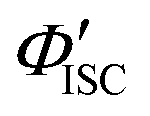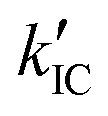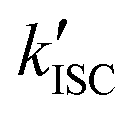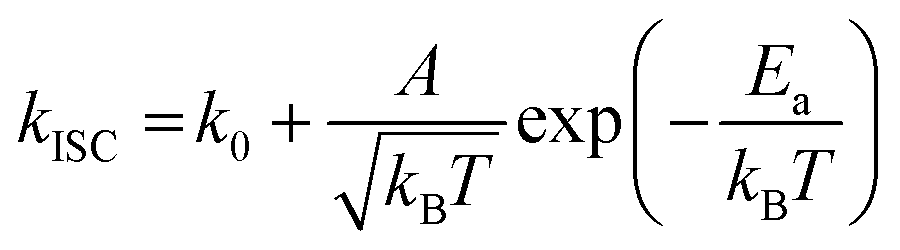DOI:
10.1039/D1TC04591A
(Paper)
J. Mater. Chem. C, 2022,
10, 4597-4606
Unravelling the origin of dual photoluminescence in Au2Cu6 clusters by triplet sensitization and photon upconversion†
Received
25th September 2021
, Accepted 9th December 2021
First published on 10th December 2021
Abstract
Ligand-protected, noble-metal clusters are promising as luminescent materials and photosensitizers because of their diverse chemical compositions and structures, as well as their easily tunable electronic and photophysical properties with single-atom precision. Many studies have shown that the photoluminescence (PL) properties of these clusters can be significantly enhanced by alloying the metal core or by increasing the rigidity of the ligand environment. However, the fundamental nature of their PL (whether fluorescent or phosphorescent) and the elementary process for enhancing their PL have not been fully clarified. In this study, we established that Au2Cu6(S-Adm)6(PPh3)2 (S-Adm = 1-adamantanethiolate) clusters exhibit not only fluorescence but also phosphorescence at room temperature. Further, we discovered that Au2Cu6 serves as a triplet sensitizer, and through the analysis of the photon-upconversion phenomenon, we determined the rate constants and quantum yields of all radiative and non-radiative processes involving the excited singlet and triplet states of the clusters. The temperature dependence of the PL and the theoretical calculations indicated that thermally activated intersystem crossing occurs through the spin–vibronic coupling mechanism mediated by higher excited states (S2 and/or T2) where direct spin–orbit coupling is possible.
Introduction
Ligand-protected noble-metal clusters have attracted significant attention because of their diverse chemical compositions and structures, as well as their potential applications as photosensitizers in photocatalytic reactions,1,2 singlet oxygen generation,3,4 solar cells,5,6 and photon upconversion.7 To realize these applications, it is important to profoundly understand the underlying photophysics of the clusters, including their photoabsorption and photoluminescence (PL) properties. As is well known, dual PL and multiple PL decay are frequently observed in ligand-protected gold, silver, and their alloy clusters at room temperature;8–12 however, their origin remains unclear. The exact origin of the PL is difficult to identify because of the presence of multiple emission sites in the clusters, such as the metal core and surface staple,7–12 as well as the contribution of electronic states with different spin multiplicities (i.e., excited singlet and triplet states) to these emission sites.7,13–15 Therefore, it would be desirable to determine the rate constants and quantum yields of all radiative and non-radiative processes involving the excited singlet and triplet states of the cluster, so that the complex behavior of the excited-state relaxation processes of the clusters can be accurately understood.
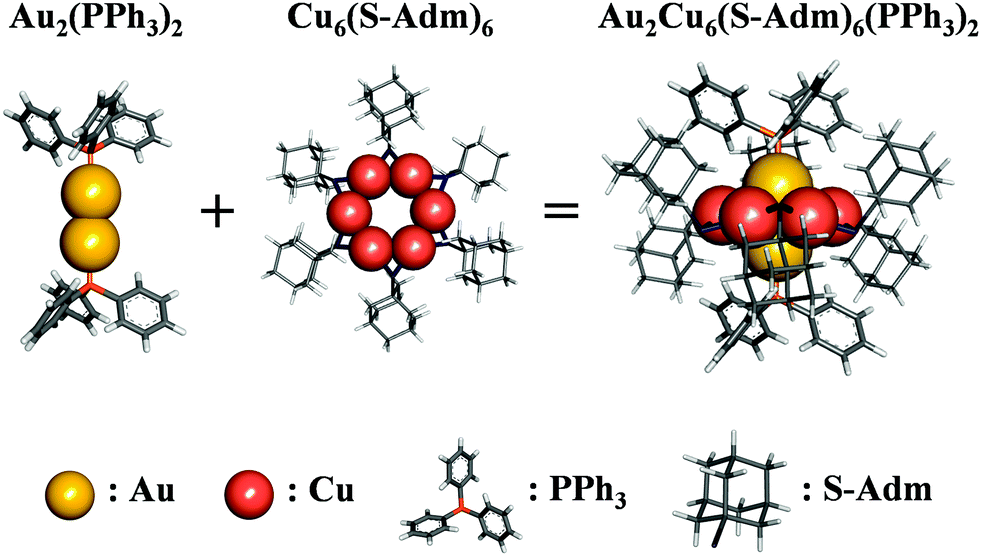
As shown in Scheme 1, the Au2Cu6(S-Adm)6(PPh3)2 (where S-Adm = 1-adamantanethiolate, hereinafter referred to as Au2Cu6) cluster, first synthesized by Zhu et al.,16 possesses a structure in which the Cu6(S-Adm)6 ring surrounds a PPh3-terminated Au dimer. This cluster exhibits a strong emission peak around 660 nm in dichloromethane (DCM), which is assigned to the ligand-to-metal charge-transfer (LMCT) fluorescence transition.16,17 Additionally, to tailor the PL characteristics, the excited-state charge-transfer property of the cluster can be perturbed using phosphine ligands that functionalize the phenyl groups with electron-donating or electron-withdrawing groups.16 Such flexibility in controlling the electronic and photophysical properties suggests that Au2Cu6-based clusters are promising as luminescent materials and photosensitizers.
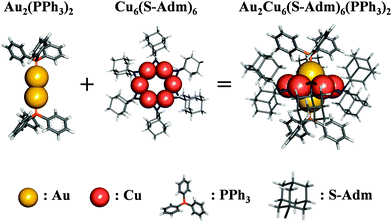 |
| | Scheme 1 Chemical structure of Au2Cu6. | |
In this study, we established that the Au2Cu6 cluster exhibits not only fluorescence but also phosphorescence at room temperature and can sensitize molecular triplets. Thus far, various triplet sensitizers, such as heavy-metal (e.g., Os, Ir, Ru, and Pt) complexes18–23 and semiconductor nanocrystals,24–37 have been developed and applied to triplet fusion upconversion (TF-UC). Recently, we presented atomically precise metal clusters as a new addition to the library of triplet sensitizers.7 Herein, we report yet another example of triplet sensitization and TF-UC by metal clusters. Through a detailed analysis of the TF-UC phenomenon occurring in the Au2Cu6 sensitizer-perylene emitter pair, we successfully determined the rate constants and quantum yields of all radiative and non-radiative processes involving the excited singlet and triplet states of the Au2Cu6 cluster. The temperature dependence of the dual PL revealed that thermally activated intersystem crossing (ISC) occurs in this cluster, and the ISC mechanism is discussed based on the results of density functional theory (DFT) and time-dependent DFT (TD-DFT) calculations.
Experimental
Chemicals
The following special-grade reagents were purchased from FUJIFILM Wako Pure Chemicals Co., Ltd: hydrogen tetrachloroaurate(III) tetrahydrate (HAuCl4·4H2O), copper(I) chloride (CuCl), triphenylphosphine (PPh3), anthracene, DCM, and tetrahydrofuran (THF). Platinum(II) octaethylporphine (PtOEP), N,N′-bis(2,5-di-tert-butylphenyl)-3,4,9,10-perylenedicarboximide (PDI), and 1-adamantanethiol (HS-Adm) were purchased from Sigma Aldrich. Sodium borohydride (NaBH4), perylene, and 9,10-diphenylanthracene (DPA) were purchased from Tokyo Chemical Industry Co., Ltd. All the chemicals were used as received without further purification. Ultrapure water (≥18.2 MΩ cm) was produced using a Direct-Q UV distillation system.
Synthesis
Au2Cu6(S-Adm)6(PPh3)2 (Scheme 1) was synthesized following a method previously reported by Zhu and coworkers.16 Briefly, 19.8 mg of CuCl (0.2 mmol) was dispersed in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 acetonitrile and methanol mixed solution, after which 136 mg (0.8 mmol) of 1-adamantanethiol was added. The mixture was stirred until a clear colorless solution was obtained. Thereafter, a solution of 198 mg (0.4 mmol) of Au(PPh3)Cl dissolved in 5 mL of toluene and 1 mL of 60 mg mL−1 of NaBH4 aq. were added simultaneously. After stirring for 60 h, the solution was centrifuged to obtain a purple precipitate. The precipitate solution was washed with toluene three or four times and subsequently extracted with DCM to obtain pure Au2Cu6(S-Adm)6(PPh3)2.
:1 acetonitrile and methanol mixed solution, after which 136 mg (0.8 mmol) of 1-adamantanethiol was added. The mixture was stirred until a clear colorless solution was obtained. Thereafter, a solution of 198 mg (0.4 mmol) of Au(PPh3)Cl dissolved in 5 mL of toluene and 1 mL of 60 mg mL−1 of NaBH4 aq. were added simultaneously. After stirring for 60 h, the solution was centrifuged to obtain a purple precipitate. The precipitate solution was washed with toluene three or four times and subsequently extracted with DCM to obtain pure Au2Cu6(S-Adm)6(PPh3)2.
The solution containing the product was diluted with a large amount of DCM (∼10−8 M) and subjected to electrospray ionization mass spectrometry (JEOL, JMS-T100LP AccuTOF LC-Plus). The mass spectrum was monitored in the positive-ion mode. A NaI methanol solution was employed for the calibration. Fig. S1 (ESI†) displays the positive-ion mode electrospray ionization mass spectrum of the product. The peaks around 2303 m/z correspond to Au2Cu6(S-Adm)6(PPh3)2. The isotope pattern of this peak is consistent with that obtained theoretically.
Absorption and PL measurements
Ultraviolet-visible (UV-vis) absorption spectra were recorded on a Lambda 650 spectrometer (PerkinElmer). The PL spectra were recorded using a SpectraPro 2300i polychromator coupled with a liquid-nitrogen-cooled charge-coupled device camera (Spec-10:100B/LN, Roper Scientific). For the PL spectrum measurements, a linearly polarized continuous-wave (CW) diode laser with a wavelength of 532 or 640 nm (Edmund Optics) was employed as the excitation source. The temperature dependence of the PL spectrum and lifetime was measured using a variable temperature cell holder (CoolSpeK, Unisoku Co., Ltd). In measuring the PL lifetime of the clusters, a 478 or 634 nm-picosecond pulsed laser (pulse width: 40–80 ps, PiL048X or PiL063X, Advanced Laser Diode System) was employed as the excitation source. All the signals, including the synchronization of the pulse laser and an avalanche photodiode (APD, SPCM-AQRH-61, PerkinElmer), were fed into a time-correlated single-photon-counting (TC-SPC) card (TimeHarp 260, PicoQuant) operated in time-tagged and time-resolved modes.38,39 Data acquisition and fluorescence decay analyses were performed using the SymPhoTime 64 (PicoQuant) software. To determine the fluorescence lifetime, single or double exponentials were fitted to the fluorescence curves by maximum likelihood estimation. For the excitation spectrum measurement, white light from an Xe arc lamp through an optical fiber was supplied into a monochromator (CM110, Spectral Products) and spectrally divided into monochromatic light. The solution sample was irradiated with the monochromatic light, thus obtained, while scanning the wavelength of the monochromatized light, and the emission from the sample was detected by the APD. A silicon photodiode (SM1PD2A-CAL-SP, Thorlabs Japan Inc.) with an amplifier (PDA200C, Thorlabs Japan Inc.) was used to record the light intensity at each wavelength and to correct the signal intensity of the excitation spectrum. To monitor only the emission band of interest, appropriate filters were placed in front of the APD. The experimental setup for the upconverted emission decay measurements has been thoroughly described elsewhere.7 Briefly, a 532 nm-CW laser, periodically blocked at 500 Hz by an optical chopper (MC2000B controller, MC1F10HP blade, Thorlabs), was employed as the excitation source. Based on the scattered light decay measurements under this condition, the time resolution was estimated to be 4.5 μs, which was sufficiently shorter than the decay time of the delayed (or upconverted) fluorescence of the perylene emitter (i.e., several hundred μs). The upconverted emission signal was detected by an APD (APD410A/M, Thorlabs) and monitored synchronously by an oscilloscope (GDS1062A, Chip One Stop). The decay profiles were averaged 256 times and stored in the oscilloscope.
Electrochemical measurement
To examine the electrochemical properties of the cluster, differential pulse-voltammetry measurements were conducted in an acetonitrile solution containing tetrabutyl ammonium perchlorate using ECstat-301 (EC Frontier Co., Ltd). For the experiment, glassy carbon was employed as the working electrode, Ag/Ag+ as the reference electrode, and a Pt wire as the counter electrode, with a scan rate of 50 mV s−1. A ferrocenium/ferrocene (Fc/Fc+) redox couple was used as an internal reference, and the measured potentials were converted to normal hydrogen electrode (NHE) potentials by +0.69 V or the energy level of the highest occupied molecular orbital (HOMO) relative to the vacuum level by EHOMO (V, vs. NHE) = −EHOMO (eV, vs. vacuum level) − 4.44.40 For perylene, DPA, and anthracene, the HOMO and lowest unoccupied molecular orbital (LUMO) energies were obtained from the literature.41,42
Transient absorption spectroscopy of Au2Cu6
Subnanosecond transient absorption (TA) spectroscopy measurements of the Au2Cu6(S-Adm)6(PPh3)2 cluster with and without perylene in deaerated THF were performed using a picosecond TA spectroscopy system (picoTAS, UNISOKU Co., Ltd) based on the randomly interleaved pulse-train method.43 The pump source was a picosecond Nd:YAG laser (355 nm, EKSPLA PL-2210A, 1 kHz, full width at half maximum (FWHM) = 25 ps) equipped with an optical parametric generator (EKSPLA PG403, 410–700 nm, 50 μJ per pulse @500 nm), and the probe light source was a supercontinuum radiation source (SMHP-20.2, Leukos, 20 MHz, FWHM = 50–100 ps, 410–2000 nm). The temporal resolution of the system was estimated to be 80–100 ps with a 10–90% rise time. The TA measurements were performed using a quartz cell with an optical path length of 2 mm.
Quantum chemical calculations
DFT and TD-DFT calculations were implemented with the Gaussian 16 program (ES64L-G16, RevB.01).44 To reduce the central processing unit cost, all the S-Adm ligands in the clusters were replaced with 1-tert-butylthiolate (S-CMe3) ligands. The geometric structure of Au2Cu6(S-CMe3)6(PPh3)2 in the S0, S1, or T1 state was optimized using the BP86 functional45 with the basis sets of def2-SV(P) for Cu and Au atoms46 and 6-31G(d,p) for H, C, P, and S atoms;47 the SDD pseudopotentials with scalar relativistic effects were included.48 Furthermore, harmonic vibrational frequency analysis was performed for all the optimized structures, and it was confirmed that there were no imaginary frequencies. Using these optimized structures, the vertical transition energies of S0 → Sn (n = 1–200), S1 → S0, and T1 → S0 were calculated using the TD-B3LYP functional,49–51 with a polarizable continuum model (solvent = THF). In this calculation, the basis sets of def2-SV(P) for the Cu and Au atoms and 6-31G(d,p) for H, C, P, and S atoms were employed, and the SDD pseudopotentials were also implemented. Note that the theoretical levels chosen in this study have previously been shown to yield relatively accurate results in reproducing the electronic structure of ligand-protected noble metal clusters.7,52 The vertical transition energies from S2 to S0 at the S1 equilibrium geometry and the transition energy from T2 to S0 at the T1 equilibrium geometry were also calculated at the same theoretical level. The molecular structures and frontier molecular orbitals were constructed using GaussView 6.0 and Avogadro 1.2.0, respectively.
Results and discussion
Photoabsorption and emission properties
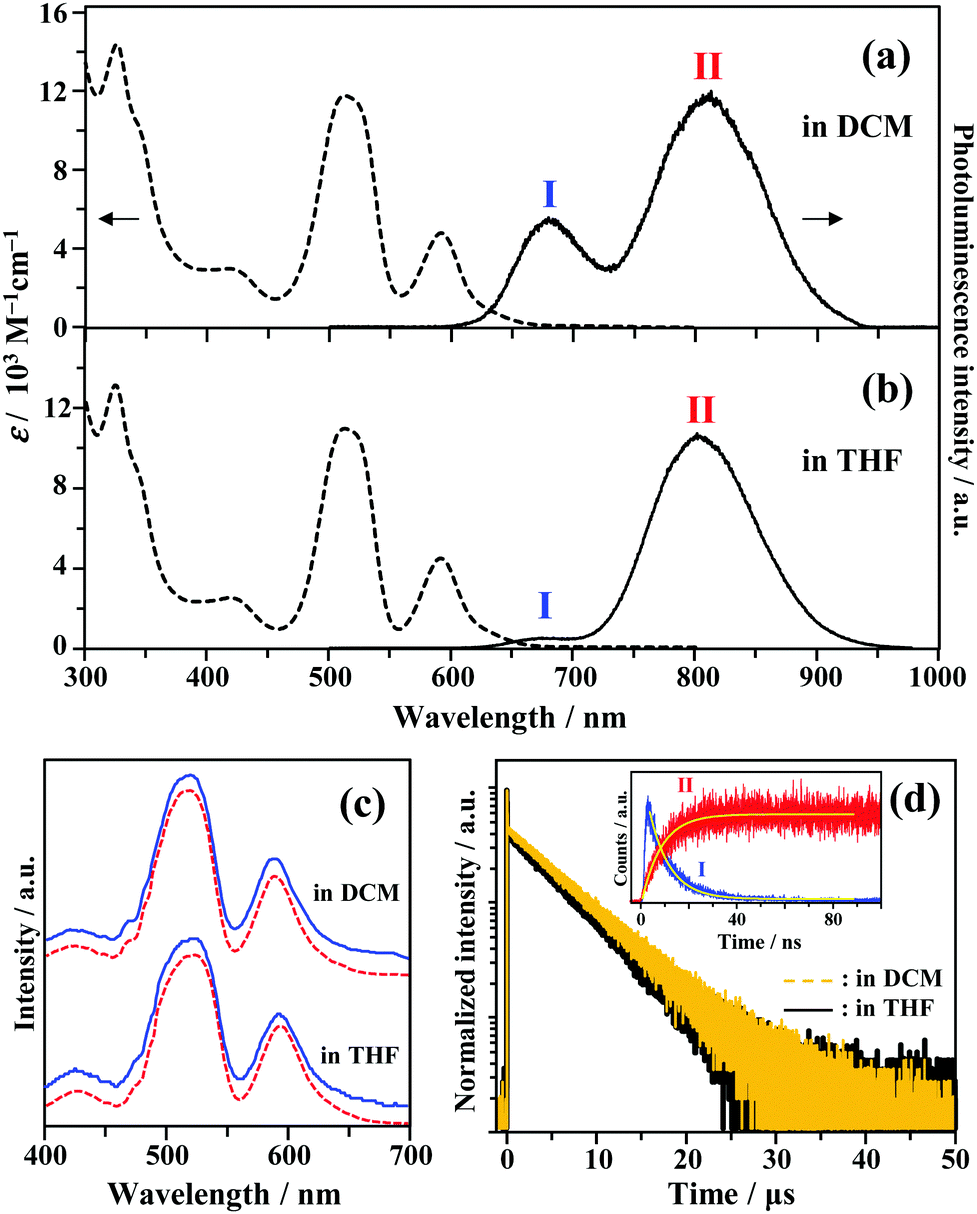
The UV-vis absorption spectra of Au2Cu6 are shown in Fig. 1. Two relatively sharp peaks around 520 nm and 590 nm are observed, which are consistent with the features of the reported absorption spectra of Au2Cu6 in DCM.16 Conversely, in the reported emission spectrum of Au2Cu6 in DCM, only a single LMCT fluorescence band (band I) around 660 nm is observed.16,17 However, Fig. 1a and b show another emission band (band II) at relatively long wavelengths in DCM and THF. The excitation spectra were measured using appropriate filters to selectively monitor only the emission of band I or II, as shown in Fig. 1c. The excitation spectra were virtually identical and matched the absorption spectra, confirming that both emission bands originated from Au2Cu6. Band II was not observed in the literatures presumably because the detector used was not sensitive to photons with wavelengths longer than 750 nm. As shown in Fig. 1d, two decay components having very different lifetimes were observed in the PL decay curve. The fast decay component originated from band I and the slow component from band II, as confirmed by the selective observation of the PL decay curves (Fig. S2 (ESI†) and the inset of Fig. 1d). Notably, the emission decay of band I and the emission rise of band II showed practically identical single exponential kinetics, both having a lifetime of 9 ns. As listed in Table 1, the emission lifetimes of band I were 19.0 and 9.0 ns in DCM and THF, respectively, while those of band II were 5.2 and 4.9 μs, respectively. Judging from these results, the band-I emission corresponds to fluorescence,16,17 while the newly observed emission (band II) can be tentatively assigned to phosphorescence. This suggests the formation of an excited triplet state in the Au2Cu6 cluster. Although the fluorescence quantum yields (Φf) of Au2Cu6 in DCM and THF were both quite small, there was a pronounced solvent effect on the fluorescence radiative rate constant (kf), with a 4.9-fold increase in DCM compared to that in THF. This solvent effect is attributable to the strong LMCT nature of the fluorescence transition,16,17 as mentioned above. The emission quantum yields of band II (ΦPL), which is considered to be phosphorescence, were determined by relative estimation (see ESI†) to be 0.17 (DCM) and 0.21 (THF). However, notably, the value of ΦPL corresponds to the product of the ISC yield (ΦISC) and the phosphorescence quantum yield (Φp), i.e., ΦPL = ΦISC·Φp. The determination of these quantum yields will be discussed later. As shown in Fig. S3 (ESI†), dynamic light scattering measurements suggest that the photostability of Au2Cu6 in DCM is low and that the clusters dissociate rapidly upon laser irradiation. Therefore, all the results shown below were obtained using THF as the solvent, in which the photostability of the Au2Cu6 cluster is significantly higher than that in DCM.
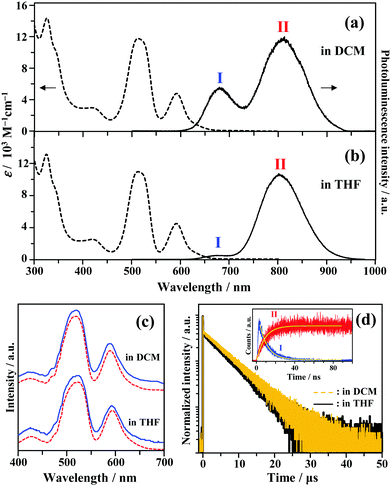 |
| | Fig. 1 Absorption and emission spectra of Au2Cu6 (10 μM) in (a) dichloromethane (DCM) and (b) tetrahydrofuran (THF). (c) Excitation spectra obtained by analysis at the emission wavelength of λ ≤ 700 nm (band I, solid line) or λ ≥ 800 nm (band II, dashed line). (d) Photoluminescence decay curves of Au2Cu6 in deaerated DCM and THF (λex = 478 nm), where both bands I and II were monitored. The inset shows the decay and rise of the selectively observed band (bands I [λ ≤ 700 nm] and II [λ ≥ 800 nm]) emissions for Au2Cu6 in deaerated THF. | |
Table 1 Photoabsorption and photoluminescence parameters of Au2Cu6 in deaerated dichloromethane (DCM) and tetrahydrofuran (THF).a
| Solvent |
λ
abs/nm (ε/M−1cm−1) |
λ
em
/nm band-I band-II |
τ
f/ns band-I |
Φ
f band-I |
k
f/s−1 |
τ
p/μs band-II |
Φ
PL band-II |
|
Note that all the data were obtained by preparing solution samples in a light-shielded environment and measuring them immediately afterwards.
The values in parentheses are in eV.
|
| DCM |
593 (4268) |
684 (1.813) |
814 (1.523) |
19.0 |
5.3 × 10−2 |
2.8 × 106 |
5.2 |
0.17 |
| THF |
591 (4483) |
674 (1.840) |
811 (1.529) |
9.0 |
5.1 × 10−3 |
5.7 × 105 |
4.9 |
0.21 |
Quenching experiments and triplet sensitization
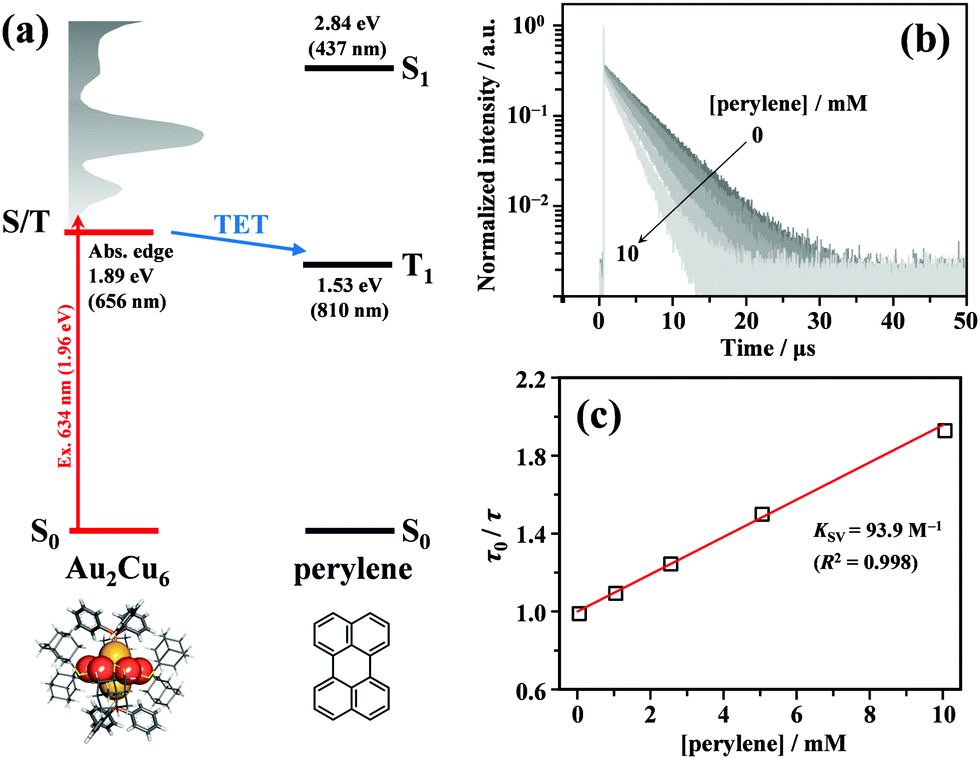
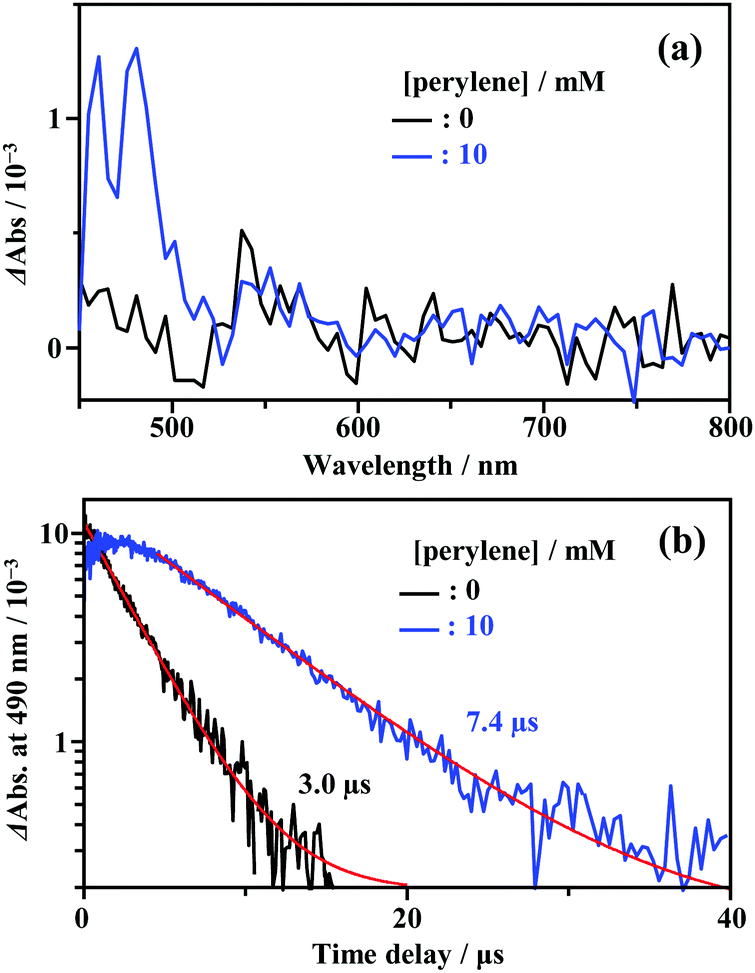
As shown in Fig. 2, the energy of the S1 state of perylene (2.84 eV) was sufficiently higher than the excitation photon energy (1.96 eV). Furthermore, the LUMO energy of perylene was higher than that of the cluster by more than 1 eV (Fig. S4, ESI†). Therefore, neither singlet energy transfer nor photoinduced electron transfer could occur. In contrast, the T1 state of perylene (1.53 eV) was lower in energy than the absorption edge of the cluster (1.89 eV); therefore, triplet energy transfer (TET) could occur in this pair. Energetically, there is also the possibility of hole transfer from the cluster to perylene (Fig. S4b, ESI†); however, no evidence suggesting this process was obtained in the present experiments. This is probably due to the fact that the HOMO localized in the Au2 and Cu6S6 portions is almost completely sterically shielded by the six adamantane and two PPh3 ligands, hindering hole transfer. As shown in Fig. 2b, the lifetime of the long-lived component (band II) gradually decreased with an increase in the perylene concentration from 0 to 10 mM. The Stern–Volmer plot of the observed lifetimes is shown in Fig. 2c. The plot was well reproduced with a linear fit, and the slope yielded a Stern–Volmer constant (Ksv) of 93.9 M−1. To confirm that this quenching was caused by TET, TA spectroscopy measurements were performed with and without perylene. Without perylene, the Au2Cu6 cluster exhibited an absorption band at 450–490 nm, attributed to the triplet–triplet (T–T) absorption, and ground-state bleaching at 520 and 590 nm. Both bands exhibited decay times of approximately 3 μs (Figs. S5a and b, ESI†), which is shorter than the phosphorescence lifetime of 4.9 μs. This difference is attributed to the higher cluster concentration (20×) required for the TA measurement compared to that for the emission-lifetime measurement, owing to the weak TA signal. At such a high concentration, T–T annihilation (TTA) may occur between the clusters, shortening their lifetime. With perylene (10 mM), the decay time of the TA band at the 450–490 nm region was significantly lengthened, as shown in Fig. S5c (ESI†), and a new TA peak around 490 nm was observed with a time delay of 20 μs, as shown in Fig. 3a. Since this peak was not observed without perylene, it can be attributed to the T–T absorption of perylene.53,54 The decay time of this T–T absorption was determined to be 7.4 μs, which was considerably shorter than the triplet lifetime of perylene in deaerated THF (∼0.7 ms) because of the TTA between the perylene molecules and the quenching by the cluster sensitizer. These results demonstrate that the band-II emission was phosphorescence and that its quenching was caused by TET from the cluster in the excited triplet state to perylene in the S0 state (i.e., the triplet sensitization of perylene [ET = 1.53 eV] by Au2Cu6). From the Stern–Volmer analysis (see ESI†), the rate constant of this TET process (kTET) was determined to be 1.9 × 107 s−1. When 9,10-diphenylanthracene (DPA) (ET = 1.77 eV)55 was employed as the triplet quencher, the TET rate constant was dramatically reduced to 2.3 × 105 s−1 (Fig. S6 and Table S1, ESI†). When anthracene (ET = 1.84 eV)56 was used, no phosphorescence quenching (i.e., TET) occurred. Therefore, we inferred that the T1 state of Au2Cu6 is located around 1.7–1.8 eV, within 0.2 eV below the absorption edge of S1 (1.89 eV). The triplet perylene sensitization ability of Au2Cu6 suggests that TF-UC can occur in this sensitizer-emitter pair, which will be discussed in the next subsection.
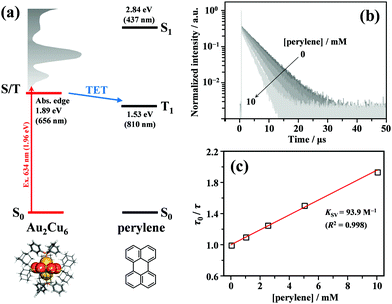 |
| | Fig. 2 (a) Energy level diagram of the Au2Cu6 and perylene pair. (b) PL decay curves of Au2Cu6 (10 μM) with [perylene] = 0–10 mM in deaerated THF, where both band-I and -II were monitored. To prevent the high concentration of perylene from absorbing the excitation light, an excitation wavelength of λex = 634 nm was used in this quenching experiment. (c) Stern–Volmer plot of phosphorescence decay times (i.e., PL lifetimes of band-II) at different perylene concentrations. KSV and R2 represent the Stern–Volmer constant and coefficient of determination. | |
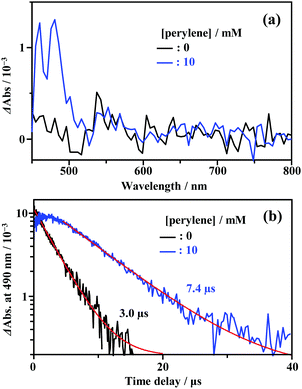 |
| | Fig. 3 (a) Transient absorption (TA) spectra at the time delay of 20 μs and (b) decay time profiles of the TA signal at 490 nm, measured for Au2Cu6 (270 μM) with 0 and 10 mM perylene in deaerated THF at 298 K (excitation wavelength, λex = 532 nm). | |
TF-UC in the Au2Cu6–perylene pair
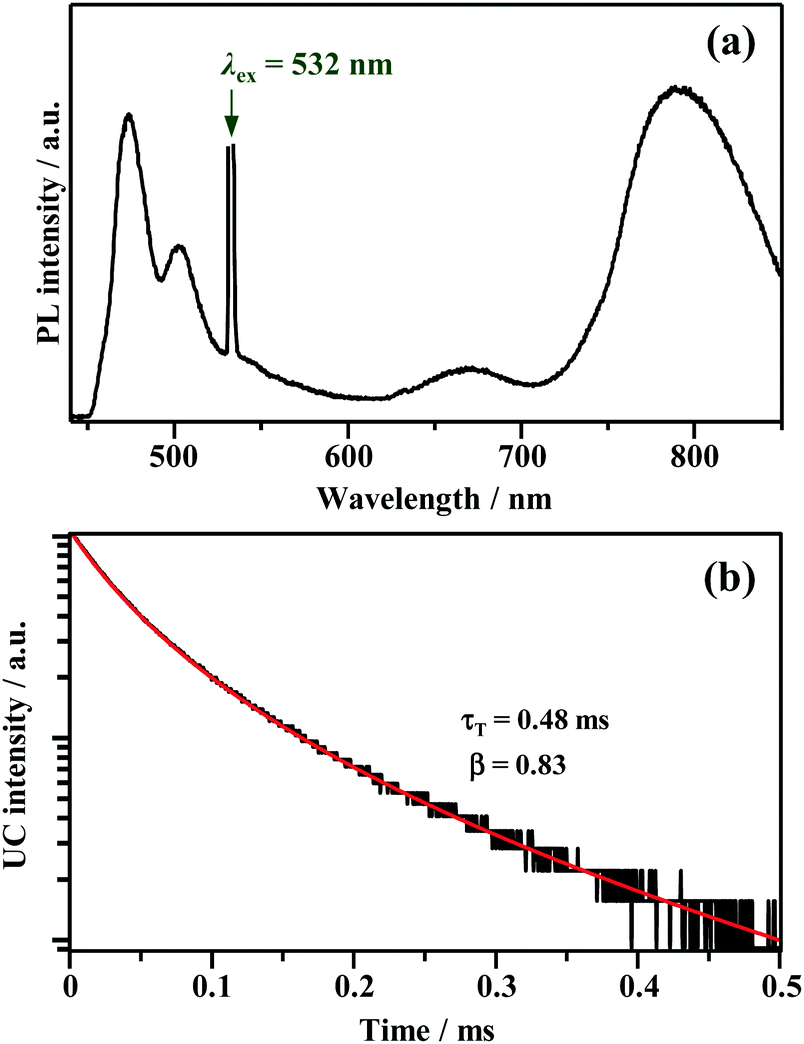
Fig. 4a shows the PL spectrum obtained when a solution of Au2Cu6 (11 μM) with 5 mM perylene was excited with a 532 nm-CW laser. Along with the fluorescence and phosphorescence of the cluster at 600–900 nm, perylene fluorescence was observed in the wavelength region shorter than the excitation wavelength. As shown in Fig. 4b, the time profile of this fluorescence signal exhibited sub-millisecond order decay. Since the upconverted emission intensity (I) is proportional to the square of the triplet emitter (3E*) concentration, its kinetics, I(t), is given by| | 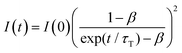 | (1) |
where τT is the lifetime of 3E*, and β is a dimensionless parameter between 0 and 1.57 Details are provided in ESI.† By fitting the decay curve using eqn (1), τT = 0.48 ms and β = 0.83 (Fig. S7, Tables S2 and S3, ESI†) were obtained. Notably, the value of τT is five orders of magnitude higher than that of the prompt perylene fluorescence (5.0 ns in THF7), indicating that a delayed fluorescence via TTA occurred between the triplet perylene molecules, i.e., TF-UC, triggered by the photoexcitation of the Au2Cu6 sensitizer.
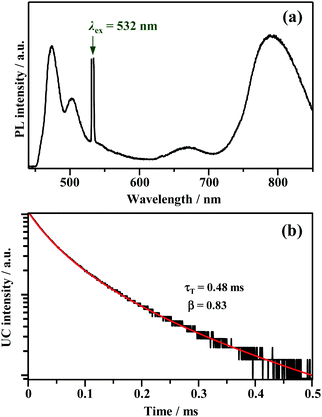 |
| | Fig. 4 (a) PL spectrum obtained for the deaerated THF solution of Au2Cu6 (11 μM) and perylene (5 mM), and (b) decay curve of the upconverted emission of perylene obtained by monitoring only the λ ≤ 492 nm region using short-pass filters. The fitting of the decay curve was performed using eqn (1). The 532 nm-CW laser was used in these measurements. Note that the upconverted emission of perylene was also observed when using a 640 nm-CW laser. | |
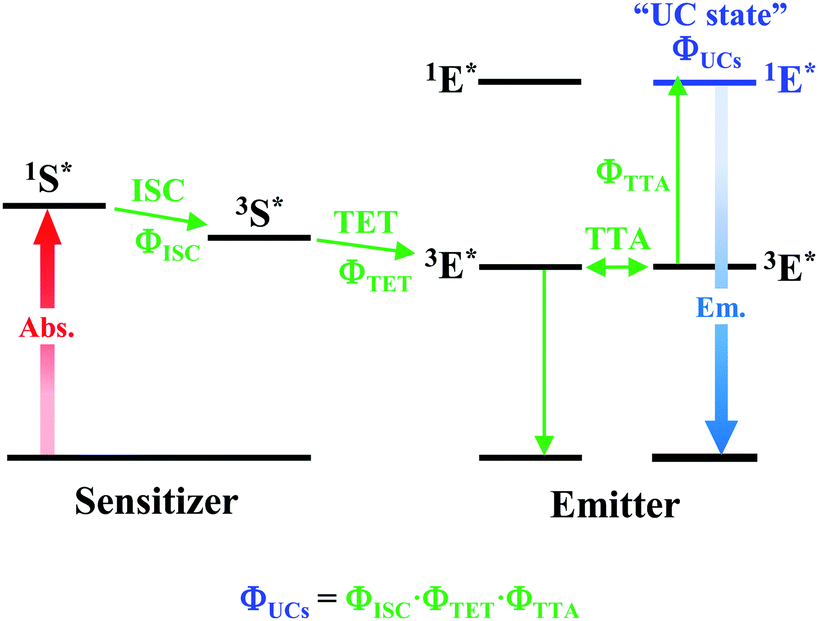
As depicted in Fig. 5, when a relatively high concentration of 3E* is generated via the sensitizer-emitter TET process, TTA can occur efficiently between the 3E* molecules because of their very long lifetimes. This process causes one emitter molecule to return to the S0 state and the other to transition to the S1 state (i.e., the UC state, 1E*), from which UC fluorescence is generated.20,58,59 The TF-UC efficiency (ΦUC), conventionally determined by relative estimation using eqn (S1) (ESI†), corresponds to the number of observed UC photons divided by the number of photons absorbed by the sensitizer (#1S*). Therefore, ΦUC is not necessarily a quantity unique to the TF-UC system, as it is strongly affected by several loss factors depending on the sample and experimental conditions, as mentioned below. Conversely, the formation yield of the UC state (ΦUCs), i.e., the number of upconverted states generated (#1E*) per #1S*, is unique to the system; thus, the following relationship holds:60
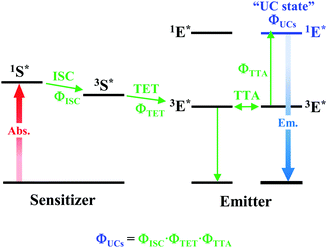 |
| | Fig. 5 Schematic diagram of TF-UC occurring in the system of a sensitizer–emitter pair. | |
Note that the theoretical maximum value of ΦUCs is 0.50 (50%) because ΦTTA ≤ 0.50. As can be seen in eqn (2), ΦUCs is proportional to the ΦISC of the sensitizer, and the ΦISC of the cluster can be estimated by evaluating ΦUCs.7 Experimentally, the value of ΦUCs is determined by appropriately correcting for the loss of UC photons due to the reabsorption of UC photons by the sensitizer and emitter molecules (i.e., optical outcoupling yield, Φout)61 and quenching of 3E* by the sensitizer (i.e., emitter quenching yield, Φq) against ΦUC.57 This is expressed in the following equation:
| | 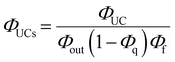 | (3) |
where
Φf represents the fluorescence quantum yield of the emitter.
60 Therefore, if the values of
ΦTET and
ΦTTA are known, the
ΦISC value can be obtained using
eqn (2). The
ΦTET can be determined by Stern–Volmer analysis, as shown in
Fig. 2c, Fig. S6, and Table S1 (ESI
†). However, since the
ΦTTA strongly depends on the emitter concentration, solvent type, intensity of the excitation laser, and so on, great care must be taken when using the values obtained from the literature. Moreover, since TF-UC measurements are usually performed at high emitter concentrations (≥1 mM), the value of
Φf is most likely to be considerably different from that of the dilute solutions of the emitter.
62 To avoid uncertainty in the values of
ΦTTA and
Φf, which are quantities related only to the emitter, we, herein, propose the following relative method.
Using a representative metal porphyrin sensitizer (e.g., PtOEP) having a known ΦISC (denoted herein as ΦrISC) and the same emitter as a reference system, we evaluate its ΦrUCs at the same emitter concentration, in the same solvent, and under the same measurement conditions (e.g., optical setup and excitation wavelength). Note that yields marked with “r” on the right shoulder indicate those of the reference system thereafter. Applying the ratio in eqn (2) to the reference and target sample, ΦTTA can be cancelled out as follows:
| | 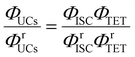 | (4) |
Using eqn (3), the left-hand side of eqn (4) can be expressed and experimentally determined by the following equation:
| | 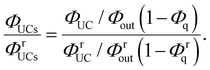 | (5) |
By combining eqn (4) and (5), we can estimate the ΦISC of the sensitizer of interest by the following equation:
| | 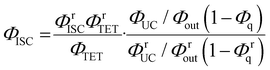 | (6) |
Notably, this equation does not include the parameters of the emitter, ΦTTA and Φf.
The value of ΦTET at a given perylene concentration can be estimated using the Ksv value (93.9 M−1) obtained from the Stern–Volmer plot shown in Fig. 2c. For example, ΦTET = 0.32 under the emitter concentration condition ([perylene] = 5 mM) in Fig. 4a, where the prominent UC emission was observed. In this study, PtOEP with ΦISC = ∼1.0 was used as the reference sensitizer,63 and a series of parameters that need to be substituted into eqn (6) were evaluated for the pair of PtOEP sensitizer and perylene emitter in deaerated THF (Table 2). The parameters of the Au2Cu6–perylene system were evaluated following the same process under the same experimental conditions, and the ΦISC of the cluster was finally determined. As summarized in Tables 2 and 3, through this method, the ΦUCs for the Au2Cu6–perylene system was estimated to be 0.0049 (0.49%), resulting in an estimated ΦISC of 0.47 for the Au2Cu6 cluster. Thus, the phosphorescence quantum yield, Φp, was determined to be 0.44 from the relationship, ΦPL = ΦISC·Φp = 0.21 (see Table 1), and the ISC quantum yield from T1 to S0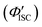 was 0.56. All the quantum yields and rate constants determined for the radiative and non-radiative processes are summarized in Table 3. The results show that the Au2Cu6 cluster has relatively high quantum yields of ISC and phosphorescence at room temperature and, in contrast, very small fluorescence quantum yields. Notably, Au2Cu6 possesses a very long fluorescence radiative lifetime (1.75 μs) and a small ISC rate of 5.3 × 107 s−1, which are strikingly different from those of many transition-metal complexes that exhibit ultrafast ISC.64–66
was 0.56. All the quantum yields and rate constants determined for the radiative and non-radiative processes are summarized in Table 3. The results show that the Au2Cu6 cluster has relatively high quantum yields of ISC and phosphorescence at room temperature and, in contrast, very small fluorescence quantum yields. Notably, Au2Cu6 possesses a very long fluorescence radiative lifetime (1.75 μs) and a small ISC rate of 5.3 × 107 s−1, which are strikingly different from those of many transition-metal complexes that exhibit ultrafast ISC.64–66
Table 2 Measured and corrected upconversion quantum yields and their related parameters
|
λ
ex
/nm |
Sensitizer |
Emitter |
[E]/mM |
Φ
TET
|
Φ
UC
|
Φ
out
|
1 − Φqd |
Φ
UCs
|
|
The 532 nm excitation light intensity, Iex (W cm−2), was set to the intensity region where the ΦUC becomes constant (Iex = 8.04 and 3.09 W cm−2 for the reference and sample systems, respectively).
PDI (Φf = 0.97 in toluene) was used as the reference dye, and the values were calculated by the relative method using eqn (S1) (ESI).
Calculated using eqn (S8) (ESI).
Calculated using eqn (S7) (ESI).
Calculated using eqn (3).
|
| 532 |
Au2Cu6 |
Perylene |
5.0 |
0.32 |
0.0018 |
0.60 |
0.65 |
0.0049 |
| PtOEP |
0.99 |
0.0052 |
0.64 |
0.27 |
0.032 |
Table 3 Quantum yields and rate constants for the radiative and nonradiative processes of the Au2Cu6(S-Adm)6(PPh3)2 cluster in deaerated THF at room temperature
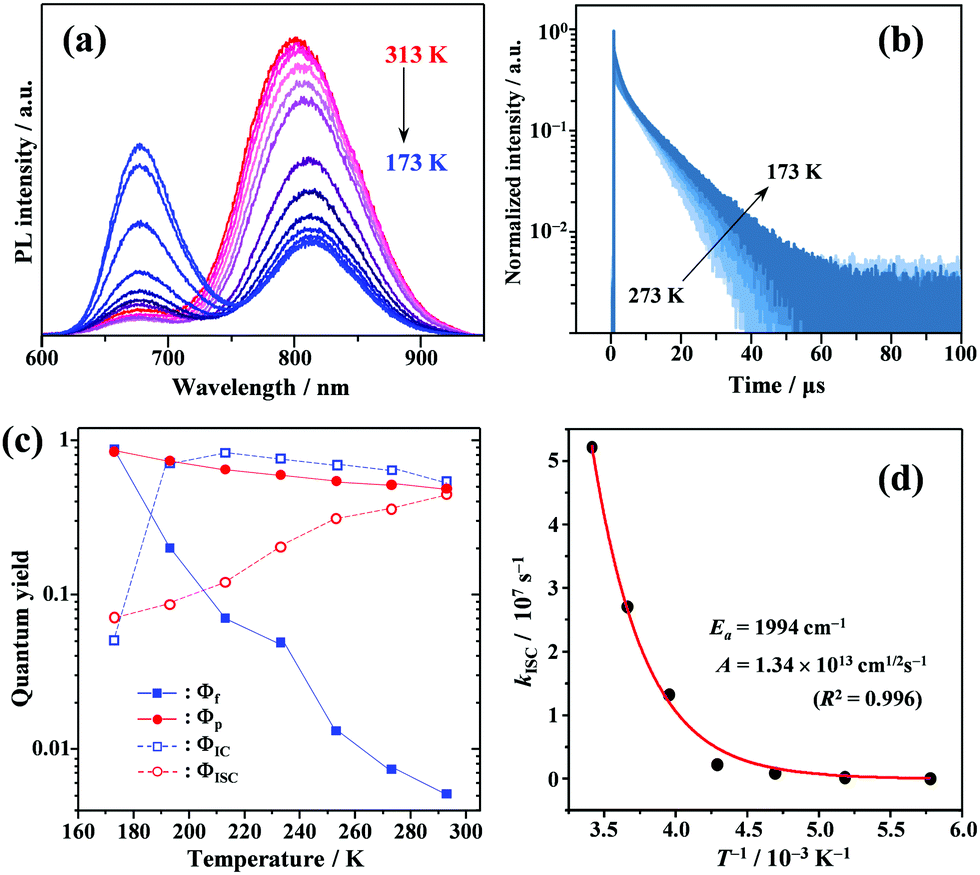
Temperature effects on photophysical processes
To gain a profound understanding of the radiative and non-radiative processes in Au2Cu6, we measured the temperature (T) dependence of the PL spectrum (Fig. 6a) and lifetime (Fig. 6b). The results revealed the T-dependence of the quantum yield of all the radiative and non-radiation processes (Fig. 6c and Tables S4, S5, ESI†). The fluorescence intensity, lifetime, and quantum yield increased with decreasing temperature, accompanied by a concomitant decrease in the phosphorescence intensity, suggesting that the ISC process is suppressed at relatively low temperatures. Indeed, the ΦISC and rate constant (kISC), which were estimated from the T-dependence of Φp and ΦPL (Table S5, ESI†) by assuming that the singlet and triplet radiative rate constants (kf and kp, respectively) were temperature-independent,67 exhibited a remarkable decline with decreasing temperature. This fact indicates that the ISC proceeds via a thermally activated pathway. Thus, we assume that kISC can be expressed by an Arrhenius-type equation (eqn (7)):68,69| | 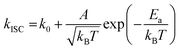 | (7) |
where k0 and Ea represent the temperature-independent rate constant and the activation energy for the ISC process, respectively. The Arrhenius-type plot shown in Fig. 6d was fitted well by eqn (7), yielding A = 1.34 × 1013 cm1/2s−1 and Ea = 1994 cm−1 (0.25 eV). Note that k0 is very small (<103 s−1) and can be neglected compared to the second term. The rate constants of the internal conversion (IC) process also decreased with a decrease in the temperature from 293 to 193 K (Table S5, ESI†), although the decrease was not as pronounced as that in the case of the ISC. However, an abrupt decrease was observed at temperatures below 193 K (kBT = ∼134 cm−1), suggesting that the IC process may be highly promoted by low-frequency vibrational modes (<140 cm−1).
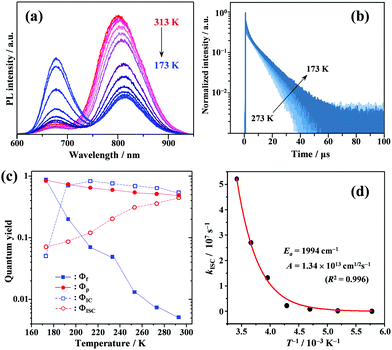 |
| | Fig. 6 Temperature dependence of (a) PL; (b) PL decay; and (c) quantum yields of fluorescence (Φf), phosphorescence (Φp), internal conversion (ΦIC), and ISC (ΦISC) of Au2Cu6 (20 μM) in deaerated THF. Note that both band-I and -II emissions were monitored in the lifetime measurements. (d) Arrhenius-type plot of the estimated ISC rate constant versus the inverse of the absolute temperature for Au2Cu6 in deaerated THF. The solid line represents the best fit of the data to eqn (7). | |
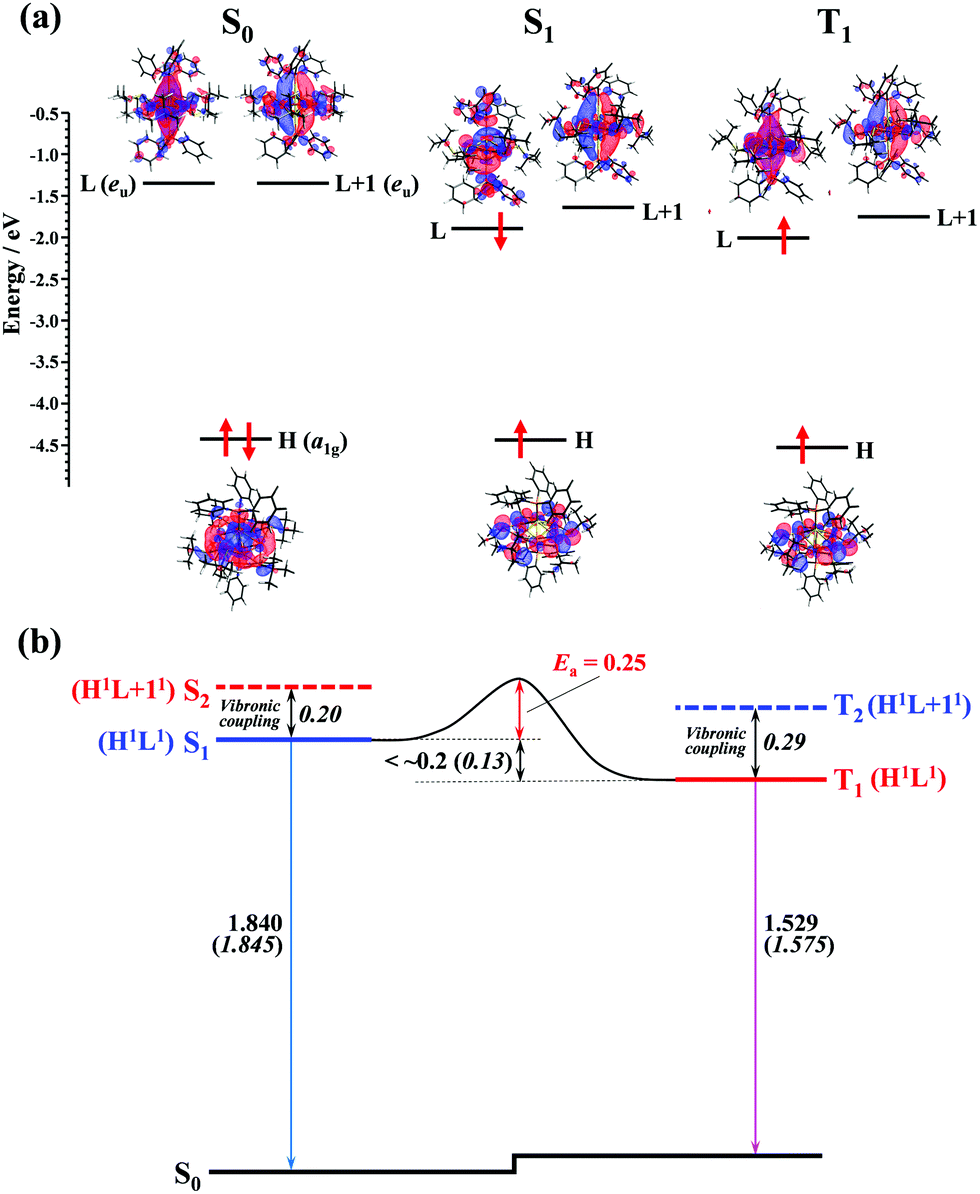
Theoretical consideration
To gain insights into the mechanism of the thermally activated ISC with the small rate constant (ca. 5 × 107 s−1), DFT and TD-DFT calculations for the ground and excited states of the Au2Cu6(S-CMe3)6(PPh3)2 cluster were performed. In the S0 structure with D3d symmetry, the LUMO (L) and LUMO+1 (L+1), which are mainly derived from π-like orbitals spreading along the P–Au–Au–P bond axis, were degenerate and orthogonal to each other (Fig. 7a). Therefore, S1 and S2 or T1 and T2 are degenerate in the S0 structure (Table S6, ESI†). However, in the equilibrium structure of the S1 and T1 states (Fig. S9, ESI†), the symmetry lowering (Fig. S10, ESI†) induced the weak splitting between L and L+1 (i.e., the S1 and S2 [or T1 and T2] states split energetically), as shown in Fig. 7. The calculated vertical transition energies from S1 to S0 (1.845 eV) and T1 to S0 (1.575 eV) agree well with the energies of the fluorescence (1.840 eV) and phosphorescence (1.529 eV) emission peaks (Table 1 and Fig. 7b). The shape of the HOMO (H), localized mainly on the Cu6S6 ring, is the same between the S1 and T1 states, and the S1 and T1 states have the same H1L1 electronic configuration. In the S1 state, although the shape of the LUMO is somewhat different from that of T1, there is no orthogonality between the LUMO (precisely, singly occupied molecular orbital on the high energy side) of S1 and that of T1. According to the El-Sayed rules,70 therefore, the direct spin–orbit coupling (SOC) between S1 and T1 is considered to be inactive. In fact, the temperature-independent term (k0) corresponding to the S1 → T1 direct tunneling was negligibly small. Conversely, the S2 and T2 states have the H1L + 11 electronic configuration (Fig. 7b and Table S6, ESI†), thereby allowing the direct SOC between S2 and T1, as well as between S1 and T2. It is also theoretically suggested that the S2 and T2 states are energetically close to the S1 and T1 states (<0.3 eV). Therefore, ISC should be partially allowed via the spin–vibronic coupling, mediated by the S2 and T2 states.70 Since the SOC constant of the Au atom is considerably larger than those of the light elements (S, P, C, and H atoms) that constitute the organic ligands, the L and L+1, which are mainly derived from the Au dimer moiety, are expected to contribute significantly to the S2–T1 and S1–T2 SOC matrix elements. Currently, there is no experimental evidence of the strong coupling (i.e., avoided crossing) between the S1 and T1 potential energy surfaces. This may be responsible for the activation barrier in the ISC process. The reverse ISC from T1 to S1 is unlikely to occur because the activation energy is as large as 0.5 eV.71 Consequently, no thermally activated delayed fluorescence was observed in this cluster.
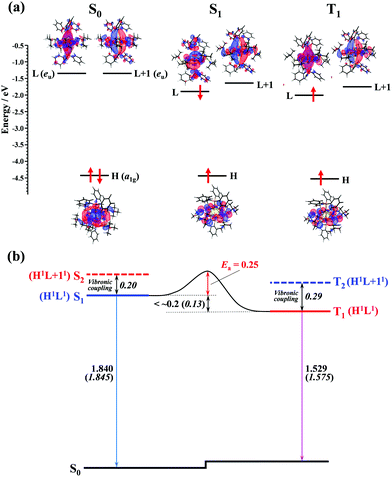 |
| | Fig. 7 (a) Computed energy-level diagram for the optimized structures of S0, S1, and T1 (lower panel). The electronic configuration of each electronic state is shown with upward (α-spin) and downward (β-spin) arrows in the notations HOMO (H), LUMO (L), and LUMO+1 (L+1). (b) Schematic representation of the ISC mechanism of the Au2Cu6 cluster. All values are in eV. Numbers in italics are the calculated values, while other numbers are estimated from the experiments. | |
Conclusions
This study demonstrates that Au2Cu6 clusters exhibit both fluorescence and phosphorescence at room temperature. Additionally, the quantum yields and rate constants of all radiative and non-radiative processes in this cluster were determined by analyzing the TF-UC phenomenon induced when Au2Cu6 is employed as a triplet sensitizer. Temperature-dependent fluorescence/phosphorescence measurements and theoretical calculations suggested that thermally activated ISC occurs via the spin–vibronic coupling mechanism mediated by the SOC-allowed S2 and T2 states. Metal clusters composed of heavy-metal elements, such as Au and Ag atoms, possess considerably larger SOC constants than those of organic molecules and relatively small S–T energy gaps, which inevitably leads to considerably stronger S–T mixing. Consequently, it is highly probable that excited states with a triplet nature are involved in various excited-state relaxation processes, including PL phenomena. Therefore, the approach reported herein, based on triplet sensitization by metal clusters, will be very useful not only for elucidating the origin of PL in clusters but also for expanding their application as photosensitizers.
Author contributions
Daichi Arima: experiment, data acquisition, analysis, and drafted the initial manuscript. Yoshiki Niihori: experiment, revision, and funding acquisition. Masaaki Mitsui: supervision, writing – revision and editing, theoretical calculations, and funding acquisition.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work is partly supported by the Grant-in-Aids for Young Scientists, no. 20K15110 and Scientific Research (C), no. 20K05653. Theoretical calculations were performed at the Research Center for Computational Science, Okazaki, Japan. M. M. thanks Mr Yasuki Yoshie and Mr Syuichi Mine (Otsuka Electronics Co., Ltd) for renting the dynamic light scattering apparatus, and Mr Kido Okamoto and Dr Tatsuo Nakagawa (Unisoku Co., Ltd) for renting the variable temperature cell holder and performing transient absorption spectroscopy measurements.
References
- F. F. Schweinberger, M. J. Berr, M. Döblinger, C. Wolff, K. E. Sanwald, A. S. Crampton, C. J. Ridge, F. Jäckel, J. Feldmann, M. Tschurl and U. Heiz, J. Am. Chem. Soc., 2013, 135, 13262 CrossRef CAS PubMed.
- Y. Du, H. Sheng, D. Astruc and M. Zhu, Chem. Rev., 2020, 120, 526 CrossRef CAS PubMed.
- H. Kawasaki, S. Kumar, G. Li, C. Zeng, D. R. Kauffman, J. Yoshimoto, Y. Iwasaki and R. Jin, Chem. Mater., 2014, 26, 2777 CrossRef CAS.
- M. Agrachev, W. Fei, S. Antonello, S. Bonacchi, T. Dainese, A. Zoleo, M. Ruzzi and F. Maran, Chem. Sci., 2020, 11, 3427 RSC.
- Y.-S. Chen, H. Choi and P. V. Kamat, J. Am. Chem. Soc., 2013, 135, 8822 CrossRef CAS PubMed.
- M. A. Abbas, T.-Y. Kim, S. U. Lee, Y. S. Kang and J. H. Bang, J. Am. Chem. Soc., 2016, 138, 390 CrossRef CAS PubMed.
- Y. Niihori, Y. Wada and M. Mitsui, Angew. Chem., Int. Ed., 2021, 60, 2822 CrossRef CAS PubMed.
- S. Link, A. Beeby, S. FitzGerald, M. A. El-Sayed, T. G. Schaaff and R. L. Whetten, J. Phys. Chem. B, 2002, 106, 3410 CrossRef CAS.
- M. Zhou and Y. Song, J. Phys. Chem. Lett., 2021, 12, 1514 CrossRef CAS PubMed.
- Z. Lei, Z.-J. Guan, X.-L. Pei, S.-F. Yuan, X.-K. Wan, J.-Y. Zhang and Q.-M. Wang, Chem. – Eur. J., 2016, 22, 11156 CrossRef CAS PubMed.
- P. Ai, M. Mauro, A. A. Danopoulos, A. M. Castro and P. Braunstein, J. Phys. Chem. C, 2019, 123, 915 CrossRef CAS.
- M. Bodiuzzama, E. Khatun, K. S. Sugi, G. Paramasivam, W. A. Dar, S. Antharjanam and T. Pradeep, J. Phys. Chem. C, 2020, 124, 23426 CrossRef.
- Y. Song, Y. Li, M. Zhou, X. Liu, H. Li, H. Wang, Y. Shen, M. Zhu and R. Jin, Sci. Adv., 2021, 7, eabd2091 CrossRef CAS PubMed.
- S. Takano, H. Hirai, T. Nakashima, T. Iwasa, T. Taketsugu and T. Tsukuda, J. Am. Chem. Soc., 2021, 143, 10560 CrossRef CAS PubMed.
- A. P. Veenstra, L. Monzel, A. Baksi, J. Czekner, S. Lebedkin, E. K. Schneider, T. Pradeep, A.-N. Unterreiner and M. M. Kappes, J. Phys. Chem. Lett., 2020, 11, 2675 CrossRef CAS PubMed.
- X. Kang, X. Li, H. Yu, Y. Lv, G. Sun, Y. Li, S. Wang and M. Zhu, RSC Adv., 2017, 7, 28606 RSC.
- X. Kang, S. Wang, Y. Song, S. Jin, G. Sun, H. Yu and M. Zhu, Angew. Chem., Int. Ed., 2016, 128, 3675 CrossRef.
- T. N. Singh-Rachford and F. N. Castellano, Coord. Chem. Rev., 2010, 254, 2560 CrossRef CAS.
- J. Zhao, W. Wu, J. Sun and S. Guo, Chem. Soc. Rev., 2013, 42, 5323 RSC.
- N. Yanai and N. Kimizuka, Acc. Chem. Res., 2017, 50, 2487 CrossRef CAS PubMed.
- L. Nienhaus, M. Wu, V. Bulovic, M. A. Baldo and M. G. Bawendi, Dalton Trans., 2018, 47, 8509 RSC.
- S. Garakyaraghi and F. N. Castellano, Inorg. Chem., 2018, 57, 2351 CrossRef CAS PubMed.
- Y. Jiang, P. Lv, J.-Q. Pan, Y. Li, H. Lin, X.-W. Zhang, J. Wang, Y.-Y. Liu, Q. Wei, G.-C. Xing, W.-Y. Lai and W. Huang, Adv. Funct. Mater., 2019, 29, 1806719 CrossRef.
- M. Wu, D. N. Congreve, M. W. B. Wilson, J. Jean, N. Geva, M. Welborn, T. Van Voorhis, V. Bulovic, M. G. Bawendi and M. A. Baldo, Nat. Photonics, 2016, 10, 31 CrossRef CAS.
- C. Mongin, S. Garakyaraghi, N. Razgoniaeva, M. Zamkov and F. N. Castellano, Science, 2016, 351, 369 CrossRef CAS PubMed.
- Z. Huang and M. L. Tang, J. Am. Chem. Soc., 2017, 139, 9412 CrossRef CAS PubMed.
- Z. Huang, X. Li, M. Mahboub, K. M. Hanson, V. M. Nichols, H. Le, M. L. Tang and C. J. Bardeen, Nano Lett., 2015, 15, 5552 CrossRef CAS PubMed.
- K. Okumura, K. Mase, N. Yanai and N. Kimizuka, Chem. – Eur. J., 2016, 22, 7721 CrossRef CAS PubMed.
- Z. Huang and M. L. Tang, J. Phys. Chem. Lett., 2018, 9, 6198 CrossRef CAS PubMed.
- X. Luo, R. Lai, Y. Li, Y. Han, G. Liang, X. Liu, T. Ding, J. Wang and K. Wu, J. Am. Chem. Soc., 2019, 141, 4186 CrossRef CAS PubMed.
- S. He, X. Luo, X. Liu, Y. Li and K. Wu, J. Phys. Chem. Lett., 2019, 10, 5036 CrossRef CAS PubMed.
- Y. Han, X. Luo, R. Lai, Y. Li, G. Liang and K. Wu, J. Phys. Chem. Lett., 2019, 10, 1457 CrossRef CAS PubMed.
- Y. Han, S. He, X. Luo, Y. Li, Z. Chen, W. Kang, X. Wang and K. Wu, J. Am. Chem. Soc., 2019, 141, 13033 CrossRef CAS PubMed.
- P. Xia, E. K. Raulerson, D. Coleman, C. S. Gerke, L. Mangolini, M. L. Tang and S. T. Roberts, Nat. Chem., 2020, 12, 137 CrossRef CAS PubMed.
- K. Mase, K. Okumura, N. Yanai and N. Kimizuka, Chem. Commun., 2017, 53, 8261 RSC.
- Z. Xu, T. Jin, Y. Huang, K. Mulla, F. A. Evangelista, E. Egap and T. Lian, Chem. Sci., 2019, 10, 6120 RSC.
- Y. Han, S. He and K. Wu, ACS Energy Lett., 2021, 6, 3151 CrossRef CAS.
- M. Mitsui, Y. Kawano, R. Takahashi and H. Fukui, RSC Adv., 2012, 2, 9921 RSC.
- M. Mitsui, Y. Takakura, K. Hirata, Y. Niihori, Y. Fujiwara and K. Kobayashi, J. Phys. Chem. B, 2021, 125, 9950 CrossRef CAS PubMed.
-
M. Lira-Cantu and G. Korotcenkov, The Future of Semiconductor Oxides in Next-Generation Solar Cells, Elsevier, 1st edn, 2017 Search PubMed.
- E. S. Pysh and N. C. Yang, J. Am. Chem. Soc., 1963, 85, 2124 CrossRef CAS.
-
L. Meites and P. Zuman, CRC Handbook Series in Organic Electrochemistry, CRC Press, Boca Raton, FL (USA), 1977, vol. I–V Search PubMed.
- T. Nakagawa, K. Okamoto, H. Hanada and R. Katoh, Opt. Lett., 2016, 41, 1498 CrossRef PubMed.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson and H. Nakatsuji, et al., Gaussian 16, Revision B.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. People, J. Chem. Phys., 1982, 77, 3654 CrossRef CAS.
- D. Andrae, U. Häußermann, M. Dolg, H. Stoll and H. Preuß, Theor. Chim. Acta, 1990, 77, 123 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372 CrossRef CAS.
- C. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS PubMed.
- E. Runge and E. K. U. Gross, Phys. Rev. Lett., 1984, 52, 997 CrossRef CAS.
- Y. Niihori, N. Takahashi and M. Mitsui, J. Phys. Chem. C, 2020, 124, 5880 CrossRef CAS.
- R. Bensasson and E. J. Land, Trans. Faraday Soc., 1971, 67, 1904 RSC.
- S.-A. Yamamoto, K. Kikuchi and H. Kokubun, J. Photochem., 1977, 7, 177 CrossRef CAS.
- H.-C. Chen, C.-Y. Hung, K.-H. Wang, H.-L. Chen, W. S. Fann, F.-C. Chien, P. Chen, T. J. Chow, C.-P. Hsu and S.-S. Sun, Chem. Commun., 2009, 4064 RSC.
- K. Tanaka, K. Inafuku and Y. Chujo, Chem. Commun., 2010, 46, 4378 RSC.
- E. M. Gholizadeh, L. Frazer, R. W. MacQueen, J. K. Gallahera and T. W. Schmidt, Phys. Chem. Chem. Phys., 2018, 20, 19500 RSC.
- T. W. Schmidt and F. N. Castellano, J. Phys. Chem. Lett., 2014, 5, 4062 CrossRef CAS PubMed.
- P. Bharmoria, H. Bildirir and K. Moth-Poulsen, Chem. Soc. Rev., 2020, 49, 6529 RSC.
- Y. Zhou, F. N. Castellano, T. W. Schmidt and K. Hanson, ACS Energy Lett., 2020, 5, 2322 CrossRef CAS.
- V. Gray, A. Dreos, P. Erhart, B. Albinsson, K. Moth-Poulsen and M. Abrahamsson, Phys. Chem. Chem. Phys., 2017, 19, 10931 RSC.
- N. Nishimura, J. R. Allardice, J. Xiao, Q. Gu, V. Gray and A. Rao, Chem. Sci., 2019, 10, 4750 RSC.
- W. Staroske, M. Pfeiffer, K. Leo and M. Hoffmann, Phys. Rev. Lett., 2007, 98, 197402 CrossRef CAS PubMed.
- W. Gawelda, A. Cannizzo, V.-T. Pham, F. van Mourik, C. Bressler and M. Chergui, J. Am. Chem. Soc., 2007, 129, 8199 CrossRef CAS PubMed.
- A. Cannizzo, F. van Mourik, W. Gawelda, G. Zgrablic, C. Bressler and M. Chergui, Angew. Chem., Int. Ed., 2006, 45, 3174 CrossRef CAS PubMed.
- A. Cannizzo, A. M. Blanco-Rodríguez, A. Nahhas, J. Šebera, S. Záliš, A. Vlček, Jr. and M. Chergui, J. Am. Chem. Soc., 2008, 130, 8967 CrossRef CAS PubMed.
-
N. J. Turro, V. Ramamurthy and J. Scaiano, Modern Molecular Photochemistry of Organic Molecules, University Science Books, Sausalito, CA, 2012 Search PubMed.
- S. J. Milder and B. S. Brunschwig, J. Phys. Chem., 1992, 96, 2189 CrossRef CAS.
- A. C. Durrell, G. E. Keller, Y.-C. Lam, J. Sýkora, A. Vlček, Jr. and H. B. Gray, J. Am. Chem. Soc., 2012, 134, 14201 CrossRef CAS PubMed.
- T. J. Penfold, E. Gindensperger, C. Daniel and C. M. Marian, Chem. Rev., 2018, 118, 6975 CrossRef CAS PubMed.
- M. J. Leitl, V. A. Krylova, P. I. Djurovich, M. E. Thompson and H. Yersin, J. Am. Chem. Soc., 2014, 136, 16032 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tc04591a |
|
| This journal is © The Royal Society of Chemistry 2022 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 and
Masaaki
Mitsui
and
Masaaki
Mitsui
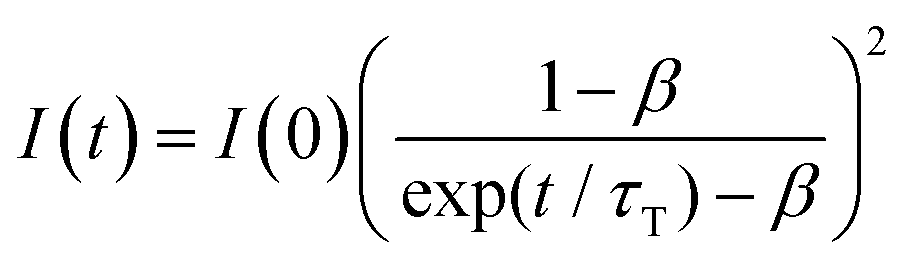
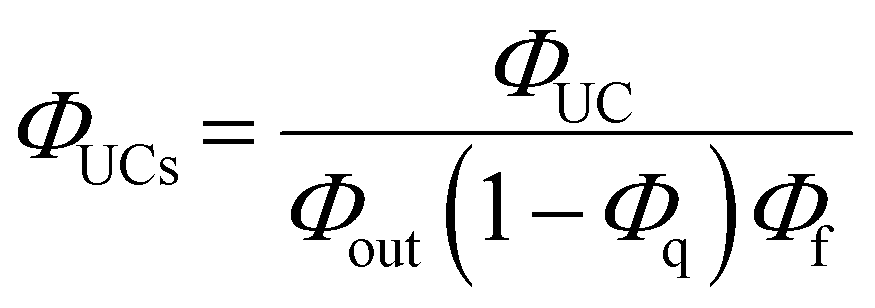
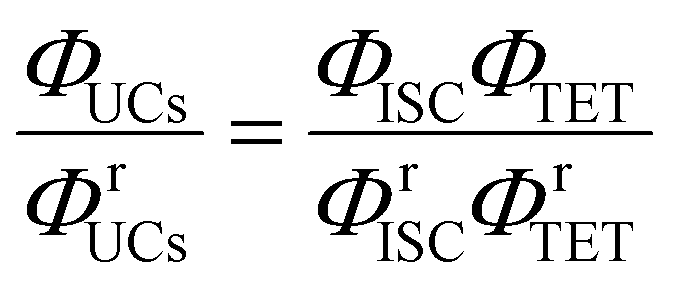
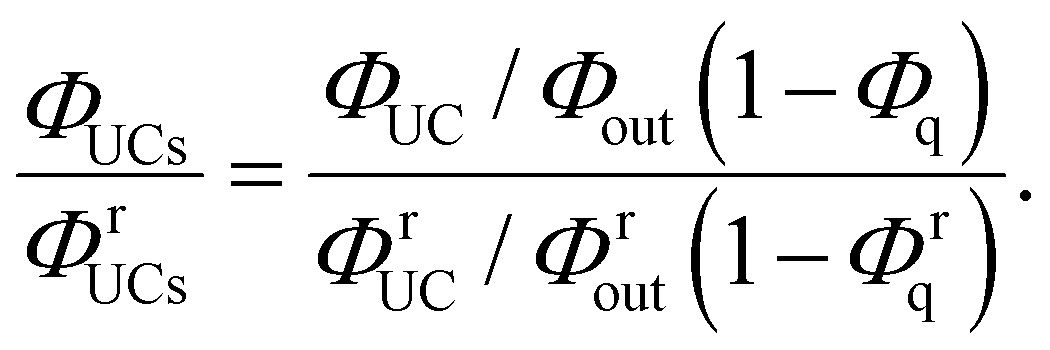
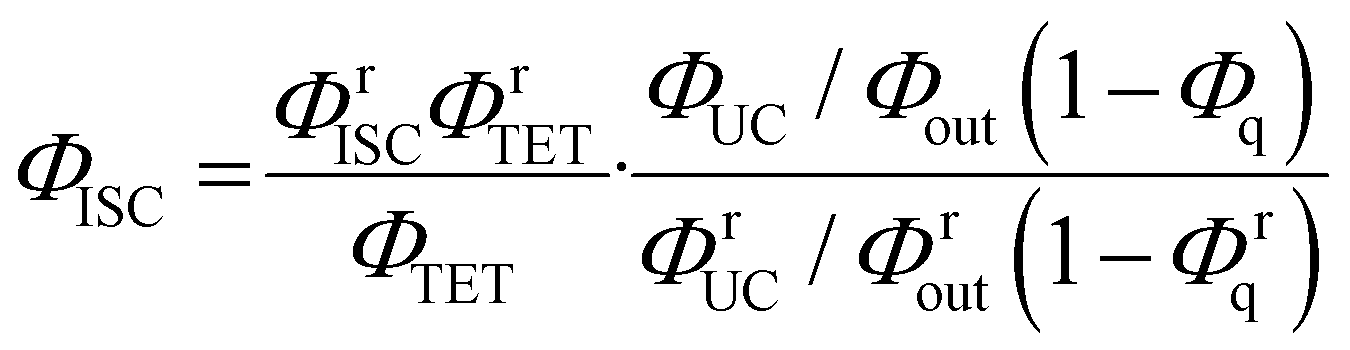
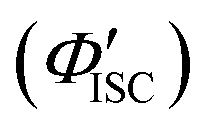 was 0.56. All the quantum yields and rate constants determined for the radiative and non-radiative processes are summarized in Table 3. The results show that the Au2Cu6 cluster has relatively high quantum yields of ISC and phosphorescence at room temperature and, in contrast, very small fluorescence quantum yields. Notably, Au2Cu6 possesses a very long fluorescence radiative lifetime (1.75 μs) and a small ISC rate of 5.3 × 107 s−1, which are strikingly different from those of many transition-metal complexes that exhibit ultrafast ISC.64–66
was 0.56. All the quantum yields and rate constants determined for the radiative and non-radiative processes are summarized in Table 3. The results show that the Au2Cu6 cluster has relatively high quantum yields of ISC and phosphorescence at room temperature and, in contrast, very small fluorescence quantum yields. Notably, Au2Cu6 possesses a very long fluorescence radiative lifetime (1.75 μs) and a small ISC rate of 5.3 × 107 s−1, which are strikingly different from those of many transition-metal complexes that exhibit ultrafast ISC.64–66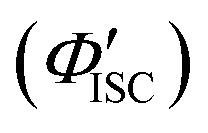 was calculated by 1 − Φp.
d Radiative rate constant from S1 to S0 (i.e., fluorescence transition) was calculated by Φfτf−1. The value in parentheses represents the radiative lifetime of S1.
e Calculated by ΦISCτf−1.
f Calculated by ΦICτf−1.
g Radiative rate constant from T1 to S0 (i.e., phosphorescence transition) was calculated by Φpτp−1.
h Calculated by
was calculated by 1 − Φp.
d Radiative rate constant from S1 to S0 (i.e., fluorescence transition) was calculated by Φfτf−1. The value in parentheses represents the radiative lifetime of S1.
e Calculated by ΦISCτf−1.
f Calculated by ΦICτf−1.
g Radiative rate constant from T1 to S0 (i.e., phosphorescence transition) was calculated by Φpτp−1.
h Calculated by 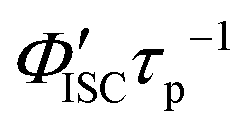 .
.