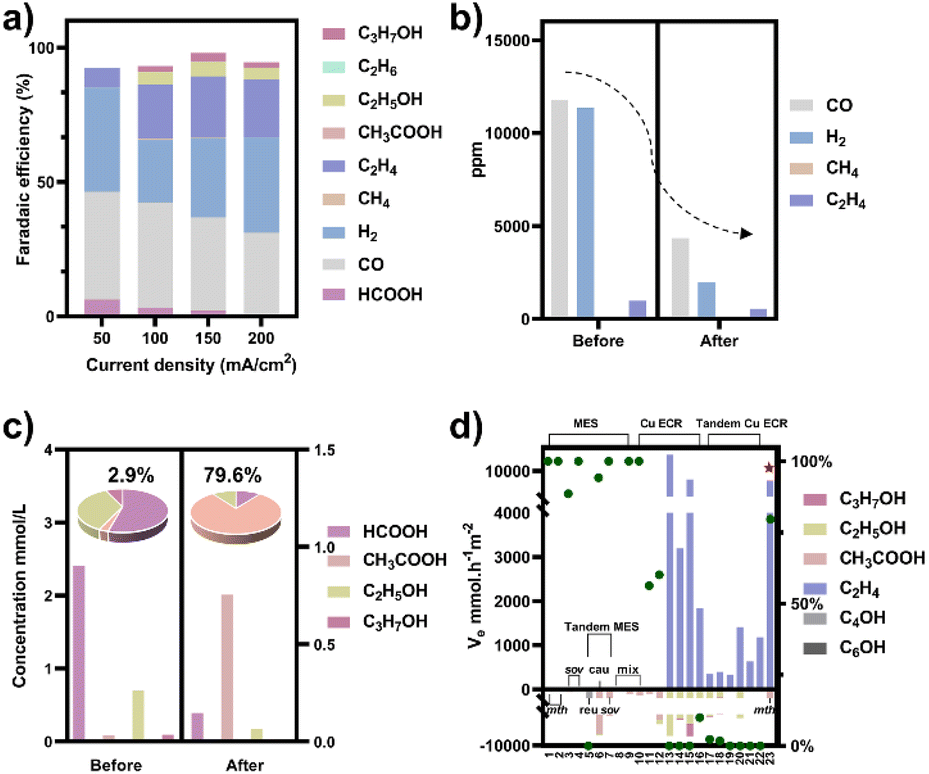
To obtain a copper electrocatalyst functioning in the integrated electro-bio tandem reactor, typical Cu(OH)2 precursors were prepared (Fig. S9†),46 which showed a rod structure (Fig. 3a and b), with a main length of 500 nm (Fig. S10†). According to the X-ray diffraction (XRD) (Fig. 3c) results and X-ray photoelectron spectroscopy (XPS) results (Fig. 3d), the crystal phase and element valence coincided with that of Cu(OH)2. Then, via a calcination process, a typical oxide-derived copper electrocatalyst was obtained. The morphology was globular in SEM and TEM images, with a size distribution of 100–150 nm (Fig. 3e, f and S11†). From the XRD and XPS spectra, the crystalline phase was primarily CuO (Fig. 3g and h). The typical high-resolution transmission electron microscopy (HRTEM) image showed widely spread lattice fringes corresponding to CuO (111) throughout the sample, indicating a high population of the CuO (111) facet (Fig. S12†). The N2 adsorption–desorption measurement revealed a Brunauer–Emmett–Teller surface area of 6.67 m2 g−1. The average pore diameter was 22.98 nm (Fig. S13†). Additionally, the CO2 adsorption capacity was tested to be 1.22 cm3 g−1 (Fig. S14†). The CuO catalyst was electroreduced in situ on the gas diffusion layer (GDL), and the morphology displayed nanoparticles adhering to each other (Fig. S15†). The distribution of Cu and O was uniform in the energy-dispersive X-ray spectroscopy (EDS) elemental maps. The typical HRTEM image showed lattice fringes corresponding to Cu2O(111) and Cu(111), with the particles inside being metallic Cu, whereas the surface was dominated by Cu2O (Fig. S16†). The characteristic peak of Cu2O was also observed in the Raman spectrum (Fig. S17†).50 These observations were in good agreement with the XRD analysis (Fig. S18†), where diffraction peaks for both metallic Cu and Cu2O phases were identified. According to the XPS spectra, the valence states of Cu was CuI and Cu0 (Fig. S19†). We individually evaluated the CO2 electroreduction performance of the nano-copper catalyst in a flow cell. The catholyte choice balanced electrocatalysis and fermentation since some substances (reductant, vitamins) and the required pH (near neutral) that are vital for microbial growth or activity can hamper the CO2RR.33 Thus, neutral 0.1 M KCl was chosen as the model catholyte. The LSV curves with CO2 or N2 in the gas phase were obtained in a flow cell (Fig. S20†). The current density showed a much larger current density in CO2 than in N2, indicating the catalytic activity for CO2 reduction. The FE of ethylene reached 22.5% at the current density of 100 mA cm−2 (Fig. 4a). The stability of CO2 electrolysis using the nano-Cu catalyst was investigated by conducting continuous CO2 electrolysis under a constant current mode at 100 mA cm−2 (Fig. S21†). The results showed a stable potential profile over 2 hours. The faradaic efficiencies of products (C2H4, CO, H2, HCOOH) remained almost identical throughout the two-hour electrolysis period. Given the consumption of protons during electrolysis, pH was measured after electrolysis (Fig. S22†). To obtain mechanistic insights, the charge transfer resistance of the Cu electrocatalyst was investigated. A small charge transfer resistance demonstrated fast charge transfer from the catalyst surface to the adsorbed CO2 molecule in the electrolyte, forming the *CO2 intermediate (Fig. S23†).51 Additionally, a Tafel slope of 193 mV dec−1 was observed in 0.1 M KCl (pH = 6.29) for ethylene formation, implying the effect of pH and a single irreversible electron transfer at the rate-determining step (Fig. S24†).52,53 Considering the pH dependence of C2 product formation, CO dimerization was suggested to be the rate-limiting step.54,55 The presence of adsorbed *CO on Cu was demonstrated by the appearance of Raman peaks located at 357 and 2120 cm−1, which correspond to Cu–CO stretching and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching, respectively (Fig. S25†).56 The partial density of states (PDOSs) was calculated to determine the d-band centers of the electrocatalyst (Fig. S26†). Cu (111) has a lower d-band position (−2.69 eV relative to the Fermi level), leading to weaker binding between the intermediate and the catalyst surface due to the occupancy of antibonding states, while the Cu2O (111) plane (−2.29 eV) should have the highest binding with CO.57 The flow cell was then used to construct an electro-bio tandem reactor, as described in detail in the ESI (Fig. S27 and S28).† Cu nanoparticle-based electrocatalysis and microbial fermentation can occur sequentially in the same flow cell due to the separated and switchable compartment. The gas composition was analyzed by GC after electrolysis for 10 minutes at a current density of 100 mA cm−2. The calculated FE of ethylene was 21.2%. Concurrently, HCOOH (2.4 mmol L−1), CO (11777 ppm), and H2 (11362 ppm) accumulated in the liquid or gas phase. Following that, the microbe-containing defined medium (without yeast extract or any other carbon source) was pumped into the electrolytic cell to clear and recycle the non-target products. Given the superiority of HCOOH, CO, and H2 as donors, as well as the potential effect of the induced biofilm on the electrocatalyst, no additional voltage was applied. In the gas phase after 12 hours, 63.1% CO and 82.8% H2 had disappeared (Fig. 4b). The tandem reaction was stopped although CO and H2 could be further transformed based on the metabolic experiment results. Simultaneously, the liquid was collected for further NMR analysis before and after the tandem experiment (Fig. S29†). A decrease in HCOOH concentration and an increase in acetate concentration were observed (Fig. 4c). The calculated FE of acetate reached up to 43.7% based on the electrons supplied in the electrocatalytic process. The faradaic selectivity of acetate in the catholyte increased to 79.6% (Fig. 4c inset), which was the highest for Cu-based ECR (Table S4†) (The ESI† describes the calculation method in detail). HCOOH, CO, and H2, containing lost carbon and electrons, were recycled to acetate with an electronic conversion rate of 628.9 mmol h−1 m−2, ranking first for M. thermoacetica and comparable to S. ovata or mixed culture in MES (the ESI† describes the calculation method in detail, including current density and faradaic efficiency parameters).
O stretching, respectively (Fig. S25†).56 The partial density of states (PDOSs) was calculated to determine the d-band centers of the electrocatalyst (Fig. S26†). Cu (111) has a lower d-band position (−2.69 eV relative to the Fermi level), leading to weaker binding between the intermediate and the catalyst surface due to the occupancy of antibonding states, while the Cu2O (111) plane (−2.29 eV) should have the highest binding with CO.57 The flow cell was then used to construct an electro-bio tandem reactor, as described in detail in the ESI (Fig. S27 and S28).† Cu nanoparticle-based electrocatalysis and microbial fermentation can occur sequentially in the same flow cell due to the separated and switchable compartment. The gas composition was analyzed by GC after electrolysis for 10 minutes at a current density of 100 mA cm−2. The calculated FE of ethylene was 21.2%. Concurrently, HCOOH (2.4 mmol L−1), CO (11777 ppm), and H2 (11362 ppm) accumulated in the liquid or gas phase. Following that, the microbe-containing defined medium (without yeast extract or any other carbon source) was pumped into the electrolytic cell to clear and recycle the non-target products. Given the superiority of HCOOH, CO, and H2 as donors, as well as the potential effect of the induced biofilm on the electrocatalyst, no additional voltage was applied. In the gas phase after 12 hours, 63.1% CO and 82.8% H2 had disappeared (Fig. 4b). The tandem reaction was stopped although CO and H2 could be further transformed based on the metabolic experiment results. Simultaneously, the liquid was collected for further NMR analysis before and after the tandem experiment (Fig. S29†). A decrease in HCOOH concentration and an increase in acetate concentration were observed (Fig. 4c). The calculated FE of acetate reached up to 43.7% based on the electrons supplied in the electrocatalytic process. The faradaic selectivity of acetate in the catholyte increased to 79.6% (Fig. 4c inset), which was the highest for Cu-based ECR (Table S4†) (The ESI† describes the calculation method in detail). HCOOH, CO, and H2, containing lost carbon and electrons, were recycled to acetate with an electronic conversion rate of 628.9 mmol h−1 m−2, ranking first for M. thermoacetica and comparable to S. ovata or mixed culture in MES (the ESI† describes the calculation method in detail, including current density and faradaic efficiency parameters).
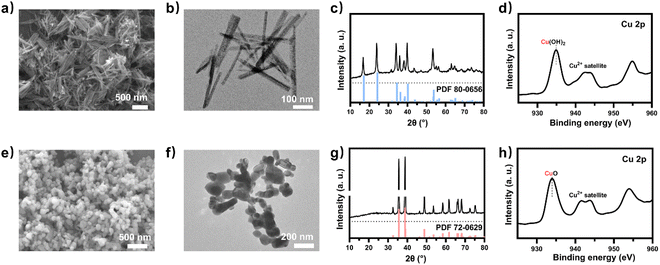 |
| | Fig. 3 (a) SEM image and (b) TEM image of Cu(OH)2. (c) XRD revealed peaks assignable to the pattern of Cu(OH)2 (reference peaks from JCPDS Data Card No. 80-0656 given as drop lines). (d) XPS spectra of Cu 2p for Cu(OH)2. (e) SEM image and (f) TEM image of the model electrocatalyst CuO. (g) XRD revealed peaks assignable to the pattern of CuO (reference peaks from JCPDS Data Card No. 72-0629 given as drop lines). (h) XPS spectra of Cu 2p for CuO. | |
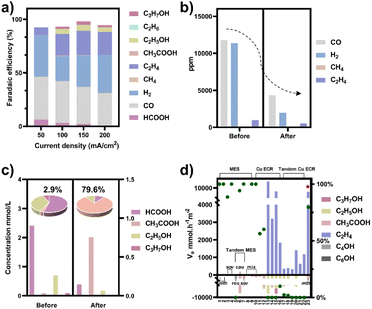 |
| | Fig. 4 (a) CO2 electroreduction performance of the nano-Cu catalyst in a flow cell when the catholyte was 0.1 M KCl. (b) The composition of gas-phase products before and after the tandem experiment. (c) The concentration of liquid-phase products before and after the tandem experiment (HCOOH (left axis), C2+ products (right axis)). The experiment under each condition was repeated three times and the average values were used. The faradaic selectivity of acetate in the total liquid products (fan chart, inset). (d) The rate of electron conversion (ve) from CO2 to C2 products in the neutral electrolyte (pH around 7) (histogram, left axis) and faradaic selectivity of acetate in liquid products (green dots, right axis) for microbial electrosynthesis (MES), Cu-based electrochemical CO2 reduction (ECR), and tandem ECR. Pentagram: this work; number: the serial number of the reference, which has been organized in Tables S4 and S5.† Bacterial abbreviations: Moorella thermoacetica (mth), Sporomusa ovata (sov), Ralstonia eutropha (reu), Clostridium autoethanogenum (cau), mixed culture (mix). | |
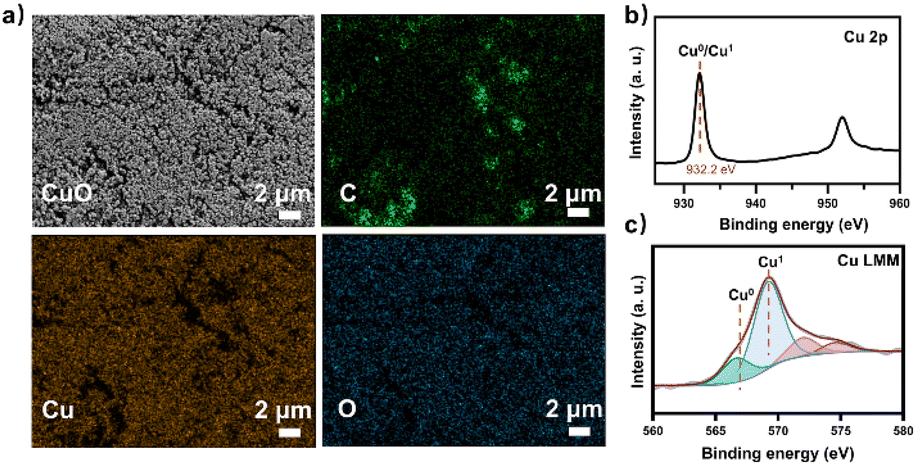
At the same time, the electronic conversion rate of ethylene was 7912.5 mmol h−1 m−2 (Table S5†). The total electron conversion rate to ethylene and acetate (8113.8 mmol h−1 m−2) was the highest for MES in the neutral electrolyte. It should be noted that the copper catalyst currently used could be substituted for more efficient ones (such as the ones in Fig. 4d Cu ECR). On the one hand, the higher the yield of multicarbon products in electrocatalysis, the lower the proportion of non-target products, and a better total performance was expected by balancing with the fermentation process. On the other hand, other microbes could be used to selectively broaden the range of liquid products. Subsequently, 13C isotope labeling was used to track the carbon source (Fig. S30†). The satellite peaks in the spectrum were due to the high abundance of 13C in the acetate, consistent with the literature.58 When the tandem experiment was finished, the cathode was used to characterize the surface state. The clean surface in SEM images demonstrated no visible biofilm formation on the electrodes. The elements Cu and O were evenly distributed on the electrocatalyst (Fig. 5a). Element C came from the exposed carbon substrates. Cu XPS (Fig. 5b) and LMM auger peaks (Fig. 5c) were obtained for surface state analysis, and both Cu0 and CuI states were present. Furthermore, due to the potential toxicity of copper ions,59 bacterial activity was estimated after the experiment. The SEM images revealed a complete bacterial structure without holes.60 Additionally, copper ions, which may accumulate on the surface of bacteria, were not detected in the EDS maps (Fig. S31†). We also tested bacterial activity by immediately inoculating them into a fresh heterotrophic medium. There was obvious growth after 48 hours, with an OD600 of 0.46 (Fig. S32†). This demonstrates high bacterial activity after the experiments and encourages long-term use.
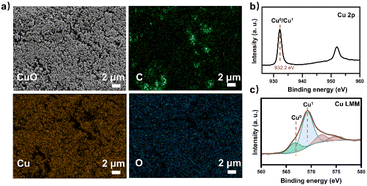 |
| | Fig. 5 The GDL cathode loaded with the nano-Cu catalyst was analyzed after the tandem experiment. (a) SEM image and EDX maps of the catalyst on GDL showed uniform Cu and O distribution. All scale bars are 2 μm. (b) Cu 2p XPS spectra and (c) Cu LMM Auger spectra of the nano-Cu catalyst. | |
4. Conclusions
In this proof-of-concept work, an electro-bio tandem reactor was successfully assembled with copper nanoparticles and microbial biocatalysts, where ethylene was the target gas product, and byproducts such as HCOOH, CO, and H2 were consumed and recycled to acetate in liquid with the highest faradaic selectivity of 79.6% for Cu-based ECR. The C–C bond was formed in tandem during electrolysis and biological metabolic processes, resulting in the highest electron conversion rate to ethylene and acetate in MES. The copper electrocatalyst was prepared in water without the use of any organic solvents. Simultaneously, a dilute neutral electrolyte was used, and the parallel strategy facilitated the acceleration of the entire process and separation to obtain the desired multicarbon products. Thus, such a parallel tandem mode is considered green and selective, and more efficient electrocatalysts and microbes could be configured to proceed with greenhouse gas conversion and utilization.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
J. L. thanks for the support from program B for the Outstanding Ph.D. candidate of Nanjing University. W. Z. would like to acknowledge the support from the National Natural Science Foundation of China (22176086), the Natural Science Foundation of Jiangsu Province (BK20210189), the State Key Laboratory of Pollution Control and Resource Reuse (PCRR-ZZ-202106), the Fundamental Research Funds for the Central Universities (021114380183, 021114380189, 021114380199), the Research Funds from Frontiers Science Center for Critical Earth Material Cycling of Nanjing University, and Research Funds for Jiangsu Distinguished Professor. Y. Z. would like to acknowledge the support from the Research startup fund of NJUPT and the Natural Science Foundation of Jiangsu Province (SBK2022044384).
References
- J. Fu, P. Li, Y. Lin, H. Du, H. Liu, W. Zhu and H. Ren, Eco-Environ. Health, 2022, 1, 259–279 CrossRef
 .
.
- X. Wang, P. Li, Y. Cao, W. Hong, Z. Geng, Z. An, H. Wang, B. Sun, W. Zhu and Y. Zhou, Chem. J. Chin. Univ., 2022, 43, 16 Search PubMed .
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, eaav3506 CrossRef CAS PubMed .
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 CrossRef CAS PubMed .
- J. Liu, Y. Cai, R. Song, S. Ding, Z. Lyu, Y.-C. Chang, H. Tian, X. Zhang, D. Du, W. Zhu, Y. Zhou and Y. Lin, Mater. Today, 2021, 48, 95–114 CrossRef CAS .
- X. Zuo, K. Chang, J. Zhao, Z. Xie, H. Tang, B. Li and Z. Chang, J. Mater. Chem. A, 2016, 4, 51–58 RSC .
- R. Song, W. Zhu, J. Fu, Y. Chen, L. Liu, J. Zhang, Y. Lin and J. Zhu, Adv. Mater., 2019, 32, 1903796 CrossRef PubMed .
- J. Li, S. U. Abbas, H. Wang, Z. Zhang and W. Hu, Nano-Micro Lett., 2021, 13, 216 CrossRef CAS PubMed .
- J. Liu, J. Fu, Y. Zhou, W. Zhu, L.-P. Jiang and Y. Lin, Nano Lett., 2020, 20, 4823–4828 CrossRef CAS PubMed .
- X. Li, J. Wang, X. Lv, Y. Yang, Y. Xu, Q. Liu and H. B. Wu, Nano-Micro Lett., 2022, 14, 134 CrossRef CAS PubMed .
- L.-X. Liu, X. Li, Y. Cai, H. Du, F. Liu, J.-R. Zhang, J. Fu and W. Zhu, Nanoscale, 2022, 14, 13679–13688 RSC .
- P. De Luna, R. Quintero-Bermudez, C.-T. Dinh, M. B. Ross, O. S. Bushuyev, P. Todorović, T. Regier, S. O. Kelley, P. Yang and E. H. Sargent, Nat. Catal., 2018, 1, 103–110 CrossRef CAS .
- A. Loiudice, P. Lobaccaro, E. A. Kamali, T. Thao, B. H. Huang, J. W. Ager and R. Buonsanti, Angew. Chem., Int. Ed., 2016, 55, 5789–5792 CrossRef CAS PubMed .
- Y. Chen, Z. Fan, J. Wang, C. Ling, W. Niu, Z. Huang, G. Liu, B. Chen, Z. Lai, X. Liu, B. Li, Y. Zong, L. Gu, J. Wang, X. Wang and H. Zhang, J. Am. Chem. Soc., 2020, 142, 12760–12766 CrossRef CAS PubMed .
- Y. Zhou, F. Che, M. Liu, C. Zou, Z. Liang, P. De Luna, H. Yuan, J. Li, Z. Wang, H. Xie, H. Li, P. Chen, E. Bladt, R. Quintero-Bermudez, T.-K. Sham, S. Bals, J. Hofkens, D. Sinton, G. Chen and E. H. Sargent, Nat. Chem., 2018, 10, 974–980 CrossRef CAS PubMed .
- Z. Q. Liang, T. T. Zhuang, A. Seifitokaldani, J. Li, C. W. Huang, C. S. Tan, Y. Li, P. De Luna, C. T. Dinh, Y. Hu, Q. Xiao, P. L. Hsieh, Y. Wang, F. Li, R. Quintero-Bermudez, Y. Zhou, P. Chen, Y. Pang, S. C. Lo, L. J. Chen, H. Tan, Z. Xu, S. Zhao, D. Sinton and E. H. Sargent, Nat. Commun., 2018, 9, 3828 CrossRef PubMed .
- C.-T. Dinh, T. Burdyny, M. G. Kibria, A. Seifitokaldani, C. M. Gabardo, F. P. García de Arquer, A. Kiani, J. P. Edwards, P. De Luna, O. S. Bushuyev, C. Zou, R. Quintero-Bermudez, Y. Pang, D. Sinton and E. H. Sargent, Science, 2018, 360, 783–787 CrossRef CAS PubMed .
- Q. Zhu, X. Sun, D. Yang, J. Ma, X. Kang, L. Zheng, J. Zhang, Z. Wu and B. Han, Nat. Commun., 2019, 10, 3851 CrossRef PubMed .
- D. R. Lovley and K. P. Nevin, Curr. Opin. Biotechnol., 2013, 24, 385–390 CrossRef CAS PubMed .
- P.-L. Tremblay and T. Zhang, Front. Microbiol., 2015, 6, 201 Search PubMed .
- N. J. Claassens, C. A. R. Cotton, D. Kopljar and A. Bar-Even, Nat. Catal., 2019, 2, 437–447 CrossRef CAS .
- C. G. Morales-Guio, E. R. Cave, S. A. Nitopi, J. T. Feaster, L. Wang, K. P. Kuhl, A. Jackson, N. C. Johnson, D. N. Abram, T. Hatsukade, C. Hahn and T. F. Jaramillo, Nat. Catal., 2018, 1, 764–771 CrossRef CAS .
- H. Zhang, X. Chang, J. G. Chen, W. A. Goddard, B. Xu, M.-J. Cheng and Q. Lu, Nat. Commun., 2019, 10, 3340 CrossRef PubMed .
- D. Chen, L. Zhang, J. Du, H. Wang, J. Guo, J. Zhan, F. Li and F. Yu, Angew. Chem., Int. Ed., 2021, 60, 24022–24027 CrossRef CAS PubMed .
- D. Meng, M. Zhang, D. Si, M. Mao, Y. Hou, Y. Huang and R. Cao, Angew. Chem., 2021, 133, 25689–25696 CrossRef .
- F. Urbain, V. Smirnov, J.-P. Becker, A. Lambertz, F. Yang, J. Ziegler, B. Kaiser, W. Jaegermann, U. Rau and F. Finger, Energy Environ. Sci., 2016, 9, 145–154 RSC .
- Gurudayal, D. Perone, S. Malani, Y. Lum, S. Haussener and J. W. Ager, ACS Appl. Energy Mater., 2019, 2, 4551–4559 CrossRef CAS .
- T. Zhang, J. C. Bui, Z. Li, A. T. Bell, A. Z. Weber and J. Wu, Nat. Catal., 2022, 5, 202–211 CrossRef CAS .
- C. Liu, B. C. Colón, M. Ziesack, P. A. Silver and D. G. Nocera, Science, 2016, 352, 1210–1213 CrossRef CAS PubMed .
- T. Haas, R. Krause, R. Weber, M. Demler and G. Schmid, Nat. Catal., 2018, 1, 32–39 CrossRef CAS .
- H. Li, P. H. Opgenorth, D. G. Wernick, S. Rogers, T.-Y. Wu, W. Higashide, P. Malati, Y.-X. Huo, K. M. Cho and J. C. Liao, Science, 2012, 335, 1596–1596 CrossRef CAS PubMed .
- K. Rabaey and R. A. Rozendal, Nat. Rev. Microbiol., 2010, 8, 706–716 CrossRef CAS PubMed .
- R. M. Rodrigues, X. Guan, J. A. Iñiguez, D. A. Estabrook, J. O. Chapman, S. Huang, E. M. Sletten and C. Liu, Nat. Catal., 2019, 2, 407–414 CrossRef CAS .
- V. Müller, Trends Biotechnol., 2019, 37, 1344–1354 CrossRef PubMed .
- C. Kim, K. M. Cho, K. Park, J. Y. Kim, G. Yun, F. M. Toma, I. Gereige and H. Jung, Adv. Funct. Mater., 2021, 31, 2102142 CrossRef CAS .
- M. Balk, J. Weijma, M. W. Friedrich and A. J. M. Stams, Arch. Microbiol., 2003, 179, 315–320 CrossRef CAS PubMed .
- J. Dolfing, B. Jiang, A. M. Henstra, A. J. M. Stams and C. M. Plugge, Appl. Environ. Microbiol., 2008, 74, 6126–6131 CrossRef CAS PubMed .
- S. Menon and S. W. Ragsdale, Biochemistry, 1996, 35, 12119–12125 CrossRef CAS PubMed .
- J. Chen, S. Huang, J. Seravalli, H. Gutzman, D. J. Swartz, S. W. Ragsdale and K. A. Bagley, Biochemistry, 2003, 42, 14822–14830 CrossRef CAS PubMed .
- S. L. Daniel, T. Hsu, S. I. Dean and H. L. Drake, J. Bacteriol., 1990, 172, 4464–4471 CrossRef CAS PubMed .
- E. Blanchet, F. Duquenne, Y. Rafrafi, L. Etcheverry, B. Erable and A. Bergel, Energy Environ. Sci., 2015, 8, 3731–3744 RSC .
- H. L. Drake and S. L. Daniel, Res. Microbiol., 2004, 155, 869–883 CrossRef PubMed .
- N. J. Claassens, I. Sánchez-Andrea, D. Z. Sousa and A. Bar-Even, Curr. Opin. Biotechnol., 2018, 50, 195–205 CrossRef CAS PubMed .
- K. Lentz and H. G. Wood, J. Biol. Chem., 1955, 215, 645–654 CrossRef CAS PubMed .
- L. G. Ljungdhal, Ann. Rev. Microbiol., 1986, 40, 415–450 CrossRef PubMed .
- J. Lv, M. Jouny, W. Luc, W. Zhu, J. Zhu and F. Jiao, Adv. Mater., 2018, 30, 1803111 CrossRef PubMed .
- A. Poehlein, F. R. Bengelsdorf, C. Esser, B. Schiel-Bengelsdorf, R. Daniel and P. Dürre, Genome Announce., 2015, 3, e01159-15 CrossRef PubMed .
- K. P. Nevin, S. A. Hensley, A. E. Franks, Z. M. Summers, J. Ou, T. L. Woodard, O. L. Snoeyenbos-West and D. R. Lovley, Appl. Environ. Microbiol., 2011, 77, 2882–2886 CrossRef CAS PubMed .
- D. R. Martin, L. L. Lundie, R. Kellum and H. L. Drake, Curr. Microbiol., 1983, 8, 337–340 CrossRef CAS .
- Y. Zhao, X. Chang, A. S. Malkani, X. Yang, L. Thompson, F. Jiao and B. Xu, J. Am. Chem. Soc., 2020, 142, 9735–9743 CAS .
- W. Zhang, C. Huang, Q. Xiao, L. Yu, L. Shuai, P. An, J. Zhang, M. Qiu, Z. Ren and Y. Yu, J. Am. Chem. Soc., 2020, 142, 11417–11427 CrossRef CAS PubMed .
- A. T. Chu and Y. Surendranath, J. Am. Chem. Soc., 2022, 144, 5359–5365 CrossRef CAS PubMed .
- X. Liu, P. Schlexer, J. Xiao, Y. Ji, L. Wang, R. B. Sandberg, M. Tang, K. S. Brown, H. Peng, S. Ringe, C. Hahn, T. F. Jaramillo, J. K. Nørskov and K. Chan, Nat. Commun., 2019, 10, 32 CrossRef CAS PubMed .
- A. J. Garza, A. T. Bell and M. Head-Gordon, ACS Catal., 2018, 8, 1490–1499 CrossRef CAS .
- F. Calle-Vallejo and M. T. M. Koper, Angew. Chem., 2013, 125, 7423–7426 CrossRef .
- J. Gao, H. Zhang, X. Guo, J. Luo, S. M. Zakeeruddin, D. Ren and M. Grätzel, J. Am. Chem. Soc., 2019, 141, 18704–18714 CrossRef CAS PubMed .
- Y. Shi, Y. Wang, C. Dong, T. T. T. Nga, D. Wei, J. Wang, X. Zhao, M. Wang, K. Zhang, M. Li, F. Dong and S. Shen, Adv. Energy Mater., 2023, 13, 2203896 CrossRef CAS .
- H. Zhang, H. Liu, Z. Tian, D. Lu, Y. Yu, S. Cestellos-Blanco, K. K. Sakimoto and P. Yang, Nat. Nanotechnol., 2018, 13, 900–905 CrossRef CAS PubMed .
- H. Babich and G. Stotzky, Ecotoxicol. Environ. Saf., 1983, 7, 576–587 CrossRef CAS PubMed .
- M. Hartmann, M. Berditsch, J. Hawecker, M. F. Ardakani, D. Gerthsen and A. S. Ulrich, Antimicrob. Agents Chemother., 2010, 54, 3132–3142 CrossRef CAS PubMed .
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 a,
Xiaoxiao
Guo‡
a,
Zhaoyuan
Lyu
b,
Rong-Bin
Song
a,
Xiaoxiao
Guo‡
a,
Zhaoyuan
Lyu
b,
Rong-Bin
Song