
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licenceπ-Electronic ion pairs: building blocks for supramolecular nanoarchitectonics viaiπ–iπ interactions
Yohei
Haketa†
 ,
Kazuhisa
Yamasumi†
and
Hiromitsu
Maeda
*
,
Kazuhisa
Yamasumi†
and
Hiromitsu
Maeda
*
Department of Applied Chemistry, College of Life Sciences, Ritsumeikan University, Kusatsu 525-8577, Japan. E-mail: maedahir@ph.ritsumei.ac.jp
First published on 5th October 2023
Abstract
The pairing of charged π-electronic systems and their ordered arrangement have been achieved by iπ–iπ interactions that are derived from synergetically worked electrostatic and dispersion forces. Charged π-electronic systems that provide ion pairs as building blocks for assemblies have been prepared by diverse strategies for introducing charge in the core π-electronic systems. One method to prepare charged π-electronic systems is the use of covalent bonding that makes π-electronic ions and valence-mismatched metal complexes as well as protonated and deprotonated states. Noncovalent ion complexation is another method used to create π-electronic ions, particularly for anion binding, producing negatively charged π-electronic systems. Charged π-electronic systems afford various ion pairs, consisting of both cationic and anionic π-systems, depending on their combinations. Geometries and electronic states of the constituents in π-electronic ion pairs affect the photophysical properties and assembling modes. Recent progress in π-electronic ion pairs has revealed intriguing characteristics, including the transformation into radical pairs through electron transfer and the magnetic properties influenced by the countercations. Furthermore, the assembly states exhibit diversity as observed in crystals and soft materials including liquid-crystal mesophases. While the chemistry of ion pairs (salts) is well-established, the field of π-electronic ion pairs is relatively new; however, it holds great promise for future applications in novel materials and devices.
 Yohei Haketa | Yohei Haketa received his PhD degree in 2011 from Ritsumeikan University, under the guidance of Prof. Hiromitsu Maeda, focusing on the assemblies of π-conjugated anion receptors. He was selected as a Research Fellow of the Japan Society for the Promotion of Science (JSPS) in 2009–2012. After working in Asahi Kasei Corporation as a researcher (2012–2015), he joined the group of Prof. Hiromitsu Maeda at Ritsumeikan University as a postdoctoral fellow in 2015 and started an academic career as an assistant professor in 2017, and, in 2019, he became a lecturer. |
 Kazuhisa Yamasumi | Kazuhisa Yamasumi received his PhD degree in 2020 from Kyushu University under the guidance of Prof. Hiroyuki Furuta, focusing on isomeric porphyrin analogues. His PhD research was focused on the synthesis of infrared dyes based on the expanded or π-extended isomeric porphyrin analogues. He joined the group of Prof. Hiromitsu Maeda at Ritsumeikan University as a postdoctoral fellow in 2020. His current research focuses on π-electronic ion-pairing assemblies. |
 Hiromitsu Maeda | Hiromitsu Maeda received his PhD degree in 2004 from Kyoto University, under the guidance of Prof. Hiroyuki Furuta (Kyushu University) and Prof. Atsuhiro Osuka, after spending three months in the Sessler group, the University of Texas at Austin, in 2001. In 2004, he started an academic career in Department of Bioscience and Biotechnology, Faculty of Science and Engineering, Ritsumeikan University. In 2008, he was transferred to College of Pharmaceutical Sciences, wherein he was promoted to a professor in 2014. In 2016, he moved to Department of Applied Chemistry, College of Life Sciences. He has been awarded several prizes, including ChemComm Emerging Investigator Lectureship (2012) and Fellow of the RSC (2015). |
1. Introduction
Various functional materials that are applied in organic electronics can be fabricated by π-electronic systems that have electronic states depending on their core structures and substituents.1 Electronic properties of such materials are modulated by the arrangement of constituents via noncovalent interactions.2 Understanding and utilization of these interactions offer valuable insights for designing captivating assembled structures and materials. Noncovalent interactions, such as π–π, hydrogen-bonding and van der Waals interactions, are facilitated by combining fundamental intermolecular forces, including (i) electrostatic, induction and dispersion forces as long-range forces originating from coulombic forces and (ii) exchange-repulsion and charge-transfer (CT) forces as short-range forces caused by interorbital interaction.3 Electronically neutral π-electronic systems form stacking assemblies by dispersion forces, the main components of π–π interactions.The introduction of charge into π-electronic systems drastically changes the assembling behaviour based on electrostatic forces. Electrostatic and dispersion forces play a significant role in the arrangement of charged π-electronic systems in their assemblies.4,5 The assemblies feature stacking structures, with the fundamental units being π-stacked ion pairs (π-sips). These π-sips represent characteristic forms of paired oppositely charged π-electronic systems (π-electronic ion pairs) (Fig. 1a).6π-Sips form various ion-pairing assemblies with the modes of charge-by-charge assemblies, wherein oppositely charged species are alternately stacked; moreover, they provide charge-segregated assemblies, in which identically charged species are stacked via dissociation and recombination (Fig. 1a). The assembling modes are modulated by the geometries and electronic states of charged π-electronic systems in ion pairs. Various π-electronic ion pairs can be formed by combining π-electronic cations and anions; ten kinds of cations and ten kinds of anions ideally afford 100 kinds of ion pairs. Chemistry of π-electronic ion-pairing assemblies started in 2010 with the fabrication of charge-by-charge assemblies comprising π-electronic receptor–anion complexes and π-electronic cations in the form of single crystals, supramolecular gel and liquid crystals (vide infra, Section 3.1).7,8 In 2021, cooperative interactions involving electrostatic and dispersion forces were proposed as iπ–iπ interactions via detailed experimental and theoretical investigations, including single-crystal X-ray analysis and energy decomposition analysis (EDA) for solid-state ion-pairing assemblies.9 As previously defined, the iπ in “iπ–iπ interactions” represents π-electronic ions and charged π-electronic systems. iπ–iπ Interactions induce stacking of oppositely charged π-electronic systems owing to the enhanced contribution of attractive electrostatic and dispersion forces, resulting in the formation of π-sips and their assemblies. For identically charged π-electronic systems, iπ–iπ interactions can be modulated depending on the balance of electrostatic and dispersion forces, facilitating the formation of charge-segregated assemblies. The electronic states of charged π-electronic systems are closely related with their HOMO (highest occupied molecular orbital) and LUMO (lowest unoccupied molecular orbital), which are essential in redox processes, photophysical properties and reactivity. Stacked states of oppositely charged π-electronic systems with relatively high HOMO levels for anions and low LUMO levels for cations can induce charge- and electron-transfer behaviours. Diverse charged π-electronic systems have been reported as potential components of π-electronic ion pairs, including commercially available reagents. This review article summarizes the ion pairs comprising π-electronic cations and anions and their properties for potential applications as functional materials.
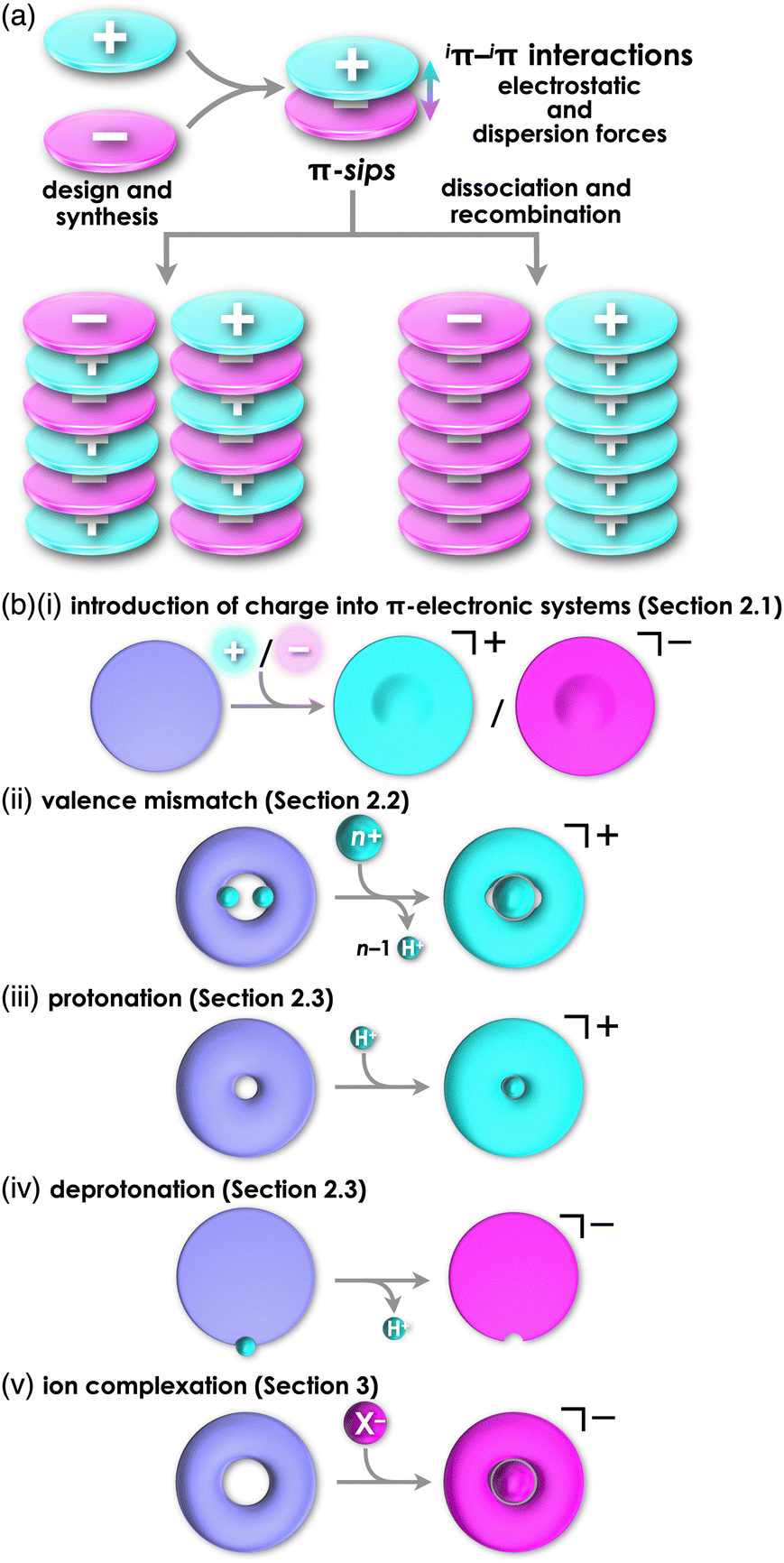 | ||
| Fig. 1 (a) Conceptual diagrams for the formation of π-sips (top) and charge-by-charge (bottom left) and charge-segregated assemblies (bottom right) comprising charged π-electronic species and (b) formation of charged π-electronic systems via “covalent approaches” by (i) introducing charges directly into the core, (ii) valence mismatch by metal complexation, denoted by cation formation as a representative and (iii) protonation and (iv) deprotonation at the basic and acidic sites, respectively, along with (v) ion complexation, as “noncovalent approach” in contrast to (i–iv), denoted by anion binding as a representative. | ||
Charged π-electronic systems as the components of π-electronic ion pairs can be obtained by diverse methods. A charge can be introduced by covalent and noncovalent approaches (Fig. 1b). In Section 2, several strategies using “covalent approaches” for the preparation of π-electronic charged species are displayed, such as the use of aromaticity and valence mismatch (Fig. 1b(i,ii)). Protonated and deprotonated species of π-electronic systems, using the coordination bond between a proton and a basic site for the former, are also included (Fig. 1b(iii,iv)). Section 3 demonstrates the formation of pseudo-π-electronic ions through the complexation of π-electronic systems and inorganic ions by noncovalent interactions (Fig. 1b(v)). This process results in the creation of ion pairs and their assemblies, including the charged π-electronic systems discussed in Section 2. Although various pseudo-π-electronic cations and anions are formed using this method, anion-complexation strategies are the main focus. In Section 4, pairs of the charged π-electronic systems shown in Section 2 are summarized as solution-state π-sips and assemblies in crystalline states and mesophases. The photophysical properties of π-electronic ion pairs are also displayed. However, radical ions derived from electronically neutral closed-shell π-systems are not included in this review.
2. π-Electronic systems with charge introduced by covalent approaches: components of ion pairs with non-π-electronic counterions
Diverse strategies, classified as covalent approaches, are available for the production of charged π-electronic systems (Fig. 1b(i–iv)). The introduced charge in π-electronic systems can be delocalized for stabilization. Aromaticity achieved by charge introduction further contributes to the stabilization of charged species. Valence mismatch between π-electronic ligands and metal ions also provides charged π-electronic systems. Furthermore, protonation and deprotonation of electronically neutral π-electronic systems are facile approaches to forming positively and negatively charged π-electronic systems.2.1. Introduction of charge into π-electronic systems
Carbocations and carbanions are generally less stable species, whose reactivity can be reduced by delocalizing the charges through extended π-systems (Fig. 1b(i)). For example, the triphenylmethyl cation (tritylium) 1+ (Fig. 2a(i)), which can be used as a Lewis acid in various organic synthesis, is more stable than the tert-butyl cation.10 The stability of cations is further improved by introducing electron-donating groups, as seen in crystal violet (2+ as a cation part, Fig. 2a(ii)).11 Moreover, the π-extension of tritylium can be achieved by reducing the steric repulsion between the ortho-CH units to form planar structures. Bridging two aryl groups with an atom results in the formation of planar structures as observed in rhodamine B (3a+ as a cation part, Fig. 2b(i)), which possesses a planar tricyclic π-unit that is characteristic of xanthene dyes.12,13 Rhodamine B, represented as 3a+-Cl−, exhibits strong absorption and fluorescence bands in the visible region at 554 and 627 nm, respectively, in MeOH. These properties make the dye suitable for fluorescence imaging applications.13a Furthermore, replacement of the oxygen with other heteroatoms, like phosphorus in 3b+ (Fig. 2b(ii)), alters the electronic states and photophysical properties. The absorption and emission maxima of 3b+ in a phosphate-buffered saline (PBS) buffer (pH = 7.4) are observed at 712 and 740 nm, respectively.13c Notably, various positively charged dyes, as seen in tritylium, crystal violet and rhodamine B, are commercially available.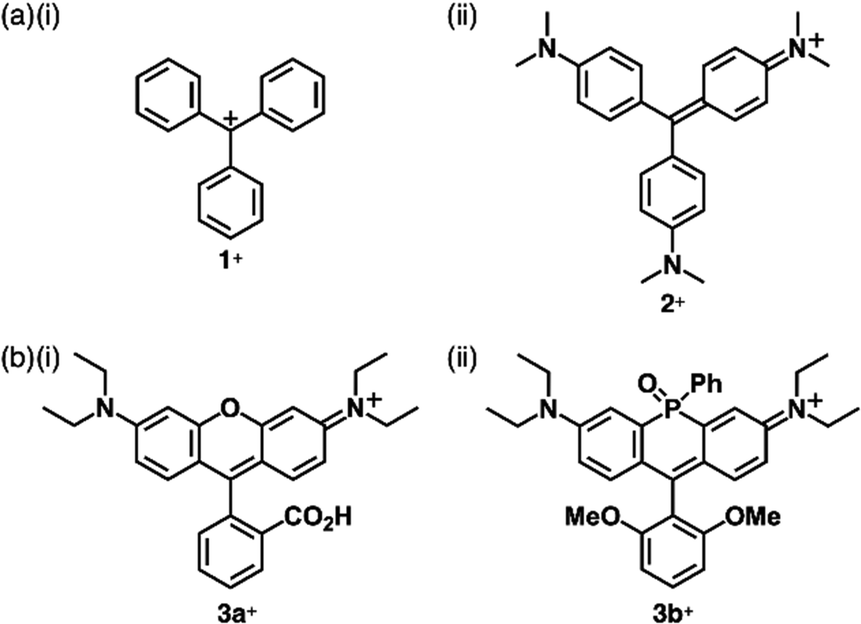 | ||
| Fig. 2 Representative charged triarylmethane dyes (a) 1+ and 2+ and (b) 3a,b+. | ||
Bridging the ortho-CH units of the three aryl groups of tritylium with three N or O linkers affords planar trianguleniums such as triazatriangulenium (TATA+)14 (Fig. 3a) and trioxatriangulenium (TOTA+)154a+ (Fig. 3b(i)). A variety of triangulenium derivatives, including less planar helicene structures,14a have been reported thus far for the use in various applications such as phase transition catalysis, bioimaging and DNA intercalation. In particular, the planar charged π-electronic systems are suitable for efficient electron-accepting properties and stacking in various supramolecular assemblies.16 For example, TATA+ bearing ether chains with a terminal hydroxy unit has been used as a visible-light absorbing photosensitizer in metal-catalyzed hydrogen production in water.16a Conversely, TOTA+ with aliphatic alkyl chains at one terminal (4b+, Fig. 3b(ii)) formed counteranion-depending nanoscale assemblies comprising stacked 4b+ (Fig. 3b(ii)).15c Planarization can also be applied to anion species: bridging triarylmethyl anion with three electron-withdrawing carbonyl groups afforded the trioxotriangulene anion 5− (Fig. 3c(i)).175− is thermodynamically stabilized owing to the charge-delocalized closed-shell structure, forming the ion pair with tetrabutylammonium (TBA+). Single-crystal X-ray analysis of TBA+-5− revealed that TBA+ and planar 5− were alternately stacked to form a columnar structure (Fig. 3c(ii)).
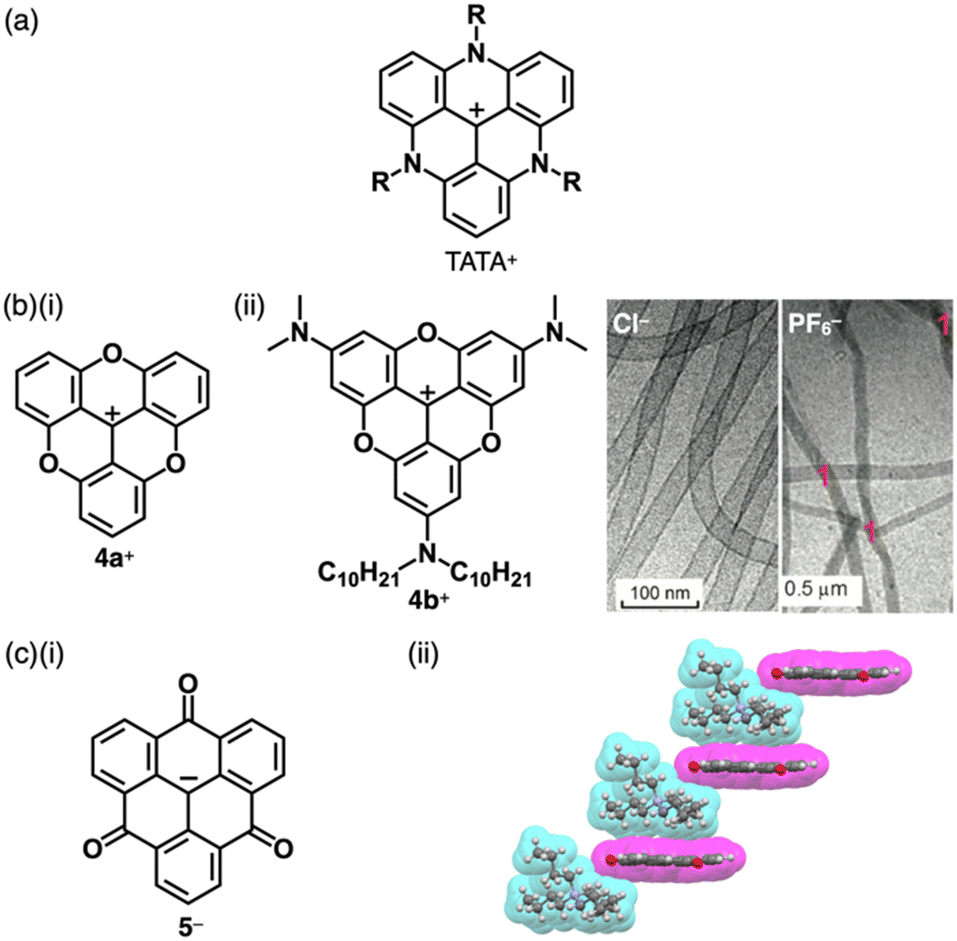 | ||
| Fig. 3 (a) General structure of TATA+, (b) (i) TOTA+4a+ and (ii) alkyl-substituted TOTA+4b+ and cryo-TEM images of nanostructures of 4a+-X− (X− = Cl− (left) and PF6− (right)) formed in water (10 vol% CH3CN) (redrawn from ref. 15c. Copyright 2014 Wiley) and (c) (i) trioxotriangulene anion 5− and (ii) single-crystal X-ray structure of TBA+-5− (redrawn from a cif file: 1303470). Atom colour code in (c) and the following figures: grey, white, blue and red in the ball-and-stick models refer to carbon, hydrogen, nitrogen and oxygen, respectively, whereas cyan and magenta in the space-filling models refer to cation and anion, respectively. | ||
Allyl ions and their extended ionic polymethines are stabilized by introducing two aryl groups at both ends (Fig. 4a).18 Various derivatives were synthesized as polymethine dyes with terminal aryl groups such as pyrylium, quinolinium18e (6a–c+, Fig. 4b(i)) and benzothiazolium18b (7a–c+, Fig. 4b(ii)). As polymethine dyes exhibit sharp absorption bands in the visible-to-near infrared (NIR) region depending on the length of the polymethine chains and the terminal π-electronic systems, cationic dyes are applied as fluorescent probes, laser dyes and photovoltaic materials. Generally, the polymethine dyes exhibit red-shifted absorption bands depending on the length of the linker polymethine units, as seen in the absorption maxima of 425, 558 and 660 nm for 7a–c+, respectively. The photophysical properties are affected by aggregation due to the significant exciton coupling caused by the large transition dipole moments. Therefore, assemblies of polymethine dyes have attracted great attention to date.18a,19 Furthermore, negatively charged π-electronic systems (e.g., 8−, Fig. 4c) are stabilized by the introduction of electron-withdrawing terminal π-units, although applications are limited owing to decreased stability.20
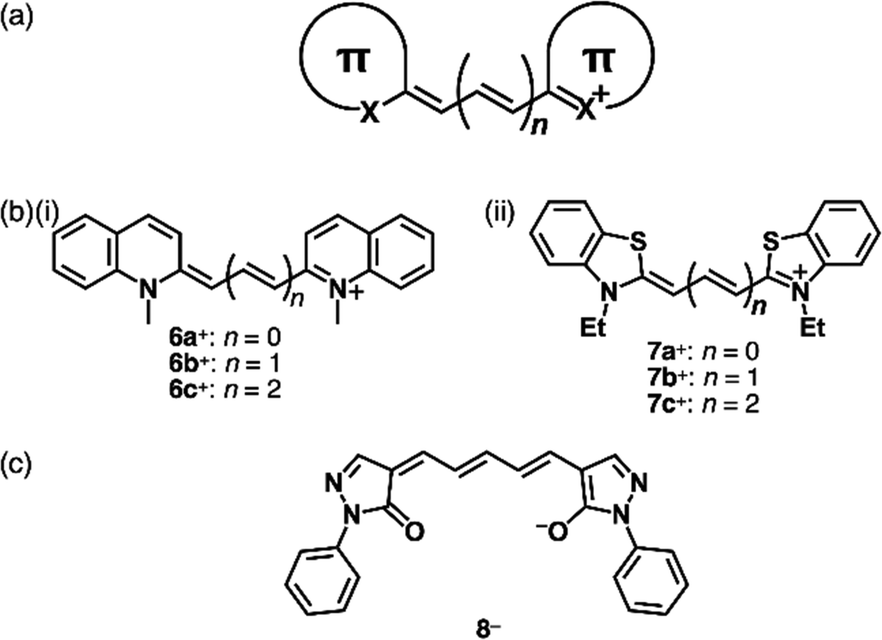 | ||
| Fig. 4 (a) General structure of polymethine dyes, (b) positively charged polymethine dyes based on (i) N-methylquinolium (6a–c+) (ii) benzothiazolium (7a–c+) and (c) negatively charged polymethine dye based on deprotonated oxonol (8−). | ||
Modifications at the centre of the polymethine chains affect the rigidity and modulate the photophysical properties.18 The positive charge can be compensated by the introduction of negatively charged substituents at the polymethine chains, as seen in zwitterionic squarylium 9 (Fig. 5a) and croconium dyes.21 These dyes are converted to the corresponding cations by introducing alkyl groups at the anionic site as seen in 10+ (Fig. 5a).22 In the crystal structures of ion pairs 10+-X− (X− = OTf− and B(C6F5)4−), the 10+ units are stacked by themselves. Notably, the stacked 10+ units exhibit antiparallel orientation to cancel their dipoles (Fig. 5b).23 Furthermore, 10+ shows sharp absorption bands in the NIR region, which are shifted depending on the assembling conditions (vide infra, Section 4.1).
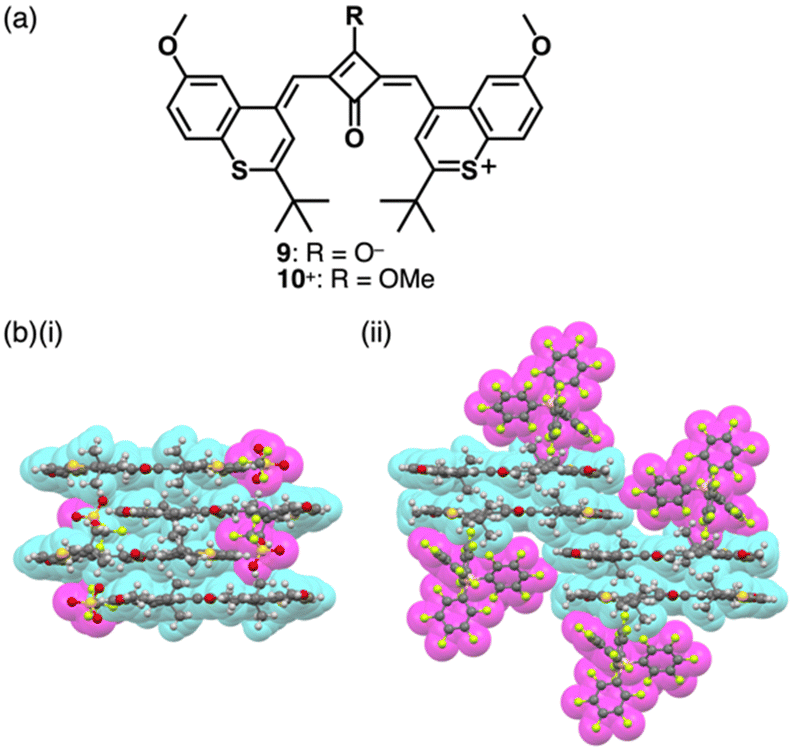 | ||
| Fig. 5 (a) Zwitterionic squarylium dye 9 and cationic squarylium dye 10+ and (b) single-crystal X-ray structures of (i) 10+-OTf− and (ii) 10+-B(C6F5)4− (redrawn from cif files: 2214017 and 2214019). Atom colour code in (b) and the following figures: pink, yellow green and dark yellow in the ball-and-stick models refer to boron, fluorine and sulfur, respectively. | ||
Charged units can be stabilized by aromaticity.24 A smallest class of aromatic compounds is cyclopropenium, a positively charged 2π electronic system (Fig. 6a(i)),25 which is highly reactive and participates in various reactions. The stability of cyclopropenium is increased by the introduction of electron-donating units, as seen in 11+ (Fig. 6a(ii)). Cycloheptatrienyl cation, tropylium (Tr+), a 6π electronic system, is a relatively stable π-electronic cation (Fig. 6b).26 In contrast, cyclopentadienide (Cp−) as a negatively charged 6π electronic system is stable enough for various metallocenes (Fig. 6c(i)).27 Delocalization of negative charges by π-extension can stabilize anions as seen in the tris(7H-dibenzo[c,g]fluorenylidenemethyl)methyl anion (Kuhn's anion) 12− (Fig. 6c(ii)), a genuine hydrocarbon charged π-electronic anion including Cp− units.28 Kuhn's anion has been synthesized as ion pairs with various cations, including π-electronic cations. Owing to the anion's easily oxidizable nature, the ion pairs must be handled under an inert atmosphere and light shading conditions to prevent unwanted reactions or degradation.
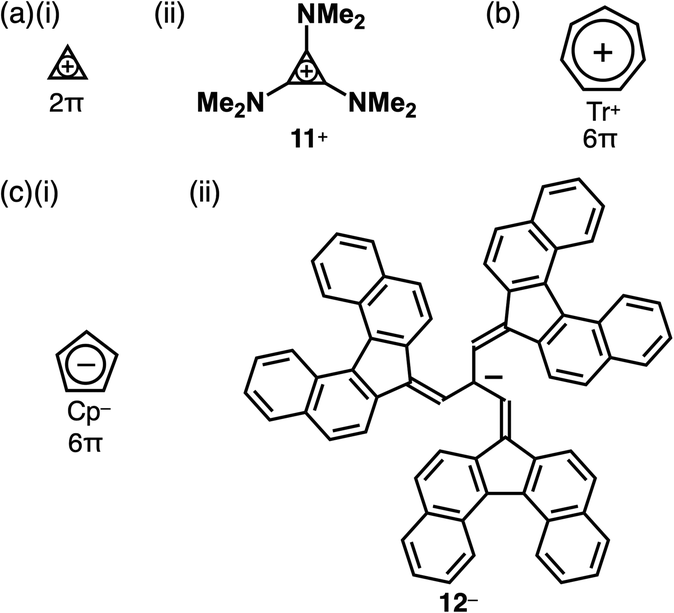 | ||
| Fig. 6 Representative charged π-electronic systems stabilized by aromaticity: (a) (i) cyclopropenium and (ii) tris(dimethylamino)-substituted derivative 11+, (b) tropylium and (c) (i) cyclopentadienide and (ii) Kuhn's anion 12−. | ||
The stability of Cp− can be enhanced by the introduction of electron-withdrawing units. For example, various ion pairs have incorporated pentamethoxycarbonylcyclopentadienide 13− (Fig. 7a(i)).29 The crystal structure of TMA+-13− (TMA+ = tetramethylammonium) revealed that dihedral angles between methoxycarbonyl CO2C planes and the core cyclopentadienide unit were within a range of 15.7°–85.9°, exhibiting a nonplanar structure (Fig. 7a(ii)).30 Replacement of the ester groups with cyano groups as less bulky electron-withdrawing groups allows the production of planar anions that are available for more effective stacking with charged π-electronic systems. Since the synthesis of pentacyanocyclopentadienide (PCCp−)31 by Webster in 1965 (Fig. 7b(i)),31a the anion has been used as the building block of solid-state coordination networks in combination with metal ions.32 Exchange of the countercation with other cations via ion-pair metathesis as an ion-exchange protocol based on hard and soft acids and bases (HSAB) theory (details are summarized in Section 4) afforded various ion pairs, demonstrating unique assembled structures.33,34 Single-crystal X-ray analysis of (C4H9)(CH3)3N+-PCCp− exhibited a columnar structure comprising stacking of PCCp− with a distance of 3.42 Å (Fig. 7b(ii)). Notably, (C4H9)(CH3)3N+ is located proximally to PCCp−, constructing a charge-segregated assembly. Furthermore, a charge-segregated assembly in the form of a hexagonal columnar (Colh) structure was observed in the liquid crystal mesophase of (C12H25)3CH3N+-PCCp− (Fig. 7b(iii)), and its hole mobility was estimated to be 0.4 cm2 V−1 s−1 using field-induced time-resolved microwave conductivity (FI-TRMC) technique.33
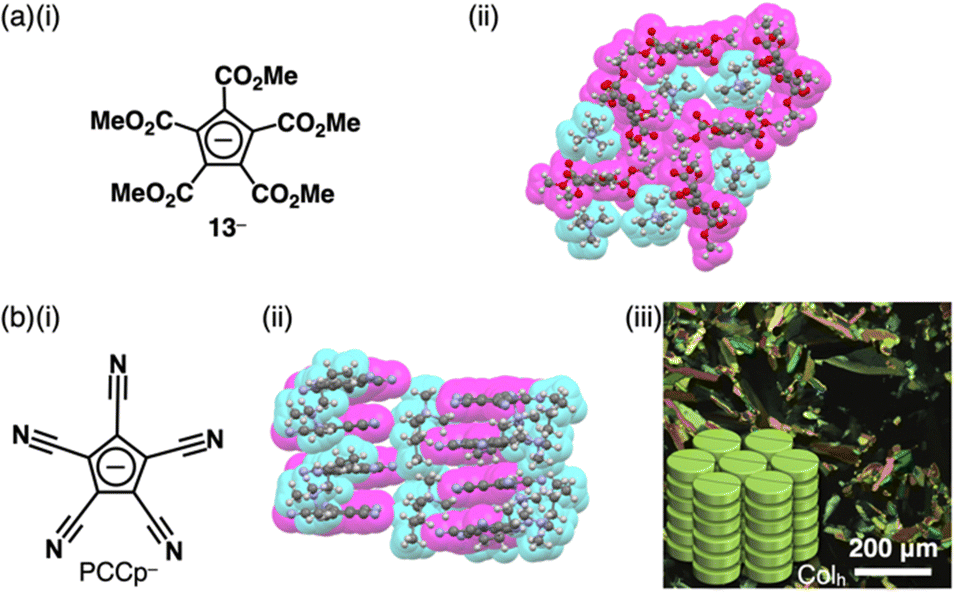 | ||
| Fig. 7 (a) (i) Pentamethoxycarbonylcyclopentadienide 13− and (ii) single-crystal X-ray structure of TMA+-13− and (b) (i) pentacyanocyclopentadienide (PCCp−), (ii) single-crystal X-ray structure of (C4H9)(CH3)3N+-PCCp− and (iii) POM of liquid crystal mesophase of (C12H25)3CH3N+-PCCp− at 70 °C upon cooling (redrawn from ref. 33. Copyright 2016 Wiley). The crystal structures are redrawn from cif files: 1137646 and 1431736. | ||
Replacement of carbons with heteroatoms in aromatic π-electronic systems affords charged aromatic π-electronic systems (Fig. 8). Notably, the replacement of a CH unit in benzene with substituted nitrogen and oxygen/sulfur produces pyridinium, pyrylium and thiopyrylium, respectively (14a–c+ as triphenyl-substituted derivatives, Fig. 8a).35 Taking advantage of the electron-deficient nature and moderate reactivity, 2,4,6-triarylpyryliums (e.g., 14b+) are used as photocatalysts and building blocks of extended π-electronic systems.35b Some charged species, such as pyridinium, are stable under ambient conditions. Other charged π-electronic systems can be stabilized by π-extensions as seen in peripherally heteroatom-containing phenanthro[2,3,4,5-pqrab]perylenes as π-extended pyridinium, pyrylium and thiopyrylium 15a–d+ reported by Feng, Müllen and coworkers (Fig. 8b(i)),36 whose electronic properties clearly depend on the introduced heteroatoms. Red-shifted UV/vis absorption and emission maxima occur at 512/531, 573/594 and 600/643 nm for 15a,b,d+ corresponding to BF4−, Br− and BF4− ion pairs, respectively, in CH2Cl2 (Fig. 8b(ii)). Dodecyl-substituted 15c+ as a Br− ion pair forms liquid crystal mesophases based on the Colh structure (Fig. 8b(iii)). Conversely, the internal carbon of naphthalene can be replaced with nitrogen, resulting in quinolizinium as the smallest N-doped polycyclic aromatic hydrocarbon (PAH).37 Electron-deficient planar charged π-electronic systems with appropriate sizes and substituents can be applied to DNA intercalation. Representative π-extended N-doped PAHs include 9-phenylbenzo[1,2]quinolizino[3,4,5,6-fed]-phenanthridinylium (PQP+) cations 16a,b+ (Fig. 9a), synthesized from a 2,4,6-triphenylpyrylium salt.38 PQP+ forms charge-segregated assemblies viaiπ–iπ interactions.38b In particular, PQP+ ion pairs 16b+-X− (X− = Cl− and BF4−) form counteranion-dependent ion-pairing assemblies with distinct morphologies (Fig. 9b).38a TEM images of the assemblies exhibit nanoribbons and nanotubes for the Cl− and BF4− ion pairs, respectively, suggesting that even small counteranions have important roles in the arrangement of charged π-electronic units and resulting organized structures.
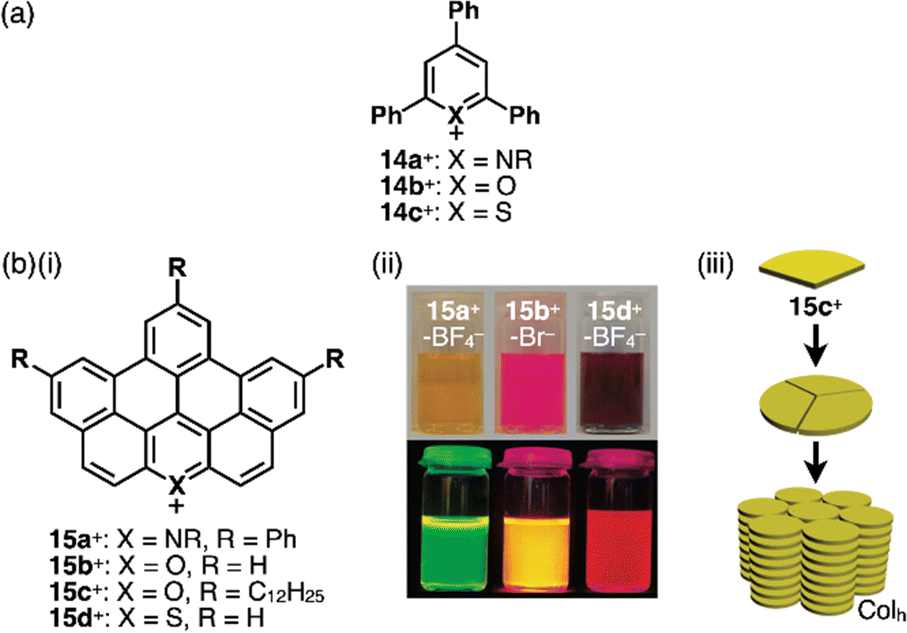 | ||
| Fig. 8 (a) Triphenylpyridinium and triphenylpyryliums 14a–c+ and (b) (i) π-extended pyridinium and pyryliums 15a–d+, (ii) photographs of CH2Cl2 solutions of 15a+-BF4−, 15b+-Br− and 15d+-BF4− (top: under visible light, bottom: under UV) and (iii) packing diagram of 15c+-Br− (redrawn from ref. 36. Copyright 2009 ACS). | ||
 | ||
| Fig. 9 (a) PQP+16a,b+ and (b) TEM images of aggregates in MeOH (1 mM): (i) 16b+-Cl− and (ii) 16b+-BF4− (redrawn from ref. 38a. Copyright 2007 Wiley). | ||
Porphyrin, consisting of four pyrrole rings bridged by methine units, is an 18π electronic aromatic macrocycle.39 Porphyrin and its analogues play important roles in biological systems such as metabolism and photosynthesis. The large π-electronic system of porphyrin contributes to charge delocalization and, thus, stabilization of charged species. Various stacking assemblies can be formed from porphyrin derivatives.19,40 Therefore, porphyrin is an excellent building block for charged π-electronic systems as components of π-electronic ion-pairing assemblies. In addition, the redox properties of porphyrin can be modulated by peripheral substituents. The introduction of substituted N and O/S at one of the porphyrin meso positions affords positively charged heteroatom-containing porphyrins 17a–c+ (Fig. 10a(i)).41 The inner NH protons of 17a,c+ are located at the nitrogens in the trans positions, whereas those of 17b+ are located at those in the cis positions, far from the introduced meso-oxygen.41b–d This arrangement suggests charge delocalization and resulting polarized structures attributed to the introduced atoms. In the crystal structures of 17a–c+-BArF− (BArF− = B(3,5-(CF3)2C6H3)4−), the cations are stacked by themselves on the π-plane, while further stacking is inhibited by BArF− located on the π-plane (Fig. 10a(ii)). Furthermore, tetraaryl-5,15-diazaporphyrin NiII complex 182+ as a dication in PF6− and SbCl6− ion pairs can be obtained by oxidizing the corresponding neutral form with AgPF6 or magic blue (Fig. 10b).42 Moreover, a similar approach to the one mentioned in the previous paragraph can be applied to heterocyclic systems, where heteroatoms are converted to other heteroatoms with different valences. Heteroporphyrins 19a,b2+, whose inner nitrogens are replaced with chalcogens, form various charged π-electronic systems (Fig. 10c(i)).43 In the crystal structure of the ClO4− ion pair, 19a2+ appears highly planar and forms a columnar structure, slip-stacking by itself on both sides (Fig. 10c(ii)). However, the thiophene units in 19b2+ are inclined by 22.8° and 3.7° in the crystal structure.43c
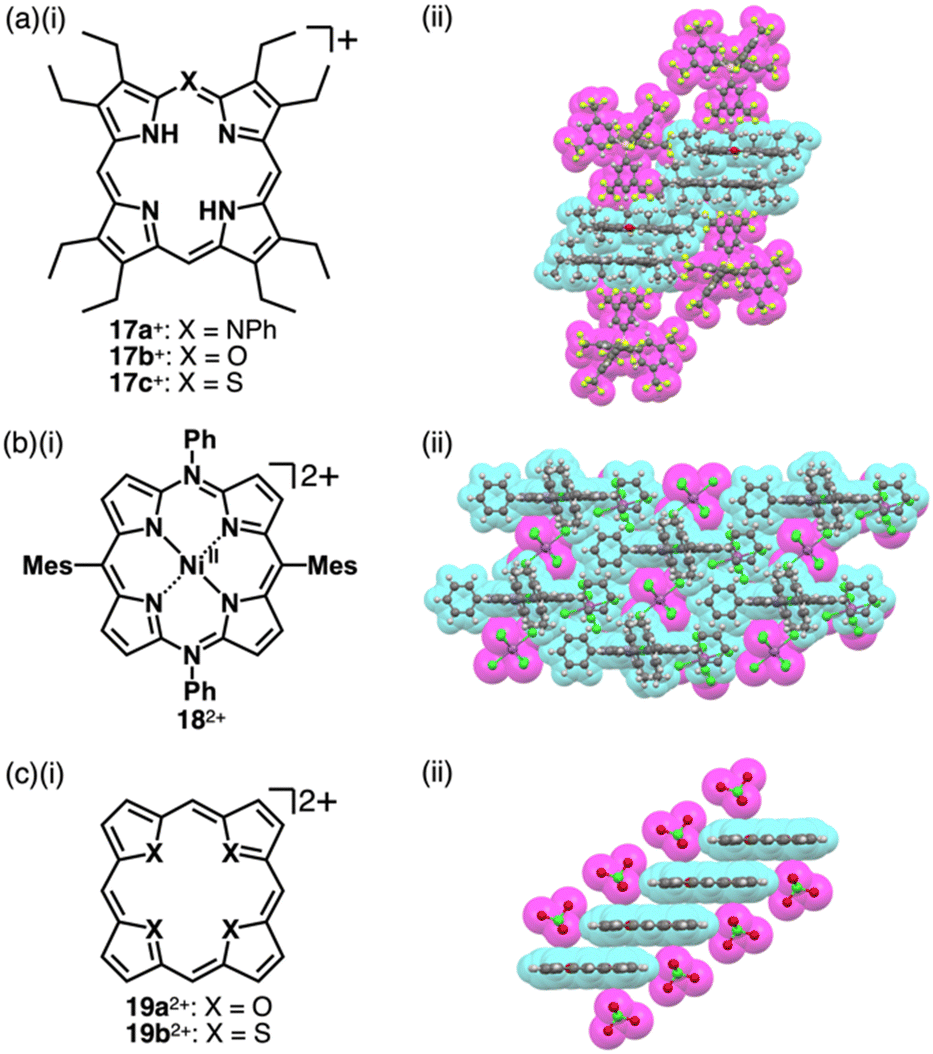 | ||
| Fig. 10 (a) (i) Heteroatom-containing porphyrins 17a–c+ and (ii) single-crystal X-ray structure of 17a+-BArF−, (b) (i) tetraaryl-5,15-diazaporphyrin NiII complex 182+ and (ii) single-crystal X-ray structure of 182+-2SbCl6− and (c) (i) core-modified porphyrins 19a,b2+ and (ii) single-crystal X-ray structure of 19a2+-2ClO4−. The crystal structures are redrawn from cif files: 2033992, 1437728 and 1166405. Atom colour codes in (b) (c) (ii) and the following figures: green, blue-green and purple in the ball-and-stick models refer to chlorine, nickel and antimony, respectively. | ||
2.2. Charged π-electronic systems derived from valence mismatch
Charges in complexes comprising multiple units can be controlled by the valences of the components. Therefore, if the number of positive charges of metal ions is not matched with that of the total negative charges of ligands, the resulting complexes behave as charged species (Fig. 1b(ii)). Metal complexes with such a valence mismatch are used in Magnus’ green salt: a salt comprising [Pt(NH3)4]2+ and [PtCl4]2−, alternately arranged to form an anisotropic quasi 1D linear array in the solid state (Fig. 11).44,45 The 1D structure of the aligned PtII complexes in Magnus’ green salt is mainly constructed by the electrostatic force between the positively and negatively charged planar PtII complexes. The fascinating ion-pairing structure has stimulated numerous studies regarding the use of 1D arrays as electrically conductive wires. Importantly, the correlation between the geometries of the constituents and the assembled structure has been clearly demonstrated. This observation has prompted us to design and synthesize charged π-electronic species for fabricating various functional ion-pairing assemblies through iπ–iπ interactions.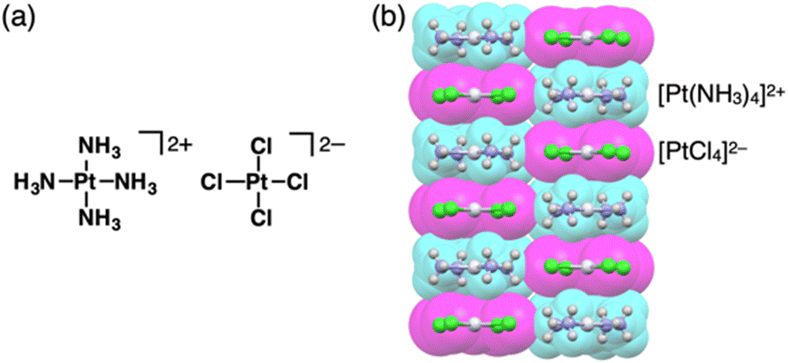 | ||
| Fig. 11 Magnus’ green salt: (a) ion-pairing form and (b) single-crystal X-ray structure (redrawn from a cif file: 2009952). Atom colour code in (b) and the following figures: light grey in the ball-and-stick models refers to platinum. | ||
As π-electronic ligands in preparation for charged π-electronic species by valence mismatch, porphyrins serve as dianionic ligands owing to the two inner NH groups. Therefore, complexation with trivalent metal ions provides π-electronic cations accompanied by an anion for charge compensation. Versatile porphyrin complexes with trivalent metal ions such as CrIII, MnIII, FeIII and CoIII have been prepared thus far.39 Most of the metal complexes have axial ligands, which compensate for the positive charge and hamper the stacking of the resulting π-electronic systems. 20+, a porphyrin FeIII complex with bulky triisopropylsilyloxy substituents, can be obtained in the neutral form coordinated by a Cl− ligand.46 Here, the Cl− is exchanged with other anions by ion-pair metathesis with Ag+ salts of weakly coordinating anions (ClO4−, OTf− and SbF6−) (Fig. 12a). Hexabromocarborane anion (CH6B11Br6−)47 is too bulky to approach the axial coordination site owing to the steric hindrance, resulting in π-electronic cations without axial ligands. Boron complexes of subporphyrins usually exhibit bowl-like structures attributed to the structural distortion caused by axial coordination at the boron centre.48 Treatment of methoxide-coordinated subporphyrin boron complex with Et3Si[CH6B11Br6] afforded a planar subporphyrin cation 21+ as an ion pair with CH6B11Br6− (Fig. 12b).49 Conversely, porphyrin AuIII complexes, whose metal centre is in a d8 electronic state, generally have no axial ligands (e.g., 22a–e+, Fig. 13a).6,50–53 Thus, porphyrin AuIII complexes readily form ion pairs in combination with desired anions and can be adopted as building blocks for π-electronic ion-pairing assemblies.6,52
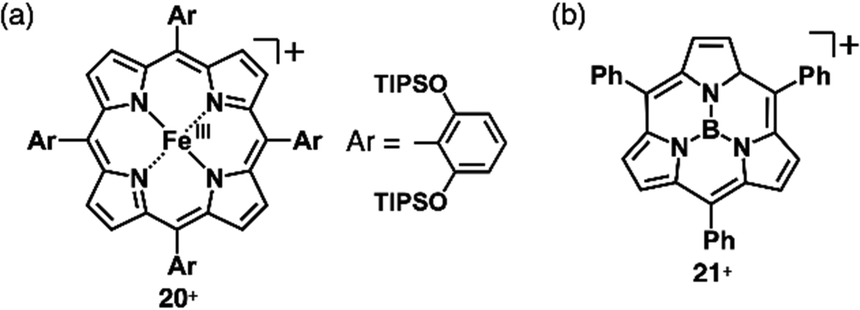 | ||
| Fig. 12 Positively charged porphyrin and analogue formed by valence mismatch: (a) porphyrin FeIII complex 20+ and (b) subporphyrin boron complex 21+. | ||
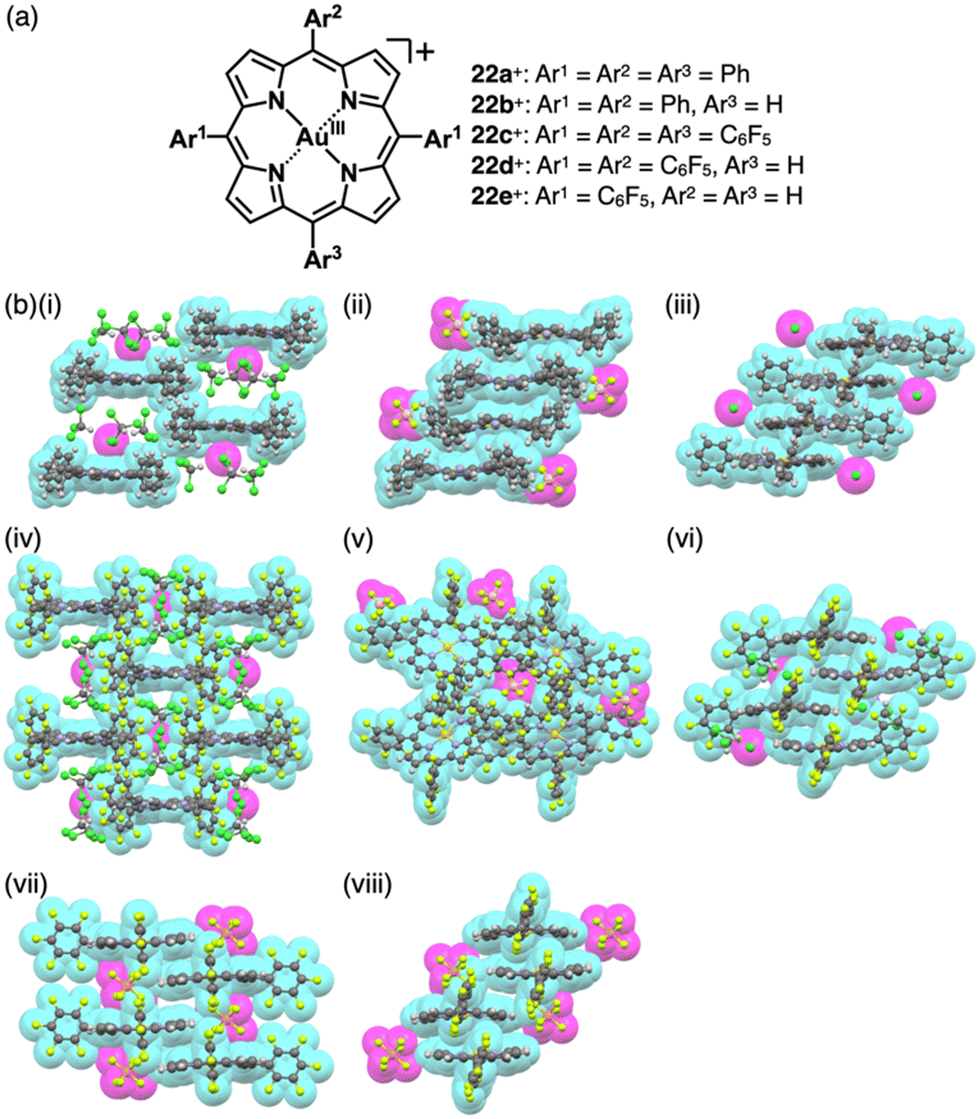 | ||
| Fig. 13 Positively charged porphyrins formed by valence mismatch: (a) meso-arylporphyrin AuIII complexes 22a–e+ and (b) single-crystal X-ray structures of (i) 22a+-Cl−, (ii) 22a+-BF4−, (iii) 22b+-Cl−, (iv) 22c+-Cl−, (v) 22c+-BF4−, (vi) 22d+-Cl−, (vii) 22d+-PF6− and (viii) 22e+-PF6− (redrawn from cif files: 1877987, 1877988, 2226630, 1904175, 1904176, 1904174, 1904178 and 1904177). Atom colour codes in (b) and the following figures: orange and yellow in the ball-and-stick models refer to phosphorus and gold, respectively. | ||
Porphyrins can be complexed with AuIII using different procedures. One well-known method involves using KAuCl4 in refluxing AcOH.51a Other synthetic approaches, such as the uses of HAuCl4·4H2O/AgOTf/NaOAc51d and Au(tht)2BF4 (tht = tetrahydrothiophene)/2,6-lutidine,51c can be applied depending on the reactivity of the porphyrins. Counteranions of the porphyrin AuIII complexes after the AuIII complexation are mainly derived from the corresponding AuIII reagents. To exchange the counteranions to Cl−, porphyrin AuIII complex ion pairs can be passed through an ion-exchange resin (Amberlite). Moreover, the Cl− ions can be exchanged with inorganic salts containing the desired anions through ion-pair metathesis, resulting in various ion pairs. The high stability of these ion pairs allows for purification using silica gel column chromatography.52
Single-crystal X-ray analysis revealed the exact structures of porphyrin AuIII complexes with no axial ligands. 22a+-Cl− in a pseudo-polymorph shows a columnar structure based on a charge-by-charge assembly including Cl− with four co-crystallized CHCl3 molecules (Fig. 13b(i)).51b,52a The proximal Au⋯Cl− distance was 3.12 Å, comparable to the sum of the ionic radii of Au3+ and Cl−, suggesting the formation of a contact ion pair. In addition, the line passing through both Au and Cl has an angle of 80.2° to the mean plane of 22a+. The non-90° angle indicates that Cl− is not directly coordinated to the core AuIII in proximity to 22a+. Instead, it exists as a π-electronic cation, and the interaction is primarily based on electrostatic interactions. In contrast to the charge-by-charge structure of 22a+-Cl−, charge-segregated assemblies are formed in 22a+-BF4− and 22a+-PF6− (Fig. 13b(ii)), exhibiting columnar structures of stacked 22a+ with stacking distances of 3.73/3.88 and 3.87/3.89 Å, respectively.52a Conversely, ion pairs of meso-triphenyl-substituted 22b+ with Cl− form charge-segregated assemblies with stacking distances of 3.50 and 3.74 Å (Fig. 13b(iii)), whereas 22b+-PF6− exhibits a stacked dimer of 22b+ with a stacking distance of 3.46 Å. In both cases, counteranions are located at either side of 22b+.52b
The assembling behaviours of the ion pairs are affected by the introduction of electron-withdrawing C6F5 groups (22c–e+, Fig. 13a).52b The Cl− in 22c+-Cl− is in contact with the β-CH units of two porphyrins, resulting in layered structures separated by the bulky C6F5 groups (Fig. 13b(iv)). Therefore, the Au⋯Cl distance (7.08 Å) is much longer than those of 22a+-Cl− (3.00 and 3.12 Å). Meanwhile, in 22c+-BF4−, the C6F5 groups are arranged around BF4− with interionic π⋯F short contacts (Fig. 13b(v)). In 22d+-Cl−, Cl− is separated from the AuIII centre by CH2Cl2 (Fig. 13b(vi)) and is located proximally to the meso-CH of 22d+. The arrangements of the cations and PF6− in 22d+-PF6− and 22e+-PF6− are similar to that of 22d+-Cl−, although the interplanar distances are slightly longer (3.50–3.84 Å) than that of 22d+-Cl− (Fig. 13b(vii,viii)).52b
A similar approach can be applied to divalent metal complexes with monoanionic ligands such as thiaporphyrins (tetraphenylthiaporphyrin NiII complex 23+ as a representative structure, Fig. 14a).39,54 In 1989, Latos-Grażyński, Balch and coworkers reported 23+ and CuII and FeII complexes as Cl− ion pairs.54 In the crystal structure of 23+-Cl−, the Ni⋯Cl− distance is 2.28 Å, indicating the axial coordination. The thiophene unit of 23+ is inclined by ∼25°, leading to a less planar structure. Furthermore, ion pair 23+-PF6−, prepared by ion-pair metathesis of 23+-Cl− with AgPF6, forms the solid-state stacked dimer structure, wherein the S⋯N distance is 3.77 Å owing to chalcogen bonding (Fig. 14b).55
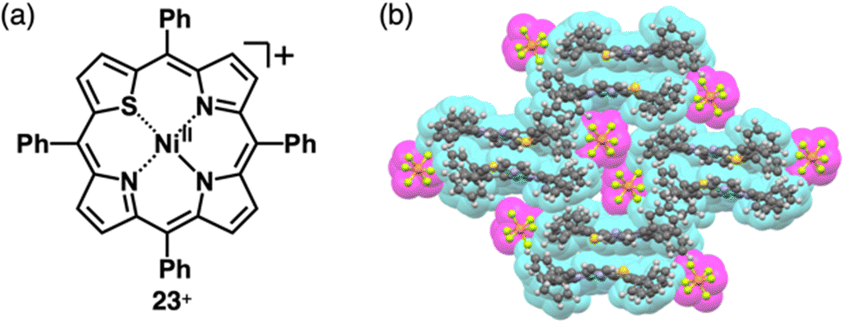 | ||
| Fig. 14 (a) Thiaporphyrin NiII complex 23+ and (b) single-crystal X-ray structure of 23+-PF6− (redrawn from a cif file: 2167302). | ||
Similarly, anionic species can be obtained by complexing divalent metals with trianionic ligands such as corrole. The introduction of electron-withdrawing substituents at appropriate positions is required for stable π-electronic anions. Gross et al. reported meso-C6F5-substiuted corrole NiII complex 24− as a monoanionic species in the TBA+ ion pair (Fig. 15a(i)).56 Here, the stability of the corrole anion was increased by introducing bromo units at the β-positions. Single-crystal X-ray analysis of TBA+-24− revealed that planar 24− and TBA+ were alternately arranged, forming a columnar structure (Fig. 15a(ii)). Similarly, the octaethylporphyrin LiI complex 25−, consisting of monovalent metal and dianionic ligand, behaves as a π-electronic anion (Fig. 15b(i)).5725− was synthesized as a [Li(thf)4]+ (thf = tetrahydrofuran (THF)) ion pair by LiI metallation of octaethylporphyrin in THF. In [Li(thf)4]+-25−, the components are alternately arranged, and the 25− units are inclined to each other (Fig. 15b(ii)). Moreover, [Li(thf)4]+-25− is sensitive to moisture and undergoes demetallation by hydrolysis.
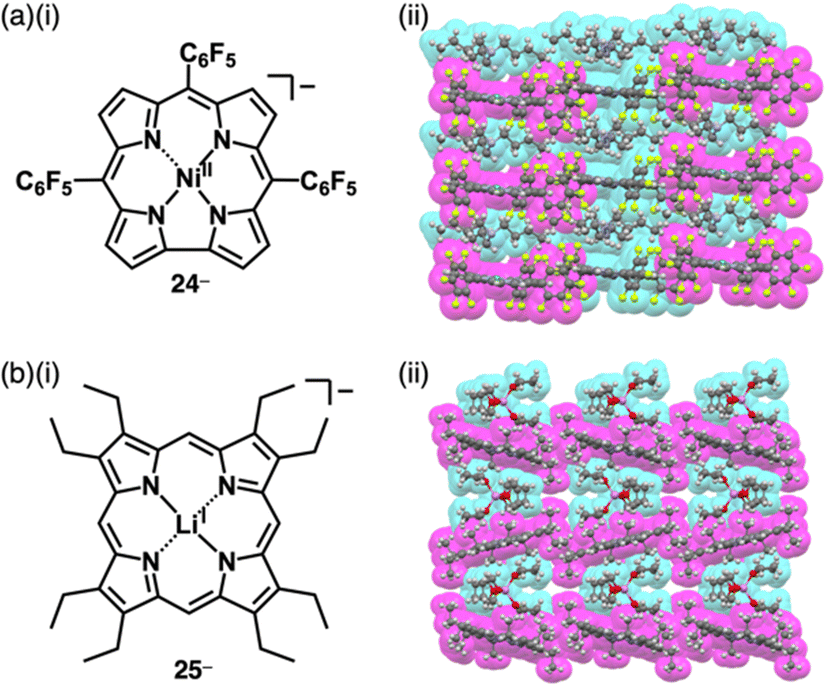 | ||
| Fig. 15 (a) (i) Corrole NiII complex 24− and (ii) single-crystal X-ray structure of TBA+-24− and (b) (i) porphyrin LiI complex 25− and (ii) single-crystal X-ray structure of [Li(thf)4]+-25− ([Li(thf)4]+ is highlighted with cyan owing to the coordination of THF to Li+). The crystal structures are redrawn from cif files: 2122003 and 1184624. Atom colour code in (b) (ii) and following figures: light purple in the ball-and-stick models refers to lithium. | ||
A variety of charged metal complexes based on neutral π-electronic ligand molecules, such as terpyridine, with another ligand for the partial charge compensation can be used to produce positively charged π-electronic species (e.g.,26+, Fig. 16a).58 Terpyridine PtII complexes with a variety of ligands at PtII afford planar monocationic species, demonstrating aggregation behaviour using iπ–iπ and PtII⋯PtII interactions.59 Yam et al. reported a terpyridine PtII complex with an aliphatic alkynyl ligand 26a+-X− with different anions (X− = Cl−, OTf−, BF4− and PF6−), exhibiting luminescence and gelation properties (Fig. 16b(i)).59b The OTf− ion pair formed coil-shaped helical fibres, whereas other ion pairs formed disordered fibres (Fig. 16b(ii)). Furthermore, in 2012, the groups of Che and Yam independently reported a class of monocationic cyclometalated AuIII alkynyl complexes 27a,b+ (Fig. 17a).60 The positive charges of the AuIII are partially cancelled by phenylbipyridyl and alkynyl ligands, affording charge-segregated assemblies comprising stacked AuIII complexes (Fig. 17b). The short AuIII⋯AuIII distances of 3.495 and 3.692 Å in 27a+-PF6− and 27b+-PF6− suggest favourable AuIII⋯AuIII interactions as confirmed by theoretical calculations. In 27a+-PF6−, contributions of iπ–iπ and AuIII⋯AuIII interactions facilitate the formation of belt-like assemblies.60a Tuning of metal centre valences also results in anionic species.61 Lalinde et al. prepared stable anionic PtII complexes with diphenylpyridine and alkynyl ligands (e.g., 28−, Fig. 18).61a Here, TBA+ was selected for charge compensation of anionic PtII complexes. Single-crystal X-ray analysis clearly elucidated the planar geometries of the anionic PtII complexes, which were further used as guest species for cationic PtII tweezers, exhibiting more efficient binding behaviour (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K = 13.5 in CH2Cl2) compared to an uncharged PtII complex.62
K = 13.5 in CH2Cl2) compared to an uncharged PtII complex.62
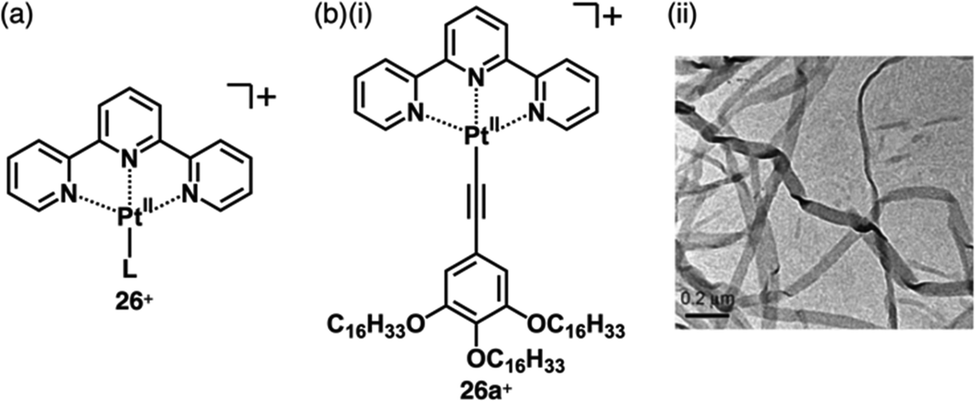 | ||
| Fig. 16 (a) General structure of positively charged terpyridine PtII complexes 26+ and (b) (i) terpyridine PtII complex with aliphatic chains and (ii) TEM image of the DMSO gel of 26a+-OTf− (redrawn from ref. 59b. Copyright 2009 Wiley). | ||
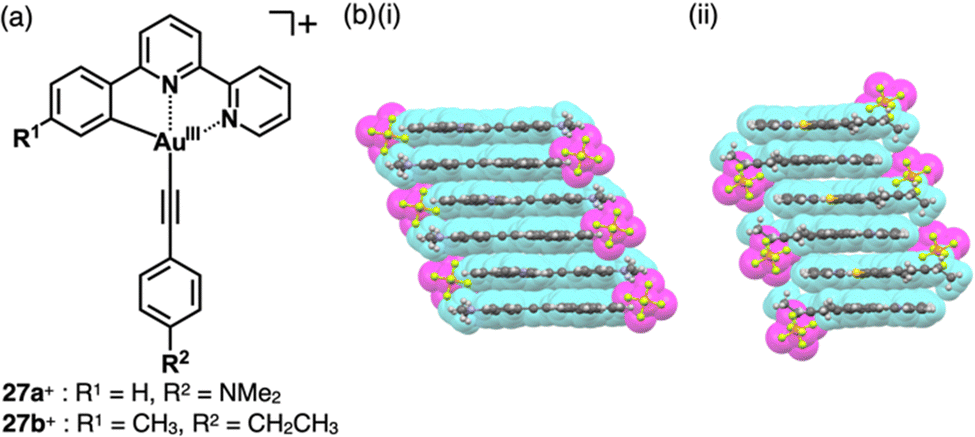 | ||
| Fig. 17 (a) Positively charged phenylbipyridine AuIII complexes 27a,b+ and (b) single-crystal X-ray structures of (i) 27a+-PF6− and (ii) 27b+-PF6− (redrawn from cif files: 841764 and 897336). | ||
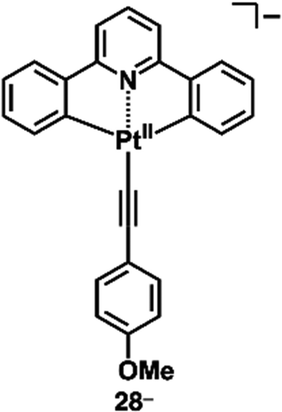 | ||
| Fig. 18 Negatively charged diphenylpyridine PtII complex 28−. | ||
2.3. Charged π-electronic systems formed by protonation and deprotonation
Reversible proton attachment and detachment can be considered covalent approach methods via the modulation of covalent linkages (Fig. 1b(iii,iv)). Proton (H+) is the simplest cation, and protonation of electronically neutral π-electronic systems produces π-electronic cations. The charged π-electronic systems are often deprotonated, reverting neutral π-electronic systems. Thus, π-electronic systems with high proton affinity, achieved by multiple hydrogen bonds, are required to form π-electronic cations suitable for ion pairing.63,64 Protonated azacalix[3](2,6)pyridine 29a+ has a cavity with three imine nitrogen sites, exhibiting high basicity (pKa = 23.1 in CD3CN) (Fig. 19a(i)).63a,b A planar structure with a bound proton in the cavity was investigated by single-crystal X-ray analysis of 29a+-PF6− (Fig. 19a(ii)). A columnar structure of slip-stacked 29a+ is formed with PF6− located at the side of the columns. The basicity is further enhanced by introducing electron-donating pyrrolidine units at the pyridine 4-positions in 29b+ (pKa = 28.1 in CD3CN)), facilitating its application as organocatalysis for Michael addition reactions.63b Replacement of the pyridyl units with triazine results in a further electron-deficient π-electronic cation azacalix[3]triazine 30+ (Fig. 19b(i)), maintaining high basicity (pKa = 16.7 for the ion pair with 30+-Cl− in DMSO).63d The protonated species 30+ interacts with a counteranion, affecting the pKa values (17.1 and 16.2 for the NO3− and PF6− ion pairs, respectively, in CD3CN). Single-crystal X-ray analysis of 30+-PF6− reveals the planar structure of 30+ with anion–π interactions between PF6− and triazine units, resulting in the formation of discrete assemblies in the form of PF6−-30+-PF6−-30+-30+-PF6−-30+-PF6− (Fig. 19b(ii)). Protonated triquinoline 31+ (Fig. 19c) possesses a planar rigid skeleton with a proton-binding site, as evidenced by the persistence of >10% of the inner NH 1H NMR signal after one week in CD3OD.64 These highly basic macrocycles have been utilized as base catalysts in organic synthesis, and can serve as π-electronic cations in ion-pairing assemblies when paired with suitable anions.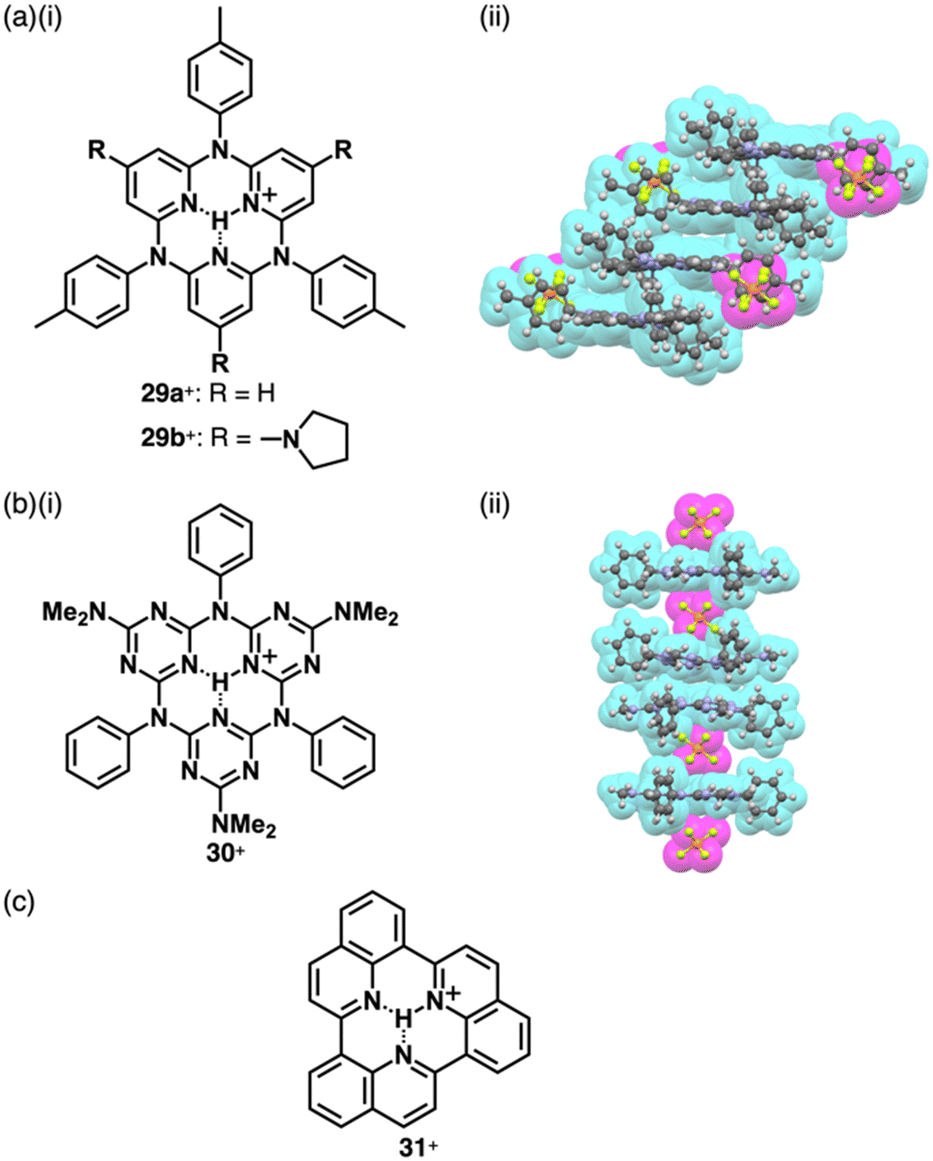 | ||
| Fig. 19 (a) (i) Protonated azacalix[3](2,6)pyridine 29a,b+ and (ii) single-crystal X-ray structure of 29a+-PF6− (redrawn from a cif file: 298963), (b) (i) protonated azacalix[3]triazine 30+ and (ii) single-crystal X-ray structure of 30+-PF6− (redrawn from a cif file: 2072959) and (c) protonated triquinoline 31+. | ||
Covalent linkage between charged substituents and π-electronic systems more drastically affects the electronic states of charged π-electronic systems. A ubiquitous negatively charged moiety is O−, formed by the deprotonation of a hydroxy group. A π-electronic anion precursor is phenol, whose pKa is 10.00 in an aqueous solution.65 π-Extended phenol derivatives, such as fluorescein (pKa = 6.7 for the phenolic OH),66 whose dianion form is uranine 322− (Fig. 20a), are also commercially available as Na+ salts. Fluorescein derivatives that exhibit pH-dependent photophysical properties are used as pH indicators. Phenoxides are further stabilized by introducing electron-withdrawing groups as seen in picric acid (pKa of 0.40),65 providing 33− upon deprotonation (Fig. 20b).
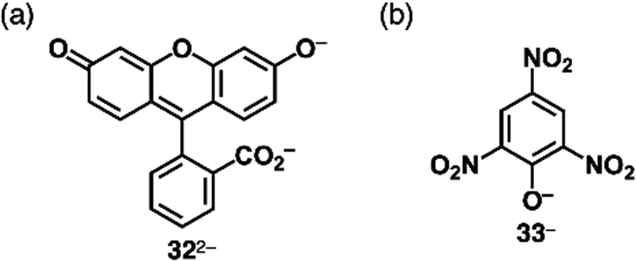 | ||
| Fig. 20 (a) Uranine 322− and (b) deprotonated picric acid 33−. | ||
4-Nitrophenol (pKa = 7.14), whose acidity is weaker than picric acid, can be used as a building unit of π-electronic anions. Introduction of pyrrole at the ortho-positions of the deprotonated site affords the intramolecular N–H⋯O− interactions, which stabilize the π-electronic anions 34a–c− (Fig. 21a(i)).67 The pKa value of 34b (7.2) is higher than that of 34a (6.2), comparable to 4-nitrophenol. In the solid state, TBA+-34a,b− form planar structures, stabilized by intramolecular N–H⋯O hydrogen bonding. Planar 34a,b− are stacked with counter TBA+, providing charge-by-charge columnar assemblies (Fig. 21a(ii)). Introduction of aliphatic alkyl chains at the aryl groups in TBA+-34c− induces liquid crystal mesophases with a Colh structure based on charge-by-charge assembly, as observed in the crystal structure of TBA+-34b− (Fig. 21a(iii)).67b Efforts to introduce electron-withdrawing fluorine at the pyrrole β-positions in search of more stable π-electronic anions yields the desired species. However, handling and purification of this species is challenging owing to its easy deprotonation.67d In contrast, introducing thiophene units instead of pyrrole rings affords the π-electronic anion 35a−, with a pKa of 6.7 (Fig. 21b(i)).68 Detailed theoretical studies and single-crystal X-ray analysis of TBA+-35a− revealed intramolecular S⋯O− chalcogen bonding (Fig. 21b(ii)). Similarly, TBA+-35b− shows alternate stacking of planar 35b− and TBA+ (Fig. 21b(i,iii)). Furthermore, introducing quinone methide units as electron-withdrawing groups and extended π-electronic system effectively stabilizes deprotonated pyrrole units (Fig. 22).69 Pyrrole-bridged quinone undergoes deprotonation by treatment with tetraalkylammonium hydroxide, inducing a red-shifted absorption band of ∼150 nm for 36− (Fig. 22a). In TPA+-36− (TPA+: tetrapropylammonium), TPA+ is located on the π-plane of 36−, inhibiting the stacking of 36− (Fig. 22b).
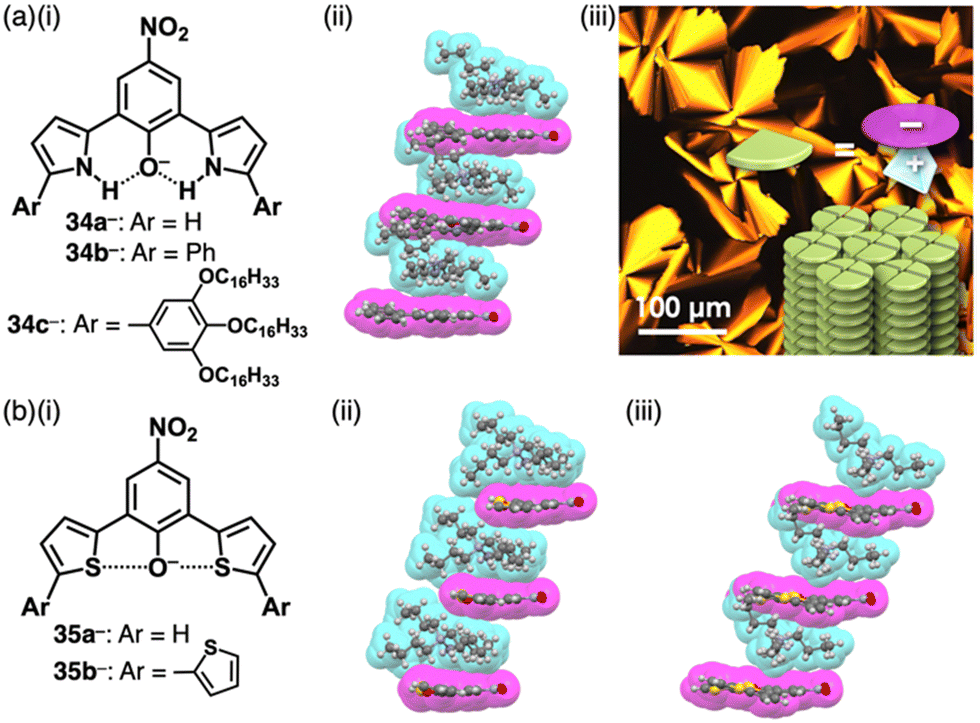 | ||
| Fig. 21 (a) (i) Deprotonated dipyrrolylnitrophenols 34a–c−, (ii) single-crystal X-ray structure of TBA+-34b− and (iii) POM image of TBA+-34c− at 65 °C upon cooling (redrawn from ref. 67b. Copyright 2018 Wiley) and (b) (i) deprotonated dithienylnitrophenols 35a,b− and single-crystal X-ray structures of (ii) TBA+-35a− and (iii) TBA+-35b−. The crystal structures are redrawn from cif files: 1830529, 2253150 and 2253154. | ||
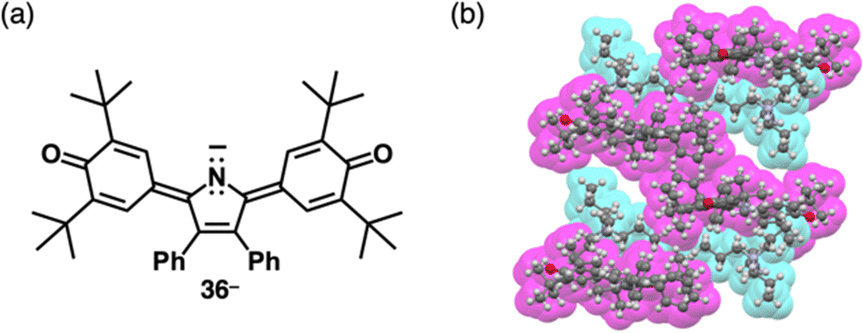 | ||
| Fig. 22 (a) Deprotonated pyrrole-bridged quinone 36− and (b) single-crystal X-ray structure of TBA+-36− (redrawn from a cif file: 2076443). | ||
The delocalization of the negative charge is essential to obtain stable π-electronic anions. Deprotonated species of meso-hydroxy-substituted porphyrins afford stable π-electronic anionic species 37− and 37a–d− (NiII, ZnII, PdII and PtII complexes, respectively) (Fig. 23a).9,70 The pKa value of 37a is estimated to be 7.89, higher than those of dipyrrolylnitrophenols 34a,b. The deprotonated structures of 37a–d− as TBA+ ion pairs exhibit the charge-by-charge columnar structures (Fig. 23b(i)). Furthermore, the hydroxy group serving as a directing group enables the selective introduction of phenyl groups at adjacent positions. Treatment of 37a with Pd(OAc)2, rac-BINAP and Cs2CO3 yields the Pd-BINAP-complex; further treatment with PhMgBr affords β-phenyl- and diphenyl-substituted 38a,b (Fig. 23a).70b The pKa values of 8.18 and 8.25 for 38a,b, respectively, are higher than that of 37a, confirming the electron-donating nature of the phenyl rings. Upon deprotonation, the ion pairs TBA+-38a,b− form charge-by-charge ion-pairing assemblies in the solid state (Fig. 23b(ii)). The introduction of suitable aryl units allows for the delocalization of the negative charge, resulting in π-extended anionic species with higher stability.
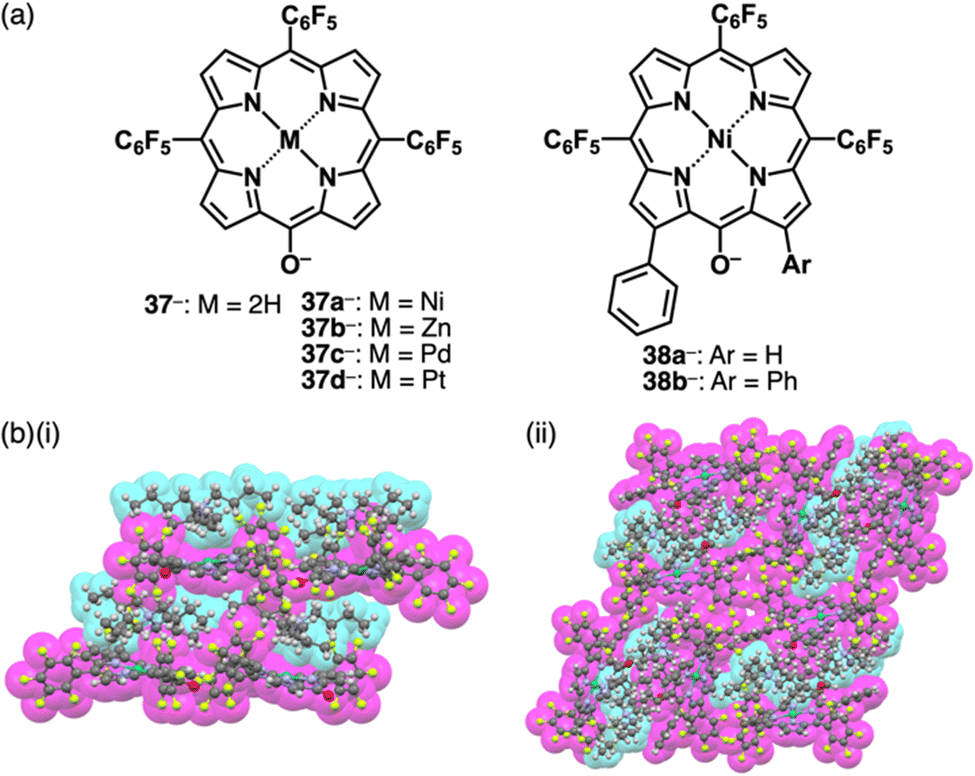 | ||
| Fig. 23 (a) Deprotonated meso-hydroxyporphyrins 37− and 37a–d− (M = Ni, Zn, Pd and Pt) and β-monophenyl 38a− and diphenyl-substituted 38b− and (b) single-crystal X-ray structures of (i) TBA+-37a− and (ii) TBA+-38b− (redrawn from cif files: 1536339 and 1901215). | ||
3. π-Electronic systems with charge introduced by noncovalent interactions: ion complexes that sparked π-electronic ion-pairing chemistry
Charge in π-electronic systems can be introduced by noncovalent interactions with inorganic ions, especially, anions (Fig. 1b(v)). Anion binding71 of anion-responsive π-electronic systems (receptors) leads to the formation of pseudo-π-electronic anions, which are incorporated into ion-pairing assemblies along with π-electronic cations, some of which are discussed in Section 2. Anion binding by π-electronic receptors transforms hard inorganic anions to soft “pseudo-π-electronic” anions, resulting in charge delocalization within the host π-electronic systems (Fig. 24(i)). These formed anion complexes, acting as pseudo-π-electronic anions, can be stacked with π-electronic cations through iπ–iπ interactions, primarily involving electrostatic and dispersion forces (Fig. 24(ii)). Various pseudo-π-electronic anions can be generated based on the guest anions, with their countercations serving as the counter species of the resulting anion complexes. The stacked pairs of anion complexes and countercations, termed pseudo-π-sips, are fundamental building blocks of ion-pairing assemblies. The focus of this section is on the anion complexes of anion-responsive π-electronic molecules, as the chemistry of ion-pairing assemblies originates from the structures assembled with alternately stacked anion complexes and π-electronic cations.5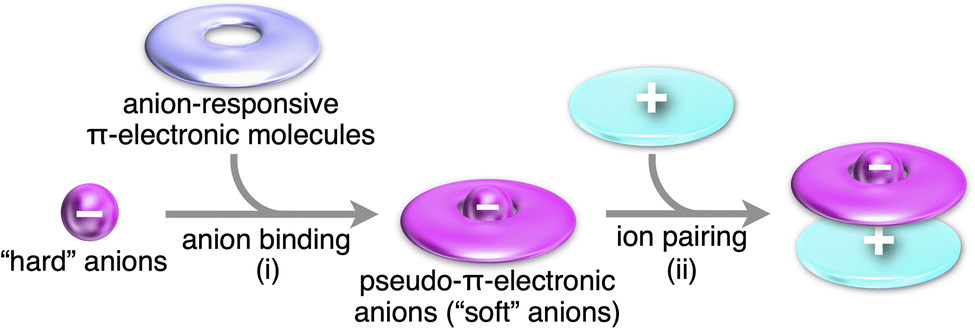 | ||
| Fig. 24 Conceptual scheme for the formation of π-electronic ion pairs through (i) anion binding and (ii) ion pairing. Pseudo-π-electronic anions as soft anions are obtained by the binding of a hard anion by π-electronic receptors. The pseudo-π-electronic anions form ion pairs in combination with counter π-electronic cations. | ||
3.1. Charged π-electronic systems formed by ion binding
Various types of anion-responsive molecules have been synthesized as colourimetric selective anion sensors and anion transporters in membranes.71 However, only a few reports discuss planar receptor–anion complexes. The design of anion receptors using π-electronic systems is essential for the formation of planar receptor–anion complexes. Building subunits such as pyrrole and triazole have polarized structures with an H unit that acts as a hydrogen-bonding donor.As pyrrole-based anion receptors that show planar structures, dipyrrolyldiketone BF2 complexes (PBs, 39a–h as examples) have two pyrrole units attached to the electron-withdrawing 1,3-propanedione unit (Fig. 25).5a,72 The pyrrole NH units can be directed to the propanedione CH side by rotating the pyrrole rings, and, in the resulting conformation, the two NH and one CH units serve as anion-binding sites with high binding constants for various anions (Ka = 15000 M−1 for 39a to bind Cl− in CH2Cl2).72c Peripheral modifications at the pyrrole and boron units facilitate further functionalization. A previous review article summarizes representative results in anion-binding and assembling behaviours.5a α-Phenyl derivative 39d (Fig. 25b) shows enhanced anion-binding ability owing to the additional o-CH as supportive interaction sites.72b Single-crystal X-ray analysis of TPA+-39d·Cl− clearly demonstrates the planar geometry of 39d·Cl−. Importantly, the planar 39d·Cl− and counter TPA+ are alternately stacked, forming a charge-by-charge columnar structure. The discovery of ion-pairing assemblies with alternately stacked anion complexes and π-electronic cations sparked the idea of incorporating planar countercations (vide infra, Section 3.2).
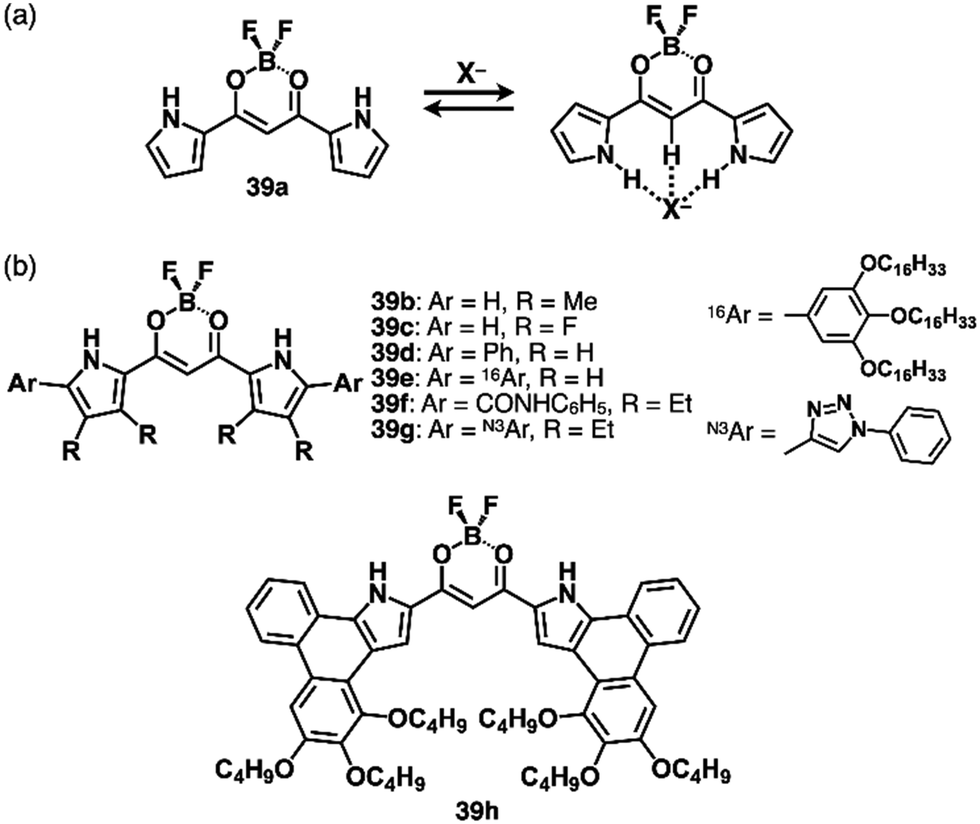 | ||
| Fig. 25 (a) Anion-binding behaviour of dipyrrolyldiketone BF2 complex (PB) 39a and (b) PB derivatives 39b–h. | ||
Efficient anion-binding behaviours of PB have been applied for inducing further functionalities (Fig. 25b). Modifications of the α-positions such as α-amide-substituted 39f exhibit higher anion-binding behaviour (Ka ≥ 108 M−1 for Cl− in CH2Cl2) to form solid-state ion-pairing assemblies.72g Moreover, click reactions of an α-diethynyl-substituted receptor72f and arylazides afford triazole-substituted receptors including 39g.72l The triazole-CH functions as an anion-binding site, forming planar receptor–anion complexes in ion-pairing assemblies. Covalently linked PB dimers show high anion-binding affinities by forming helical structures as building blocks of ion-pairing assemblies.72d,e,73 OFF–ON fluorescence switching by anion binding has been demonstrated in hexaaryl-substituted PB derivatives based on anion-dependent intramolecular photo-induced electron transfer (PET), demonstrating potential logic gate systems.72j,k More efficient anion-binding and ion-pairing behaviours can be achieved by including the PB unit in the macrocycles owing to pyrrole-inverted preorganized structures.72h
Shape-persistent macrocycles with preorganized anion-binding structures based on π-electronic subunits facilitate the preparation of planar receptor–anion complexes.74 Flood et al. synthesized a triazolophane anion receptor 40 (Fig. 26), containing polarized aryl-CH units as interaction sites, providing an anion-binding cavity.75 By exclusively employing aryl-CH units, 40 demonstrates a highly efficient anion-binding capability (Ka = 4700000 M−1 for Cl− in CH2Cl2). Moreover, 40 forms stacked [2+1]-type anion complexes with larger anions, such as I−, through the effective π–π stacking of the aromatic units present in 40.
 | ||
| Fig. 26 Macrocyclic anion receptors: triazolophane 40 and cyanostar (CS) 41. | ||
Cyanostar (CS) 41, serving as another planar π-electronic anion receptor, incorporates five electron-withdrawing cyano groups, making the inner CH units highly efficient hydrogen-bonding donors (Fig. 26).76 Owing to the presence of an internal anion-binding cavity with five CH units, CS exhibits high binding constants for larger anions, including BF4−, PF6− and ClO4− (logKa ∼ 5). [2+1]-Type anion complexes for larger anions based on effective π–π stacking of CS serve as key structures for fabricating ion-pairing assemblies.
3.2. π-Electronic ion-pairing assemblies comprising anion receptors
Since the initial discovery of the solid-state charge-by-charge assembly of TPA+-39d·Cl−, as illustrated in Section 3.1, there has been a trend of introducing more planar countercations instead of the bulky ammonium cations for the formation of charge-by-charge assemblies. In our preliminary trial for the use of planar countercations, tris(diethylamino)-substituted TOTA+15b was incorporated in the solid-state charge-by-charge assembly with the anion complex of 39d. Because of the difficulty in excluding I− from the starting ion pair TOTA+-Cl− by anion exchange protocol, other suitable π-electronic cations have been explored to fabricate ion-pairing assemblies. For example, 3TATA+-Cl−, which can be synthesized from the Cl− salt a tritylium derivative, was used for an alternative π-electronic cation.7 Planar 39d·Cl− and 3TATA+ formed a charge-by-charge assembly viaiπ–iπ interactions, with a shorter stacking distance than that of TPA+-39d·Cl− (Fig. 27a). Furthermore, 3TATA+-39e·Cl− with aliphatic alkyl chains forms a supramolecular gel comprising micrometre-scale fibrous morphologies (Fig. 27b). X-ray diffraction analysis of the xerogel revealed the formation of a Colh structure with a diffraction peak at 0.74 nm, which is nearly twice of π–π stacking distances. This distance was assigned as the repeating distance of identically charged species in the charge-by-charge assembly. In addition, 3TATA+-39e·Cl− forms a liquid crystal mesophase based on the Colh packing of charge-by-charge assembly.7
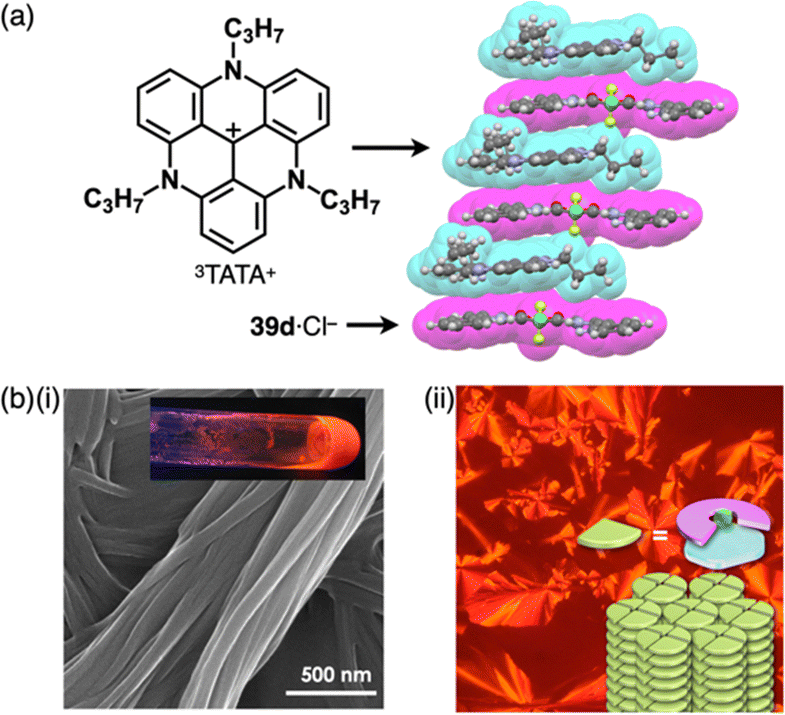 | ||
| Fig. 27 (a) Single-crystal X-ray structure of 3TATA+-39d·Cl− (redrawn from a cif file: 745780) and (b) (i) photograph and SEM of the supramolecular gel and (ii) POM and packing model of the liquid crystal mesophase of 3TATA+-39e·Cl− (redrawn from ref. 7. Copyright 2010 Wiley). | ||
Moreover, porphyrin AuIII complexes have been used as countercations of π-electronic ion-pairing assemblies comprising PB-based anion complexes. For example, in the crystal structure of 22a+-39a·Cl−, a stacked 22a+ dimer was sandwiched by planar 39a·Cl− (Fig. 28a).77 In contrast, in 22a+-39b·Cl−, 22a+ and planar 39b·Cl− are alternately stacked with a π–π stacking distance (Fig. 28b). 22a+-39c·Cl− forms a similar charge-by-charge assembly. Furthermore, charge-by-charge assemblies of 3TATA+-39f·Cl−, 3TATA+-39g·Cl− and 22a+-39g·Cl− have been observed (Fig. 28c–e). Effective stacking of the planar receptor–anion complex and countercation is supported by EDA, suggesting a cooperative contribution of electrostatic and dispersion forces in the stacked ion pairs of 3TATA+-39g·Cl− and 22a+-39g·Cl−.72l Furthermore, π-extended PB 39h exhibits a planar [1+1]-type Cl− complex, showing the stacking with 3TATA+ in the solid state (Fig. 28f).72i
 | ||
| Fig. 28 Single-crystal X-ray structures of (a) 22a+-39a·Cl−, (b) 22a+-39b·Cl−, (c) 3TATA+-39f·Cl−, (d) 3TATA+-39g·Cl−, (e) 22a+-39g·Cl− and (f) 3TATA+-39h·Cl− (redrawn from cif files: 1969168, 1969169, 1546127, 213826, 213827 and 1900650). | ||
Recent studies based on the crystal analysis have revealed a variety of charge-by-charge assemblies depending on the constituting receptor–anion complexes and π-electronic countercations. 42 forms a planar [2+1]-type anion complex 422·Cl− in the crystal structure of 3TATA+-422·Cl−, where the layers of identically charged species are alternately stacked (Fig. 29a).78 Furthermore, introducing naphthalenediolate units instead of two fluorine moieties at the boron unit affects the assembling modes.79 In the assembly of the naphthalene-2,3-diolate boron complex as a 3TATA+ ion pair 3TATA+-43a·Cl−, the stacked 3TATA+ dimer is sandwiched by the naphthyl unit of 43a·Cl−. Conversely, in 3TATA+-43b·Cl−, alternately stacked 3TATA+ and the receptor–Cl− complex unit in 43b·Cl− are observed (Fig. 29b). Square planar dipyrrolyldiketone PtII complexes that have phenylpyridine as another ligand part are also suitable for stacking assemblies (Fig. 30a).80 In the crystals of PtII complex ion pairs 22c+-44a·Cl− and 22a+-44b·Cl−, AuIII complexes and anion complexes are alternately stacked and form columnar structures (Fig. 30b). These solid-state charge-by-charge assemblies, comprising the anion complexes of PB and PtII complexes, demonstrate the potential to induce diverse assembling modes and resulting properties based on neutral receptor units.
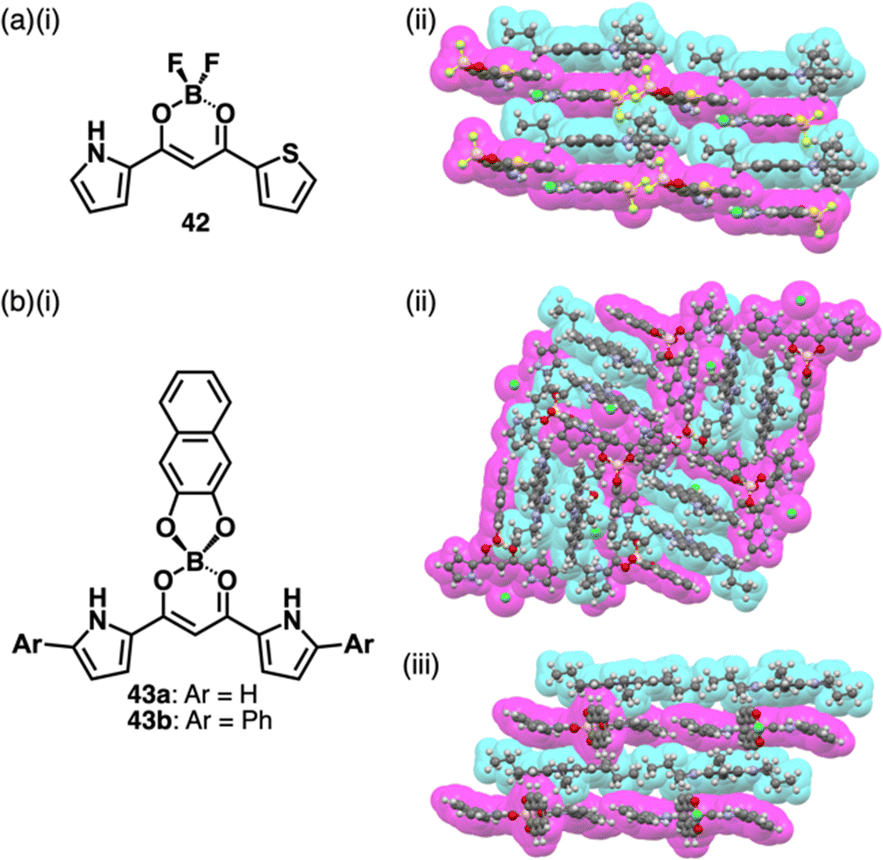 | ||
| Fig. 29 (a) (i) Pyrrolylthienyldiketone BF2 complex 42 and (ii) single-crystal X-ray structure of 3TATA+-422·Cl− and (b) (i) naphthalenediolate boron complexes 43a,b and (ii) single-crystal X-ray structures of 3TATA+-43a·Cl− and (iii) 3TATA+-43b·Cl−. The crystal structures are redrawn from cif files: 1840326, 2227398 and 2227399. | ||
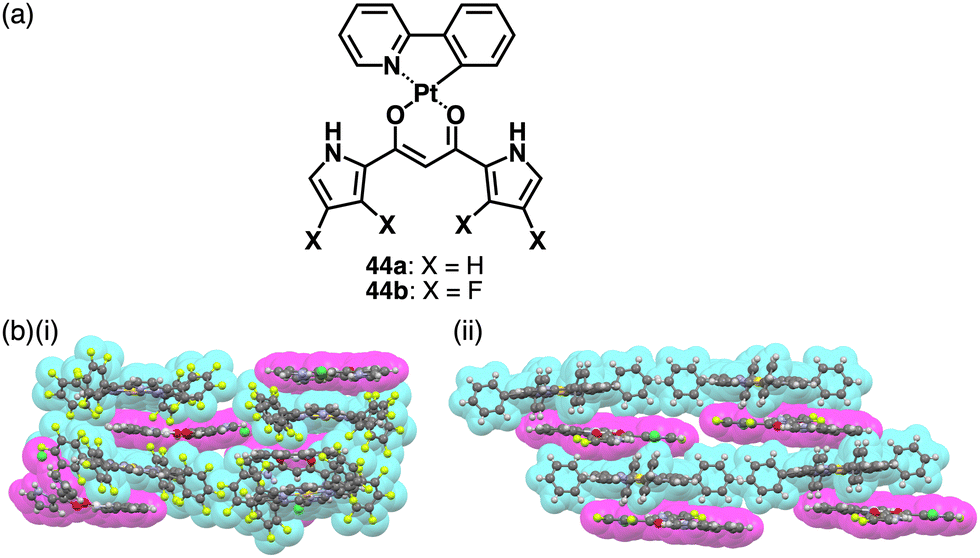 | ||
| Fig. 30 (a) Dipyrrolyldiketone PtII complexes 44a,b and (b) single-crystal X-ray structures of (i) 22c+-44a·Cl− and (ii) 22a+-44b·Cl− (redrawn from cif files: 2063433 and 2063434). | ||
In the ion-pairing assemblies of CS, the 4a+-412·BF4− ion pair is stacked with another ion pair, forming a charge-by-charge assembly, exhibiting a distinct red-shifted emission band derived from 4a+, in a concentrated solution (0.2 mM) as well as crystalline and film states.76b Various cationic dyes can be introduced as the countercations of ClO4−, and the ion pairs form charge-by-charge assemblies in the crystalline state, as shown in 45a+-412·ClO4− (Fig. 31a and b).76c Usually, crystal-state photophysical properties, different from those in solution, are affected by exciton coupling depending on the arrangement of π-electronic molecules. In contrast, the cationic dyes within the charge-by-charge assemblies with CS anion complexes are spatially and electronically isolated, and the exciton coupling is suppressed. Flood et al. proposed charge-by-charge assemblies as small-molecule ionic isolation lattices (SMILES). Ensuring that the frontier molecular orbitals of the cationic dyes are located inside those of the anion complex of CS is crucial. Using SMILES, the polymer resin containing 45a+-412·ClO4− exhibits an emissive solid-state material (Fig. 31c).76c Furthermore, 45b+-412·PF6− forms SMILES-based nanoparticles, exhibiting bright emission derived from 45b+ upon excitation of 412·PF6− owing to the effective energy transfer (φET = 80%) between the oppositely charged π-electronic systems.76d A similar phenomenon was observed in the nanoparticles of 46b+-412·PF6−.76e In particular, the energy transfer efficiency of 46b+-412·PF6− reaches 100% owing to the larger overlap of emission of 412·PF6− and the absorption bands of the cationic dye. However, the fluorescence quantum yield of SMILES is low (12.8%), owing to Förster resonance energy transfer to dark trap states or reabsorption. The deactivation path is suppressed by doping fluorescent dyes into SMILES comprising cationic dyes with higher excitation energies, drastically improving the fluorescent quantum yields (65% and 29% for 45a+-ClO4−-doped and pristine SMILES comprising 46a+-PF6−, respectively).76f
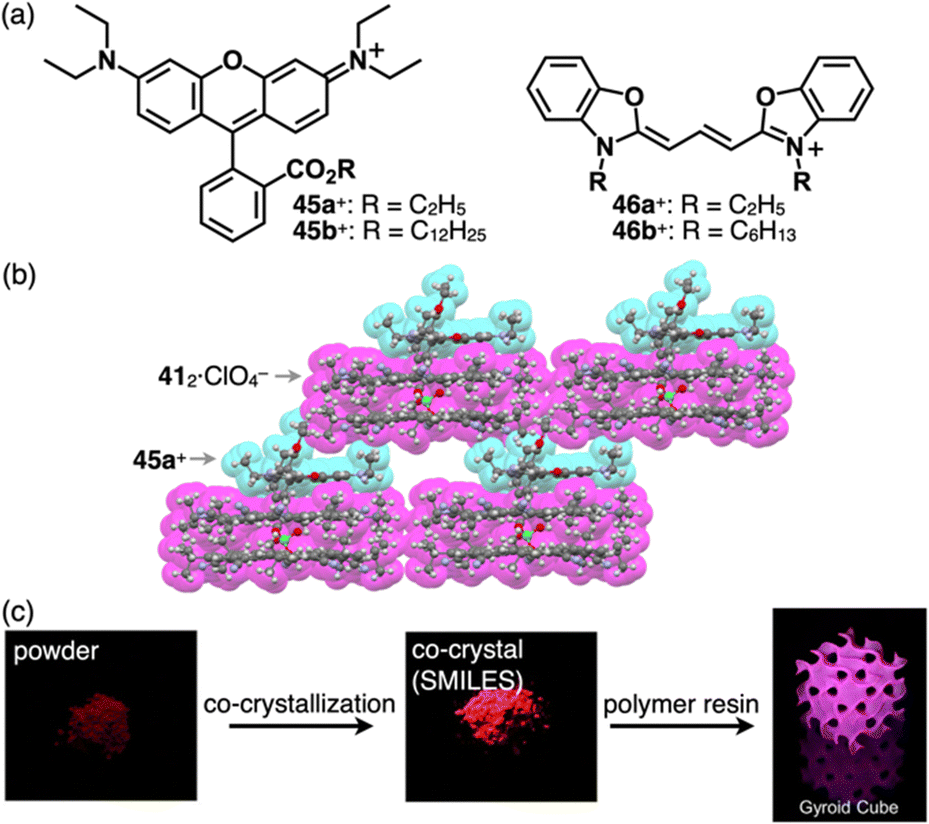 | ||
| Fig. 31 (a) Rhodamines 45a,b+ and cyanines 46a,b+, (b) single-crystal X-ray structure of 45a+-412·ClO4− (redrawn from a cif file: 1895155) and (c) photographs of 45a+ (left), 45a+-412·ClO4− (centre) and 45a+-412·ClO4− contained in polymer resin (right) (redrawn from ref. 76c. Copyright 2020 Elsevier). | ||
4. π-Electronic ion pairs: pairs of π-electronic cations and anions
The π-electronic cations described in Section 2 are introduced as the counter species for the anion complexes of anion-responsive π-electronic molecules (Section 3). The combination of π-electronic cations with π-electronic anions, as summarized in Section 2, provides a variety of ion pairs and assemblies. π-Electronic ion pairs are prepared by ion-pair metathesis from desired π-electronic ions with corresponding inorganic counterions.5b Ion-pair metathesis, a key procedure for producing π-electronic ion pairs, is derived from the hard and soft acids and bases (HSAB) theory, which requires hard ions such as Cl− and Na+ for desired cation and anions, respectively, in starting materials (Fig. 32). For categorizing and summarizing π-electronic ion pairs, this section focuses on the types of π-electronic anions, which are relatively difficult to prepare than π-electronic cations owing to their reactivity.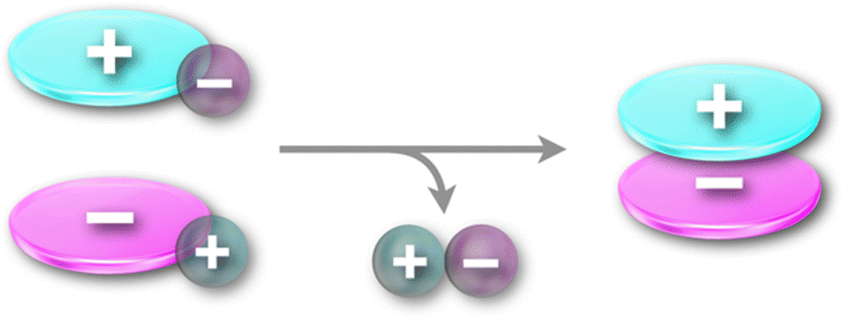 | ||
| Fig. 32 Ion-pair metathesis for the preparation of π-electronic ion pairs. | ||
4.1. π-Electronic ion pairs comprising cyclopentadienide-based π-electronic anions
Photophysical properties and ion-pairing assemblies of π-electronic ion pairs were investigated based on cyclopentadienide 13−.81 In 1963, ion pairs of 13− were prepared by ion-pair metathesis of K+-13− and various π-electronic cations, such as Tr+ and 47–49+ as BF4− salts (Fig. 33a).81a The ion pairs exhibit countercation-dependent CT bands. The CT band of Tr+-13− is blue-shifted in polar solvents (474 and 439 nm in CH2Cl2 and MeOH, respectively) with a smaller solvent dependence than Tr+-I− (571 and 403 nm, respectively).81b Alternately arranged Tr+ and 13− form a charge-by-charge assembly in the crystal structure (Fig. 33b).81d The CT absorption band of an N-methylpyridinium ion pair 49a+-13− at −170 °C in a MeOH/EtOH glassy solution was observed (λabs ∼ 340 nm as a shoulder); compared to other ion pairs with Br−, N3−, SCN− and I−, it was red-shifted, whereas the emission band was blue-shifted.81c Therefore, the Stokes shift of 49a+-13− (1.1 × 104 cm−1) is smaller than those of the other ion pairs (>1.55 × 104 cm−1), ascribed to the smaller structural relaxation in the excited state. Ion pairs of dicationic 50a2+ and 13−, 50a2+-13−-X− (X− = OTf− and BF4−) and 50a2+-213−, were prepared by the ion-pair metathesis of 50a2+-2X− and K+-13−.81e Similarly, 50b2+ provided ion pairs with 13− in the presence and absence of BF4− by the ion-pair metathesis between 50b2+-2BF4− and K+-13−. The ion pairs of 50a2+ exhibit concentration-dependent CT bands (λabs = 406 and 424 nm at 1.65 and 3.30 M, respectively, in MeCN for 50a2+-213−). In addition, 1H NMR signals of 50a2+-2OTf− are shifted upfield in the presence of K+-13−, suggesting an interaction between 13− and 50a2+. In the crystal structure of 50a2+-13−-BF4−, the dication and 13− are alternately stacked on the cyclopropenyl unit rather than the pyridinium unit, forming a charge-by-charge assembly with BF4−, located between the columns.81e The CO2Me groups in 13− are distorted from the cyclopentadienide core owing to their steric repulsion.81d,e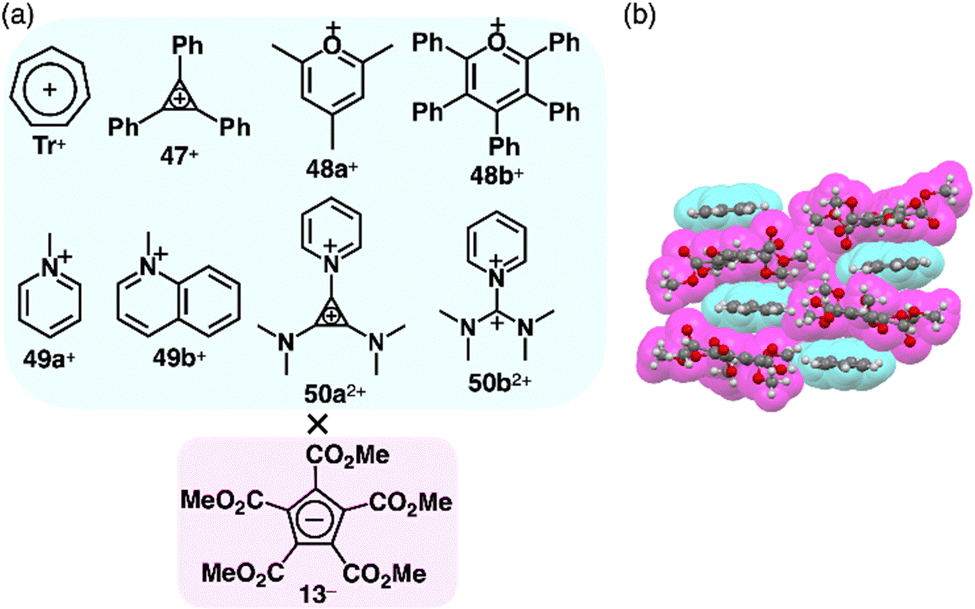 | ||
| Fig. 33 (a) π-Electronic ion pairs of pentamethoxycarbonyl-substituted cyclopentadienide 13− with π-electronic cations Tr+, 47–49+ and 50a,b2+ and (b) single-crystal X-ray structure of Tr+-13− (redrawn from a cif file: 1141949). | ||
The planar structure of PCCp− is suitable for more effective stacking of oppositely charged species in combination with π-electronic cations (Fig. 34a). An ion pair of PCCp− with trityl cation 1+ was prepared as the precursor of triethylsilyl PCCp− by the ion-pair metathesis of 1+-Br− and Ag+-PCCp−.821+ and PCCp− afforded a columnar structure comprising identically charged species in a crystalline state (Fig. 34b(i)). PCCp− is stacked with another PCCp− at an interplanar distance of 3.42 Å to form a pair, which is slip-stacked with other pairs at the cyano groups.82 In 2+-PCCp−, 2+ and PCCp− are alternately stacked at the two aminophenyl units of 2+ (Fig. 34b(ii)).33 Meanwhile, the other aminophenyl group is stacked with the corresponding part of the neighbouring 2+, resulting in a partially charge-segregated assembly. Meanwhile, the propeller geometries of triarylmethyliums inhibits effective stacking, resulting in the limited contact with the oppositely charged species. On the other hand, PCCp− in π-electronic ion pairs with more planar π-electronic cations such as Tr+ and 3TATA+ is effectively stacked with the countercations. In Tr+-PCCp−, Tr+ and PCCp− are alternately stacked to afford a charge-by-charge assembly (Fig. 34b(iii)). In contrast, in 3TATA+-PCCp−, 3TATA+ and PCCp− are stacked with the respective identically charged species to form stacked dimers, resulting in a two-by-two charge-by-charge assembly (Fig. 34b(iv)).33 The ion pair of PCCp− with 51+, an isoelectronic cation of TATA+ with a B–N unit in place of a central C–C unit, was also prepared by ion-pair metathesis of 51+-OTf− and Na+-PCCp−.83 The crystal structure resembles that of 3TATA+-PCCp−, although each component is slip-stacked with the identically charged species (Fig. 34b(v)). Notably, 3TATA+ and 51+ are stacked with PCCp− in relatively short contact (3.29 and 3.25 Å, respectively).
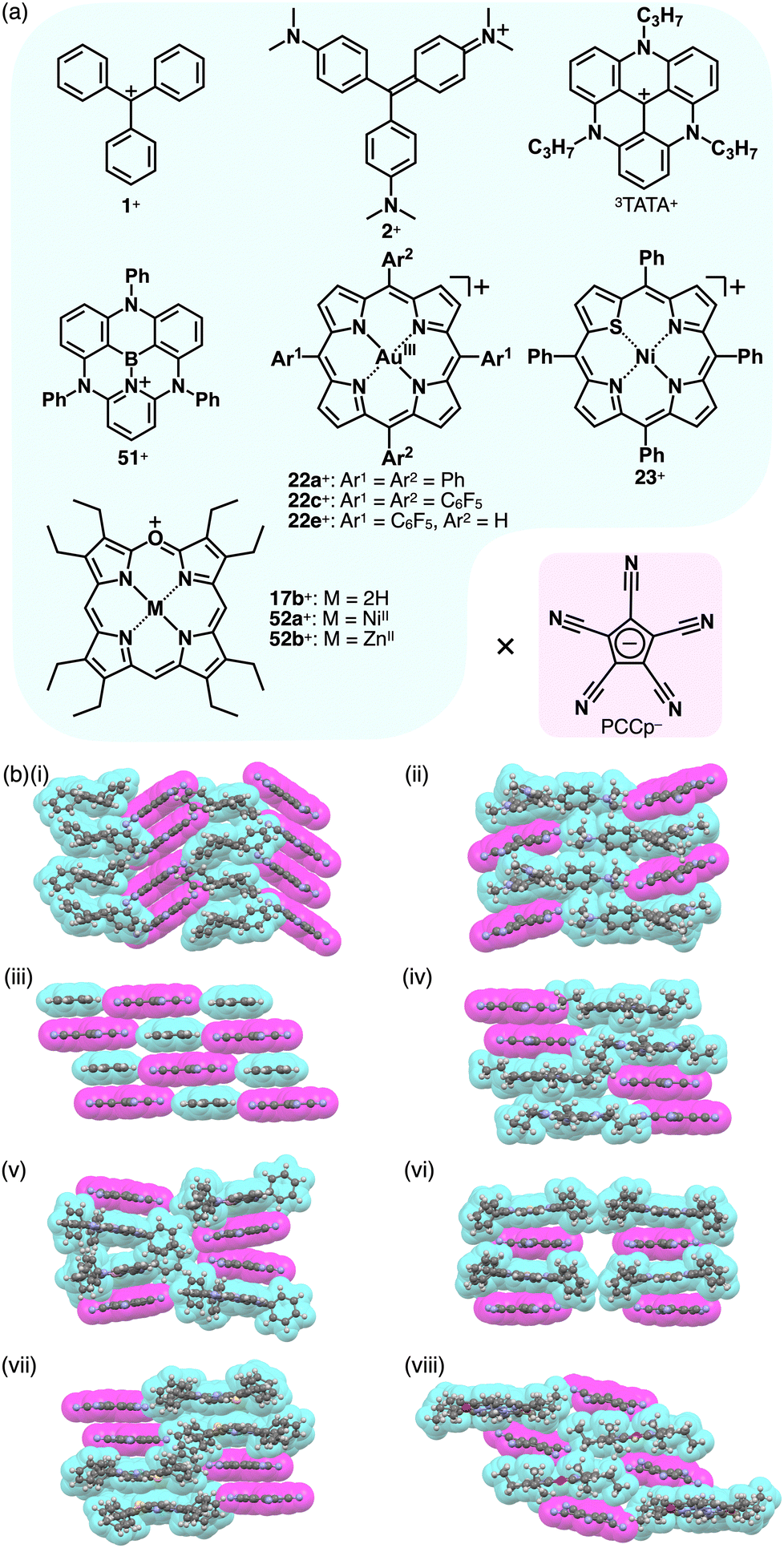 | ||
| Fig. 34 (a) Combinations of π-electronic ion pairs: PCCp− with 1+, 2+, 3TATA+, BN-TATA 51+, 22a,c,e+, 23+, 17b+ and 52a,b+ and (b) single-crystal X-ray structures of (i) 1+-PCCp−, (ii) 2+-PCCp−, (iii) Tr+-PCCp−, (iv) 3TATA+-PCCp−, (v) 51+-PCCp−, (vi) 22a+-PCCp−, (vii) 23+-PCCp− and (viii) 52b+-PCCp− (redrawn from cif files: 227124, 1431734, 1431733, 1431735, 2063989, 1877990, 2167304 and 2144318). Atom colour code in (viii): violet in the ball-and-stick models refers to zinc. | ||
Porphyrin AuIII complexes, which are discussed in Sections 2.2 and 3.2, have been adopted as the components of π-electronic ion pairs, as seen in 22a+-PCCp−.52 The ion pair forms a solid-state charge-by-charge assembly with the interplanar distances of 3.37 and 3.40 Å (Fig. 34b(vi)).52a22e+-PCCp− is also included in a similar charge-by-charge assembly, where the two free meso-positions are in contact with PCCp− CN units.52b Conversely, 22c+-PCCp− forms a one-by-two charge-by-charge assembly with another 22c+ unit located between the columns. Asymmetric heteroatom-containing porphyrin analogues with chalcogen elements has been adopted as the countercation of PCCp−. 21-Thiaporphyrin NiII complex ion pair 23+-PCCp− was prepared by ion-pair metathesis of 23+-Cl− and Na+-PCCp−.55 In the crystal structure of 23+-PCCp−, the thiophene unit of 23+ is inclined by 24.5°. The 23+ units are stacked by themselves in antiparallel orientation to form a dimer via chalcogen bonding between the thiophene and adjacent pyrrole units at short S⋯N distances of 3.26 and 3.33 Å. In addition, PCCp− forms a stacked dimer, providing a two-by-two charge-by-charge assembly (Fig. 34b(vii)).55 Similarly, in the PCCp− ion pair with oxaporphyrin NiII complex 52a+, the constituent ions form the respective stacked dimers with interplanar distances of 3.31 and 3.28 Å, respectively.84 Besides, PCCp− ion pairs of free-base 17b+ and ZnII complex 52b+ form one-by-one charge-by-charge assemblies. In contrast to the almost parallel arrangement observed in 17b+ and PCCp−, in the ion pair 52b+-PCCp−, the components are not arranged in parallel owing to the coordination of PCCp− CN to the ZnII centre (Fig. 34b(viii)).
The introduction of aliphatic alkyl chains into charged π-electronic species induces various soft materials based on ion-pairing assemblies. The porphyrin AuIII complex bearing hexadecyloxy chains at the meso-aryl moieties 22f+ provides supramolecular gels and discotic columnar liquid crystals, whose assembled structures depend on the counteranions (Fig. 35).52a22f+-PCCp− forms a supramolecular octane gel (10 mg mL−1), owing to the entanglement of fibrous morphologies with diameters of 1–3 μm and lengths exceeding 100 μm (Fig. 35b(i)). Conversely, 22f+-PCCp− exhibits a liquid crystal mesophase (36–293 °C) based on the Colh structure of the charge-by-charge assembly, as observed in the crystal structure of 22a+-PCCp− (mentioned earlier) (Fig. 35b(ii)). Meanwhile, 22f+-Cl− forms a Colh structure with charge-segregated assembly, excluding the nonplanar anion from the stacked structure of 22f+. These findings suggest that the size of PCCp− complements the core part of porphyrin AuIII complexes, facilitating the formation of charge-by-charge assemblies. Notably, the charge-by-charge assembly of 22g+-PCCp−, which possesses icosyloxy chains, shows anisotropic alignment after shearing. The alignment persists after thermal phase transitions, indicating a robust packing state of charge-by-charge assemblies.52a
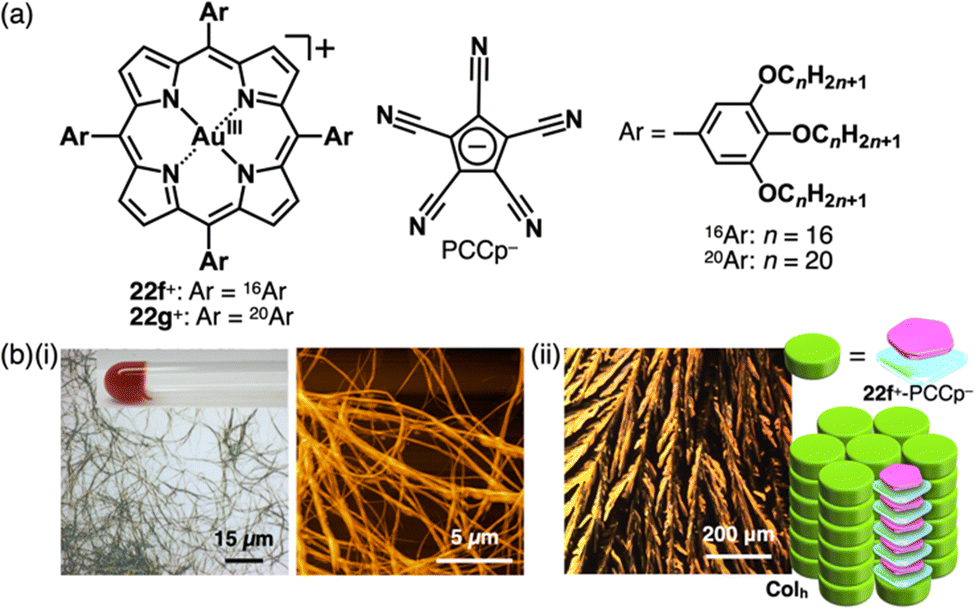 | ||
| Fig. 35 (a) Ion pair of aliphatic porphyrin AuIII complex 22f+ with PCCp− and (b) (i) optical microscope and atomic force microscope images of 22f+-PCCp− octane gel (10 mg mL−1) and (ii) mesophase properties of 22f+-PCCp−: POM and a proposed assembled model at 280 °C upon cooling from the isotropic liquid state (redrawn from ref. 52a. Copyright 2019 Elsevier). | ||
In the previous paragraphs in this section, the π-electronic ion pairs of PCCp− form the charge-by-charge assemblies, some of which comprise the stacked dimers of PCCp− and those of the cations. As seen in the ion pairs with bulky cations, stacking structures of PCCp− can be realized by combining π-electronic cations that aggregate independently. For instance, in the crystal structure of the ion pair comprising PCCp− with pentamethylimidazolium 53+, PCCp− is stacked with another PCCp−, leading to a charge-segregated assembly (Fig. 36).85 In this arrangement, 53+ is located between the PCCp− columns without participating in the stacking. Despite the absence of long alkyl chains, the crystal of 53+-PCCp− undergoes a phase transition and transforms to the mesophase in a temperature range of 234–240 °C. Interestingly, the ion pair, especially when doped with Na+-PCCp−, exhibits ionic conductivities.
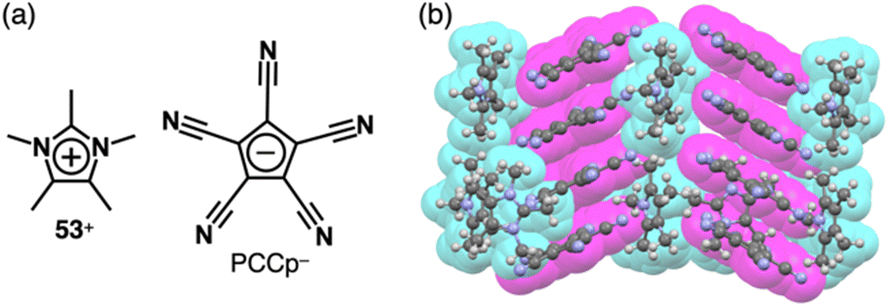 | ||
| Fig. 36 (a) Ion pair of pentamethylimidazolium 53+ with PCCp− and (b) single-crystal X-ray structure of 53+-PCCp− (redrawn from a cif file: 1868291). | ||
Charge-segregated assemblies based on stacked structures of PCCp− and those of π-electronic cations can be constructed by introducing the cationic species that induce specific interactions. The dipole of cationic squarylium 10+ can be used for stacking in the PCCp− ion pair (Fig. 37a).23 The photophysical properties of 10+-PCCp− vary with the stacking modes, forming J- and H-aggregates in 1% CH2Cl2/n-hexane and 1% acetone/water, respectively, while a monomeric state is observed in CH2Cl2 (Fig. 37b). Additionally, J-aggregates are formed in chiral media (0.33% to 1% CH2Cl2/α- and β-pinene) and exhibit CD signals, indicating the formation of chiral assemblies. These findings highlight the sensitivity of ion-pairing behaviour to the surrounding environment, offering potential applications in chiral recognition and sensing. 10+-PCCp− gives two crystal pseudo-polymorphs: (i) a charge-segregated assembly (10+-PCCp−CS) (Fig. 37c(i)) and (ii) a partially charge-segregated assembly (10+-PCCp−SS) (Fig. 37c(ii)), where 10+ is partially slip-stacked; hence, the direct contact of identically charged species is smaller than that of 10+-PCCp−CS. The EDA of the 10+-PCCp−CS packing structure indicates that electrostatic force in a stacked pair of 10+ (c2–c3 in Fig. 37d), oriented in an antiparallel fashion, is smaller than that in a separated pair of 10+ (c1–c3 in Fig. 37d). The peculiar electrostatic forces contributing to the characteristic assembling behaviour are ascribed to dipole–dipole interactions between identically charged species. Solid-state absorption spectra of the 10+ ion pairs exhibit counteranion-dependent bands derived from the exciton coupling between stacked 10+ units. Flash-photolysis (FP) TRMC measurements of the crystalline states of 10+-PCCp−CS and 10+-PCCp−SS reveal that the former exhibits higher electric conductivity (φ∑μ = 3.0 × 10−8 m2 V−1 s−1, where φ is the photo-carrier generation yield and ∑μ is the sum of hole and electron mobilities) than the latter (φ∑μ = 1.0 × 10−8 m2 V−1 s−1), confirming the effective electric conducting properties in the charge-segregated assembly (Fig. 37e). Moreover, 10+-PCCp−CS exhibits anisotropic electric conductivity along the stacking direction (b axis in Fig. 37c(i)).23
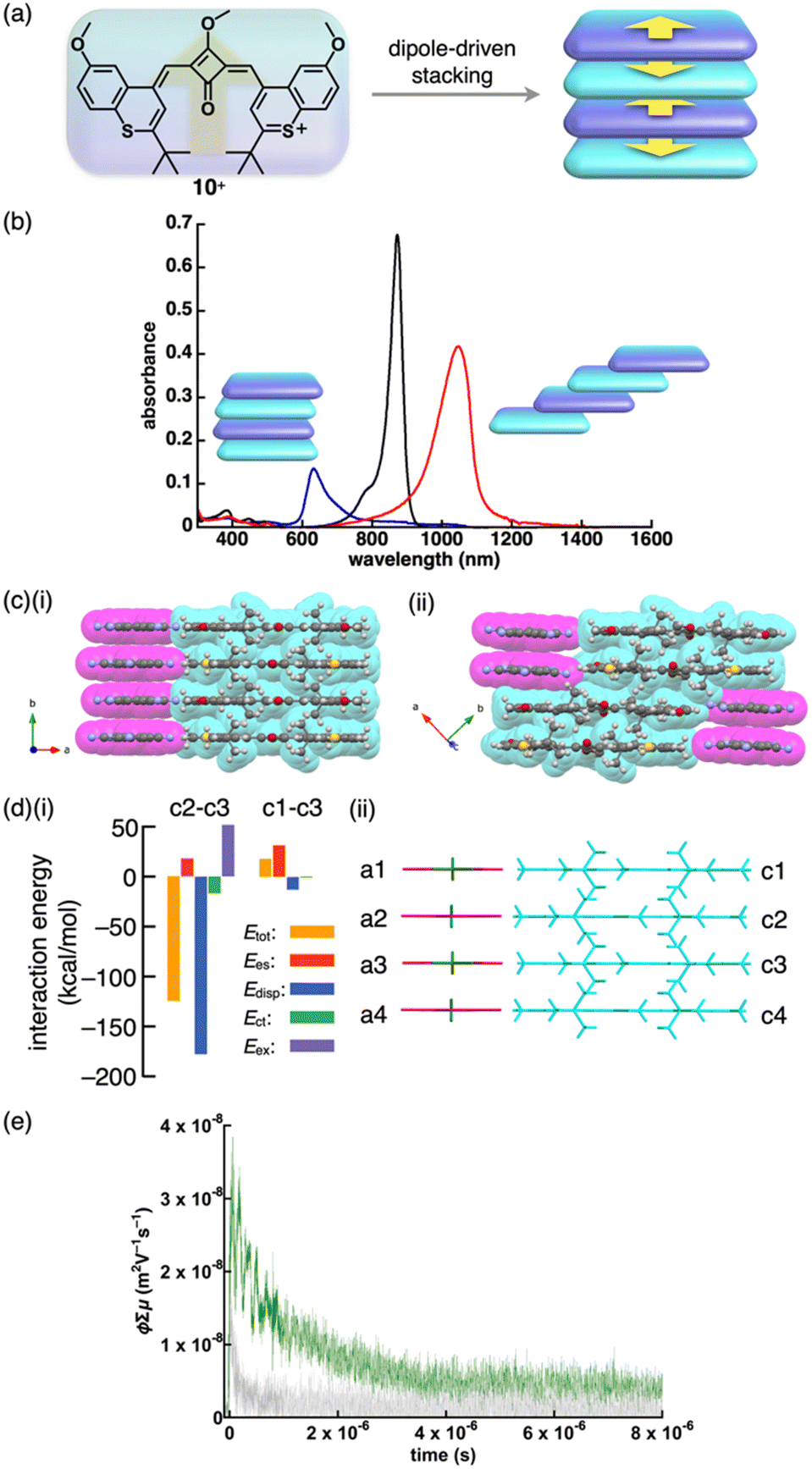 | ||
| Fig. 37 (a) Conceptual illustration of 10+ with dipole, which induces stacking of identically charged species, (b) UV/vis absorption spectra of 10+-PCCp− in CH2Cl2 (black), dispersed in 1% CH2Cl2/n-hexane (red) and 1% acetone/water (blue), (c) single-crystal X-ray structures of (i) 10+-PCCp−CS and (ii) 10+-PCCp−SS (redrawn from cif files: 2214020 and 2214021), (d) (i) decomposition of the total intermolecular interaction energies of 10+-PCCp−CS for (ii) the single-crystal X-ray structure with labels and (e) photoconductivity transients observed upon excitation at 355 nm, 9.1 × 1015 photons cm−2 pulse−1 for distinctive single crystals of 10+-PCCp− as distinctive pseudo-polymorphs of 10+-PCCp−CS (green) and 10+-PCCp−SS (light grey) along the long axes of crystals (redrawn from ref. 23. Copyright 2023 Wiley). | ||
4.2. π-Electronic ion pairs comprising azolate-based π-electronic anions
The replacement of CHs with electronegative nitrogens in cyclopentadienides enhances their stability, allowing for the production of various π-electronic anions, even with electron-donating groups. Azolium–azolate ion pairs, investigated by Shreeve et al., serve as energetic ionic liquids, finding applications as propellants owing to their explosive nature.86,87 Ionic liquid ion pairs, such as ethylmethylimidazolium (EMIm+) with 1,2,4-triazolate and tetrazolate, were synthesized using EMIm+-OH−.87a Their glass transition temperatures vary based on the constituents. Additionally, the π-electronic ion pair of butylmethylimidazolium 3,5-dinitro-1,2,4-triazolate BMIm+-57− was prepared by ion-pair metathesis of BMIm+-Cl− and K+-57−.87b Surprisingly, the melting point of BMIm+-57− is much lower than those of the ion pairs with TMA+, TEA+ and TBA+. Assembling behaviours of various azolium-azolate ion pairs with high phase transition temperatures were revealed by single-crystal X-ray analysis. Azolates interact with hydrogen-bonding donors, resulting in characteristic assembling modes. Ion pairs of nitroazolates with 1,3-dimethylimidazolium 54a+ were prepared in metal-free conditions by the deprotonation of azoles with imidazolium bicarbonate or imidazolium carboxylate followed by decarboxylation.87h The ion pair of 54a+ and nitroimidazolate 55− forms solid-state charge-by-charge assembly (Fig. 38a). In contrast, 56+-57− shows the stacking of identically charged species, forming a charge-segregated assembly, where the pyridinium unit of 56+ is stacked with the pyridinium units of adjacent 56+ (Fig. 38b).87e56+ is stacked with 58− on the pyridinium unit, resulting in a charge-by-charge assembly of 56+-58− (Fig. 38c). The anion unit in 54a+-58− is partially stacked with identically charged species, resulting in an intermediate assembling mode (Fig. 38c).87h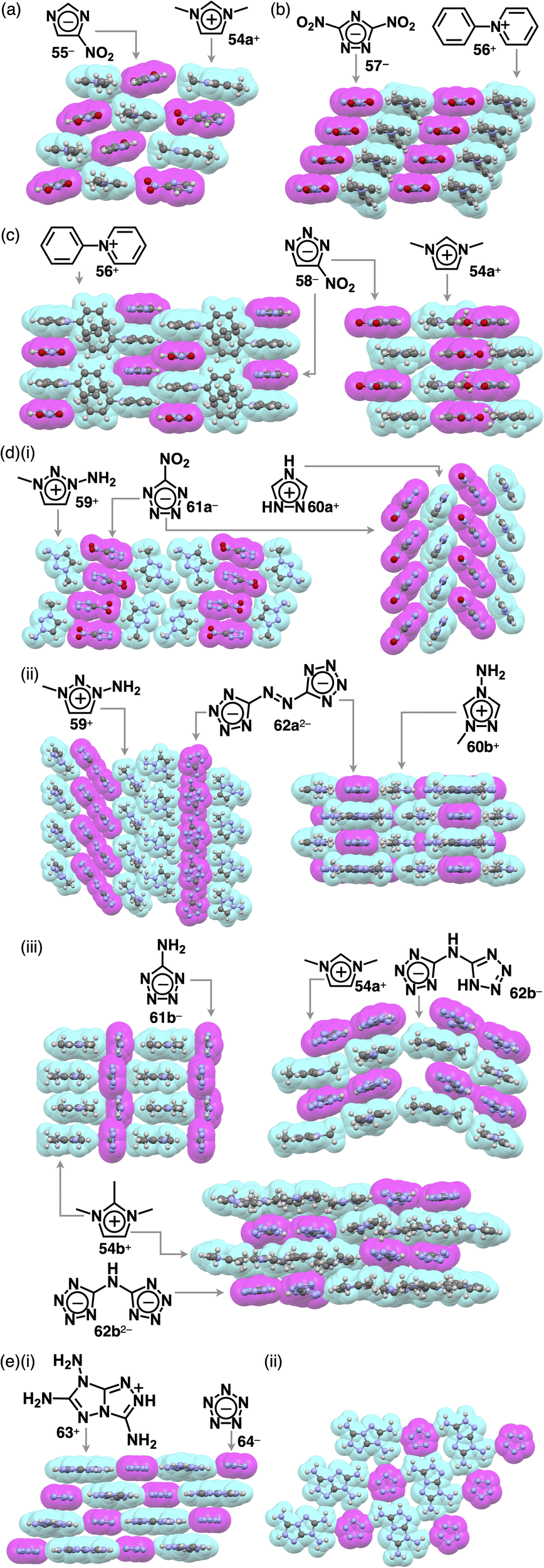 | ||
| Fig. 38 Single-crystal X-ray structures of ion pairs comprising (a) imidazolate 55−, (b) 1,2,4-triazolate 57−, (c) 1,2,3-triazolate 58−, (d) tetrazolate (i) 61a−, (ii) 62a2− and (iii) 61b−, 62b− and 62b2− and (e) pentazolate 64− as (i) side and (ii) top views (redrawn from cif files: 902237, 733829, 733828, 902240, 837299, 280499, 837300, 265160, 855087, 913585, 913586 and 1973518). | ||
Charge-segregated assemblies are obtained for several tetrazolate ion pairs. In 59+-61a−, 61a− is stacked with another anion, whereas 59+ is assembled via hydrogen bonding (Fig. 38d(i)).87g In the crystal structure of 60a+-61a−, the tetrazole unit of the anion is stacked with the nitro group of the adjacent anion as well as the cation, resulting in a partially charge-segregated structure (Fig. 38d(i)).87c
The ion pairs of a tetrazolate dimer 62a2− were synthesized using ion-pair metathesis of triazolium iodide with Ag2SO4, followed by another metathesis with a Ba2+ salt of a tetrazolate dimer.87d,g The resulting 259+-62a2− ion pair shows 59+ located beside 62a2− owing to the presence of hydrogen bonding between the amino group of 59+ and a tetrazole N of 62a2− (Fig. 38d(ii)). In addition, 62a2− is slip-stacked with adjacent 62a2−, giving a tilted anionic columnar structure.87g Conversely, in the crystal structure of 260b+-62a2−, two 60b+ units are located at both sides of the diazo group of 62a2− (Fig. 38d(ii)).87d The ion pair comprising 60b+ and 62a2− forms a columnar structure as they are stacked alternately in a short interplanar distance of 3.04 Å. In the case of 54b+-61b−, 61b− arranged at the side of adjacent 61b− through hydrogen-bonding interaction results in a belt-like structure of 61b− and a stacked structure of 54b+ (Fig. 38d(iii)).87f Ion pairs 54a+-62b− and 54b+-62b− were prepared by treating bis(tetrazolyl)amine, the protonated form of 62b−, with AgNO3, followed by ion-pair metathesis with 54a,b+-I−.87i A single crystal of 254b+-62b2− was obtained by recrystallizing 54b+-62b−, the corresponding monoanionic ion pair, from the MeOH/EtOAc solution. In the crystal structures of 54a+-62b− and 254b+-62b2−, 62b− and 62b2− form hydrogen-bonding dimers, exhibiting alternate stacking between the anion dimers and the coexisting two or four cation units, respectively. Additionally, in 54a+-62b−, the 62b− dimer is partially stacked with another dimer, while in 254b+-62b2−, the 62b2− dimer is surrounded by 54b+.87i Furthermore, pentazolate, cyclopentadienide analogue, with all CHs replaced by nitrogen, was also reported as π-electronic ion pairs. In 63+-64−, all the amino groups of 63+ and nitrogen of 64− participated in hydrogen bonding, providing a two-dimensional hydrogen-bonding network further stacked viaiπ–iπ interactions (Fig. 38e).87j Hydrogen-bonding interaction often affects the assembling modes of ion-pairing assemblies, resulting in segregated columnar structures consisting of identically charged species.
4.3. π-Electronic ion pairs comprising polymethine-based anions
Monomethine-based π-electronic anions 65− and 662− were found to form π-electronic ion-pairing assemblies when combined with π-electronic cations (Fig. 39a).88–90 The ion pairs of triphenyl(thio)pyryliums 67a,b+ with tetracyanoallyl anion 65− were synthesized by ion-pair metathesis of the ClO4− salts of the cations with Na+-65−.88a In CHCl3, 67a,b+-65− exhibits CT bands at 570 and 595 nm, respectively (Fig. 39b(i)). The CT bands exhibited negative solvatochromism, which is well-known for zwitterionic systems without electronically neutral resonance structures such as Reichardt's dyes.91 The difference in the redox potentials of the cations correlates with the excitation energies of the CT bands. Variable-concentration UV/vis absorption spectra afford association constants of ∼104 and ∼105 M−1 for 67a+-65− and 67b+-65−, respectively.88a In the 67a+-65− ion pair, 67a+ is stacked with another 67a+ on the pyrylium core and a phenyl group in an antiparallel arrangement, resulting in a stacked dimer, which is further stacked with 65− on both faces (Fig. 39b(ii)).88d This configuration leads to an intensified CT band in the polarized absorption spectrum of 67a+-65− aligned with the crystal growth direction, corresponding to the stacking direction (c axis in Fig. 39b(ii)).88a In contrast, the ion pair 67b+-65− exhibits the CT fluorescence at 740 nm in the crystalline state, while it shows no CT fluorescence in solution (measured up to ∼800 nm), emitting light from the monomeric form of 67b+.88b Interestingly, in a frozen polar solvent (MeOH-EtOH (1:1)), its spectral feature is similar to that of 67b+-ClO4−, whereas in a frozen low polar solvent (2-methyl-THF-toluene (9:1)), CT fluorescence is observed at 585 nm.88b The photoconductivity of 67b+-65− is much larger than that of 67b+-ClO4− owing to the interionic CT absorption.88c67b+-65− exhibits weak ESR signals even under dark conditions, whereas 67b+-ClO4− is ESR silent. The ESR signal intensity of 67b+-65− under photoirradiation is decreased upon cooling. The carrier mobility μ of 67b+-65− was estimated to be 10−5–10−6 cm2 V−1 s−1, assuming that the carrier concentration under dark conditions was similar to the spin concentration determined by ESR.88c
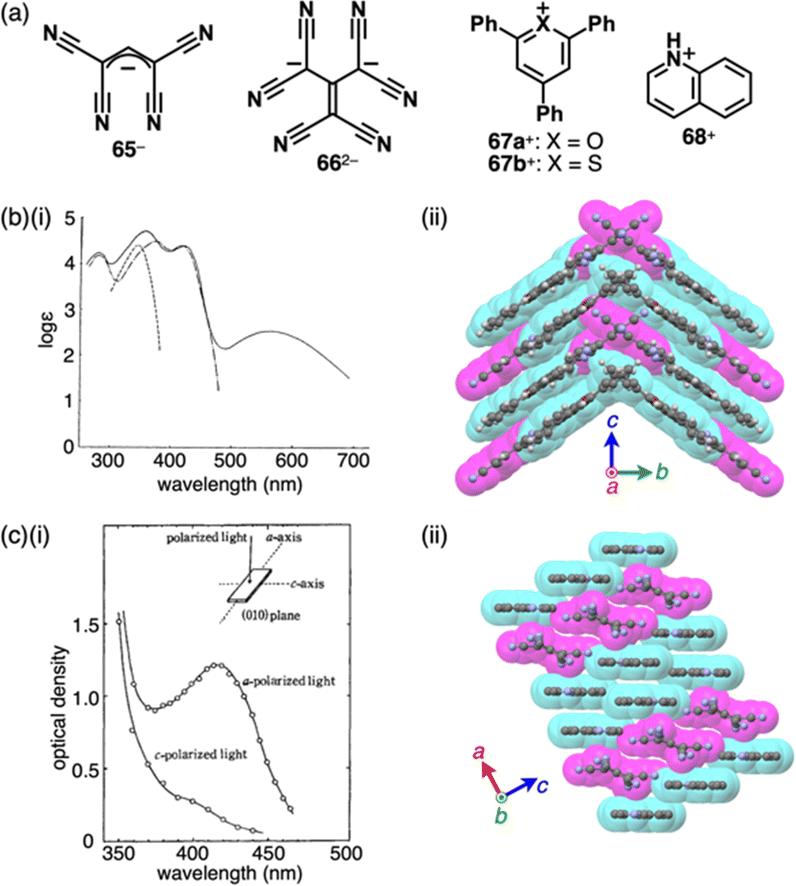 | ||
| Fig. 39 Monomethine-based cyanocarbon anions and the countercations, (b) (i) UV/vis absorption spectra of 67a+-65− (solid line), 67a+-ClO4− (chain line) and Na+-65− (dashed line) in CHCl3 (redrawn from ref. 88a. Copyright 1970 CSJ) and (ii) single-crystal X-ray structure of 67a+-65− and (c) (i) polarized absorption spectra of 268+-662− along with the direction of the long and short axes (redrawn from ref. 89a. Copyright 1969 CSJ) and (ii) single-crystal X-ray structure of 268+-662−. The long axes of single crystals of 67a+-65− and 268+-662− corresponded to the c and a axes in (ii), respectively. The crystal structures are redrawn from cif files: 1240563 and 1245498. | ||
π-Electronic ion pair of hexacyanotrimethylenemethandiide 662− with quinolinium 68+ exhibits a broad absorption band in CHCl3/EtOH (Fig. 39c(i)).89a Moreover, the ion pairs with pyridinium and N-methylacridinium cations display CT absorption bands that are red-shifted according to the increased electron affinity of the cations. In 268+-662−, two cations are located in the antiparallel orientation with an interplanar distance of 3.68 Å. The stacked cation dimer and one dianion are arranged alternately with a short N⋯N distance of 2.99 Å, forming a two-by-one columnar structure.89b Similar absorption band is observed in the polarized absorption spectrum of the single crystal, indicating CT complexation (Fig. 39c(ii)).
4,4′-Bipyridines with different alkyl chains (702+, n = 1–8) were used to form ion pairs with 69− (Fig. 40a).90a When shorter alkyl chains (n = 1–5) are present in the 702+ ion pairs, the solid-state properties vary depending on alkyl chain lengths, as indicated by differences in melting points, XRD spacing and the intensity of CT-like absorption bands. These observations strongly suggest the formation of CT complexes in these ion pairs. Additionally, in the crystal structures of 71–73+-69−, the anion 69− is stacked by itself, giving charge-segregated assemblies (Fig. 40b).90b Particularly, the stacking direction in 73+-69− is almost perpendicular to the mean plane of the constituent charged π-systems (Fig. 40b(iii)).
 | ||
| Fig. 40 (a) Polymethine-based charged π-electronic systems for ion pairs 702+-269− and 71–73+-69− and (b) single-crystal X-ray structures of (i) 71+-69−, (ii) 72+-69− and (iii) 73+-69− (redrawn from cif files: 1935496, 1935498 and 1935501). | ||
π-Electronic ion pairs of further extended oppositely charged polymethines 74–76− and 77–80+ were investigated for the effects on the spectral and assembling features (Fig. 41a).92–94 Tatikolov et al. prepared the ion pairs of oppositely charged polymethine dyes. The absorption spectrum of 77+-74a− in CH2Cl2 is almost identical to the summed spectra of 77+-Br− and TBA+-74a−.92 Similarly, the absorption maxima of 78b+-75b− are similar to those of the components.93 In contrast, 78a+-75a−, whose components show overlapped absorptions in toluene at ∼560–580 nm, exhibits a characteristic absorption band at ∼520 nm, indicating interchromophoric interactions (Fig. 41b). Theoretical calculations supported the notably blue-shifted absorption bands of 78a+-75a−, while showing negligible changes in the case of 78b+-75b−.93 Photovoltaic cell based on 77+-74a− shows significantly higher external quantum efficiency compared to that based on Na+-74a− (Fig. 41c).92 Focusing on assembled structures, in 79+-76− forming the charge-segregated assembly, 79+ and 76− are independently stacked in brickwork-like and columnar structures, respectively (Fig. 41d(i)).94 In contrast, the stacking of identical species is hindered in 80+-74b− owing to the introduction of a phenyl group at the centre of the polymethine chains, resulting in partial stacking between the oppositely charged species at their terminal π-electronic systems (Fig. 41d(ii)).
 | ||
| Fig. 41 (a) Extended polymethine-based π-electronic anions 74–76− and the countercations 77–80+, (b) UV/vis absorption spectra of 78a+-75a− (A), 1,3-bis(dimethylamino)propenium-75a− (B) and 78a+-I− (C) in toluene (redrawn from ref. 93. Copyright 2001 ACS), (c) external quantum efficiency (EQE) of bulk heterojunction solar cells fabricated with ion pairs 77+-74a− (blue, red) and Na+-74a− (green, yellow) blended with [60]PCBM in a 2:1 weight ratio (for comparison, the EQE spectra measured with and without white light bias are shown) (redrawn from ref. 92. Copyright 2009 ACS) and (d) single-crystal X-ray structures of (i) 79+-76− and (ii) 80+-74b− (redrawn from cif files: 1910541 and 1910529). | ||
4.4. π-Electronic ion pairs comprising anions formed by deprotonation
In Sections 4.2 and 4.3, π-electronic anions that form different types of π-electronic ion pairs are discussed. These anions are obtained through deprotonation of the corresponding π-electronic systems. Similarly, π-electronic dianions can be prepared by removing protons attached to the π-electronic systems, resulting in the formation of ion pairs and assemblies. By employing ion-pair metathesis between three different deprotonated anions (eosine 2Na+-81a2−, erythrosine 2Na+-81b2− and rose bengal 2Na+-822−) and three different cations (crystal violet 2+-Cl− (Section 2.1), malachite green 83+-Cl− and methylene blue 84+-Cl−) in aqueous solutions (Fig. 42a), nine types of π-electronic ion pairs (22+-81a2−, 22+-81b2−, 22+-822−, 283+-81a2−, 283+-81b2−, 283+-822−, 284+-81a2−, 284+-81b2− and 284+-822−) were obtained.95 These ion pairs exhibit distinct electrical resistivity at 20 °C in compressed disk states, with resistivity values ranging from 2.5 × 109 Ω cm for 284+-81a2− to 3.1 × 1014 Ω cm for 283+-822−. | ||
| Fig. 42 (a) Xanthene-based π-electronic anions 81a,b2− and 822−, the countercations 83,84+, 85a,d2+ and 85b+ and neutral 85c and (b) (i) 1H NMR and (ii) ESR spectra of 85a2+-81a2− in DMSO-d6 at 320 and 290 K and in DMSO at 295 K, respectively (redrawn from ref. 96c. Copyright 1992 ACS). | ||
Ion-pairing behaviours in aqueous solutions were investigated by water-soluble charged π-electronic systems such as 81a2−.96 Anionic fluorescence dyes in diluted aqueous solutions show fluorescence quenching by adding π-electronic cations.96a The absorption spectra of mixed solutions comprising different dyes such as 2Na+-81a2− and 2+-Cl− are different from the sum of those of the components, suggesting the ion pairing in aqueous solution. Fluorescence quantum yields of the ion pairs in organic solvents are considerably lower than those in aqueous solution.96a In particular, a new absorption band appears upon increasing the concentration of 2Na+-822− in an aqueous solution containing 85a2+-2Cl− (∼10−5 M) (Fig. 42a).96b Variable concentration measurements revealed an association constant of 9000 M−1 at 298 K. In addition, similar measurements for the methylviologen derivatives whose charges are partially compensated by negatively charged substituents, 85b+ and 85c, suggest that the contributions of electrostatic attractions to the enthalpy changes are −1.1 to −1.3 kcal mol−1 in aqueous solutions.
In the crystal structure of 85a2+-81a2−, 81a2− is stacked with 85a2+ in the interplanar distance of 3.43 Å.96c The resulting π-sip forms a slip-stacked arrangement with another π-sip through contact between the xanthene units of two 81a2− units that are arranged in the antiparallel orientation. The stacked tetrad is slip-stacked with another tetrad using the remaining part of the xanthene unit of 81a2−, resulting in the formation of a columnar structure. In 85d2+-81a2−, the dianion 81a2− is stacked with another 81a2− in the interplanar distance of 3.76 Å in an antiparallel fashion. The stacked dianion dimer and one dication are alternately stacked with an interplanar distance of 3.58 Å, while another dication is positioned between the columns comprising one dication and two dianion. For 85a2+-81a2−, partial dissociation is observed in DMF with an association constant of 91000 M−1. In this solvent, the stacked ion pair is further converted to 85a2+-[81a2−-85a2+-81a2−]. Intriguingly, the 1H NMR signals assigned to 85a2+ of 85a2+-81a2− in DMSO-d6 broaden upon increasing temperature and disappear at 320 K, and the ESR is observed in DMSO at 295 K, indicating the presence of a thermally accessible CT state 85a˙+-81a˙− (Fig. 42b).96c
Deprotonated dipyrrolylnitrophenols 34a,b− (Fig. 21a) were used for the components of π-electronic ion-pairing assemblies.67c π-Electronic cations, 3TATA+ and 22a+, and π-electronic anions, 34a,b−, are alternately stacked with the stacking distances in the range of 3.27–3.47 Å, forming charge-by-charge assemblies (Fig. 43a and b).67c35a− exhibits charge-by-charge assemblies with 3TATA+ and 22d+ in the solid state (Fig. 43c).68
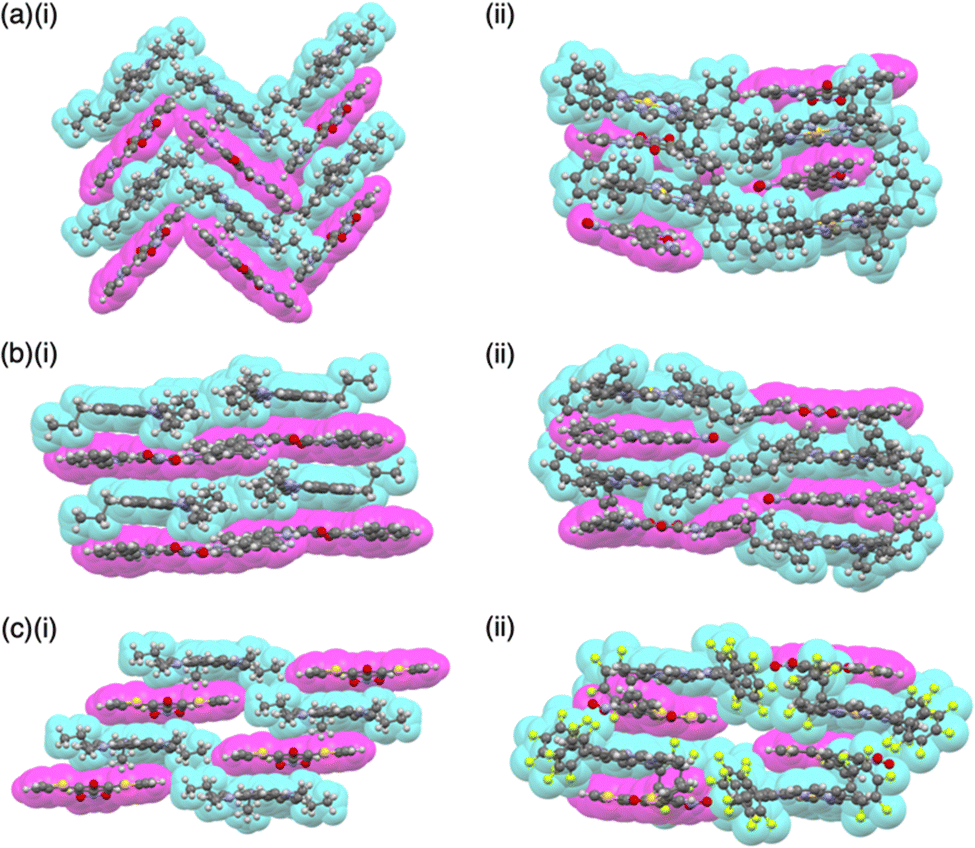 | ||
| Fig. 43 Single-crystal X-ray structures of diarylnitrophenoxides: (a) (i) 3TATA+-34a− and (ii) 22a+-34a−, (b) (i) 3TATA+-34b− and (ii) 22a+-34b− and (c) (i) 3TATA+-35a− and (ii) 22d+-35a−(redrawn from cif files: 2061520, 2061524, 2061521, 2061525, 2253152 and 2253153). | ||
As described in Section 2.3, quinone methide units effectively stabilize π-electronic anions.69 In addition, as represented by Yang's diradical,97 combinations of multiple quinone methide units can provide open-shell species, whose spin states depend on the bridging unit as spin coupler; however, the ground state of 36− is closed-shell state.69 Modifications of the deprotonated pyrrole-based quinoidal systems induce remarkable electronic properties derived from the molecular structures and interactions with coexisting cations. Quinoidal systems exhibit unique diradical properties, with their singlet and triplet states dependent on the linker units acting as spin couplers. The PB core, which has been employed for anion binding (Section 3), possesses a partially cross-conjugated structure that functions as a singlet coupler, resembling a polarized carbonyl unit (Fig. 44a). PB-based quinone methide 86 (Fig. 44b(i)), which is an oxidized form of PB, was synthesized as a benzo-fused derivative to enhance stability.9886 was deprotonated by TBAOH and NaOH to provide 86− in the forms of TBA+ and Na+ ion pairs, the latter of which was further converted to a 3TATA+ ion pair by ion-pair metathesis (Fig. 44b(i)). In the crystal structure of 3TATA+-86−, 3TATA+ and 86− are alternately stacked with their π-systems, creating the charge-by-charge assembly (Fig. 44b(ii)). Upon further deprotonation of the ion pairs of 86− with TBA+ and 3TATA+ by TBAOH, the dianion 862− was formed, resulting in the ion pairs 2TBA+-862− and 3TATA+-TBA+-862−, respectively (Fig. 44c). Unlike 86 and the monoanion 86−, the dianion 862− in 2TBA+-862− exhibits sharp and broad 1H NMR signals at −50 °C and r.t., respectively, in CD2Cl2 and displays the ESR signals with a g value of 2.003 at r.t. in mesitylene (Fig. 44c(i)). These observations indicate a ground-state singlet diradical character. Additionally, the ESR signals of 3TATA+-TBA+-862− differ from those of 2TBA+-862−, displaying countercation-dependent magnetic properties (Fig. 44c(ii)). The ground-state singlet diradical nature is further confirmed by the enhancement of the variable-temperature ESR for 3TATA+-TBA+-862−, yielding an estimated ΔEST of −6.4 kcal mol−1. The interaction between 862− and 3TATA+ is also observed in single-crystal X-ray analysis of 3TATA+-TBA+-862−, wherein TBA+ is situated on the PB unit of 862− with reduced contact of 3TATA+ and 862− (Fig. 44d).98
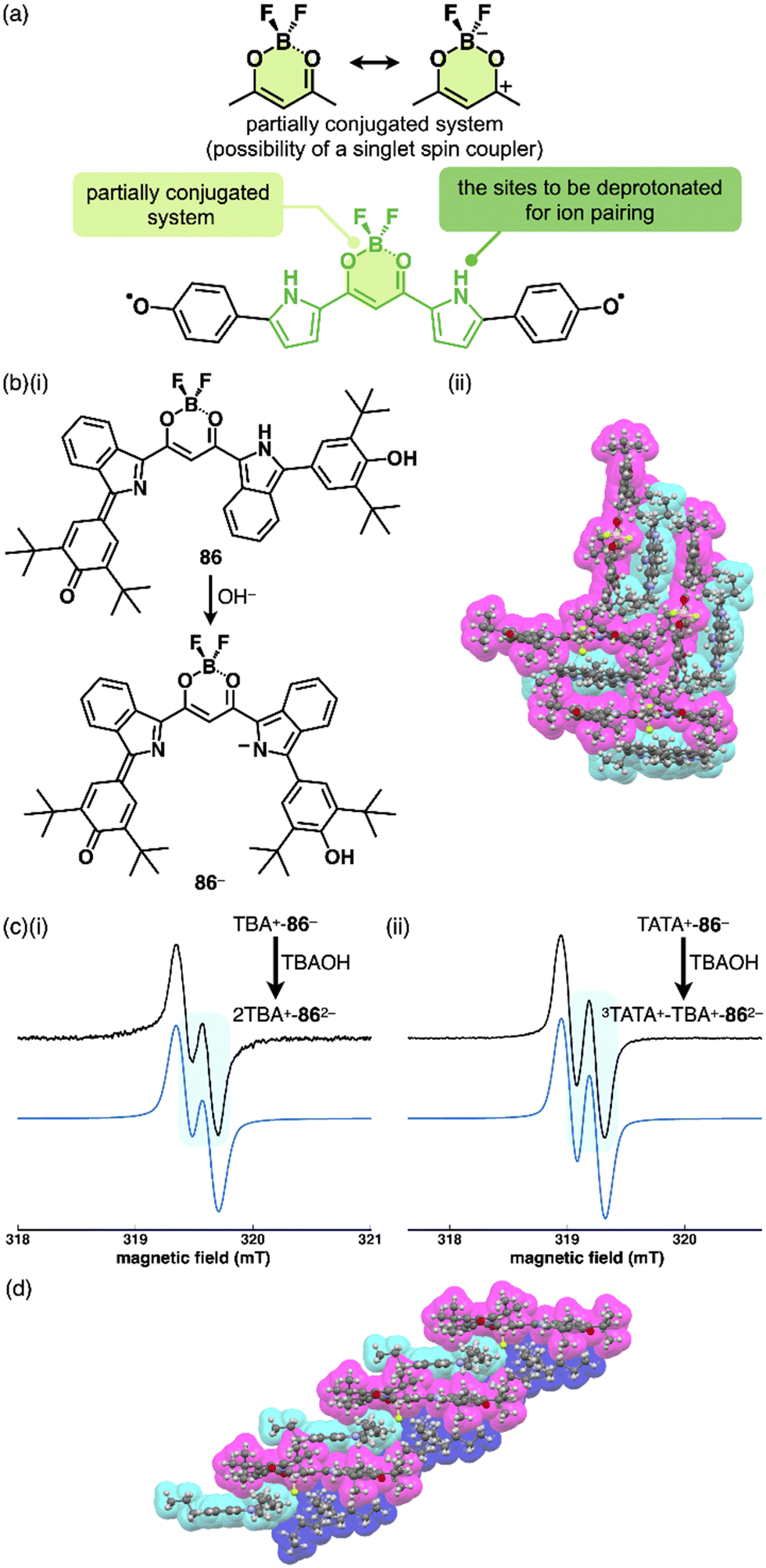 | ||
| Fig. 44 (a) Resonance structures of diketone-boron complex (top) and conceptual diagram of PB-based quinoidal molecule represented in the diradical tautomer of the parent form (bottom), (b) (i) formation of quinoidal dipyrrolyldiketone boron complex in the form of monoanion 86− by deprotonation of 86 and (ii) single-crystal X-ray structure of 3TATA+-86−, (c) ESR spectra of (i) 2TBA+-862− and (ii) 3TATA+-TBA+-862− in mesitylene (1 mM) (black line) and corresponding simulation patterns (blue line) and (d) single-crystal X-ray structure of 3TATA+-TBA+-862− (redrawn from ref. 98. Copyright 2023 ACS). The crystal structures are redrawn from cif files: 2225435 and 2225436. | ||
Moreover, deprotonated hydroxyporphyrins are also adopted in π-electronic ion pairs. Assembling behaviours of 3TATA+ ion pairs of anionic porphyrin free base 37− and NiII and PdII complexes 37a,c− were investigated (Fig. 45a).9 The anions are stacked with 3TATA+ to form charge-by-charge assemblies in the crystal structures. In 3TATA+-37a−, each component is stacked with identically charged species, resulting in a partially charge-segregated mode (Fig. 45b). The absorption spectrum of 3TATA+-37a− in the solid state exhibits blue-shifted and red-shifted bands derived from 37a− and 3TATA+, respectively, compared to those for the monomeric states of 37a− (TBA+-37a−) and 3TATA+ (3TATA+-Cl−) in CH2Cl2 (Fig. 45c). This shift is attributed to exciton coupling arising from the stacking of identically charged species in the parallel (H-like) arrangement for 37a− and a head-to-tail (J-like) arrangement for 3TATA+. Additionally, solid-state transient absorption spectra of 3TATA+-37a− reveal the generation of radical 37a˙ by the PET between the π-electronic systems. Furthermore, EDA of the packing structure of 3TATA+-37a− indicates a significant contribution of electrostatic and dispersion forces in stabilizing the stacked structures of the constituent π-electronic ions.9
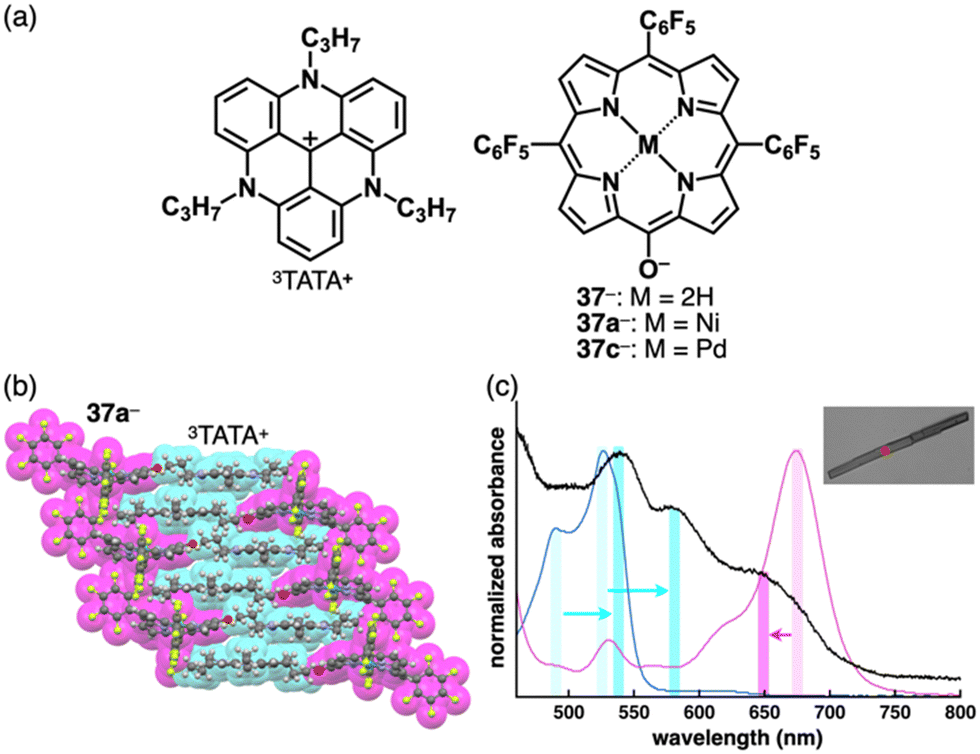 | ||
| Fig. 45 (a) Ion pairs 3TATA+-37− and 3TATA+-37a,c−, (b) single-crystal X-ray structure of 3TATA+-37a− (redrawn from a cif file: 1906678) and (c) UV/vis absorption spectrum of the single crystal of 3TATA+-37a− (black) along with those of the solution states (1 × 10−5 M in CH2Cl2) of TBA+-37a− (magenta) and 3TATA+-Cl− (cyan) (inset: optical microscopy image of a single crystal of 3TATA+-37a−) (redrawn from ref. 9. Copyright 2021 RSC). | ||
Remarkable electronic properties emerge from π-electronic ion pairs and their assemblies comprising charged species with large π-systems. Promising π-electronic ion pairs can be produced by introducing charged porphyrins as cations and anions, including 37a− for the latter. Modifications of the electronic states and resulting assembled structures can be achieved by introducing substituents, as seen in AuIII complexes 22a,c+ that possess four C6H5 and C6F5 moieties, respectively, at the meso positions.52 Combining oppositely charged porphyrins with distinct electronic states according to introduced substituents provides diverse ion pairs that can be used for functional materials. Thus, in addition to 37a−, meso-phenyl-substituted 87− was prepared as an electron-rich “activated” anion (Fig. 46a).6 The “activated” cation 22d+, with three meso-C6F5 units for efficient stacking, has been a focal point for creating an activated ion pair with 87−. Several porphyrin ion pairs, including 22a+-37a−,52a22a+-87− and 22d+-37a− as well as the activated ion pair 22d+-87−, were prepared by ion-pair metathesis from charged porphyrin starting materials, resulting in solid-state ion-pairing assemblies. In the case of 22a+-37a−, stacking arrangement exhibits a zigzag pattern owing to the steric effect of meso-phenyl groups.52a In contrast, the assembly of 22d+-87− involves a charge-segregated mode, with four benzene molecules included for each ion pair (Fig. 46b(i)). Nevertheless, π-sip-based charge-by-charge assembly modes in 22a+-87− results in iπ–iπ interaction between porphyrin ions, as the steric hindrance of the porphyrin anion is relaxed (Fig. 46b(ii)). The packing diagram in 22d+-37a− demonstrates a 1D-chain structure of π-sip assembled in a four-fold helical symmetry, with 22d+ and 37a− arranged in a stacking distance of 3.57 Å (Fig. 46b(iii)).6,99
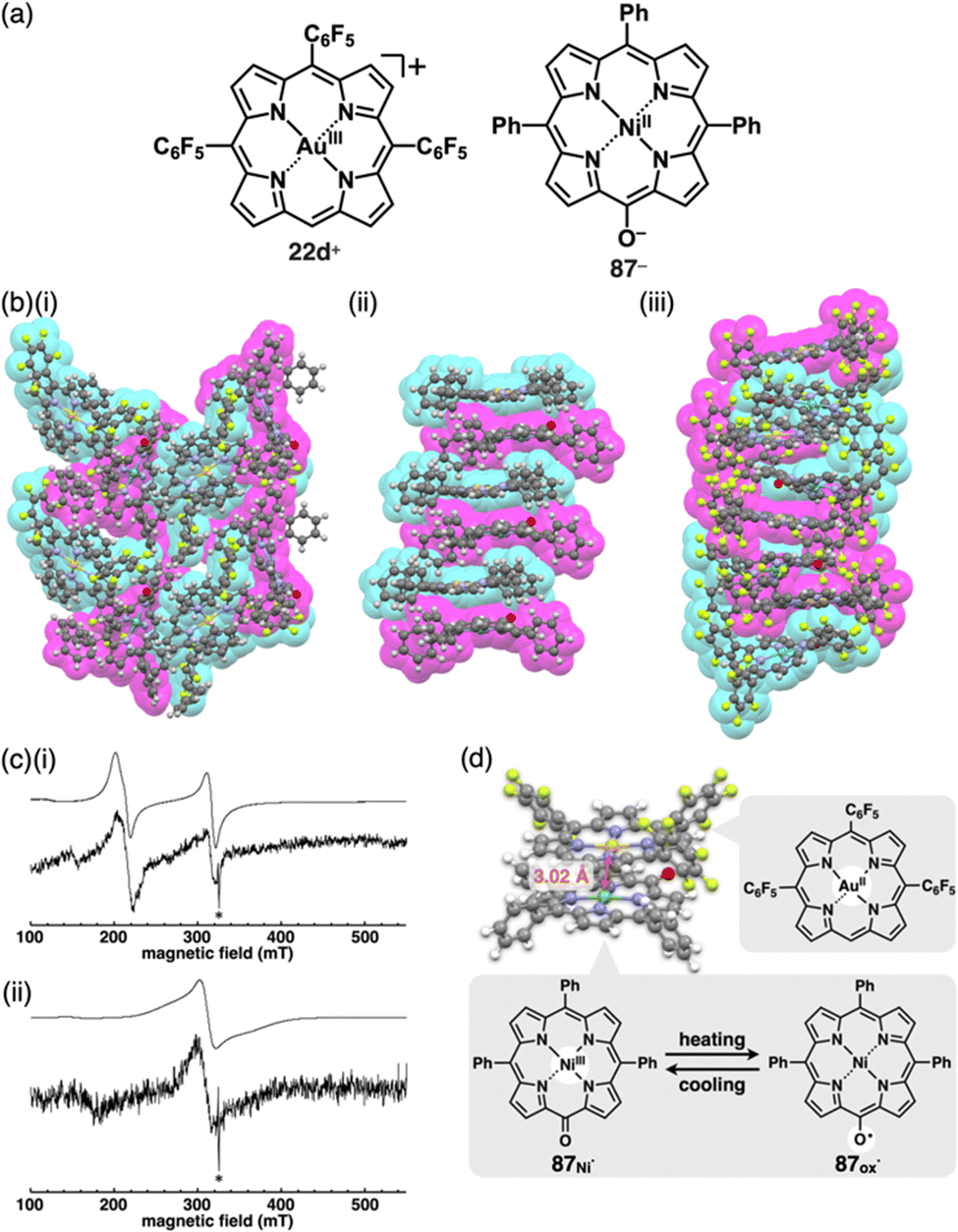 | ||
| Fig. 46 (a) Ion pair of charged porphyrins, 22d+-87−, as an activated ion pair, (b) single-crystal X-ray structures of (i) 22d+-87−, (ii) 22a+-87− and (iii) 22d+-37a− (redrawn from cif files: 2201074, 2201075 and 2201076), (c) simulated (top) and observed (bottom) ESR spectra of 22d+-87− in toluene at (i) 10 and (ii) 140 K and (d) closely stacked structure (π-srp) of 22d˙ and 87˙ with the spin–spin distance and the change in spin distribution of 87˙ (87Ni˙ and 87ox˙) as a result of temperature change (redrawn from ref. 6. Copyright 2022 ACS). Signals marked with * in (c) are ascribable to a trace amount of dissociated 87˙, whereas white circles in (d) represent spin distributions. | ||
Charged porphyrins form stacked dimers in solution, as detected by 1H NMR chemical shifts derived from shielding and deshielding effects. Concentration-dependent 1H NMR of 22d+-37a− in CD2Cl2 provides the heterodimerization constant (Kdim = 2.8 × 105 M−1) at 20 °C. On the other hand, the UV/vis absorption spectrum of 22d+-37a− in the π-sip is almost the sum of the spectra of constituent 22d+ and 37a−. In contrast, the activated ion pair 22d+-87− exhibits a UV/vis absorption spectrum that differs from the corresponding summed spectra in less polar solvents. The spectrum observed in toluene is consistent with the combined spectra of the reduced AuII complex 22d˙ and the oxidized NiII complex oxyradical 87ox˙, indicating an electron transfer from 87− to 22d+ to generate a π-stacked radical pair (π-srp). Notably, the ESR spectrum in frozen toluene at 10 K (Fig. 46c(i)) displays the π-srp as a pair of 22d˙ and NiIII complex 87˙ with a short spin–spin distance of 3.02 Å (Fig. 46d top). VT ESR spectra suggest spin transfer in 87˙ and antiferromagnetic interaction in π-srp (Fig. 46c(ii) and d bottom).6 The less activated porphyrin ion pairs exhibit π-sips that correlate with the redox potentials of their constituent components based on the substituents. Further investigations on the PET for ion pairs, including triaryl-substituted 22b+ and 22d+, revealed varying electron-transfer rates, leading to the formation of excited-state radical species. These findings suggest application potential of these ion pairs in photocatalysis owing to their tuneable electronic state.6,53
5. Conclusions
π-Electronic ion pairs, comprising countercharged π-electronic systems, hold immense potential in generating structured assembled arrangements and displaying unique properties. Assembling modes of charged components are modulated by their geometries and electronic states. Thus, the design and synthesis of charged π-electronic systems are crucial for the fabrication of ion-pairing assemblies (Fig. 47). The presence of charges within the core π-electronic units makes their preparation challenging owing to the inherent reactivity arising from electron-deficient and -rich states. Furthermore, the specific combinations of π-electronic ions are crucial for determining the nature of the assemblies and the resulting properties. Less stable π-electronic ions can be stabilized by combining specific counterions via the formation of π-sips based on iπ–iπ interactions. Hence, diversity is a central aspect in ion-pairing chemistry.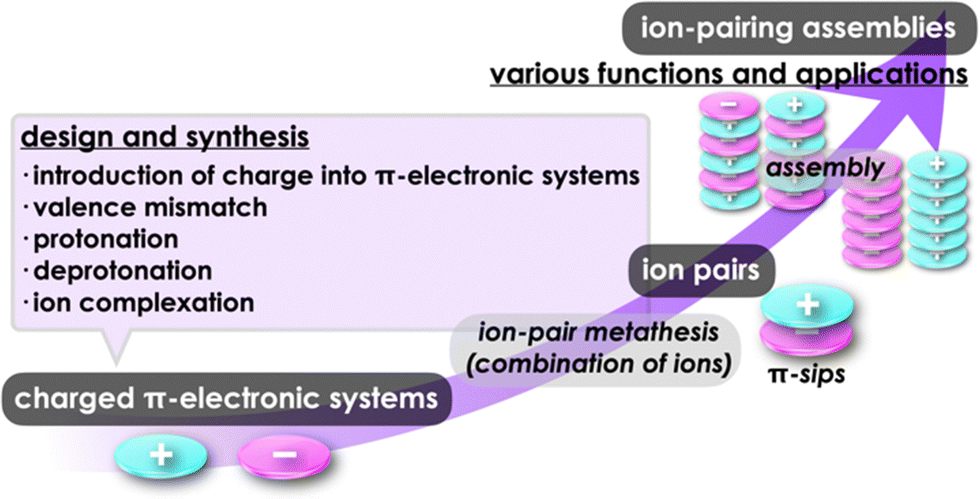 | ||
| Fig. 47 Formation of ion-pairing assemblies through design and synthesis of charged π-electronic systems, ion-pair metathesis and assembly. | ||
The assemblies of ion pairs reveal their distinct characteristics not only in solution but also in bulk states like crystals and soft materials. The ability to control assembling modes, whether charge-by-charge or charge-segregated, is crucial for exhibiting specific electronic properties based on the arrangements. The electronic properties of π-electronic ion pairs and their assemblies are distinct from those of electronically neutral π-systems. The proximally located oppositely charged π-electronic systems induce the polarized columnar structures, most polarizations of which are cancelled by those of neighbouring columns. Spontaneous polarization with uniform alignment of polarized columns facilitates the development of ferroelectric materials.100 The manipulation of redox potentials between π-electronic cations and anions enables electron transfer in both excited and ground states. The resulting radical pairs, especially under photoirradiation, can serve as catalysts, with cation- and anion-derived radicals acting as reducing and oxidizing species, respectively.101 Furthermore, columnar structures comprising identically charged species offer potential applications in efficient semiconductive materials. The electrostatic repulsion between identically charged species can be overcome by dispersion forces originating from π-electronic systems and dipole–dipole interactions in polarized charged species. These strategies are easily attainable through simple modifications for π-electronic systems, in contrast to inorganic ionic species. This review article showcases the production of diverse π-electronic ion pairs, demonstrating possibilities for future property investigations and applications. The authors have made significant strides in understanding their intriguing behaviours of these ion pairs through thorough comprehensive studies in synthesis, combination, assembly and electronic and photophysical properties in recent years.102 A significant portion of ion-pairing chemistry remains unexplored and awaits exploration through ongoing research endeavours.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The recent contributions of our group reported herein were mainly supported by JSPS KAKENHI Grant Numbers JP18H01968 and JP22H02067 for Scientific Research (B), JP20H05863 for Transformative Research Areas (A) “Condensed Conjugation” and the Ritsumeikan Global Innovation Research Organization (R-GIRO) project, 2017–2022 and 2022–2027.Notes and references
- Selected books on supramolecular assemblies for electronic materials: (a) Supramolecular Soft Matter: Applications in Materials and Organic Electronics, ed. T. Nakanishi, Wiley, 2011 Search PubMed; (b) Functional Supramolecular Architectures, ed. P. Samorì and F. Cacialli, Wiley, 2011 Search PubMed; (c) Unimolecular and Supramolecular Electronics I: Chemistry and Physics Meet at Metal-Molecule Interfaces, Topics in Current Chemistry, ed. R. M. Metzger, Springer, 2012 Search PubMed; (d) Supramolecular Materials for Opto-Electronics, ed. N. Koch, RSC, 2015 Search PubMed.
- (a) K. Müller-Dethlefs and P. Hobza, Chem. Rev., 2000, 100, 143 CrossRef PubMed; (b) A. S. Mahadevi and G. N. Sastry, Chem. Rev., 2016, 116, 2775 CrossRef CAS PubMed.
- A. J. Stone, The Theory of Intermolecular Forces, Oxford University Press, 2013 Search PubMed.
- (a) C. F. J. Faul and M. Antonietti, Adv. Mater., 2003, 15, 673 CrossRef CAS; (b) C. F. J. Faul, Acc. Chem. Res., 2014, 47, 3428 CrossRef CAS PubMed.
- (a) Y. Haketa and H. Maeda, Bull. Chem. Soc. Jpn., 2018, 91, 420 CrossRef CAS; (b) Y. Haketa, K. Urakawa and H. Maeda, Mol. Syst. Des. Eng., 2020, 5, 757 RSC; (c) K. Yamasumi and H. Maeda, Bull. Chem. Soc. Jpn., 2021, 94, 2252 CrossRef CAS.
- H. Tanaka, Y. Kobayashi, K. Furukawa, Y. Okayasu, S. Akine, N. Yasuda and H. Maeda, J. Am. Chem. Soc., 2022, 144, 21710 CrossRef CAS PubMed.
- Y. Haketa, S. Sasaki, N. Ohta, H. Masunaga, H. Ogawa, N. Mizuno, F. Araoka, H. Takezoe and H. Maeda, Angew. Chem., Int. Ed., 2010, 49, 10079 CrossRef CAS PubMed.
- Several π-electronic ion-pairing assemblies had been reported as listed in the following references (e.g., ref. 81d,e,82,87c–e,88d,89b,96c) before the report of ref. 7. However, to the best of our knowledge, no reports clearly have shown the chemistry of π-electronic ion-pairing assemblies.
- Y. Sasano, H. Tanaka, Y. Haketa, Y. Kobayashi, Y. Ishibashi, T. Morimoto, R. Sato, Y. Shigeta, N. Yasuda, T. Asahi and H. Maeda, Chem. Sci., 2021, 12, 9645 RSC.
- (a) D. F. Duxbury, Chem. Rev., 1993, 93, 381 CrossRef CAS; (b) V. Nair, S. Thomas, S. C. Mathew and K. G. Abhilash, Tetrahedron, 2006, 62, 6731 CrossRef CAS.
- E. Q. Adams and L. Rosenstein, J. Am. Chem. Soc., 1914, 36, 1452 CrossRef CAS.
- Reviews of fluorescent dyes: (a) R. N. Dsouza, U. Pischel and W. M. Nau, Chem. Rev., 2011, 111, 7941 CrossRef CAS PubMed; (b) Y. Geng, Z. Wang, J. Zhou, M. Zhu, J. Liu and T. D. James, Chem. Soc. Rev., 2023, 52, 3873 RSC.
- Reviews of rhodamines: (a) M. Beija, C. A. M. Afonso and J. M. G. Martinho, Chem. Soc. Rev., 2009, 38, 2410 RSC; (b) T. Ikeno, T. Nagano and K. Hanaoka, Chem. – Asian J., 2017, 12, 1435 CrossRef CAS PubMed; (c) E. Reglska, P. Hindenberg and C. Romero-Nieto, Eur. J. Inorg. Chem., 2019, 1519 CrossRef; (d) S. G. Keller, M. Kamiya and Y. Urano, Molecules, 2020, 25, 5964 CrossRef CAS PubMed.
- Reviews and first two reports of TATA+: (a) J. Bosson, J. Gouin and J. Lacour, Chem. Soc. Rev., 2014, 43, 2824 RSC; (b) A. Winter and U. S. Schubert, Mater. Chem. Front., 2019, 3, 2308 RSC; (c) B. W. Laursen and F. C. Krebs, Angew. Chem., Int. Ed., 2000, 39, 3432 CrossRef CAS; (d) B. W. Laursen and F. C. Krebs, Chem. – Eur. J., 2001, 7, 1773 CrossRef CAS PubMed.
- A review and selected reports of TOTA+: (a) A. C. Shaikh, J. M. Veleta, J. Moutet and T. L. Gianetti, Chem. Sci., 2021, 12, 4841 RSC; (b) B. W. Laursen, F. C. Krebs, M. F. Nielsen, K. Bechgaard, J. B. Christensen and N. Harrit, J. Am. Chem. Soc., 1998, 120, 12255 CrossRef CAS; (c) D. Shi, C. Schwall, G. Sfintes, E. Thyrhaug, P. Hammershøj, M. Cárdenas, J. B. Simonsen and B. W. Laursen, Chem. – Eur. J., 2014, 20, 6853 CrossRef CAS PubMed.
- Examples of TATA+: (a) R. Gueret, L. Poulard, M. Oshinowa, J. Chauvin, M. Dahmane, G. Dupeyre, P. P. Lainé, J. Fortage and M.-N. Collomb, ACS Catal., 2018, 8, 3792 CrossRef CAS; (b) P.-Y. Ho, S.-C. Cheng, F. Yu, Y.-Y. Yeung, W.-X. Ni, C.-C. Ko, C.-F. Leung, T.-C. Lau and M. Robert, ACS Catal., 2023, 13, 5979 CrossRef CAS.
- G. Allinson, R. J. Bushby, J.-L. Paillaud and M. Thornton-Pett, J. Chem. Soc., Perkin Trans. 1, 1995, 385 RSC.
- Reviews of cyanines: (a) A. Mishra, R. K. Behera, P. K. Behera, B. K. Mishra and G. B. Behera, Chem. Rev., 2000, 100, 1973 CrossRef CAS PubMed; (b) M. Henary, S. Paranijpe and E. A. Owens, Heterocycl. Commun., 2013, 19, 1 CrossRef CAS; (c) J. L. Bricks, A. D. Kachkovski, Y. L. Slominski, A. O. Gerasov and S. V. Popov, Dyes Pigm., 2015, 121, 238 CrossRef CAS; (d) W. Sun, S. Guo, C. Hu, J. Fan and X. Peng, Chem. Rev., 2016, 116, 7768 CrossRef CAS PubMed; (e) H. A. Shindy, Dyes Pigm., 2017, 145, 505 CrossRef CAS; (f) K. Ilina and M. Henary, Chem. – Eur. J., 2021, 27, 4230 CrossRef CAS PubMed; (g) H. Mustroph, Dyes Pigm., 2022, 208, 110783 CrossRef CAS.
- F. Würthner, T. E. Kaiser and C. R. Saha-Möller, Angew. Chem., Int. Ed., 2011, 50, 3376 CrossRef PubMed.
- H. Mustroph, Phys. Sci. Rev., 2021, 7, 37 Search PubMed.
- S. Khopkar and G. Shankarling, Dyes Pigm., 2019, 170, 107645 CrossRef CAS.
- (a) Y. Xu, A. Malkovskiy, Q. Wang and Y. Pang, Org. Biomol. Chem., 2011, 9, 2878 RSC; (b) Y. Xu, A. Malkovskiy and Y. Pang, Chem. Commun., 2011, 47, 6662 RSC; (c) Y. Xu, B. Li, L. Xiao, J. Ouyang, S. Sun and Y. Pang, Chem. Commun., 2014, 50, 8677 RSC; (d) B. Li, W. Li, Y. Xu, J. Li, J. Tu and S. Sun, Chem. Commun., 2015, 51, 14652 RSC.
- K. Yamasumi, K. Ueda, Y. Haketa, Y. Hattori, M. Suda, S. Seki, H. Sakai, T. Hasobe, R. Ikemura, Y. Imai, Y. Ishibashi, T. Asahi, K. Nakamura and H. Maeda, Angew. Chem., Int. Ed., 2023, 62, e202216013 CrossRef CAS PubMed.
- D. J. M. Lyons, R. D. Crocker, M. Blümel and T. V. Nguyen, Angew. Chem., Int. Ed., 2017, 56, 1466 CrossRef CAS PubMed.
- (a) K. Komatsu and T. Kitagawa, Chem. Rev., 2003, 103, 1371 CrossRef CAS PubMed; (b) J. S. Bandar and T. H. Lambert, Synthesis, 2013, 2485 CAS; (c) R. M. Wilson and T. H. Lambert, Acc. Chem. Res., 2022, 55, 3057 CrossRef CAS PubMed.
- F. T. Zahra, A. Saeed, K. Mumtaz and F. Albericio, Molecules, 2023, 28, 4095 CrossRef CAS PubMed.
- Metallocenes: Synthesis Reactivity Applications, ed. A. Togni and R. L. Halterman, Wiley, 1998 Search PubMed.
- R. Kuhn and D. Rewicki, Angew. Chem., Int. Ed. Engl., 1967, 6, 635 CrossRef CAS.
- O. Diels, Ber. Dtsch. Chem. Ges., 1942, 75, 1452 CrossRef.
- M. I. Bruce, P. A. Humphrey, B. W. Skelton and A. H. White, Aust. J. Chem., 1984, 37, 2441 CrossRef CAS.
- (a) O. W. Webster, J. Am. Chem. Soc., 1965, 87, 1820 CrossRef CAS; (b) T. Sakai, S. Seo, J. Matsuoka and Y. Mori, J. Org. Chem., 2013, 78, 10978 CrossRef CAS PubMed.
- A selected report: J. Bacsa, R. J. Less, H. E. Skelton, Z. Soracevic, A. Steiner, T. C. Wilson, P. T. Wood and D. S. Wright, Angew. Chem., Int. Ed., 2011, 29, 8279 CrossRef PubMed.
- Y. Bando, Y. Haketa, T. Sakurai, W. Matsuda, S. Seki, H. Takaya and H. Maeda, Chem. – Eur. J., 2016, 23, 7843 CrossRef PubMed.
- PCCp− as a guest species can control the states of self-assembled cage-shaped nanocubes: (a) Y.-Y. Zhan, T. Kojima, T. Nakamura, T. Takahashi, S. Takahashi, Y. Haketa, Y. Shoji, H. Maeda, T. Fukushima and S. Hiraoka, Nat. Commun., 2018, 9, 4530 CrossRef PubMed; (b) Y.-Y. Zhan, T. Kojima, K. Ishii, S. Takahashi, Y. Haketa, H. Maeda, S. Uchiyama and S. Hiraoka, Nat. Commun., 2019, 10, 1440 CrossRef PubMed.
- (a) S. Sowmiah, J. M. S. S. Esperança, L. P. N. Revelo and C. A. M. Afonso, Org. Chem. Front., 2018, 5, 453 RSC; (b) Y. Li, H. Wang and X. Li, Chem. Sci., 2020, 11, 12249 RSC; (c) E. Hola and J. Ortyl, Eur. Polym. J., 2021, 150, 110365 CrossRef CAS.
- D. Wu, W. Pisula, M. C. Haberecht, X. Feng and K. Müllen, Org. Lett., 2009, 11, 5686 CrossRef CAS PubMed.
- A. Granzhan and H. Ihmels, Synlett, 2016, 1775 CAS.
- (a) D. Wu, L. Zhi, G. J. Bodwell, G. Cui, N. Tsao and K. Müllen, Angew. Chem., Int. Ed., 2007, 46, 5417 CrossRef CAS PubMed; (b) D. Wu, W. Pisula, V. Enkelmann, X. Feng and K. Müllen, J. Am. Chem. Soc., 2009, 131, 9620 CrossRef CAS PubMed.
- (a) The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, 1999 Search PubMed; (b) Handbook of Porphyrin Science, ed. K. M. Kadish, K. M. Smith and R. Guilard, World Scientific, 2010 Search PubMed.
- Selected reviews: (a) J. A. A. W. Elemans, R. van Hameren, R. J. M. Nolte and A. E. Rowan, Adv. Mater., 2006, 18, 1251 CrossRef CAS; (b) A. D’Urso, M. E. Fragalá and R. Purrello, Chem. Commun., 2012, 48, 8165 RSC; (c) H. Lee, H. Park, D. Y. Ryu and W.-D. Jang, Chem. Soc. Rev., 2023, 52, 1947 RSC.
- A review and selected reports of heteroatom-containing porphyrins: (a) Y. Matano, Chem. Rev., 2017, 117, 3138 CrossRef CAS PubMed; (b) A. Takiguchi, S. Kang, N. Fukui, D. Kim and H. Shinokubo, Angew. Chem., Int. Ed., 2021, 60, 2915 CrossRef CAS PubMed; (c) A. Takiguchi and H. Shinokubo, Chem. Lett., 2022, 51, 590 CrossRef CAS; (d) A. Takiguchi, N. Inai, S. Kang, M. Hagai, S. Lee, T. Yanai, D. Kim and H. Shinokubo, Chem. Commun., 2022, 58, 5956 RSC.
- T. Satoh, M. Minoura, H. Nakano, K. Furukawa and Y. Matano, Angew. Chem., Int. Ed., 2016, 55, 2235 CrossRef CAS PubMed.
- A review and selected reports of heteroporphyrins: (a) T. Chatterjee, V. S. Shetti, R. Sharma and M. Ravikanth, Chem. Rev., 2017, 117, 3254 CrossRef CAS PubMed; (b) E. Vogel, W. Haas, B. Knipp, J. Lex and H. Schmickler, Angew. Chem., Int. Ed. Engl., 1988, 27, 406 CrossRef; (c) E. Vogel, P. Röhrig, M. Sicken, B. Knipp, A. Herrmann, M. Pohl, H. Schmickler and J. Lex, Angew. Chem., Int. Ed. Engl., 1989, 28, 1651 CrossRef; (d) E. Vogel, J. Dörr, A. Herrmann, J. Lex, H. Schmickler, P. Walgenbach, J. P. Gisselbrecht and M. Gross, Angew. Chem., Int. Ed. Engl., 1993, 32, 1597 CrossRef; (e) M. Bröring, H.-J. Dietrich, J. Dörr, G. Hohlneicher, J. Lex, N. Jux, C. Pütz, M. Roeb, H. Schmickler and E. Vogel, Angew. Chem., Int. Ed., 2000, 39, 1105 CrossRef.
- (a) G. Magnus, Poggendorff's Ann. Phys., 1828, 90, 239 CrossRef; (b) G. Magnus, Ann. Chim. Phys., 1829, 40, 110 Search PubMed.
- M. Atoji, J. W. Richardson and R. E. Rundle, J. Am. Chem. Soc., 1957, 79, 3017 CrossRef CAS.
- M. Fang, S. R. Wilson and K. S. Suslick, J. Am. Chem. Soc., 2008, 130, 1134 CrossRef CAS PubMed.
- Z. Xie, J. Manning, R. W. Reed, R. Mathur, P. D. W. Boyd, A. Benesi and C. A. Reed, J. Am. Chem. Soc., 1996, 118, 2922 CrossRef CAS.
- Y. Inokuma, J. H. Kwon, T. K. Ahn, M.-C. Yoo, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2006, 45, 961 CrossRef CAS PubMed.
- E. Tsurumaki, S. Y. Hayashi, F. S. Tham, C. A. Reed and A. Osuka, J. Am. Chem. Soc., 2011, 133, 11956 CrossRef CAS PubMed.
- A review of cationic AuIII complexes: R. Kumar and C. Nevado, Angew. Chem., Int. Ed., 2017, 56, 1994 CrossRef CAS PubMed.
- (a) E. B. Fleischer and A. Laszlo, Inorg. Nucl. Chem. Lett., 1969, 5, 373 CrossRef CAS; (b) R. Timkovich and A. Tulinsky, Inorg. Chem., 1977, 16, 962 CrossRef CAS; (c) J.-C. Chambron, V. Heitz and J.-P. Sauvage, New J. Chem., 1997, 21, 237 CAS; (d) H. Lv, B. Yang, J. Jing, Y. Yu, J. Zhang and J.-L. Zhang, Dalton Trans., 2012, 41, 3116 RSC.
- (a) Y. Haketa, Y. Bando, Y. Sasano, H. Tanaka, N. Yasuda, I. Hisaki and H. Maeda, iScience, 2019, 14, 241 CrossRef CAS PubMed; (b) H. Tanaka, Y. Haketa, N. Yasuda and H. Maeda, Chem. – Asian J., 2019, 14, 2129 CrossRef CAS PubMed.
- H. Tanaka, Y. Okayasu, Y. Kobayashi and H. Maeda, Chem. – Eur. J., 2023, 29, e202203957 CrossRef CAS PubMed.
- L. Latos-Grażyński, J. Lisowski, M. M. Olmstead and A. L. Balch, Inorg. Chem., 1989, 28, 1183 CrossRef.
- M. Fujita, H. Tanaka, N. Yasuda and H. Maeda, Chem. Commun., 2022, 58, 9870 RSC.
- Q.-C. Chen, S. Fite, N. Fridman, B. Tumanskii, A. Mahammed and Z. Gross, ACS Catal., 2022, 12, 4310 CrossRef CAS.
- J. Arnold, J. Chem. Soc., Chem. Commun., 1990, 976 RSC.
- A review: V. W.-W. Yam, V. K.-M. Au and S.-L. Leung, Chem. Rev., 2015, 115, 7589 CrossRef CAS PubMed.
- Selected articles: (a) V. W.-W. Yam, K. M.-C. Wong and N. Zhu, J. Am. Chem. Soc., 2002, 124, 6506 CrossRef CAS PubMed; (b) A. Y.-Y. Tam, K. M.-C. Wong and V. W.-W. Yam, Chem. – Eur. J., 2009, 15, 4775 CrossRef CAS PubMed; (c) S. Y.-L. Leung, A. Y.-L. Tam, C.-H. Tao, H.-S. Chow and V. W.-W. Yam, J. Am. Chem. Soc., 2012, 134, 1047 CrossRef CAS PubMed; (d) K. Zhang, M. C.-L. Yeung, S. Y.-L. Leung and V. W.-W. Yam, Chem, 2017, 2, 825 CrossRef CAS.
- (a) W. Lu, K. T. Chan, S.-X. Wu, Y. Chen and C.-M. Che, Chem. Sci., 2012, 3, 752 RSC; (b) V. K.-M. Au, W. H. Lam, W.-T. Wong and V. W.-W. Yam, Inorg. Chem., 2012, 51, 7537 CrossRef CAS PubMed.
- Selected reports of anionic metal complexes: (a) J. R. Berenguer, E. Lalinde and J. Torroba, Inorg. Chem., 2007, 46, 9919 CrossRef CAS PubMed; (b) L. Ricciardi, M. L. Deda, A. Ionescu, N. Godbert, I. Aiello and M. Ghedini, Dalton Trans., 2017, 46, 12625 RSC.
- Y. Tanaka, K. M.-C. Wong and V. W.-W. Yam, Angew. Chem., Int. Ed., 2013, 52, 14117 CrossRef CAS PubMed.
- Azacalix[3]pyridine derivatives and analogue: (a) T. Kanbara, Y. Suzuki and T. Yamamoto, Eur. J. Org. Chem., 2006, 3314 CrossRef CAS; (b) N. Uchida, A. Taketoshi, J. Kuwabara, T. Yamamoto, Y. Inoue, Y. Watanabe and T. Kanbara, Org. Lett., 2010, 12, 5242 CrossRef CAS PubMed; (c) N. Uchida, J. Kuwabara, A. Taketoshi and T. Kanbara, J. Org. Chem., 2012, 77, 10631 CrossRef CAS PubMed; (d) H. Gong, C. Zhang, T. Ogaki, H. Inuzuka, D. Hashizume and D. Miyajima, Angew. Chem., Int. Ed., 2021, 60, 16377 CrossRef CAS PubMed.
- S. Adachi, M. Shibasaki and N. Kumagai, Nat. Commun., 2019, 10, 3820 CrossRef PubMed.
- E. Raamat, K. Kaupmees, G. Ovsjannikov, A. Trummal, A. Kütt, J. Saame, I. Koppel, I. Kaljurand, L. Lipping, T. Rodima, V. Pihl, I. A. Koppel and I. Leito, J. Phys. Org. Chem., 2013, 26, 162 CrossRef CAS.
- S. A. Smith and W. A. Pretorius, Water SA, 2002, 28, 395 CAS.
- (a) H. Maeda, A. Fukui, R. Yamakado and N. Yasuda, Chem. Commun., 2015, 51, 17572 RSC; (b) H. Maeda, Y. Takeda, Y. Haketa, Y. Morimoto and N. Yasuda, Chem. – Eur. J., 2018, 24, 8910 CrossRef CAS PubMed; (c) N. Fumoto, Y. Haketa, H. Tanaka, N. Yasuda and H. Maeda, Org. Lett., 2021, 23, 3897 CrossRef CAS PubMed; (d) M. Yokoyama and H. Maeda, Chem. Lett., 2023, 52, 598 CrossRef CAS.
- M. Yokoyama, Y. Okayasu, Y. Kobayashi, H. Tanaka, Y. Haketa and H. Maeda, Org. Lett., 2023, 25, 3676 CrossRef CAS PubMed.
- S. Sugiura and H. Maeda, Chem. Commun., 2021, 57, 6983 RSC.
- (a) Y. Sasano, N. Yasuda and H. Maeda, Dalton Trans., 2017, 46, 8924 RSC; (b) Y. Sasano, Y. Haketa, H. Tanaka, N. Yasuda, I. Hisaki and H. Maeda, Chem. – Eur. J., 2019, 25, 6712 CrossRef CAS PubMed.
- Selected books and reviews: (a) Supramolecular Chemistry of Anion, ed. A. Bianchi, K. Bowman-James and E. García-España, Wiley, 1997 Search PubMed; (b) Anion Sensing, Topics in Current Chemistry, ed. I. Stibor, Springer, 2005, vol. 255 Search PubMed; (c) J. L. Sessler, P. A. Gale and W.-S. Cho, Anion Receptor Chemistry, RSC, 2006 RSC; (d) Anion Recognition in Supramolecular Chemistry, Topics in Heterocyclic Chemistry, ed. P. A. Gale and W. Dehaen, Springer, 2010, vol. 24 Search PubMed; (e) Anion Coordination Chemistry, ed. K. Bowman-James, A. Bianchi and E. García-España, Wiley, 2011 CrossRef; (f) P. D. Beer and P. A. Gale, Angew. Chem., Int. Ed., 2001, 40, 486 CrossRef CAS; (g) R. M. Duke, E. B. Veale, F. M. Pfeffer, P. E. Krugerc and T. Gunnlaugsson, Chem. Soc. Rev., 2010, 39, 3936 Search PubMed; (h) Y. Zhou, J. F. Zhang and J. Yoon, Chem. Rev., 2014, 114, 5511 CrossRef CAS PubMed; (i) P. Molina, F. Zapata and A. Caballero, Chem. Rev., 2017, 117, 9907 CrossRef CAS PubMed.
- Selected original reports: (a) H. Maeda and Y. Kusunose, Chem. – Eur. J., 2005, 11, 5661 CrossRef CAS PubMed; (b) H. Maeda, Y. Haketa and T. Nakanishi, J. Am. Chem. Soc., 2007, 129, 13661 CrossRef CAS PubMed; (c) H. Maeda, M. Terasaki, Y. Haketa, Y. Mihashi and Y. Kusunose, Org. Biomol. Chem., 2008, 6, 433 RSC; (d) Y. Haketa and H. Maeda, Chem. – Eur. J., 2011, 17, 1485 CrossRef CAS PubMed; (e) Y. Haketa, Y. Bando, K. Takaishi, M. Uchiyama, A. Muranaka, M. Naito, H. Shibaguchi, T. Kawai and H. Maeda, Angew. Chem., Int. Ed., 2012, 51, 7967 CrossRef CAS PubMed; (f) R. Yamakado, T. Sakurai, W. Matsuda, S. Seki, N. Yasuda, S. Akine and H. Maeda, Chem. – Eur. J., 2016, 22, 626 CrossRef CAS PubMed; (g) A. Kuno, N. Tohnai, N. Yasuda and H. Maeda, Chem. – Eur. J., 2017, 82, 11166 Search PubMed; (h) S. Kaname, Y. Haketa, N. Yasuda and H. Maeda, Org. Lett., 2018, 20, 3268 CrossRef CAS PubMed; (i) S. Sugiura, W. Matsuda, W. Zhang, S. Seki, N. Yasuda and H. Maeda, J. Org. Chem., 2019, 84, 8886 CrossRef CAS PubMed; (j) S. Sugiura, Y. Kobayashi, N. Yasuda and H. Maeda, Chem. Commun., 2019, 55, 8242 RSC; (k) S. Sugiura and H. Maeda, Org. Biomol. Chem., 2020, 18, 4433 RSC; (l) H. Maeda, T. Nishimura, Y. Haketa, H. Tanaka, M. Fujita and N. Yasuda, J. Org. Chem., 2022, 87, 7818 CrossRef CAS PubMed.
- Helical structures controlled by ion pairing with chiral counteracation can be applied for the evaluation of conformational changes and optical properties under hydrostatic pressure: T. Kinoshita, Y. Haketa, H. Maeda and G. Fukuhara, Chem. Sci., 2021, 12, 6691 RSC.
- A review: A. H. Flood, Beilstein J. Org. Chem., 2016, 12, 611 CrossRef CAS PubMed.
- A review and the first report: (a) Y. Hua and A. H. Flood, Chem. Soc. Rev., 2010, 39, 1262 RSC; (b) Y. Li and A. H. Flood, Angew. Chem., Int. Ed., 2008, 47, 2649 CrossRef CAS PubMed.
- (a) S. Lee, C.-H. Chen and A. H. Flood, Nat. Chem., 2013, 5, 704 CrossRef CAS PubMed; (b) B. Qiao, B. E. Hirsch, S. Lee, S. Lee, M. Pink, C.-H. Chen, B. W. Laursen and A. H. Flood, J. Am. Chem. Soc., 2017, 139, 6226 CrossRef CAS PubMed; (c) C. R. Benson, L. Kacenauskaite, K. L. VanDenburgh, W. Zhao, B. Qiao, T. Sadhukhan, M. Pink, J. Chen, S. Borgi, C.-H. Chen, B. J. Davis, Y. C. Simon, K. Raghavachari, B. W. Laursen and A. H. Flood, Chem, 2020, 6, 1978 CrossRef CAS; (d) J. Chen, S. M. A. Fateminia, L. Kacenauskaite, N. Bærentsen, S. G. Stenspil, J. Bredehoeft, K. L. Martinez, A. M. Flood and B. W. Laursen, Angew. Chem., Int. Ed., 2021, 60, 9450 CrossRef CAS PubMed; (e) J. Chen, S. G. Stenspil, S. Kaziannis, L. Kacenauskaite, N. Lenngren, M. Kloz, A. H. Flood and B. W. Laursen, ACS Appl. Nano Mater., 2022, 5, 13887 CrossRef CAS; (f) L. Kacenauskaite, S. G. Stenspil, A. H. Olsson, A. H. Flood and B. W. Laursen, J. Am. Chem. Soc., 2022, 144, 19981 CrossRef CAS PubMed.
- H. Tanaka, Y. Haketa, Y. Bando, R. Yamakado, N. Yasuda and H. Maeda, Chem. – Asian J., 2020, 15, 494 CrossRef CAS PubMed.
- A. Kuno, M. Fujiwara, Y. Haketa and H. Maeda, Chem. – Asian J., 2019, 14, 1777 CrossRef CAS PubMed.
- N. Koda, Y. Haketa, M. Yokoyama, N. Yasuda and H. Maeda, Org. Lett., 2023, 25, 1120 CrossRef CAS PubMed.
- A. Kuno, G. Hirata, H. Tanaka, Y. Kobayashi, N. Yasuda and H. Maeda, Chem. – Eur. J., 2021, 27, 10068 CrossRef CAS PubMed.
- (a) E. L. Goff and R. B. LaCount, J. Am. Chem. Soc., 1963, 85, 1354 CrossRef; (b) T. G. Beaumont and K. M. C. Davis, J. Chem. Soc. B, 1968, 1010 RSC; (c) G. Briegleb, J. Trencséni and W. Herre, Chem. Phys. Lett., 1969, 3, 146 CrossRef CAS; (d) M. I. Bruce, P. A. Humphrey, B. W. Skelton and A. H. White, Aust. J. Chem., 1986, 39, 165 CrossRef CAS; (e) G. Maas, H. M. Weber, R. Exner and J. Salbeck, J. Phys. Org. Chem., 1990, 3, 459 CrossRef CAS.
- C. Richardson and C. A. Reed, Chem. Commun., 2004, 706 RSC.
- H. Gotoh, S. Nakatsuka, H. Tanaka, N. Yasuda, Y. Haketa, H. Maeda and T. Hatakeyama, Angew. Chem., Int. Ed., 2021, 60, 12835 CrossRef CAS PubMed.
- A. Takiguchi, H. Tanaka, H. Maeda and H. Shinokubo, Bull. Chem. Soc. Jpn., 2022, 95, 796 CrossRef CAS.
- K. Goossens, L. Rakers, B. Heinrich, G. Ahumada, T. Ichikawa, B. Donnio, T. J. Shin, C. W. Bielawski and F. Glorius, Chem. Mater., 2019, 31, 9593 CrossRef CAS.
- Reviews: (a) R. P. Singh, R. D. Verma, D. T. Meshri and J. M. Shreeve, Angew. Chem., Int. Ed., 2006, 45, 3584 CrossRef CAS PubMed; (b) M. Smiglak, A. Metlen and R. D. Rogers, Acc. Chem. Res., 2007, 40, 1182 CrossRef CAS PubMed; (c) E. Sebastiao, C. Cook, A. Hu and M. Murugesu, J. Mater. Chem. A, 2014, 2, 8153 RSC; (d) Q. Zhang and J. M. Shreeve, Chem. Rev., 2014, 114, 10527 CrossRef CAS PubMed; (e) M. E. Easton, H. Choudhary and R. D. Rogers, Chem. – Eur. J., 2019, 25, 2127 CrossRef CAS PubMed.
- Selected reports: (a) W. Ogihara, M. Yoshizawa and H. Ohno, Chem. Lett., 2004, 33, 1022 CrossRef CAS; (b) A. R. Katritzky, S. Singh, K. Kirichenko, J. D. Holbrey, M. Smiglak, W. M. Reichert and R. D. Rogers, Chem. Commun., 2005, 868 RSC; (c) H. Xue, Y. Gao, B. Twamley and J. M. Shreeve, Inorg. Chem., 2005, 44, 5068 CrossRef CAS PubMed; (d) C. Ye, J.-C. Xiao, B. Twamley and J. M. Shreeve, Chem. Commun., 2005, 2750 RSC; (e) M. Smiglak, C. C. Hines, T. B. Wilson, S. Singh, A. S. Vincek, K. Kirichenko, A. R. Katritzky and R. D. Rogers, Chem. – Eur. J., 2010, 16, 1572 CrossRef CAS PubMed; (f) G.-H. Tao, M. Tang, L. He, S.-P. Ji, F.-D. Nie and M. Huang, Eur. J. Inorg. Chem., 2012, 3070 CrossRef CAS; (g) Q.-H. Lin, Y.-C. Li, Y.-Y. Li, Z. Wang, W. Liu, C. Qi and S.-P. Pang, J. Mater. Chem., 2012, 22, 666 RSC; (h) M. Smiglak, C. C. Hines, W. M. Reichert, J. L. Shamshina, P. A. Beasley, P. D. McCrary, S. P. Kelley and R. D. Rogers, New J. Chem., 2013, 37, 1461 RSC; (i) L.-L. Dong, L. He, H.-Y. Liu, G.-H. Tao, F.-D. Nie, M. Huang and C.-W. Hu, Eur. J. Inorg. Chem., 2013, 5009 CrossRef CAS; (j) C. Yang, L. Chen, W. Wu, C. Zhang, C. Sun, Y. Du and B. Hu, ACS Appl. Energy Mater., 2021, 4, 146 CrossRef CAS.
- Ion pairs with tetracyanoallyl anion: (a) H. Yasuba, T. Imai and K. Okamoto, Bull. Chem. Soc. Jpn., 1970, 43, 3101 CrossRef CAS; (b) T. Tamamura, M. Yokoyama, S. Kusabayashi and H. Mikawa, Bull. Chem. Soc. Jpn., 1974, 47, 442 CrossRef CAS; (c) T. Tamamura, H. Yasuba, K. Okamoto, T. Imai, S. Kusabayashi and H. Mikawa, Bull. Chem. Soc. Jpn., 1974, 47, 448 CrossRef CAS; (d) T. Tamamura, T. Yamane, N. Yasuoka and N. Kasai, Bull. Chem. Soc. Jpn., 1974, 47, 832 CrossRef CAS.
- Ion pairs with hexacyanotrimethylenemethandiide: (a) S. Sakanoue, T. Tamamura, S. Kusabayashi, H. Mikawa, N. Kasai, M. Kakudo and H. Kuroda, Bull. Chem. Soc. Jpn., 1969, 42, 2407 CrossRef CAS; (b) S. Sakanoue, N. Yasuoka, N. Kasai and M. Kakudo, Bull. Chem. Soc. Jpn., 1971, 44, 1 CrossRef.
- Ion pairs with 1,3-bis(dicyanomethylidene)indan anion: (a) E. Saito, T. Yasuhara, Y. Tanaka, R. Yamakado, S. Okada, T. Nohara, A. Masuhara and T. Yoshida, ECS Trans., 2018, 88, 301 CrossRef CAS; (b) Y. Tanaka, K. Ichijo, S. Kodama, S. Aoyama, T. Yoshida, R. Yamakado and S. Okada, Cryst. Growth Des., 2019, 19, 5811 CrossRef CAS.
- C. Reichardt, Chem. Rev., 1994, 94, 2319 CrossRef CAS.
- P.-A. Bouit, D. Rauh, S. Neugebauer, J. L. Delgado, E. D. Piazza, S. Rigaut, O. Maury, C. Andraud, V. Dyakonov and N. Martin, Org. Lett., 2009, 11, 4806 CrossRef CAS PubMed.
- I. Baraldi, F. Momicchioli, G. Ponterini, A. S. Tatikolov and D. Vanossi, J. Phys. Chem. A, 2001, 105, 4600 CrossRef CAS.
- Z. Li, S. Mukhopadhyay, S.-H. Jang, J.-L. Brédas and A. K.-Y. Jen, J. Am. Chem. Soc., 2015, 137, 11920 CrossRef CAS PubMed.
- S. Kusabayashi, A. Taniguchi and H. Mikawa, Bull. Chem. Soc. Jpn., 1966, 39, 1344 CrossRef CAS.
- (a) E. D. Owen and Q. Sultana, J. Appl. Chem. Biotechnol., 1972, 22, 1043 CrossRef CAS; (b) I. Willner, Y. Eichen, A. Doron and S. Marx, Isr. J. Chem., 1992, 32, 53 CrossRef CAS; (c) I. Willner, Y. Eichen, M. Rabinovitz, R. Hoffman and S. Cohen, J. Am. Chem. Soc., 1992, 114, 637 CrossRef CAS.
- N. C. Yang and A. J. Castro, J. Am. Chem. Soc., 1960, 82, 6208 CrossRef CAS.
- S. Sugiura, T. Kubo, Y. Haketa, Y. Hori, Y. Shigeta, H. Sakai, T. Hasobe and H. Maeda, J. Am. Chem. Soc., 2023, 145, 8122 CrossRef CAS PubMed.
- The space group of the single crystal of 22d+-38a− is triclinic P1, which induces polarization caused by inhomogeneous stacking.
- A review on supramolecular ferroelectrics: A. S. Tayi, A. Kaeser, M. Matsumoto, T. Aida and S. I. Stupp, Nat. Chem., 2015, 7, 281 CrossRef CAS PubMed.
- S. Takizawa, T. Okuyama, S. Yamazaki, K. Sato, H. Masai, T. Iwai, S. Murata and J. Terao, J. Am. Chem. Soc., 2023, 145, 15049 CrossRef CAS PubMed.
- Very recent examples: R. Sengupta, H. Hashimoto, Y. Haketa, H. Sakai, T. Hasobe and H. Maeda, Org. Lett., 2023, 25, 6040 CrossRef CAS PubMed.
Footnote |
| † These authors equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2023 |