
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Eu3+ doped ZnAl layered double hydroxides as calibrationless, fluorescent sensors for carbonate†
Alysson F.
Morais
 *abe,
Ivan G. N.
Silva
a,
Bruno J.
Ferreira
a,
Alexandre C.
Teixeira
a,
Sreeprasanth P.
Sree
bc,
Huayna
Terraschke
d,
Fernando A.
Garcia
a,
Eric
Breynaert
be and
Danilo
Mustafa
*a
*abe,
Ivan G. N.
Silva
a,
Bruno J.
Ferreira
a,
Alexandre C.
Teixeira
a,
Sreeprasanth P.
Sree
bc,
Huayna
Terraschke
d,
Fernando A.
Garcia
a,
Eric
Breynaert
be and
Danilo
Mustafa
*a
aInstituto de Física da Universidade de São Paulo, 05508-090 – São Paulo, SP, Brazil. E-mail: dmustafa@if.usp.br
bCenter for Surface Chemistry and Catalysis, KU Leuven B-3001, Leuven, Belgium. E-mail: alysson.morais@kuleuven.be
cDepartment of Materials Engineering, KU Leuven 3001, Leuven, Belgium
dInstitut für Anorganische Chemie, CAU Kiel, 24118, Kiel, Germany
eNMR/X-Ray platform for Convergence Research (NMRCoRe), KU Leuven 3001, Leuven, Belgium
First published on 30th October 2023
Abstract
The photoluminescence properties (PL) of Eu3+ hosted in the hydroxide layers of layered double hydroxides (LDHs) enables calibrationless quantification of anions in the interlayers. The concept is demonstrated during the nitrate-to-carbonate ion exchange in Zn2+/Al3+/Eu3+ LDHs and can be implemented as a remote optical sensor to detect intrusion of anions such as Cl− or CO32−.
Corrosion of concrete rebar is one of the dominant mechanisms limiting the lifetime of reinforced concrete constructions.1,2 Fresh concrete contains a mixture of highly alkaline materials, ensuring a high pH concrete pore water (pH 12–13) environment. This high pH induces the formation of a passivating layer on the surface of the steel rebar, thus reducing corrosion. Carbonation of concrete, i.e., the uptake of carbon dioxide from the air, can cause the passivating layer in steel rebars to shrink. In this mechanism, CO2 reacts with the hydroxides in the concrete, converting them into metal carbonates and decreasing the local pH. When the low pH front reaches the steel rebar and the steel is depassivated, corrosion kicks thus jeopardising the lifetime of the construction.3–6 Aside its impact in the rebar passivation layer, carbonation also increases the sensitivity of concrete to other corrosion mechanisms such as chloride attack, and thus is a key determinant of the service life of concrete structures.7
The progress of carbonation fronts in concrete structures is typically visualised by spraying a phenolphthalein solution on concrete cores freshly drilled out of the concrete cover. In contact with pristine concrete, phenolphthalein turns bright pink while its colour fades or completely disappears when carbonation has occurred, thus indicating the pH has been lowered below pH 10.8,9 This procedure is, however, detrimental to the concrete structure. For critical infrastructure, e.g., bridges, dams, and nuclear containments, a reliable, in situ, non-destructive method to characterize the evolution of the carbonation front as a function of the distance to the rebar would be advantageous, allowing improved planning of maintenance actions. A sensor allowing non-destructive remote detection of the progress of the carbonation front could be created from lanthanide-doped LDHs combined with fibre-based detection of their PL.
Layered double hydroxides are materials formed by the stacking of positively charged 2D networks (or layers) of edge-sharing octahedra of divalent and trivalent metal cations bridged by hydroxyl groups. The stacking of these layers is mediated by anionic species intercalated between the hydroxide layers.10–14 As result of the high variability in cation and anion composition, LDHs are promising materials for use as anion exchangers, adsorbents, catalysts, for controlled release of pharmaceutical components, and as solid electrolytes for battery applications.15–22 Recently, even nanotubular, luminescent, and mixed ligand luminescent LDHs have been reported.10,23–25 Introduction of trivalent europium (Eu3+) in the hydroxide layers of LDHs produces luminescent LDH phases whose PL depend on the anionic spacer.25–27 At the same time, the anions in LDHs are exchangeable based on their affinity to the LDH host, carbonate (CO32−) standing high in the LDH affinity series.28–30 The combination of high affinity to carbonate and the anion dependent PL of LDHs can then turn these materials into a class of high affinity, calibrationless carbonate sensors with potential application for the in situ, non-destructive assessment of the evolution of the carbonation front in reinforced concrete.
Here we investigate and report on the structural and luminescent properties of Eu-doped Zn2+/Al3+ LDHs. The changes in the PL of these LDHs have been followed in the equilibrium state and in situ during anion exchange reactions where pristine LDHs initially intercalated with nitrate (LDH-NO3−) were dispersed in sodium carbonate solutions at different concentrations. These anion exchange reactions led to LDHs where nitrate was completely or partially exchanged for carbonate, which could be qualitatively and quantitatively assessed by, respectively, powder X-ray diffraction (PXRD) and elemental analysis. LDHs with remarkably different luminescence properties have been obtained after the anion exchange reactions, from which the local changes in the Eu3+ coordination could be inferred. Extended X-ray absorption fine structure (EXAFS31) spectroscopy shows Eu3+ is hosted in the hydroxide layers of the LDHs, in positions normally occupied by Al3+. Further, insights on the coordination changes caused by anion exchange were given by EXAFS data analysis.
In brief, the inclusion of Eu3+ in the hydroxide layers of LDHs demands no special or modified synthetic routes as either Eu(NO3)3 or EuCl3, salts highly soluble in water, can readily serve as precursors for Eu3+ in synthetic procedures involving controlled hydrolysis.10,32 By this strategy, Eu3+ can be doped in the hydroxide layers of Zn/Al LDHs up to Eu/(Eu + Al) ratios of about 15% without segregation of Eu3+ in additional crystalline phases.25 Further, analysis of Eu LIII-edge EXAFS data confirms the successful incorporation of Eu3+ in the hydroxide layers of these LDHs in replacement of Al3+.25–27 For the present work, nitrate intercalated Eu3+-doped Zn2+/Al3+ LDHs (LDH-NO3−) were prepared by coprecipitation of Zn2+, Al3+ and Eu3+ at constant pH (see ESI†). This procedure successfully yields pristine NO3−-intercalated LDHs with a characteristic basal distance of 8.6 Å, as revealed by the average between the interplanar distances calculated for the (003) and (006) Bragg reflections observed at, respectively, 10.4° and 20.4° 2θ in their PXRD pattern (Fig. 1).33
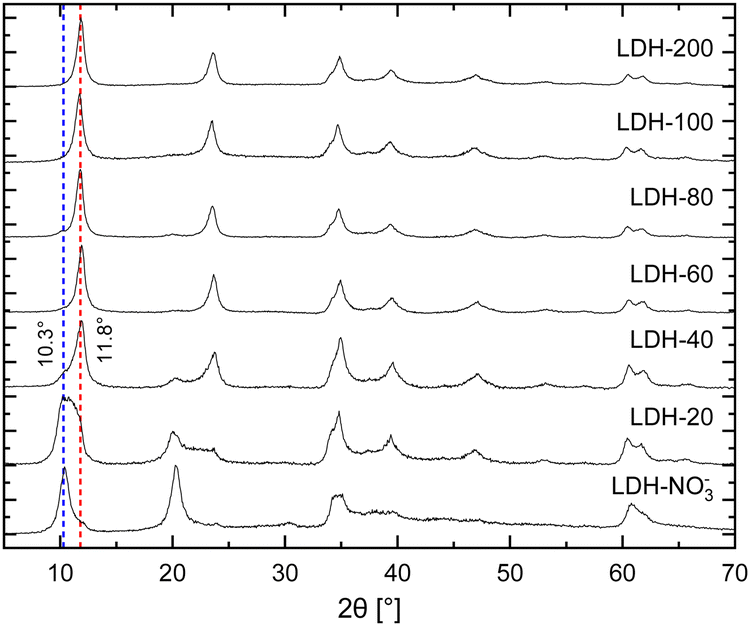 | ||
| Fig. 1 PXRD patterns of pristine LDH-NO3− and the anion exchanged samples LDH-A (A = 20, 40, 60, 80, 100 and 200 standing for the initial carbonate concentration in mmol L−1 in the anion exchange solution). The NO3−- and CO32−-intercalated phases can be identified by the characteristic basal reflections at 10.3 and 11.8° 2θ, respectively. | ||
Carbonate sequestration with release of nitrate when pristine LDH-NO3− is dispersed in a Na2CO3 solution is evident both from elemental analysis (Table S1, ESI†) and PXRD (Fig. 1). CO32−-intercalated LDHs feature a shorter basal distance as compared to LDH-NO3−, making a basal reflection to appear at 11.8° 2θ for the CO32−-intercalated LDHs.14,34
Table S1 (ESI†) shows the elemental composition of LDH-NO3− before and after anion exchange. Nitrate was exchanged for carbonate by dispersing LDH-NO3− in solutions of different initial carbonate concentrations (samples dubbed LDH-A, A = 20, 40, 60, 80, 100 and 200 standing for the initial carbonate concentration, in mmol L−1, in the anion exchange solution). More details on the anion exchange procedure are provided in the ESI.† In line with PXRD, the carbon content in the samples increases with the increase in the CO32− concentration in the exchange solution, confirming that LDHs are effective materials for carbonate uptake. At the same time, the nitrogen content decreases, showing that carbonate is fixed in the samples at the expense of the nitrate anions. The effectiveness of the anion exchange in the samples can be experimentally quantified by the equivalent fraction X = (1 + 0.5 × mol% NO3−/mol% CO3−)−1, which defines the LDH composition as in the general formula [Zn2Al0.95Eu0.05(OH)6][(NO3−)1−X(CO32−)X/2]·mH2O. From the point of view of elemental analysis, carbonate saturation in the LDHs is reached for initial CO32− concentrations between 80 (X = 0.95) and 100 mmol L−1 (X = 1.0). Interestingly, as seen from PXRD, CO32− already dominates the interlayer space of the LDHs for concentrations above 60 mmol L−1. Above this concentration, the structural properties of the LDHs hinder any tentative to discriminate the presence or absence of nitrate by using PXRD.
The PL spectra obtained for LDH-A (A = 20, 40, 60, 80, 100 and 200) are shown on Fig. 2A. In the spectral region from 580 to 710 nm, the spectra show the emission bands of Eu3+, indicated by the term symbols of its 4f electronic states. Eu3+ is a lanthanide element and, as such, features a set of narrow emission bands arising from its 4f–4f electronic transitions.35 Appearing in the spectra shown on Fig. 2A are the 5D0 → 7F1 and 5D0 → 7F1 transitions. While the intensity I(5D0 → 7F1) is known to be largely independent on the host Eu3+ is embedded in, the intensity I(5D0 → 7F2) is very sensitive to the geometry and chemical environment around the luminescent centre.35–37 This makes I(5D0 → 7F1) an internal luminescence standard useful when comparing luminescence spectra of different Eu-containing samples, turning Eu-containing LDH into calibrationless molecular sensors.
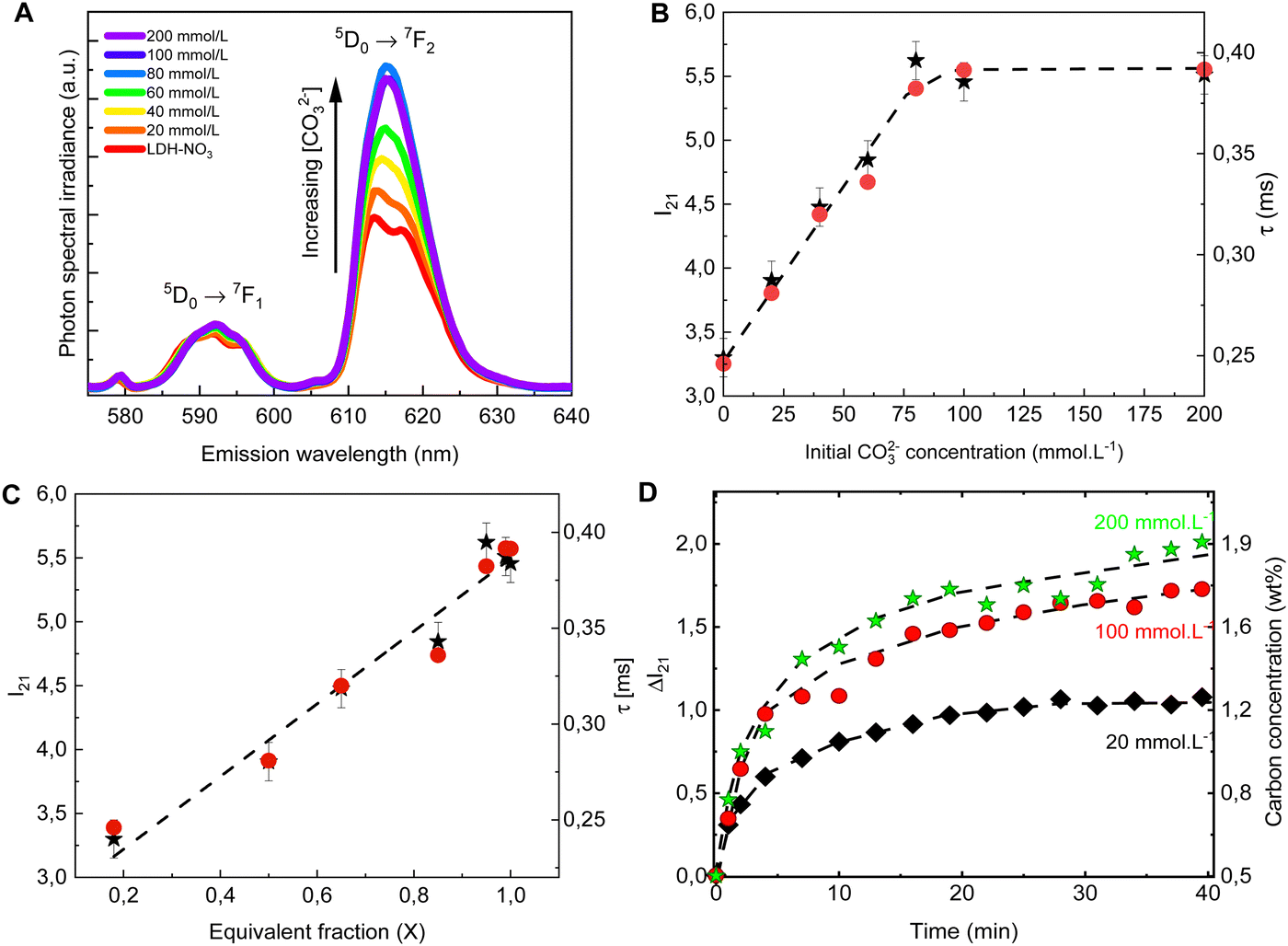 | ||
| Fig. 2 Emission spectra (A), I21 = I(5D0 → 7F2)/I(5D0 → 7F1) intensity ratio (B, ★, left scale) and luminescence decay lifetime (B, ●, right scale) of the anion exchanged LDHs after equilibration at different concentrations of CO32−. The spectra are normalized by the integral intensity of the (Eu3+)5D0 → 7F1 emission band. (C) I21 intensity ratio (★, left scale) and luminescence decay lifetime (●, right scale) as a function of the carbonate equivalent fraction (X) in the LDH samples. (D) Changes in the I21 intensity ratio over time in the in situ experiments with different initial carbonate concentrations. The right scale shows the carbon concentration calculated from eqn (1). The dashed lines are guides to the eye. | ||
In the PL spectra shown on Fig. 2A, all spectral profiles have been normalized by the integral intensity I(5D0 → 7F1). With this internal normalization, the (Eu3+)5D0 → 7F2 emission has been observed to increase (Fig. 2B and C) with the CO32− concentration in the anion exchange solution. While the PXRD patterns of LDH-60 and LDH-80 are very similar, the I21 = I(5D0 → 7F2)/I(5D0 → 7F1) intensity ratio of these two samples are strikingly different, as seen from Fig. 2B, thus showcasing the sensitivity of the luminescence properties of Eu-containing LDH to determine differences in the carbonate content between two different LDH hosts. Also the decay lifetime (Fig. 2B. C and Table S2, Fig. S1, ESI†) of the (Eu3+)5D0 → 7F2 transition has been observed to increase with the uptake of carbonate from the anion exchange solution. With carbonate concentrations exceeding 100 mmol L−1 these changes cease, indicating the saturation of the anion sites in the LDHs, which is consistent with the stoichiometry derived from elemental analysis (Table S1 and Fig. S2, ESI†).
Both the luminescence decay lifetime and the I(5D0 → 7F2)/I(5D0 → 7F1) intensity ratio in the Eu-containing LDHs strongly correlate with the CO32− load in the samples, as shown in Fig. 2C and Fig. S2 (ESI†). The I(5D0 → 7F2)/I(5D0 → 7F1) intensity ratio is shown to feature a linear relationship with the CO32− concentration in the interlayer gallery (Fig. S2, ESI†). This relationship can then be extrapolated for an ideal LDH phase containing only NO3− (no CO32−), giving the value: I(5D0 → 7F2)/I(5D0 → 7F1) = 2.8 ± 0.7, from which the CO32− loading in the LDHs can be written as:
| wt% C = 0.75 × (I21 − 2.8), | (1) |
To follow the PL of LDHs in situ during a CO32− uptake experiment, a setup comprising a portable fluorimeter equipped with an optical fibre to monitor the emission of the sample was assembled. A commercial 10 W UV LED lamp (Fig. S3, ESI†) was used as excitation source. Carbonate solutions with three different concentrations were added to three LDH suspensions, with the LDHs used in the different suspensions originated from the same batch. For these in situ experiments, the initial carbonate concentrations were chosen to be well below (20 mmol L−1), similar to (100 mmol L−1) and well beyond (200 mmol L−1) the concentration seen by elemental analysis to cause saturation of the anionic sites in the LDHs. As evident from Fig. 2D, the carbonate uptake by the LDHs is followed by an increase in time of the I(5D0 → 7F2)/I(5D0 → 7F1) intensity ratio. Eqn (1) was used to calculate the carbonate concentration in the solid phase based on the measured PL. The carbonate load in the solid phase linearly increases in time in the initial steps of the exchange reaction. Then, as equilibrium is reached, a plateau starts to form, with the stabilization of the carbonate loading in the solid phase. Similar carbonate uptake profiles are observed for the LDHs dispersed in the solutions with initial carbonate concentration of 100 and 200 mmol L−1, again a consequence of the saturation of the LDHs.
As the PL data of Eu-containing compounds is sensitive to the presence of water and to the coordination geometry around the luminescent activator, it is clear from Fig. 2A that the local environment around europium in the LDHs is changed when nitrate is exchanged for carbonate. To characterize these changes, the Eu LIII EXAFS spectra of LDH-NO3 and a carbonate-intercalated LDH-CO3 were fitted and analysed together with the PL data (see Fig. 3 and Fig. S4, Table S3, ESI†). A detailed description of the EXAFS fitting procedure is available in the ESI.† 6 Zinc atoms are found at 4.00 and 3.93 Å from Eu in LDH-NO3 and LDH-CO3, respectively, in full accordance with Eu3+ being hosted in the hydroxide layers of the LDHs, replacing Al3+. In both samples, the first coordination shell around Eu comprises 7 oxygen atoms at 2.42 Å, with an additional oxygen appearing at 3.03 Å in LDH-NO3 and at a slightly shorter distance, at 2.85 Å, in LDH-CO3. For LDH-NO3, this additional oxygen coordination is likely to belong to coordination water, as already reported elsewhere.25–27 For LDH-CO3, a different origin is more likely, based on the much longer (Eu3+)5D0 → 7F2 luminescence decay lifetime (τ) observed for this sample as compared to LDH-NO3 (see Fig. 2B). The (Eu3+)5D0 → 7F2 luminescence decay lifetime in Eu-containing compounds is strongly dependent on the number of OH quenchers around the Eu3+ activators, the lifetime increasing when OH quenchers are removed from the first coordination shell of Eu3+.38–40 The increase in τ as observed in Fig. 2B and C would then be consistent with the replacement of the coordination water around Eu upon carbonate adsorption.
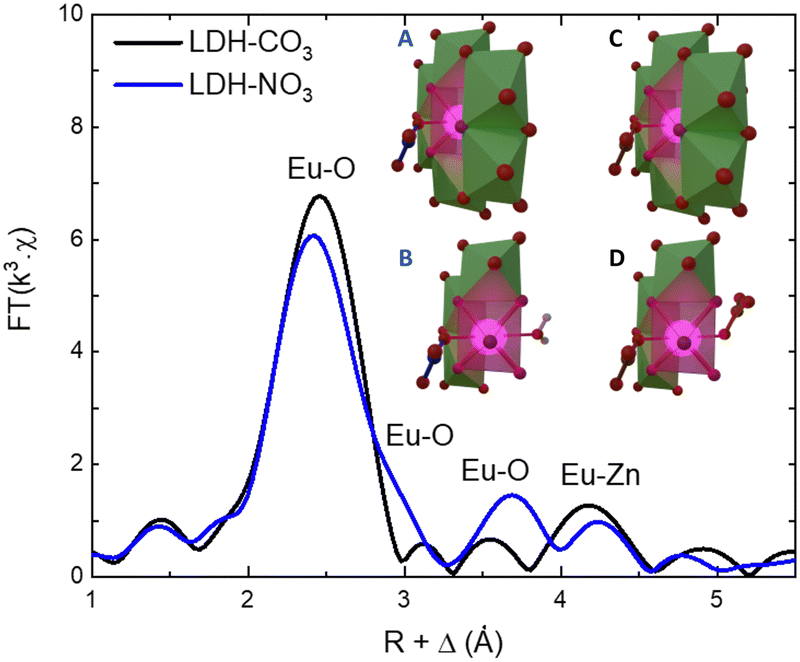 | ||
| Fig. 3 Phase-corrected Fourier transform of the Eu LIII-edge EXAFS data for LDH-NO3 and a carbonate-intercalated Zn2+/Al3+/Eu3+ LDH (LDH-CO3). The inset illustrates the coordination structure of Eu3+ in LDH-NO3 (A) and (B) and LDH-CO3 (C) and (D). For better visualization of the Eu3+ coordination with the interlayers, B and D present a cut in the structure where some Zn-centred polyhedrons have been supressed. Europium is coordinated to 8 oxygen atoms, of which 6 are attributed to OH groups in the hydroxide layers. In LDH-NO3 one additional coordinated oxygen come from interlayer water and another from interlayer nitrate (B), whereas in LDH-CO3 the 2 additional coordinated oxygen come from CO32− anions (D).25–27 | ||
In summary, the here reported experiments show that the introduction of Eu3+ in the hydroxide layers of LDHs enables the optical assessment of the degree of carbonate uptake by the LDH matrix. Since the intensity of the magnetic dipole transition (Eu3+)5D0 → 7F1 is largely independent on the host, it can serve as an internal standard during the analysis of PL spectra, turning Eu3+-doped LDHs into calibrationless carbonate sensors with potential application for in situ, non-destructive assessment of the progress of the carbonation front in concrete. This would enable continuous, remote quality control of critical infrastructure built from reinforced concrete.
This research was funded by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP, 2015/19210-0, 2022/01314-8, 2019/25665-1 and 2018/13837-0) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES, 1723707 and 88887.371434/2019-00). The authors acknowledge CNPEM-LNLS for concession of beamtime (Proposals No. 20190148 and 20180133). E. B. and A. F. M. acknowledge support from the European Research Council through an Advanced Research Grant under the European Union's Horizon 2020 research and innovation programme (No. 834134 WATUSO). A. F. M. acknowledges support from the European Union’s Horizon Europe programme through a Marie Skłodowska-Curie postdoctoral fellowship (No. 101063656, H2E). Christine E. A. Kirschhock is kindly acknowledged for the illustration in the inset of Fig. 3. NMRCoRe acknowledges the Flemish government, department EWI for financial support as International Research Infrastructure (I001321N: Nuclear Magnetic Resonance Spectroscopy Platform for Molecular Water Research).
Conflicts of interest
There are no conflicts to declare.Notes and references
- Portland Cement Association, Types and causes of concrete deterioration, IS356, Skokie, Illinois, 2002.
- Y. Zhou, B. Gencturk, K. Willam and A. Attar, J. Mater. Civ. Eng., 2015, 27, 04014245 CrossRef.
- B. Huet, V. L’Hostis, F. Miserque and H. Idrissi, Electrochim. Acta, 2005, 51, 172–180 CrossRef CAS.
- G. Roventi, T. Bellezze, G. Giuliani and C. Conti, Cem. Concr. Res., 2014, 65, 76–84 CrossRef CAS.
- D. N. Huntzinger, J. S. Gierke, S. K. Kawatra, T. C. Eisele and L. L. Sutter, Environ. Sci. Technol., 2009, 43, 1986–1992 CrossRef CAS PubMed.
- C. Andrade and M. Á. Sanjuán, Dev. Built Environ., 2021, 8, 0–10 Search PubMed.
- M. Kue Lee, S. Hwa Jung and H. O. Byung, ACI Mater. J., 2013, 110, 559–566 Search PubMed.
- H. Bui, F. Delattre and D. Levacher, Appl. Sci., 2023, 13, 2533 CrossRef CAS.
- J. Il Choi, Y. Lee, Y. Y. Kim and B. Y. Lee, Constr. Build. Mater., 2017, 154, 451–461 CrossRef.
- A. F. Morais, I. G. N. N. Silva, S. P. Sree, F. M. De Melo, G. Brabants, H. F. Brito, J. A. Martens, H. E. Toma, C. E. A. A. Kirschhock, E. Breynaert and D. Mustafa, Chem. Commun., 2017, 53, 7341–7344 RSC.
- A. Teixeira, A. Morais, I. Silva, E. Breynaert and D. Mustafa, Crystals, 2019, 9, 153 CrossRef.
- R. Gao, D. Yan, D. G. Evans and X. Duan, Nano Res., 2017, 10, 3606–3617 CrossRef CAS.
- D. Yan, J. Lu, L. Chen, S. Qin, J. Ma, M. Wei, D. G. Evans and X. Duan, Chem. Commun., 2010, 46, 5912–5914 RSC.
- S. Radhakrishnan, K. Lauwers, C. V. Chandran, J. Trébosc, S. Pulinthanathu Sree, J. A. Martens, F. Taulelle, C. E. A. Kirschhock and E. Breynaert, Chem. – Eur. J., 2021, 27, 15944–15953 CrossRef CAS PubMed.
- L. Desigaux, M. Ben Belkacem, R. Peggy, C. Taviot-gue, B. Pitard, F. Leroux and J. Cellier, Nano Lett., 2005, 6, 199–204 CrossRef PubMed.
- Y. Zhao, Q. Wang, T. Bian, H. Yu, H. Fan, C. Zhou, L.-Z. Wu, C.-H. Tung, D. O’Hare and T. Zhang, Nanoscale, 2015, 7, 7168–7173 RSC.
- G. Rathee, A. Awasthi, D. Sood, R. Tomar, V. Tomar and R. Chandra, Sci. Rep., 2019, 9, 1–14 CrossRef CAS PubMed.
- Q. Wang and D. Ohare, Chem. Rev., 2012, 112, 4124–4155 CrossRef CAS PubMed.
- D. Chaillot, S. Bennici and J. Brendlé, Environ. Sci. Pollut. Res., 2021, 28, 24375–24405 CrossRef CAS PubMed.
- M. Daud, A. Hai, F. Banat, M. B. Wazir, M. Habib, G. Bharath and M. A. Al-Harthi, J. Mol. Liq., 2019, 288, 110989 CrossRef CAS.
- J. Wen, K. Yang, J. Huang and S. Sun, Mater. Des., 2021, 198, 109298 CrossRef CAS.
- R. Gao, D. Yan and X. Duan, Cell Reports Phys. Sci., 2021, 2, 100536 CrossRef CAS.
- A. F. F. Morais, F. O. O. Machado, A. C. C. Teixeira, I. G. N. G. N. Silva, E. Breynaert and D. Mustafa, J. Alloys Compd., 2019, 771, 578–583 CrossRef CAS.
- D. Nanclares, A. F. Morais, T. Calaça, I. G. N. N. Silva and D. Mustafa, RSC Adv., 2021, 11, 24747–24751 RSC.
- A. F. Morais, D. Nanclares, I. G. N. N. Silva, A. Duarte, F. A. Garcia, E. Breynaert and D. Mustafa, Nanoscale, 2021, 13, 11781–11792 RSC.
- A. F. Morais, I. G. N. Silva, B. C. Lima, F. A. Garcia and D. Mustafa, ACS Omega, 2020, 5, 23778–23785 CrossRef CAS PubMed.
- I. G. N. Silva, A. F. Morais, B. C. Lima, F. A. Garcia and D. Mustafa, Appl. Clay Sci., 2020, 199, 105861 CrossRef CAS.
- S. Miyata, Clays Clay Miner., 1983, 31, 305–311 CrossRef CAS.
- S. H. Huang, S. J. Liu and J. Y. Uan, J. Mater. Chem. C, 2019, 7, 11191–11206 RSC.
- I. Yanase, Y. Horiuchi and H. Kobayashi, Mater. Res. Bull., 2019, 110, 207–213 CrossRef CAS.
- S. J. A. Figueroa, J. C. Mauricio, J. Murari, D. B. Beniz, J. R. Piton, H. H. Slepicka, M. F. De Sousa, A. M. Espíndola and A. P. S. Levinsky, J. Phys. Conf. Ser., 2016, 712, 12022 CrossRef.
- X. Gao, M. Hu, L. Lei, D. O’Hare, C. Markland, Y. Sun and S. Faulkner, Chem. Commun., 2011, 47, 2104–2106 RSC.
- S. Marappa, S. Radha and P. V. Kamath, Eur. J. Inorg. Chem., 2013, 2122–2128 CrossRef CAS.
- S. Marappa and P. V. Kamath, Ind. Eng. Chem. Res., 2015, 54, 11075–11079 CrossRef CAS.
- K. Binnemans, Coord. Chem. Rev., 2015, 295, 1–45 CrossRef CAS.
- B. M. Walsh, in International School of Atomic and Molecular Spectroscopy, ed. B. Di Bartolo and O. Forte, Springer Netherlands, Dordrecht, 2006, pp. 403–433 Search PubMed.
- H. F. Brito, O. M. L. Malta, M. C. F. Cunha Felinto and E. E. de S. Teotonio, The chemistry of metal enolates. Part 1, John Wiley & Sons, Ltd, Chichester, UK, 2010, pp. 131–184 Search PubMed.
- R. M. Supkowski and W. D. W. Horrocks, Inorganica Chim. Acta, 2002, 340, 44–48 CrossRef CAS.
- W. DeW Horrocks Jr and D. R. Sudnick, Acc. Chem. Res., 1981, 384–392 CrossRef.
- S. Gago, M. Pillinger, R. A. Sá Ferreira, L. D. Carlos, T. M. Santos and L. S. Gonçalves, Chem. Mater., 2005, 17, 5803–5809 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cc03066k |
| This journal is © The Royal Society of Chemistry 2023 |