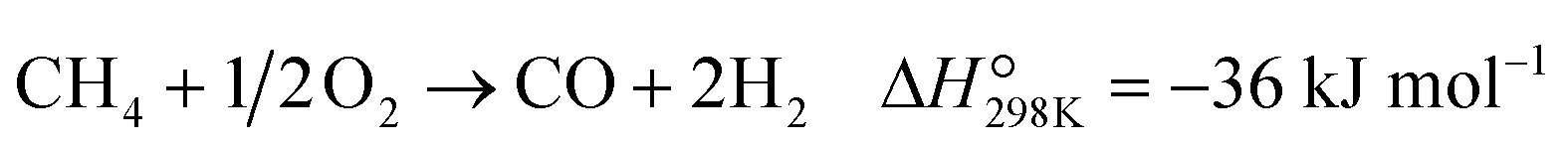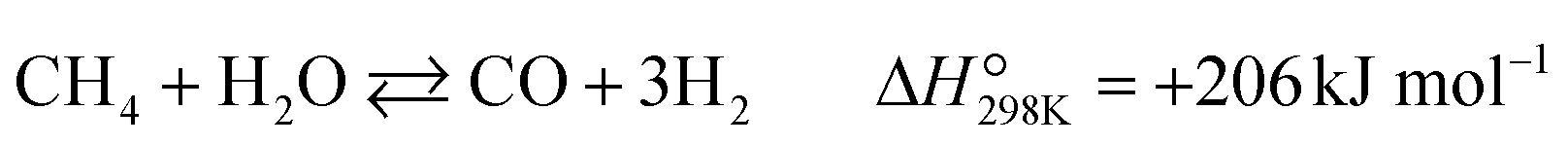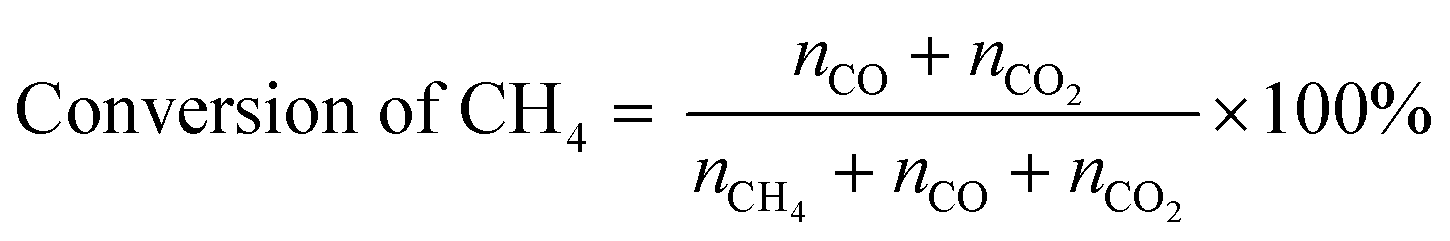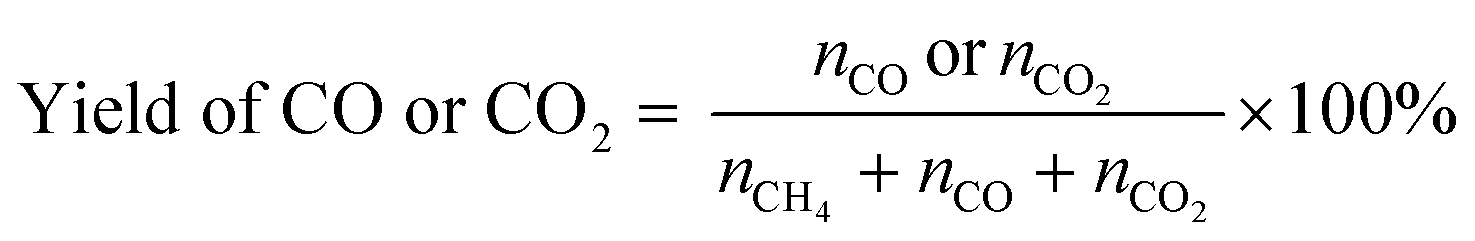Mordenite-stabilised rhenium catalyst for partial oxidation of methane to syngas†
Received
16th January 2023
, Accepted 18th June 2023
First published on 19th June 2023
Abstract
Partial oxidation of methane (POM) to syngas, a mixture of CO and H2, has remained a challenge for utilising natural gas efficiently. Rhenium (Re) shows unique catalytic activity in this reaction, but it is easily deactivated by oxidation. This work shows that mordenite (MOR)-supported Re catalyst is significantly more active and resistant to oxidising conditions than typical oxide-supported Re catalysts. POM with Re/MOR produces CO in 52% yield and H2 in 55% yield at a low temperature of 600 °C. Characterisation of the catalysts has indicated that MOR stabilises low-valent Re species that work for POM. The low-valent Re species produces CO from methane in the presence of O2, presumably via a direct oxidation pathway.
Introduction
Partial oxidation of methane (POM, eqn (1)) is a potential alternative to steam reforming (eqn (2)) for the production of syngas, a mixture of CO and H2.1–4 It is a mildly exothermic reaction (ΔH° = −36 kJ mol−1), and thus the process is free from the heat supply essential for steam reforming (ΔH° = +206 kJ mol−1). The economical process will facilitate the use of syngas for the production of fundamental chemicals.5,6 However, POM has remained a challenge in the chemical industry. Specifically, we should avoid the formation of a hot spot in the catalyst bed and decrease the reaction temperature below 650 °C to enable using inexpensive stainless steel reactors.7–12| | 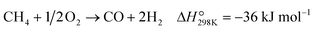 | (1) |
| | 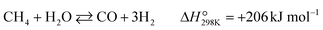 | (2) |
| | 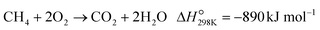 | (3) |
| | 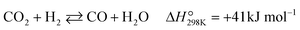 | (4) |
The POM to syngas commonly passes through the so-called indirect pathway, which consists of complete oxidation of CH4 (eqn (3)), steam reforming (eqn (2)) and reverse water-gas shift reaction (eqn (4)).3 This reaction pathway suffers from two major issues. First, the complete oxidation generates severe heat at the inlet of the catalyst bed, elevating the temperature very high. Second, steam reforming and reverse water-gas shift reactions are reversible thermodynamically. Reduction in operating temperature of the POM process shifts the equilibrium to the left side (eqn (2) and (4)), thus decreasing yields of CO and H2. Accordingly, the indirect pathway poses a significant limitation on the process.
A fundamental solution is to change the reaction mechanism. An ideal case is the direct POM to syngas via neither CO2 nor H2O,13–15 but developing a catalyst that follows this mechanism is under investigation.16–18 We focus on catalysis of Re to alter the reaction mechanism from the indirect pathway. Previous works used Re as an additive for supported metal catalysts to improve their activity.19,20 However, recently, we found that low-valent Re oxides in Ru–Re/Al2O3 can be main active species that produce CO not via CO2.21 The role of Ru is to preserve the low oxidation state of Re. Although the catalyst generates H2 by steam reforming, the direct CO formation is advantageous over the complete oxidation of CH4.
While the activity of low-valent Re species is intriguing, Re is easily oxidised to a heptavalent state under oxidising reaction conditions. Moreover, Re2O7, the predominant species of heptavalent Re, is volatile at a high temperature of POM. Torniainen et al. reported that a Re-coated monolith catalyst was quickly deactivated in the POM due to the formation of high-valent Re species and their sublimation.22 Chen et al. reported low reducibility of Re species on Al2O3;23 in other words, Re/Al2O3 is susceptible to oxidation. Thus, it is a challenge to preserve low-valent Re species during the oxidation of CH4.
In this work, we show that a mordenite-type zeolite (MOR) support facilitates the reduction of Re, thus making Re species more resistant against oxidation than typical oxide supports. Re/MOR can sustain the activity in POM under relatively oxidising atmosphere in the absence of noble metals that was needed in previous works.
Experimental
Catalyst preparation
Re/MOR was synthesised by an impregnation method with ammonium perrhenate (NH4ReO4, Sigma-Aldrich Co., Ltd. 99.9%) as a precursor. A 21.6 mg of NH4ReO4 was dissolved in 25 mL of deionised water by stirring for 10 min. After adding a proton-type MOR (0.485 g; Si/Al = 15, HSZ-660HOA, Tosoh), the mixture was stirred at room temperature for 24 h. The theoretical loading amount of Re is 3.0 wt%. The sample was evaporated at 50 °C at 80 hPa, dried in air at 110 °C overnight, and calcined in air. The calcination condition was as follows: increasing temperature from room temperature to 500 °C over 2 h and keeping 500 °C for 3 h. Energy-dispersive X-ray analysis (EDX; EDX-720, Shimadzu) with an absolute calibration method indicated that the loading amount of Re was 3.0 wt% after the calcination, viz. no detectable loss of Re.
The same procedure was adopted for other supports, Al2O3 (JRC-ALO-8, Catalysis Society of Japan (CSJ)), SiO2 (JRC-SIO-9A, CSJ) and MgO (JRC-MGO-4-500A, CSJ).
Catalyst characterisation
X-ray diffraction (XRD) measurements were performed on a Cu Kα radiation diffractometer (Ultima IV, Rigaku) equipped with a semiconductor array detector (D/teX Ultra 2). Transmission electron microscopy (TEM) images of the samples were observed by a JEM-2100F (JEOL) at an acceleration voltage of 200 kV. N2 adsorption isotherms were obtained at −196 °C using a BELSORP mini (BEL Japan), after degassing samples at 200 °C for 1 h. Temperature-programmed reduction with hydrogen (H2-TPR) was performed using a BELCAT (Microtrac-BEL) equipped with a molecular sieve trap and a thermal conductivity detector (TCD). A powder sample of 40 mg was put into a quartz tube and heated under He flow (50 mL min−1) at 200 °C for 30 min and cooled to room temperature. Afterward, a 5% H2/Ar mixed gas (50 mL min−1) was passed through the sample tube with increasing the temperature from 30 to 900 °C at a ramping rate of 10 °C min−1. Raman spectra were obtained with a Renishaw inVia reflex spectrometer with a 532 nm laser. The grating used was 1800 mm−1 and the exposure time was 30 s.
POM
POM was carried out using a vertical quartz fixed-bed flow reactor (i.d. = 7 mm). A powder catalyst (20 mg) was put at the middle of reactor and sustained by quartz wool. The reactor was heated to 600 °C under a N2 flow (20 mL min−1). Subsequently, the catalyst was kept at 600 °C and reduced under an H2 flow (20 mL min−1) for 1 h. After that, a 20 mL min−1 of the gas mixture consisting of 8% CH4/4% O2/88% N2 (vol.) was introduced to the reactor. An on-line gas chromatograph (GC-8A, Shimadzu) equipped with a thermal conductivity detector (TCD) was used to quantify the gaseous products. N2, O2, CH4 and CO were separated with a Molecular Sieve 5A column, and CO and CO2 were done with a Shincarbon ST column using He carrier. H2 was analysed by another GC equipped with a Molecular Sieve 5A column using Ar carrier. The conversion of CH4 and yields of CO and CO2 were determined by eqn (5) and (6), where nx is the amount of compound x that determined by GC analysis. These equations are justified due to no formation of other carbon products (<1%).| | 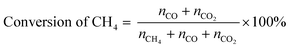 | (5) |
| | 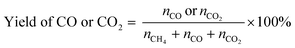 | (6) |
Steam reforming
The steam reforming reaction was performed in the same reactor for POM. A 100 mg of catalyst was charged in the reactor and purged with 20 mL min−1 of N2 at room temperature for 1 h. The reactor was heated to 600 °C under N2 flow. After reaching the temperature, H2 (20 mL min−1) was introduced to reduce the catalyst for 1 h, followed by a N2 purge for 20 min. Then, the steam reforming was performed with a wet gas mixture made by the following procedure. A gas mixture (CH4 10.0 mL min−1, N2 9.2 mL min−1) passed through a water bubbler at ca. 40 °C. The gas line for the wet gas was kept at 120 °C by a ribbon heater so that water does not condense. The steam-containing gas was introduced into the reactor.
Density functional theory (DFT) calculations
We performed DFT calculations at the MN15/def2-SVPD level of theory to predict the Raman spectra and the reduction energy of Re species.24–26 The effective core potential was employed for Re atoms.27 The software used was Gaussian 16 (rev. A.03), and the ultrafine grid (99, 590) was applied for the numerical integration. A scale factor of 0.933 was used to adjust the vibrational frequencies so that the estimation matches the experimentally observed frequencies of the Re![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O vibration for Re2O7 in the gas phase.28 The energy includes electronic and zero-point vibration energies. For the Re species in MOR, a partial structure of MOR around an acid site (consisting of 33 atoms) was extracted from its crystallographic data, and the edge oxygen atoms were terminated with hydrogen atoms to avoid dangling bonds (Table S1†).
O vibration for Re2O7 in the gas phase.28 The energy includes electronic and zero-point vibration energies. For the Re species in MOR, a partial structure of MOR around an acid site (consisting of 33 atoms) was extracted from its crystallographic data, and the edge oxygen atoms were terminated with hydrogen atoms to avoid dangling bonds (Table S1†).
Results and discussion
Characterisation of Re/MOR
The 3 wt% Re/MOR sample was prepared by a wet impregnation method, followed by calcination at 500 °C. We chose MOR among zeolites and 3 wt% of the loading amount of Re to obtain high activity and good reproducibility based on the screening tests (Fig. S1 and S2†). TEM measurement of Re/MOR showed no particles for Re species (Fig. 1a), indicating that the Re species were highly dispersed. The XRD analysis (Fig. 1d) represented that the diffraction pattern of MOR was preserved but the intensity for the peak at 13° became slightly smaller after the impregnation of Re. The decrease in the intensity of low-angle peaks is often observed when metal is incorporated into the micropores of zeolites.29 No peak for Re species was seen, which also suggests highly dispersed Re species.
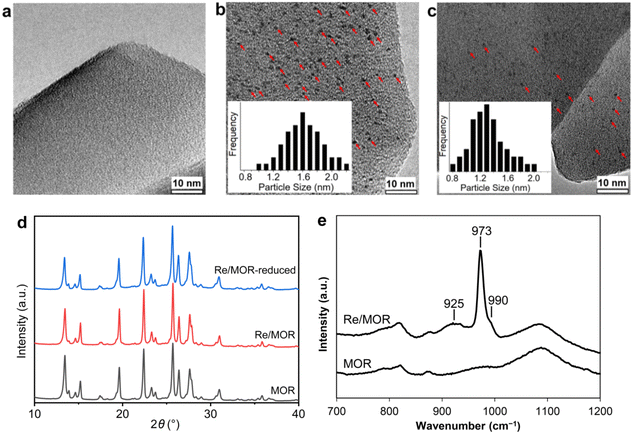 |
| | Fig. 1 Characterisation of Re/MOR. TEM images of (a) Re/MOR, (b) reduced Re/MOR and (c) spent Re/MOR after POM for 72 h. (d) XRD patterns. (e) Raman spectra. The red arrows in (b) and (c) indicate the Re species. | |
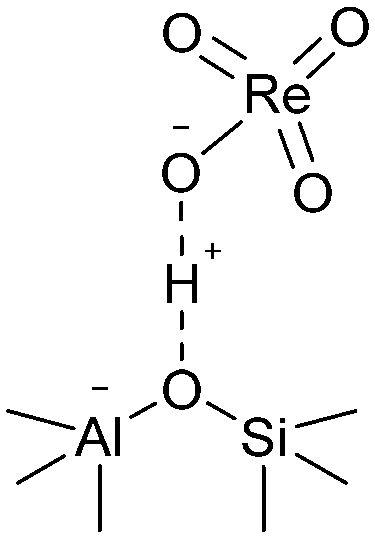
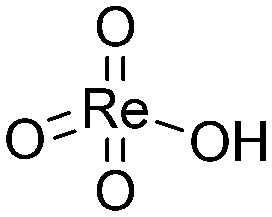
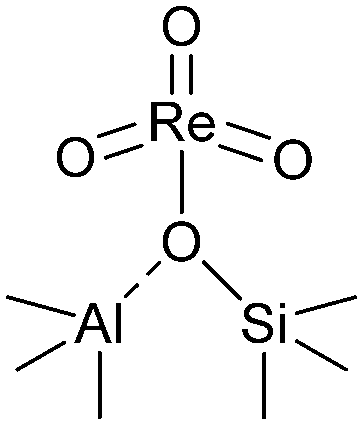
For the chemical state of the Re species, the UV-vis spectroscopy suggested 7+ oxidation state (Fig. S3†). In the Raman scattering analysis, Re/MOR gave a large peak at 973 cm−1, a weak shoulder at 990 cm−1 and a broad peak around 925 cm−1 (Fig. 1e). To assign these peaks, we estimated the vibration frequencies of possible hepta-valent Re species on MOR support by DFT calculations as listed in Table 1: (i) ReO4−, (ii) that interacting with H+ on MOR (ReO4−⋯H+), (iii) HReO4, (iv) ReO4 structure within the zeolite framework,30 and (v) Re2O7. The result indicated that the peaks at 973 cm−1 and ca. 925 cm−1 are assignable to the symmetric and asymmetric Re–O stretching of the species (ii), respectively. Regarding the predicted peak intensity, the former is several times larger than the latter, which is consistent with the actual experimental data (Fig. 1e). The minor peak at 990 cm−1 is ascribed to the symmetric Re–O stretching of the species (iii) to (v). In addition, we measured a Raman spectrum of Re species on SiO2, a support that does not give ionic interactions (Fig. S4†). The sample showed a peak at 987 cm−1, perhaps due to HReO4 or its dehydrated species interacting with O atoms on SiO2. This result implies that acid sites of MOR are involved in the formation of the Re species that shows a Raman peak at 973 cm−1. Based on these analyses, we propose that the predominant Re species is ReO4− interacting with acid sites of the MOR support.
Table 1 Raman frequencies predicted by the DFT calculationsa
| Species |
ν
s(Re–O)/cm−1 |
ν
as(Re–O)/cm−1 |
|
MN15/def2-SVPD. The scale factor was 0.933 to reproduce the experimental vibration frequencies of Re2O7 in gas phase.
Atomic coordinate: Table S1.†
For the Re species with one water-molecule coordination (Re–OH2).
|
(i) 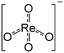 |
947 |
892 |
(ii)b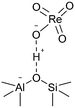 |
977 |
937 |
(iii) 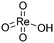 |
1004 |
964 |
(iv) 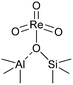 |
1004 (997)c |
955 (949)c |
(v) 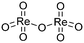 |
1011 |
968 |
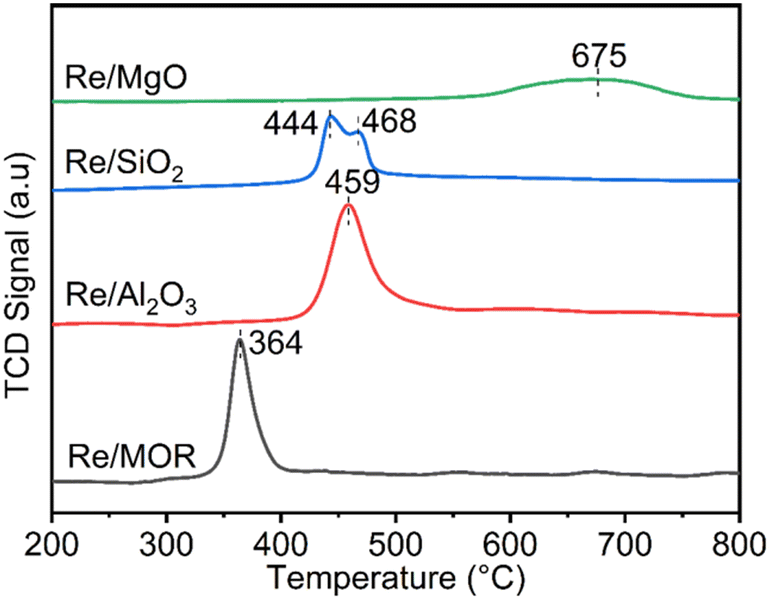
We studied the reduction of Re/MOR with H2 to prepare the active catalyst. The TPR treatment reduced the Re species of Re/MOR at 364 °C, which was clearly lower than the reduction temperature of Re/Al2O3 (459 °C), Re/SiO2 (440–470 °C) and Re/MgO (>600 °C) (Fig. 2). This result indicates that the MOR support facilitates the reduction of Re. As suggested by the Raman spectroscopy, H+ coordinates to ReO4−, which may decrease electron density of Re. Indeed, the natural population analysis31 represented the increase in positive charge of Re atom from +1.72e [ReO4− (i)] to +1.83e [ReO4−⋯H+ (ii)]. Therefore, Re becomes more electrophilic and easily reducible. In our DFT calculations, free ReO4− was reduced by H2 to tetra-valent state (see below) with ΔE = +58 kJ mol−1, while the species (ii) offered −1 kJ mol−1. As a result, the coordination of proton facilitates the reduction by ca. 60 kJ mol−1. In contrast, the high basicity of MgO results in the high reduction temperature due to the electron donating effect. The amount of hydrogen consumption for the reduction of Re/MOR was 200 μmol g−1, corresponding to an H/Re atomic ratio of 2.5. As the original oxidation state of Re was 7+, after reducing one Re atom with 2.5H, the resulting oxidation state would be 4+ to 5+ in average. We employed XPS to check the oxidation state of Re species, but the measurements gave very weak Re signals that cannot be distinguished from noise in spite of the high loading amount of Re (3 wt%) (data not shown). The concentration of Re on the external surface should be low, which also implies that most of the Re species are inside the micropores of MOR. Therefore, the Ar ion sputtering for exposing the internal Re species gained Re 4f peaks. However, the Ar treatment affected the oxidation state of Re, and the data was not useful (data not shown). The TEM imaging of the reduced catalyst exhibited 1 to 2 nm of particles assignable to Re species (Fig. 1b). Re species aggregate on the external surface in the reduction treatment, but it should be only the small fraction, given the other results.
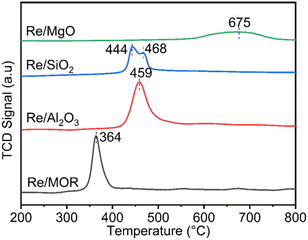 |
| | Fig. 2 TPR profiles of Re catalysts. | |
Activity of Re/MOR for POM
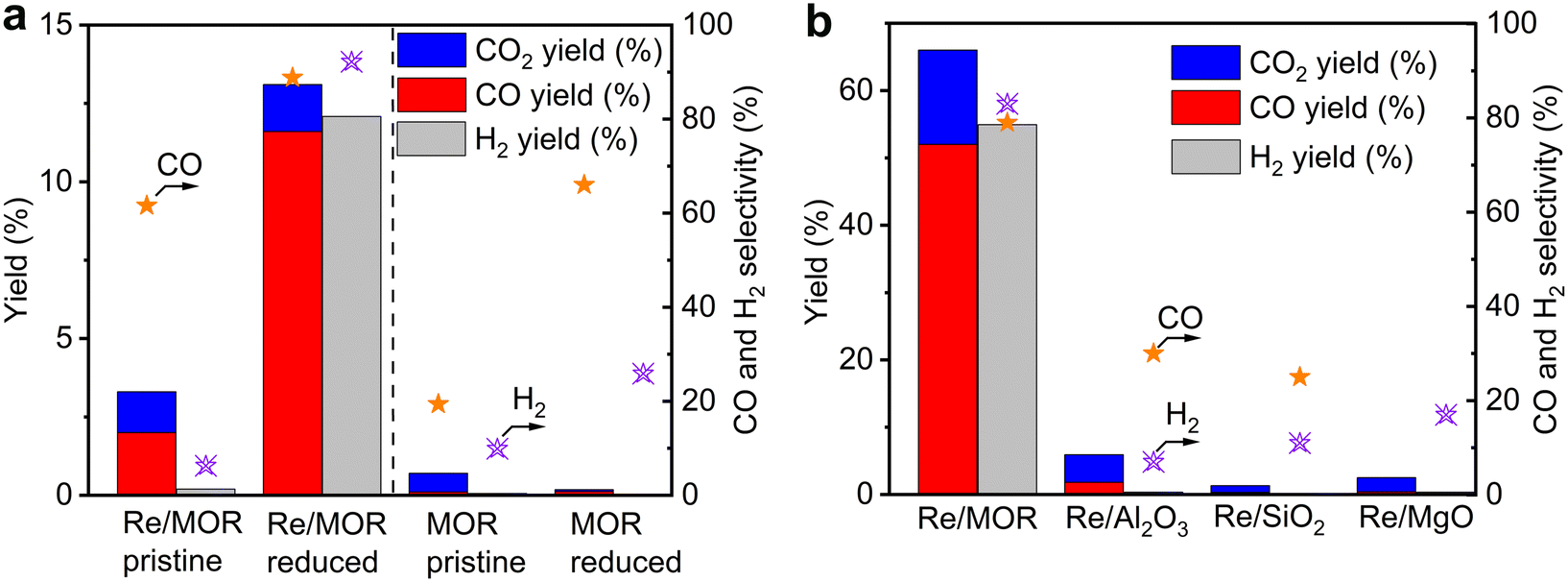
Catalytic performance of Re/MOR was evaluated in POM at a low conversion region using a small amount of O2 (O2/CH4 = 0.08) at 600 °C (Fig. 3a). Pristine Re/MOR, namely the catalyst with no reduction pretreatment, gave low conversion values of CH4 (3.3%) and O2 (38%). O2 was remaining, but the reaction produced CO in 2.0% yield with 60% selectivity. This result suggests that the catalyst produces CO via the direct partial oxidation of CH4. As discussed in the Introduction, POM can occur by the direct partial oxidation or indirect processes. In the indirect pathway, CH4 is initially converted to CO2 and H2O, and the steam reforming of CH4 produces CO. As O2 inhibits the steam reforming, CO is generated only after the complete consumption of O2.10 Accordingly, the CO formation in the presence of O2 is likely due to the direct partial oxidation rather than the indirect pathway.
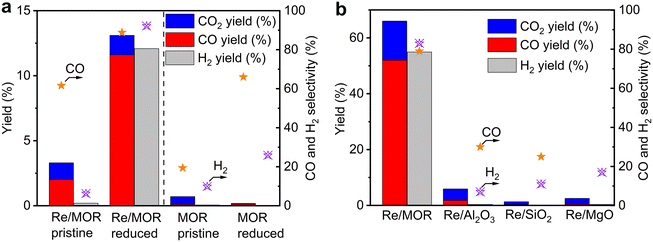 |
| | Fig. 3 POM over different catalysts. (a) Re/MOR and MOR before and after H2 reduction (600 °C). Reaction conditions: 600 °C, CH4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O2:N2 = 50:4:46, space velocity (SV) of 60000 mL g−1 h−1. (b) Effect of catalyst support. Catalysts were reduced before POM. 600 °C, CH4:O2:N2 = 8:4:88, SV of 12000 mL g−1 h−1. All the data in (a) and (b) are results at 1 h after starting the reaction. :O2:N2 = 50:4:46, space velocity (SV) of 60000 mL g−1 h−1. (b) Effect of catalyst support. Catalysts were reduced before POM. 600 °C, CH4:O2:N2 = 8:4:88, SV of 12000 mL g−1 h−1. All the data in (a) and (b) are results at 1 h after starting the reaction. | |
Re/MOR reduced by H2 at 600 °C increased CH4 conversion to 13% in POM (Fig. 3a) and produced CO (12% yield, 89% selectivity) and H2 (12% yield, 92% selectivity). An H2/CO mole ratio in the product was 2.1. In control experiments, MOR was inactive for the POM before and after H2 reduction in the absence of Re. These results indicate that low-valent Re species are active for the POM reaction.
As shown in the H2-TPR measurements, Re species are reduced on MOR more easily than those on other supports. To clarify the influence of the reducibility on POM, we tested MOR-, Al2O3-, SiO2- and MgO-supported Re catalysts in POM at an O2/CH4 ratio of 0.5, namely the stoichiometric ratio (eqn (1); Fig. 3b). The catalysts were reduced with H2 at 600 °C before the reaction. Re/MOR gave 66% CH4 conversion, 52% yield of CO (selectivity 79%) and 55% yield of H2 (selectivity 83%). Thus, Re/MOR is active under the condition. In contrast, Re/Al2O3, Re/SiO2 and Re/MgO catalysts showed low CH4 conversion (<10%) and low CO selectivity (<30%). We have previously clarified that the reduced Re/Al2O3 is active at an O2/CH4 ratio of 0.08 but the catalyst is deactivated under the more oxidizing atmosphere (O2/CH4 = 0.5).21 Based on these facts, we propose that MOR sustains the low oxidation state of Re to make it active, which is in contrast to typical oxide supports. To support this idea, we conducted the O2 temperature-programmed oxidation (O2-TPO) that directly evaluates the oxidation resistance. However, reduced Re/MOR adsorbed O2 at low temperatures during waiting stabilisation prior to elevating the temperature and released O2 at a higher temperature with no oxidation peaks. The adsorbed O2 was possibly used for the oxidation of Re also. Therefore, the oxidation temperature was not clarified in this method.
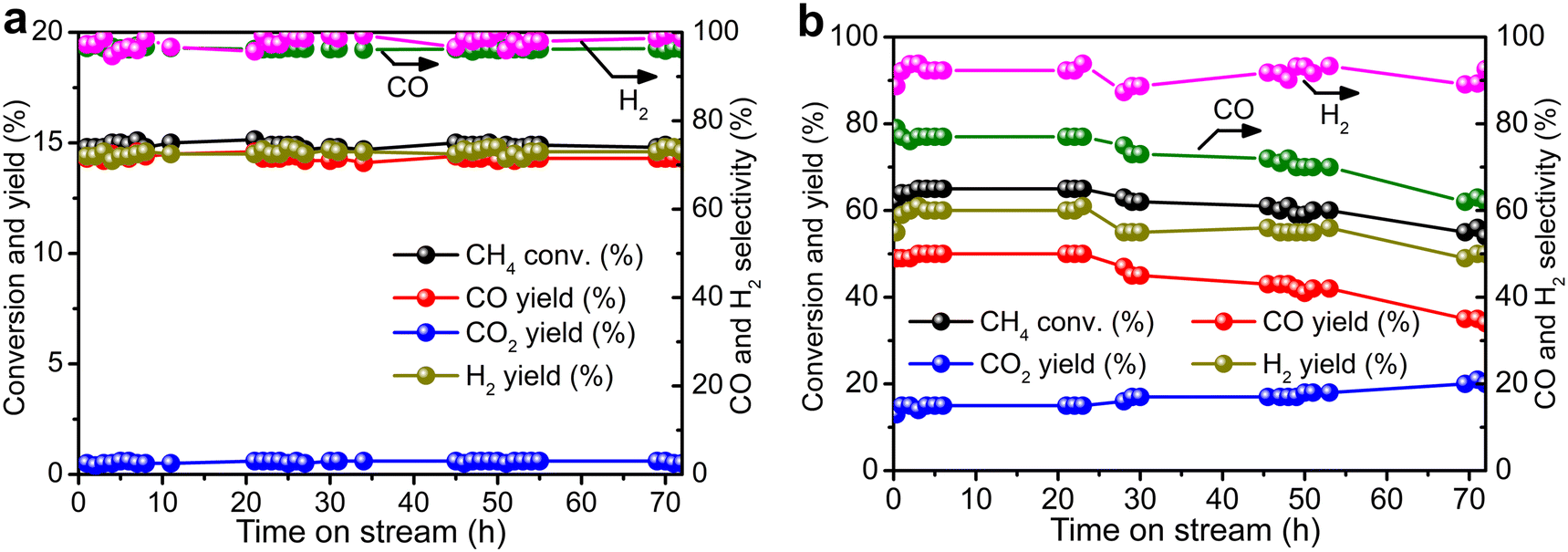
Durability tests of Re/MOR were performed at 600 °C for 72 h at O2/CH4 ratios of 0.08 (Fig. 4a) and 0.5 (Fig. 4b). When the O2/CH4 ratio was 0.08, CH4 was steadily converted to CO (96–97% selectivity) and H2 (≥95% selectivity) at a CH4 conversion of 15% (Fig. 4a). An EDS analysis showed no loss of Re species after the reaction. However, when the O2/CH4 ratio was increased to 0.5, CH4 conversion started decreasing from 64% to 55% after 24 h (Fig. 4b). For the cause of deactivation, TEM showed no sintering of observable Re species (before reaction 1.6 nm, Fig. 1b; after reaction 1.3 nm, Fig. 1c), but EDS represented that the residual Re amount decreased from 3.0 wt% to 2.2 wt%. The oxidising condition (O2/CH4 = 0.5) may produce Re2O7 to cause sublimation.
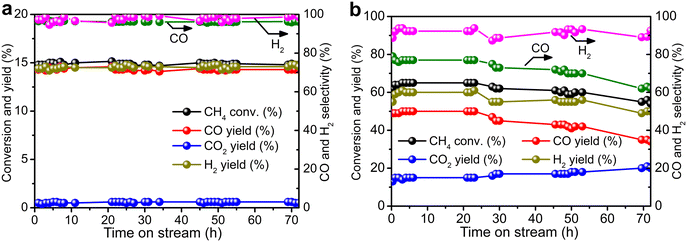 |
| | Fig. 4 Durability tests of Re/MOR. (a) CH4:O2:N2 = 50:4:46. (b) CH4:O2:N2 = 8:4:88. Conditions: 600 °C, SV of 12000 mL g−1 h−1. | |
POM reaction pathway
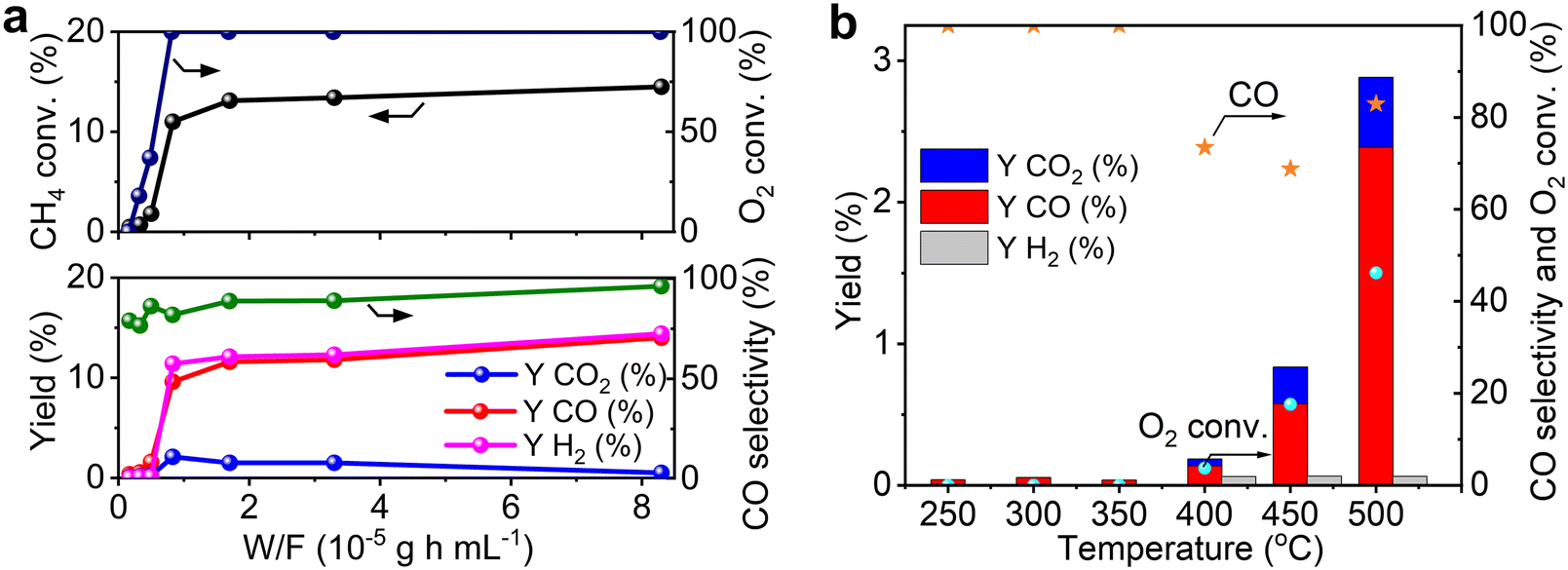
The effect of contact time (W/F, defined as the weight of catalyst per total flow rate of gases) was studied to understand the reaction pathway of POM over Re/MOR catalyst (Fig. 5a and Table S2†). At a short contact time (0.17 × 10−5 g h mL−1), CH4 and O2 conversion values were 0.47% and ca. 0%, respectively. CO was a main product (0.37% yield and 80% selectivity), but H2 was obtained in a very low yield (0.06%). Therefore, the Re catalyst oxidises CH4 to CO and H2O predominantly (eqn (7)). By increasing the contact time to 0.50 × 10−5 g h mL−1, conversion of CH4 and O2 gradually increased to 1.8% and 37%, respectively, and CO yield proportionally increased to 1.6% with keeping 80% selectivity. Almost no H2 formation was observed in this region of contact time. A further increase in contact time to 0.83 × 10−5 g h mL−1 completely consumed O2, and then CH4 conversion, CO yield and H2 yield suddenly increased to 11%, 9.5% and 11%, respectively. This fashion is an evidence for the steam reforming of CH4 in the absence of O2. Eventually, the reaction provided a CH4 conversion of 15%, a CO yield of 14% and an H2 yield of 14% at 8.3 × 10−5 g h mL−1. The product composition is similar to that predicted by the chemical equilibrium of steam reforming and reverse water-gas shift reactions (eqn (2) and (4); CH4 conversion 16%, CO yield 15%, H2 yield 16%). This agreement also suggests the presence of the reforming reactions in the absence of O2. We verified this hypothesis by performing steam reforming of methane using the reduced Re/MOR. The reaction afforded 9.1% yield of CO with 99% selectivity at a contact time of 8.3 × 10−5 g h mL−1. In addition, it is known that Re converts CO2 to CO under H2 atmosphere.19 Hence, we conclude that Re/MOR catalyses the direct partial oxidation of CH4 to CO in the presence of O2 (eqn (7)) and steam reforming and reverse water-gas shift reactions after completely consuming O2 (eqn (2) and (4)).| | 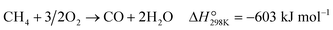 | (7) |
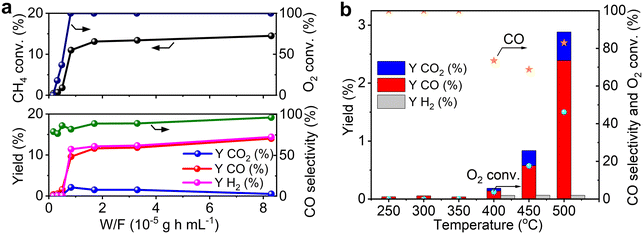 |
| | Fig. 5 Effect of contact time (a) and reaction temperature (b) on the POM over reduced Re/MOR. CH4:O2:N2 = 50:4:46. Detailed reaction data for (a) are available in Table S2.† | |
To further study the reaction pathway, POM reaction was operated at different temperatures (Fig. 5b). The reactions at a temperature range of 250 to 350 °C produced small amounts of CO at very low conversion values of CH4 and O2. Neither CO2 nor H2 was detected. An increase in the temperature to 400 °C maintained CO as the major product (0.19% yield, 73% selectivity) but gave small amounts of CO2 (0.05% yield) and H2 (0.06% yield). After the reaction temperature was increased to 500 °C, the yield of CO increased to 2.4% (83% selectivity), but that of H2 was still only 0.06%. In all cases O2 was remaining, specifically <5 to 46% conversion. Accordingly, both contact-time and temperature-dependence experiments show that the Re catalyst converts CH4 to CO and H2O by direct partial oxidation.
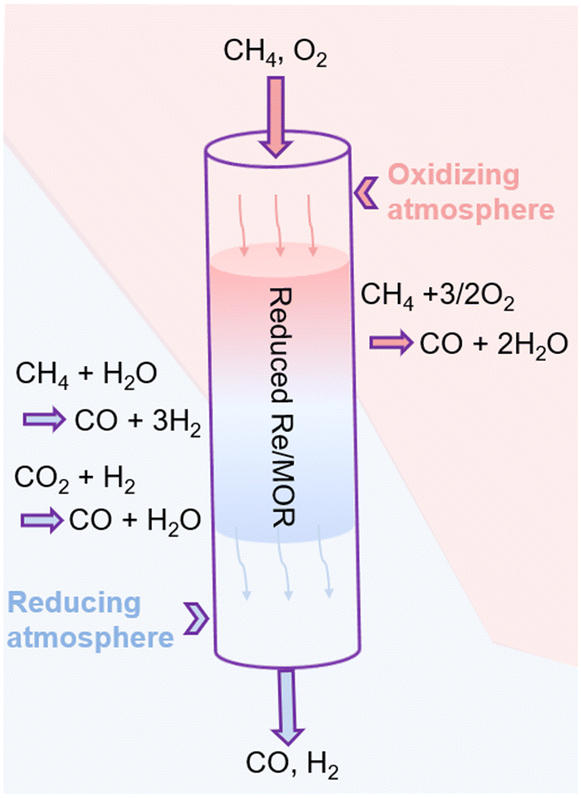
Based on all the results, we propose the catalysis of Re as shown in Fig. 6. First, the hepta-valent Re is reduced to a lower oxidation state in the pretreatment process. The reduced Re/MOR converts CH4 to CO (ca. 80% selectivity) and H2O in the presence of O2 at the inlet of catalyst bed. Wang et al. have reported that Re/SiO2 can oxidise methane with O2 to formaldehyde.32 The decomposition of formaldehyde should produce CO and H2 (HCHO → CO + H2), but almost no H2 is detected in our case at a short contact time or at a low reaction temperature. Instead, we assume that formaldehyde is further oxidised to formic acid in our system, and the species is decomposed to CO (HCOOH → CO + H2O). Unfortunately, the infrared spectroscopy did not detect formic acid or formate (Fig. S5†), so the decomposition of intermediates is perhaps rapid. O2 may oxidise the low-valent Re species, but the oxidised species are again reduced with CH4. Therefore, Re/MOR can maintain its activity at a low O2/CH4 ratio (Fig. 4a). Meanwhile, the reduction rate is unlikely very fast as the catalyst is gradually deactivated under a more oxidising condition (Fig. 4b). After completely consuming O2 by the above mentioned scheme, the low-valent Re species start steam reforming of methane to CO and H2. The H2 produced is used for the reduction of CO2 to CO, as CO2 amount was gradually decreased in the contact time experiments (Fig. 5a).
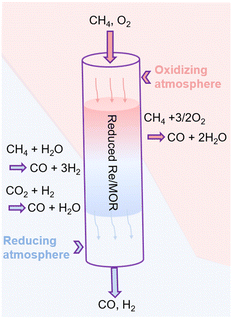 |
| | Fig. 6 Scheme of the catalysis of Re for POM over Re/MOR. | |
Conclusions
Re/MOR shows a good catalytic performance in POM under a stoichiometric condition (O2/CH4 = 0.5) at 600 °C for 24 h: CH4 conversion 66%, CO yield 52% (selectivity 79%) and H2 yield 55% (selectivity 83%). The activity is significantly higher than those of typical oxide-supported Re catalysts such as Re/Al2O3. The better performance is due to stabilising a low oxidation state of Re, which is the main active phase. Specifically, MOR protonates hepta-valent Re species (ReO4−), the electron-withdrawing effect of which advantages the reduction of the Re species by about 60 kJ mol−1. For the reaction mechanism of POM, Re/MOR produces CO in the presence of O2, which suggests a direct partial oxidation reaction. After consuming O2 completely, steam reforming and reverse water-gas shift reactions occur. Accordingly, the equilibrium issue still partially limits the reaction performance, but the direct formation of CO is notable. For longer-term durability, the slight sublimation of high-valent Re species should be suppressed by stabilising the low-valent state further. We hope these findings will facilitate designing more stable and active Re catalysts for the POM without complete oxidation.
Author contributions
L. L.: investigation, visualization, writing – original draft. A. S.: investigation. K. K.: investigation. A. F.: funding acquisition, supervision, resources, writing – review & editing. H. K.: funding acquisition, formal analysis, supervision, project administration, resources, visualization, writing – review & editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by Grant-in-Aid for Challenging Research (Exploratory) from Japan Society for the Promotion of Science (No. 21K18838) and by Japan Science and Technology Agency (JST) CREST (No. JPMJCR15P4). The authors thank the Technical Division of the Institute for Catalysis of Hokkaido University for assistance in the XPS measurements.
References
- R. Ma, B. Xu and X. Zhang, Catal. Today, 2019, 338, 18–30 CrossRef CAS.
- A. I. Osman, Chem. Eng. Technol., 2020, 43, 641–648 CrossRef CAS.
- A. H. Elbadawi, L. Ge, Z. Li, S. Liu, S. Wang and Z. Zhu, Catal. Rev.: Sci. Eng., 2021, 63, 1–67 CrossRef CAS.
- A. Masudi, N. W. C. Jusoh and O. Muraza, Catal. Sci. Technol., 2020, 10, 1582–1596 RSC.
- X. Pan, F. Jiao, D. Miao and X. Bao, Chem. Rev., 2021, 121, 6588–6609 CrossRef CAS PubMed.
- Z. Zhao, J. Jiang and F. Wang, J. Energy Chem., 2021, 56, 193–202 CrossRef CAS.
- A. C. Chien and J. A. van Bokhven, Catal. Sci. Technol., 2015, 5, 3518–3524 RSC.
- Z. Luo, D. A. Kriz, R. Miao, C.-H. Kuo, W. Zhong, C. Guild, J. He, B. Willis, Y. Dang, S. L. Suib and P. Nandi, Appl. Catal., A, 2018, 554, 54–63 CrossRef CAS.
- R. K. Singha, S. Ghosh, S. S. Acharyya, A. Yadav, A. Shukla, T. Sasaki, A. M. Venezia, C. Pendem and R. Bal, Catal. Sci. Technol., 2016, 6, 4601–4615 RSC.
- Y. Hou, S. Ogasawara, A. Fukuoka and H. Kobayashi, Catal. Sci. Technol., 2017, 7, 6132–6139 RSC.
- Y. Hou, S. Nagamatsu, K. Asakura, A. Fukuoka and H. Kobayashi, Commun. Chem., 2018, 1, 41 CrossRef.
- Y. Tang, V. Fung, X. Zhang, Y. Li, L. Nguyen, T. Sakata, K. Higashi, D. Jiang and F. F. Tao, J. Am. Chem. Soc., 2021, 143, 16566–16579 CrossRef CAS PubMed.
- Y. Boucouvalas, Z. Zhang and X. E. Verykios, Catal. Lett., 1996, 40, 189–195 CrossRef CAS.
-
A. M. Gaffney and D. R. Corbin, US Pat., 6409940, 2002 Search PubMed.
- S. Naito, H. Tanaka, S. Kado, T. Miyao, S. Naito, K. Okumura, K. Kunimori and K. Tomishige, J. Catal., 2008, 259, 138–146 CrossRef CAS.
- K. Urasaki, S. Kado, A. Kiryu, K. Imagawa, K. Tomishige, R. Horn, O. Korup and Y. Suehiro, Catal. Today, 2018, 299, 219–228 CrossRef CAS.
- S. Yasuda, R. Osuga, Y. Kunitake, K. Kato, A. Fukuoka, H. Kobayashi, M. Gao, J. Hasegawa, R. Manabe, H. Shima, S. Tsutsuminai and T. Yokoi, Commun. Chem., 2020, 3, 129 CrossRef CAS PubMed.
- Y. Kim, S. Kang, D. Kang, K. R. Lee, C. K. Song, J. Sung, J. S. Kim, H. Lee, J. Park and J. Yi, Angew. Chem., Int. Ed., 2021, 60, 25411–25418 CrossRef CAS PubMed.
- K. Bawornruttanaboonya, N. Laosiripojana, A. S. Mujumdar and S. Devahastin, AIChE J., 2018, 64, 1691–1701 CrossRef CAS.
- C. Cheephat, P. Daorattanachai, S. Devahastin and N. Laosiripojana, Appl. Catal., A, 2018, 563, 1–8 CrossRef CAS.
- L. Li, N. H. Md Dostagir, A. Shrotri, A. Fukuoka and H. Kobayashi, ACS Catal., 2021, 11, 3782–3789 CrossRef CAS.
- P. Torniainen, X. Chu and L. Schmidt, J. Catal., 1994, 146, 1–10 CrossRef CAS.
- L. Chen, Y. Ni, J. Zang, L. Lin, X. Luo and S. Cheng, J. Catal., 1994, 145, 132–140 CrossRef CAS.
- H. S. Yu, X. He, S. L. Li and D. G. Truhlar, Chem. Sci., 2016, 7, 5032–5051 RSC.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- D. Rappoport and F. Furche, J. Chem. Phys., 2010, 133, 134105 CrossRef PubMed.
- D. Andrae, U. Haeussermann, M. Dolg, H. Stoll and H. Preuss, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- I. Beattie, T. Gilson and P. Jones, Inorg. Chem., 1996, 35, 1301–1304 CrossRef CAS PubMed.
- J. Janas, J. Gurgul, R. P. Socha and S. Dzwigaj, Appl. Catal., B, 2009, 91, 217–224 CrossRef CAS.
- Y. Wu, S. Holdren, Y. Zhang, S. C. Oh, D. T. Tran, L. Emdadi, Z. Lu, M. Wang, T. J. Woehl and M. Zachariah, J. Catal., 2019, 372, 128–141 CrossRef CAS.
- A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS.
- Y. Wang, K. Otsuka and H. Wan, React. Kinet. Catal. Lett., 2003, 79, 127–133 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional reaction and spectroscopic data and atomic coordinate for DFT calculations. See DOI: https://doi.org/10.1039/d3cy00077j |
|
| This journal is © The Royal Society of Chemistry 2023 |

 a,
Kazuya
Kato
a,
Atsushi
Fukuoka
a,
Kazuya
Kato
a,
Atsushi
Fukuoka